

Abstract
G protein-coupled receptors mediate cell signaling and regulate the majority of sensory and physiological processes in the human body. Recent breakthroughs in cryo-electron microscopy and X-ray free electron lasers have accelerated structural studies of difficult-to-crystallize receptors and their signaling complexes, and have opened up new opportunities in understanding conformational dynamics and visualizing the process of receptor activation with unprecedented spatial and temporal resolution. Here, we summarize major milestones and challenges associated with the application of these techniques and outline future directions in their development with a focus on membrane protein structural biology.
Introduction
G protein-coupled receptors (GPCRs) are ubiquitous cellular gatekeepers that share the characteristic architecture of a seven-transmembrane alpha-helical bundle (7TM) and are involved in the regulation of virtually every physiological process in the human body. Due to their biomedical relevance, GPCRs are targeted by a major share of therapeutic drugs and pose as attractive targets for structure-based drug design. In humans, there are over 800 receptors that belong to 5 classes: A, B, C, Frizzled, and Adhesion. Structural studies of GPCRs have been enabled about a decade ago by multiple breakthroughs in protein engineering [1-3], high-throughput nanovolume crystallization in a native-like lipidic cubic phase (LCP) matrix [4-6], and micro-crystallography [7,8]. Despite the enormous progress achieved in structural biology of GPCRs, obtaining structures of new receptors still represents a challenging task. Since GPCRs have evolved to be highly dynamic to perform their respective functions, their crystallization requires the stabilization in a specific conformational state. Moreover, GPCR crystals that grow in LCP are often too small for high-resolution structure determination even at modern microfocus synchrotron beamlines. Finally, our understanding of signal transduction mechanisms is incomplete without detailed knowledge about the structural dynamics of GPCRs and without structures of receptors in complex with their signaling partners, which typically are even less stable and more difficult to crystallize. These challenges of GPCR structural biology call for new tools and approaches. The recently emerged techniques of serial femtosecond crystallography (SFX) using X-ray free-electron lasers (XFELs) and high-resolution cryoelectron microscopy (cryoEM) are starting to tackle some of the most difficult problems (Figure 1).
Figure 1. Schematic diagrams of cryoEM and LCP-SFX experiments.
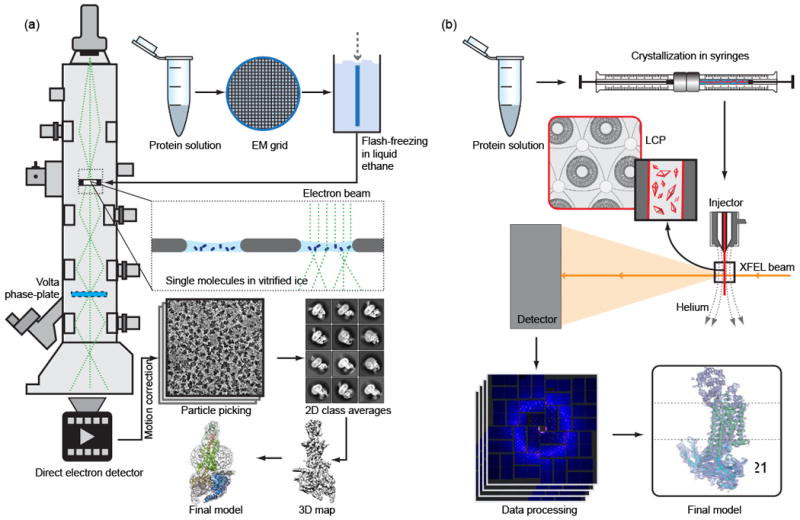
(a) For cryoEM, the purified monodisperse protein solution is deposited on EM grids, blotted and flash-frozen in liquid ethane. The grids are then cryo-transferred into the electron microscope and thousands of images are collected by a direct-electron detector. After performing motion correction, individual particles are picked and 2D classification and 3D classification is applied. Finally, a 3D map is reconstructed, which is used to fit and refine a structure model. Images from Ref. 44 have been re-used in this illustration with permission from Macmillan Publishers Ltd. (b) For LCP-SFX, purified protein is reconstituted in LCP, and crystallization is set up in syringes. After microcrystals have grown, samples from several syringes are consolidated and transferred into a viscous media injector. Tens to hundreds of thousands of diffraction images are collected from microcrystals intersecting the XFEL beam in random orientations. After data processing with specialized software, the structure is solved and refined by standard crystallographic approaches.
XFELs generate extremely intense X-ray pulses of tens of femtoseconds duration with nine-to-ten orders of magnitude higher peak brilliance than third-generation synchrotrons. Such unique characteristics of XFELs prompted a new approach for crystallographic data collection called serial femtosecond crystallography (SFX) [9]. Unlike traditional crystallography, where a complete dataset is collected from a single large (or a few small) crystals, SFX data are acquired from tens to hundreds of thousands of crystals intersecting the XFEL beam in random orientations. Although each crystal is destroyed by the beam, the short pulse duration allows outrunning radiation damage and obtaining structural information from intact molecules at room temperature without the necessity of cryocooling [10]. The extremely high brightness of each XFEL pulse provides sufficient signal for the detection of high-resolution diffraction patterns from micrometer- [11] and even submicrometer-sized crystals [12]. Lastly, but arguably most importantly, the femtosecond pulse duration makes time-resolved crystallography a reality, illuminating proteins in action rather than producing static “snapshots” [13-16]. Even though time-resolved crystallography is being successfully conducted at synchrotron sources, it is mostly limited to light-induced reversible reactions at time scales longer than 100 ps, whereas femtosecond XFEL pulses provide access to irreversible transitions and fundamental chemical processes like isomerization and electron transfer [17].
In parallel to these ground-breaking XFEL developments, cryoEM of biological macromolecules has undergone a ‘resolution revolution’ [18]. The advent of direct-electron detectors with improved quantum efficiencies allowed for the correction of beam-induced motions of the specimen in vitrified ice [19]. This advancement resulted in overcoming previously perceived resolution barriers of 5-6 Å in single molecule density reconstructions. Over the last few years, near-atomic resolution maps, with which the conformations of individual amino acids could be assigned, were routinely achieved, allowing the investigation of biomolecules without the need for crystallization. In combination with the development of the Volta phase-plate, resulting in a significantly improved contrast of weak-phase objects, ever smaller molecules can be studied by cryoEM [20], immensely expanding the general scope of the technique.
In this review, we describe the progress in structural biology of GPCRs during the last 4-5 years brought about by the advancements in SFX and cryoEM (Figure 2), as well as discuss challenges associated with current applications and new opportunities related to future developments of these techniques.
Figure 2. Timeline of major milestones (right) and published GPCR structures (left) achieved with XFELs and cryoEM.
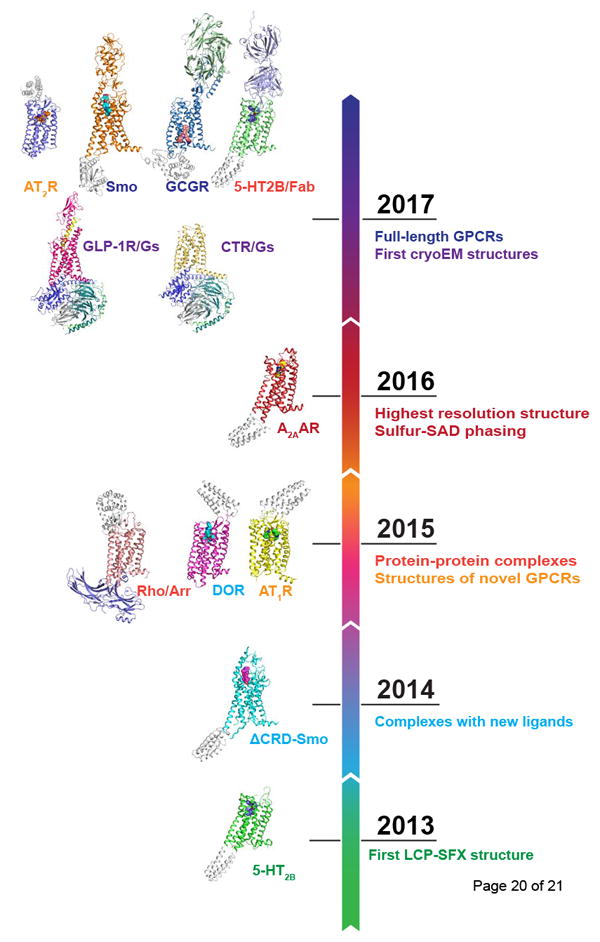
Major milestones of GPCR structural studies at XFELs
Structure determination of GPCRs at XFELs has been realized by the development of special viscous media injectors [21] and new sample preparation protocols [22,23]. Such injectors allowed streaming microcrystals grown in LCP across an XFEL beam for SFX data collection. Notably, the viscous media injector greatly reduced sample consumption compared with commonly used liquid media injectors and has been shown to be suitable for the delivery of crystals of soluble proteins embedded in LCP [24] or other viscous matrices [25-27].
The LCP-SFX approach (Figure 1) was first introduced in 2013 with the high-resolution room temperature structure determination of the human serotonin 2B (5-HT2B) receptor in complex with the anti-migraine medication ergotamine [11]. Compared with the structure solved by traditional cryocrystallography [28] the room temperature XFEL structure displayed a distinct distribution of thermal motions and conformations of residues that likely more accurately represent the receptor structure and dynamics in native cellular environments. LCP-SFX was subsequently applied to solve structures of the human smoothened receptor in complex with the teratogen cyclopamine [21] and of the human δ-opioid receptor bound to a bifunctional peptidic painkiller [29]. In both cases, microcrystals have shown substantially better diffraction at XFELs than their larger cryocooled counterparts at synchrotrons, enabling unambiguous placement of the corresponding ligands into the electron density.
The next important milestone was reached in 2015 (Figure 2), when the first novel GPCR structure of the human angiotensin II receptor type 1 was determined by LCP-SFX [30]. Angiotensin II is a peptide hormone that plays a major role in the renin–angiotensin-aldosterone system, and is involved in the regulation of the plasma sodium concentration and arterial blood pressure. Signaling responses to angiotensin II are mediated by type 1 and 2 angiotensin receptors (AT1R and AT2R). While AT1R is primarily involved in blood pressure regulation, the function of AT2R is much less understood, although with increasing evidence that this receptor may serve as a potential target for non-opioid treatment of neuropathic pain [31]. The 2.9 Å resolution AT1R structure in complex with an angiotensin receptor blocker was followed by the 2.8 Å AT2R structure bound to an AT2R-selective ligand [32]. The structures uncovered new insights into the distinct functions of the two angiotensin receptors and provided reliable templates to facilitate structure-based drug design with improved selectivity.
One of the most dramatic examples demonstrating the advantage of XFELs was the structure determination of a major signaling complex between visual rhodopsin and arrestin (Figure 3), which, at the time, was intractable by means of traditional crystallography [33] and cryoEM [34]. Arrestin binds to activated and phosphorylated receptors, blocking G protein interaction and redirecting signaling to numerous G protein-independent pathways. It has been shown that biased ligands that direct signaling through either predominately G proteins or arrestins may have pharmacological benefits compared to balanced ligands [35]. The crystals of the rhodopsin-arrestin complex, which could not be optimized to grow beyond ~20 μm, diffracted to 7-8 Å resolution at a synchrotron, while yielding a 3.3 Å (anisotropic) structure by LCP-SFX. The structure revealed conformational re-arrangements in arrestin and rhodopsin and the details of their interactions. Recently, re-processed data improved resolution and, in combination with extensive biochemical data, revealed combinations of phosphorylation codes for arrestin recruitment by GPCRs and possibly other proteins [36].
Figure 3. Examples of GPCR structures determined by LCP-SFX and cryoEM.
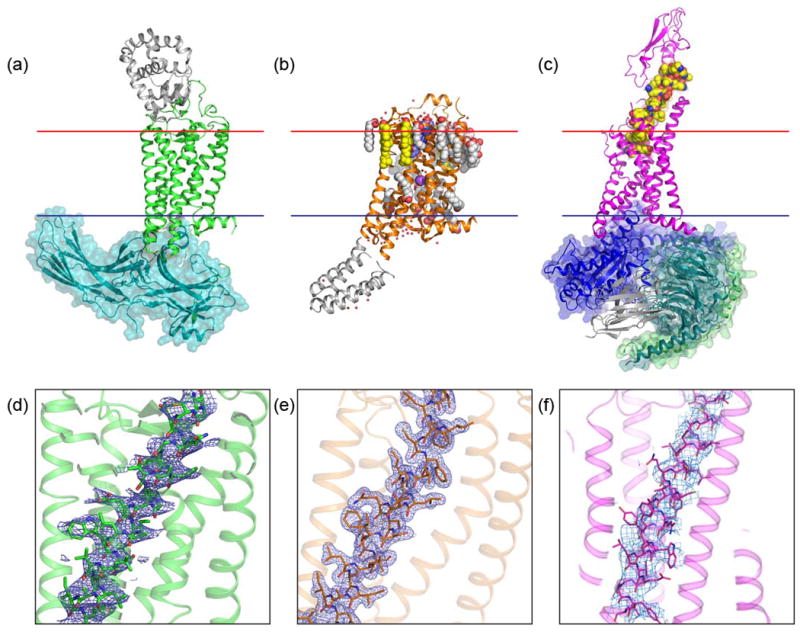
(a) Structure of the rhodopsin-arrestin complex (PDB ID 5W0P) solved using LCP-SFX at 3.0 Å (anisotropic) resolution. (b) The 1.9 Å resolution structure of the adenosine A2A receptor (PDB ID 5K2C) solved by the sulfur single anomalous dispersion (S-SAD) method using data collected by LCP-SFX. (c) The 4.1 Å resolution structure of the GLP-1 receptor in complex with its native agonist peptide, Gs protein and a stabilizing nanobody (PDB ID 5VAI) obtained by single-molecule cryoEM. All structures are shown in cartoon representation with fusion partners and the nanobody colored in gray. Transparent surface is shown for signaling partners, arrestin, and Gs protein. Ligands, lipids and ions are shown as van der Waals spheres, water molecules as small red spheres. The membrane boundaries are shown as red (extracellular) and blue (intracellular) lines. (d) – (f) Electron density of transmembrane helix III is shown as a blue mesh for corresponding structures in (a) – (c). In (d) and (e) 2Fo-Fc density is contoured at 1s level, in (f) the density is contoured at the authors’ recommended level of 0.055 [44].
Another example of a macromolecular complex structure determination, which has been enabled by LCP-SFX, is the 5-HT2B receptor with a selective Fab antibody fragment bound to its extracellular loops (ECLs) [37]. Monoclonal antibodies (mAbs) provide an attractive alternative to small molecules therapies [38], however, the generation of mAbs against the extracellular side of class A GPCRs is challenging due to a small area of solvent-exposed epitopes. The structure of the 5-HT2B/Fab complex sheds light on the mechanisms of extracellular recognition of GPCRs by antibodies.
Finally, LCP-SFX has accelerated the structure determination of full-length receptors from non-class-A GPCRs. These receptors contain large extracellular domains (ECDs) crucial for ligand recognition and signal transduction. While initial efforts were focused on the structure determination of individual domains, the structure of the full-length receptors remained elusive due to difficulties in crystallization. Recently, extensive efforts aimed at stabilization of multidomain receptors by antibodies and designed ligands have culminated in the high-resolution structure determination of the full-length class B glucagon receptor (GCGR) and the class Frizzled smoothened receptor (Smo) at XFEL and synchrotron sources [39,40]. As in the previous examples, the room temperature XFEL structures were of higher resolution and had overall superior quality with respect to their electron density maps. While most GPCR structures were solved by molecular replacement, the recent demonstration of de novo phasing of a GPCR structure using the anomalous signal from sulfur atoms present in most proteins [41] opened up opportunities for structural studies of novel membrane protein families at XFELs.
First high-resolution cryoEM structures of GPCRs
The first application of direct-electron detectors combined with motion correction in cryoEM to a membrane protein was the structure elucidation of the TRPV1 channel at 3.4 Å resolution [42]. This work demonstrated that cryoEM is able to overcome difficulties traditionally associated with structural studies of membrane proteins, such as low expression yields and limited stability in detergent micelles, the reasons why many membrane proteins are often not suitable for crystallization.
Another major breakthrough in cryoEM of membrane proteins was the 3.4 Å resolution structure of γ-secretase (~170 kDa) [43], a medically important protease being the source of abnormally folded amyloid-beta fibers in Alzheimer’s disease. It was the first study showing that a sub-200 kDa membrane protein with no symmetry applied in the reconstruction could be resolved to near-atomic resolution. Over the last 5 years, approximately 16% of all < 4 Å structures submitted to the Electron Microscopy Data Bank (EMDB) were derived from membrane proteins, including a variety of ion channels, transporters, enzymes, and receptors. These stats highlight a higher success rate of cryoEM for membrane proteins compared to crystallography, in which membrane proteins contribute less than 2% of all entries.
Due to the relatively small size of GPCRs and their inherent dynamic nature, their structure determination by cryoEM has been extremely challenging. At last, in 2017, structures of two class B receptors, the calcitonin receptor (CTR) [44] and the glucagon-like peptide-1 receptor (GLP-1R) [45] in complex with their cognate G proteins have been published (Figure 3). While structures of GPCRs in complex with downstream partners are highly sought after, they are particularly difficult to solve by crystallography. In contrast, cryoEM was able to readily overcome this hurdle. Both structures were obtained in complex with their native peptide agonist, Gs protein, and a stabilizing nanobody. Using such ~150 kDa complexes did not only help with the orientation determination by increasing the molecular weight (and therefore improving the contrast), but also conformationally locked the receptor in the active state to minimize structural heterogeneity. Additionally, a Volta phase-plate was instrumental for the structure determination of the CTR/Gs complex [44].
In summary, these structures highlight the potential of cryoEM to observe relatively small isolated GPCR complexes in detergent micelles at nearly-atomic resolution. One of the most remarkable features of both studies is the use of full-length, wild-type GPCRs, which has not been possible with crystallography with an exception of the visual rhodopsin. Furthermore, both studies provided important insights into the activation mechanism of class B receptors, expanding our understanding of GPCR signaling.
Current challenges, limitations and future perspectives
As outlined above, recent breakthroughs in SFX and cryoEM have greatly advanced our structural understanding of GPCRs and other membrane proteins. While both techniques produce structural models, they have different requirements and limitations (Table 1). One of the most important advantages of cryoEM is its ability to obtain structural information from single molecules, while SFX still requires crystals, albeit much smaller than traditional crystallography. The downside of cryoEM, however, is the requirement of cryocooling the sample, which helps to reduce but does not completely overcome radiation damage. XFELs, on the other hand, can reveal structures at room temperature without detectable radiation damage effects [12]. Another important limitation for cryoEM is the minimal particle size. Although a near-atomic resolution structure of 64 kDa hemoglobin has recently been reported [20], the structure determination of < 100 kDa molecules has not yet been routinely achieved. Both cryoEM and SFX methods rely on collecting large amounts of data with typical data acquisition times of a few hours for SFX vs. a few days for cryoEM. The available SFX beamtime is, however, severely limited by the extreme cost and scarcity of large-scale XFEL facilities. In contrast, cryoEM instruments, while not inexpensive, are nonetheless affordable to major universities, research institutes, and core facilities, contributing to a much broader accessibility of this method to the structural biology community worldwide.
Table 1.
Typical experimental conditions, challenges, and opportunities for cryoEM and SFX
| cryoEM | SFX | |
|---|---|---|
| Temperature | Cryogenic | Room |
| Sample state | Isolated molecules | Crystals |
| Protein size, kDa | > 100 (smallest 64) | No limit |
| Radiation damage | Yes | No |
| Resolution, Å | 3 – 5 (best 1.8) | 1.5 – 3 (best 1.2) |
| Protein consumption, μg | 1 – 10 | 100 – 500 |
| Final protein concentration, mg/mL | 0.5 – 5 | 10 – 50 |
| Data collection time, hrs | 24 – 96 | 2 – 6 |
| Accessibility | Core facilities, in-house instruments | Large facilities: LCLS, SACLA, EuXFEL, PAL, SwissFEL |
| Protein dynamics | Equilibrium states in a population | Room temperature fluctuations, molecular movies, ultrafast dynamics, non-equilibrium states |
| Opportunities for GPCRs | Signaling complexes, homo- and heterodimers, receptors with large ECDs | Small crystals, room temperature structures, SBDD, receptor activation movies |
For structural studies, resolution is the single most important parameter that defines the amount and accuracy of information, which can be deduced for a given structure. Most recent cryoEM structures have been determined at 3–5 Å resolution (best resolution 1.8 Å [46]), while most SFX structures are in the 1.5-3.0 Å resolution range (best resolution 1.2 Å [47]). Of the 5,908 single particle structures deposited in EMDB by March 2018, only 1% (total of 62) contain structural information beyond 3 Å. The majority (82%) of these 62 maps are virus(-like) molecules, MDa-sized complexes or other high symmetry assemblies. The remaining entries consist of 11 maps, of which 10 reconstructions applied symmetry, only 6 maps are derived from proteins smaller than 400 kDa and 3 represent structures of membrane proteins. Of the above 11, the latter 3 membrane proteins are also the only ones that have not previously been solved by X-ray crystallography. These statistics illustrate the need for further technological developments before the accuracy of cryoEM maps will become on par with crystallography and can be routinely used for applications, such as structure-based drug design.
The success of both cryoEM and SFX experiments strongly depends on sample quality, although sample requirements for these techniques are quite different. CryoEM requires single particles embedded in thin vitrified ice in random orientations and at a high concentration. The preferred orientation of molecules in ice and the degradation of delicate protein complexes during the grid preparation process pose bottlenecks in cryoEM. Typical sample preparation starts with a few microliters of purified monodisperse protein solution at 0.5-5 mg/mL concentration (Figure 1). The quality of vitrified sample depends on many factors and typically involves extensive screening of several parameters such as the grid type, buffer composition, and protein concentration. The molecule itself has to be sufficiently large and rigid to allow for orientation determination. In contrast, an ideal sample for LCP-SFX contains highly ordered micrometer-sized crystals grown in LCP at a high density. The starting point is a few dozen microliters of purified monodisperse protein solution at 10-50 mg/mL concentration. Apart from the higher protein consumption, the protein for LCP-SFX has to be stabilized in a predominately single conformational state to support crystallization, which, in the case of GPCRs, typically requires extensive protein engineering.
Finally, apart from static structures both cryoEM and SFX methods can also provide information about conformational dynamics of macromolecules. CryoEM is capable of revealing multiple equilibrium conformational states in a population of single molecules [48]. XFELs, due to their femtosecond pulse duration, allow for recording molecular movies using a pump-probe technique [13,14,49], enabling the capturing of ultrafast conformational transitions and transient states.
Conclusions
With recent breakthroughs in cryoEM and XFELs, the structural biology field is experiencing a resurgence. Most current limitations will soon be addressed if not completely resolved by the developments of instrumentation and data analysis, leading to faster data collection, higher resolution and lower molecular sizes accessible to cryoEM. At the same time, XFELs will continue tackling ever smaller crystals, which may eventually approach single molecules of a typical protein size [17].
In the case of GPCRs, we would expect an increasing number of cryoEM structures of signaling complexes with different G protein types, arrestins, kinases and other partners, as well as structures of homo- and heterodimeric receptors and receptors with large ECDs. In fact, while this review was in preparation, two additional cryoEM structures of GPCR signaling complexes, obtained with a Volta phase-plate, have been published [50,51]. We anticipate that XFELs will further contribute to the high-resolution structural coverage of the whole GPCR superfamily, help with structure-based drug design efforts, and, most importantly, should produce detailed molecular movies of signal transduction by GPCRs.
Highlights.
Recent advancements in cryoEM and XFELs accelerated structural studies of GPCRs
XFELs enabled room-temperature damage-free structures from micrometer-sized crystals
CryoEM demonstrated its potential for elucidating GPCR signaling complexes
Both cryoEM and XFELs promise to shed light on structural dynamics of GPCRs
Acknowledgments
This work was supported by the National Institutes of Health [grant number R01 GM108635], the National Science Foundation [grant number 1231306], and the Russian Science Foundation [project number 16-14-10273]. C.G. acknowledges the Panofsky Fellowship from SLAC National Accelerator Laboratory and Stanford University for financial support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chun E, Thompson AA, Liu W, Roth CB, Griffith MT, Katritch V, Kunken J, Xu F, Cherezov V, Hanson MA, et al. Fusion partner toolchest for the stabilization and crystallization of G protein-coupled receptors. Structure. 2012;20:967–76. doi: 10.1016/j.str.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SGF, Thian FS, Kobilka TS, Choi H-J, Yao X-J, Weis WI, Stevens RC, et al. GPCR Engineering Yields High-Resolution Structural Insights into β2-Adrenergic Receptor Function. Science. 2007;318:1266–1273. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- 3.Serrano-Vega MJ, Magnani F, Shibata Y, Tate CG. Conformational thermostabilization of the β1-adrenergic receptor in a detergent-resistant form. Proc Natl Acad Sci USA. 2008;105:877–882. doi: 10.1073/pnas.0711253105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Caffrey M, Cherezov V. Crystallizing membrane proteins using lipidic mesophases. Nat Protoc. 2009;4:706–31. doi: 10.1038/nprot.2009.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cherezov V. Lipidic cubic phase technologies for membrane protein structural studies. Curr Opin Struct Biol. 2011;21:559–66. doi: 10.1016/j.sbi.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stevens RC, Cherezov V, Katritch V, Abagyan R, Kuhn P, Rosen H, Wüthrich K. The GPCR Network: a large-scale collaboration to determine human GPCR structure and function. Nat Rev Drug Discov. 2013;12:25–34. doi: 10.1038/nrd3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith J, Fischetti R, Yamamoto M. Micro-crystallography comes of age. Curr Opin Struct Biol. 2012;22:602–612. doi: 10.1016/j.sbi.2012.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cherezov V, Hanson MA, Griffith MT, Hilgart MC, Sanishvili R, Nagarajan V, Stepanov S, Fischetti RF, Kuhn P, Stevens RC. Rastering strategy for screening and centring of microcrystal samples of human membrane proteins with a sub-10 μm size X-ray synchrotron beam. J R Soc Interface R Soc. 2009;6:S587–S597. doi: 10.1098/rsif.2009.0142.focus. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chapman HN, Fromme P, Barty A, White TA, Kirian RA, Aquila A, Hunter MS, Schulz J, DePonte DP, Weierstall U, et al. Femtosecond X-ray protein nanocrystallography. Nature. 2011;470:73–77. doi: 10.1038/nature09750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neutze R, Wouts R, van der Spoel D, Weckert E, Hajdu J. Potential for biomolecular imaging with femtosecond X-ray pulses. Nature. 2000;406:752–757. doi: 10.1038/35021099. [DOI] [PubMed] [Google Scholar]
- 11•.Liu W, Wacker D, Gati C, Han GW, James D, Wang D, Nelson G, Weierstall U, Katritch V, Barty A, et al. Serial Femtosecond Crystallography of G Protein-Coupled Receptors. Science. 2013;342:1521–1524. doi: 10.1126/science.1244142. This paper describes the first application of the LCP-SFX method and its validation by comparing structures of serotonin 2B receptor solved by traditional crystallography and LCP-SFX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gati C, Oberthuer D, Yefanov O, Bunker RD, Stellato F, Chiu E, Yeh S-M, Aquila A, Basu S, Bean R, et al. Atomic structure of granulin determined from native nanocrystalline granulovirus using an X-ray free-electron laser. Proc Natl Acad Sci USA. 2017;114:2247–2252. doi: 10.1073/pnas.1609243114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kupitz C, Basu S, Grotjohann I, Fromme R. Serial time-resolved crystallography of photosystem II using a femtosecond X-ray laser. Nature. 2014;513:261–265. doi: 10.1038/nature13453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pande K, Hutchison CDM, Groenhof G, Aquila A, Robinson JS, Tenboer J, Basu S, Boutet S, DePonte DP, Liang M, et al. Femtosecond structural dynamics drives the trans/cis isomerization in photoactive yellow protein. Science. 2016;352:725–729. doi: 10.1126/science.aad5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stagno JR, Liu Y, Bhandari YR, Conrad CE, Panja S, Swain M, Fan L, Nelson G, Li C, Wendel DR, et al. Structures of riboswitch RNA reaction states by mix-and-inject XFEL serial crystallography. Nature. 2016;541:242–246. doi: 10.1038/nature20599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16••.Nango E, Royant A, Kubo M, Nakane T, Wickstrand C, Kimura T, Tanaka T, Tono K, Song C, Tanaka R, et al. A three-dimensional movie of structural changes in bacteriorhodopsin. Science. 2016;354:1552–1557. doi: 10.1126/science.aah3497. This study presents a time-resolved movie of conformational changes in a prototypical 7TM membrane protein bacteriorhodopsin triggered by a flash of light and captured using LCP-SFX. [DOI] [PubMed] [Google Scholar]
- 17.Spence JCH. XFELs for structure and dynamics in biology. IUCrJ. 2017;4:322–339. doi: 10.1107/S2052252517005760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuhlbrandt W. The Resolution Revolution. Science. 2014;343:1443–1444. doi: 10.1126/science.1251652. [DOI] [PubMed] [Google Scholar]
- 19.Li X, Mooney P, Zheng S, Booth CR, Braunfeld MB, Gubbens S, Agard DA, Cheng Y. Electron counting and beam-induced motion correction enable near-atomic- resolution single-particle cryo-EM. Nat Methods. 2013;10:584–590. doi: 10.1038/nmeth.2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khoshouei M, Radjainia M, Baumeister W, Danev R. Cryo-EM structure of haemoglobin at 3.2 Å determined with the Volta phase plate. Nat Commun. 2017;8:16099. doi: 10.1038/ncomms16099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21•.Weierstall U, James D, Wang C, White TA, Wang D, Liu W, Spence JCH, Bruce Doak R, Nelson G, Fromme P, et al. Lipidic cubic phase injector facilitates membrane protein serial femtosecond crystallography. Nat Commun. 2014;5:3309. doi: 10.1038/ncomms4309. This paper describes the development of the first LCP injector and its application to solving the structure of the smoothened receptor in complex with cyclopamine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22•.Liu W, Ishchenko A, Cherezov V. Preparation of microcrystals in lipidic cubic phase for serial femtosecond crystallography. Nat Protoc. 2014;9:2123–2134. doi: 10.1038/nprot.2014.141. This paper provides detailed sample preparation protocols for LCP-SFX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ishchenko A, Cherezov V, Liu W. Preparation and Delivery of Protein Microcrystals in Lipidic Cubic Phase for Serial Femtosecond Crystallography. J Vis Exp. 2016;115:e54463. doi: 10.3791/54463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fromme R, Ishchenko A, Metz M, Chowdhury SR, Basu S, Boutet S, Fromme P, White TA, Barty A, Spence JCH, et al. Serial femtosecond crystallography of soluble proteins in lipidic cubic phase. IUCrJ. 2015;2:545–551. doi: 10.1107/S2052252515013160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Conrad CE, Basu S, James D, Wang D, Schaffer A, Roy-Chowdhury S, Zatsepin NA, Aquila A, Coe J, Gati C, et al. A novel inert crystal delivery medium for serial femtosecond crystallography. IUCrJ. 2015;2:421–430. doi: 10.1107/S2052252515009811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kovácsová G, Grünbein ML, Kloos M, Barends TRM, Schlesinger R, Heberle J, Kabsch W, Shoeman RL, Doak RB, Schlichting I. Viscous hydrophilic injection matrices for serial crystallography. IUCrJ. 2017;4:400–410. doi: 10.1107/S2052252517005140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sugahara M, Mizohata E, Nango E, Suzuki M, Tanaka T, Masuda T, Tanaka R, Shimamura T, Tanaka Y, Suno C, et al. Grease matrix as a versatile carrier of proteins for serial crystallography. Nat Methods. 2014;12:61–63. doi: 10.1038/nmeth.3172. [DOI] [PubMed] [Google Scholar]
- 28.Wacker D, Wang C, Katritch V, Han GW, Huang X-P, Vardy E, McCorvy JD, Jiang Y, Chu M, Siu FY, et al. Structural features for functional selectivity at serotonin receptors. Science. 2013;340:615–619. doi: 10.1126/science.1232808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29•.Fenalti G, Zatsepin NA, Betti C, Giguere P, Han GW, Ishchenko A, Liu W, Guillemyn K, Zhang H, James D, et al. Structural basis for bifunctional peptide recognition at human d-opioid receptor. Nat Struct Mol Biol. 2015;22:265–268. doi: 10.1038/nsmb.2965. This paper describes the structure of the d-opioid receptor (DOR) bound to a bi-functional peptide solved using LCP-SFX The structure revealed the recognition mode of a peptide at DOR and provided a template for designing promising painkillers with reduced tolerance and dependency. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30••.Zhang H, Unal H, Gati C, Han GW, Liu W, Zatsepin NA, James D, Wang D, Nelson G, Weierstall U, et al. Structure of the Angiotensin Receptor Revealed by Serial Femtosecond Crystallography. Cell. 2015;161:833–844. doi: 10.1016/j.cell.2015.04.011. This paper presents the structure of the first novel GPCR obtained by LCP-SFX The structure of the major blood pressure regulator, the human angiotensin II receptor type 1 (AT1R), revealed binding modes of common angiotensin receptor blockers and provided insights into the AT1R structure-function relationship. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rice A, Smith M. Angiotensin II Type 2-Receptor: New Clinically Validated Target in the Treatment of Neuropathic Pain. Clin Pharmacol Ther. 2015;97:128–130. doi: 10.1002/cpt.29. [DOI] [PubMed] [Google Scholar]
- 32••.Zhang H, Han GW, Batyuk A, Ishchenko A, White KL, Patel N, Sadybekov A, Zamlynny B, Rudd MT, Hollenstein K, et al. Structural basis for selectivity and diversity in angiotensin II receptors. Nature. 2017;544:327–332. doi: 10.1038/nature22035. This study reveals the structure of the human angiotensin II receptor type 2 (AT2) by LCP-SFX, shedding light on selectivity and functional diversity in angiotensin II receptors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33••.Kang Y, Zhou XE, Gao X, He Y, Liu W, Ishchenko A, Barty A, White TA, Yefanov O, Han GW, et al. Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature. 2015;523:561–567. doi: 10.1038/nature14656. In this landmark study, the first structure of a signaling complex between GPCR and arrestin was elucidated using LCP-SFX The structure revealed the structural determinants of arrestin recognition by GPCRs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shukla AK, Westfield GH, Xiao K, Reis RI, Huang L-Y, Tripathi-Shukla P, Qian J, Li S, Blanc A, Oleskie AN, et al. Visualization of arrestin recruitment by a G-protein-coupled receptor. Nature. 2014;512:218–222. doi: 10.1038/nature13430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Violin JD, Crombie AL, Soergel DG, Lark MW. Biased ligands at G-protein-coupled receptors: promise and progress. Trends Pharmacol Sci. 2014;35:308–316. doi: 10.1016/j.tips.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 36••.Zhou XE, He Y, de Waal PW, Gao X, Kang Y, Van Eps N, Yin Y, Pal K, Goswami D, White TA, et al. Identification of Phosphorylation Codes for Arrestin Recruitment by G Protein-Coupled Receptors. Cell. 2017;170:457–469. doi: 10.1016/j.cell.2017.07.002. In this work, the structure of rhodopsin-arresting complex determined by LCP-SFX was further improved and used to identify phosphorylation codes for arresting recruitment by GPCRs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ishchenko A, Wacker D, Kapoor M, Zhang A, Han GW, Basu S, Patel N, Messerschmidt M, Weierstall U, Liu W, et al. Structural insights into the extracellular recognition of the human serotonin 2B receptor by an antibody. Proc Natl Acad Sci USA. 2017;114:8223–8228. doi: 10.1073/pnas.1700891114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Golay J, Introna M. Mechanism of action of therapeutic monoclonal antibodies: promises and pitfalls of in vitro and in vivo assays. Arch Biochem Biophys. 2012;526:146–153. doi: 10.1016/j.abb.2012.02.011. [DOI] [PubMed] [Google Scholar]
- 39.Zhang X, Zhao F, Wu Y, Yang J, Han GW, Zhao S, Ishchenko A, Ye L, Lin X, Ding K, et al. Crystal structure of a multi-domain human smoothened receptor in complex with a super stabilizing ligand. Nat Commun. 2017;8:15383. doi: 10.1038/ncomms15383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40•.Zhang H, Qiao A, Yang D, Yang L, Dai A, de Graaf C, Reedtz-Runge S, Dharmarajan V, Zhang H, Han GW, et al. Structure of the full-length glucagon class B G-protein-coupled receptor. Nature. 2017;546:259–264. doi: 10.1038/nature22363. This paper describes the first full-length class B glucagon receptor structure in complex with a negative allosteric modulator and a stabilizing Fab antibody fragment captured in an inactive state by LCP-SFX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41•.Batyuk A, Galli L, Ishchenko A, Han GW, Gati C, Popov PA, Lee M-Y, Stauch B, White TA, Barty A, et al. Native phasing of X-ray free-electron laser data for a G protein–coupled receptor. Sci Adv. 2016;2:e1600292. doi: 10.1126/sciadv.1600292. In this work, the first successful de novo phasing of LCP-SFX data by sulfur-SAD was demonstrated, leading to a 1.9 Å resolution bias-free room-temperature structure of the adenosine A2A receptor bound to an antagonist. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liao M, Cao E, Julius D, Cheng Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature. 2013;504:107–112. doi: 10.1038/nature12822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bai X, Yan C, Yang G, Lu P, Ma D, Sun L, Zhou R, Scheres SHW, Shi Y. An atomic structure of human γ-secretase. Nature. 2015;525:212–217. doi: 10.1038/nature14892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44•.Liang Y-L, Khoshouei M, Radjainia M, Zhang Y, Glukhova A, Tarrasch J, Thal DM, Furness SGB, Christopoulos G, Coudrat T, et al. Phase-plate cryo-EM structure of a class B GPCR-G-protein complex. Nature. 2017;546:118–123. doi: 10.1038/nature22327. This study reports on the first GPCR structure solved by cryoEM at a near-atomic resolution, revealing the details of interactions between the calcitonin receptor and Gs protein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45••.Zhang Y, Sun B, Feng D, Hu H, Chu M, Qu Q, Tarrasch JT, Li S, Sun Kobilka T, Kobilka BK, et al. Cryo-EM structure of the activated GLP-1 receptor in complex with a G protein. Nature. 2017;546:248–253. doi: 10.1038/nature22394. Here, the structure of the GLP-1 receptor with a fully resolved extracellular domain and peptide agonist and in complex with Gs protein and stabilizing nanobody is obtained by cryoEM, shedding light on the mechanism of signal transduction by class B GPCRs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Merk A, Bartesaghi A, Banerjee S, Falconieri V, Rao P, Davis MI, Pragani R, Boxer MB, Earl LA, Milne JLS, et al. Breaking Cryo-EM Resolution Barriers to Facilitate Drug Discovery. Cell. 2016;165:1698–1707. doi: 10.1016/j.cell.2016.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Masuda T, Suzuki M, Inoue S, Song C, Nakane T, Nango E, Tanaka R, Tono K, Joti Y, Kameshima T, et al. Atomic resolution structure of serine protease proteinase K at ambient temperature. Sci Rep. 2017;7:45604. doi: 10.1038/srep45604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhao J, Benlekbir S, Rubinstein JL. Electron cryomicroscopy observation of rotational states in a eukaryotic V-ATPase. Nature. 2015;521:241–245. doi: 10.1038/nature14365. [DOI] [PubMed] [Google Scholar]
- 49.Tenboer J, Basu S, Zatsepin N, Pande K, Milathianaki D, Frank M, Hunter M, Boutet S, Williams GJ, Koglin JE, et al. Time-resolved serial crystallography captures high-resolution intermediates of photoactive yellow protein. Science. 2014;346:1242–1246. doi: 10.1126/science.1259357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50•.Tate CG, García-Nafría J, Lee Y, Bai X, Carpenter B. Cryo-EM structure of the adenosine A2A receptor coupled to an engineered heterotrimeric G protein. bioRxiv. 2018 doi: 10.1101/267674. In this study, a new cryoEM structure of the adenosine A2A receptor in complex with agonist NECA and engineered mini-Gs protein was obtained and compared with the previously determined crystal structure. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51•.Liang Y-L, Khoshouei M, Glukhova A, Furness SGB, Zhao P, Clydesdale L, Koole C, Truong TT, Thal DM, Lei S, et al. Phase-plate cryo-EM structure of a biased agonist-bound human GLP-1 receptor–Gs complex. Nature. 2018;555:121–125. doi: 10.1038/nature25773. This cryoEM structure of a biased agonist-bound human GLP-1 receptor-Gs complex provides insights in the molecular mechanisms of biased signaling. [DOI] [PubMed] [Google Scholar]