

Abstract
Myogenesis is a highly regulated process that involves the conversion of progenitor cells into multinucleated myofibers. Besides proteins and miRNAs, long noncoding RNAs (lncRNAs) have been shown to participate in myogenic regulatory circuitries. Here, we characterize a murine chromatin‐associated muscle‐specific lncRNA, Charme, which contributes to the robustness of the myogenic program in vitro and in vivo. In myocytes, Charme depletion triggers the disassembly of a specific chromosomal domain and the downregulation of myogenic genes contained therein. Notably, several Charme‐sensitive genes are associated with human cardiomyopathies and Charme depletion in mice results in a peculiar cardiac remodeling phenotype with changes in size, structure, and shape of the heart. Moreover, the existence of an orthologous transcript in human, regulating the same subset of target genes, suggests an important and evolutionarily conserved function for Charme. Altogether, these data describe a new example of a chromatin‐associated lncRNA regulating the robustness of skeletal and cardiac myogenesis.
Keywords: CRISPR/Cas9, epigenetic control, heart development, lncRNAs, myogenesis
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; RNA Biology; Vascular Biology & Angiogenesis
Introduction
Myogenesis is a highly regulated multistep process, in which the conversion of progenitor cells into multinucleated and functional myofibers can be easily reproduced in vitro. Besides the well‐characterized protein factors and miRNAs (Buckingham & Rigby, 2014), also long noncoding RNAs (lncRNAs) have been shown to participate in myogenic regulatory circuitries (Rinn & Chang, 2012; Fatica & Bozzoni, 2014; Ballarino et al, 2015). lncRNAs, through their ability to interact with nucleic acids as well as with proteins (Guttman & Rinn, 2012; Rinn & Chang, 2012; Batista & Chang, 2013; Cipriano & Ballarino, 2018), act as scaffolds for the formation of specific functional complexes where different proteins and RNA and even DNA molecules can be tethered together (Wutz et al, 2002; Clemson et al, 2009; Tsai et al, 2010; Ariel et al, 2014; Hacisuleyman et al, 2014; Ribeiro et al, 2017). Nuclear lncRNAs have been described to control the transcriptional myogenic program through diverse modes of action (Ballarino et al, 2016): as enhancer‐associated transcripts (Mousavi et al, 2014; Mueller et al, 2015; Ounzain et al, 2015), as guides for the recruitment of PRCs and MLL epigenetic regulators or DNA methyltransferases (DNMT; Wang et al, 2015, 2016), and also as allosteric inhibitors (Han et al, 2014; Wang et al, 2016). Important functions have also been ascribed to cytoplasmic lncRNAs through their ability to control mRNA stability and translation (Wang et al, 2013), to modulate miRNA function acting as competing endogenous RNA (Cesana et al, 2011; Salmena et al, 2011; Han et al, 2015), or to encode for micropeptides (Anderson et al, 2015; Nelson et al, 2016). Interestingly, de‐regulation of these molecules was often associated with skeletal (Ballarino et al, 2015) and cardiac (Han et al, 2014; Ounzain & Pedrazzini, 2015; Uchida & Dimmeler, 2015; Wang et al, 2016) diseases.
We recently identified a novel nuclear lncRNA, Charme (Chromatin architect of muscle expression), conserved in human and with restricted expression in skeletal and cardiac muscles (Ballarino et al, 2015). Here, we show that the depletion of Charme produces the downregulation of myogenic genes, many of which involved in human cardiomyopathies (Becker et al, 2011; Harvey & Leinwand, 2011). Through a combination of ChIRP and FISH approaches, we identified a Charme/chromatin interaction that is required for the correct expression of a subset of relevant myogenic genes. Notably, the functional knockout of Charme in mice produced morphological alterations of both skeletal and cardiac muscles and resulted in a shorter life span. The structural and functional conservation of Charme in human further corroborates its relevance in the control of proper muscle differentiation and homeostasis.
Results
Charme is a chromatin‐associated long noncoding RNA
Previous transcriptome analysis from murine C2C12 myoblasts and myotubes discovered novel lncRNAs with muscle restricted expression (Ballarino et al, 2015). Among them, lnc‐405, here named Charme (Chromatin architect of muscle expression; Figs 1A and EV1A), is a novel isoform of the 5430431A17Rik transcript of particular interest for (i) its specific expression in both skeletal and cardiac muscles, (ii) its predominant nuclear localization, and (iii) the presence of an orthologue transcript in human.
Figure 1. Charme is a novel long noncoding transcript associated with myogenesis.
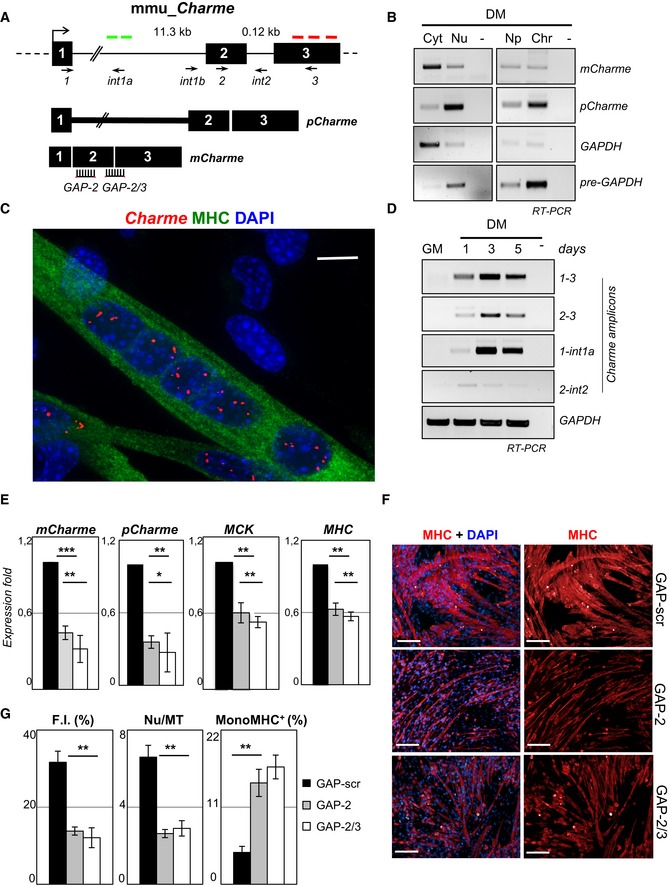
- Genomic structure of the Charme locus. The position of PCR primers, LNA GAPmers (GAP‐2, GAP‐2/3), and in situ probes (green—intronic; or red—exonic) used in this study are shown together with the produced Charme isoforms.
- Semiquantitative RT–PCR (sqRT–PCR) quantification of pCharme and mCharme in cytoplasmic (Cyt), nuclear (Nu), nucleoplasmic (Np), or chromatin (Chr) fractions from 2‐day differentiated myotubes. The quality of fractionation was tested with mature (GAPDH) and precursor (pre‐GAPDH) RNAs.
- Co‐staining of MHC protein (green) and Charme RNA (red) in fully differentiated myotubes. DAPI, 4′,6‐diamidino‐2‐phenylindole. MHC, myosin heavy chain. Scale bar = 10 μm.
- sqRT–PCR quantification of Charme amplicons in growth (GM) and differentiated (DM) conditions. GAPDH mRNA serves as control. −, RT‐minus control.
- Real‐time RT–PCR (qRT–PCR) quantification of mCharme and pCharme, MCK, and MHC in differentiated myotubes treated with GAP‐2, GAP‐2/3, or GAP‐scr as negative control. Data were normalized to GAPDH mRNA and represent mean ± SD of triplicates.
- Immunofluorescence staining for MHC on C2C12 cells treated with GAP‐scr, GAP‐2, or GAP‐2/3 (right) and merged with the DAPI staining (left). Scale bar = 100 μm.
- Quantification of myotubes formation (F.I., Nu/MT, MonoMHC+) on cells treated with GAP‐2, GAP‐2/3, or GAP‐scr. Bars represent mean ± SD of triplicates of randomly chosen microscope fields.
Figure EV1. Genomic features and expression analysis of the murine Charme locus.
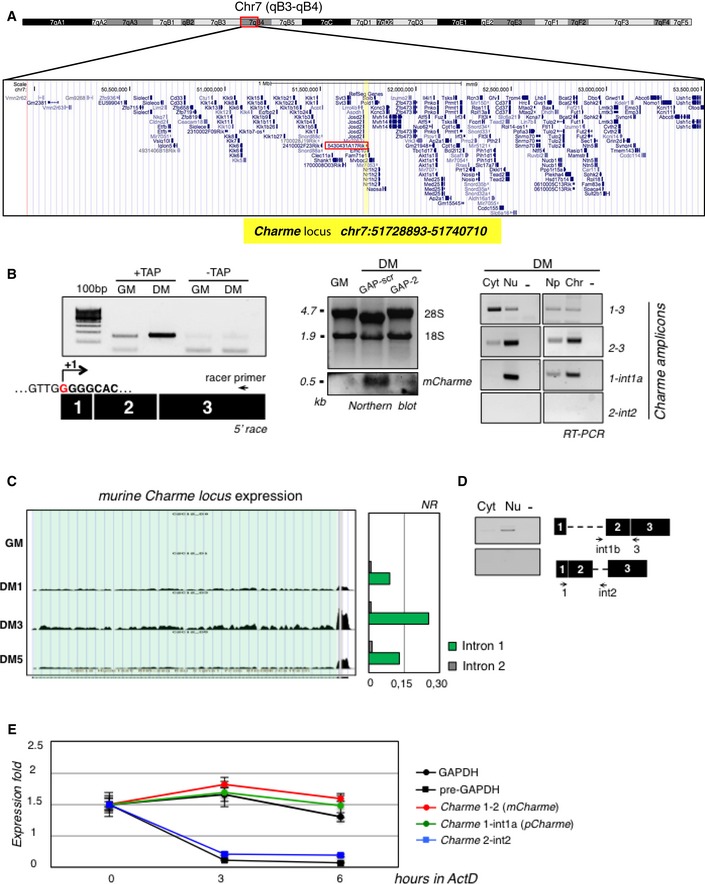
- UCSC visualization showing the chromosome position and the genomic coordinates of Charme (highlighted in yellow) in the mm9 mouse genome. Red box indicates the genomic position of the previously annotated 5430431A17Rik transcript.
- Left: 5′‐RACE analyses of Charme in proliferating (GM) and differentiating (DM) conditions. The position of the Racer primer is indicated. RNA treated to the same experimental regime, but with tobacco acid pyrophosphatase digestion omitted (−TAP), did not generate a product. The PCR products were sequenced, and the identified Charme transcription start site is reported (+1). Middle: Northern blot analysis of Charme on total RNA from proliferating (GM), differentiating (DM, GAP‐scr), and Charme‐depleted (DM, GAP‐2) myotubes. The reduced level of the signal upon Charme depletion confirms the specificity of the observed band. 18S rRNA and 28S rRNA serve as a loading control. Right: RT–PCR quantification of Charme in cytoplasmic (Cyt), nuclear (Nu), nucleoplasmic (Np), or chromatin (Chr) fractions from differentiated myotubes. The quality of fractionation is shown in Fig 1B. Three biological replicates were analyzed, and a representative experiment is shown. −, RT‐minus control.
- RNA‐seq coverage visualization of Charme locus during the time‐course of C2C12 differentiation. A quantification of the intronic reads is represented in the histogram aside. Normalized reads (NR) were obtained by dividing the total number of intronic reads for their respective lengths.
- RT–PCR validation of Charme intron 1 retention performed on RNA from cytoplasmic or nuclear samples.
- Assessment of Charme isoform stability. RNA half‐lives were calculated upon 0, 3, and 6 h of actinomycin D (ActD) treatment; GAPDH and pre‐GAPDH serve as controls. Data represent the means ± SD from three independent experiments.
Source data are available online for this figure.
The expression of Charme was studied by 5′‐RACE, Northern blot, and RT–PCR analyses (Figs 1B and EV1B): It is predominantly localized in the nucleus, mainly associated with the chromatin, with a minor fraction being present in the cytoplasm. In C2C12 cells, RNA FISH showed a well‐defined punctate localization of Charme in the nuclei of myofibers defined as multinucleated cells expressing the myogenic marker myosin heavy chain (MHC). Charme was instead absent in proliferating myoblasts, identified as mononucleated cells lacking MHC staining (Fig 1C). Charme expression starts at day 1 upon shift to differentiation medium (DM), reaching high levels at day 3 (Fig 1D). In agreement with RNA‐seq data, RT–PCR analyses revealed the existence of an isoform (pCharme) still retaining intron 1 and devoid of intron 2 (Figs 1A and D, and EV1C and D). Notably, actinomycin experiments indicated that this isoform was as stable as the fully spliced Charme species (mCharme; Fig EV1E). RNA FISH with intron probes detected an average of three nuclear spots which co‐localize with the exonic signals (Appendix Fig S1A and B). This pattern, whose specificity was tested by both RNaseA and RNAi sensitivity (Appendix Fig S1C), well correlates with the aneuploidy of C2C12 cells having three copies of chromosome 7, where Charme locus resides. The number, the intensity, and the focal plane distribution of the signals obtained with exonic vs. intronic probes allowed us to establish that the major fraction of Charme is retained at the sites of its own transcription as a precursor species.
Charme depletion affects myogenesis
To analyze the role of Charme in myogenesis, we treated C2C12 myotubes with two different LNA GAPmers (GAP‐2 and GAP‐2/3; Fig 1A), which provided 60–70% average reduction in both intron 1‐harboring (pCharme) and mature (mCharme) Charme transcripts (Fig 1E). Upon knockdown, 50% decrease of the mRNAs for two diagnostic markers of differentiation, myosin creatine kinase (MCK) and the myosin heavy chain (MHC), was found (Fig 1E and Table EV1) indicating quite a clear impairment of myogenesis. Morphological parameters were also specifically affected: a consistent reduction in the caliber of differentiated MHC‐positive myotubes (Fig 1F) was observed, together with a decrease of more than twofold of the fusion index (F.I.) and of the number of nuclei per fiber (Nu/MT). In parallel, the number of mononucleated (MonoMHC+) cells increased by a factor of 3 (Fig 1G). These results suggest that Charme inhibition mainly affects the ability of myogenic cells to fuse and to proceed to later stages of differentiation.
RNA‐seq analysis on cells treated with GAP‐2 or GAP‐2/3, in parallel with a scramble control (Appendix Fig S2A and B) led to the identification of a set of 826 genes differentially expressed upon Charme depletion with two different sets of LNA GAPmers (Fig 2A and Appendix Fig S2C). Of these, 302 were upregulated and 524 downregulated in comparison with the scramble condition (Fig 2A and Table EV1). Real‐time quantitative PCR on a selected number of targets confirmed the sequencing data (Appendix Fig S2D). The Gene Ontology (GO) term enrichment study performed with the FIDEA web tool (D'Andrea et al, 2013) suggested that Charme may act by activating genes involved in muscle function and contraction processes (Fig 2B). To note, we did not observe significant enrichment of any cell‐cycle‐ and/or proliferation‐related categories.
Figure 2. Charme regulates myogenesis by interacting with the nctc region.
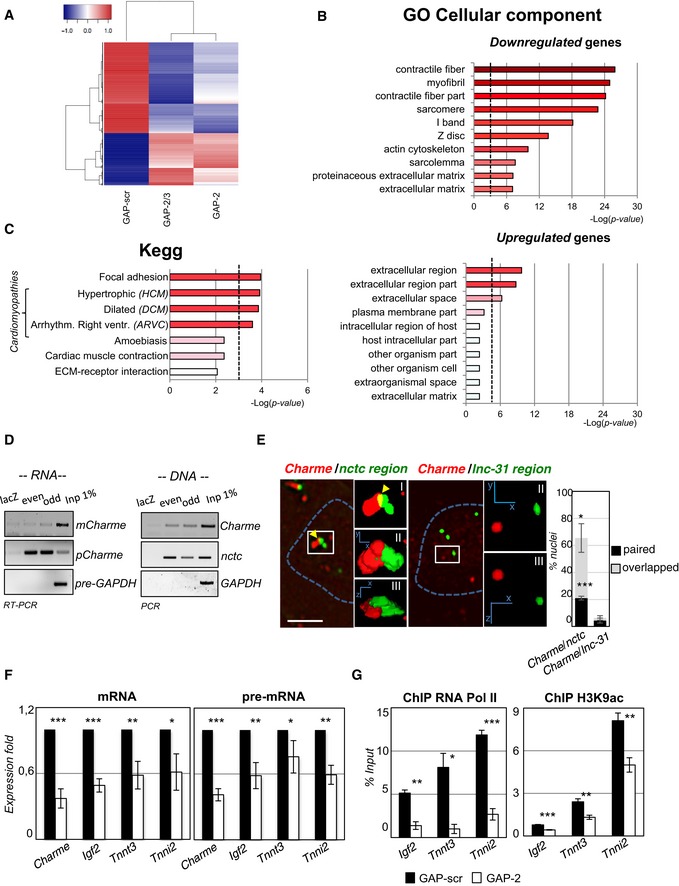
- RNA‐seq analysis of transcriptome changes upon Charme depletion. Heatmap was drawn using the heatmap3 R package and represents hierarchical clustering of the common genes differentially expressed upon Charme depletion with both GAP‐2 and GAP‐2/3 GAPmers, compared to GAP‐scr control. The expression levels correspond to mean‐centered log2‐transformed FPKM values. See also Table EV1.
- Gene Ontology (GO) term enrichment analyses performed by FIDEA (D'Andrea et al, 2013) on genes downregulated (top) or upregulated (bottom) upon Charme depletion. Bars indicate the top 10 categories of cellular components in decreasing order of significance. Threshold (P‐value < 0.01) is indicated by the dashed black line.
- KEGG pathway enrichment analysis on genes downregulated by Charme depletion.
- Quantification of recovered RNA (left) and DNA (right) upon Charme ChIRP with the pool of biotinylated probes reported in Table EV4.
- RNA/DNA FISH for Charme RNA and nctc (left) or lnc‐31 (right) loci in 2‐day differentiated myotubes. Inserts show a magnification of the spots converted into binary images (I), 3D rendering (II), or rotated on Z‐axis (III). Yellow arrows indicate signals overlapping. Right histogram reports the mean ± SD percentage of nuclei with paired or overlapped spots from two biological replicates. Scale bar = 5 μm.
- qRT–PCR quantification of Charme and Charme target genes in GAP‐scr‐ vs. GAP‐2‐transfected (2‐day differentiated) myotubes. PCR data were normalized to GAPDH and represent mean ± SD of triplicates.
- RNA Pol II (left) and H3K9ac (right) ChIP performed in GAP‐scr‐ vs. GAP‐2‐transfected myotubes. The recovered chromatin was analyzed by qPCR in parallel with an intergenic region used to normalize the two (GAP‐scr vs. GAP‐2) conditions. Data were subtracted for background and are expressed as input percentage (% Input). Histograms represent the mean ± SEM of three biological replicates.
Interestingly, Charme depletion resulted in the downregulation of genes (i.e., Myh7 and Tnnt2; Appendix Fig S2E) implicated in several types of cardiomyopathies (Fig 2C), such as familial hypertrophic (HCM), dilated (DCM), and arrhythmogenic right ventricular (ARVC) cardiomyopathies (Harvey & Leinwand, 2011; Becker et al, 2011; Maron et al, 2012).
pCharme is localized at specific chromosomal sites
To elucidate Charme mode of action, we proceeded with the identification of the sites of chromatin interaction in C2C12 cells. Chromatin isolation by RNA purification (ChIRP) analysis was carried out with two different pools of four alternating antisense biotinylated oligonucleotides (even and odd), together with four oligonucleotides against lacZ sequences (Appendix Fig S2F). Figure 2D (panel RNA) shows that a specific pull‐down of pCharme isoform was obtained when compared to the lacZ control and the GAPDH pre‐mRNA, while no enrichment was obtained for mCharme isoform. The co‐purified DNA was subjected to high‐throughput sequencing (ChIRP‐seq), and the reads from odd and even pull‐downs were compared to those of the lacZ control (Table EV2). In agreement with the strong chromatin association, the major peak corresponded to the Charme locus. Several other genomic loci were identified (Table EV2); among them, the most prominent corresponded to a region (hereafter named as nctc) located on the same Charme chromosome (chr7:149,746,850–149,747,033) and containing several genes affected by Charme depletion (Appendix Fig S2G and Table EV1). The nctc peak was validated by PCR amplification on the initial ChIRP samples (Fig 2D, panel DNA) and reproduced in independent replicates. Interestingly, this region contains a shared core muscle enhancer (CME; Alzhanov et al, 2010) required for muscle‐specific activation of Igf2 and H19 (Eun et al, 2013), which are both decreased upon Charme knockdown (Table EV1). Interestingly, the knockout of part of this enhancer was previously shown to produce the loss of Igf2 expression and defects in muscle differentiation (Kaffer et al, 2001). Therefore, for subsequent analysis we concentrated on this specific locus.
The physical association of pCharme RNA with nctc (98 Mb far from Charme locus) was validated by FISH analysis with probes against Charme RNA (red) and BAC probes against the target loci (green). A detailed quantification of the signals indicated that in more than 60% of the nuclei Charme is in close proximity with the nctc region, with 2/3 of the signals showing partial overlapping (Fig 2E). No proximity was instead found when the unrelated lnc‐31 locus (Ballarino et al, 2015), which was absent in the ChIRP‐seq dataset, was used as negative control (Fig 2E).
Notably, the surrounding 2.5 Mb of the ChIRP peak in the nctc region contains relevant myogenic genes, such as Igf2, Tnnt3, and Tnni2, that were downregulated upon Charme depletion in the RNA‐seq dataset ([Link], [Link]). qRT–PCR confirmed their downregulation upon Charme depletion both at the mRNA and pre‐mRNA levels (Fig 2F). Along this line, ChIP analyses performed in conditions of Charme downregulation showed a strong decrease in the association of the RNA polymerase II (RNA Pol II) and in the acetylation of lysine K9 histone 3 (H3K9ac) of these genes (Fig 2G). Altogether, these data indicate that the effect of Charme on these target genes is exerted at the transcriptional level.
Due to the strong accumulation of pCharme at the sites of its own transcription, we tested whether the Charme genomic locus itself could be in contact with nctc. To this purpose, FISH analyses were performed with DNA BAC probes flanking the Charme gene (red) or overlapping the nctc region (green; Figs 3 and EV2A–C). In GM conditions, when Charme is not expressed, the two districts are clearly distinguished as independent spots (Fig 3A, left panel, and Fig EV2C, GM). Notably, upon Charme expression, the two regions get close (Fig EV2C, DM1 and DM1.5, and Fig EV2D) with a consistent percentage of chromosomes showing overlapping or proximal signals at days 1 and 1.5 of differentiation (Fig 3A, right panels, and Fig EV2C). As control, no proximity was found with the unrelated lnc‐31 locus (Figs 3A and EV2C, lower panels). Notably, the pCharme‐mediated long‐range interaction was significantly reduced when Charme was downregulated by 50% through GAP‐2 treatment (Figs 3B and EV2E). Overall, these in vitro data suggest an important role of pCharme in a long‐range physical interaction between two chromatin districts distantly located, but present on the same chromosome.
Figure 3. pCharme contributes to the maintenance of chromatin contacts with the nctc region.
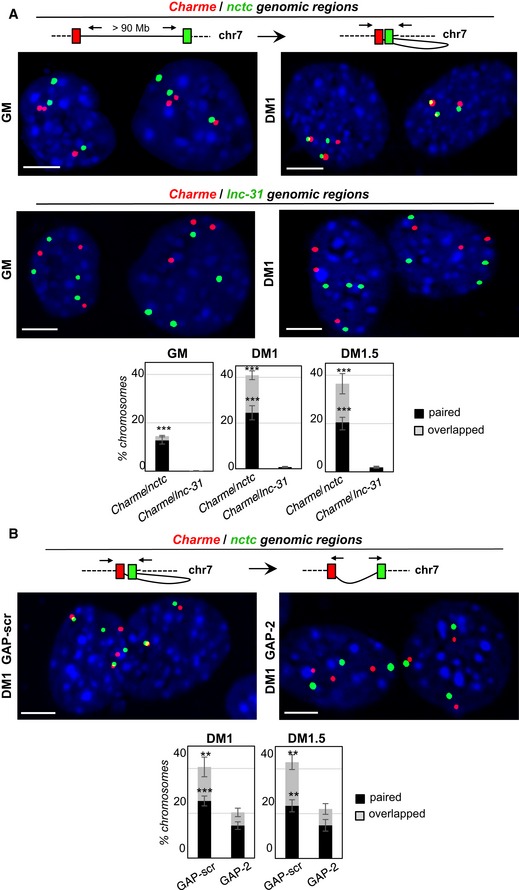
- Double DNA FISH for Charme and nctc (top) or Charme and lnc‐31 (bottom) loci in growth (GM) and differentiated (DM1) conditions. Histograms represent the percentages ± SD of chromosome 7 showing paired and overlapped signals in GM, DM1, and DM1.5 days of differentiation from two biological replicates.
- DNA/DNA FISH for Charme and nctc loci in GAP‐scr‐ vs. GAP‐2‐transfected myotubes (DM1). Percentage of Charme locus ± SD from three biological replicates showing paired and overlapped signals with nctc is indicated in the histograms below.
Figure EV2. Chromatin contacts and functional characterization of the murine Charme transcript.
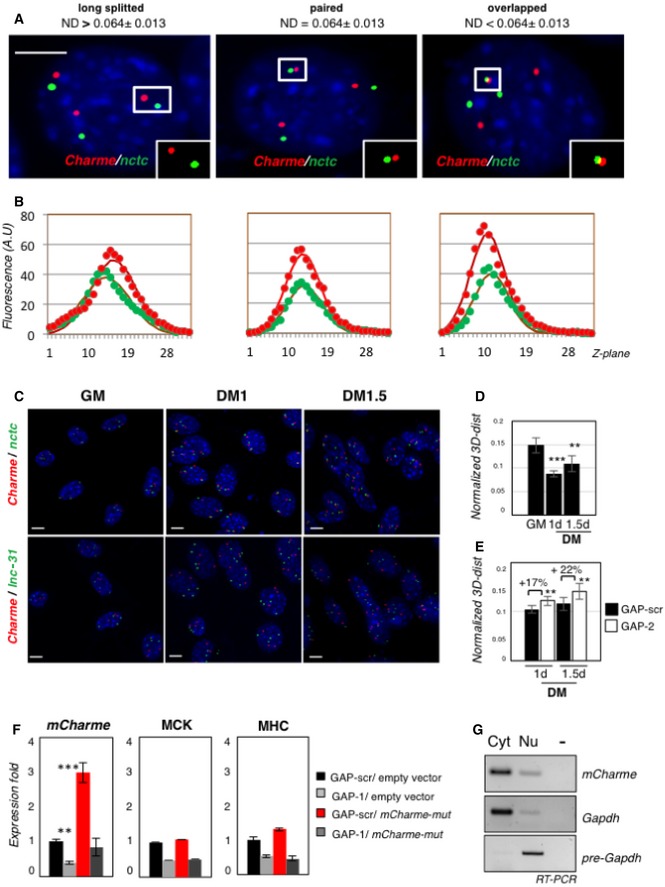
- Examples of proximal Charme/nctc DNA FISH spots on the same Z‐section. Red and green signals correspond to Charme or nctc chromatin regions, respectively. White boxes show binarized images of single focal planes containing long‐splitted, paired, or overlapped patterns. On top of the panels, the 3D interallelic distance interval (ND) for each category is shown. ND values were obtained by normalization to the diameter of the nuclei. The number of green/red DNA spots per nucleus (n = 3) reflects the C2C12 aneuploidy with three copies of chromosome 7. Scale bar = 5 μm.
- Plotting of Z‐stack intensity distribution for each spot in the insets of (A). Gaussian fit curve (full line) for the data of fluorescence intensity (filled circles) in each channel is plotted along Z‐planes (Z‐step = 200 nm). The strong correspondence of green and red channel distribution indicates near co‐localization and co‐planarity of the signals.
- Full field view of the DNA FISH studies on the nuclear distribution of Charme and nctc (top) or Charme and lnc‐31 (bottom) loci. Scale bar = 5 μm.
- Normalized 3D distances between Charme and nctc loci at the indicated time points (GM, DM 1–1.5 days). Interallelic distances were normalized to nuclei diameter.
- Normalized 3D distances between Charme and nctc loci in GAP‐scr or GAP‐2 treated cells at the indicated differentiation times (DM 1–1.5 days). Interallelic distances were normalized to nuclei diameter. Mean ± SD ND values are shown. **P < 0.01, ***P < 0.001, unpaired Student's t‐test.
- Quantitative real‐time RT–PCR analyses of mCharme, MHC, and MCK mRNAs in C2C12 cells transfected with GAP‐scr or GAP‐1 in combination with a mCharme mutant (mCharme‐mut) in the GAP‐1 targeting site. Transfections with the empty vector were used as negative control. PCR data were normalized to GAPDH mRNA.
- sqRT–PCR quantification of mCharme in cytoplasmic (Cyt) and nuclear (Nu) fractions from C2C12 cells transfected with mCharme‐mut. The quality of fractionation was tested with mature (GAPDH) and precursor (pre‐GAPDH) RNAs. −, RT‐minus control.
Source data are available online for this figure.
A further evidence for Charme acting in the nucleus at the sites of its own transcription derives from experiments where we tried to rescue the phenotype of Charme depletion with a cDNA construct expressing a GAPmer‐resistant mCharme. This construct failed to recover the expression of myogenic markers (Fig EV2F) and produced a transcript with a cytoplasmic‐restricted localization (Fig EV2G). The lack of rescue with cytoplasmic RNA further supports the finding that the active Charme species is the nuclear one; moreover, these results are in line with the accepted notion that for chromatin‐associated lncRNAs acting in cis, rescue phenotypes are not obtained with exogenous gene overexpression (Goff & Rinn, 2015; Wang et al, 2015).
Charme functional knockout in mice affects the myogenic process
In consideration of the interesting function of Charme observed in vitro, the study of its function in vivo was strongly demanded. Therefore, the CRISPR/Cas9 methodology was used to generate a Charme null allele in mice. In order to avoid any structural alteration of the locus, we inserted at the beginning of exon 2 the strong poly(A) polyadenylation signal of the β‐globin gene (Levitt et al, 1989) followed by two MAZ sites, known to promote efficient cleavage and transcriptional termination (Yonaha & Proudfoot, 2000; Gromak et al, 2006; Fig 4A). The correct genomic editing was confirmed in a founder mouse, and the Charme null allele was backcrossed for four generations to obtain homozygous animals (Fig EV3A). Charme −/− mice are viable, fertile, and produced in Mendelian ratios. Analysis of skeletal muscles of 1‐month‐old mutant and control animals indicated that the insertion of the poly(A)/MAZ cassette resulted in a very effective depletion of both pCharme and mCharme isoforms (Fig 4B). The histological analysis of skeletal muscles from Charme −/− mice revealed a slight but significant reduction in the area of the fiber sections (Fig 4C), which is the most common parameter used to assess correct tissue morphology. Moreover, as a further evidence for delayed differentiation, a significant decrease in the mRNAs of myogenic markers myosin creatine kinase (MCK) and myosin heavy chain (MHC) was observed (Fig 4D). These features appeared consistent with the reduction in the fiber caliber of differentiating myotubes and the decrease in myoblast fusion observed in vitro in C2C12 cells (Fig 1G). Moreover, the Charme targets located in the nctc region identified in C2C12 cells were also specifically affected in murine muscle tissues (Fig 4E).
Figure 4. Generation and skeletal muscle characterization of Charme −/− mice.
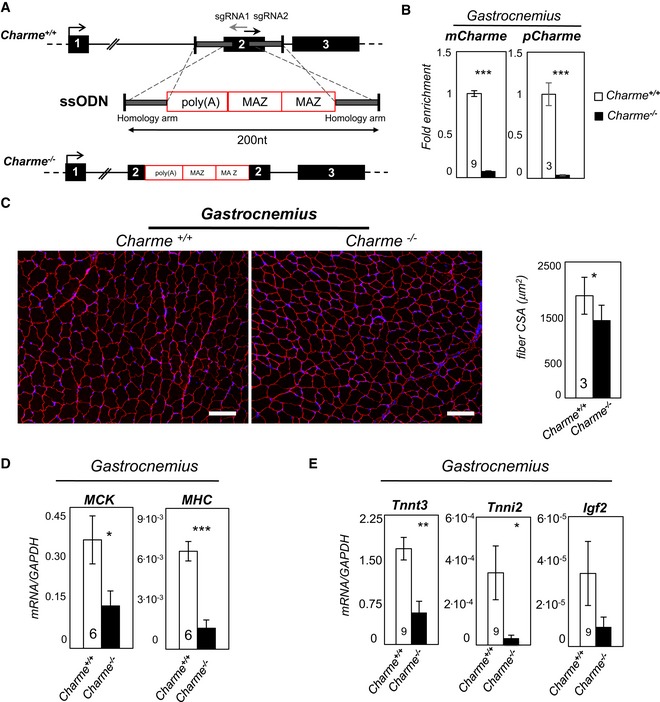
- Top: Charme locus is shown together with the position of sgRNA1 and sgRNA2 divergent single guides. Bottom: design of the single‐stranded oligodeoxynucleotide (ssODN) containing the two homology arms (gray line) and the 100‐nt‐long poly(A)/2×MAZ cassette.
- qRT–PCR quantification of mCharme and pCharme expression in skeletal homogenates from Charme +/+ and Charme −/− mice. The number of mice tested is the same for the two groups, and it is indicated in the white bars. Data were normalized to GAPDH mRNA and represent mean ± SEM.
- Dystrophin staining analyzed by immunofluorescence on Charme +/+ and Charme −/− skeletal (gastrocnemius) biopsies. The mean ± SD of cross‐sectional area (μm2) is reported in the histograms besides. In total, N = 667 and 1,039 fibers per condition were analyzed. Original magnification = ×10. The number of mice tested is the same for the two groups, and it is indicated in the white bars. Scale bar = 100 μm.
- qRT–PCR quantification of MHC and MCK transcripts in skeletal (gastrocnemius) homogenates from Charme +/+ and Charme −/− mice. The number of mice tested is the same for the two groups, and it is indicated in the white bars. Data were normalized to GAPDH mRNA and represent mean ± SEM.
- qRT–PCR quantification of Tnnt3, Tnni2, and Igf2 transcripts in skeletal (gastrocnemius) homogenates from Charme +/+ and Charme −/− mice. Data were normalized to GAPDH mRNA and represent mean ± SEM. The number of mice tested is the same for the two groups, and it is indicated in the white bars.
Figure EV3. Generation and functional characterization of Charme −/− mice.
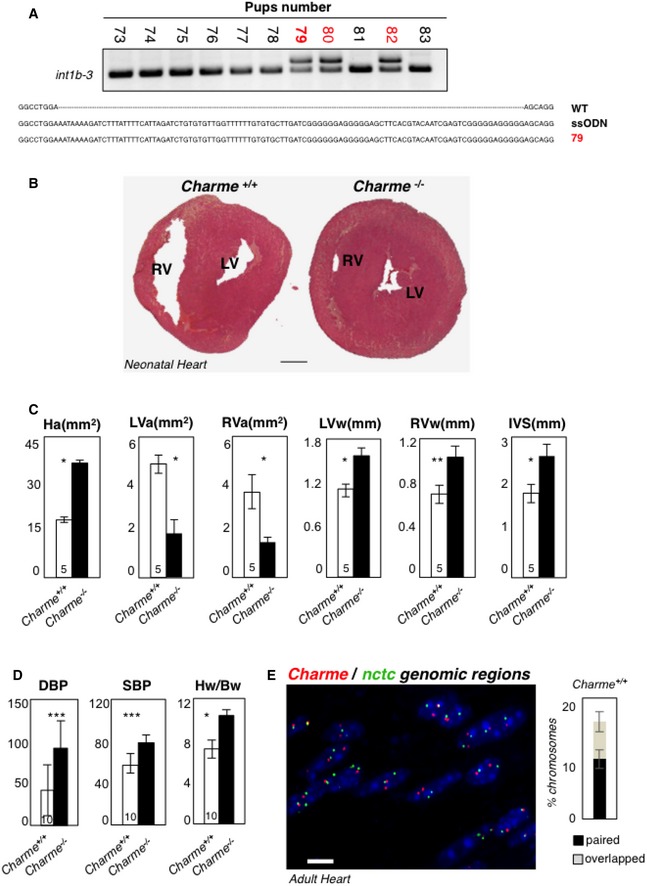
- DNA extracted from F0‐generation mice was analyzed by PCR amplification for genotyping. The position of PCR primers is indicated in Fig 1A. Electrophoretic analyses of PCR products and multiple sequence alignment (performed by Muscle‐3.8) indicated the insertion of the entire poly(A)/2×MAZ cassette on a single allele in the #79, #80, and #82 pups (red). The #79 was selected as founder for successive breeding.
- Hematoxylin and eosin staining of transverse sections of the myocardium in 2‐day‐old Charme +/+ (left) and Charme −/− (right) mice. Left (LV) and right (RV) ventricles are indicated. Scale bar = 0.5 mm.
- Morphometric analyses of heart area (Ha), left and right ventricle areas (LVa and RVa), left and right ventricle walls (LVw and RVw), and interventricular septum (IVS) in Charme +/+ and Charme −/− mice.
- Systolic (SBP) and diastolic (DBP) blood pressures and heart/body weight ratio (Hw/Bw) in Charme +/+ and Charme −/− mice. Values represent mean ± SEM of replicates. The numbers of mice tested for each group are indicated in the white bars. Mean ± SD ND values are shown. **P < 0.01, ***P < 0.001, unpaired Student's t‐test.
- DNA/DNA FISH in adult cardiac tissues. Charme +/+ adult cardiac tissues exhibit spatial proximity of Charme and nctc genomic regions. The percentages of chromosome 7 showing paired and overlapped signals are 12% and 7%, respectively, in a total of 412 nuclei analyzed. Scale bar = 10 μm.
Source data are available online for this figure.
Charme −/− mice exhibit an altered cardiac phenotype
Since Charme also displays a cardiac restricted expression (Ballarino et al, 2015), we analyzed the effects of Charme knockout in heart. A peculiar phenotype was detected when analyzing the heart of Charme −/− mice where the expression of both pCharme and mCharme isoforms was almost abolished by the poly(A) site insertion (Fig 5A). Morphometric analyses, performed with hematoxylin/eosin staining in both adult (Fig 5B) and neonatal (Fig EV3B) cardiac sections, showed in all the mice tested an increase in the heart area (Ha), a definite decrease in ventricular chamber dimensions (LVa and RVa), and an increase in the thickness of the ventricular free walls (LVw and RVw) and interventricular septum (IVS; Fig EV3C). Moreover, as expected from this altered morphology, higher blood pressures (DBP and SBP) and heart‐to‐body weight ratio (Hw/Bw) were also detected in Charme −/− animals (Fig EV3D). Notably, compared to wild‐type littermates, which have a life expectancy of 24–38 months, none of the available Charme −/− mice (n = 5, P < 0.001) survived for more than 1 year, suggesting a midlife lethality caused by Charme ablation. In analogy to skeletal muscles, the histological analysis performed on Charme −/− cardiac tissues revealed a reduction in cell area (Fig 5C) together with the downregulation of terminal differentiation markers (Fig 5D). To note, the expression of Igf2, the only gene of the nctc region expressed in the cardiac tissue, was also found to be downregulated. Notably, in line with the in vitro data, DNA/DNA FISH experiments performed in neonatal hearts proved that the co‐localization between Charme and the nctc regions is also occurring in vivo while it is impaired in Charme −/− animals (Fig 5E). Interestingly, the proximity was also found to persist in adult hearts (Fig EV3E).
Figure 5. Charme −/− mice exhibit an altered cardiac phenotype.
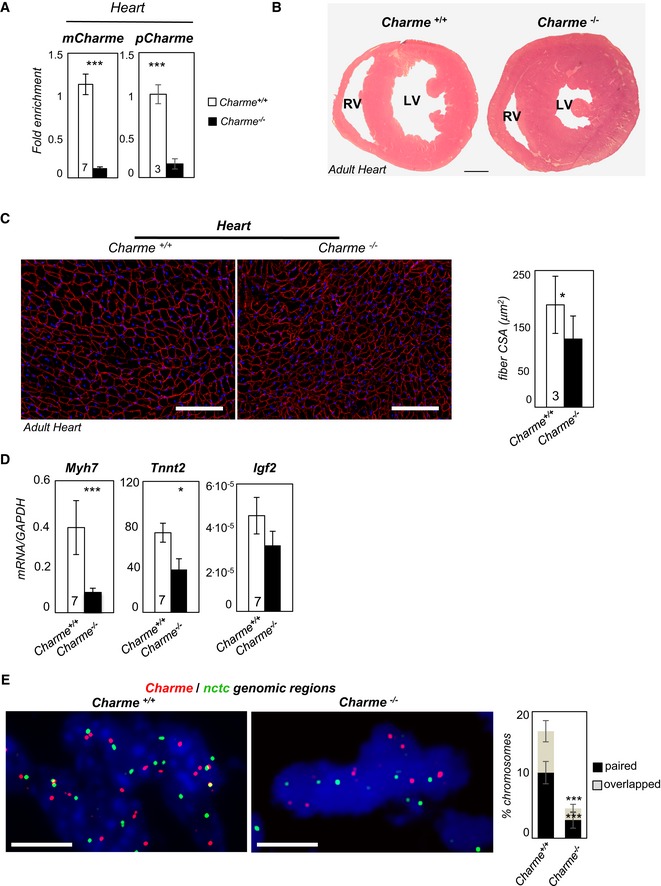
- qRT–PCR quantification of mCharme and pCharme expression in cardiac homogenates from Charme +/+ and Charme −/− mice. The number of mice tested (the same for the two groups) is indicated in the white bars. Data were normalized to GAPDH mRNA and represent mean ± SEM.
- Hematoxylin and eosin staining of transverse sections of the myocardium in 1‐month‐old Charme +/+ (left) and Charme −/− (right) mice. Left (LV) and right (RV) ventricles are indicated. Scale bar = 1 mm.
- Dystrophin staining analyzed by immunofluorescence on Charme +/+ and Charme −/− cardiac tissues. The mean ± SD of cross‐sectional area (μm2) is reported in the histograms besides. In total, N = 1,491 and 1,724 fibers per condition were analyzed. Original magnification = ×20. The number of mice tested is the same for the two groups, and it is indicated in the white bars. Scale bar = 100 μm.
- qRT–PCR quantification of Myh7, Tnnt2, and Igf2 transcripts in cardiac homogenates from 1‐month‐old Charme +/+ and Charme −/− mice. Data were normalized to GAPDH mRNA and represent mean ± SEM. The number of mice tested (the same for the two groups) is indicated in the white bars.
- DNA/DNA FISH for Charme and nctc loci in Charme +/+ and Charme −/− neonatal (1 day old) cardiac tissues are shown. Percentage of Charme locus ± SD showing paired and overlapped signals with nctc is indicated in the histogram. N = 395 and 315 chromosome pairs per condition were analyzed. Scale bar = 5 μm.
Identification of a functionally conserved human Charme transcript
An evolutionary conservation analysis performed by examining RNA‐seq (Legnini et al, 2017) and FANTOM5 (Noguchi et al, 2017) data revealed the existence of a Charme orthologue transcript (hs‐Charme) produced by the human syntenic locus (Fig EV4A), with a structure very similar to the murine counterpart (Figs 6A and EV4B–D, and Table EV3). Notably, the hs‐Charme locus displays an overall ~ 45% sequence identity (Fig EV4E and Appendix Table S1), which is a high value for a lncRNA genomic region (Pang et al, 2006). Similarly to the murine counterpart (Ballarino et al, 2015), the expression of the early myogenic transcriptional factor MyoD was found to precede that of hs‐Charme (Fig EV5A, left panel; and Table EV3) and a specific peak of MyoD ChIP‐seq (MacQuarrie et al, 2013) was identified, as in mouse, at the level of MyoD canonical E‐boxes upstream to the TSS (Fig EV5B and C). Altogether, these data suggest a common regulatory control of MyoD on mouse and human Charme RNA expression. Indeed, both hs‐pCharme and hs‐mCharme were found expressed in human primary myoblasts upon induction of differentiation; moreover, both transcripts resulted in delayed expression in myoblasts from Duchenne muscular dystrophy patients (Figs 6B and EV5A, right panel; and Table EV3), where MyoD expression is strongly impaired and cells are blocked in a more proliferative state (Delaporte et al, 1984; Cazzella et al, 2012). Similarly to the murine species, hs‐pCharme is mainly accumulated in the nucleus as an unprocessed species still containing intron 1 while the fully spliced isoform (hs‐mCharme) is mainly localized in the cytoplasm compartment (Fig 6C and D). In human primary myoblasts, GAPmer‐directed knockdown produced a strong downregulation of both hs‐pCharme and hs‐mCharme transcripts as well as the myogenic markers identified in mouse (Fig 6E). Altogether, these data suggest a conserved role of Charme in mammalian myogenesis.
Figure EV4. Genomic features and expression analysis of the human Charme locus.
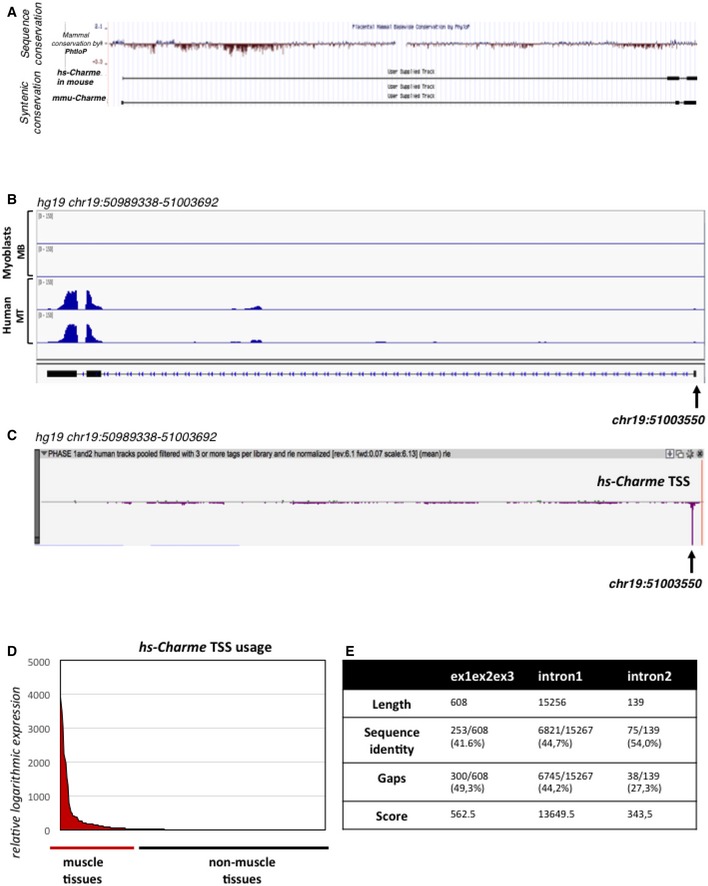
- UCSC visualization of the merged between human (hg19, top) and mouse (mm9, bottom) Charme genomic coordinates in the mm9 mouse genome. The analysis shows the mammal genomic sequence conservation (PhyloP) and the Charme transcript synteny between mouse and human.
- IGV (Robinson et al, 2011; Thorvaldsdóttir et al, 2013) visualization of the reads coverage from a RNA‐seq experiment performed in human myoblasts (Legnini et al, 2017; GSE70389) in proliferating (MB) and differentiating (MT) conditions; the arrow shows the TSS coordinate. The aligned reads were assembled into the transcript whose exon/intron structure is shown below.
- Zenbu (Severin et al, 2014) visualization of the hs‐Charme TSS peak from FANTOM5 (Phase 1 and 2) CAGE datasets across 1,829 samples; the arrow shows the TSS coordinate which corresponds to the one identified by RNA‐seq as reported in (B). The genomic coordinates are reported below.
- The diagram shows the hs‐Charme expression in muscle vs. non‐muscle tissues. The entire dataset was exported from FANTOM5 (Phase 1 and 2) CAGE datasets across 1,829 samples (see also Table EV3, sheet 1).
- Table represents the values obtained by analyzing the local sequence alignment between the human and murine Charme transcripts. Data were produced by using the implementation of the Smith–Waterman algorithm available at http://www.ebi.ac.uk/Tools/psa/emboss_water/. See also Table EV4.
Figure 6. Expression profiling and characterization of the human Charme upon in vitro differentiation of primary myoblasts.
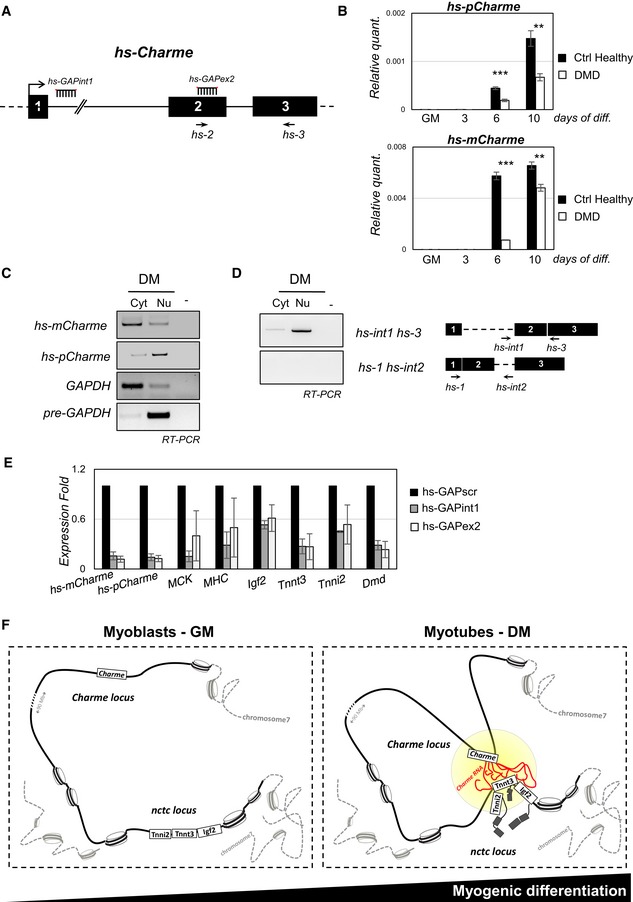
- The human Charme genomic locus is indicated together with the position of PCR primers (hs‐2, hs‐3) and LNA GAPmers (hs‐GAP‐int1, hs‐GAP‐ex2) used in this study.
- RT–PCR quantification of the human hs‐pCharme (upper panel) and hs‐mCharme (lower panel) in control healthy or dystrophic (DMD) primary myoblasts in proliferating (GM) and differentiating (3, 6, 10) conditions. Data were normalized to GAPDH mRNA and represent mean ± SD. Three biological replicates were analyzed.
- sqRT–PCR quantification of hs‐pCharme and hs‐mCharme in cytoplasmic (Cyt) and nuclear (Nu) RNA fractions from differentiated myotubes. The quality of fractionation was tested with precursor (pre‐GAPDH) and mature (GAPDH) RNAs.
- Analysis by PCR amplification of hs‐pCharme intron 1 retention. The position of the oligos used for the analyses is depicted aside.
- qRT–PCR quantification of hs‐mCharme, hs‐pCharme, MCK, MHC, Igf2, Tnnt3, Tnni2, and Dmd mRNAs in primary cells treated with hs‐GAP‐int1, hs‐GAP‐ex2, or hs‐GAP‐scr GAPmers at day 5 of differentiation. PCR data were normalized to GAPDH mRNA and represent mean ± SD of duplicates.
- Proposed model for pCharme mode of action: In proliferating myoblasts, Charme is not expressed and its chromatin locus is spatially distant from the nctc region; in differentiated myotubes, pCharme stabilizes the physical proximity of the two loci allowing their co‐regulated expression.
Figure EV5. MyoD is an upstream transcriptional regulator of both murine and human Charme transcripts.
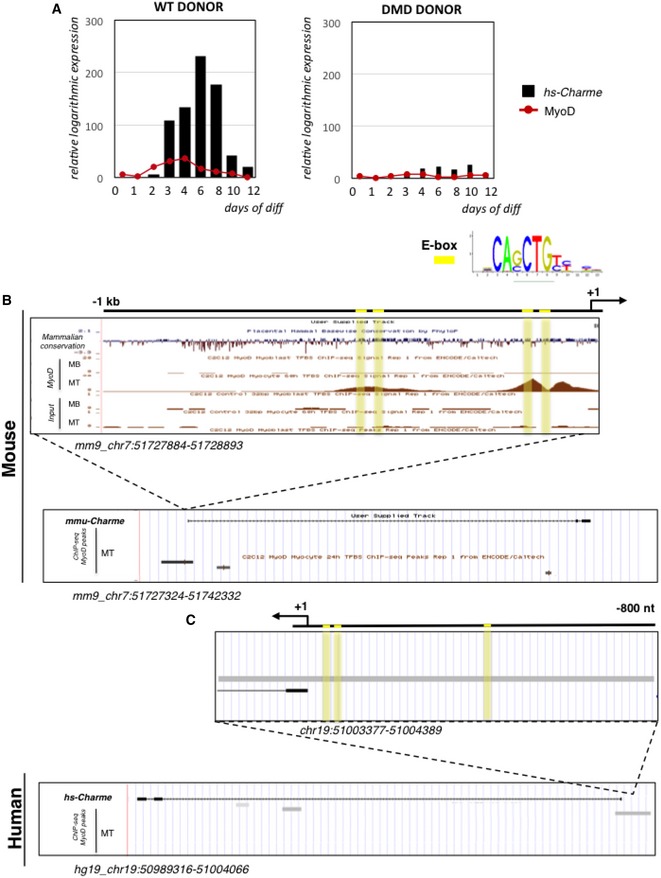
- hs‐Charme and MyoD expression during a time‐course of in vitro differentiation of WT (left panel) and DMD (right panel) myoblast into myotubes. The expression values were exported from FANTOM5 (Phase 1 and 2) CAGE datasets and are reported in Table EV3 (sheet 2). Data represent the relative logarithmic expression (rle) of the tag‐per‐million (TPM) values of the TSS.
- UCSC visualization of MyoD ChIP‐seq binding peaks in myoblast (MB) and myotubes (MT) from murine C2C12 cells.
- UCSC visualization of MyoD ChIP‐seq binding peaks in human myotubes (MT). The identity of each track is indicated along the left side, while the genomic coordinates are indicated below. E‐boxes are highlighted in yellow.
Discussion
In this study, we show the characterization of Charme, a chromatin‐associated lncRNA induced upon myogenic differentiation. In vitro Charme knockdown causes defects in myoblast fusion and downregulation of several crucial myogenic genes. Among them, several have been previously shown to be involved in familial hypertrophic (HCM), dilated (DCM), and arrhythmogenic right ventricular (ARVC) cardiomyopathies (Maron et al, 2012). Interestingly, an orthologue transcript, hs‐Charme, was identified in human myoblasts. Similarly to its murine counterpart, hs‐Charme is induced upon differentiation, and its depletion affects the same subset of myogenic genes. These data support the notion of an important conserved function of Charme in mammalian myogenesis.
Through ChIRP analyses, we found that Charme associates with several different genomic loci containing a number of the myogenic genes affected by its depletion. One of these regions, nctc, seemed particularly interesting because it is localized in cis with respect to the Charme locus and harbors a shared core muscle enhancer (CME; Alzhanov et al, 2010), previously shown to control muscle‐specific activation of the neighboring Igf2 and H19 promoters (Eun et al, 2013). Moreover, the knockout of part of the nctc region was performed by several groups and resulted in the loss of Igf2 expression and in defective myogenesis (Kaffer et al, 2001; Alzhanov & Rotwein, 2016). The nctc region also contains genes involved in muscle activity and metabolism, such as muscle troponins (Tnni2 and Tnnt3). Interestingly, all these genes are affected by Charme depletion, indicating a direct connection between the lncRNA expression and the muscle‐specific transcriptional output of the region. We also show that Charme, retained at the sites of its own transcription as an incompletely spliced form, is required for the maintenance of the long‐distance chromosomal interaction with the nctc locus and for consolidating the expression of the myogenic genes therein included. These data are in line with recent studies indicating the importance of the tridimensional chromatin folding and with the evidence that the eukaryotic genome is organized in a hierarchy of well‐defined, highly compartmentalized, and extremely dynamic structures within the nucleus, where chromosome territories are segmented into functional compartments (Rowley & Corces, 2016). These regions can be considered as discrete regulatory units where genes far apart in terms of linear distance but close in space are activated or repressed in a coordinated fashion during cell differentiation (Zaug & Cech, 1980; Guerrier‐Takada et al, 1983).
Notably, Charme depletion in vivo produced similar molecular and morphological phenotypes as the one observed in vitro. In fact, the same genes affected in C2C12 cells were downregulated in the skeletal muscles of Charme −/− mice and the decrease in fiber calibers observed in adult muscles correlated well with the reduction in the caliber of differentiating myotubes observed in vitro. Charme −/− mice showed also a clear cardiac phenotype, characterized by a conspicuous decrease in fiber size, and resulting in an overall heart remodeling and alteration of morphological and functional parameters. Also in this district, Charme was shown to contribute to the physical proximity with the nctc genomic region and to affect the cardiac expression of Igf2. Since Charme depletion provides a clear heart phenotype already at neonatal stages, the observed remodeling of the cardiac tissue must represent outcomes of defects starting early during embryonic development and persisting in adult life. The reduction in life span observed in adult mice indicated the persistence of effects of Charme depletion even at very advanced ages. It is important to note that this is the first time that such a cardiac phenotype has been directly ascribed to a lncRNA ablation in the absence of any stressful condition (Guttman et al, 2010; Devaux et al, 2015). Moreover, the chronic in vivo phenotype, where the overall alteration of tissue homeostasis and cardiac structure leads to late lethality, correlates well with the role of Charme as a fine‐tuning regulator.
Since Charme is interacting with several other regions besides the nctc locus, it is important to underline that these other domains will likely contribute to the overall phenotype observed in vitro and in vivo. For the nctc locus, which has been prioritized in this study, a clear correlation between the expression of the myogenic genes therein included and Charme activity has been assessed (Fig 6F). Further characterization of the other Charme targets will be necessary to complete the genome‐wide clarification of its function in skeletal and cardiac muscles.
Materials and Methods
Cell culture conditions and transfection
C2C12 murine myoblasts were cultured as previously described (Agostini et al, 2013) and differentiated in 0.5% fetal bovine serum (FBS). Human control healthy and DMD myoblasts from the Telethon Neuromuscular Biobank were cultured as previously described (Faghihi et al, 2008) and differentiated with human skeletal muscle differentiation medium (PromoCell). RNAi experiments were performed with 75 nM of LNA GAPmers (Exiqon) using Lipofectamine 2000 (Invitrogen). GAPmers were designed against Charme exon 2 (GAP‐2) or EX2/EX3 splice junction (GAP‐2/3) sequences using the Exiqon web tool. Sequences are reported in Table EV4.
RNA analyses
Total RNA from myoblasts/myotubes was isolated using QIAzol Lysis Reagent (Qiagen)/chloroform extraction followed by spin‐column purification (RNeasy Mini Kit; Qiagen) and quantification by NanoDrop (Thermo Scientific). Northern blot analysis was performed on 12 μg of total RNA from mouse myoblasts/myotubes. Total RNA was denatured in glyoxal loading dye (Ambion) for 30 min at 50°C and loaded on 1.2% agarose gel. Electrophoresis was carried out for 2 h at 60 V. RNA was transferred overnight on Hybond‐N+ membrane (GE Healthcare) by capillarity in 10× SSC. The transferred RNA was UV‐cross‐linked (1,200 × 100 μJ/cm2), and the membrane was washed in 50 mM Tris (pH 8) for 20 min at 45°C. Pre‐hybridization and hybridization were performed in NorthernMax buffer (Ambion) at 68°C (30 min and overnight, respectively). For the hybridization, 500 ng of DIG‐labeled probes (DIG‐RNA Labeling Mix; Roche) was produced by in vitro transcription with T7 RNA polymerase (Promega) using a PCR‐amplified Charme template (see Table EV4 for oligo sequences). The membrane was washed twice in 2× SSC/0.1% SDS and twice in 0.2× SSC/0.1% SDS at 68°C. The membrane was finally processed for DIG detection using the DIG Luminescence Detection Kit (Roche), according to the manufacturer's instructions. Nucleoplasm/chromatin/cytoplasmic fractionation was performed using the NE‐PER Kit (Pierce‐Thermo Scientific) following the manufacturer's specifications. 5′‐RACE was performed using FirstChoice RLM‐RACE kit (Ambion) on 5 μg of total RNA and by PCR amplification with Charme 4 and Charme 3 reverse primers. For semiquantitative and quantitative PCR analyses, RNA (0.5–1.0 μg) was reverse‐transcribed using SuperScript III RT (Life Technologies) according to the manufacturer's instructions and amplified by PCR using MyTaq (Bioline) enzyme. Real‐Time PCRs (qPCRs) were performed using QuantiTect SYBR Green (Qiagen).
RNA‐seq and functional enrichment analysis
RNA‐seq data were analyzed using RAP (D'Antonio et al, 2015) using FASTQC (http://www.bioinformatics.babraham.ac.uk), NGS QC Toolkit (Patel & Jain, 2012), Bowtie 2 (Langmead & Salzberg, 2012), TopHat2 (Kim et al, 2013), and Cufflinks and Cuffdiff (Trapnell et al, 2012, 2013). Default parameters were used for the analyses except for library type (fr‐firststrand), mate inner distance (set to 30), standard deviation of inner distance (set to 60), and minimum read length (set to 30). Reads were aligned using mm9 assembly. Genes deregulated in both GAP‐2 and GAP‐2/3 conditions were selected choosing Cuffdiff q‐value < 0.1, abs(log2 fold change) > 0.5, and variation of gene expression in the same direction. Functional enrichment analysis was performed by FIDEA (D'Andrea et al, 2013) using the genes expressed in the scramble condition as background.
RNA‐seq data from GSE70389 (Legnini et al, 2017) were analyzed as described above. The read coverage and the Cufflinks tracks were loaded on IGV genome browser (Robinson et al, 2011; Thorvaldsdóttir et al, 2013); see also Fig EV4B.
Chromatin isolation by RNA purification (ChIRP) sequencing
ChIRP‐seq was performed according to the procedure described by Chang (Chu et al, 2011) using nuclear extract from 107 differentiated myotubes/each sample. Charme pull‐down was performed with different sets of oligos (Appendix Fig S2F) or lacZ as control. Input and purified DNA were sequenced with HiSeq 2500 (40 million single‐end, 50‐base pair‐long reads/sample). Reads were pre‐processed with Trimmomatic (Bolger et al, 2014) and Cutadapt (Martin, 2011) to trim the 5′ and 3′ adapters and to remove the first five nucleotides, whose average quality score was lower than that of the other nucleotides. Low‐complexity reads were then removed using the DUST algorithm included in the PRINSEQ suite (Fatica & Bozzoni, 2014; Morlando et al, 2014; complexity score threshold = 7). The procedure adopted by Chu and colleagues (Chu et al, 2011) was used to find the EVEN and ODD common peaks (mm9 assembly, MACS14 bw = 300 (Zhang et al, 2008), fold enrichment/input > 2, average coverage > 10, and Pearson correlation > 0.3). BEDTools (Quinlan & Hall, 2010) was used to identify the overlapping lacZ peaks (obtained with MACS14 using the input as background). Peaks with fold enrichment/input 25 times lower than that of the overlapping lacZ peaks and with more than 50% of the length covered by repeats (Chen, 2002; http://www.repeatmasker.org) were excluded. HOMER (Heinz et al, 2010) was employed to annotate peaks using GENCODE M4 (Harrow et al, 2012). WindowBed tool was used to find all downregulated genes which are closer than 2.5 Mb to Charme ChIRP peaks.
Immunofluorescence and DNA/RNA FISH
MHC staining and dystrophin staining were performed as previously described (Cazzella et al, 2012). Samples were imaged on inverted microscope Zeiss AxioObserver A1 equipped with Axiocam MRm R camera and Plan‐Neofluar EC 10 × /0.3 M27 and LD 20 × /0.4 Korr objectives. Images were acquired with AxioVision Rel.4.8 software.
Fluorescent in situ hybridization (FISH) was carried out on cells that adhered to collagen (Corning) using fluorescent (Fluorescein or Cy3)‐conjugated nick‐translated BAC DNA (DNA FISH) or synthetic DNA oligonucleotides (RNA FISH). Three‐dimensional (3D) DNA FISH on C2C12 was performed with some modifications of an already described procedure (Cremer et al, 2008). Briefly, cells were fixed in 4% PFA (20 min at 4°C) and pre‐processed by freeze‐thawing permeabilization to ensure the preservation of nuclear structures. After denaturation, FISH probes were hybridized O.N. at 37°C and samples washed, DAPI‐stained, and mounted in ProLong Diamond reagent (Thermo Fisher Scientific). For DNA FISH on histological muscle sections, after permeabilization with Triton buffer (0.5% Triton X‐100/PBS), a mild pepsin digestion (pepsin 0.1%/0.1 M HCl) was applied before denaturation in order to reduce the autofluorescence deriving from protein components. For RNA FISH, fixed cells were permeabilized in 0.5% Triton (10 min at 4°C) and incubated O.N. at 37°C with fluorescent DNA probes diluted in hybridization buffer (10% dextran sulfate, 2× SSC, 10% formamide, 2 mM ribonucleoside‐vanadyl complex). The specificity of RNA FISH was tested by pre‐incubating the cells with 200 μg/ml RNaseA for 1 h at 37°C, or by GAPmer transfection. Sequential hybridizations were used for DNA/RNA FISH. Samples were imaged on inverted microscope (Olympus IX73) equipped with a Confocal Imager (CREST X‐LIGHT) spinning disk, a CoolSNAP MYO CCD camera (Photometrics), and a Lumencor Spectra X LED illumination. Images were acquired with 60× NA1.35 oil objective (UPLANSApo) and MetaMorph (Molecular Devices) using 300 (Cy3), 500 (Fluorescein), or 100 (DAPI) ms as exposure time.
Immunofluorescence and RNA and DNA FISH post‐acquisition analyses
The fusion index (F.I.) indicates the percentage of nuclei in multinucleated myotubes (defined by at least two nuclei) divided by total number of nuclei in a microscope field; the average number of nuclei inside individual myotubes (Nu/MT) was determined by dividing the total number of nuclei by the number of myotubes; and the fraction of mononucleated MHC‐positive cells (MonoMHC+) was calculated as the mean percentage of nuclei in MHC‐positive cells divided by total number of nuclei in each field. The number of nuclei was estimated by the average of nuclei counted in three independent and randomly chosen microscope fields. Dystrophin staining was used to evidence the border of the muscle fibers. The single‐channel dystrophin images were merged in dystrophin/DAPI images with ImageJ software and then used to quantify the fiber number manually. The fiber cross‐sectional area analysis was performed on dystrophin immunofluorescence by using a semi‐automatic tool (Briguet et al, 2004; Mula et al, 2013). The immunofluorescence images were binarized to obtain a mask of fiber boundaries and then applied to originals for the automatic measure of the areas. Statistical analysis of fiber cross‐sectional area (CSA) was performed using unpaired t‐test, and the differences between means were considered significant at P ≤ 0.05.
For DNA/RNA FISH staining analysis, the Z‐stack confocal microscopy images were taken automatically (0.2‐μm path) and merged with maximum‐intensity projection or processed for image analysis. A Laplacian‐of‐Gaussian filter was applied to 3D volume after image acquisition to enhance the spots signal over background (Raj et al, 2008). Intensity threshold was manually adjusted using MetaMorph or ImageJ software. Signal from cells hybridized without probes was considered as background.
Spatial proximity analysis of long‐splitted, paired, or overlapped patterns in DNA FISH was performed by computing the 3D interallelic distance on Z‐stack acquisitions using ImageJ (Spot Distance; Schober et al, 2008). Changes in the C2C12 (elliptic) shape of nuclei occurring along differentiation were taken into account as in Cremer et al (1984) before the calculation of the Normalized Distances mean (ND; ND = Interallelic Distances/d, d = major axis + minor axis/2). Values above or below the paired ND range were attributed to long‐splitted or overlapped patterns, respectively (Fig EV2A). The co‐planarity and the co‐localization of the DNA and RNA spots were established by recording the main grayscale value (expressed as arbitrary units) along Z‐planes using ImageJ (Plot Z‐axis profile). A Gaussian function (determination coefficient > 0.9) was used to fit the fluorescent intensities and to extrapolate the main Z‐plane value, which corresponds to the maximum fluorescence value of green and red channels in DNA FISH staining (Fig EV2B). The percentage of nuclei showing spatial proximity of genomic loci was calculated by manual screening on binarized images on a total of about 500 nuclei showing at least one pair of spots with paired or overlapped patterns. To determine spot intensity (fluorescence intensity) in RNA FISH analysis, we measured the mean gray value of nonbinarized images within a selection that circumscribes the spot; this value was divided by the diameter of the spot (d = 2*((3/4*V)/π)1/3)) by applying the formula of a sphere with the same volume of the spot.
Chromatin immunoprecipitation (ChIP)
ChIP experiments were performed on chromatin extracts according to the manufacturer's protocol (MAGnify ChIP; Life Technologies) by O.N. incubation with 2 μg of immobilized monoclonal anti‐RNA Pol II (Millipore), anti‐acetyl‐histone H3 (Lys9; Millipore), or rabbit IgG antibodies. A standard curve was generated for each primer pair testing 5‐point dilutions of input sample. Fold enrichment was quantified using qRT–PCR (SYBR Green; Qiagen) and calculated as a percentage of input chromatin (% Inp). Data from GAP‐scr vs. GAP‐2 conditions were normalized to an unrelated genomic region and represent the mean of three experiments ± SEM.
Generation of Charme −/− mice
Charme −/− mice were generated in the C57BL/6 background using a CRISPR genome‐editing system (Yang et al, 2014). A pair of divergent single guide RNAs (sgRNAs) inside exon 2 was designed using the http://crisp.mit.edu resource and cloned into the pX330 vector (Addgene). The sgRNAs were amplified together with the T7 promoter to generate templates for in vitro transcription. For homologous recombination, a ssODN (200 nt) containing two homology arms (50 nt each), a short poly(A) site (49 nt), and two MAZ sites (Levitt et al, 1989; Yonaha & Proudfoot, 2000) was synthetized by IDT. In vitro transcribed Cas9 mRNA and sgRNAs were injected into the cytoplasm of fertilized eggs of C57BL/6 mice in M2 medium (Sigma‐Aldrich, MO, USA). Successful knockout was validated by PCR analyses (see Table EV4 for oligo sequences). The wild‐type allele yielded an amplicon of 235 bp, whereas the mutated allele yielded an amplicon of 335 bp. One mouse was selected to mate with the wild‐type C57BL/6 strain to obtain F1‐generation mice. Heterozygous F1 offspring were interbred to establish the Charme −/− strain. Molecular and histological analyses were assessed in F4‐generation mice.
Offspring genotyping
DNA extraction from tail biopsies was performed using RBC Real Genomics DNA Extraction Kit (RBC Bioscience) according to the manufacturer's protocol. The poly(A)/MAZ insertion was detected by PCR amplifications with oligo pairs int1b‐3 (Table EV4).
Histological analysis
Mice were sacrificed at different ages, and the morphological changes in the skeletal and cardiac muscles were observed under light microscopy. Muscle tissues were fixed in 4% formaldehyde, and sections were stained with hematoxylin and eosin.
Study approval
Animals were treated in respect of housing, nutrition, and care according to the guidelines of Good Laboratory Practice (GLP). MB and RT have successfully completed the Module1 as co‐coordinated by the Administration of Animal Facilities in the premises of EMBL. The EMBL course meets the requirements of European legislation on basic training of researchers.
Data availability
Data that support the findings of this study have been deposited in GEO with the primary accession code GSE94498 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE94498).
Author contributions
IB conceived the study, wrote the manuscript, and secured funding; MB designed the study, wrote the manuscript, and conducted ChIRP and ChIP analyses; ACi performed most of the experiments and the computational analysis of RNA‐seq; CC and DO'C developed the Charme −/− F0 mice; RT generated and characterized the Charme −/− mice; TS conducted the RNA and DNA FISH experiments; FD performed the Northern blot analyses; ACo performed the computational analysis of ChIRP‐seq; MM provided expertise on myogenic field; and CN and AM provided expertise and feedback on the experiments in mice.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
The authors acknowledge O. Sthandier and M. Marchioni for technical support, L. Bisceglie for support in ChIRP analysis, and the EuroBioBank and Telethon Network of Genetic Biobanks (GTB12001F) for providing biological samples. This work was partially supported by grants from ERC‐2013 (AdG 340172–MUNCODD), Telethon (GGP16213), Human Frontiers Science Program Award RGP0009/2014, Parent Project Italia, AFM‐Telethon (17835), Epigen‐Epigenomics Flagship Project, and AriSLA full grant 2014 “ARCI” to I.B.; Telethon GGP14066 to A.M.; Fondazione Roma to I.B. and A.M.; ERC grant agreement no. GA 310206 (FP7/2007‐2013) to D.O'C.; and Sapienza University (prot. RM11715C7C8176C1) to M.B.
The EMBO Journal (2018) 37: e99697
References
- Agostini F, Zanzoni A, Klus P, Marchese D, Cirillo D, Tartaglia GG (2013) catRAPID omics: a web server for large‐scale prediction of protein‐RNA interactions. Bioinformatics 29: 2928–2930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alzhanov DT, McInerney SF, Rotwein P (2010) Long range interactions regulate Igf2 gene transcription during skeletal muscle differentiation. J Biol Chem 285: 38969–38977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alzhanov D, Rotwein P (2016) Characterizing a distal muscle enhancer in the mouse Igf2 locus. Physiol Genomics 48: 167–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson DM, Anderson KM, Chang C‐L, Makarewich CA, Nelson BR, McAnally JR, Kasaragod P, Shelton JM, Liou J, Bassel‐Duby R, Olson EN (2015) A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell 160: 595–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariel F, Jegu T, Latrasse D, Romero‐Barrios N, Christ A, Benhamed M, Crespi M (2014) Noncoding transcription by alternative RNA polymerases dynamically regulates an auxin‐driven chromatin loop. Mol Cell 55: 383–396 [DOI] [PubMed] [Google Scholar]
- Ballarino M, Cazzella V, D'Andrea D, Grassi L, Bisceglie L, Cipriano A, Santini T, Pinnarò C, Morlando M, Tramontano A, Bozzoni I (2015) Novel long noncoding RNAs (lncRNAs) in myogenesis: a miR‐31 overlapping lncRNA transcript controls myoblast differentiation. Mol Cell Biol 35: 728–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballarino M, Morlando M, Fatica A, Bozzoni I (2016) Non‐coding RNAs in muscle differentiation and musculoskeletal disease. J Clin Invest 126: 2021–2030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista PJ, Chang HY (2013) Long noncoding RNAs: cellular address codes in development and disease. Cell 152: 1298–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker JR, Deo RC, Werdich AA, Panàkovà D, Coy S, MacRae CA (2011) Human cardiomyopathy mutations induce myocyte hyperplasia and activate hypertrophic pathways during cardiogenesis in zebrafish. Dis Model Mech 4: 400–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briguet A, Courdier‐Fruh I, Foster M, Meier T, Magyar JP (2004) Histological parameters for the quantitative assessment of muscular dystrophy in the mdx‐mouse. Neuromuscul Disord 14: 675–682 [DOI] [PubMed] [Google Scholar]
- Buckingham M, Rigby PWJ (2014) Gene regulatory networks and transcriptional mechanisms that control myogenesis. Dev Cell 28: 225–238 [DOI] [PubMed] [Google Scholar]
- Cazzella V, Martone J, Pinnarò C, Santini T, Twayana SS, Sthandier O, D'Amico A, Ricotti V, Bertini E, Muntoni F, Bozzoni I (2012) Exon 45 skipping through U1‐snRNA antisense molecules recovers the Dys‐nNOS pathway and muscle differentiation in human DMD myoblasts. Mol Ther 20: 2134–2142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesana M, Cacchiarelli D, Legnini I, Santini T, Sthandier O, Chinappi M, Tramontano A, Bozzoni I (2011) A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell 147: 358–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen N (2002) Using RepeatMasker to identify repetitive elements in genomic sequences. Hoboken, NJ: John Wiley & Sons Inc; [Google Scholar]
- Chu C, Qu K, Zhong FL, Artandi SE, Chang HY (2011) Genomic maps of long noncoding RNA occupancy reveal principles of RNA‐chromatin interactions. Mol Cell 44: 667–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipriano A, Ballarino M (2018) The ever‐evolving concept of the gene: the use of RNA/protein experimental techniques to understand genome functions. Front Mol Biosci 5: 2928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemson CM, Hutchinson JN, Sara SA, Ensminger AW, Fox AH, Chess A, Lawrence JB (2009) An architectural role for a nuclear noncoding RNA: NEAT1 RNA is essential for the structure of paraspeckles. Mol Cell 33: 717–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremer T, Baumann H, Nakanishi K, Cremer C (1984) Correlation between interphase and metaphase chromosome arrangements as studied by laser‐UV‐microbeam experiments In Chromosomes today, Bennett MD, Gropp A, Wolf U. (eds), pp 203–212. Dordrecht, the Netherlands: Springer; [Google Scholar]
- Cremer M, Grasser F, Lanctôt C, Müller S, Neusser M, Zinner R, Solovei I, Cremer T (2008) Multicolor 3D fluorescence in situ hybridization for imaging interphase chromosomes. Methods Mol Biol 463: 205–239 [DOI] [PubMed] [Google Scholar]
- D'Andrea D, Grassi L, Mazzapioda M, Tramontano A (2013) FIDEA: a server for the functional interpretation of differential expression analysis. Nucleic Acids Res 41: W84–W88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Antonio M, De Meo PD, Pallocca M, Picardi E, D'Erchia AM, Calogero RA, Castrignanò T, Pesole G (2015) RAP: RNA‐Seq analysis pipeline, a new cloud‐based NGS web application. BMC Genom 16: S3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaporte C, Dehaupas M, Fardeau M (1984) Comparison between the growth pattern of cell cultures from normal and Duchenne dystrophy muscle. J Neurol Sci 64: 149–160 [DOI] [PubMed] [Google Scholar]
- Devaux Y, Zangrando J, Schroen B, Creemers EE, Pedrazzini T, Chang C‐P, Dorn GW, Thum T, Heymans S, Cardiolinc Network (2015) Long noncoding RNAs in cardiac development and ageing. Nat Rev Cardiol 12: 415–425 [DOI] [PubMed] [Google Scholar]
- Eun B, Sampley ML, Van Winkle MT, Good AL, Kachman MM, Pfeifer K (2013) The Igf2/H19 muscle enhancer is an active transcriptional complex. Nucleic Acids Res 41: 8126–8134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faghihi MA, Modarresi F, Khalil AM, Wood DE, Sahagan BG, Morgan TE, Finch CE, St Laurent GIII, Kenny PJ, Wahlestedt C (2008) Expression of a noncoding RNA is elevated in Alzheimer's disease and drives rapid feed‐forward regulation of β‐secretase. Nat Med 14: 723–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatica A, Bozzoni I (2014) Long non‐coding RNAs: new players in cell differentiation and development. Nat Rev Genet 15: 7–21 [DOI] [PubMed] [Google Scholar]
- Goff LA, Rinn JL (2015) Linking RNA biology to lncRNAs. Genome Res 25: 1456–1465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromak N, West S, Proudfoot NJ (2006) Pause sites promote transcriptional termination of mammalian RNA polymerase II. Mol Cell Biol 26: 3986–3996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrier‐Takada C, Gardiner K, Marsh T, Pace N, Altman S (1983) The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell 35: 849–857 [DOI] [PubMed] [Google Scholar]
- Guttman M, Garber M, Levin JZ, Donaghey J, Robinson J, Adiconis X, Fan L, Koziol MJ, Gnirke A, Nusbaum C, Rinn JL, Lander ES, Regev A (2010) Ab initio reconstruction of cell type‐specific transcriptomes in mouse reveals the conserved multi‐exonic structure of lincRNAs. Nat Biotechnol 28: 503–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman M, Rinn JL (2012) Modular regulatory principles of large non‐coding RNAs. Nature 482: 339–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacisuleyman E, Goff LA, Trapnell C, Williams A, Henao‐Mejia J, Sun L, McClanahan P, Hendrickson DG, Sauvageau M, Kelley DR, Morse M, Engreitz J, Lander ES, Guttman M, Lodish HF, Flavell R, Raj A, Rinn JL (2014) Topological organization of multichromosomal regions by the long intergenic noncoding RNA Firre. Nat Struct Mol Biol 21: 198–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han P, Li W, Lin C‐H, Yang J, Shang C, Nurnberg ST, Jin KK, Xu W, Lin C‐Y, Lin C‐J, Xiong Y, Chien H‐C, Zhou B, Ashley E, Bernstein D, Chen P‐S, Chen H‐SV, Quertermous T, Chang C‐P (2014) A long noncoding RNA protects the heart from pathological hypertrophy. Nature 514: 102–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Yang F, Cao H, Liang Z (2015) Malat1 regulates serum response factor through miR‐133 as a competing endogenous RNA in myogenesis. FASEB J 29: 3054–3064 [DOI] [PubMed] [Google Scholar]
- Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, Barnes I, Bignell A, Boychenko V, Hunt T, Kay M, Mukherjee G, Rajan J, Despacio‐Reyes G, Saunders G, Steward C et al (2012) GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res 22: 1760–1774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey PA, Leinwand LA (2011) The cell biology of disease: cellular mechanisms of cardiomyopathy. J Cell Biol 194: 355–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK (2010) Simple combinations of lineage‐determining transcription factors prime cis‐regulatory elements required for macrophage and B cell identities. Mol Cell 38: 576–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaffer CR, Grinberg A, Pfeifer K (2001) Regulatory mechanisms at the mouse Igf2/H19 locus. Mol Cell Biol 21: 8189–8196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL (2012) Fast gapped‐read alignment with Bowtie 2. Nat Methods 9: 357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legnini I, Di Timoteo G, Rossi F, Morlando M, Briganti F, Sthandier O, Fatica A, Santini T, Andronache A, Wade M, Laneve P, Rajewsky N, Bozzoni I (2017) Circ‐ZNF609 is a circular RNA that can be translated and functions in myogenesis. Mol Cell 66: 22–37.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitt N, Briggs D, Gil A, Proudfoot NJ (1989) Definition of an efficient synthetic poly(A) site. Genes Dev 3: 1019–1025 [DOI] [PubMed] [Google Scholar]
- MacQuarrie KL, Yao Z, Fong AP, Diede SJ, Rudzinski ER, Hawkins DS, Tapscott SJ (2013) Comparison of genome‐wide binding of MyoD in normal human myogenic cells and rhabdomyosarcomas identifies regional and local suppression of promyogenic transcription factors. Mol Cell Biol 33: 773–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maron BJ, Maron MS, Semsarian C (2012) Genetics of hypertrophic cardiomyopathy after 20 years: clinical perspectives. J Am Coll Cardiol 60: 705–715 [DOI] [PubMed] [Google Scholar]
- Martin M (2011) Cutadapt removes adapter sequences from high‐throughput sequencing reads. EMBnet.journal 17: 10–12 [Google Scholar]
- Morlando M, Ballarino M, Fatica A, Bozzoni I (2014) The role of long noncoding RNAs in the epigenetic control of gene expression. ChemMedChem 9: 505–510 [DOI] [PubMed] [Google Scholar]
- Mousavi K, Zare H, Koulnis M, Sartorelli V (2014) The emerging roles of eRNAs in transcriptional regulatory networks. RNA Biol 11: 106–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller AC, Cichewicz MA, Dey BK, Layer R, Reon BJ, Gagan JR, Dutta A (2015) MUNC, a long noncoding RNA that facilitates the function of MyoD in skeletal myogenesis. Mol Cell Biol 35: 498–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mula J, Lee JD, Liu F, Yang L, Peterson CA (2013) Automated image analysis of skeletal muscle fiber cross‐sectional area. J Appl Physiol 114: 148–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson BR, Makarewich CA, Anderson DM, Winders BR, Troupes CD, Wu F, Reese AL, McAnally JR, Chen X, Kavalali ET, Cannon SC, Houser SR, Bassel‐Duby R, Olson EN (2016) A peptide encoded by a transcript annotated as long noncoding RNA enhances SERCA activity in muscle. Science 351: 271–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi S, Arakawa T, Fukuda S, Furuno M, Hasegawa A, Hori F, Ishikawa‐Kato S, Kaida K, Kaiho A, Kanamori‐Katayama M, Kawashima T, Kojima M, Kubosaki A, Manabe R‐I, Murata M, Nagao‐Sato S, Nakazato K, Ninomiya N, Nishiyori‐Sueki H, Noma S et al (2017) FANTOM5 CAGE profiles of human and mouse samples. Sci Data 4: 170112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ounzain S, Micheletti R, Arnan C, Plaisance I, Cecchi D, Schroen B, Reverter F, Alexanian M, Gonzales C, Ng SY, Bussotti G, Pezzuto I, Notredame C, Heymans S, Guigó R, Johnson R, Pedrazzini T (2015) CARMEN, a human super enhancer‐associated long noncoding RNA controlling cardiac specification, differentiation and homeostasis. J Mol Cell Cardiol 89: 98–112 [DOI] [PubMed] [Google Scholar]
- Ounzain S, Pedrazzini T (2015) The promise of enhancer‐associated long noncoding RNAs in cardiac regeneration. Trends Cardiovasc Med 25: 592–602 [DOI] [PubMed] [Google Scholar]
- Pang KC, Frith MC, Mattick JS (2006) Rapid evolution of noncoding RNAs: lack of conservation does not mean lack of function. Trends Genet 22: 1–5 [DOI] [PubMed] [Google Scholar]
- Patel RK, Jain M (2012) NGS QC toolkit: a toolkit for quality control of next generation sequencing data. PLoS One 7: e30619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A, Tyagi S (2008) Imaging individual mRNA molecules using multiple singly labeled probes. Nat Methods 5: 877–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro DM, Zanzoni A, Cipriano A, Delli Ponti R, Spinelli L, Ballarino M, Bozzoni I, Tartaglia GG, Brun C (2017) Protein complex scaffolding predicted as a prevalent function of long non‐coding RNAs. Nucleic Acids Res 46: 917–928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinn JL, Chang HY (2012) Genome regulation by long noncoding RNAs. Annu Rev Biochem 81: 145–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP (2011) Integrative genomics viewer. Nat Biotechnol 29: 24–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowley MJ, Corces VG (2016) The three‐dimensional genome: principles and roles of long‐distance interactions. Curr Opin Cell Biol 40: 8–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP (2011) A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell 146: 353–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schober H, Kalck V, Vega‐Palas MA, Van Houwe G, Sage D, Unser M, Gartenberg MR, Gasser SM (2008) Controlled exchange of chromosomal arms reveals principles driving telomere interactions in yeast. Genome Res 18: 261–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severin J, Lizio M, Harshbarger J, Kawaji H, Daub CO, Hayashizaki Y3; FANTOM Consortium , Bertin N, Forrest AR (2014) Interactive visualization and analysis of large‐scale sequencing datasets using ZENBU. Nat Biotechnol 32: 217–219 [DOI] [PubMed] [Google Scholar]
- Thorvaldsdóttir H, Robinson JT, Mesirov JP (2013) Integrative Genomics Viewer (IGV): high‐performance genomics data visualization and exploration. Brief Bioinform 14: 178–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L (2012) Differential gene and transcript expression analysis of RNA‐seq experiments with TopHat and Cufflinks. Nat Protoc 7: 562–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L (2013) Differential analysis of gene regulation at transcript resolution with RNA‐seq. Nat Biotechnol 31: 46–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai M‐C, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, Shi Y, Segal E, Chang HY (2010) Long noncoding RNA as modular scaffold of histone modification complexes. Science 329: 689–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida S, Dimmeler S (2015) Long noncoding RNAs in cardiovascular diseases. Circ Res 116: 737–750 [DOI] [PubMed] [Google Scholar]
- Wang J, Gong C, Maquat LE (2013) Control of myogenesis by rodent SINE‐containing lncRNAs. Genes Dev 27: 793–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Zhao Y, Bao X, Zhu X, Kwok YK‐Y, Sun K, Chen X, Huang Y, Jauch R, Esteban MA, Sun H, Wang H (2015) LncRNA Dum interacts with Dnmts to regulate Dppa2 expression during myogenic differentiation and muscle regeneration. Cell Res 25: 335–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Zhang X‐J, Ji Y‐X, Zhang P, Deng K‐Q, Gong J, Ren S, Wang X, Chen I, Wang H, Gao C, Yokota T, Ang YS, Li S, Cass A, Vondriska TM, Li G, Deb A, Srivastava D, Yang H‐T et al (2016) The long noncoding RNA Chaer defines an epigenetic checkpoint in cardiac hypertrophy. Nat Med 22: 1131–1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wutz A, Rasmussen TP, Jaenisch R (2002) Chromosomal silencing and localization are mediated by different domains of Xist RNA. Nat Genet 30: 167–174 [DOI] [PubMed] [Google Scholar]
- Yang H, Wang H, Jaenisch R (2014) Generating genetically modified mice using CRISPR/Cas‐mediated genome engineering. Nat Protoc 9: 1956–1968 [DOI] [PubMed] [Google Scholar]
- Yonaha M, Proudfoot NJ (2000) Transcriptional termination and coupled polyadenylation in vitro . EMBO J 19: 3770–3777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaug AJ, Cech TR (1980) In vitro splicing of the ribosomal RNA precursor in nuclei of tetrahymena. Cell 19: 331–338 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nussbaum C, Myers RM, Brown M, Li W, Liu XS (2008) Model‐based analysis of ChIP‐Seq (MACS). Genome Biol 9: R137 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Data Availability Statement
Data that support the findings of this study have been deposited in GEO with the primary accession code GSE94498 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE94498).