

Abstract
Heterozygous somatic mutations in spliceosome genes (U2AF1, SF3B1, ZRSR2, or SRSF2) occur in >50% of myelodysplastic syndrome (MDS) patients. These mutations occur early in disease development, suggesting that they contribute to MDS pathogenesis and may represent a unique genetic vulnerability for targeted therapy. Here we show that RNA splicing perturbation by expression of the U2AF1(S34F) mutant causes accumulation of R loops, a transcription intermediate containing RNA:DNA hybrids and displaced single-stranded DNA, and elicits an ATR response. ATR inhibitors (ATRi) induced DNA damage and cell death in U2AF1(S34F)-expressing cells, and these effects of ATRi were enhanced by splicing modulating compounds. Moreover, ATRi-induced DNA damage was suppressed by overexpression of RNaseH1, an enzyme that specifically removes the RNA in RNA:DNA hybrids, suggesting that the ATRi sensitivity of U2AF1(S34F)-expressing cells arises from R loops. Taken together, our results demonstrate that ATR may represent a novel therapeutic target in MDS patients carrying the U2AF1(S34F) mutation and potentially other malignancies harboring spliceosome mutations.
Keywords: U2AF1, SRSF2, Myelodysplastic Syndrome, R loop, RNA:DNA hybrids, ATR, targeted therapy
Introduction
Myelodysplastic syndrome (MDS), a heterogeneous group of clonal hematopoietic stem cell disorders, is the most common adult myeloid malignancy (1). MDS is characterized by peripheral blood cytopenias and progenitor expansion, and can transform to chemo-resistant secondary acute myeloid leukemia (sAML) (2). Somatic heterozygous mutations in the spliceosome genes such as U2AF1, SF3B1, SRSF2, and ZRSR2 occur in over 50% of MDS patients and are mutually exclusive (3–11). We and others have identified U2AF1 (U2 small nuclear RNA auxiliary factor 1) mutations in 11% of MDS patients, making it one of the most commonly mutated genes in this disease (3,4). U2AF1 mutations are typically present in the founding clones of MDS tumors, suggesting that U2AF1 mutations are important in disease initiation. Although the mechanism by which U2AF1 mutations contribute to MDS pathogenesis remains unclear, the occurrence of U2AF1 mutations early in disease development suggests that these are likely driver mutations (9,10), raising the possibility that they generate specific oncogenic stresses.
Bone marrow cells from patients and genetically-engineered mouse models carrying spliceosome mutations displayed RNA splicing abnormalities (12–24). Expression of MDS-associated U2AF1, SF3B1, ZRSR2, or SRSF2 mutants in cell lines also altered RNA splicing. These studies suggest that cells harboring spliceosome mutations have altered splicing programs, which may contribute to MDS pathogenesis. Intriguingly, hotspot mutations in different spliceosome proteins (U2AF1, S34F/Y or Q157P/Q; SF3B1, K700E; SRSF2, P95H) altered splicing of distinct groups of transcripts, raising an important question as to how these spliceosome mutations converge on similar disease mechanisms. We recently demonstrated that RNA splicing perturbation by either pharmacologic modulation or expression of the U2AF1 S34F (U2AF1S34F) mutant increased levels of R loops, a transcription intermediate containing an RNA:DNA hybrid and displaced single-stranded DNA (ssDNA) (25). Although R loops have physiological functions, aberrant levels and distributions of R loops are associated with genomic instability (26–28). Since RNA splicing normally occurs in a transcription-coupled manner, splicing perturbations may interfere with transcription elongation and increase R loop formation (29). The associations of RNA splicing perturbation, R loop accumulation, and genomic instability prompted us to investigate whether the spliceosome mutations in MDS generate a common vulnerability that can be exploited therapeutically.
Replication Protein A (RPA), a ssDNA-binding heterotrimeric complex, has diverse functions in DNA replication, DNA repair and other cellular processes (30). During responses to DNA damage and replication problems, RPA functions as a key sensor of ssDNA at sites of DNA damage and stalled DNA replication forks. RPA-coated ssDNA (RPA-ssDNA) acts as a platform to recruit the ATR checkpoint kinase and its regulators and substrates (31). We recently found that RPA is present at R loops and is important for R loop suppression through its interaction with RNaseH1, an enzyme that specifically removes the RNA moiety within RNA:DNA hybrids (25). Given the role of RPA as a master sensor of genomic stress arising from diverse sources, our results raised the possibility that the RPA at R loops may enable ATR to respond to aberrant R loops or the genomic instability that they induce.
Here, we report that cells expressing mutant splicing factors accumulated R loops and elicited an R loop-associated ATR response. ATR inhibition using specific ATR inhibitors (ATRi) induced more DNA damage in cells expressing the U2AF1S34F mutant than in cells expressing wild-type U2AF1 (U2AF1WT), killing U2AF1S34F-expressing cells preferentially. The spliceosome modulator E7107, which specifically targets the SF3B complex, induced further R loop accumulation and an ATR response in U2AF1S34F-expressing cells, rendering cells more sensitive to ATRi. Consequently, combination of E7107 and ATRi (E7107+ATRi) induced significantly higher levels of DNA damage in U2AF1S34F-expressing cells compared to U2AF1WT-expressing cells, resulting in an increase in apoptosis. Finally, expression of RNaseH1 attenuated the E7107+ATRi-induced DNA damage in U2AF1S34F-expressing cells, suggesting that the DNA damage induced by E7107 and ATRi arises from R loops. These results suggest that ATR plays an important role in suppressing the R loop-associated genomic instability in U2AF1S34F-expressing cells and maintaining cell viability. Altogether, our results provide a preclinical rationale to test ATR inhibitors in MDS and other myeloid malignancies driven by the U2AF1S34F mutation. Furthermore, they provide a basis to characterize other spliceosome mutations and possibly exploit the R loop-associated vulnerability induced by splicing perturbations.
Materials & Methods
Cell culture
The HeLa cells used in this study were obtained from Dr. Stephen Elledge’s laboratory, and have been analyzed by RNA-seq. The K562 cells were obtained from ATCC and have been analyzed by RNA-seq. The OCI-AML3 cells were obtained from DSMZ without any further authentication. All cell lines used in this study were tested for Mycoplasma and passaged for less than 2 months after thawing. HeLa cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2mM Glutamine, and 1% penicillin/streptomycin. The HeLa-derived cell lines that inducibly express GFP-tagged nuclear RNaseH1 were generated by lentiviral infection and neomycin selection. All HeLa-derived cell lines were cultured in medium supplemented with G418 (600 μg/ml). RNaseH1-GFP expression was induced by doxycycline (200 ng/ml) for 48 h. Viruses expressing indicated Flag-tagged wildtype or mutant U2AF1 and SRSF2 containing an IRES-GFP were used to infect HeLa cells (22). The plasmids also contain an IRES-GFP, which was used to sort for transduced cells. K562 cells stably expressing Flag-tagged U2AF1WT and U2AF1S34F containing an P2A-mCherry were grown in RPMI 1640 medium supplemented with 10% FBS, 1X Gluta-Max and 1X penicillin/streptomycin. OCI-AML3 cells carrying doxycycline inducible Flag-tagged wildtype or mutant U2AF1 were cultured in α-MEM supplemented with 20% FBS, 1X Gluta-Max and 1X penicillin/streptomycin.
Inhibitors and antibodies
Cells were treated with inhibitors: pladienolide B (EMD Millipore), E7107 (H3 Biomedicine), ATR inhibitors (VE-821, VE-822, SelleckChem; AZ-20), ATMi (KU55933, SelleckChem). Antibodies used in this study include AQR (A302–547A-T, Bethyl), pChk2 (2661, Cell Signaling), Flag (F7425, Sigma), GAPDH, (ABS16, Millipore), GFP (A11122, ThermoFisher Scientific), γH2AX (9718S, Cell Signaling), Ku70 (GTX70271, Genetex), PCNA (sc-56, Santa Cruz Biotechnology), pRPA (A300–246A, Bethyl), U2AF1 (ab197591, abcam), and S9.6 (Protein A purified from hybridoma S9.6 by Antibodies Incorporated).
RNA extraction, reverse transcription and alternative RNA splicing assay
RNA material was extracted from cells with RNeasy Plus Mini Kit (Qiagen) according to the manufacturer’s instructions. Total RNA (2 μg) was used for reverse transcription to cDNA using the High-Capacity RNA-to-cDNA™ Kit (Applied Biosystems), followed by PCR using Platinum® Blue PCR Supermix (Invitrogen). Primer sequences for indicated genes are listed in Supplementary Table 1. PCR products were visualized using gel imager G:Box (Syngene) after separation in agarose gel electrophoresis or SDS-PAGE. The intensity of each segregated band was quantified using ImageJ.
Cell viability assay
Cell viability measurement for HeLa-derived, OCI-AML3, and K562-derived cells was performed using Cell TiterGlo and CellTiter 96®AQueous One Solution Cell Proliferation Assay Kit (Promega), respectively.
Cell apoptosis assay
Cell apoptosis assay was performed according to manufacturer’s protocol (BD Biosciences). Briefly, cells were washed once with PBS, followed by addition of fluorophore-conjugated Annexin V and DAPI or 7-AAD. After a 15-minute incubation at room temperature, cells were analyzed using flow cytometry analysis on a Fortessa X-20 (BD Biosciences).
Isolation and ex vivo culture of primary human CD34+ hematopoietic cells
Deidentified umbilical cord blood units (UCB) was obtained from New York Blood Center and was deemed by the center as research-grade. Informed consent was given previously from the donor to the New York Blood Center. The authors in this study have no contacts with donors. The use of UCB was conducted under IRB approval from Massachusetts General Hospital (2014P000092/MGH) and were processed within 48 hours after collection. The UCB was diluted at 1:1 ratio at room temperature in 1X PBS (Gibco) supplemented with 2% FBS and 1X Penicillin/Streptomycin. Total mononuclear cells (MNCs) were fractionated by gradient centrifugation in Ficoll-Paque Plus (GE Healthcare). After removal of platelet cells and erythrocytes, CD34+ cells were enriched from the total MNCs using autoMACS-positive selection system and CD34 MicroBead Kit UltraPure Human (Miltenyi Biotec) according to the manufacturer’s instructions. The isolated CD34+ cells were recovered overnight in X-Vivo-15 media (Lonza) supplemented by 50 ng/mL of each cytokine (human interleukin-3, stem cell factor, thrombopoietin and FLT-3 ligand) at 37°C in the 5% CO2 incubator. All cytokines were purchased from Peprotech. Isolated CD34+ cells were recovered and assessed by flow cytometry analysis using an APC-conjugated CD34 antibody (BD Biosciences). CD34+ cells were cultured in 24-well plates (2 × 105 cells/well) in X-Vivo-15 media supplemented with cytokines and 4 μg/μL of Polybrene (Millipore). Cells were infected with lentiviral vectors carrying U2AF1WT or U2AF1S34F (1180 × g, 90 minutes) at 30°. After 24h recovery, CD34+ cells were transferred into 96-well plates (5 × 104 cells/well) and treated with indicated VE-821 concentration for 48h. GFP-positive cells were sorted and immediately processed for γH2AX immunofluorescence. For R-loop staining, CD34+ cells were infected with lentiviral vectors carrying U2AF1WT or U2AF1S34F for 48h. GFP-positive cells were sorted and cultured for another 72h prior to immunofluorescence. For apoptosis assay, GFP-sorted cells were treated with ATRi for 48h followed by Annexin V staining to measure apoptotic cell population.
Immunofluorescence
For immunofluorescence in HeLa cells, samples were prepared as previously described (25). For immunofluorescence using phospho-H2AX antibody in K562 and primary CD34+ cells, cells were pelleted at 500 rpm for 5–8 minutes, then washed once with PBS. Cells were then fixed with 3% paraformaldehyde/2% sucrose for 15 minutes and cytospun onto coated glass slides. Subsequently, cells were permeabilized with 1x PBS containing 0.5% Triton X-100, and treated in blocking buffer (1x PBS, 3% BSA, 0.05% Tween-20, and 10% milk) prior to primary antibody incubation for 2 h at room temperature. After the incubation in primary antibody, cells were washed three times with 1x PBS containing 0.05% Tween-20 and incubated with anti-rabbit secondary antibody for 1 h. To visualize nuclei, cells were stained with DAPI after the final wash in PBS. The images were captured using a Nikon 90i microscope and analyzed using ImageJ software.
Results
Expression of U2AF1S34F induces an R loop-associated ATR response
We recently showed that RPA localizes to R loops and interacts with RNaseH1, an enzyme that specifically hydrolyzes RNA:DNA hybrids to promote R loop resolution (25). In the absence of R loop suppressor AQR, R loop levels were elevated, and the RPA32 colocalized with R loops was phosphorylated at Ser33 (RPA32 pS33; p-RPA) (25), suggesting that a kinase is active at R loops. Similar to AQR depletion, expression of U2AF1S34F increased R loops (25), prompting us to test whether U2AF1S34F also activates the kinase that phosphorylates RPA. We generated HeLa cells stably expressing either U2AF1WT or U2AF1S34F at levels below that of endogenous U2AF1 (Supplementary Fig. S1A). Even at levels lower than U2AF1WT and endogenous U2AF1, U2AF1S34F exerted dominant effects and induced R loop accumulation (25). Compared with cells expressing U2AF1WT, U2AF1S34F-expressing cells exhibited higher levels of p-RPA in both immunofluorescence and western blot (Fig. 1A-C). Similar to that in HeLa cells, expression of U2AF1S34F in the leukemia cell line K562 also induced p-RPA (Fig. 1D). To test whether p-RPA arises from U2AF1S34F-induced R loops, we generated a U2AF1S34F-expressing HeLa cell line that inducibly produces GFP-tagged nuclear RNaseH1 (Supplementary Fig. S1B-C). Induction of RNaseH1 in U2AF1S34F-expressing cells reduced p-RPA staining to a level similar to that in U2AF1WT-expressing cells (Fig. 1A-B), showing that U2AF1S34F induces p-RPA in an R loop-dependent manner. Next, we tested whether U2AF1S34F-induced RPA phosphorylation is mediated by the ATR kinase, which interacts with RPA (31,32). The p-RPA in U2AF1S34F-expressing HeLa and K562 cells was reduced by an inhibitor of ATR (ATRi; VE-821) in a dose-dependent manner, but not by an inhibitor of the related kinase ATM (ATMi; KU55933) (Fig. 1C-D, Supplementary Fig. S1D) (33,34). In contrast to ATR, ATM was not activated in U2AF1S34F-expressing cells as indicated by the lack of Chk2 phosphorylation (Supplementary Fig. S1E). However, Chk2 was phosphorylated in U2AF1S34F-expressing cells after Camptothecin (CPT) treatment and the phosphorylation was abolished by ATMi (Supplementary Fig. S1E), showing that the ATM pathway is intact in these cells and the ATMi is effective. Together, these results suggest that U2AF1S34F expression induces R loop accumulation and activates ATR but not ATM in an R loop-dependent manner.
Figure 1. U2AF1S34F expressing cells induce R loop-associated ATR-mediated RPA32 phosphorylation.
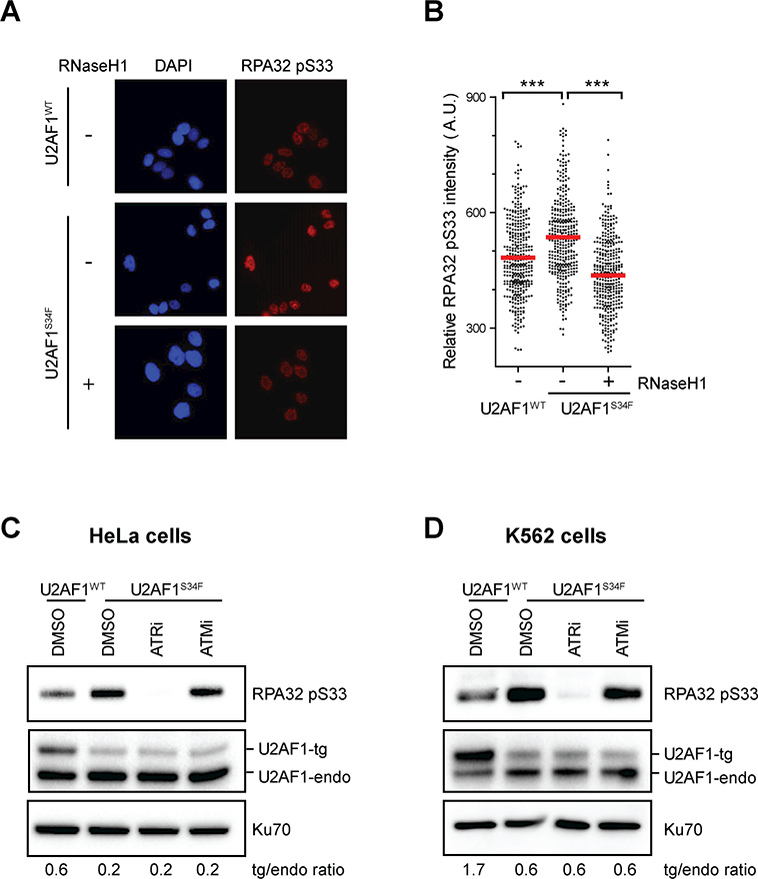
(A and B) HeLa cells stably expressing either U2AF1WT or U2AF1S34F were cultured for 24h. RNaseH1 expression was induced by the addition of doxycycline as indicated. Individual cells were analyzed by immunofluorescence using an RPA32 phospho-specific S33 (RPA32 pS33) antibody. Representative images are shown in A; in B, intensities of RPA32 pS33 staining in individual cells were analyzed by immunofluorescence (n>350). Red bars represent the mean RPA32pS33 intensities of the indicated cell populations. ***, p≤0.001. (C and D) HeLa and K562 cells stably expressing either U2AF1WT or U2AF1S34F were cultured for 48 hours, followed by treatment with either ATRi or ATMi (10 μM) for 1h. Levels of indicated proteins were analyzed by western blot and the ratios of transgenic (tg): endogenous (endo) U2AF1 are shown.
U2AF1S34F induces alternative splicing independently of R loops
The U2AF1-S34F mutation in MDS and AML leads to alternative splicing of a subset of genes, principally by promoting exon skipping and utilization of cryptic 3’ splice sites (4,12,35). The relationship between R loops and aberrant splicing in U2AF1S34F-expressing cells is not known. To test whether U2AF1S34F-induced R loops contribute to aberrant splicing, we examined the splice isoform changes in five transcripts (MED24, KMT2D/MLL, BCOR, PICALM, and H2AFY) that were previously identified in MDS patient bone marrow and shown to be caused by U2AF1S34F (Fig. 2A-E) (35). As previously reported, U2AF1S34F expression altered the splicing of these transcripts. However, induction of RNaseH1 in U2AF1S34F-expressing cells did not affect levels of the alternative splice isoforms. These results suggest that U2AF1S34F-induced alternative RNA splicing occurs independently of R loops and associated ATR response. The protein levels of several known suppressors of R loops were similar in U2AF1WT- and U2AF1S34F-expressing cells (Supplementary Fig. 2), suggesting that alternative splicing of R loop suppressors is unlikely the cause of U2AF1S34F-induced R loops. Thus, the dominant effects of U2AF1S34F on the spliceosome may induce alternative splicing and R loop accumulation independently.
Figure 2. RNaseH1 overexpression does not affect alternative splicing in U2AF1S34F-expressing cells.
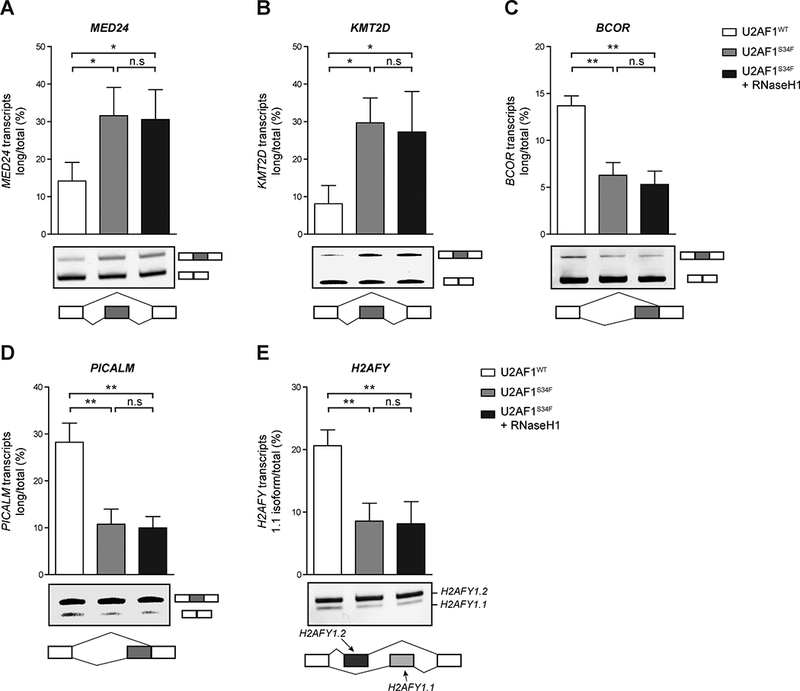
Quantification of exon skipping (MED24, KMT2D) in A and B, alternative splice site utilization (BCOR, PICALM) in C and D, or mutually exclusive exons (H2AFY) in E were detected and measured by gel electrophoresis (n=3 replicates). Representative images of the gel image (middle panel), and a corresponding diagram of the event measured (bottom panel) are shown. n.s., non-significant; *, p≤0.05; **, p≤0.01; ***, p≤0.001.
U2AF1S34F and other MDS-associated spliceosome mutants sensitize cells to ATRi
The ATR response triggered by U2AF1S34F expression prompted us to investigate whether ATR is functionally important in U2AF1S34F-expressing cells. We treated U2AF1WT- and U2AF1S34F-expressing HeLa cells with ATRi or ATMi for 48 hours, and then measured DNA damage levels using γH2AX as a marker (Fig. 3A). Without altering the cell cycle significantly, ATRi, but not ATMi, induced γH2AX in U2AF1S34F-expressing cells (Fig. 3A, Supplementary Fig. S3A). Furthermore, ATRi induced higher levels of γH2AX in U2AF1S34F-expressing cells than in U2AF1WT-expressing cells (Fig. 3A). ATRi also reduced the viability of U2AF1S34F-expressing cells more than that of U2AF1WT-expressing cells (Fig. 3B). In contrast, U2AF1WT- and U2AF1S34F-expressing cells exhibited similar sensitivity to ATMi (Fig. 3B). To confirm the results from HeLa cells, we inducibly expressed U2AF1WT and U2AF1S34F in the AML cell line OCI-AML3 (Supplementary Fig. S3B) (36). Neither U2AF1WT nor U2AF1S34F rendered OCI-AML3 cells sensitive to ATMi (Fig. 3C). However, compared with U2AF1WT OCI-AML3 cells, U2AF1S34F OCI-AML3 cells were more susceptible to growth inhibition by ATRi (Fig. 3C). These results show that ATR but not ATM promotes the survival of U2AF1S34F-expressing cells by suppressing DNA damage.
Figure 3. Cells expressing spliceosome mutations confer sensitivity to ATR inhibitors.
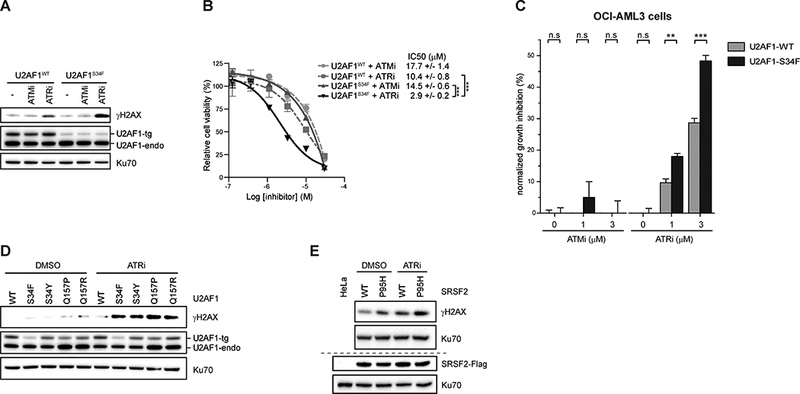
(A) HeLa cells stably expressing either U2AF1WT or U2AF1S34F were treated with either ATMi or ATRi (5 μM) for 48h. Levels of indicated proteins were analyzed by western blot. (B) HeLa cells stably expressing either U2AF1WT or U2AF1S34F were either treated with indicated ATMi or ATRi for 4 days. Relative percent of cell survival was measured by CellTiter-Glo Solution. ***, p≤0.001. (C) U2AF1WT or U2AF1S34F expression in OCI-AML3 cells were induced by addition of doxycycline (250 ng/mL) for 48h. Cells were then treated with either DMSO or indicated ATRi doses for 5 days. Cell growth was measured by CellTiter96®AQueous One Solution. **, p≤0.01, ***, p≤0.001. (D) HeLa cells stably expressing either wild type or different mutants of U2AF1 were treated with ATRi (5 μM) for 48h. Levels of indicated proteins were analyzed by western blot. (E) HeLa cells stably expressing either wild type or P95H mutant of SRSF2 were treated with DMSO or ATRi (5 μM) for 48h. The levels of indicated proteins were analyzed by western blot.
The increased ATRi sensitivity of U2AF1S34F-expressing cells led us to ask whether other spliceosome mutations also sensitize cells to ATRi. To examine the effects of other MDS-associated U2AF1 mutations on the ATRi response, we generated HeLa cell lines that stably express the S34Y, Q157P, or Q157R mutant alleles at similar levels (Fig. 3D). In all the cell lines expressing these U2AF1 mutants, ATRi induced higher levels of γH2AX than in U2AF1WT-expressing cells (Fig. 3D). To test whether mutations in splicing factors other than U2AF1 also sensitize cells to ATRi, we generated stable cells expressing wild-type SRSF2 (SRSF2WT) and its MDS-associated mutant derivative, SRSF2P95H. SRSF2P95H-expressing cells exhibited higher levels of γH2AX than SRSF2WT-expresing cells in the absence of ATRi (Fig. 3E). ATRi treatment further increased γH2AX levels in SRSF2P95H-expressing cells (Fig. 3E). Collectively, these results suggest that the MDS-associated U2AF1 and SRSF2 mutations create a dependency on ATR to suppress DNA damage.
RNA splicing modulators induce R loops and associated ATR response
Having established that U2AF1S34F induces R loops and associated ATR response, we investigated whether perturbation of RNA splicing by splicing modulators could also activate ATR through induction of R loops. E7107, a splicing modulator that targets the SF3B complex, induces splicing alterations such as intron retention (37–39). As previously described, we used the monoclonal antibody S9.6 that specifically recognizes DNA:RNA hybrids to detect nuclear R loops by immunofluorescence (25,40). E7107-treated HeLa cells showed an increase in S9.6 staining compared to control cells treated with DMSO (Fig. 4A). We also observed colocalization of p-RPA and S9.6 staining in E7107-treated cells but not control cells (Fig. 4B). Notably, p-RPA and S9.6 staining increased together in individual E7107-treated cells (Fig. 4C), suggesting that they reflect two associated events induced by E7107.
Figure 4. RNA splicing perturbation by pharmacologic compounds induce R loop-associated ATR-mediated RPA32 phosphorylation.
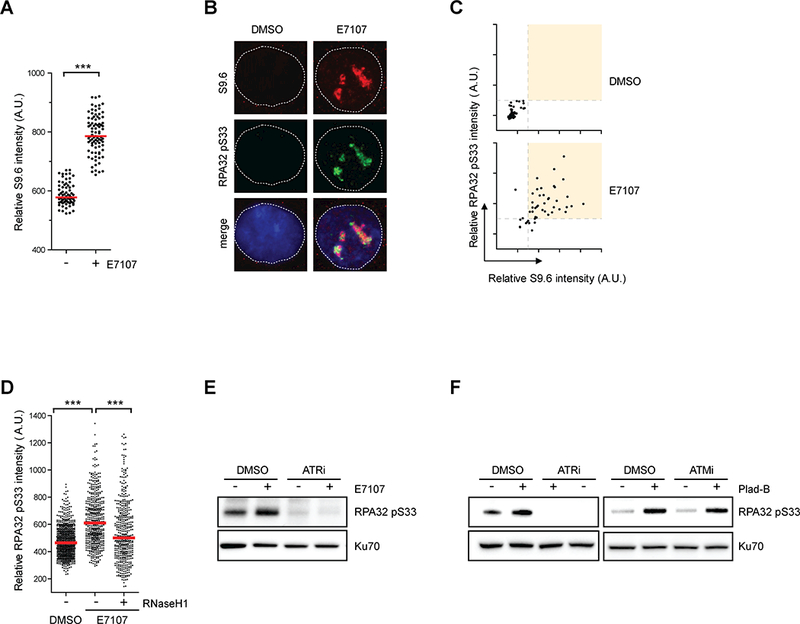
(A) HeLa cells were treated with DMSO or E7107 (1 nM) for 24h. Individual cells were analyzed by immunofluorescence using the S9.6 antibody. Intensities of S9.6 staining in individual cells were analyzed by immunofluorescence (n>70). Red bars represent the median S9.6 intensities of the indicated cell populations. ***, p≤0.001. (B-C) HeLa cells were treated with DMSO or E7107 (2 nM) for 24h. In B, representative images individual cells stained with S9.6 and p-RPA. In C, intensities of S9.6 and p-RPA staining of individual cells were plotted in 2D. (D) HeLa cells inducibly expressing RNaseH1 were treated with DMSO or E7107 (1 nM) for 24h. RNaseH1 expression was induced by addition of doxycycline as indicated. Intensities of RPA32 pS33 staining in individual cells were analyzed by immunofluorescence (n>460). Red bars represent the median RPA32 pS33 intensities of the indicated cell populations. ***, p≤0.001. (E) HeLa cells were treated with DMSO or E7107 (2 nM) for 24h, followed by ATRi treatment (10 μM, 1h). Indicated proteins were analyzed by western blot. (F) HeLa cells were either treated with DMSO or 1 nM Plad-B. After 24h treatment, ATR or ATM inhibitors (10 μM) were added for 1h. Levels of indicated proteins were analyzed by western blot.
To test whether p-RPA arises from E7107-induced R loops, we generated a stable cell line that inducibly expresses GFP-tagged nuclear RNaseH1 (Supplementary Fig. S4A). Expression of RNaseH1 in E7107-treated cells significantly suppressed p-RPA (Fig. 4D). Furthermore, when HeLa cells were first treated with E7107 and then exposed to ATRi, the p-RPA in E7107-treated cells was reduced by ATRi in a concentration-dependent manner (Fig 4E, Supplementary Fig. S4B). Consistent with these results, Pladienolide B (Plad-B), another splicing modulator that induces R loops (25), also increased p-RPA levels (Fig. 4F). The p-RPA in Plad-B treated cells was eliminated by ATRi but not ATMi (Fig. 4F). Collectively, these results suggest that RNA splicing perturbation by splicing modulating compounds leads to accumulation of R loops and elicits an ATR response.
RNA splicing modulators sensitize cells to ATRi
Given the induction of R loops and ATR response by splicing modulators, we asked whether splicing modulators sensitize cells to ATRi. We treated HeLa cells with E7107 for 24 hours in the presence or absence of ATRi or ATMi (Fig. 5A). Only ATRi but not ATMi induced γH2AX in E7107-treated cells in a dose-dependent manner (Fig. 5A, Supplementary Fig. S5A). Similarly, in Plad-B treated cells, γH2AX was only induced by ATRi but not ATMi (Fig. 5B). In addition to VE-821, two other ATR inhibitors, AZ20 (ATRi #2) and VE-822 (ATRi #3), also induced γH2AX in Plad-B treated cells (Fig. 5C). These results suggest that splicing modulators create a dependence on ATR to suppress DNA damage.
Figure 5. Cells treated with splicing modulators confer sensitivity to ATR inhibitors.

(A) HeLa cells were treated with either ATMi or ATRi (10 μM) in the presence or absence of E7107 (1 nM) for 24h. Levels of indicated proteins were analyzed by western blot. (B-C) HeLa cells were treated with either ATM or ATR inhibitors (10 μM, ATRi: VE-821, ATRi #2: AZ-20, and ATRi #3: VE-822) in the presence or absence of Plad-B (1 nM) for 24h. Levels of indicated proteins were analyzed by western blot. In D-E, individual cells were analyzed by immunofluorescence using a γH2AX antibody. Representative images are shown in D; in E, intensities of γH2AX staining in individual cells were analyzed by immunofluorescence (n>300). Red bars represent the mean γH2AX intensities of the indicated cell populations. ***, p≤0.001. (F) HeLa cells were treated either with ATMi or ATRi in the presence or absence of 0.5 nM Plad-B for 4 days. Relative percent of cell survival was measured by CellTiter-Glo® Solution.
To understand how ATR suppresses DNA damage in cells treated splicing modulators, we analyzed individual cells for γH2AX by immunofluorescence. A significant fraction of the cells treated with Plad-B and ATRi, but not those treated with Plad-B alone, ATRi alone, or Plad-B and ATMi, displayed γH2AX staining (Fig. 5D-E). The majority of the γH2AX-positive cells induced by Plad-B and ATRi showed discrete γH2AX foci, suggesting formation of DNA double-strand breaks (DSBs) (Fig. 5D). A subset of the cells treated with Plad-B and ATRi displayed strong pan-nuclear γH2AX staining and TUNEL (Terminal deoxynuleotidyl transferase-mediated dUTP Nick End Labeling) signals, indicating severe chromosome fragmentation and apoptosis (Fig. 5D-E; Supplementary Fig. S5B-C). Consistent with the Plad-B results, a significant fraction of E7107 treated cells displayed γH2AX staining in the presence of ATRi (Supplementary Fig. S5D).
The collision between R loops and replication forks is a potential source of DNA damage in cells treated with splicing modulators and ATRi. Consistent with this possibility, E7107 reduced S-phase cells (Supplementary Fig. S5E), indicating a checkpoint response to replication problems. To test whether DNA damage arises during DNA replication in cells treated with both E7101 and ATRi, we compared the effects of these compounds on S-phase and non-S-phase cells (Supplementary Fig. S5F). We used PCNA to distinguish S-phase and non-S-phase cells, and γH2AX to measure DNA damage. E7107 and ATRi induced much more DNA damage in S-phase cells than in non-S-phase cells, supporting the idea that the DNA damage induced by E7107 and ATRi primarily arises from the collision between R loops and replication forks (Supplementary Fig. S5F).
Next, to test whether ATRi affects the survival of cells treated with splicing modulators, we treated HeLa cells with ATRi in the presence or absence of 0.5 nM Plad-B (Fig. 5F). ATMi was tested in parallel with ATRi in this experiment. Plad-B only increased the sensitivity of cells to ATRi but not ATMi. Thus, splicing modulators induce R loops and generate replication problems in S-phase cells, possibly through the collision between R loops and replication forks. Upon the inhibition of ATR, the collision between R loops and replication forks likely give rise to high levels of DSBs, which ultimately drive cells into apoptosis.
E7107 potentiates the ATRi sensitivity of U2AF1S34F-expressing cells
We next tested whether splicing modulators can exacerbate R loop accumulation in cells expressing U2AF1S34F and increase their ATRi sensitivity. We treated U2AF1WT- and U2AF1S34F-expressing cells with E7107 for 24 hours and analyzed R loop levels. The baseline levels of R loops are higher in U2AF1S34F-expressing cells than in U2AF1WT-expressing cells (Fig. 6A, lane 1 vs. 3). After E7107 treatment, R loop levels were increased in both U2AF1WT- and U2AF1S34F-expressing cells (Fig. 6A, lane 1 vs. 2, and 3 vs. 4). Importantly, the levels of R loops were much higher in U2AF1S34F-expressing cells than in U2AF1WT-expressing cells (Fig. 6A, lane 2 vs. 4), suggesting that the U2AF1S34F mutant potentiates the induction of R loops by E7107. The increased R loop accumulation in E7107-treated U2AF1S34F-expressing cells was accompanied by an increase in p-RPA (Fig. 6B). Suppression of R loops by RNaseH1 in E7107-treated U2AF1S34F-expressing cells reduced p-RPA levels (Fig. 6B), suggesting that the increased of p-RPA reflects an R loop-associated ATR response. The combination of E7107 and ATRi (E7107+ATRi) induced higher levels of γH2AX in U2AF1S34F-expressing cells than in U2AF1WT-expressing cells (Fig. 6C). The γH2AX levels in U2AF1S34F-expressing cells treated with E7107+ATRi were also higher than those in cells treated with either E7107 or ATRi alone. Importantly, induction of RNaseH1 in U2AF1S34F-expressing cells treated with E7107 and ATRi significantly reduced γH2AX levels (Fig. 6D), showing that the DNA damage induced by E7107 and ATRi arises from R loops. Consistent with the increase in γH2AX, ATRi reduced the viability of U2AF1S34F-expressing cells more than that of U2AFWT-expressing cells in the presence of E7107 (Fig. 6E). The combination of E7107 and ATRi increased apoptosis more efficiently in U2AFS34F-expressing cells than in U2AF1WT-expressing cells (Fig. 6F-G, and Supplementary Fig. S6). Therefore, splicing modulators increase R loop levels in cells expressing U2AF1S34F, rendering them more sensitive to ATRi.
Figure 6. Combination of splicing modulator drugs and ATR inhibitors sensitizes U2AF1S34F-expressing cells.
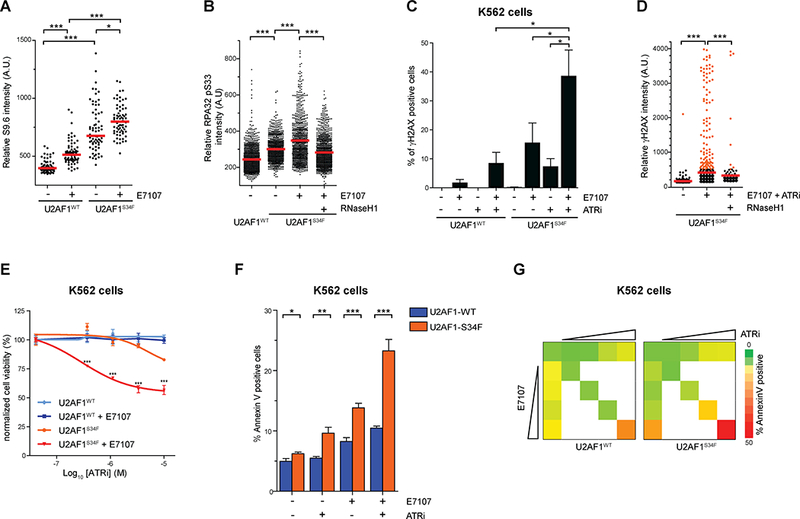
(A) HeLa cells stably expressing U2AF1WT or U2AF1S34F mutant treated with either DMSO or 1 nM E7107 for 24h. Individual cells were analyzed by immunofluorescence using an S9.6 antibody. Intensities of S9.6 staining in individual cells were analyzed by immunofluorescence. Red bars represent the median S9.6 intensities of the indicated cell populations. *, p≤0.05; ***, p≤0.001. (B) HeLa cells stably expressing U2AF1WT or U2AF1S34F mutant treated with either DMSO or 1 nM E7107 for 24h. RNaseH1 expression was induced by the addition of doxycycline simultaneously with E7107 treatment. Individual cells were analyzed by immunofluorescence using an RPA32 pS33 antibody. Intensities of RPA32 pS33 staining in individual cells were analyzed by immunofluorescence (n>800). Red bars represent the median RPA32 pS33 intensities of the indicated cell populations. ***, p≤0.001. (C) U2AF1WT and U2AF1S34F expressing K562 cells were treated with either E7107 (1 nM) or ATRi (3 μM) alone or in combination for 72h. Individual cells were analyzed by immunofluorescence using γH2AX antibody. The percentage of γH2AX positive cells was quantified (n=3 replicates). *, p≤0.05. (D) HeLa cells stably expressing U2AF1S34F mutant was treated with DMSO or combined E7107 (1 nM) and ATRi (3 μM) for 24h. RNaseH1 expression was induced by addition of doxycycline as indicated throughout drug exposure. Intensities of γH2AX staining in individual cells were analyzed by immunofluorescence (n>720). Red bars represent the mean γH2AX intensities of the indicated cell populations. ***, p≤0.001. (E) U2AF1WT- or U2AF1S34F-expressing K562 cells were treated with DMSO or E7107 (2 nM) with varying ATRi concentration for 2 days. Relative cell survival was measured by CellTiter96®AQueous One Solution. (F) U2AF1WT- or U2AF1S34F-expressing K562 cells were treated with 2 nM E7107, 3 μM ATRi, or both for 72h. Percent cell death was analyzed by Annexin V staining. *, p<0.05; **, p<0.005; ***, p<0.0001. (G) U2AF1WT- or U2AF1S34F-expressing K562 cells were treated with increasing doses of E7107 (0 – 4 nM, 2 fold-dilution) or ATRi (0 – 12 μM, 2 fold-dilution) individually or both for 72h. Percent cell death was analyzed by Annexin V staining. Color-coding denotes the percent of apoptotic cells (red [50% Annexin V positive] to green [0% Annexin V staining]).
ATRi induces DNA damage in primary human CD34+ hematopoietic cells expressing U2AF1S34F
Since spliceosome mutations often occur in the founding clones of MDS, we next asked whether U2AF1S34F expression in primary human hematopoietic progenitor cells sensitizes them to ATR inhibition. CD34+ hematopoietic progenitor cells were isolated from human umbilical cord blood (Fig. 7A). Using γ-irradiated CD34+ progenitor cells, we first confirmed that the formation of DSBs in these cells can be reliably monitored with γH2AX immunofluorescence (Supplementary Fig. S7). Next, we expressed GFP-tagged U2AF1WT and U2AF1S34F in CD34+ progenitor cells by lentiviral infection. The CD34+ cells expressing U2AF1WT or U2AF1S34F were isolated by cell sorting and R-loop staining was performed. Consistent with our observation in HeLa and K562 cells, U2AF1S34F-expressing CD34+ cells displayed higher levels of R loops compared to U2AF1WT-expressing CD34+ cells (Fig. 7B). Next, we treated the two CD34+ cell populations with ATRi and used γH2AX to measure the induction of DNA damage. ATR inhibition in U2AF1WT-expressing CD34+ cells did not significantly alter γH2AX levels (Fig. 7C, lanes 1 vs 2). However, ATRi caused a significant increase of γH2AX in U2AF1S34F-expressing CD34+ cells compared to DMSO-treated U2AF1S34F-expressing cells (Fig. 7C, lanes 3 vs 4) and ATRi-treated U2AF1WT-expressing cells (Fig. 7C, lanes 2 vs 4). Finally, we used Annexin V staining to compare the ATRi induced cell death in the two CD34+ cell populations. ATR inhibition, in a dose-dependent manner, induced higher levels of cell death in U2AF1S34F-expressing CD34+ cells than in U2AF1WT-expressing CD34+ cells (Fig. 7D). Collectively, these results demonstrate that expression of U2AF1S34F in human primary CD34+ hematopoietic progenitor cells indeed induces R loops, rendering them susceptible to ATR inhibition.
Figure 7. U2AF1S34F-expression in primary CD34+ hematopoietic progenitor cells confer sensitivity to ATR inhibition.
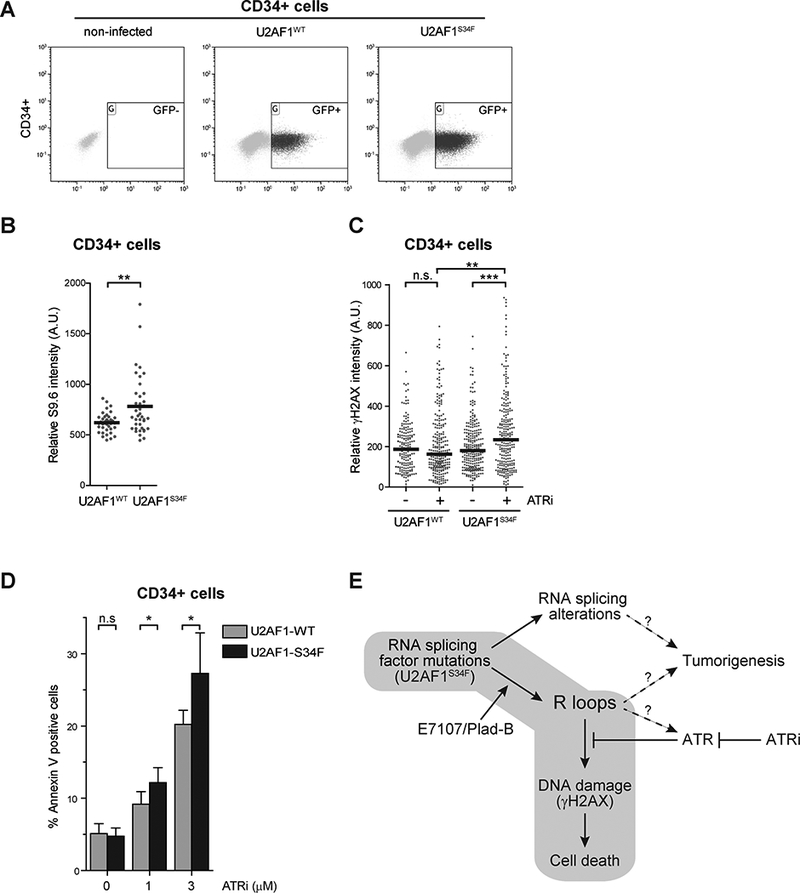
(A) A representative FACS plots of GFP-sorted cell populations primary CD34+ hematopoietic progenitor cells from human umbilical cord blood that were isolated and infected with lentiviruses encoding either U2AF1WT or U2AF1S34F. (B) Cells were isolated, cultured for an additional 72 hours, and subjected to immunofluorescence analysis using an S9.6 antibody. Intensities of S9.6 staining in individual cells were analyzed by immunofluorescence. Black bars represent the median S9.6 intensities of the indicated cell populations. **, p≤0.01. (C) Cells were either treated with DMSO or ATRi (1 μM) for 2 days, followed by cell sorting, and cytospin. Intensities of γH2AX staining in individual cells were analyzed by immunofluorescence from three biological replicates. Black bars represent the mean γH2AX intensities of the indicated cell populations (n>180). **, p≤0.01; ***, p≤0.001; n.s., non-significant. (D) Primary CD34+ hematopoietic progenitor cells from human umbilical cord blood were isolated similar to A. After 24h recovery, cells were seeded and treated with indicated concentrations of ATRi for 48h. Percent cell death was analyzed by Annexin V staining. *, p≤0.05; n.s., non-significant. (E) RNA splicing perturbation in U2AF1S34F cells potentially induces two independent pathways. First, cells expressing U2AF1S34F mutant induces RNA splicing changes that may promote tumorigenesis. Second, U2AF1S34F cells also accumulate R loops, which elicit an ATR-mediated response. Inhibition of ATR increases DNA damage and ultimately cell death. The addition of spliceosome modulators, Pladienolide B (Plad-B) or E7107, further enhances R loop accumulation and an ATR-mediated response.
Discussion
Growing evidence has shown that defects of RNA processing factors, including spliceosome components, trigger R loop accumulation and R loop-associated genomic instability (26–28). We previously reported that expression of the MDS-associated U2AF1S34F mutant caused accumulation of R loops (25). Here, we present evidence that the R loop accumulation in U2AF1S34F-expressing cells elicits an ATR response, which is consistent with a recent report (41). We further show that R loop accumulation is not required for the splicing alterations induced by U2AF1S34F expression. Whether splicing alterations contribute to R loop accumulation is still unclear. Since splicing factor mutations often arise in the founding clones of MDS, both R loops and splicing alterations may contribute to initiation of the disease (Fig. 7E). R loops can not only induce genomic instability but also alter gene expression. Although R loop accumulation alone may not be sufficient to promote wide-spread genomic instability in MDS, which is characterized by a low mutation burden (42,43), aberrant R loops may potentiate specific oncogenic events. Importantly, aberrant R loops become a significant source of DNA damage when ATR is inhibited. The ATRi-induced and R loop-associated DNA damage in U2AF1S34F-expressing cells is sufficient to reduce cell viability. Our results collectively suggest a strategy to therapeutically target the MDS cells harboring U2AF1 and SRSF2 mutations. Future studies will be necessary to test the ability of other MDS-associated splicing factor mutations to induce R loops and R loop-associated ATRi sensitivity.
RPA is a crucial sensor of DNA replication stress and DNA damage in eukaryotic cells (30). At stalled replication forks and resected DSBs, RPA-ssDNA acts as a key platform to recruit the ATR kinase and its regulators and substrates. Like replication forks and repair intermediates, R loops contain ssDNA. We recently showed that the RPA associated with R loops is phosphorylated at S33 (25). Here, we show that RPA is phosphorylated by ATR at R loops. How ATR is activated at R loops remains to be elucidated. The most straightforward possibility is that the RPA-ssDNA in R loops recruits the ATR-ATRIP complex, which in turn phosphorylates RPA. It is also possible that R loops need to be processed by certain repair and recombination factors to trigger the ATR response. Additional proteins are likely required to activate ATR at R loops. In S phase, R loops may activate ATR by stalling replication forks (44). Regardless of how ATR is activated at R loops, its function is needed to suppress R loop-associated DNA damage. Consistent with this notion, the combination of ATRi and E7107 induced DNA damage primarily in S-phase cells. In the absence of ATR, R loops may lead to collapse of DNA replication forks or be aberrantly processed by nucleases. These events could give rise to DSBs, driving cells into apoptosis. Therefore, ATR inhibition abrogates an important protective mechanism at R loops, leaving R loops to generate DNA damage and promote cell death (Fig. 7E). The synthetic lethal relationship between R loop accumulation and ATR inhibition may have broad implications in cancer therapy.
Heterozygous somatic mutation in the spliceosome genes such as U2AF1, SF3B1, SRSF2, or ZRSR2 occur in over 50% of MDS patients (3–10). These spliceosome mutations occur in the founding clones of MDS, implicating RNA splicing perturbation in disease initiation. Intriguingly, the splicing alternations observed in U2AF1 mutant cells are distinct from those in SF3B1 and SRSF2 mutant cells (13–24). In addition, we and others showed that U2AF1S34F/Y and U2AF1Q157P/R mutants induced alterative splicing in different transcripts (17,42). These findings raise an important question as to how mutually exclusive spliceosome mutations converge on similar MDS phenotypes. Here, we show that ATRi treatment of cells expressing the MDS-associated U2AF1 and SRSF2 mutants induced DNA damage. These results raise the possibility that aberrant R loop accumulation may be a common vulnerability in MDS cells harboring spliceosome mutants. In addition, it is tempting to speculate that various spliceosome mutations in MDS may promote disease initiation through R loop induction. In future studies, it will be important to test whether other MDS-associated spliceosome mutants also induce R loops, and whether these R loops have common functional effects in myeloid progenitor cells. Furthermore, it will be important to go beyond cell models and directly test these hypotheses in MDS patients and genetic models. Regardless of whether and how R loops contribute to MDS pathogenesis, they may create a common dependency of MDS cells on ATR for survival, rendering ATR a potential therapeutic target in MDS patients carrying spliceosome mutations. Although our current results are limited to U2AF1 and SRSF2 mutations, our study provides a rationale to characterize other spliceosome mutations and a possibly way to target them in therapy.
Both splicing modulators and ATR inhibitors have already advanced into clinical trials for different cancers (45–47). Splicing modulator therapy is being pursued in MDS patients with spliceosome mutations who have failed first-line therapies (NCT02841540). ATR inhibitors (VX-970 and AZD6738) are currently being tested in several clinical trials in patients with advanced solid malignancies (46). Our results suggest that ATR inhibitors may also benefit MDS patients with U2AF1 and SRSF2 mutations. Furthermore, a recent pan-cancer analysis suggested that as many as 119 splicing factor genes are recurrently mutated and may contribute to oncogenesis in not only in MDS but also in other cancers, including AML, CLL, lung adenocarcinoma, head and neck squamous cell carcinoma, endometrial carcinoma, bladder urothelial carcinoma, breast adenocarcinoma, and colorectal carcinoma (48,49). In future studies, it will be important to address whether all these cancer-associated splicing factor mutations induce R loops. If aberrant R loop accumulation is common in cancer cells harboring spliceosome mutations, ATR inhibitors may have therapeutic potential in a broad spectrum of cancers. Notably, combinations of splicing modulators and ATRi killed U2AF1S34F-expressing cells more effectively than splicing modulators or ATRi alone. Our results suggest that the mechanisms of action for ATRi and splicing modulators are distinct. While splicing modulators promote R loop accumulation, ATRi allows R loops to induce DNA damage (Fig. 7E). These findings suggest that splicing modulators can be used to potentiate the effects of ATRi in the presence of spliceosome mutations, making these drugs more effective in cancer cells at tolerable concentrations. Together, our studies establish a scientific rationale to test ATR inhibitors and splicing modulators in MDS patients carrying U2AF1 and SRSF2 mutations, providing a therapeutic strategy that could potentially be applied to other cancers with spliceosome mutations.
Supplementary Material
Statement of Significance.
This study provides pre-clinical evidence that patients with MDS or other myeloid malignancies driven by spliceosome mutations may benefit from ATR inhibition to exploit the R loop-associated vulnerability induced by perturbations in splicing.
Acknowledgements
We thank Drs. R. Crouch, R. Bradley for reagents, members of the Zou, Dyson, and Graubert labs for helpful discussions. E7107 was kindly provided by S. Buonamici (H3 Biomedicine). H.D. Nguyen is supported by NIH T32 postdoctoral training grant (T32 DK007540). P.N.G. Reddy is supported by a fellowship grant from the American Society of Hematology. L. Zou is the James & Patricia Poitras Endowed Chair in Cancer Research, and was supported by a Jim & Ann Orr Massachusetts General Hospital Research Scholar Award. This work is supported by grants from the NIH (GM076388 and CA197779) to L. Zou, (CA171963) to T. Graubert and M. Walter, the V Foundation to L. Zou and T. Graubert, and the Edward P. Evans Foundation to T. Graubert and M. Walter.
Financial Support: H.D. Nguyen is supported by NIH T32 postdoctoral training grant (T32 DK007540). L. Zou is the James & Patricia Poitras Endowed Chair in Cancer Research, and is supported by a Jim & Ann Orr Massachusetts General Hospital Research Scholar Award. P.N.G. Reddy is supported by a fellowship grant from the American Society of Hematology. This work is supported by grants from the NIH (GM076388 and CA197779) to L. Zou, (CA171963) to T. Graubert and M. Walter, the V Foundation to L. Zou and T. Graubert, and the Edward P. Evans Foundation to T. Graubert and M. Walter.
Abbreviations list
- U2AF1
U2 small nuclear RNA auxiliary factor 1
- SRSF2
Serine/arginine-rich splicing factor 2
- ATR
ataxia telangiectasia and Rad3 related
- RPA
replication protein A
Footnotes
The conflict of interest statement: The authors do not have conflicts of interest to disclose.
References
- 1.Ma X Epidemiology of Myelodysplastic Syndromes. Am J Med. 2012;125:S2–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Troy JD, Atallah E, Geyer JT, Saber W. Myelodysplastic syndromes in the United States: an update for clinicians. Ann Med. 2014;46:283–9. [DOI] [PubMed] [Google Scholar]
- 3.Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478:64–9. [DOI] [PubMed] [Google Scholar]
- 4.Graubert TA, Shen D, Ding L, Okeyo-Owuor T, Lunn CL, Shao J, et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat Genet. 2011;44:53–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D, et al. Somatic SF3B1 Mutation in Myelodysplasia with Ring Sideroblasts. N Engl J Med. 2011;365:1384–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Visconte V, Makishima H, Jankowska A, Szpurka H, Traina F, Jerez A, et al. SF3B1, a splicing factor is frequently mutated in refractory anemia with ring sideroblasts. Leukemia. 2012;26:542–5. [DOI] [PubMed] [Google Scholar]
- 7.Damm F, Kosmider O, Gelsi-Boyer V, Renneville A, Carbuccia N, Hidalgo-Curtis C, et al. Mutations affecting mRNA splicing define distinct clinical phenotypes and correlate with patient outcome in myelodysplastic syndromes. Blood. 2012;119:3211–8. [DOI] [PubMed] [Google Scholar]
- 8.Thol F, Kade S, Schlarmann C, Löffeld P, Morgan M, Krauter J, et al. Frequency and prognostic impact of mutations in SRSF2, U2AF1, and ZRSR2 in patients with myelodysplastic syndromes. Blood. 2012;119:3578–84. [DOI] [PubMed] [Google Scholar]
- 9.Walter MJ, Shen D, Shao J, Ding L, White BS, Kandoth C, et al. Clonal diversity of recurrently mutated genes in myelodysplastic syndromes. Leukemia. 2013;27:1275–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Papaemmanuil E, Gerstung M, Malcovati L, Tauro S, Gundem G, Van Loo P, et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood. 2013;122:3616–27; quiz 3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 2014;28:241–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saez B, Walter MJ, Graubert TA. Splicing factor gene mutations in hematologic malignancies. Blood. 2017;129:1260–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Darman RB, Seiler M, Agrawal AA, Lim KH, Peng S, Aird D, et al. Cancer-Associated SF3B1 Hotspot Mutations Induce Cryptic 3’ Splice Site Selection through Use of a Different Branch Point. Cell Rep. 2015;13:1033–45. [DOI] [PubMed] [Google Scholar]
- 14.Kesarwani AK, Ramirez O, Gupta AK, Yang X, Murthy T, Minella AC, et al. Cancer-associated SF3B1 mutants recognize otherwise inaccessible cryptic 3′ splice sites within RNA secondary structures. Oncogene. 2017;36:1123–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alsafadi S, Houy A, Battistella A, Popova T, Wassef M, Henry E, et al. Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nat Commun. 2016;7:10615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DeBoever C, Ghia EM, Shepard PJ, Rassenti L, Barrett CL, Jepsen K, et al. Transcriptome Sequencing Reveals Potential Mechanism of Cryptic 3’ Splice Site Selection in SF3B1-mutated Cancers. PLOS Comput Biol. 2015;11:e1004105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ilagan JO, Ramakrishnan A, Hayes B, Murphy ME, Zebari AS, Bradley P, et al. U2AF1 mutations alter splice site recognition in hematological malignancies. Genome Res. 2015;25:14–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brooks AN, Choi PS, de Waal L, Sharifnia T, Imielinski M, Saksena G, et al. A Pan-Cancer Analysis of Transcriptome Changes Associated with Somatic Mutations in U2AF1 Reveals Commonly Altered Splicing Events. PLoS One. 2014;9:e87361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Przychodzen B, Jerez A, Guinta K, Sekeres MA, Padgett R, Maciejewski JP, et al. Patterns of missplicing due to somatic U2AF1 mutations in myeloid neoplasms. Blood. 2013;122:999–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shao C, Yang B, Wu T, Huang J, Tang P, Zhou Y, et al. Mechanisms for U2AF to define 3′ splice sites and regulate alternative splicing in the human genome. Nat Struct Mol Biol. 2014;21:997–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yip BH, Steeples V, Repapi E, Armstrong RN, Llorian M, Roy S, et al. The U2AF1S34F mutation induces lineage-specific splicing alterations in myelodysplastic syndromes. J Clin Invest. 2017;127:2206–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim E, Ilagan JO, Liang Y, Daubner GM, Lee SC-W, Ramakrishnan A, et al. SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell. 2015;27:617–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang J, Lieu YK, Ali AM, Penson A, Reggio KS, Rabadan R, et al. Disease-associated mutation in SRSF2 misregulates splicing by altering RNA-binding affinities. Proc Natl Acad Sci U S A. 2015;112:E4726–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Komeno Y, Huang Y-J, Qiu J, Lin L, Xu Y, Zhou Y, et al. SRSF2 Is Essential for Hematopoiesis, and Its Myelodysplastic Syndrome-Related Mutations Dysregulate Alternative Pre-mRNA Splicing. Mol Cell Biol. 2015;35:3071–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nguyen HD, Yadav T, Giri S, Saez B, Graubert TA, Zou L. Functions of Replication Protein A as a Sensor of R Loops and a Regulator of RNaseH1. Mol Cell. 2017;65:832–847.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Santos-Pereira JM, Aguilera A. R loops: new modulators of genome dynamics and function. Nat Rev Genet. 2015;16:583–97. [DOI] [PubMed] [Google Scholar]
- 27.Skourti-Stathaki K, Proudfoot NJ. A double-edged sword: R loops as threats to genome integrity and powerful regulators of gene expression. Genes Dev. 2014;28:1384–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sollier J, Cimprich KA. Breaking bad: R-loops and genome integrity. Trends Cell Biol. 2015;25:514–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coulon A, Ferguson ML, de Turris V, Palangat M, Chow CC, Larson DR. Kinetic competition during the transcription cycle results in stochastic RNA processing. Elife. 2014;3:e03939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maréchal A, Zou L. RPA-coated single-stranded DNA as a platform for post-translational modifications in the DNA damage response. Cell Res. 2015;25:9–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–8. [DOI] [PubMed] [Google Scholar]
- 32.Shiotani B, Nguyen HD, Håkansson P, Maréchal A, Tse A, Tahara H, et al. Two distinct modes of ATR activation orchestrated by Rad17 and Nbs1. Cell Rep. 2013;3:1651–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reaper PM, Griffiths MR, Long JM, Charrier J-D, MacCormick S, Charlton PA, et al. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat Chem Biol. 2011;7:428–30. [DOI] [PubMed] [Google Scholar]
- 34.Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NMB, Orr AI, et al. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004;64:9152–9. [DOI] [PubMed] [Google Scholar]
- 35.Shirai CL, Ley JN, White BS, Kim S, Tibbitts J, Shao J, et al. Mutant U2AF1 Expression Alters Hematopoiesis and Pre-mRNA Splicing In Vivo. Cancer Cell. 2015;27:631–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shirai CL, White BS, Tripathi M, Tapia R, Ley JN, Ndonwi M, et al. Mutant U2AF1-expressing cells are sensitive to pharmacological modulation of the spliceosome. Nat Commun. 2017;8:14060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kotake Y, Sagane K, Owa T, Mimori-Kiyosue Y, Shimizu H, Uesugi M, et al. Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nat Chem Biol. 2007;3:570–5. [DOI] [PubMed] [Google Scholar]
- 38.Folco EG, Coil KE, Reed R. The anti-tumor drug E7107 reveals an essential role for SF3b in remodeling U2 snRNP to expose the branch point-binding region. Genes Dev. 2011;25:440–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee SC-W, Dvinge H, Kim E, Cho H, Micol J-B, Chung YR, et al. Modulation of splicing catalysis for therapeutic targeting of leukemia with mutations in genes encoding spliceosomal proteins. Nat Med. 2016;22:672–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boguslawski SJ, Smith DE, Michalak MA, Mickelson KE, Yehle CO, Patterson WL, et al. Characterization of monoclonal antibody to DNA.RNA and its application to immunodetection of hybrids. J Immunol Methods. 1986;89:123–30. [DOI] [PubMed] [Google Scholar]
- 41.Chen L, Chen J-Y, Huang Y-J, Gu Y, Qiu J, Qian H, et al. The Augmented R-Loop Is a Unifying Mechanism for Myelodysplastic Syndromes Induced by High-Risk Splicing Factor Mutations. Mol Cell. 2018;69:412–425.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Okeyo-Owuor T, White BS, Chatrikhi R, Mohan DR, Kim S, Griffith M, et al. U2AF1 mutations alter sequence specificity of pre-mRNA binding and splicing. Leukemia. 2015;29:909–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Walter MJ, Shen D, Ding L, Shao J, Koboldt DC, Chen K, et al. Clonal architecture of secondary acute myeloid leukemia. N Engl J Med. 2012;366:1090–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hamperl S, Bocek MJ, Saldivar JC, Swigut T, Cimprich KA. Transcription-Replication Conflict Orientation Modulates R-Loop Levels and Activates Distinct DNA Damage Responses. Cell. 2017;170:774–786.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brierley CK, Steensma DP. Targeting Splicing in the Treatment of Myelodysplastic Syndromes and Other Myeloid Neoplasms. Curr Hematol Malig Rep. 2016;11:408–15. [DOI] [PubMed] [Google Scholar]
- 46.Rundle S, Bradbury A, Drew Y, Curtin NJ. Targeting the ATR-CHK1 Axis in Cancer Therapy. Cancers (Basel). 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee SC-W, Abdel-Wahab O. Therapeutic targeting of splicing in cancer. Nat Med. 2016;22:976–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dvinge H, Kim E, Abdel-Wahab O, Bradley RK. RNA splicing factors as oncoproteins and tumour suppressors. Nat Rev Cancer. 2016;16:413–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Seiler M, Peng S, Agrawal AA, Palacino J, Teng T, Zhu P, et al. Somatic Mutational Landscape of Splicing Factor Genes and Their Functional Consequences across 33 Cancer Types. Cell Rep. 2018;23:282–296.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.