

Abstract
The RAS proteins are the most frequently mutated oncogenes in cancer, with highest frequency found in pancreatic, lung, and colon tumors. Moreover, the activity of RAS is required for the proliferation and/or survival of these tumor cells and thus represents a high-value target for therapeutic development. Direct targeting of RAS has proven challenging for multiple reasons stemming from the biology of the protein, the complexity of downstream effector pathways and upstream regulatory networks. Thus, significant efforts have been directed at identifying downstream targets on which RAS is dependent. These efforts have proven challenging, in part due to confounding factors such as reliance on two-dimensional adherent monolayer cell cultures that inadequately recapitulate the physiologic context to which cells are exposed in vivo. To overcome these issues, we implemented a High Throughput Screening (HTS) approach using a spheroid-based 3-dimensional culture format, thought to more closely reflect conditions experienced by cells in vivo. Using isogenic cell pairs, differing in the status of KRAS, we identified Proscillaridin A as a selective inhibitor of cells harboring the oncogenic KRasG12V allele. Significantly, the identification of Proscillaridin A was facilitated by the 3D screening platform and would not have been discovered employing standard 2D culturing methods.
Keywords: Ras, Spheroid, Synthetic Lethal, 3D, Proscillaridin A
Introduction
The RAS proteins are a family of small GTP-binding proteins comprised of HRAS, NRAS and 2 splice forms of KRAS (KRAS4A and KRAS4B), that function as a switch cycling between “ON” (GTP-bound) or “OFF” (GDP-bound) conformations. They mediate signals from the extracellular environment into intracellular signaling pathways and function as master regulators in almost every aspect of cellular behavior including cell proliferation, differentiation and cell death. Given these functions, the involvement of RAS proteins in pathological conditions, such as cancer, is no surprise. Indeed, the RAS genes are the most frequently mutated oncogenes, with oncogenic mutations found in approximately 30% of all cancers (1, 2). Examples include mutations of KRAS found in pancreatic carcinomas (>90%), lung adenocarcinomas (>30%) and colorectal tumors (>40%). Mutations in HRAS are found mostly in bladder (>15%) and head and neck squamous cell carcinoma (>10%). NRAS mutations are found mostly in melanoma (>30%) and multiple myeloma (18%) (3). Mutations in RAS genes cluster most frequently to codons 12, 13 and 61, all within the G-domain of the protein that is involved in nucleotide binding and hydrolysis (4). This results in an oncogenic version of the protein that is preferentially in the “ON” state.
Given the oncogenic role of RAS proteins and prevalence of mutations, they have been of great interest as therapeutic targets for decades. However, efforts to target RAS proteins directly have proven challenging (4). The majority of efforts have focused on attacking the catalytic G-domain as well as interference with functionally required post-translational modifications (PTMs). In the case of attacking the G-domain, the use of nucleotide analogs is problematic given the affinities of RAS proteins to GTP, the intracellular concentration of GTP and kinetics of GTP hydrolysis. Likewise, the identification of small molecules that can specifically bind to RAS proteins has proven extremely difficult. In the case of PTMs, processing of the RAS C-terminus involves farnesylation, palmitoylation, methylation and proteolysis, all processes that could potentially be targeted. Several approaches to target these processes have been attempted, again with little success (4).
Other efforts have focused on identifying indirect targets through which RAS proteins drive tumorigenesis or on which RAS proteins are dependent. Efforts to develop inhibitors of downstream effector pathways have provided a number of targets that are currently in various stages of development. Examples include the MAPK and PI3K pathways, which are well documented as required for RAS-driven transformation and tumorigenesis. Attempts to target these pathways have focused on development of kinase inhibitors against effectors of these pathways including RAF, MEK and PI3K. However, inhibition of either of these pathways alone seems to be insufficient for multiple reasons including powerful feedback mechanisms, the activation of alternate signaling pathways and the co-opting of new effectors (4). As an alternative to targeting downstream effectors, synthetic-lethality (SL) approaches have been utilized to identify targets that are non-essential when inhibited in normal cells, but are required for the viability of tumor cells expressing an oncogenic allele of KRAS (5, 6). Initially, the approaches taken were candidate based, relying on the known functions of select effectors. Examples include the small G-protein Rac1, the cyclin-dependent kinase CDK4, NF-κB and cyclin D1 (reviewed in (4)). More recently, efforts to identify synthetic lethal interactions have relied on unbiased loss-of-function approaches using RNAi libraries to knock down specific mRNAs in cells harboring an oncogenic RAS mutation (4). Such efforts typically rely on lentiviral or retroviral libraries to introduce shRNA into RAS mutant cell lines. Examples include the identification of STK33 – a calcium/calmodulin-dependent serine/threonine kinase (7), TBK1 – a non-canonical IκB kinase (8), Polo-like kinase 1 (9), Wilm’s tumor 1 (WT1) and Snail2 (10, 11). These efforts have proven extremely challenging and in some cases the synthetic lethality of hits could not be reproduced (12–14). Another approach taken towards identification of oncogenic RAS-specific dependencies involves high throughput screening (HTS) of small molecule libraries. The scope of these efforts has been much more limited when compared to genetic screens described above, albeit a number of compounds have been identified including sulfinyl cytidine, triphenyltetrazolium, erastin and tolperisone (15–19). Further development of many of these leads is awaiting identification and confirmation of their respective targets in relevant tumor models.
One potential limitation of previous screening efforts was the reliance on 2-dimensional (2D) cell culture conditions, in which cells were directly plated onto plastic. These conditions vastly oversimplify the conditions to which cancer cells are exposed in vivo and are likely to be a major confounding factor. Indeed, cells grown on polystyrene in 2D lose many of the characteristics they possess under physiological conditions. Moreover, there is extensive data showing that cells behave differently when grown in 2D versus 3D conditions, mainly due to different cell-cell and cell-matrix interactions (20–22). To overcome many of the challenges presented by first generation screening efforts, we developed a 3D screening approach that is amenable to HTS small molecule screening using assay conditions that more closely reflect the conditions experienced by cells in vivo. Using this approach, we identified a number of cardiac glycosides that exhibit preferential inhibition of pancreatic ductal adenocarcinoma cells carrying oncogenic KRAS mutations.
Materials and Methods
KRAS Cell Lines
Human pancreatic epithelial carcinoma cells were purchased from the ATCC (American Type Culture Collection, Manassas, VA). These include BxPC-3 (ATCC#CRL-1687: Human pancreatic epithelial carcinoma), AsPC1 (ATCC#CRL1682: Human pancreatic adenocarcinoma), E6/E7 (ATCC#CRL4036: human pancreatic ductal cells–hTERT-HPNE-E6/E7 transformed), HPAFII (ATCC#CRL1997: Human pancreatic epithelial adenocarcinoma), PANC1 (ATCC#1469: Human pancreatic duct epithelioid carcinoma). Cell lines were authenticated by short tandem repeat (STR) DNA profiling (DDC Medical) and were tested every 3 months for mycoplasma contamination and confirmed free of contamination.
To create an isogenic pair, the BxPC-3 Pancreatic Ductal Adenocarcinoma (PDAC) cell line, which is wild type for KRAS [30], was transfected with an expression plasmid for wild type KRAS (BxPC-3KRASWT) or KRASG12V (BxPC-3KRASG12V) and selected in hygromycin to generate stable clones expressing these alleles. Expression of the introduced alleles was confirmed by isolation of mRNA from the cells, reverse transcription and DNA sequencing.
3D Cell Culture and 3D Luminescent Proliferation Assay
Cells were originally grown and passaged using a 1:3 or 1:6 subcultivation ratio 2 or 3 times per week in standard tissue culture flasks using ATCC guidelines for culture methods. Upon harvest for adaptation to 3D spheroids, flasks were decanted, washed with 1X PBS (part#14190, Thermo Fisher, Waltham, MA) and subsequently lifted using TryPLE (part#12604, Thermo Fisher). Cells were then suspended to the appropriate concentration for dispensing into Corning 384-well format 3D spheroid culture plates (part#3830, Corning Inc., NY). Cells were dispensed utilizing a Matrix Wellmate plate dispenser (ThermoFisher, Waltham, MA) at 2500 cells per well in 20 μL. Plates were centrifuged (1250 RPM, 5 min) and incubated for 1 day at 37°C, 95% relative humidity, 5% CO2. This allowed for spheroid formation, which was verified using a bright field microscope (Thermo Fisher). Upon verification of spheroids, test compounds or controls were transferred into the spheroid test plates using an automated BioMEK NXP Pintool. Plates were incubated for an additional 24 hours (for a total of 48 hours) under the same atmosphere and then treated with 20 μL per well of CellTiter-Glo 3D (Part#G9683, Promega Corp., Madison, WI). Following a 30 minute incubation at RT, luminescence was quantified on an EnVision plate reader (PerkinElmer Life Sciences, Waltham, MA).
Luminescent Apoptosis Assay
BxPC-3-KRASG12V and BxPC-3-KRASWT cells were seeded at the density of 2500 cells in 20 μL media per well into Corning 384-well spheroid plates for 3D analysis (part#3830, Corning Inc., NY) or white TC treated 384-well plates for 2D analysis (part#789163-T, Greiner Bio-One, Monroe, NC) and incubated for 24 hours at 37°C, 95% relative humidity, 5% CO2. Test compounds or vehicle (final 0.2% DMSO) were added followed by immediate addition of Real Time-Glo Annexin V apoptosis and Necrosis reagent (part# JA1011, Promega Corp., Madison, WI). Luminescence signal was monitored overtime up to 24 hrs using ViewLux plate reader (PerkinElmer Life Sciences, Waltham, MA).
Confocal Microscopy
BxPC-3-KRASG12V or BxPC-3-KRASWT cells were grown as described above. 48 hours post-seeding, spheroids were stained with Hoechst stain and incubated overnight. The stained spheroids were transferred to a flat, clear bottom plate, and cells were imaged on a GE IN Cell Analyzer 6000. To confirm the spheroidicity, multiple Z-stack images were taken at 10 μm increments and aligned in Image J to generate a composite intensity projection biased by color scale.
Screening Libraries
To aid in target validation of the 3D KRAS assays, sub-libraries of pharmacologically active compounds, such as the Scripps-curated Spectrum Collection (2400 compounds from MicroSource Discovery Systems, Inc., Gaylordsville, CT), a collection of small molecules with pharmacologic activity against a broad range of targets, were implemented as they are ideally suited in early stage testing.
Screening data acquisition, normalization, representation and analysis
All data files were uploaded into the Scripps institutional HTS database (Symyx Technologies, Santa Clara, CA) for plate QC and hit identification. Activity for each well was normalized on a per-plate basis using the following equation:
| Eq (1) |
Where “High Control” represent wells containing cells with media only; while “Low Control” represents wells containing cells and DMSO and finally the “Test Wells” contain cells with test compounds. The Z’ and S:B (signal:background) were calculated using the High Control and Low Control wells. In each case, a Z’ value greater than 0.5 was required for a plate to be considered acceptable (23)
ATP1A1 Knockdown
BxPC-3-KRASG12V or BxPC-3-KRASWT cells were transfected with either non-targeting control siRNA (Qiagen, SI03650318) or human pooled ATP1A1 siRNAs (GE Dharmacon, M-006111-02). After 24 hrs, cells were trypsinized, counted and seeded into (a) 96-well Corning Spheroid Microplates (10,000 cells/well, 100 μL) and (b) 384-well Corning Spheroid Microplates (2,500 cells/well, 20 μL) for the spheroid experiments. Spheroid formation and culture conditions proceeded as described above. 48h after seeding, (a) spheroids were collected from 96-well spheroid microplates and prepared for immunoblotting, and (b) CellTiter-Glo 3D was added 1:1 to spheroids in 384-well format and luminescence was measured on a PerkinElmer EnVision plate reader. For 2D experiments, cells were seeded into (a) 10 cm2 dishes for immunoblots and (b) 384-well Corning CulturPlates (2500 cells/well, 20 μL). 48h after seeding, (a) cells were collected and prepared for immunoblotting, and (b) CellTiter-Glo was added 1:1 to cells in 384-well format and luminescence measured as stated above. Percent inhibition was calculated using Eq (1).
Immunoblotting
Spheroids were collected, washed twice with ice-cold PBS, and lysed on ice in RIPA buffer with dissolved protease inhibitors. Lysates were centrifuged at 14,000 RPM (4°C) and supernatant protein concentration was quantified by modified Bradford assay (Bio-Rad Laboratories, Hercules, CA). Blots were probed with antibodies for ATP1A1 (D4Y7E, Cell Signaling Technology (CST), Danvers, MA), ATP1B1 (D6U8Q, CST), AKT (11E7, CST), phospho-AKT S473 (D9E, CST), ERK1/2 (137F5, CST), phospho-ERK1/2 (D13.14.4E, CST), tubulin (T5168, Sigma Aldrich, St. Louis, MO), or vinculin (V4505, Sigma Aldrich) and appropriate secondary antibodies (CST). Blots were developed using Amersham ECL and ECL Prime chemiluminescent developers (GE Healthcare Life Sciences, Boston, MA) and were exposed to X-ray film.
Statisitical analysis
The statistical tests were done using GraphPad Prism. The test used, number of samples and significance are indicated in the respective figure legends.
Results
Development of the 3D spheroid-based screening assay
To identify small molecules that are synthetic lethal to oncogenic KRAS and overcome many of the challenges faced in previous screening efforts, we sought to conduct a screening campaign using a primary screening platform that more closely reflects conditions experienced by tumor cells in vivo. Towards this goal, we developed a 3D spheroid-based primary screening assay that could be applied in a HTS small molecule screening campaign. In addition, to validate the specificity of validated hits against mutant KRAS, we developed an isogenic pair of cell lines that differ in the status of KRAS. Specifically, we employed the BxPC-3 pancreatic epithelial tumor cells that are wild type for KRAS and generated stable clones expressing KRAS wild type (BxPC-3-KRASWT) or mutant (BxPC-3-KRASG12V) alleles. From several stable clones isolated, we confirmed expression of wild type or mutant alleles by DNA sequencing (not shown) and selected clones expressing similar levels of KRAS protein compared to BxPC3 parental cell line. (Figure 1A–B).
Figure 1. Characterization of the BxPC-3 isogenic cell pair.
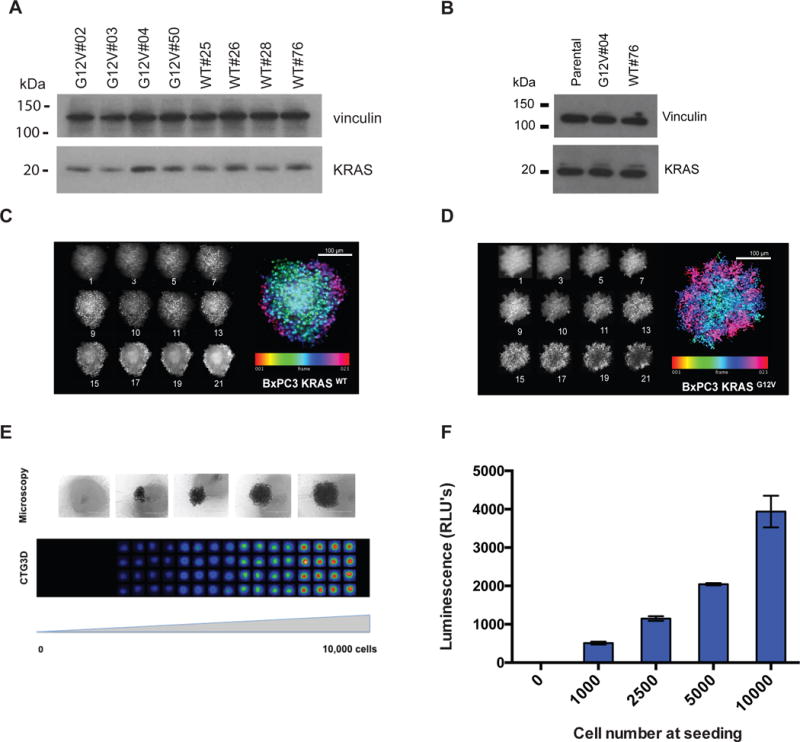
(A) Analysis of KRAS expression levels in BxPC-3-KRASG12V and BxPC-3-KRASWT stable cell lines. Individual clones were isolated and evaluated for the expression of KRAS by western blotting analysis using anti-KRAS or anti-Vinculin (loading control) antibodies. (B) Analysis of KRAS expression levels in selected BxPC-3-KRASG12V and BxPC-3-KRASWT stable cell lines and BxPC-3-parental cell line. Confirmation of spheroidicity of (C) BxPC-3-KRASWT or (D) BxPC-3-KRASG12V cells by confocal imaging. Z-stack images were taken at 10 μm increments from the equator of Hoechst-stained spheroids of BxPC-3-KRASG12V and BxPC-3-KRASWT on a GE IN Cell 6000 Analyzer (10× objective, f=1.18AU). Maximum intensity projection along the z-axis of the 12 individual planes aligned in Image J to generate an intensity projection biased by color scale are shown in the left panel. (E–F) Determination of cell viability assay conditions using CellTiter-Glo 3D (CTG3D). BxPC-3 cells were seeded at increasing numbers in a 384-well spheroid plate, grown for 24 hours and treated with CTG3D to assess viability. Relative luminescence of cells was determined at 48 hours post-seeding, using a ViewLux microplate imager (PerkinElmer). Error Bars = S.D. The data shown represent the mean of 3 independent replicates with triplicate data points.
Cells harvested from 2D monolayer culture were subsequently tested for their ability to form multi-cellular spheres over a period of 24 hours. Both BxPC-3-KRASWT and BxPC-3-KRASG12V cells formed spheroids as determined using light microscopy and confirmed by confocal microscopy of Hoescht-stained spheroids. Multiple Z-stack images were collected at 10μm increments and aligned in Image J to generate a composite intensity projection biased by color scale (Figure 1C–D). We next determined the linearity of spheroid growth using 3D CellTiter-Glo reagent, which allows for determination of cell number based on ATP levels. Linearity of detection was confirmed in the range of 1000 to 10,000 cells seeded (Figure 1E–F).
Execution of a 3D screen to identify selective inhibitors of mutated KRAS
We first determined the HTS readiness of the assay in 384-well format by examining the results from two separate experiments performed on two separate days (N=4 plates) using DMSO transfers for test wells. We were able to determine the averages of sample field %CV at 6.6% ± 1.6%, S:B at 89.14 ± 2.75, a Z score of 0.80 ± 0.05 and a Z’ score of 0.87 ± 0.07 (Figure 2A–B). We then screened the BxPC-3-KRASG12V spheroids in the primary assay against the Spectrum Collection (MicroSource) at a final assay concentration of 12.4 μM. The assay statistics yielded an average Z’= 0.82 ± 0.07, S:B= 247.8 ± 7.8. Utilizing a hit cut-off of 3 standard deviations plus average of all samples tested, we found 55 hits that had a percent response greater than 46.81%, equating to a 2.3% hit rate (Figure 2C). In order to identify selective inhibitors of mutant KRAS spheroids and eliminate non-specific hits, we employed a counter screen against BxPC-3-KRASWT spheroids. For the pilot screen using the BxPC-3-KRASWT cells the overall assay statistics yielded an average Z’= 0.72 ± 0.04, S:B= 258.4 ± 8.3. Utilizing a standard activity hit cut-off of 3 standard deviations plus average of all samples tested, we found 63 hits that had a percent response greater than 50.69% equating to a 2.6% hit rate (Figure 2D). Of the hits identified, 51 hits inhibited both cell types to a similar extent, while 4 hits show selectivity towards BxPC-3-KRASG12V spheroids and 12 towards the BxPC-3-KRASWT spheroids (Figure 3A and Appendix 1).
Figure 2. Spectrum Library Screen of BxPC-3-KRASG12V and BxPC-3-KRASWT cells in 3D and 2D formats.
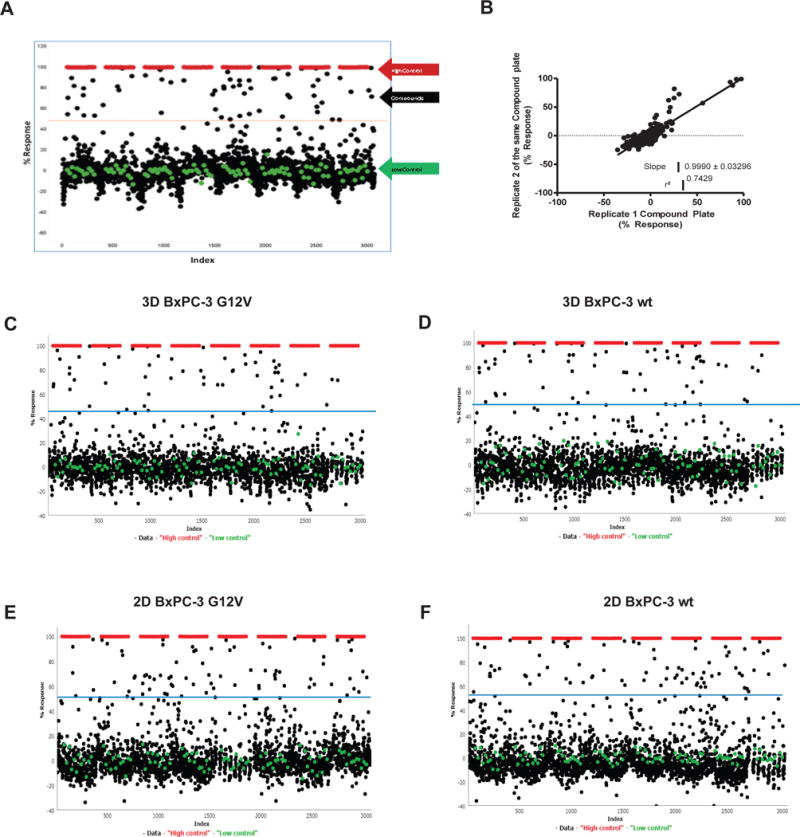
(A–B) 2400 compounds from the Spectrum Library were screened in duplicate on 3D against BxPC-3-KRASG12V to validate the 3D assay. (A) The activity of the compounds was plotted (duplicate data but showing single point percent response), with high control, low control and hit cutoff (dashed line) shown. (B) Correlation plot for the two replicate screening datasets. (C–F) Activity of 2,400 compounds on BxPC-3-KRASWT and BxPC-3-KRASG12V cells in 3D and 2D formats (singlicate showing single point percent response along with high control, low control and hit cutoff shown): (C) 3D format BxPC-3-KRASG12V, (D) 3D format BxPC-3-KRASWT, (E) 2D format BxPC-3-KRASG12V, (F) 2D BxPC-3 KRASWT.
Figure 3. Comparison of performance of compounds in BxPC-3-KRASG12V and BxPC-3-KRASWT cells in 3D and 2D formats.
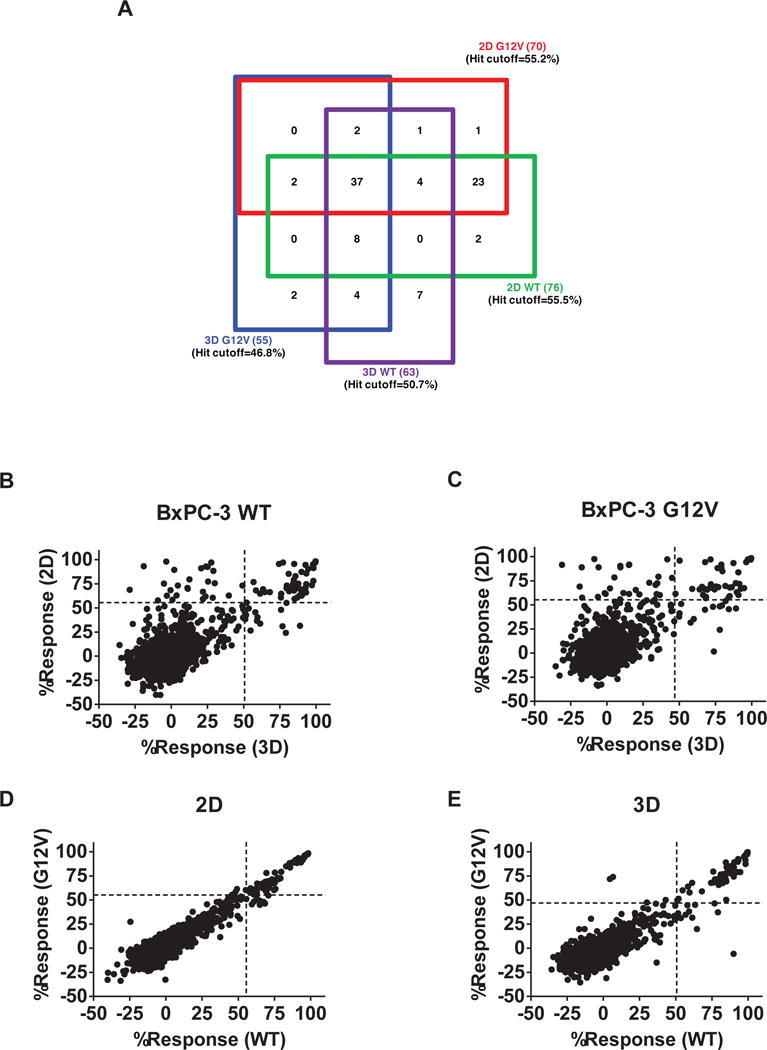
Primary screening results of Spectrum library against BxPC-3-KRASWT and BxPC-3-KRASG12V in 3D and 2D formats. (A) Four-way Venn diagram of active compounds identified from the four screens. A hit was identified as any compound with % inhibition > the corresponding screen hit cutoff. The numbers in parentheses are the numbers of hits specific for that cell line. The numbers in the boxes represent the number of compounds found to active in those overlapping assays. (B–E) Correlation plots of the % inhibition values of compounds in each of the screens: (B) BxPC-3-KRASWT, 2D vs. 3D. (C) BxPC-3-KRASG12V, 2D vs. 3D. (D) BxPC-3-KRASG12V vs. BxPC-3-KRASWT, 2D. (E) BxPC-3-KRASG12V vs. BxPC-3-KRASWT, 3D.
Comparison of 2D and 3D format assays
To compare the 3D screening format to a traditional 2D monolayer assay, we carried out a comprehensive analysis and compared the performance of the BxPC-3-KRASG12V and BxPC-3-KRASWT cell-based assays under 2D conditions. Again, we used the Spectrum library at a final assay concentration of 12.4 μM. The assay statistics gave an average Z’= 0.82 ± 0.05, S:B= 130.5 ± 5.70. Utilizing a standard activity hit cut-off of 3 standard deviations plus average of all samples tested, we found 70 hits that had average inhibition rates greater than 55.22%, equating to a 2.9% hit rate (Figure 2E). For the BxPC-3-KRASWT cells, the overall assay statistics gave an average Z’= 0.87 ± 0.03, S:B= 128.2 ± 3.3. Utilizing a standard activity hit cut-off of 3 standard deviations plus average of all samples tested, we found 76 hits that had average inhibition rates greater than 55.52%, equating to a 3.2% hit rate (Figure 2F). Of the hits identified, 66 hits inhibited both cell types to a similar extent, while 4 hits show selectivity towards BxPC-3-KRASG12V cells and 10 towards the BxPC3-KRASWT cells (Figure 3A and Appendix 1).
Comparing the performance of the cells between 2D and 3D formats suggests that over all, the cells in the 3D assay format are generally more resistant to cytotoxicity (Figure 3B–C). These results are in agreement with the IC50 values determined for a number of well-characterized anti-neoplastic agents, which when tested in 2D and 3D assay formats indicate that in 3D format the IC50’s of these agents tend to be higher (Supplemental Figure 1A–D). Importantly, when comparing the responses of the BxPC3-KRASG12V cells to BxPC3-KRASWT cells in 2D format, compounds that showed significant inhibition (> 3X S.D.) did not show a preference towards WT over mutant or vice versa. (Figure 3D). In contrast, when comparing the responses in 3D formatted assays, several compounds show preferential inhibition towards one of the cell types (Figure 3E).
Finally, as shown in Figure 3A, a comparison of the 2D vs 3D responses of the BxPC3-KRASG12V cells initially identified two compounds as hits in 3D format but not in the 2D format. We pursued these two hits further, testing for their IC50 using 10-point, three-fold serial dilutions done in triplicate in both the BxPC3-KRASG12V and BxPC3-KRASWT cells. Unfortunately, only one compound reproduced activity at the original test concentration and neither compound elicited a meaningful concentration response curve, indicating these hits identified from a single data point are false positives (Supplemental Figure 2).
Characterization of top hits
At the completion of the pilot assays we chose to pursue 15 analogs that appeared to be most active against the KRAS mutant in 3D format. To confirm the activity of these and to determine specificity towards mutant KRAS, we retrieved hit compounds from the original source plates and assessed their activity against the BxPC-3-KRASG12V or BxPC-3-KRASWT cells, respectively. Each hit was tested at a single dose (~12.4 μM), in triplicate. From the top 15 hits, 14 hits were confirmed as having average inhibition values of >48% (Figure 4A). Assessing the activity of the 15 initial hits in the counter screen (BxPC-3-KRASWT cells) confirmed several of the compounds were selective towards the BxPC-3-KRASG12V mutant cells. In particular, two cardiotonic glycosides, compound SR-838893 and SR-841251 (Proscillaridin A), displayed the largest difference in response rates between the isogenic cell pair (Figure 4A and Appendix 1). As Proscillaridin A displayed the greatest selectivity against the BxPC-3-KRASG12V cells, we focused on this hit for further characterization. First, we determined the IC50 of Proscillaridin A against the BxPC-3-KRASG12V or BxPC-3-KRASWT spheroids. While BxPC-3-KRASG12V cells displayed an IC50 = 240 nM, treatment of the BxPC-3-KRASWT cells with Proscillaridin A did not yield a dose-response and the response remained at 30–40% inhibition at all tested doses (Figure 4B). We next assessed the activity of Proscillaridin A against a panel of pancreatic tumor cell lines (AsPC-1, HPAF-II, PANC-1- all carrying KRASG12V mutations) and immortalized pancreatic ductal cells (hTERT-HPNE E6/E7 - wild type for KRAS). All cell lines were confirmed to form spheroids under the same conditions used for the BxPC3 cells (not shown). Proscillaridin A displayed inhibitory activity against all the pancreatic tumor cell lines, with IC50’s similar to BxPC-3-KRASG12V cells, in the mid-nanomolar range (Figure 4C). However, hTERT-HPNE E6/E7 did not respond to Proscillaridin A treatment, similar to the BxPC-3-KRASWT and parental BxPC3 cells (Figure 4D).
Figure 4. Validation of specificity of select inhibitors toward KRAS mutant cells.
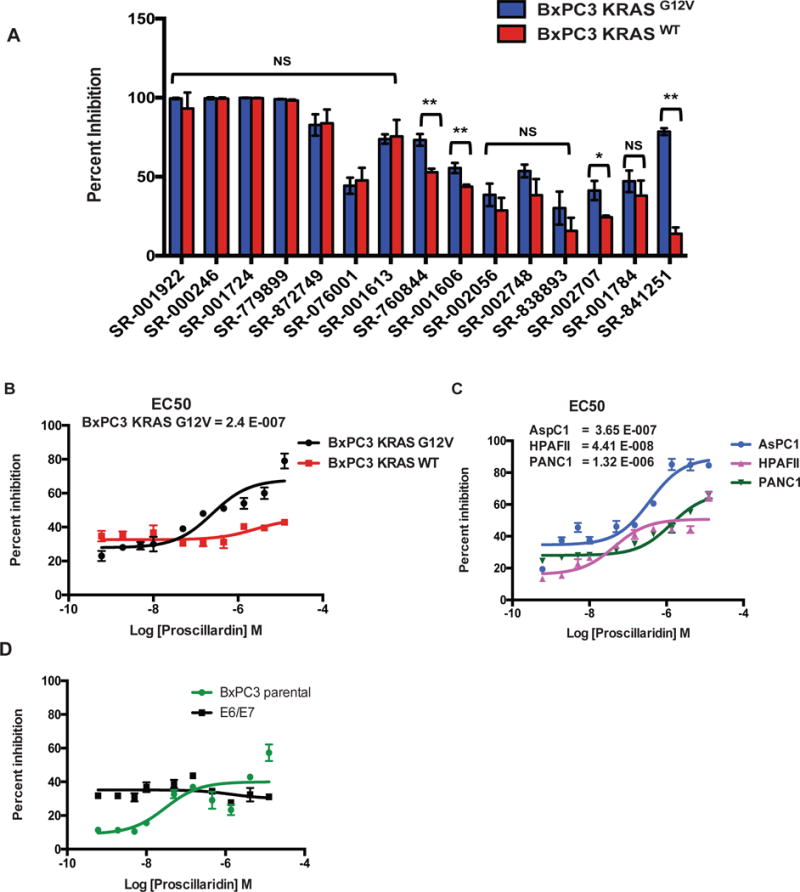
(A) Top 15 compounds from the Spectrum library screen were analyzed at 12.4 μM in triplicate against BxPC-3-KRASG12V and BxPC-3-KRASWT cells in 3D format. Statistical significance was determined by unpaired t-test. NS = Non significant, * = p < 0.001, ** = p < 0.0001. (B–D) Concentration-response curves of Proscillaridrin A (SR-841251) on different 3D cell models: (B) BxPC-3-KRASG12V and BxPC-3-KRASWT; (C) Pancreatic ductal adenocarcinoma cell lines AspC1, HPAF11, and PANC-1. (D) Parental BxPC3 or hTERT-HPNE-E6/E7 immortalized pancreatic ductal cells. The data shown represent the mean of 3 independent experiments with triplicate data points in each. Error bars = S.D.
To compare the activity profile of Proscillaridin A in standard 2D culture conditions versus the 3D spheroid assay, we compared the responses of BxPC-3-KRASG12V or BxPC-3-KRASWT cells grown in 2D or 3D conditions. Proscillaridin A displayed strong selectivity towards BxPC-3-KRASG12V spheroids in 3D, resulting in >90% inhibition compared to <10% inhibition of the BxPC-3-KRASWT spheroids (Figure 5A). This selectivity was lost under 2D conditions where treatment of either BxPC-3-KRASG12V or BxPC-3-KRASWT cells with Proscillaridin A at a single dose resulted in about 50% inhibition with no apparent selectivity (Figure 5B). Since the difference in response to drug treatment could be a reflection of different cellular proliferation rates, we compared the proliferation of BxPC-3-KRASG12V and BxPC-3-KRASWT cells grown in 2D or 3D. The BxPC-3-KRASG12V cells displayed a slightly elevated growth rate, compared to BxPC-3-KRASWT cells, both in 2D and 3D growth conditions (Figure 5C–D). Since this difference in growth rates is consistent between the 2D and 3D culture conditions, the selectivity of Proscillaridin A under 3D conditions is unlikely to result from different cell growth rates.
Figure 5. Characterization of select inhibitors in 2D and 3D formats.
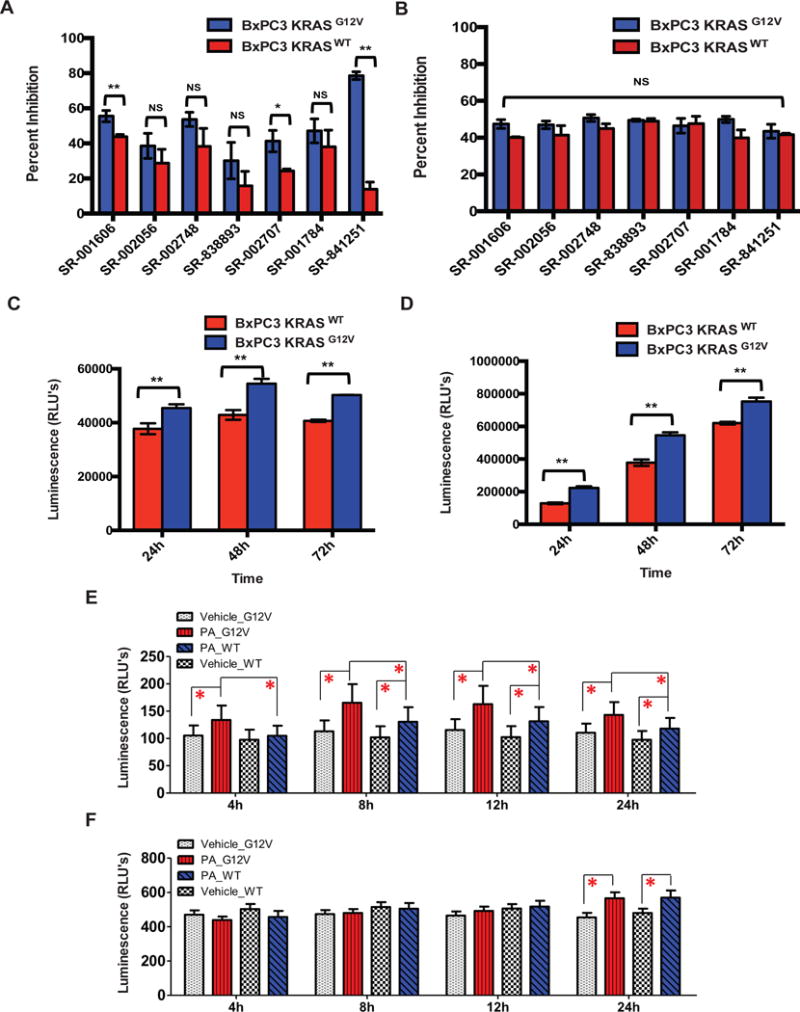
(A–B) Validation and specificity of select hits towards KRAS mutant cells in 3D (A) or 2D (B) formats. The top seven compounds from the Spectrum library screen were analyzed at ~ 12.4 μM in triplicate on BxPC-3-KRASG12V and BxPC-3-KRASWT cells in 3D and 2D formats. (C–D) Evaluation of BxPC-3-KRASG12V and BxPC-3-KRASWT cell growth rates in 3D (C) and 2D (D) formats. Cells were plated at 2500 cells/well in 3D and 2D formats and the growth rate was evaluated at 24, 48, and 72 hour time points using CTG3D or CTG, respectively. Statistical significance was determined by unpaired t-test. NS = Non significant, * = p < 0.001, ** = p < 0.0001. The data shown represent the mean of 3 independent experiments with triplicate data points in each. Error bars = S.D. (E–F) Effects of Proscillaridin A on apoptosis of BxPC-3-KRASG12V and BxPC-3-KRASWT cells. Proscillaridin A (PA) was tested at 12.4 μM against BxPC-3-KRASG12V and BxPC-3-KRASWT in 3D (E) and 2D (F) formats, and monitored at different treatment time points by RT-Glo Annexin V. Statistical significance was determined by unpaired t-test. All points represent the mean of 8 independent replicates. Error bars = S.D., *P <0.05.
To determine whether Proscillaridin A has an impact on cell viability, we employed the RT-Glo Annexin V apoptosis assay that measures the exposure of phosphatidylserine (PS) on the outer leaflet of the cell membrane during the apoptotic process, through annexin V binding detected with a luminescence signal. To ascertain the effect of Proscillaridin A in this assay we tested drug-treated versus vehicle-treated BxPC-3-KRASG12V and BxPC-3-KRASWT cells, in both 3D and 2D formats. In the 3D format, Proscillaridin A induced apoptosis at earlier time points and at higher rates in BxPC-3-KRASG12V cells compared to the BxPC-3-KRASWT cells. The rate of apoptosis induced by Proscillaridin A became significant in comparison to the vehicle group (*P < 0.05) at ~4hr for BxPC-3-KRASG12V cells, and ~8hrs for BxPC-3-KRASWT cells. The higher apoptotic rate of BxPC-3-KRASG12V cells was sustained throughout the experiment (Figure 5E). In contrast, the effects of Proscillaridin A treatment on BxPC-3-KRASG12V and BxPC-3-KRASWT cells grown in the 2D format did not become apparent until ~24h and similar rates of apoptosis were observed for both cell types (Figure 5F).
To determine whether the activity of Proscillaridin A, a well-characterized inhibitor of the Na+/K+ -ATPase pump, could be explained by on-target activity, we examined the consequences of interfering with activity of the Na+/K+-ATPase pump by knocking down ATP1A1, the alpha subunit of the transporter. Control or ATP1A1 siRNAs were transfected into both BxPC-3-KRASG12V and BxPC-3-KRASWT cells, and after seeding into spheroid plates, ATP1A1 knockdown and viability were assessed after 48 hours. The knockdown of ATP1A1 by siRNA was confirmed by western blotting (Figure 6A) and significantly reduced the viability of BxPC-3-KRASG12V spheroids, but did not have a significant impact on the viability of BxPC-3-KRASWT spheroids (Figure 6C). Interestingly, this selective effect was not replicated in 2D, where ATP1A1 knockdown (Figure 6B) did not reduce the viability of either KRASWT or KRASG12V cells (Figure 6D). These findings suggest the activity of Proscillaridrin A is mediated through the inhibition of the Na+/K+-ATPase transporter.
Figure 6. Assessing the Na+/K+-ATPase as the potential target of Proscillaridin A.
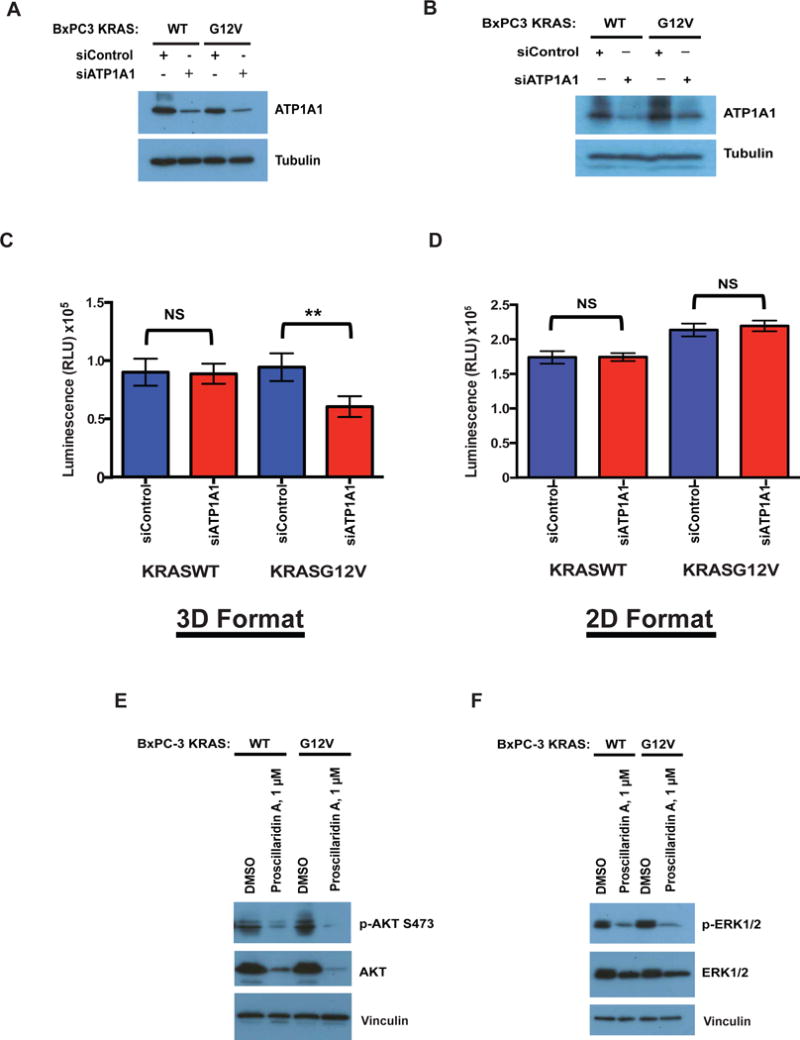
(A–D) BxPC-3-KRASG12V and BxPC-3-KRASWT were transfected with siRNA targeting ATP1A1 or control siRNA and knockdown of the ATP1A1 subunit was confirmed by western blotting in cells grown in (A) 3D format or (B) 2D format. Viability of BxPC-3-KRASG12V and BxPC-3-KRASWT cells transfected with siRNA targeting ATP1A1 or control siRNA grown in grown in (C) 3D format or (D) 2D format was determined at 48 hours post-transfection using CTG3D or CTG, respectively. Statistical significance was determined by unpaired t-test. NS = Non significant, ** = p < 0.0001. The data shown represent the mean of 3 independent experiments with triplicate data points in each. Error bars = S.D. Levels of (E) pAKT, AKT and (F) pERK1/2, ERK1/2 were determined in BxPC-3-KRASG12V and BxPC-3-KRASWT cells grown in 3D format. Vinculin was used as a loading control.
Finally, we examined whether the effects of Proscillaridin A on two main effectors of survival and proliferation, AKT and ERK1/2, could further explain the 3D-selective effects against mutant KRAS spheroids. Interestingly, Proscillaridin A treatment significantly decreased total AKT and ERK1/2 in both KRASWT and KRASG12V spheres relative to DMSO-treated controls, but there does not appear to be a selective reduction in the KRASG12V cells (Figure 6E–F).
Discussion
In this report we describe a new spheroid-based 3D screening platform optimized for HTS applications. We validated this screening platform and executed a proof of principle screen to identify small molecules that preferentially inhibit the viability of pancreatic adenocarcinoma cells harboring an oncogenic KRAS mutation. These efforts led to identification of a cardiotonic glycoside, Proscillaridin A, as a potent and selective inhibitor of KRAS mutant cells. Importantly, assessment of Proscillaridin A in traditional 2D screening formats suggests that this molecule would not have been identified as a selective hit in a 2D assay, illustrating the utility of the spheroid-based 3D platform to uncover new biology.
A major challenge for modern drug screening is presented by the compromise between numerous factors including cost, efficiency and accuracy. Indeed, the majority of cell-based screening platforms in current use rely on 2D monolayers which are easy to implement and cost-effective (24). However, these traditional monolayer models have proven of limited value in predicting clinical response to novel agents (25). In this respect, 3D-based cell culture offers a model that is thought to more closely reflect the environment experienced by cells in vivo (26). In particular, 3D spheroid models are thought to better recapitulate features experienced by tumor cells in vivo such as cell-cell interactions, cell-matrix interactions, hypoxia, heterogeneity of tumors, drug penetration and drug resistance (27–31). Several examples illustrate this point (32). For example, previous studies demonstrate that treatment with 5-FU has different outcomes depending on the assay format. While tumor cells grown as a monolayer on polystyrene are highly sensitive to 5-FU, cells gown as 3D spheroids are more resistant. This is thought to be a reflection of the higher and more uniform proliferation of cells grown in 2D (33). Another example includes the upregulation in expression of drug metabolizing proteins in liver cells grown in 3D, which more closely reflects the expression of these proteins in vivo, compared to 2D (34). Whether or not the BxPC-3 isogenic cell spheroid-based approach we have developed is indeed a more accurate predictor of an in vivo response remains to be established, and future studies aimed at validating the selectivity of Proscillaridin A towards KRAS mutant tumors in vivo are required. Regardless, the fact remains that this approach is useful in uncovering new biology that would have otherwise not been identified in traditional 2D formats, namely the selective activity of Proscillaridin A against BxPC-3-KRASG12V spheroids.
Proscillaridin A is a cardiac glycoside (CG) that inhibits the Na+/K+-ATPase, which functions as a transporter of K+ and Na+ into and out of the cell, respectively. This function is required for the maintenance of osmotic and ionic balance in all mammalian cells (35). Several cardiac glycosides (also known as cardiotonic steroids) have been identified as potent anti-neoplastic agents (36). Indeed, this class of molecules is highly represented in the top hits of our screens, with members demonstrating variable levels of potency and selectivity. Proscillaridin A itself has been previously identified in screens for anti-neoplastic compounds against glioblastoma, osteosarcoma and colon cancer cell lines (37–39). The reason(s) underlying Proscillaridin A’s selectivity towards KRAS mutated tumor cells in the 3D spheroid format are unclear. The finding that Proscillaridin A induces apoptosis earlier and at a higher rate in the BxPC-3-KRASG12V spheroids compared to BxPC-3-KRASWT spheroids suggests that ATP1A1 is required for cell survival, although the exact molecular mechanisms remain unknown. It is also possible that due to an increased proliferation rate, the BxPC-3-KRASG12V cells are more dependent on the function of the Na+/K+-ATPase pump for proliferation. However, although the BxPC-3-KRASG12V cells show a slightly increased proliferation rate compared to the BxPC-3-KRASWT cells, this difference in proliferation is also observed when the cells are grown in 2D format and Proscillaridin A is not selective in this format.
Of note, while selectivity towards an oncogenic mutation has not been previously reported for Proscillaridin A, other CGs were reported to display selectivity in other contexts. In particular, digoxin, digitoxin and ouabain were identified in a screen for synthetic lethal interaction with STK11 mutant cancer cell lines (40). This study suggests the selectivity of the CGs could be attributed to increased cellular stress in the STK11 mutant cells. Specifically, the authors propose that CG treatment leads to increased levels of reactive oxygen species (ROS) and that cells deficient in STK11 are impaired in their stress responses. Thus it is possible that the stress responses of the BxPC-3-KRASG12V cells are impaired compared to BxPC-3-KRASWT cells.
Finally, although BxPC3 cells are wildtype for KRAS, they harbor several other mutations (https://cansar.icr.ac.uk/cansar/cell-lines/BxPC-3/mutations/) including an in-frame deletion in the BRAF which results in activation of the kinase (41). This suggests that other effector pathways downstream of KRAS might underly the sensitivity to Proscillaridin. Clearly, further studies will be required to determine the basis for Proscillaridin’s selectivity against BxPC-3-KRASG12V cells.
Supplementary Material
Acknowledgments
We thank Pierre Baillargeon and Lina Deluca at Scripps for their help with compound management. This work was supported in part by the National Cancer Institute of the National Institutes of Health under Award Number R33CA206949 (T.S.) and CA124495 (J.K.)
Footnotes
Conflict of interest declaration
The authors declare no conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- 1.Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer. 2011;11(11):761–74. doi: 10.1038/nrc3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3(1):11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- 3.Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012;72(10):2457–67. doi: 10.1158/0008-5472.CAN-11-2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gysin S, Salt M, Young A, McCormick F. Therapeutic strategies for targeting ras proteins. Genes Cancer. 2011;2(3):359–72. doi: 10.1177/1947601911412376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Singh A, Settleman J. Oncogenic K-ras “addiction” and synthetic lethality. Cell Cycle. 2009;8(17):2676–7. doi: 10.4161/cc.8.17.9336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chan DA, Giaccia AJ. Harnessing synthetic lethal interactions in anticancer drug discovery. Nat Rev Drug Discov. 2011;10(5):351–64. doi: 10.1038/nrd3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scholl C, Frohling S, Dunn IF, Schinzel AC, Barbie DA, Kim SY, et al. Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell. 2009;137(5):821–34. doi: 10.1016/j.cell.2009.03.017. [DOI] [PubMed] [Google Scholar]
- 8.Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462(7269):108–12. doi: 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Luo J, Emanuele MJ, Li D, Creighton CJ, Schlabach MR, Westbrook TF, et al. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009;137(5):835–48. doi: 10.1016/j.cell.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vicent S, Chen R, Sayles LC, Lin C, Walker RG, Gillespie AK, et al. Wilms tumor 1 (WT1) regulates KRAS-driven oncogenesis and senescence in mouse and human models. J Clin Invest. 2010;120(11):3940–52. doi: 10.1172/JCI44165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Y, Ngo VN, Marani M, Yang Y, Wright G, Staudt LM, et al. Critical role for transcriptional repressor Snail2 in transformation by oncogenic RAS in colorectal carcinoma cells. Oncogene. 2010;29(33):4658–70. doi: 10.1038/onc.2010.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Babij C, Zhang Y, Kurzeja RJ, Munzli A, Shehabeldin A, Fernando M, et al. STK33 kinase activity is nonessential in KRAS-dependent cancer cells. Cancer research. 2011;71(17):5818–26. doi: 10.1158/0008-5472.CAN-11-0778. [DOI] [PubMed] [Google Scholar]
- 13.Luo T, Masson K, Jaffe JD, Silkworth W, Ross NT, Scherer CA, et al. STK33 kinase inhibitor BRD-8899 has no effect on KRAS-dependent cancer cell viability. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(8):2860–5. doi: 10.1073/pnas.1120589109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weiwer M, Spoonamore J, Wei J, Guichard B, Ross NT, Masson K, et al. A Potent and Selective Quinoxalinone-Based STK33 Inhibitor Does Not Show Synthetic Lethality in KRAS-Dependent Cells. ACS Med Chem Lett. 2012;3(12):1034–8. doi: 10.1021/ml300246r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bittker JA, Weiwer M, Lewis T, Shimada K, Yang WS, MacPherson L, et al. Screen for RAS-Selective Lethal Compounds and VDAC Ligands - Probe. 2010;2 [PubMed] [Google Scholar]
- 16.Bittker JA, Weiwer M, Shimada K, Yang WS, MacPherson L, Dandapani S, et al. Screen for RAS-Selective Lethal Compounds and VDAC Ligands - Probe. 2010;1 [PubMed] [Google Scholar]
- 17.Torrance CJ, Agrawal V, Vogelstein B, Kinzler KW. Use of isogenic human cancer cells for high-throughput screening and drug discovery. Nat Biotechnol. 2001;19(10):940–5. doi: 10.1038/nbt1001-940. [DOI] [PubMed] [Google Scholar]
- 18.Guo W, Wu S, Liu J, Fang B. Identification of a small molecule with synthetic lethality for K-ras and protein kinase C iota. Cancer research. 2008;68(18):7403–8. doi: 10.1158/0008-5472.CAN-08-1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shaw AT, Winslow MM, Magendantz M, Ouyang C, Dowdle J, Subramanian A, et al. Selective killing of K-ras mutant cancer cells by small molecule inducers of oxidative stress. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(21):8773–8. doi: 10.1073/pnas.1105941108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fennema E, Rivron N, Rouwkema J, van Blitterswijk C, de Boer J. Spheroid culture as a tool for creating 3D complex tissues. Trends Biotechnol. 2013;31(2):108–15. doi: 10.1016/j.tibtech.2012.12.003. [DOI] [PubMed] [Google Scholar]
- 21.Cukierman E, Pankov R, Stevens DR, Yamada KM. Taking cell-matrix adhesions to the third dimension. Science. 2001;294(5547):1708–12. doi: 10.1126/science.1064829. [DOI] [PubMed] [Google Scholar]
- 22.Pampaloni F, Reynaud EG, Stelzer EH. The third dimension bridges the gap between cell culture and live tissue. Nat Rev Mol Cell Biol. 2007;8(10):839–45. doi: 10.1038/nrm2236. [DOI] [PubMed] [Google Scholar]
- 23.Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. Journal of biomolecular screening. 1999;4(2):67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 24.Antoni D, Burckel H, Josset E, Noel G. Three-dimensional cell culture: a breakthrough in vivo. Int J Mol Sci. 2015;16(3):5517–27. doi: 10.3390/ijms16035517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zanoni M, Piccinini F, Arienti C, Zamagni A, Santi S, Polico R, et al. 3D tumor spheroid models for in vitro therapeutic screening: a systematic approach to enhance the biological relevance of data obtained. 2016;6:19103. doi: 10.1038/srep19103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamada KM, Cukierman E. Modeling tissue morphogenesis and cancer in 3D. Cell. 2007;130(4):601–10. doi: 10.1016/j.cell.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 27.Powell K. Adding depth to cell culture. Science. 2017;356(6333):96–8. [Google Scholar]
- 28.Clevers H. Modeling Development and Disease with Organoids. Cell. 2016;165(7):1586–97. doi: 10.1016/j.cell.2016.05.082. [DOI] [PubMed] [Google Scholar]
- 29.Madoux F, Tanner A, Vessels M, Willetts L, Hou S, Scampavia L, et al. A 1536-Well 3D Viability Assay to Assess the Cytotoxic Effect of Drugs on Spheroids. SLAS Discov. 2017;22(5):516–24. doi: 10.1177/2472555216686308. [DOI] [PubMed] [Google Scholar]
- 30.Zhang J-H, Chung TDY, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. Journal of Biomolecular Screening. 1999;4(2):67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 31.Longati P, Jia X, Eimer J, Wagman A, Witt M-R, Rehnmark S, et al. 3D pancreatic carcinoma spheroids induce a matrix-rich, chemoresistant phenotype offering a better model for drug testing. BMC Cancer. 2013;13(1):95. doi: 10.1186/1471-2407-13-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Verjans ET, Doijen J, Luyten W, Landuyt B, Schoofs L. Three-dimensional cell culture models for anticancer drug screening: Worth the effort? J Cell Physiol. 2017 doi: 10.1002/jcp.26052. [DOI] [PubMed] [Google Scholar]
- 33.Tung YC, Hsiao AY, Allen SG, Torisawa YS, Ho M, Takayama S. High-throughput 3D spheroid culture and drug testing using a 384 hanging drop array. Analyst. 2011;136(3):473–8. doi: 10.1039/c0an00609b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramaiahgari SC, den Braver MW, Herpers B, Terpstra V, Commandeur JN, van de Water B, et al. A 3D in vitro model of differentiated HepG2 cell spheroids with improved liver-like properties for repeated dose high-throughput toxicity studies. Arch Toxicol. 2014;88(5):1083–95. doi: 10.1007/s00204-014-1215-9. [DOI] [PubMed] [Google Scholar]
- 35.Lingrel JB, Kuntzweiler T. Na+,K(+)-ATPase. J Biol Chem. 1994;269(31):19659–62. [PubMed] [Google Scholar]
- 36.Alevizopoulos K, Calogeropoulou T, Lang F, Stournaras C. Na+/K+ ATPase inhibitors in cancer. Curr Drug Targets. 2014;15(10):988–1000. [PubMed] [Google Scholar]
- 37.Delebinski CI, Georgi S, Kleinsimon S, Twardziok M, Kopp B, Melzig MF, et al. Analysis of proliferation and apoptotic induction by 20 steroid glycosides in 143B osteosarcoma cells in vitro. Cell Prolif. 2015;48(5):600–10. doi: 10.1111/cpr.12208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Denicolai E, Baeza-Kallee N, Tchoghandjian A, Carre M, Colin C, Jiglaire CJ, et al. Proscillaridin A is cytotoxic for glioblastoma cell lines and controls tumor xenograft growth in vivo. Oncotarget. 2014;5(21):10934–48. doi: 10.18632/oncotarget.2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Felth J, Rickardson L, Rosen J, Wickstrom M, Fryknas M, Lindskog M, et al. Cytotoxic effects of cardiac glycosides in colon cancer cells, alone and in combination with standard chemotherapeutic drugs. J Nat Prod. 2009;72(11):1969–74. doi: 10.1021/np900210m. [DOI] [PubMed] [Google Scholar]
- 40.Kim N, Yim HY, He N, Lee CJ, Kim JH, Choi JS, et al. Cardiac glycosides display selective efficacy for STK11 mutant lung cancer. Sci Rep. 2016;6:29721. doi: 10.1038/srep29721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Foster SA, Whalen DM, Ozen A, Wongchenko MJ, Yin J, Yen I, et al. Activation Mechanism of Oncogenic Deletion Mutations in BRAF, EGFR, and HER2. Cancer Cell. 2016;29(4):477–93. doi: 10.1016/j.ccell.2016.02.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.