

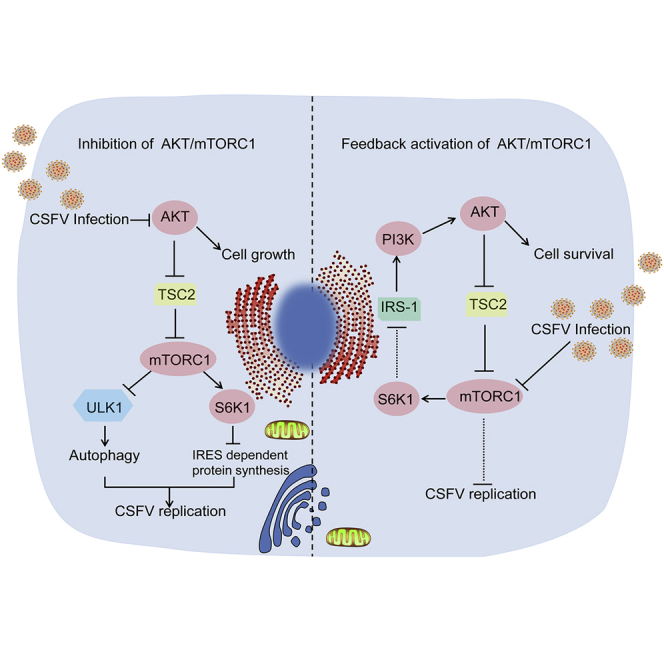
Summary
Classical swine fever virus (CSFV) can utilize diverse host signaling pathways for its replication; however, the cross talk between mammalian target of rapamycin (mTOR) and CSFV remains unknown. Here, we describe the potential role of mTOR complex 1 (mTORC1) in promoting CSFV replication via virus-induced hypophosphorylation of the Akt/mTORC1/S6 pathway, especially at an early stage of viral infection. Conversely, activation of mTORC1 inhibited the replication of CSFV. Furthermore, we revealed the underlying mechanisms of mTORC1 pathway in mediating CSFV replication; in addition, our data also showed that CSFV-induced transient inhibition of mTORC1 elicited a negative feedback activation of PI3K/Akt/mTORC1pathway, likely contributing to maintain the dynamic balance between viral replication and host cell survival. This study has provided strong evidence showing how CSFV utilizes mTORC1 pathway for viral replication at an early stage in the viral replicative cycle and how the mTORC1 rescues itself by eliciting a feedback loop to limit viral replication and maintain cell survival.
Subject Areas: Pathophysiology, Molecular Biology, Virology
Graphical Abstract
Highlights
-
•
Akt/mTORC1 pathway negatively regulates the replication of CSFV
-
•
CSFV induces autophagy for viral replication in an mTORC1/ULK1-dependent manner
-
•
CSFV enhances the translation of viral proteins in an mTORC1/S6K1/eIF3-dependent manner
-
•
Feedback activation of Akt/mTORC1 equilibrates viral replication and cell survival
Pathophysiology; Molecular Biology; Virology
Introduction
Classical swine fever virus (CSFV) is a key member of the genus Pestivirus of the family Flaviviridae. It has a single positive-stranded RNA genome of approximately 12.3 kb, containing a single large open reading frame encoding 3,898 amino acids, which are processed by viral and host proteases to produce 12 mature proteins, four structural proteins (C, Erns, E1, and E2) and eight non-structural proteins (NSPs) (Npro, p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B) (BD et al., 2007). CSFV or its proteins can hijack host signaling pathways and biosynthetic systems for viral replication via interaction between viral and host proteins. CSFV-E2 protein has been reported to promote viral replication by interacting with annexin 2 or by activating the MEK2/ERK1/2 signal transduction cascade (Wang et al., 2016, Yang et al., 2015), and E2 can also negatively regulate viral replication by interacting with thioredoxin 2 (Trx2) and activating nuclear factor (NF)-κB pathway (Li et al., 2015b). CSFV-NS5A promotes viral replication and spreading by interaction with heat shock protein 70 (HSP70) or by localization within the endoplasmic reticulum (ER) where it induces oxidative stress in vascular endothelial cells (Zhang et al., 2015, He et al., 2012). CSFV-NS5A can also inhibit internal ribosome entry site (IRES)-mediated translation by interaction with eukaryotic elongation factor 1A (eEF1A) (Li et al., 2015a), whereas CSFV-NS3 interacts with TRAF6 and degrades it to promote CSFV replication via the NF-κB signaling pathway (Lv et al., 2017), and CSFV-Npro interacts with interferon regulatory factor 3 (IRF-3) or IRF-7 and blocks type I interferon induction (Bauhofer et al., 2007, Fiebach et al., 2011).
Mammalian target of rapamycin (mTOR), of the phosphatidylinositol kinase-related kinase family, usually acts as a crucial nutritional and cellular energy checkpoint sensor and mediator for cell growth, proliferation, and survival. Indeed, the mTOR complex 1 (mTORC1) kinase is the central node of a serial signaling pathway (Saxton and Sabatini, 2017). Increasing studies have revealed that mTOR not only works as a master sensor of cellular homeostatic perturbations but also significantly contributes to viral infections (McNulty et al., 2013, Mannova and Beretta, 2005). For example, hepatitis C virus (HCV) infection upregulates the activity of mTOR and blocking of mTOR potently inhibits HCV RNA replication (Stohr et al., 2016). A similar mechanism is also operative during infection with human cytomegalovirus and human immunodeficiency virus (HIV) (Martin et al., 2012, Cinti et al., 2017). In contrast, however, hepatitis B virus (HBV) or hepatitis E virus (HEV) downregulates the activity of mTOR pathway, and blocking mTOR facilitates their replication (Guo et al., 2007, Zhou et al., 2014). In addition, inhibition of mTORC1 enhances the protein translation of chikungunya virus (CHIKV) by activating the Mnk/eukaryotic initiation factor-4E (eIF4E) pathway (Joubert et al., 2015). Intriguingly, after infection of host cells with porcine reproductive and respiratory syndrome virus (PRRSV), mTOR has been shown to be upregulated initially but later downregulated, resulting in viral replication in a 4E-binding protein 1 (4E-BP1)-associated manner (Zhang and Wang, 2010). These ongoing studies have shown that the Akt/mTOR pathway is involved in the replication of an increasing number of viruses through regulation of cell metabolic process, autophagy, and protein synthesis, but its role in the regulation of CSFV replication is unknown.
Autophagy is an intracellular degradation process that maintains the metabolic balance and homeostasis of cells by clearance and recycling of intercellular constituents (Jiang et al., 2015). Previous studies have suggested that autophagy not only exerts a protective function in cellular survival under stress but also plays a role to facilitate or inhibit viral replication in various ways (Shintani and Klionsky, 2004, Levine and Kroemer, 2008, Deretic, 2010). Viruses such as Zika (ZIKV), HCV, bovine viral diarrhea (BVDV), and dengue (DENV) have been found to hijack cellular autophagy to facilitate their replication (Liang et al., 2016, Dreux et al., 2009, Fu et al., 2014, Datan et al., 2016), whereas herpes simplex virus type 1 (HSV-1) inhibits autophagy to facilitate its replication (Orvedahl et al., 2007). Chen et al. (Pei et al., 2014) have reported that CSFV developed autophagic machinery by interaction of its E2 protein with autophagosomes to favor viral growth and survival of host cells and that inhibition of autophagy significantly reduced viral proliferation. Cellular autophagy is regulated by multiple cellular signaling pathways, including Atg6/Beclin-1, which forms a complex with Vps34, thereby recruiting other proteins to the complex and eventually promoting autophagy. However, the anti-apoptotic protein Bcl-2 inhibits autophagy by binding Beclin-1. Two ubiquitin-like conjugation processes also contribute to the autophagy by generating membrane-bound protein complexes. ULK1/Atg1, the key serine/threonine kinase in the core autophagy pathway, is activated in response to nutrient deprivation and serves as a critical initiator of starvation-induced autophagy. Normally, mTORC1 is an upstream kinase that can inactivate ULK1 by phosphorylating a single site, Ser757 (Egan et al., 2011). Recent studies have shown that several viruses can induce autophagy in an mTOR-dependent manner (Shrivastava et al., 2012, Yin et al., 2017, Zhou et al., 2017), although some cell types undergo autophagy without involvement of the ULK1 complex (Cheong et al., 2011), presumably, at least in part, by direct signaling to the downstream Vps34/Beclin1 complex.
The mTOR pathway also plays a key role in regulating the translation of cellular and viral proteins through its downstream effectors such as p70S6 kinase1 (S6K1) and 4E-BPs. Normally in cap-dependent translation systems, mTOR directly activates S6K1 and 4E-BPs through phosphorylation, following which the activated 4E-BPs release the binding to eIF4E and promote the interaction of eIF4E with the cap structure of messenger RNA (mRNA). Alternatively, the activated mTOR binds to the eIF3 complex, resulting in the dissociation of S6K1 from eIF3 and activation of S6K1, which leads to the binding of eIF3 to the 40S subunit and stimulation of cap-dependent translation (Schepetilnikov et al., 2013, Hashem et al., 2013). However, some RNA viruses without cap-structured 5′ untranslated regions (UTR), including CSFV, have developed other mechanisms for their replication bypassing the cap-dependent translation (Leen et al., 2016, Matsuda and Mauro, 2014, Amorim et al., 2014, Hashem et al., 2013). For example, an IRES located between nt 40 and 350 at the 5′ UTR has been shown to mediate translation of the viral genome. This has a high degree of RNA secondary structure and recruits ribosomal 40S subunits in close proximity to the initiation codon in a cap-independent manner (Fletcher and Jackson, 2002, Friis et al., 2012, Fletcher et al., 2002), IRES-mediated translation has a great advantage for viruses containing an IRES because viral proteins can be generated efficiently even in cells that undergo apoptosis or nutritional starvation (Hashem et al., 2013, Kolupaeva et al., 2000). Recent findings have suggested that activated mTOR exhibits anti-viral activity in HCV, which processes a non-cap construction at the 5′ end of the viral mRNA by inducing an association between eIF3 and eIF4G (Muraoka et al., 2012, Schepetilnikov et al., 2013). Moreover, HCV and CSFV mRNAs contain related IRESs that promote 5′-end-independent initiation of translation, requiring only a subset of the eIFs needed for canonical initiation on cellular mRNAs (Schepetilnikov et al., 2013). A cryoelectron microscopic reconstruction of a 40S ribosome complex containing eIF3 and the CSFV-IRES has been presented (Hashem et al., 2013), in which eIF3 is completely displaced from its ribosomal position in the 43S complex and instead interacts through its ribosome binding surface exclusively with the apical region of domain III of the IRES. Holz et al. (Holz et al., 2005) then demonstrated that eIF3 family members, as S6K1 interactors, represented a potential important connection between the mTOR pathway and an integral component of the translational pre-initiation apparatus, with the activity of S6K1 negatively regulating its binding to eIF3. Mutants that eliminate S6K1 activity constitutively bind eIF3, whereas active S6K1 mutants are incapable of binding eIF3. However, whether the CSFV can benefit their protein synthesis through modifying the mTOR pathway remained unclear.
In this study, we have demonstrated cross talk between the mTORC1 signaling pathway and CSFV replication and have also disclosed the underlying mechanism by which mTORC1 contributes to viral replication. We also found that inhibition of the mTORC1 pathway induced by CSFV elicited a negative feedback activation of the PI3K/Akt/mTORC1 pathway, which contributed to maintaining the dynamic balance between viral replication and host cell survival. Collectively, our results provide strong evidence showing that the mTORC1 pathway plays a crucial role in regulating CSFV replication and cell survival.
Results
CSFV Infection Inhibited the Activation of PI3K/AKT/mTORC1 Pathway
To investigate the impact of CSFV infection on the mTORC1 pathway, ST cells were infected with CSFV and subjected to PathScan intracellular signaling membrane array analysis. See Figure S1 for a detailed map of the proteins tested via the PathScan assay. Our results showed that the phosphorylation states of Akt (S473), mTORC1 (Ser 2448), and their downstream effectors GSK-3β (Ser9) and ribosomal protein S6 were significantly decreased in CSFV-infected cells within 24 hr post-infection (p.i.), but intriguingly, the phosphorylation level had recovered by 48 hr p.i. when compared with mock-infected controls (Figure 1A). To further investigate the impact of CSFV infection on mTORC1 activity, we examined the activity of Akt/mTORC1 pathway proteins at different time points after CSFV infection by western blot. Results were consistent with the array analysis. CSFV infection suppressed the activity of Akt/mTORC1pathway, with protein activities bottoming around 12 hr p.i., followed by recovery to the basal level at 48 hr p.i. (Figure 1B), whereas the levels of these proteins remained unchanged in mock-infected controls.
Figure 1.

CSFV Infection Inhibits the Activation of Akt/mTORC1 Pathway
(A and B) ST cells, either uninfected or infected with CSFV (MOI = 1), at the indicated time points. (A) PathScan Intracellular Signaling Membrane Array performed to analyze the phosphorylation of pathway proteins. Akt, mTORC1, S6, and GSK-3β dot blots are identified; (B) Western blots performed using antibodies against Akt, mTORC1, and S6, with β-actin as a loading control. GSK-3β, glycogen synthase kinase-3β.
(C) ST cells transfected with Erns (left panel) or NS3, NS4B, or NS5A (right panel) for 48 hr, and Western blots performed using antibodies against the above-mentioned specific viral and mTOR pathway proteins. Similar results were observed in three independent experiments.
The CSFV genome encodes four structural and eight NSPs. To delineate the role of individual viral proteins in Akt/mTORC1 regulation, we transfected ST cells with several recombinant constructs encoding individual CSFV proteins, Erns, NS3, NS4B, and NS5A, all of which are involved in viral infection, replication, and virulence. The expression of each protein was detected at 48 hr p.i. after transfection. As shown in Figure 1C, expression of Erns, NS3, and NS5A increased the activity of Akt/mTORC1pathway, whereas transduced NS4B did not exhibit significant influence. These results indicate that CSFV proteins Erns, NS3, and NS5A play key roles in suppressing the activity of Akt/mTORC1 pathway, which implied a likely role for Akt/mTORC1 in CSFV replication.
Activation of Akt/TSC2/mTORC1 Pathway Inhibits CSFV Replication and Viral Production
To understand the role of mTORC1 in CSFV replication, we inhibited mTORC1 activity by rapamycin, which works as an allosteric inhibitor of mTORC1, or activated mTORC1 with insulin, which works as a major cytokine for protein synthesis and cell growth by activating the PI3K/Akt/mTORC1 signaling pathway (Muraoka et al., 2012). As shown in Figure 2, rapamycin potently inhibited the activity of mTORC1 pathway whether or not CSFV was present. Conversely, insulin not only moderately promoted the activity of mTORC1 in the absence of CSFV but also rescued its activity level in the presence of CSFV. As expected, ultraviolet (UV)-inactivated CSFV lost its capability to inhibit the activation of the mTORC1 pathway (Figure 2A), indicating that CSFV infection is necessary for the induction of Akt/mTORC1 inhibition. Importantly, we further confirmed that rapamycin significantly increased, and insulin decreased, the expression of CSFV proteins (Core and Npro) at 24 hr p.i. (Figure 2B). In parallel, the intracellular CSFV-RNA replication and extracellular virus production were examined, with results consistent with the western blot data (Figures 2C and 2D). To exclude the influence of cell numbers on virus copies and load, we determined cell growth inhibition after exposing to diverse treatments as mentioned above. The results showed that increased virus levels were found in decreased numbers of cells in the rapamycin treatment group, whereas decreased virus levels were found in elevated cell numbers in the insulin treatment group (Figure 2E). This positively excludes the influence of cell numbers on virus production and indicates that activation of the Akt/TSC2/mTORC1 pathway effectively inhibits CSFV replication and virus production.
Figure 2.

Inhibition of Akt/mTORC1 Pathway Enhances CSFV Replication and Viral Titers
(A–E) ST cells pretreated with rapamycin (Rapa., 100 nM) or insulin (Ins.,1 μM) for 2 h were infected with CSFV (MOI = 1) for indicated times. (A) Western blots performed 12 hr p.i. using antibodies against mTORC1 and S6 (T/P). (B) Western blots performed 24 or 48 hr p.i. using antibodies to the core and Npro proteins of CSFV. (C) qRT-PCR performed to determine the intra- and extracellular viral copies at 24 or 48 hr p.i. Results are expressed as 106 copies/mL. (D) Intra- and extracellular viral titers determined at 24 or 48 hr p.i. (E) ST cells transduced for 24 hr with TSC2-shRNA and negative control and infected with CSFV (MOI = 1). Western blots performed 24 hr p.i. using antibody to TSC2 (upper). See also Figure S2. qRT-PCR performed to detect intra- and extracellular viral copies of CSFV at 24 hr p.i. (lower).
(F) Sulforhodamine B (SRB) assay performed to detect cell viability. Results are expressed as % control cell growth.
(G and H) ST cells pretreated for 2 hr with LY294002 (1 μM) (G) or SC79 (1 μM) (H) and infected with CSFV at the indicated times. qRT-PCR performed to detect total viral copies at 24, 48, or 72 hr p.i. The data are representative of three independent experiments. Bars indicate means ± SEM *p < 0.05, **p < 0.01, and ***p < 0.001 versus control. qRT-PCR, quantitative reverse-transcriptase polymerase chain reaction; SEM, standard error of the mean.
We further evaluated the impact of mTORC1 on CSFV replication by enhancing the activity of mTORC1. This was achieved by inhibiting the expression of the gene tuberous sclerosis 2 (TSC2) by TSC2-short hairpin RNA (shRNA), a physiologic inhibitor of mTORC1 (Sengupta et al., 2010). As shown in Figure 2E, the phosphorylation of S6, as the key downstream indicator of mTORC1 signaling activity, was significantly inhibited upon the potent inhibition of TSC2 (see Figure S2 for quantified protein bands of TSC2 and phospho-S6). Also as expected, the CSFV gene copies were significantly decreased at 24 hr p.i. when compared with control at both intra- and extracellular levels.
Intriguingly, as shown in Figures 2B–2D, rapamycin and insulin both had significant effects in regulating the expression of CSFV proteins, CSFV copies, and viral titers within the first 24 hr p.i, but these had disappeared by 48 hr p.i. According to the results in Figure 2F, the cells in the rapamycin-treated group were significantly fewer than those following insulin treatment at 48 hr, which might be ascribed, at least in part, to the inconsistent cell numbers following the different treatments. Alternatively, we speculated whether CSFV-induced mTORC1 inhibition might elicit the feedback activation of Akt, which would facilitate mTORC1 activation and result in loss of the ability to promote viral replication.
To determine if mTORC1-inhibition-induced Akt feedback activation is indeed involved in CSFV-infected cells, an Akt-specific inhibitor (LY294002) was used. As shown in Figure 2G, LY294002 potently enhanced CSFV RNA copy numbers. More importantly, the efficacy of LY294002 was much stronger than that of rapamycin (Figure 2C) with the effect persistent over the entire 72 hr p.i. Conversely, Akt activator SC79 significantly suppressed the CSFV RNA copies at all time intervals (Figure 2H). These results positively support our hypothesis that CSFV-induced mTORC1 inhibition elicits the feedback activation of Akt, which contributes to viral replication. Altogether, these results provide evidence for the involvement of Akt/TSC2/mTORC1 as an anti-viral mechanism within the context of CSFV infection.
CSFV Infection Induces Cellular Autophagy in an mTOR/ULK1-Dependent Manner
Autophagy has been reported to enhance the replication of CSFV; however, the underlying mechanisms remained unclear. The above-mentioned results indicated that CSFV infection dysregulated the mTORC1pathway, noting that mTOR could act as a regulator of autophagy in several cell lines. We then clarified whether CSFV infection induces autophagy in a manner dependent on mTOR. The specific autophagy markers and autophagy flux were detected in CSFV-infected cells by western blot and the stubRFP-SensGFP-LC3 lentivirus. As expected, the expression of LC3II increased from 4 hr to 12 hr p.i., and then decreased to the base level. Correspondingly, the level of SQSTM1/p62 (the substrate of autophagy) decreased between 4 hr and 12 hr p.i. in CSFV-infected cells, and then began to recover (Figure 3A). In parallel, we observed that CSFV induced the accumulation of cytoplasmic autophagosomes (RFP+ GFP+ [red fluorescent protein, green fluorescent protein, respectively]) at 12 hr p.i., following which the autophagosomes began to fuse with the lysosomes. An abundance of autolysosomes (RFP+ GFP−) had formed by 24 hr p.i., as GFP fluorescence declined upon lysosomal acidification (with reduced pH values), whereas RFP fluorescence remained stable (Figure 3B). These data show that CSFV indeed induces an integrated autophagic process.
Figure 3.

Inhibition of mTORC1 Favors Viral Replication by mTOR/ULK1 Dependent-Autophagy
(A–C) (A and C) Uninfected (Mock) ST cells or infected with CSFV (MOI = 1) for the indicated times. (A) Western blots performed from 2 to 48 hr p.i. using antibodies to SQSTM1/p62, LC3-II, and CSFV core protein. (B) ST cells stably transduced with StubRFP-SensGFP-LC3 lentivirus infected with CSFV for 12 or 24 hr. Confocal microscopy was performed to detect fluorescence (magnification, 200 or 400×; scale bar, 25 μm). (C) Western blots performed from 0 to 36 hr p.i. using antibodies to mTORC1, ULK1, SQSTM1/p62, LC3-II, and CSFV core protein.
(D) ST cells pretreated for 2 hr with rapamycin (Rapa, 100 nM) or insulin (Ins, 1 μM) and infected for 12 hr with CSFV. Western blots performed using antibodies to mTORC1, S6, ULK1, SQSTM1/p62, and LC3-II.
(E) ST cells stably transduced with StubRFP-SensGFP-LC3 lentivirus pretreated for 2 hr with rapamycin (100 nM) or insulin (1 μM), or SBI-0206965 (500 nM), and then infected with CSFV for 12 hr. Confocal microscopy was performed to detect fluorescence (magnification, 200 or 400×; scale bar, 25 μm).
(F–H) ST cells pretreated for 2 h with SBI. (500 nM) and infected with CSFV for 24 or 48 hr. (F) Western blots performed using antibodies to CSFV core protein, with β-actin as the loading control. (G) qRT-PCR performed to detect intra- and extracellular viral copies of CSFV at 24 or 48 hr p.i. (H) Viral titers determined at 24 or 48 hr p.i.
(I) ST cells treated for 48 hr with SBI or CSFV, alone or in combination. Sulforhodamine B (SRB) assay performed to detect the cell viability. Results are expressed as % control cell growth.
(J) ST cells transduced with Atg5-shRNA infected with CSFV and treated for 24 hr with rapamycin (100 nM). Western blots performed 48 hr post transfection using antibody to Atg5 (left). qRT-PCR performed to determine total viral copies of CSFV at 24 h p.i.(right). The data are representative of three independent experiments. Bars indicate means ± SEM *p < 0.05, **p < 0.01, and ***p < 0.001. vs. CSFV alone. qRT-PCR, quantitative reverse-transcriptase polymerase chain reaction; SEM, standard error of the mean.
We then examined whether the mTORC1 pathway contributes to CSFV-infection-induced autophagy. With regard to this, ULK1 is the key downstream kinase that connects the mTOR pathway and autophagy, with dephosphorylation being the activated status of ULK1 and promoting the autophagy process by recruiting Atg12 and FIP200 to form a complex. As indicated in Figure 3C, the alteration of phosphorylated mTORC1 and ULK1 negatively correlated with the process of autophagy, i.e., CSFV infection dephosphorylated mTORC1 and ULK1, thereby promoting autophagy. To further clarify whether mTORC1/ULK1 pathway regulates CSFV-induced autophagy, the process was examined in CSFV-infected ST cells with mTORC1 inhibition or activation. As shown in Figure 3D, inhibition of mTORC1 by rapamycin synergistically enhanced CSFV-induced autophagy, whereas activation of mTORC1 with insulin reversed CSFV-induced autophagy. A similar outcome was observed by transducing stubRFP-SensGFP-LC3 lentivirus into ST cells in the presence of rapamycin, insulin, or ULK1 inhibitor SBI-0206965 (which works as a specific inhibitor of ULK1 to block the formation of the Atg13/FIP200/ULK1 autophagy complex) (Egan et al., 2015, Petherick et al., 2015). Autophagy was potently increased when mTORC1 was inhibited by rapamycin, but diminished when it was activated by insulin or when the ULK1-induced autophagy complex was blocked by SBI-0206965 (Figure 3E).
Further experiments were performed to confirm that CSFV-induced autophagy promoted viral replication in a manner dependent on mTORC1/ULK1. As shown in Figures 3F–3H, after blockage of ULK-1 by SBI-0206965, not only the production of CSFV core protein but also the viral gene copies and virus titers were inhibited at 24–48 hr p.i. both intra- and extracellularly, whereas cell growth was not influenced by SBI-0206965 (500 nM) treatment with or without CSFV infection (Figure 3I), thereby excluding the possibility that cell numbers affected viral copies. Altogether, these results show that CSFV induced autophagy in an mTORC1/ULK1-dependent manner, which facilitated viral replication.
To study the importance of mTORC1-dependent autophagy in CSFV replication, we assessed viral replication in ST cells deficient in key autophagy gene Atg5 (autophagy-related gene 5). Interestingly, the autophagy-deficient cells still exhibited increased viral replication following rapamycin treatment, although the increase was reduced when compared with normal control ST cells (Figure 3J). The conclusion is that although mTORC1-dependent autophagy indeed plays a key role in viral replication, there still exist some other mechanism(s) apart from autography contributing to mTORC1-inhibition-induced viral replication in CSFV-infected cells.
CSFV Infection Improves the Translation of Viral Proteins in an mTORC1/S6K1/eIF3-Dependent Manner
Previous studies have shown that HCV-IRES and eIF3 can competitively gain access to the 40S ribosomal subunit, thereby regulating the translation of viral mRNA (Hashem et al., 2013), and that S6K1 can interact with the eIF3 complex (Schepetilnikov et al., 2013). We therefore hypothesized that CSFV-induced mTORC1 inhibition could promote association between S6K1 and the eIF3 complex, resulting in the release of eIF3 from the 40S subunit and promoting the specific interaction between CSFV-IRES and the 40S subunit, thereby facilitating translation of viral proteins and enhancing CSFV replication.
To examine how S6K1 regulates the translation of viral proteins by manipulating the activity of CSFV-IRES, S6K1 knockdown (S6K1-KD), and S6K1 over-activation (S6K1-Over; mutant Thr229/Thr389/Ser371) ST cell lines were established. As shown in Figure 4A, the phosphorylation of S6K1 was significantly decreased in S6K1-KD ST cells and increased in S6K1-Over cells. Activity of CSFV-IRES was then monitored by constructing a reporter CSFV-5′ UTR (IRES included)-encoding luciferase under the control of the genomic promoter (IRES-Luc; Figure 4B, upper). Strikingly, the luciferase activity of CSFV-IRES was increased by 33.9% in S6K1-KD cells, but decreased by 30.2% in S6K1-Over ST cells when compared with the negative control (Figure 4B, lower). Determination of viral gene copies and viral titers at 24 hr p.i. provided results consistent with the activity of CSFV-IRES: overexpression of activated S6K1 resulted in a decrease, whereas knockdown of S6K1 increased the viral gene copies and titers both intra- and extracellularly (Figures 4C and 4D).
Figure 4.

Inhibition of mTORC1 Promotes the Translation of Viral Proteins by mTOR/S6K1/eIF3-Dependent Biosynthesis
(A) ST cells stably transduced with S6K1-shRNA (S6K1-KD) and GV144-S6K1 (S6K1-Overactivation, mutant Thr229/Thr389/Ser371). Western blots performed using antibodies to S6K1.
(B) Dual-luciferase reporter assay performed to detect the activity of CSFV-IRES in S6K1-KD and S6K1-over cells transfected with pGL-IRES. A schematic of the CSFV-IRES-luciferase construct is depicted (upper).
(C and D) S6K1-KD and S6K1-over cells infected with CSFV. (C) qRT-PCR performed to determine the intra- and extracellular viral copies of CSFV at 24 hr p.i. (D) Viral titers determined at 24 hr p.i.
(E) ST cells infected with CSFV for 24 hr, coIP experiments were carried out using S6K1 (left) or eIF3A (right) antibodies. S6K1-associated eIF3A, S6K1, eIF3A-associated S6K1, or total eIF3A were analyzed by western blot. With normal rabbit IgG as a negative control.
(F–H) Cellular ribosomes isolated from ST cells infected with CSFV in the presence of Rapa. (100 nM) or Ins. (1 μM). (F) Western blots performed using antibodies to RPS-12 and S6 to detect the proteins in the sediment following centrifugation (ribosomes) and supernatant. (G) qRT-PCR performed to determine the copy number of CSFV-IRES in the sediment at 24 hr p.i. (H) Indirect immunofluorescence assay performed to determine the virus in the sediment and supernatant (magnification, 40×; scale bar, 1 μm).
(I) Schematic of the interaction between mTORC1, S6K1, eIF3, and CSFV-IRES in the presence or absence of CSFV infection. The above data are representative of three independent experiments. Bars indicate means ± SEM *p < 0.05, **p < 0.01, and ***p < 0.001. vs. control or CSFV alone. coIP, co-immunoprecipitation, qRT-PCR quantitative reverse-transcriptase polymerase chain reaction; SEM, standard error of the mean.
To clarify that the inactivated S6K1 promoted the activity of CSFV-IRES by releasing its binding with eIF3 complex, the interaction between S6K1 and eIF3 was determined in CSFV-infected cells by co-immunoprecipitation. As shown in Figure 4E, CSFV infection inhibited the phosphorylation of S6K1 and enhanced S6K1 interaction with eIF3A.
In addition, we examined whether CSFV-induced inhibition of mTORC1 releases the 40S ribosome-binding site of eIF3, thereby enhancing the specific interaction between CSFV-IRES and 40S ribosome. Ribosomes were isolated from ST cells treated with rapamycin or insulin and incubated with CSFV for 24 hr. The fraction-specific proteins of ribosomes and virus were then assessed to determine the purity of ribosomes. As shown in Figure 4F, ribosomal proteins S12 (RPS12) and S6 were detected only in the bottom pellet, whereas the viral core protein was detected exclusively in the supernatant. Importantly, inhibition of mTORC1 by rapamycin enhanced CSFV-IRES ribosomal binding and insulin released the binding between ribosome and CSFV-IRES (Figure 4G), which strongly supports the conclusion that rapamycin promoted and insulin inhibited the translation of CSFV-mRNA. To further ensure that the binding of ribosomes to IRES copies was not contaminated by residual virus particles, virus distribution in the supernatant and ribosome pellet was monitored by immunofluorescence assay. As shown in Figure 4H, abundant viruses were observed in the supernatant and virus distribution showed a pattern consistent with that found above, i.e., more viruses were observed following rapamycin treatment, and less following insulin treatment, when compared with the untreated control. However, in ribosome pellets isolated from cells receiving either treatment, only one or two virus plaques (green dots) were observed in each well, with no differences in plaque numbers among the three groups (Figure 4H). The possibility of artifactual cross virus contamination was therefore eliminated, and the data show that CSFV-induced inactivation of mTORC1/S6K1 promoted non-cap-dependent translation of viral mRNA by increasing S6K1/eIF3 binding, relieving competition between eIF3 and CSFV-IRES for a common binding site on the 40S subunit and thereby favoring translation of viral mRNA (Figure 4I).
CSFV-Mediated Inhibition of mTORC1 Equilibrates Viral Replication and Cell Survival by Eliciting the Feedback Activation of Akt Kinase
The above-mentioned studies have indicated that CSFV-induced mTORC1 inhibition failed to induce cell growth inhibition and apoptosis (Figure 2F) and, importantly, that a rebound in mTORC1 levels was followed by a decrease (Figure 1B). Considering the similar phenomenon in cancer research (Sun et al., 2005, Wang et al., 2008), we hypothesized that temporary inhibition of mTORC1 induced by CSFV might stimulate the feedback activation of Akt/mTORC1, contributing to viral replication and cell survival.
The two phosphorylation sites of Akt kinase were examined to define whether Akt could be re-activated in CSFV-infected cells. As shown in Figure 5A, phosphorylation of both Akt (Thr308) (which usually acts as an effector of mTORC1) and Akt (Ser473) (acts upon mTORC2) was potently inhibited following CSFV infection, although strikingly, the inhibition was transient, with a low at 8–12 hr p.i., rebounding to the basal level by 48 hr p.i. Importantly, the alteration of phosphor-Akt was consistent with that affecting downstream effectors, including S6 and mTORC1 (Figures 1A and 1B).
Figure 5.

Inhibition of mTORC1 Induces Feedback Activation of Akt, which Contributes to Cell Survival and Viral Replication
(A) ST cells infected with CSFV (MOI = 1) at indicated time points. Western blots performed using antibody to Akt (P/T).
(B) ST cells pre-treated for 2 hr with Akt LY294002 (0.5 μM, 1 μM, 2 μM) and infected for 48 hr with CSFV or UV-CSFV (MOI = 1). Sulforhodamine B (SRB) assay performed to determine cell viability (upper). Western blots performed 48 hr p.i. in parallel using antibodies to Akt (P/T) (lower).
(C–E) ST cells treated for 24 or 48 hr with CSFV or LY294002 (1 μM), alone or in combination. (C) Flow cytometry performed to determine cell cycle distribution using propidium iodide (PI) staining. Results are presented as % cells in G0/G1, S, or G2/M phases. (D) Flow cytometry performed to assess cell apoptosis using annexin-V/PI double staining. Cells in different stages of apoptosis/death are shown (upper right: late-stage apoptosis, lower right: early-stage apoptosis). Data represent the mean ± SD of three independent experiments. ∗∗p < 0.01, ∗∗∗p < 0.001. (E) Determination of cell apoptosis using the TUNEL Apoptosis Detection Kit. Fluorescence microscopy was used to observe apoptotic cells (green dots) (magnification, 40×; scale bar, 1 μm).
(F) ST cells infected with CSFV for various times in the presence or absence of LY294002 (1 μM). qRT-PCR performed to determine the total copy number of CSFV. Data represent the mean ± SD of three independent experiments. ∗p < 0.05, ∗∗p < 0.01. qRT-PCR: quantitative reverse-transcriptase polymerase chain reaction; SD, standard deviation.
To investigate the role of Akt/mTORC1 feedback activation in maintaining cell survival in CSFV-infected cells, ST cells were incubated with its specific inhibitor LY294002 or CSFV (active and UV-inactive) alone, or with LY294002 for 2 hr before viral infection with CSFV at 1 MOI (multiplicity of infection) for 48 h. As shown in Figure 5B, both active and UV-inactivated CSFV had little effect on cell growth (<5%), whereas LY294002 (1 μM) inhibited cell growth by 13.3% and the combination of CSFV and LY294002 caused a potent growth inhibition of up to 30.5%. In parallel, the level of Akt phosphorylation was determined at each treatment (Figure 5B, lower), showing that inhibition of PI3K/Akt feedback activation resulted in a synergetic cell growth inhibition when compared with CSFV or LY294002 alone.
Furthermore, cell cycle and cell apoptosis analyses were performed to confirm the role of Akt feedback activation in maintaining cell homeostasis. As shown in Figure 5C, CSFV induced a cell-cycle arrest in the G0/G1 phase within 24 hr (from 72.6% to 77.2%), while effectively releasing the cell cycle by 48 hr, during which time period the G0/G1 phase decreased from 84.6% to 70.2%, whereas the S phase increased from 11.0% to 23.0%, indicating a rebound of the cell cycle. Importantly, after blockage of Akt by LY294002, the cell cycle did not return to the level without Akt blockage (S phase: 14.27% versus 22.95%). In parallel, we examined cell apoptosis after infection with CSFV for 48 hr in the presence or absence of LY294002. As shown in Figure 5D, treatment with CSFV or LY294002 separately induced cell apoptosis of 0.71% or 2.06%, respectively; however, their combination induced a synergetic apoptosis of 6.16%. TUNEL assay also confirmed this synergetic efficacy (Figure 5E). All the above-mentioned results are consistent with the conclusion that CSFV-induced Akt feedback activation indeed played a key role in rescuing the cell and that blockage of Akt resulted in the synergistic inhibition of the cell cycle and induction of cell apoptosis in CSFV-infected cells.
To clarify the influence of Akt/mTORC1 feedback activation on viral replication, viral gene copy numbers were determined in the presence or absence of LY294002. Results showed that blockage of Akt activity increased the virus copies in infected cells continuously from 24 to 96 hr when compared with CSFV treatment (Figure 5F). This confirmed that Akt persistent inhibition promotes viral replication, which also indicates that CSFV-induced Akt/mTORC1 negative feedback activation indeed contributed to maintaining viral copy numbers at a stable level rather than increasing.
Discussion
As a sensor of cellular homeostasis, mTOR activity is often perturbed during viral infection. For instance, envelope protein E2 of HCV suppresses the activation of Akt/mTORC1 by inducing ER stress (Wang et al., 1999), vaccinia virus (VACV) induces the activation of PI3K/Akt/mTOR via lipid rafts, and vesicular stomatitis virus (VSV) induces hypophosphorylation of Akt, resulting in the inhibition of GSK3β and mTOR and promotion of viral replication (Dunn and Connor, 2011). In our study, we found that several CSFV proteins, Erns, NS3, and NS5A, were involved in inhibiting the activation of both mTORC1 and Akt kinase, whereas E2 had the reverse function of moderately inducing the activation of Akt/mTORC1. Accordingly, we concluded that different viral proteins were likely to hijack diverse signaling pathways to support cell survival and viral replication, and undoubtedly, Akt/mTORC1 was an important one.
The mTOR pathway has been identified as being involving in diverse biological functions, such as cell survival, autophagy, energy metabolism, and biosynthesis. Herein, we focused on exploring the mechanism by which mTORC1 contributes to the regulation of the replication of CSFV. Autophagy has been implicated in viral replication by providing intracellular membrane structures or by maintaining intracellular homeostasis (Miller and Krijnse-Locker, 2008). For example, measles virus (MeV) induces autophagy to prevent cell death and promote proliferation of progeny virions (Richetta et al., 2013), HCV induces complete autolysosome formation for viral replication by repressing the innate immune response (Ke and Chen, 2011), and human parainfluenza virus type 3 (HPIV3) infection induces the accumulation of cytoplasmic autophagosomes by direct inhibition of autolysosome formation, resulting in an increase of viral production (Ding et al., 2014). CSFV NS5A and E2 have been reported to induce autophagy during viral replication (Pei et al., 2014). Paradoxically, virus-induced autophagy can also serve as a mechanism of host defense by favoring cell survival and thereby restricting viral replication, as with CHIKV and HIV-1; both could cause the subversion of autophagy to facilitate new virion production (Kyei et al., 2009, Joubert et al., 2012). Our results clarified that CSFV infection induces a complete autophagy process that involves not only autophagosome formation but also an increasing autophagic flux in host cells for viral replication.
To further confirm that autophagy in CSFV-infected cells is induced by an mTORC1/ULK1-dependent pathway, and that it is indeed necessary for viral replication, we blocked activation of ULK1, the key hub connecting mTOR pathway and autophagy. Our results showed that viral replication was significantly inhibited following ULK1 inhibition and the resulting diminished autophagy, which strongly indicates that CSFV induces autophagy in an mTORC1/ULK1-dependent manner, with this autophagy playing a key role in promoting CSFV replication.
In addition to being a regulator of autophagy, mTORC1 also acts as a regulator of protein translation via its downstream effectors, such as 4E-binding proteins (4E-BPs) or S6K1. We have presented evidence that mTORC1 inhibition acts to increase viral replication in Atg5-silenced cells and that the inhibition efficacy of Akt/mTORC1 activator (insulin) to CSFV replication is more potent than that of autophagy blockage. We questioned, therefore, whether mTORC1 contributes to CSFV replication by other mechanism(s) than autophagy, such as involving other downstream effectors of Akt/mTORC1 pathway that may promote CSFV viral replication.
A major function of mTORC1 is to initiate cap-dependent translation by directly phosphorylating 4E-BPs and S6K1, and the inhibitory effect of mTORC1 inhibitors (rapalogs) on viral replication has been well documented in instances in which the virus possesses capped mRNA (Bose et al., 2012, McNulty et al., 2013). However, whether mTORC1 facilitates the replication of viruses without a capped mRNA, such as HCV or CSFV, remained uncertain. Some viruses have evolved alternative mechanisms to bypass cellular programs that limit the ribosomal machinery and facilitate viral protein translation, including the use of IRES, ribosome shunting, and the substitution of VPg for the mRNA cap (Leen et al., 2016), which permit efficient replication in the absence of cap-dependent initiation protein eIF4E. IRES-mediated translation has the advantage for viruses with such a domain since viral proteins can continue to be generated efficiently even in cells undergoing apoptosis or nutrient starvation.
Many of the mysteries of how mTORC1 and S6K1 exert their control over translation of CSFV mRNA are now much closer to being solved. Previous studies have found a 40S ribosome complex containing eIF3 and CSFV-IRES, in which eIF3 has been completely displaced from its ribosomal position in the 43S complex (Hashem et al., 2013). Holz et al. (46) then demonstrated that eIF3 family members, as S6K1 interactors, represented a potential important connection between the mTOR pathway and an integral component of the translational pre-initiation apparatus, with the activity of S6K1 negatively regulating its binding to eIF3. For example, the mutants that eliminate S6K1 activity constitutively bound eIF3, whereas active S6K1 mutants were incapable of this (Holz et al., 2005). Therefore, we hypothesized that CSFV-induced hypophosphorylation of S6K1 at mTORC1-specific residue T449 would promote its interaction with eIF3, thereby releasing the 40S ribosome binding site for CSFV-IRES and facilitating viral replication. Our results have demonstrated that S6K1 was indeed hypophosphorylated following CSFV-induced mTORC1 inhibition, which increased the binding between inactivated S6K1 and eIF3A (a key subunit of eIF3), whereas interaction between S6K1 and the 40S ribosome was decreased and interaction between CSFV-IRES and 40S ribosome was increased, resulting in enhancement of virus production. These data imply that translation of host cell mRNA might be inhibited and translation of viral mRNA might be enhanced when mTORC1 is inhibited by invasive viruses. Furthermore, two interesting phenomena were observed following CSFV-induced mTORC1 inhibition: on the one hand, infected cells maintained survival with no increase in cellular apoptosis (see Figures 5B–5D), and on the other, rapamycin induced a significant increase in CSFV viral replication, although this enhancement was lost by 48 hr p.i.
Inhibition of mTORC1 by rapalogs fails to induce significant cell growth inhibition and cell apoptosis in almost all kinds of tumor cells, and several mechanisms have been uncovered to counteract their anti-cancer efficacy; for example, rapalogs block S6K1-IRS1 negative feedback loop, leading to the activation of PI3K/Akt prosurvival signals (O'reilly et al., 2006), or they increase eIF4E phosphorylation through an MnK-dependent mechanism, thereby promoting cell survival and apoptotic resistance (Sun et al., 2005). Intriguingly, we found that CSFV induced temporary inhibition of Akt activity at both Ser473 and Thr308 sites within the first 12 hr, following which Akt/mTORC1 is feedback activated (Figure 5A). We then speculated whether CSFV infection induced the feedback activation would contribute to inhibiting cell apoptosis and limiting viral replication. Akt was blocked by LY294002 in ST cells, and the results indicated that CSFV rapidly accelerated cell cycle into the S phase, whereas the G0/G1 release was diminished. This provides strong evidence that Akt was reactivated and released the cell cycle. Moreover, blockage of Akt synergistically inhibits cell growth and promotes cell apoptosis when compared with CSFV or LY294002 mono-treatment. In addition to elucidating the role of Akt feedback activation in maintaining cell survival, its effect on viral replication was determined. When Akt was blocked, the virus copy numbers increased continuously from 24 hr to 96 hr when compared with CSFV infection alone. All these results provide strong evidence that CSFV-induced mTORC1 inhibition induces Akt feedback activation, thereby maintaining equilibrium between cell survival and viral replication. Indeed, feedback-like activation mechanisms have been noted in several viruses; for example, HSV-1 Us3 virus kinase stimulates Akt and induces the activation of mTOR, which avoids S6K-mediated feedback activation, thereby maintaining Akt activation and viral replication (Naghavi et al., 2013). Influenza A virus induces transient PI3K and mTORC1 activity for regulating cell survival and virus production in an autophagy-dependent manner (Datan et al., 2014). In addition, PRRSV infection has been found to increase phosphorylation of mTOR at early stage, followed by a decrease (Zhang and Wang, 2010); although the mechanism has not been elucidated, it might be associated with Akt feedback activation.
In summary, our study has determined that CSFV infection induces the hypophosphorylation of mTORC1, and furthermore, we have provided evidence for a novel mechanism by which CSFV responds to mTORC1 inhibition, resulting in a more favorable environment for viral replication via mTORC1/ULK1-dependent autophagy and mTORC1/S6K1/eIF3-mediated translation of viral mRNA. We have also provided evidence that CSFV-induced mTORC1 inhibition elicits Akt/mTORC1 negative feedback activation, thereby maintaining intracellular homeostasis of cell survival and viral replication. Our study provides new insights into the mechanisms of CSFV replication and may suggest new therapies for the treatment of classical swine fever disease.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This work was supported by the following grants: National Key Research and Development Program of China (No. 2017YFD0500103 and 2016YFD0501002), China Postdoctoral Science Foundation project (No. 2016M590264), the Bethune Project Plan B of Jilin University (No. 450060521279), and the Science and Technology Department of Jilin Province (Nos. 20160414052GH and 20160101053JC).
Author Contributions
Q.L., L.Z., F.W., Q. F., F.B., S.M., and N.L. conducted the experiments, and C.W., Y.L., and C.T. designed the experiments and wrote the paper.
All the authors have read and approved the final manuscript for publication.
Declaration of Interests
The authors declare no competing interests.
Published: May 25, 2018
Footnotes
Supplemental Information includes Transparent Methods and two figures and can be found with this article online at https://doi.org/10.1016/j.isci.2018.04.010.
Contributor Information
Yan Liu, Email: liu820512@163.com.
Changchun Tu, Email: changchun_tu@hotmail.com.
Supplemental Information
References
- Amorim R., Costa S.M., Cavaleiro N.P., Da Silva E.E., Da Costa L.J. HIV-1 transcripts use IRES-initiation under conditions where Cap-dependent translation is restricted by poliovirus 2A protease. PLoS One. 2014;9:e88619. doi: 10.1371/journal.pone.0088619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauhofer O., Summerfield A., Sakoda Y., Tratschin J.D., Hofmann M.A., Ruggli N. Classical swine fever virus Npro interacts with interferon regulatory factor 3 and induces its proteasomal degradation. J. Virol. 2007;81:3087–3096. doi: 10.1128/JVI.02032-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose S.K., Shrivastava S., Meyer K., Ray R.B., Ray R. Hepatitis C virus activates the mTOR/S6K1 signaling pathway in inhibiting IRS-1 function for insulin resistance. J. Virol. 2012;86:6315–6322. doi: 10.1128/JVI.00050-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong H., Lindsten T., Wu J., Lu C., Thompson C.B. Ammonia-induced autophagy is independent of ULK1/ULK2 kinases. Proc. Natl. Acad. Sci. USA. 2011;108:11121–11126. doi: 10.1073/pnas.1107969108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cinti A., Le Sage V., Milev M.P., Valiente-Echeverria F., Crossie C., Miron M.J., Pante N., Olivier M., Mouland A.J. HIV-1 enhances mTORC1 activity and repositions lysosomes to the periphery by co-opting rag GTPases. Sci. Rep. 2017;7:5515. doi: 10.1038/s41598-017-05410-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datan E., Shirazian A., Benjamin S., Matassov D., Tinari A., Malorni W., Lockshin R.A., Garcia-Sastre A., Zakeri Z. mTOR/p70S6K signaling distinguishes routine, maintenance-level autophagy from autophagic cell death during influenza A infection. Virology. 2014;452-453:175–190. doi: 10.1016/j.virol.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datan E., Roy S.G., Germain G., Zali N., Mclean J.E., Golshan G., Harbajan S., Lockshin R.A., Zakeri Z. Dengue-induced autophagy, virus replication and protection from cell death require ER stress (PERK) pathway activation. Cell Death Dis. 2016;7:e2127. doi: 10.1038/cddis.2015.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deretic V. Autophagy in infection. Curr. Opin. Cell Biol. 2010;22:252–262. doi: 10.1016/j.ceb.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding B., Zhang G., Yang X., Zhang S., Chen L., Yan Q., Xu M., Banerjee A.K., Chen M. Phosphoprotein of human parainfluenza virus type 3 blocks autophagosome-lysosome fusion to increase virus production. Cell Host Microbe. 2014;15:564–577. doi: 10.1016/j.chom.2014.04.004. [DOI] [PubMed] [Google Scholar]
- Dreux M., Gastaminza P., Wieland S.F., Chisari F.V. The autophagy machinery is required to initiate hepatitis C virus replication. Proc. Natl. Acad. Sci. USA. 2009;106:14046–14051. doi: 10.1073/pnas.0907344106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn E.F., Connor J.H. Dominant inhibition of Akt/protein kinase B signaling by the matrix protein of a negative-strand RNA virus. J. Virol. 2011;85:422–431. doi: 10.1128/JVI.01671-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan D., Kim J., Shaw R.J., Guan K.L. The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy. 2011;7:643–644. doi: 10.4161/auto.7.6.15123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan D.F., Chun M.G., Vamos M., Zou H., Rong J., Miller C.J., Lou H.J., Raveendra-Panickar D., Yang C.C., Sheffler D.J. Small molecule inhibition of the autophagy kinase ULK1 and identification of ULK1 substrates. Mol. Cell. 2015;59:285–297. doi: 10.1016/j.molcel.2015.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiebach A.R., Guzylack-Piriou L., Python S., Summerfield A., Ruggli N. Classical swine fever virus N(pro) limits type I interferon induction in plasmacytoid dendritic cells by interacting with interferon regulatory factor 7. J. Virol. 2011;85:8002–8011. doi: 10.1128/JVI.00330-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher S.P., Jackson R.J. Pestivirus internal ribosome entry site (IRES) structure and function: elements in the 5' untranslated region important for IRES function. J. Virol. 2002;76:5024–5033. doi: 10.1128/JVI.76.10.5024-5033.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher S.P., Ali I.K., Kaminski A., Digard P., Jackson R.J. The influence of viral coding sequences on pestivirus IRES activity reveals further parallels with translation initiation in prokaryotes. RNA. 2002;8:1558–1571. [PMC free article] [PubMed] [Google Scholar]
- Friis M.B., Rasmussen T.B., Belsham G.J. Modulation of translation initiation efficiency in classical swine fever virus. J. Virol. 2012;86:8681–8692. doi: 10.1128/JVI.00346-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Q., Shi H., Zhang H., Ren Y., Guo F., Qiao J., Jia B., Wang P., Chen C. Autophagy during early stages contributes to bovine viral diarrhea virus replication in MDBK cells. J. Basic Microbiol. 2014;54:1044–1052. doi: 10.1002/jobm.201300750. [DOI] [PubMed] [Google Scholar]
- Guo H., Zhou T., Jiang D., Cuconati A., Xiao G.H., Block T.M., Guo J.T. Regulation of hepatitis B virus replication by the phosphatidylinositol 3-kinase-akt signal transduction pathway. J. Virol. 2007;81:10072–10080. doi: 10.1128/JVI.00541-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashem Y., des Georges A., Dhote V., Langlois R., Liao H.Y., Grassucci R.A., Pestova T.V., Hellen C.U., Frank J. Hepatitis-C-virus-like internal ribosome entry sites displace eIF3 to gain access to the 40S subunit. Nature. 2013;503:539–543. doi: 10.1038/nature12658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L., Zhang Y.M., Lin Z., Li W.W., Wang J., Li H.L. Classical swine fever virus NS5A protein localizes to endoplasmic reticulum and induces oxidative stress in vascular endothelial cells. Virus Genes. 2012;45:274–282. doi: 10.1007/s11262-012-0773-2. [DOI] [PubMed] [Google Scholar]
- Holz M.K., Ballif B.A., Gygi S.P., Blenis J. mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell. 2005;123:569–580. doi: 10.1016/j.cell.2005.10.024. [DOI] [PubMed] [Google Scholar]
- Jiang X., Overholtzer M., Thompson C.B. Autophagy in cellular metabolism and cancer. J. Clin. Invest. 2015;125:47–54. doi: 10.1172/JCI73942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joubert P.E., Werneke S.W., de La Calle C., Guivel-Benhassine F., Giodini A., Peduto L., Levine B., Schwartz O., Lenschow D.J., Albert M.L. Chikungunya virus-induced autophagy delays caspase-dependent cell death. J. Exp. Med. 2012;209:1029–1047. doi: 10.1084/jem.20110996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joubert P.E., Stapleford K., Guivel-Benhassine F., Vignuzzi M., Schwartz O., Albert M.L. Inhibition of mTORC1 enhances the translation of chikungunya proteins via the activation of the MnK/eIF4E pathway. PLoS Pathog. 2015;11:e1005091. doi: 10.1371/journal.ppat.1005091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke P.Y., Chen S.S. Autophagy: a novel guardian of HCV against innate immune response. Autophagy. 2011;7:533–535. doi: 10.4161/auto.7.5.14732. [DOI] [PubMed] [Google Scholar]
- Kolupaeva V.G., Pestova T.V., Hellen C.U. Ribosomal binding to the internal ribosomal entry site of classical swine fever virus. RNA. 2000;6:1791–1807. doi: 10.1017/s1355838200000662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyei G.B., Dinkins C., Davis A.S., Roberts E., Singh S.B., Dong C., Wu L., Kominami E., Ueno T., Yamamoto A. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J. Cell Biol. 2009;186:255–268. doi: 10.1083/jcb.200903070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leen E.N., Sorgeloos F., Correia S., Chaudhry Y., Cannac F., Pastore C., Xu Y., Graham S.C., Matthews S.J., Goodfellow I.G., Curry S. A conserved interaction between a C-terminal motif in norovirus VPg and the HEAT-1 domain of eIF4G is essential for translation initiation. PLoS Pathog. 2016;12:e1005379. doi: 10.1371/journal.ppat.1005379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B., Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S., Feng S., Wang J.H., He W.R., Qin H.Y., Dong H., Li L.F., Yu S.X., Li Y., Qiu H.J. eEF1A interacts with the NS5A protein and inhibits the growth of classical swine fever virus. Viruses. 2015;7:4563–4581. doi: 10.3390/v7082833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S., Wang J., He W.R., Feng S., Li Y., Wang X., Liao Y., Qin H.Y., Li L.F., Dong H. Thioredoxin 2 is a novel E2-interacting protein that inhibits the replication of classical swine fever virus. J. Virol. 2015;89:8510–8524. doi: 10.1128/JVI.00429-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Q., Luo Z., Zeng J., Chen W., Foo S.S., Lee S.A., Ge J., Wang S., Goldman S.A., Zlokovic B.V. Zika virus NS4A and NS4B proteins deregulate Akt-mTOR signaling in human fetal neural stem cells to inhibit neurogenesis and induce autophagy. Cell Stem Cell. 2016;19:663–671. doi: 10.1016/j.stem.2016.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindenbach B.D., Thiel H.J., Rice C.M. Flaviviridae: the viruses and their replication. In: Knipe D.M., Howley P.M., editors. Fields Virology. Fifth Edition. Lippincott-Raven Publishers; 2007. pp. 1101–1152. [Google Scholar]
- Lv H., Dong W., Cao Z., Li X., Wang J., Qian G., Lv Q., Wang C., Guo K., Zhang Y. TRAF6 is a novel NS3-interacting protein that inhibits classical swine fever virus replication. Sci. Rep. 2017;7:6737. doi: 10.1038/s41598-017-06934-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannova P., Beretta L. Activation of the N-Ras-PI3K-Akt-mTOR pathway by hepatitis C virus: control of cell survival and viral replication. J. Virol. 2005;79:8742–8749. doi: 10.1128/JVI.79.14.8742-8749.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin S., Saha B., Riley J.L. The battle over mTOR: an emerging theatre in host-pathogen immunity. PLoS Pathog. 2012;8:e1002894. doi: 10.1371/journal.ppat.1002894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda D., Mauro V.P. Base pairing between hepatitis C virus RNA and 18S rRNA is required for IRES-dependent translation initiation in vivo. Proc. Natl. Acad. Sci. USA. 2014;111:15385–15389. doi: 10.1073/pnas.1413472111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNulty S., Flint M., Nichol S.T., Spiropoulou C.F. Host mTORC1 signaling regulates Andes virus replication. J. Virol. 2013;87:912–922. doi: 10.1128/JVI.02415-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller S., Krijnse-Locker J. Modification of intracellular membrane structures for virus replication. Nat. Rev. Microbiol. 2008;6:363–374. doi: 10.1038/nrmicro1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muraoka T., Ichikawa T., Taura N., Miyaaki H., Takeshita S., Akiyama M., Miuma S., Ozawa E., Isomoto H., Takeshima F., Nakao K. Insulin-induced mTOR activity exhibits anti-hepatitis C virus activity. Mol. Med. Rep. 2012;5:331–335. doi: 10.3892/mmr.2011.662. [DOI] [PubMed] [Google Scholar]
- Naghavi M.H., Gundersen G.G., Walsh D. Plus-end tracking proteins, CLASPs, and a viral Akt mimic regulate herpesvirus-induced stable microtubule formation and virus spread. Proc. Natl. Acad. Sci. USA. 2013;110:18268–18273. doi: 10.1073/pnas.1310760110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'reilly K.E., Rojo F., She Q.B., Solit D., Mills G.B., Smith D., Lane H., Hofmann F., Hicklin D.J., Ludwig D.L. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–1508. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orvedahl A., Alexander D., Talloczy Z., Sun Q., Wei Y., Zhang W., Burns D., Leib D.A., Levine B. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe. 2007;1:23–35. doi: 10.1016/j.chom.2006.12.001. [DOI] [PubMed] [Google Scholar]
- Pei J., Zhao M., Ye Z., Gou H., Wang J., Yi L., Dong X., Liu W., Luo Y., Liao M., Chen J. Autophagy enhances the replication of classical swine fever virus in vitro. Autophagy. 2014;10:93–110. doi: 10.4161/auto.26843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petherick K.J., Conway O.J., Mpamhanga C., Osborne S.A., Kamal A., Saxty B., Ganley I.G. Pharmacological inhibition of ULK1 kinase blocks mammalian target of rapamycin (mTOR)-dependent autophagy. J. Biol. Chem. 2015;290:11376–11383. doi: 10.1074/jbc.C114.627778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richetta C., Gregoire I.P., Verlhac P., Azocar O., Baguet J., Flacher M., Tangy F., Rabourdin-Combe C., Faure M. Sustained autophagy contributes to measles virus infectivity. PLoS Pathog. 2013;9:e1003599. doi: 10.1371/journal.ppat.1003599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton R.A., Sabatini D.M. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168:960–976. doi: 10.1016/j.cell.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schepetilnikov M., Dimitrova M., Mancera-Martinez E., Geldreich A., Keller M., Ryabova L.A. TOR and S6K1 promote translation reinitiation of uORF-containing mRNAs via phosphorylation of eIF3h. EMBO J. 2013;32:1087–1102. doi: 10.1038/emboj.2013.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta S., Peterson T.R., Laplante M., Oh S., Sabatini D.M. mTORC1 controls fasting-induced ketogenesis and its modulation by ageing. Nature. 2010;468:1100–1104. doi: 10.1038/nature09584. [DOI] [PubMed] [Google Scholar]
- Shintani T., Klionsky D.J. Autophagy in health and disease: a double-edged sword. Science. 2004;306:990–995. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrivastava S., Bhanja Chowdhury J., Steele R., Ray R., Ray R.B. Hepatitis C virus upregulates Beclin1 for induction of autophagy and activates mTOR signaling. J. Virol. 2012;86:8705–8712. doi: 10.1128/JVI.00616-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stohr S., Costa R., Sandmann L., Westhaus S., Pfaender S., Anggakusuma, Dazert E., Meuleman P., Vondran F.W., Manns M.P. Host cell mTORC1 is required for HCV RNA replication. Gut. 2016;65:2017–2028. doi: 10.1136/gutjnl-2014-308971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S.Y., Rosenberg L.M., Wang X., Zhou Z., Yue P., Fu H., Khuri F.R. Activation of Akt and eIF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition. Cancer Res. 2005;65:7052–7058. doi: 10.1158/0008-5472.CAN-05-0917. [DOI] [PubMed] [Google Scholar]
- Wang Z., Lu Y., Zhou P., Zhai Z., Ding M. The morphological structure of classical swine fever virus and some characteristics of its multiplication. Wei Sheng Wu Xue Bao. 1999;39:189–195. [in Chinese] [PubMed] [Google Scholar]
- Wang X., Hawk N., Yue P., Kauh J., Ramalingam S.S., Fu H., Khuri F.R., Sun S.Y. Overcoming mTOR inhibition-induced paradoxical activation of survival signaling pathways enhances mTOR inhibitors' anticancer efficacy. Cancer Biol. Ther. 2008;7:1952–1958. doi: 10.4161/cbt.7.12.6944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Chen S., Liao Y., Zhang E., Feng S., Yu S., Li L.F., He W.R., Li Y., Luo Y. Mitogen-activated protein kinase kinase 2 (MEK2), a novel E2-interacting protein, promotes the growth of classical swine fever virus via attenuation of the JAK-STAT signaling pathway. J Virol. 2016;90:10271–10283. doi: 10.1128/JVI.01407-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z., Shi Z., Guo H., Qu H., Zhang Y., Tu C. Annexin 2 is a host protein binding to classical swine fever virus E2 glycoprotein and promoting viral growth in PK-15 cells. Virus Res. 2015;201:16–23. doi: 10.1016/j.virusres.2015.02.013. [DOI] [PubMed] [Google Scholar]
- Yin H., Zhao L., Li S., Xu L., Wang Y., Chen H. Impaired cellular energy metabolism contributes to duck-enteritis-virus-induced autophagy via the AMPK-TSC2-MTOR signaling pathway. Front Cell Infect. Microbiol. 2017;7:423. doi: 10.3389/fcimb.2017.00423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H., Wang X. A dual effect of porcine reproductive and respiratory syndrome virus replication on the phosphatidylinositol-3-kinase-dependent Akt pathway. Arch. Virol. 2010;155:571–575. doi: 10.1007/s00705-010-0611-6. [DOI] [PubMed] [Google Scholar]
- Zhang C., Kang K., Ning P., Peng Y., Lin Z., Cui H., Cao Z., Wang J., Zhang Y. Heat shock protein 70 is associated with CSFV NS5A protein and enhances viral RNA replication. Virology. 2015;482:9–18. doi: 10.1016/j.virol.2015.02.014. [DOI] [PubMed] [Google Scholar]
- Zhou X., Wang Y., Metselaar H.J., Janssen H.L., Peppelenbosch M.P., Pan Q. Rapamycin and everolimus facilitate hepatitis E virus replication: revealing a basal defense mechanism of PI3K-PKB-mTOR pathway. J. Hepatol. 2014;61:746–754. doi: 10.1016/j.jhep.2014.05.026. [DOI] [PubMed] [Google Scholar]
- Zhou Y., Geng P., Liu Y., Wu J., Qiao H., Xie Y., Yin N., Chen L., Lin X., Liu Y. Rotavirus-encoded virus-like small RNA triggers autophagy by targeting IGF1R via the PI3K/Akt/mTOR pathway. Biochim. Biophys. Acta. 2017;1864:60–68. doi: 10.1016/j.bbadis.2017.09.028. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.