

Summary
Genome architecture has emerged as a critical element of transcriptional regulation, although its role in the control of cell identity is not well understood. Here we use transcription factor (TF)-mediated reprogramming to examine the interplay between genome architecture and transcriptional programs that transition cells into the myogenic identity. We recently developed new methods for evaluating the topological features of genome architecture based on network centrality. Through integrated analysis of these features of genome architecture and transcriptome dynamics during myogenic reprogramming of human fibroblasts we find that significant architectural reorganization precedes activation of a myogenic transcriptional program. This interplay sets the stage for a critical transition observed at several genomic scales reflecting definitive adoption of the myogenic phenotype. Subsequently, TFs within the myogenic transcriptional program participate in entrainment of biological rhythms. These findings reveal a role for topological features of genome architecture in the initiation of transcriptional programs during TF-mediated human cellular reprogramming.
Subject Areas: Molecular Structure, Integrative Aspects of Cell Biology, Systems Biology, Omics
Graphical Abstract
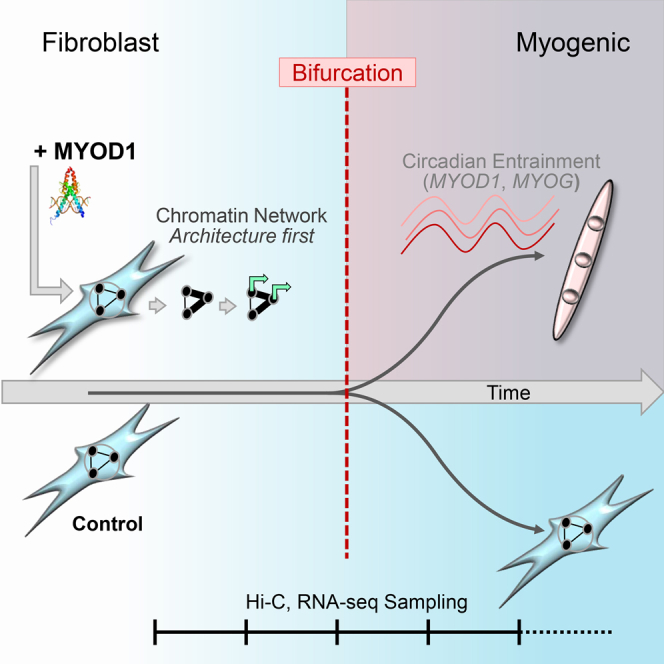
Highlights
-
•
4D Nucleome analysis of direct human fibroblast to muscle reprogramming
-
•
A space-time bifurcation marks transit to a new cell identity
-
•
Chromatin reorganization precedes significant transcriptional changes
-
•
Myogenic master regulators have a role in entraining biological rhythms
Molecular Structure; Integrative Aspects of Cell Biology; Systems Biology; Omics
Introduction
During cellular reprogramming, the mechanisms by which a small number of transcription factors (TF) (Takahashi et al., 2007), or a single TF as in Weintraub's work (Weintraub et al., 1989, Weintraub, 1993), impose new transcriptional programs that supersede established cell identities are not well understood. Unbiased technologies such as genome-wide chromosome conformation capture (Hi-C) and RNA sequencing (RNA-seq) are yielding ever higher resolution data that are essential to refining our notions of cell identity formation and maintenance. Yet these platforms are not well integrated analytically to understand the interplay between architecture and transcription. Furthermore, the dynamical nature of both architecture and transcription, during cellular reprogramming and natural biological rhythms, is challenging to capture experimentally and informatically and therefore is not well resolved. Thus the multi-platform genome-wide temporal capture of cells during reprogramming with integrated analytic approaches will be valuable for gaining insight into the mechanisms of reprogramming (Rajapakse and Groudine, 2011) and in line with the 4D Nucleome (4DN) movement (Chen et al., 2015, Dixon et al., 2015, Fortin and Hansen, 2015, Krijger et al., 2016).
Although genome architecture is a key element in transcriptional programs, its role in TF-mediated reprogramming is poorly understood, partially due to limited temporal data and analytic methods. During differentiation, architecturally defined regions can change their overall gene expression to facilitate a transcriptional program that supports a new cell state (Chen et al., 2015, Dixon et al., 2012, Dixon et al., 2015, Lieberman-Aiden et al., 2009). These regions can be defined based on Hi-C contact maps into 2 major compartments: open, transcriptionally active chromatin, classically termed compartment A, or closed, transcriptionally inactive chromatin, termed compartment B (Chen et al., 2015, Lieberman-Aiden et al., 2009). Of critical importance in analyzing genome architecture data are sophisticated approaches to extract the most prominent, biologically relevant features. Our recent technique based on spectral graph theory extracts critical architectural information from Hi-C data, showing utility in defining chromatin domains at many scales (Chen et al., 2016).
In this work, we examined the dynamical interactions between the genome architectural features and transcription in human fibroblasts undergoing MYOD1-mediated reprogramming into the myogenic lineage. Sampling across a time course during reprogramming, we captured architecture by Hi-C, transcription by RNA-seq, and protein content by proteomics. To better understand the features of genome architecture and expression in a dynamical setting, we adopt a network point of view. Nodes of the network correspond to genomic loci that can be partitioned at different scales, for example, into larger scale 1-Mb regions or smaller scale gene-level regions. The edges of the network indicate contact between two genomic loci, with edge weights given by Hi-C entries. From the network perspective, A/B compartments are identified as distinct connected nodes of a network.
To further reveal chromatin spatial organization, we use network centrality measures. Using network centrality enables identification of nodes that play influential topological roles in the network (Newman, 2010). A number of centrality measures exist, each specialized to a particular type of nodal influence. For example, degree centrality characterizes the local connectedness of a node as measured by the number of edges connecting to this node, whereas betweenness centrality is a global connectedness measure that quantifies the number of times a node acts as a bridge along the shortest path between two other nodes. Eigenvector centrality is a neighborhood connectedness property in which a node has high centrality if many of its neighbors also have high centrality. In other words, a node is important if it is connected to other important nodes. For reference, Google's PageRank algorithm uses a variant of eigenvector centrality (Lohmann et al., 2010).
By examining different centrality measures we have discovered important features in Hi-C data largely overlooked in previous studies. We also found that cells undergoing reprogramming have significant architectural reorganization before changes in transcription, navigate through a critical transition point into the myogenic lineage, and subsequently show potent activation of the myogenic program that ties into regulation of biological rhythms.
Results
Myogenic Reprogramming of Human Fibroblasts
We converted primary human fibroblasts into the myogenic lineage using the TF and master regulator MYOD1, following Weintraub's method for myogenic reprogramming (Weintraub, 1993) (Figure S1A). Fibroblasts were transduced with a lentiviral construct that expressed human MYOD1 fused with a tamoxifen-inducible ER(T) domain (L-MYOD1) (Kimura et al., 2008). With 4-hydroxytamoxifen (4-OHT) treatment, transduced cells showed nuclear translocation of L-MYOD1, morphological changes consistent with expression of key myogenic genes downstream of MYOD1 (MYOG and MYH1) (Figure S1B), and myogenic differentiation (Figures S1C and S1D). These data demonstrate the conversion of fibroblasts into the myogenic lineage by L-MYOD1 (see Figure S7).
We used this system to delineate the dynamics of architecture and transcription underlying direct cellular reprogramming. Analyses were carried out on transduced, 4-OHT-treated cells, sampling at 8-hr intervals for RNA-seq (3 replicates per time point, small RNA-seq, and single replicate per time point, Hi-C; see Figure S8) and at 24-hr intervals for proteomics (Figure 1A).
Figure 1.

Myogenic Reprogramming of Human Fibroblasts
(A) Time course of MYOD1-mediated reprogramming. The time window outlined in green corresponds to time points at which both genome architecture and transcription were captured by Hi-C (single replicates) and RNA-seq (in triplicate).
(B) Scale-adaptive Hi-C matrices and gene expression. The considered scales include 1 Mb, 100 kb, TAD, and gene level.
We evaluated up to 16 time points (−48, 0, …, 112 hr) for genome architecture (form) through Hi-C and for transcription (function) through RNA-seq. The resulting time series data were studied at different scales (Figure 1B). The scale was based on units of length along the linear genome (1 Mb, 100 kb) or by structurally/functionally defined units of the genome, such as topologically associating domains (TADs) or individual genes.
Network Representation of Genomic Time Series Data
With the aid of network representation, we captured multiple topological properties of genome architecture using the concept of network centrality (Methods). For this analysis, we interpreted 100-kb-resolution Hi-C and RNA-seq data as measurements of dynamical networks, where Hi-C contact maps depict network topologies and RNA-seq data characterize the function of nodes (Figure 2A).
Figure 2.

Network Representation of Genomic Time Series Data
(A) Mapping genomic form (Hi-C) and function (RNA-seq) to network architecture and node dynamics. Top left: Hi-C contact map (Toeplitz normalized, Methods) and RNA-seq at 100 kb resolution for chromosome 19. Top right: Network representation in which edge width indicates the Hi-C contact number and node color implies the magnitude of RNA-seq FPKM value. Bottom left: Network features given by eigenvector centrality, degree centrality, and betweenness centrality scores. The bars marked by different colors correspond to maximum centrality values. Bottom right: An illustrative network under different centrality measures.
(B) Eigenvector centrality indicates chromatin compartments, termed A and B. Top left: Hi-C contact map of chromosome 3 at 100 kb resolution. Bottom left: RNA-seq, the first principal component (PC1) of the Hi-C correlation matrix, and eigenvector centrality (in terms of its Z score). Right: Correlation between RNA-seq, PC1, and eigenvector centrality extracted from Hi-C data for all chromosomes. Eigenvector centrality is a better indicator for chromatin compartments, marked by asterisk.
(C) Betweenness centrality indicates A/B switched loci. Top left: Hi-C contact map of chromosome 19 (100 kb resolution) at time points 0 and 40 hr. Bottom left: A/B partition and betweenness centrality (in terms of its Z score) at 0 and 40 hr. The blue color represents A/B switched bins from 0 to 40 hr. The switched loci tend to have large betweenness centrality scores. Right: Significance of betweenness centrality at A/B switched loci. The p value is determined by comparing the average betweenness value at A/B switched bins with a random background distribution of other centrality values under the same number of bins. p Values are computed for all chromosomes and shown through an error bar plot in which the circle represents the p value averaged over all chromosomes and the horizontal error bar is determined by the SD of p values for all chromosomes.
We found that network centrality measures such as degree centrality, eigenvector centrality, and betweenness centrality quantified different architectural features that reflect the importance of specific genomic loci within the network. Beyond the simplest measure, degree centrality, we found that eigenvector centrality identified architecturally defined regions of active/inactive gene expression (A/B compartments). In addition, across chromosomes, eigenvector centrality yielded a higher correlation with transcriptional activity than conventionally defined A/B compartments (Figures 2B and S2), which are derived from the first principal component of a spatial correlation Hi-C matrix (Lieberman-Aiden et al., 2009).
Betweenness centrality recognized regions that switched A/B compartment assignment between time steps. The values of betweenness at A/B switched regions were significantly higher than other centrality measures (Figure 2C). We then observed that A/B switched regions tended to be at boundaries between other A/B compartments and determined that this observation held for 70% of switched regions (Figure S3). Altogether, these results suggested that betweenness centrality detected boundary regions between open and closed chromatin that had a high propensity for altered architecture between time steps. We speculate that these regions, or “bridge nodes” in the network, serve as architectural buffers between largely active and inactive transcriptional chromatin that could limit access of transcriptional machinery to undesired regions.
We then sought to determine which genes showed differential expression within A/B switched regions. In A/B switched regions between 0 and 40 hr, we identified 175 genes (Table S1) that had at least 2-fold difference in expression (Methods). From this set, 47% of genes that change from compartment A to B had concordant gene expression (decrease), whereas 67% of genes that change from compartment B to A had concordant gene expression (increase).
Architectural Changes Precede Activation of the Myogenic Program
Given the cell state trajectory, it was unclear whether MYOD1-mediated reprogramming induced rewiring of genome architecture before the role of MYOD1 in mediating muscle gene transcription, or vice versa (Kosak and Groudine, 2004, Rajapakse and Groudine, 2011). To answer this question, we focused on form and function dynamics of 22,083 genes genome-wide, where the form is depicted by inter-gene Hi-C contact maps (Methods) and the function corresponds to RNA-seq Fragments Per Kilobase of transcript per Million (FPKM) values (Figure 3A). The form-function evolution is then evaluated by determining the difference in network centrality features (extracted from inter-gene contact maps) and gene expression between successive time points. We refer to this measure as the temporal difference score (TDS; Methods). Based on TDS at successive time points (Figure 3B), we found that a significant form change at 8 hr preceded a significant function change at 16 hr.
Figure 3.

Changes in Genome Architecture Precede Activation of the Myogenic Program
(A) Genomic architecture (form) and gene expression (function) given by a Hi-C contact map and RNA-seq. Hi-C and RNA-seq are constructed at gene-level resolution.
(B) Function and form change at successive time points evaluated by temporal difference score (TDS; Methods) of RNA-seq and network centrality features of Hi-C data, respectively. The significant form change (at 8 hr) occurs before the function change (at 16 hr).
(C) Illustration of function TDS from 8 to 16 hr. Genes are divided into 2 clusters by applying K-means to their TDS values. Cluster 1 contains genes with the largest temporal change in RNA-seq. The gene expression can either decrease or increase from 8 to 16 hr.
(D) Illustration of form TDS from 0 to 8 hr. Two gene clusters are obtained by applying K-means to their TDS values. Hi-C contact maps associated with a subset of genes in cluster 1 are shown from 0 to 8 hr, where the blue color indicates the Hi-C difference between the 2 time points.
(E) Form-function change indicators for gene modules of interest during cellular reprogramming (top) and fibroblast proliferation (bottom), respectively. Here each row represents one gene module of interest, each column represents a time step, and the amount of change, as a percentage of total change over time for each module, is depicted by color. Percentage is determined by finding the number of genes with significant form-function change for each module and time step and dividing this number by the total number of significant gene changes for each module over time (row).
For deeper understanding of form-function evolution during the reprogramming process, we applied K-means clustering (with 2 clusters) on both form and function data, separately. This was done to identify subsets of genes that yielded the most significant temporal change (Figures 3C and 3D). From this analysis, we found that genes contained within each cluster of high TDS, which are responsible for function and form change, at most have 20% overlap (Table S2). This suggests that the mechanism of form evolution could be different from that of function evolution and that these two mechanisms are steered by different sets of genes. Furthermore, we investigated 4 gene modules extracted from Gene Ontology (GO): fibroblast, myotube, cell cycle, and circadian genes (Table S3). We then contrasted our reprogramming data with data on human fibroblast proliferation. Data on proliferating human fibroblasts were previously obtained using similar methods over a time course (Chen et al., 2015) after cell cycle and circadian rhythm synchronization, with collection of RNA-seq and Hi-C every 8 hr. We found that the pattern of form-function evolution during reprogramming is quite different from fibroblast proliferation (Figure 3E). Consistent with findings represented in Figure 3B, the effects of nuclear reorganization were detectable before transcription changes, that is, form preceded function. Given these results, we propose that chromatin architectural changes facilitate the orchestrated activation of transcriptional networks associated with the adoption of a new cell identity.
We then sought to identify which genes may be responsible for these form-function dynamics. Within GO fibroblast and muscle gene modules, a significant proportion (>30%) of genes had form change at 8 hr and function change at 32–40 hr (Figure S4A). For comparison, less than 5% of these genes for each module showed similar form-function changes in fibroblast proliferation data. From these sets of genes, we extracted 77 fibroblast genes and 72 muscle genes that had significant change during reprogramming but low activity in proliferation. This yielded core or “backbone” genes, which had distinct form-function evolution during reprogramming (Figures S4B–S4D). The statistical significance of temporal change of the identified genes was p <0.05 when compared with proliferation data (Methods).
Fibroblasts Navigate a Critical Transition En Route to the Myogenic Lineage
Genome dynamics during a direct transition between cell identities are poorly understood. We hypothesized that genome-wide data could be used to pinpoint a definitive transition. From our data we sought to identify the time at which cells transitioned into the myogenic state and which features of architecture and transcription define it. We therefore compared our reprogramming data with previously generated data on proliferating fibroblasts (Chen et al., 2015), where the time window of divergence between the datasets, or bifurcation, would indicate transition into a new cell identity.
To facilitate comparison, from each dataset we extracted a low-dimensional genome-wide form-function representation. This was done by first integrating centrality-based network features with transcription and then extracting the low-dimensional representation of the data using the dimension reduction technique of Laplacian eigenmaps at 1 Mb resolution (Methods). Within each dataset, the form-function representation, fitted by a minimum volume ellipsoid (MVE) (Methods), showed distinct configurations at different time points (Figure 4A). Comparing between datasets, we observed a striking divergence, or bifurcation, at 32 hr (p = 0.0048), suggesting an abrupt shift in the genomic system during a transition from a fibroblast state to a myogenic state.
Figure 4.

Fibroblasts Navigate a Critical Transition En Route to the Myogenic Lineage
(A) Cell state trajectory of MYOD1-mediated reprogramming and fibroblast proliferation (Chen et al., 2015). Ellipsoids represent low-dimensional data representations obtained by applying Laplacian eigenmaps (Methods) to network form-function features. The branching trajectory shows a critical transition, or bifurcation, at 32 hr (p < 0.01).
(B) Portrait of 4DN in the context of reprogramming and proliferation, respectively. It is described by a form-function domain (2D), constructed from 8 time points, for each chromosome. The fitted ellipsoid is obtained from the MVE estimate (Methods).
(C) Shift of form-function domains of chromosomes at 32 hr. Chromosomes 5, 12, and 13 show the most significant changes of all chromosomes.
(D) Form-function differences between cellular reprogramming and fibroblast proliferation, indicated by centroids and volumes of form-function ellipsoids for each chromosome. Top: Comparison between form change (horizontal shift) and function change (vertical shift) for each chromosome. Bottom: Variance of 4DN, given by volumes of chromosome ellipsoids under different cell dynamics.
In examining local genome dynamics, distinct transitions were also observed for myogenic genes MYOD1 and MYOG. Endogenous MYOD1 and MYOG expressions were first detected around 32 hr. In addition, the transition was identified in intra-gene Hi-C contact maps of MYOD1 and MYOG (Figure S5, Methods). Here the difference between gene-level Hi-C matrices at successive time points revealed a pattern strikingly similar to what was found in genome-wide dynamics. Taken together, our results are consistent with 3 phases for reprogramming from our data: fibroblast, bifurcation, and myogenic.
Phase Portraits Show Chromosome Architectural Changes Outpacing Transcriptional Changes
To further quantitate form-function dynamics on the chromosome level, we evaluated 2D phase portraits at 100 kb resolution. On a 2D plane, we designate one axis as a measure of form in terms of network connectivity (Methods) and the other as a measure of function in terms of average RNA-seq FPKM value. The portrait of 4DN is then described by a form-function domain, made up of 8 time points [0,56] (hr) for each chromosome (Figure 4B).
The portraits of 4DN for reprogramming and fibroblast proliferation showed similar positional patterns for chromosomes across time points. The centroid of the fitted form-function ellipsoid (MVE estimate; Methods) for each chromosome was shifted for reprogramming versus proliferation as illustrated for chromosomes 12, 5, and 13 (Figure 4C). Comparing the 32-hr critical transition point for chromosomes between datasets illustrates that the horizontal shift in form is greater than the vertical shift in function. Across time points, we found that most chromosomes undergo more form change (86.4%) than function change (13.8%) (Figure 4D). Furthermore, the area of the chromatin ellipse characterizes the variance (uncertainty) of 4DN (Figure 4D). Ellipsoids associated with reprogramming have larger volumes than those for fibroblast proliferation. Taken together, our results demonstrate a more complex dynamical behavior for reprogramming that is measurable through form-function dynamics, with notable involvement of genome architecture.
Fibroblasts Bypass a Myoblast-Like State during Myogenic Reprogramming
Intermediate stages of reprogramming are of consequence in the design of cell-based therapeutic strategies, as risks and benefits of cells that, for example, retain proliferative potential must be weighed. We therefore sought to further examine the pathway into the myogenic lineage, to determine whether the data support transit through a myoblast-like state or directly to a more differentiated myotube-like state. For this analysis, we considered TADs as functional units of the genome and identified those with significant form-function changes as playing important roles during reprogramming. Previous work showed that the boundaries of TADs remain stable between cell types (Dixon et al., 2012, Dixon et al., 2015); however, the dynamical TAD-level interactions and functional changes during cellular reprogramming are not well understood. We interpreted the genome as a network of TADs (Figure S6), where network vertices corresponded to TADs, and edge weights were given by the interaction frequency between 2 TADs from Hi-C (retaining only interactions that exceeded the 50th percentile of inter-TAD contacts; see Methods). The function associated with a TAD was characterized by the sum of RNA-seq values of the set of genes contained within the TAD-defined region.
We applied network centrality analysis (Methods) to extract the 2D representation of dynamical form-function features at the TAD scale, using previously defined TAD boundaries (Figure S6A) (Dixon et al., 2012). The TAD-TAD network was constructed based on Hi-C matrices at 100 kb resolution, which facilitated the evaluation of whole genome characteristics. The resulting configuration of chromosomes was robust over time, but TADs within a chromosome showed form-function shifts. This can also be observed by contrasting the fibroblast stage (before 4-OHT; −48 hr) with the subsequent reprogramming time points (0, …, 80 hr) (Figure S6B).
We extracted the top 10% (220) of TADs whose positions change the most; these TADs are associated with the largest deviations from the fibroblast stage due to reprogramming (Figure S6C). We found that the identified TADs had high gene density and that genes within them are highly expressed (p < 0.001; see Methods). This implies that a core set of genes might exist within these TADs that induce significant form-function changes.
With this motivation, we focused on TADs containing genes related to fibroblasts and myogenesis to determine whether cells transitioned through a myoblast-like state. Gene sets were extracted from GO (Table S3), and for myogenesis included myoblast, myotube, and skeletal muscle. We found that TADs containing fibroblast or myotube genes had significant position shifts over time with p = 0.0029 or 0.0191, respectively (Figure S6D, and Methods). By contrast, the position shifts of TADs that contained myoblast genes were not statistically significant.
A direct pathway of reprogramming is further supported by the expression analysis of 3 myogenic regulatory factors: MYF5, MYOD1, and MYOG (Weintraub et al., 1991, Bentzinger et al., 2012). It is known from the hierarchy of TFs regulating progression through natural myogenic differentiation (Bentzinger et al., 2012) that MYF5 is expressed in myoblasts, whereas MYOD1 and MYOG are upregulated in myotubes. In our data, MYF5 was not activated during reprogramming, whereas MYOD1 and MYOG were expressed after the 32-hr critical transition point (Figures S1F and S6E).
Early-Stage Chromatin Remodeling and microRNA Dynamics
We additionally sought to understand the regulatory dynamics during reprogramming, including early-stage gene expression dynamics related to chromatin remodeling, super enhancer dynamics, and microRNA (miRNA) expression. Examination of early-stage RNA-seq data [-48, 16] (hr) revealed endogenous mechanisms relevant to MYOD1 transcriptional activation including muscle stage-specific markers and chromatin remodeling factors (see Figure 5A). At 16 hr, the combined upregulation of DES, MYL4, TNNT1, and TNNT2 suggests myogenic differentiation (Gard and Lazarides, 1980, Schiaffino et al., 2015). EZH2 has been associated with both “safeguarding” the transcriptional identity of skeletal muscle stem cells and terminal differentiation of myoblasts into mature muscle (Juan et al., 2011). ARID5A, a regulator of the myotube BAF47 chromatin remodeling complex, is significantly upregulated at 8 hr (p = 7.2×10−5) and may act to enhance MYOD1 binding to target promoters (Joliot et al., 2014). NR4A3, MEF2D, SIX4, SIX1, and SOX4 expression are also increased at 8 hr, all of which have important regulatory functions during differentiation in the myogenic lineage (see Figure 5B) (Ferrán et al., 2016, Bentzinger et al., 2012, Jang et al., 2015).
Figure 5.

Increased Genomic Contacts among Myogenic Regulatory Elements Set the Stage for Reprogramming
(A) Early-phase expression dynamics of genes related to muscle cell terminal differentiation and chromatin remodeling. Genes encoding proteins involved in adult muscle function, including components of the contractile apparatus (DES, MYL4, TNNT1, TNN2), and EZH2, a repressor that is involved in myogenesis.
(B) Chromatin remodeling factors and master transcription factors act cooperatively with MYOD1 to drive proliferating human fibroblasts into muscle cells. These factors include ARID5A, part of the BAF47 muscle remodeling complex that acts in cooperation with MYOD1; MEF2D, which drives differentiation of myotubes to skeletal and cardiac muscle; NR4A3 (aka NOR1) involved in differentiation of myotubes into smooth muscle; and SIX1, SIX4, and SOX4, which control the differentiation of myotubes into muscle cells.
(C) Form and function of super enhancers and associated genes over time. Average Hi-C (read per million; RPM) contact between potential super enhancer and associated gene TSS regions over time, as defined by Hnisz et al. (2013).
(D) Top upregulated SE-P genes, log2(FPKM) (blue), and SE-P Hi-C normalized contact (red; see Methods) over time.
(E) Four muscle-specific miRNAs have significantly increased expression levels in the later time points relative to the baseline control. X axis, sampling time points; y axis, log-scale differences at other time points compared with baseline (−48 hr).
We also investigated how muscle-related super enhancer-promoter (SE-P) interactions change over time throughout MYOD1-mediated reprogramming. To capture these dynamics, we extracted the Hi-C contact between skeletal muscle super enhancer regions and the associated genes' transcription start sites (±1 kb), as determined by Hnisz et al. (2013) (618 SE-P regions; Methods). We observed that for these skeletal muscle SE-P Hi-C regions, the strongest amount of contact occurred relatively early in the reprogramming process, peaking 16–24 hr post-L-MYOD1 addition to the nucleus (p = 4.17×10−9, Figure 5C; Methods). Exact SE-P contact versus function trends were variable, but a number of important myogenesis genes, such as TNNI1, MYLPF, ACTN2, and TNNT3, show strong upregulation in function over time, with an increase in SE-P contact post-MYOD1 activation. Contact versus function trends for the top 36 upregulated genes are shown in Figure 5D (Methods).
We measured the abundance of 2,588 miRNA species with reads from small RNA-seq. Using the edgeR software (Robinson et al., 2010) for data analysis, we identified 266 miRNA species that were significantly up- or downregulated in expression levels over the time course relative to the baseline control (false discovery rate [FDR] <0.05) (Table S5). Among these significant miRNAs, miR-1-3p, miR-133a-3p, and miR-206 have been previously identified as myogenic factor-regulated, muscle-specific species (McCarthy, 2011, Rao et al., 2006). We observed that the 3 miRNAs, plus miR-133b (FDR = 0.09), significantly increased in expression levels after 4-OHT treatment (Figure 5E). Their expression patterns were highly similar to that observed in mouse C2C12 cell differentiation (Rao et al., 2006). The observation of muscle-specific miRNAs, particularly miR-206, which had 1,000-fold greater expression at later time points than baseline (Figure 5E), further supports MYOD1-mediated reprogramming of fibroblasts to myotubes. Notably, the cardiac-specific species miR-1-5p, miR-208a, and miR-208b (McCarthy, 2011) were not detected in our samples.
Linking Myogenic Genes with Entrainment of Biological Rhythms
A number of studies have explored the link between MYOD1 and circadian genes ARNTL and CLOCK, revealing that ARNTL and CLOCK bind to the core enhancer of the MYOD1 promoter and subsequently induce rhythmic expression of MYOD1 (Andrews et al., 2010, Zhang et al., 2012). Here we discovered that upon MYOD1 activation, circadian genes exhibited robust synchronization in gene expression, suggesting MYOD1 feedback onto the circadian gene network. Further inspection showed that core circadian genes (Table S3) that contain E-boxes displayed the most profound synchronization initially, starting with an uptick in gene expression just after MYOD1 activation (Figures 6A–6D). Analysis using JTK_CYCLE (Hughes et al., 2010) confirmed our observation; all E-box circadian genes were found to have a synchronized period of 24 hr, with a maximum lag of 4 hr between genes, with the exception of CRY1 (Table S6).
Figure 6.

Myogenic Genes Participate in Entrainment of Biological Rhythms
(A) Gene network interactions between circadian E-box genes, derived from Ingenuity Pathway Analysis.
(B) Core circadian gene expression over time. (B1) Dexamethasone synchronization. (B2) L-MYOD1 synchronization. Target and factor correspond to genes with E-box targets and TFs that bind to E-box genes, respectively.
(C) Hi-C contacts between 26 core circadian genes over time (see Table S3). Rows and columns correspond to core circadian genes; contacts are binary (i.e., any contact between genes at a given time are shown).
(D) Network connectivity of the largest connected component (LCC; Methods) of the studied Hi-C contact maps at different time points.
(E) Normalized gene expression (FPKM, cubic spline) highlighting oscillation dampening after the bifurcation time 32 hr (red line) and the switch to differentiation medium for select core circadian genes; MYOD1 and MYOG also shown.
(F) Normalized transcripts per million (TPM) of transcription factors that are targeted by MYOG or MYOD1 (ELF3) and that only showed oscillation after the critical transition at 32 hr (red line).
(G) Conceptual diagram of biological rhythm entrainment during MYOD1-mediated reprogramming, where the red line signifies the bifurcation event.
Consistent with a critical transition point, the subset of transcripts with oscillatory behavior was different before and after the 32 hr time point. Endogenous MYOD1 and MYOG expression began close to 32 hr, and both transcripts displayed oscillatory expression. In addition, circadian transcript oscillations dampened at 40 hr, coinciding with the switch to low-serum differentiation medium (Figure 6E). To determine which newly oscillating transcripts were potential targets of MYOD1 and MYOG, we further investigated which transcripts have MYOD1 or MYOG binding motifs in their promoters using MotifMap (Daily et al., 2011), and which were synchronized in expression with MYOD1 and MYOG. Among the oscillating transcripts that fit these criteria, we found 6 TFs that were oscillatory only after the 32-hr critical transition point, have upstream MYOG binding sites, and were synchronized in expression with MYOG. Of these 6 TFs, only ELF3 was found to have binding motifs for MYOD1, as well as synchronized expression with MYOD1 (Figure 6F). Several of the 6 oscillatory TFs targeted by MYOG or MYOD1 are associated with muscle developmental and differentiation processes, including SOX15 (Meeson et al., 2007), GATA6 (Xie et al., 2015), ISL1 (Pacheco-Leyva et al., 2016), and ELF3 (Böck et al., 2014).
Robust synchronization in the expression of circadian genes that are downstream targets of MYOD1 suggests MYOD1 feedback onto circadian gene circuits. After the 32-hr critical transition point, MYOG was associated with synchronized expression of a subset of important myogenic TFs. These findings support regulatory roles for MYOD1 and MYOG in entraining circadian and cell type-specific biological rhythms (Figure 6G).
Discussion
In this study, we analyzed MYOD1-mediated reprogramming of human fibroblasts into the myogenic lineage from a dynamical network perspective. Distinct from previous studies, we generated an enriched time series dataset including Hi-C, RNA-seq, miRNA, and proteomics data. This provides a comprehensive genome-wide form-function description over time and allows us to detect early-stage cell fate commitment changes during cellular reprogramming. We found both global and local phenomena supporting a critical transition point between cell identities during reprogramming. Capturing these dynamics may help us identify genes that are key players in other reprogramming settings and develop a more universal understanding of the process and requirements for reprogramming between any two cell types.
Our data further suggest a direct pathway of reprogramming from fibroblasts to myotubes that bypasses a myoblast intermediate and is associated with the expression of MYOD1 and MYOG, but not MYF5. Related results have been described in studies on control of the cell cycle during muscle development, in which MYOD1 and MYF5 are involved in the determination of myogenic cell fate, with a switch from MYF5 to MYOG during muscle cell differentiation (Singh and Dilworth, 2013, Zeng et al., 2016). Moreover, it has been theorized (Del Vecchio et al., 2017) that a reprogrammed biosystem with positive perturbation (i.e., overexpression of one or more specific TFs like MYOD1) would bypass the intermediate state and move directly toward the terminally differentiated state. This claim is consistent with our finding, where the intermediate and terminally differentiated states correspond to myoblast and myotube stages, respectively. Understanding the intermediates of direct reprogramming will be important in the design of potential therapeutics, as their properties must be fully evaluated to understand the risk and efficiency of reprogramming, and to optimize the scalability of cell number, taking into account the proliferative capacity at different stages.
A number of studies have explored the link between MYOD1 and circadian genes ARNTL and CLOCK, revealing that ARNTL and CLOCK bind to the core enhancer of the MYOD1 promoter and subsequently induce rhythmic expression of MYOD1 (Andrews et al., 2010, Zhang et al., 2012). We found that upon activation of L-MYOD1, the population of cells exhibits robust synchronization in circadian E-box gene expression. Among these E-box targets are the PER and CRY gene family, whose protein products are known to repress CLOCK-ARNTL function, thus repressing their own transcription. In addition, E-box target NR1D1, which is synchronized upon addition of L-MYOD1, competes with ROR proteins to repress ARNTL transcription directly. This adds another gene network connection under MYOD1 influence, indirectly acting to repress ARNTL, leading us to posit that MYOD1 can affect CLOCK-ARNTL function through E-Box elements, in addition to CLOCK-ARNTL's established activation effect on MYOD1. Furthermore, these oscillations dampen just after the 32-hr critical transition point, after which MYOG entrains the oscillations of a distinct subset of myogenic TFs. Therefore, MYOD1-mediated reprogramming and circadian synchronization are mutually coupled, consistent with other systems that modulate cell fate (Umemura et al., 2014, Dierickx et al., 2018).
Our proposed biological and computational technologies shed light on the hypothesis that nuclear reorganization occurs at the time of cell specification and both precedes and facilitates activation of the transcriptional program associated with differentiation (or reprogramming), i.e., form precedes function (Rajapakse and Groudine, 2011). The alternative hypothesis is that function precedes form, that is, nuclear reorganization occurs as a consequence of differential transcription and is a consequence of, rather than a regulator of, differentiation programs (Kosak and Groudine, 2004). Our findings support that nuclear reorganization occurs before gene transcription during cellular reprogramming, i.e., form precedes function, and that dynamical nuclear reorganization plays a key role in defining cell identity. However, our data do not establish a causal relationship, and for this, additional experiments will be necessary. For example, Hi-C and RNA-seq can be supplemented using MYOD1 chromatin immunoprecipitation sequencing to identify the regions of greatest adjacency differences between cell types that correlate with transcription and/or MYOD1 binding.
As demonstrated by our study, network centrality-based analysis allows us to study Hi-C architecture from multiple views and facilitates quantitative integration with gene expression. Accordingly, the detailed connections between network architecture and network function in the context of the genome can be used to probe genomic reorganization during normal and abnormal cell differentiation. It will also be helpful to determine whether nuclear architectural remodeling can be both temporally and molecularly separated from transcriptional regulation.
Understanding the dynamical process of cellular reprogramming is critical in regenerative medicine to improve our ability to guide cells toward repair and regeneration of tissue in injury and disease. Furthermore, identifying an architectural function for TFs that is distinct from transcription would define a new molecular function with an as yet unknown role in development and disease.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank the University of Michigan Sequencing Core, and especially Jeanne Geskes, for assistance. We thank John Hogenesch for helpful discussions on circadian rhythms. We thank Daniel Burns and Stephen Lindsly for critical reading of the manuscript and helpful discussions. We extend special thanks to James Gimlett and Srikanta Kumar at Defense Advanced Research Projects Agency (DARPA) for support and encouragement. This work is supported in part by the DARPA Biochronicity, Deep-Purple, and FunCC Programs, and the Smale Institute. We also acknowledge the seminal work of Mark Groudine and late Hal Weintraub, whose ideas continue to guide our thinking.
Author Contributions
All authors including N.C.,P. B., and S.S. participated in the discussion of the results. S.L., H.C., S.R., L.A.M., and I.R. prepared the manuscript with input from all authors. N.C. and P. B. performed computational analyses and interpreted the data.
Declaration of Interests
The authors declare no competing interests.
Published: August 31, 2018
Footnotes
Supplemental Information includes Transparent Methods, eight figures, and eight tables and can be found with this article online at https://doi.org/10.1016/j.isci.2018.08.002.
Supplemental Information
References
- Andrews J.L., Zhang X., McCarthy J.J., McDearmon E.L., Hornberger T.A., Russell B., Campbell K.S., Arbogast S., Reid M.B., Walker J.R. CLOCK and BMAL1 regulate MyoD and are necessary for maintenance of skeletal muscle phenotype and function. Proc. Natl. Acad. Sci. USA. 2010;107:19090–19095. doi: 10.1073/pnas.1014523107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentzinger C.F., Wang Y.X., Rudnicki M.A. Building muscle: molecular regulation of myogenesis. Cold Spring Harb. Perspect. Biol. 2012;4:1–16. doi: 10.1101/cshperspect.a008342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böck M., Hinley J., Schmitt C., Wahlicht T., Kramer S., Southgate J. Identification of ELF3 as an early transcriptional regulator of human urothelium. Dev. Biol. 2014;386:321–330. doi: 10.1016/j.ydbio.2013.12.028. [DOI] [PubMed] [Google Scholar]
- Chen H., Chen J., Muir L.A., Ronquist S., Meixner W., Ljungman M., Ried T., Smale S., Rajapakse I. Functional organization of the human 4d nucleome. Proc. Natl. Acad. Sci. USA. 2015;112:8002–8007. doi: 10.1073/pnas.1505822112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Hero A.O., Rajapakse I. Spectral identification of topological domains. Bioinformatics. 2016;32:2151–2158. doi: 10.1093/bioinformatics/btw221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daily K., Patel V.R., Rigor P., Xie X., Baldi P. MotifMap: integrative genome-wide maps of regulatory motif sites for model species. BMC Bioinformatics. 2011;12:495. doi: 10.1186/1471-2105-12-495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Vecchio D., Abdallah H., Qian Y., Collins J.J. A blueprint for a synthetic genetic feedback controller to reprogram cell fate. Cell Syst. 2017;4:109–120.e11. doi: 10.1016/j.cels.2016.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dierickx P., Van Laake L.W., Geijsen N. Circadian clocks: from stem cells to tissue homeostasis and regeneration. EMBO Rep. 2018;19:18–28. doi: 10.15252/embr.201745130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon J.R., Jung I., Selvaraj S., Shen Y., Antosiewicz-Bourget J.E., Lee A.Y., Ye Z., Kim A., Rajagopal N., Xie W. Chromatin architecture reorganization during stem cell differentiation. Nature. 2015;518:331–336. doi: 10.1038/nature14222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon J.R., Selvaraj S., Yue F., Kim A., Li Y., Shen Y., Hu M., Liu J.S., Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrán B., Martí-Pàmies I., Alonso J., Rodríguez-Calvo R., Aguiló S., Vidal F., Rodríguez C., Martínez-González J. The nuclear receptor NOR-1 regulates the small muscle protein, X-linked (SMPX) and myotube differentiation. Sci. Rep. 2016;6:1–11. doi: 10.1038/srep25944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin J.-P., Hansen K.D. Reconstructing a/b compartments as revealed by Hi-C using long-range correlations in epigenetic data. Genome Biol. 2015;16:180. doi: 10.1186/s13059-015-0741-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gard D.L., Lazarides E. The synthesis and distribution of desmin and vimentin during myogenesis in vitro. Cell. 1980;19:263–275. doi: 10.1016/0092-8674(80)90408-0. [DOI] [PubMed] [Google Scholar]
- Hnisz D., Abraham B.J., Lee T.I., Lau A., Saint-André V., Sigova A.A., Hoke H.A., Young R.A. Super-enhancers in the control of cell identity and disease. Cell. 2013;155:934–947. doi: 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes M.E., Hogenesch J.B., Kornacker K. Jtk_cycle: an efficient nonparametric algorithm for detecting rhythmic components in genome-scale data sets. J. Biol. Rhythms. 2010;25:372–380. doi: 10.1177/0748730410379711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang S., Kim J., Kim C., An J., Johnson A., Song P., Rhee S., Choi K. KAT5-mediated SOX4 acetylation orchestrates chromatin remodeling during myoblast differentiation. Cell Death Dis. 2015;6:1–11. doi: 10.1038/cddis.2015.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joliot V., Ait-Mohamed O., Battisti V., Pontis J., Philipot O., Robin P., Ito H., Ait-Si-Ali S. The SWI/SNF subunit/tumor suppressor BAF47/INI1 is essential in cell cycle arrest upon skeletal muscle terminal differentiation. PloS One. 2014;9:1–11. doi: 10.1371/journal.pone.0108858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juan A.H., Derfoul A., Feng X., Ryall J.G., Dell’Orso S., Pasut A., Zare H., Simone J.M., Rudnicki M.A., Sartorelli V. Polycomb ezh2 controls self-renewal and safeguards the transcriptional identity of skeletal muscle stem cells. Genes Dev. 2011;25:789–794. doi: 10.1101/gad.2027911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura E., Han J.J., Li S., Fall B., Ra J., Haraguchi M., Tapscott S.J., Chamberlain J.S. Cell-lineage regulated myogenesis for dystrophin replacement: a novel therapeutic approach for treatment of muscular dystrophy. Hum. Mol. Genet. 2008;17:2507–2517. doi: 10.1093/hmg/ddn151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosak S.T., Groudine M. Form follows function: the genomic organization of cellular differentiation. Genes Dev. 2004;18:1371–1384. doi: 10.1101/gad.1209304. [DOI] [PubMed] [Google Scholar]
- Krijger P.H.L., Di Stefano B., de Wit E., Limone F., van Oevelen C., de Laat W., Graf T. Cell-of-origin-specific 3d genome structure acquired during somatic cell reprogramming. Cell Stem Cell. 2016;18:597–610. doi: 10.1016/j.stem.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E., van Berkum N.L., Williams L., Imakaev M., Ragoczy T., Telling A., Amit I., Lajoie B.R., Sabo P.J. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmann G., Margulies D.S., Horstmann A., Pleger B., Lepsien J., Goldhahn D., Schloegl H., Stumvoll M., Villringer A., Turner R. Eigenvector centrality mapping for analyzing connectivity patterns in fMRI data of the human brain. PLoS One. 2010;5:1–8. doi: 10.1371/journal.pone.0010232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy J.J. The MyomiR network in skeletal muscle plasticity. Exerc. Sport Sci. Rev. 2011;39:150. doi: 10.1097/JES.0b013e31821c01e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeson A.P., Shi X., Alexander M.S., Williams R., Allen R.E., Jiang N., Adham I.M., Goetsch S.C., Hammer R.E., Garry D.J. Sox15 and fhl3 transcriptionally coactivate Foxk1 and regulate myogenic progenitor cells. EMBO J. 2007;26:1902–1912. doi: 10.1038/sj.emboj.7601635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman M. Oxford University Press; 2010. Networks: An Introduction. [Google Scholar]
- Pacheco-Leyva I., Matias A.C., Oliveira D.V., Santos J.M., Nascimento R., Guerreiro E., Michell A.C., van De Vrugt A.M., Machado-Oliveira G., Ferreira G. CITED2 cooperates with isl1 and promotes cardiac differentiation of mouse embryonic stem cells. Stem Cell Rep. 2016;7:1037–1049. doi: 10.1016/j.stemcr.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajapakse I., Groudine M. On emerging nuclear order. J. Cell Biol. 2011;192:711–721. doi: 10.1083/jcb.201010129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao P.K., Kumar R.M., Farkhondeh M., Baskerville S., Lodish H.F. Myogenic factors that regulate expression of muscle-specific microRNAs. Proc. Natl. Acad. Sci. USA. 2006;103:8721–8726. doi: 10.1073/pnas.0602831103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M.D., McCarthy D.J., Smyth G.K. edger: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiaffino S., Rossi A.C., Smerdu V., Leinwand L.A., Reggiani C. Developmental myosins: expression patterns and functional significance. Skelet. Muscle. 2015;5:22. doi: 10.1186/s13395-015-0046-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh K., Dilworth F.J. Differential modulation of cell cycle progression distinguishes members of the myogenic regulatory factor family of transcription factors. FEBS J. 2013;280:3991–4003. doi: 10.1111/febs.12188. [DOI] [PubMed] [Google Scholar]
- Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K., Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Umemura Y., Koike N., Matsumoto T., Yoo S.-H., Chen Z., Yasuhara N., Takahashi J.S., Yagita K. Transcriptional program of Kpna2/importin-α2 regulates cellular differentiation-coupled circadian clock development in mammalian cells. Proc. Natl. Acad. Sci. USA. 2014;111:5039–5048. doi: 10.1073/pnas.1419272111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weintraub H. The MyoD family and myogenesis: redundancy, networks, and thresholds. Cell. 1993;75:1241–1244. doi: 10.1016/0092-8674(93)90610-3. [DOI] [PubMed] [Google Scholar]
- Weintraub H., Davis R., Tapscott S., Thayer M., Krause M., Benezra R., Blackwell T.K., Turner D., Rupp R., Hollenberg S. The myoD gene family: nodal point during specification of the muscle cell lineage. Science. 1991;251:761–766. doi: 10.1126/science.1846704. [DOI] [PubMed] [Google Scholar]
- Weintraub H., Tapscott S.J., Davis R.L., Thayer M.J., Adam M.A., Lassar A.B., Miller A.D. Activation of muscle-specific genes in pigment, nerve, fat, liver, and fibroblast cell lines by forced expression of MyoD. Proc. Natl. Acad. Sci. USA. 1989;86:5434–5438. doi: 10.1073/pnas.86.14.5434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y., Jin Y., Merenick B.L., Ding M., Fetalvero K.M., Wagner R.J., Mai A., Gleim S., Tucker D., Birnbaum M.J. Phosphorylation of gata-6 is required for vascular smooth muscle cell differentiation after mTORC1 inhibition. Sci. Signal. 2015;8:1–27. doi: 10.1126/scisignal.2005482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng W., Jiang S., Kong X., El-Ali N., Ball A.R., Christopher I., Ma H., Hashimoto N., Yokomori K., Mortazavi A. Single-nucleus RNA-seq of differentiating human myoblasts reveals the extent of fate heterogeneity. Nucleic Acids Res. 2016;44:e158. doi: 10.1093/nar/gkw739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Patel S.P., McCarthy J.J., Rabchevsky A.G., Goldhamer D.J., Esser K.A. A non-canonical e-box within the MyoD core enhancer is necessary for circadian expression in skeletal muscle. Nucleic Acids Res. 2012;40:3419–3430. doi: 10.1093/nar/gkr1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.