

Abstract
Objective. Inorganic polyphosphates (polyP) play a multitude of roles in mammalian biology. PolyP research is hindered by the lack of a simple and sensitive quantification method. The aim of this study was to develop a robust method for quantifying the low levels of polyP in mammalian tissue such as cartilage, which is rich in macromolecules that interfere with its determination. Design. Native and in vitro formed tissues were digested with proteinase K to release sequestrated polyP. The tissue digest was loaded on to silica spin columns, followed by elution of bound polyP and various treatments were assessed to minimize non-polyP fluorescence. The eluent was then quantified for polyP content using fluorometry based on DAPI (4′,6-diamidino-2-phenylindole) fluorescence shift occurring with polyP. Results. Proteinase K pretreatment reduced the inhibitory effect of proteins on polyP recovery. The eluent was contaminated with nucleic acids and glycosaminoglycans, which cause extraneous fluorescence signals. These were then effectively eliminated by nucleases treatment and addition of concentrated Tris buffer. PolyP levels were quantified and recovery ratio determined using samples spiked with a known amount of polyP. This silica spin column method was able to recover at least 80% of initially loaded polyP, and detect as little as 10−10 mol. Conclusions. This sensitive, reproducible, easy to do method of quantifying polyP will be a useful tool for investigation of polyP biology in mammalian cells and tissues. Although the protocol was developed for mammalian tissues, this method should be able to quantify polyP in most biological sources, including fluid samples such as blood and serum.
Keywords: inorganic polyphosphates, DAPI, fluorometric quantification, mammalian tissue
Introduction
Inorganic polyphosphates (polyP) are linear polymers of phosphate residues linked by high-energy phosphoanhydride bonds. Studies with mammalian cells have demonstrated that polyP plays a role in coagulation,1,2 proliferation,3 cell signaling,4,5 skeletal mineralization,7 cartilage mineralization,6 and glial cell function.8 PolyP has recently been used by our group for tissue engineering applications for cartilage9 and nucleus pulposus tissue.10 Thus, the ability to quantify polyP becomes essential as knowing the levels in degenerating tissues may help optimize the dose required to achieve optimal tissue engineering outcomes. However, polyP present in mammalian cells and tissues cannot easily be quantified due to the lack of simple analytical methods that are sufficiently sensitive11 as the concentration of polyP in these have been reported to be 100-fold less than those in prokaryotic cells.11,12 Thus, there is a need for a new method to quantify such low levels of polyP.
Current polyP quantification methods for mammalian tissues have significant limitations. Mass spectrometry or chromatographic methods have been described for polyP quantification, 11,12 but these are limited to short polyP chains (chain length 5) and cannot quantify biologically relevant polyP chains longer than 15 phosphate residues, such as polyP of 45 (polyP-45) residues that play a role in cartilage biomineralization,13 and polyP of 60 to 100 residues that are present in platelets.14 Nuclear magnetic resonance techniques can quantify both the amount and chain length of polyP.15 However, this method is too cumbersome and has lower throughput, which is problematic for routine quantification of polyP in biological samples. Other approaches utilized include hydrolyzing polyP to orthophosphates by treatment with Saccharomyces cerevisiae exopolyphosphatase (PPX) or with conversion to adenosine 5′-triphosphate with Escherichia coli polyphosphate kinase and quantification by a luciferase assay.16,17 These are indirect methods and the enzymes required are not readily available. Aschar-Sobbi et al18 described a fluorometric alternative that exploits the shift in the excitation-emission peaks of 4′,6-diamidino-2-phenylindole (DAPI) when bound to polyP. However, this method requires a high purity of samples as many other biological molecules can similarly shift the fluorescence spectrum of DAPI, such as DNA and RNA, glycosaminoglycans (GAG) and proteins, and give a false signal.
Isolation of polyP from cells and tissues is also difficult due to its structural similarity with nucleic acids. Kumble and Kornberg12 attempted to accomplish this by co-extracting polyP and nucleic acids, followed by nuclease digestion and phenol-chloroform extraction to eliminate nucleic acid contaminants. Alternatively, silica column adsorption–based isolation techniques were developed16,17; however, the reported polyP content is a few magnitudes higher than that obtained by Rao et al.11 Kulakova et al.19 found this approach inaccurate and instead suggested that direct quantification (using the fluorometric quantification) without a prior isolation step may be optimal for microbial cells. Martin and Van Mooy20 refined this approach by compensating for the “matrix effect.” However, correcting for extraneous fluorescence from mammalian cells and tissues may be more challenging because of the lower polyP levels in mammalian cells.12
In this study, we describe a new silica membrane spin column–based polyP isolation method from which subnanomole quantities of polyP, whose chain lengths range from 45 to 130 residues sizes, which are present in tissues can be recovered with minimal loss, minimizes extraneous signals and can be quantified by fluorometry. The method does not require specialized analytical equipment, thus it is cost-effective and readily adaptable for use in biological assays where multiple sample analysis is required.
Materials and Methods
Preparation of PolyP Standard Solutions
PolyP solution was prepared by dissolving sodium phosphate glass (45 phosphate residues: Sigma-Aldrich, Oakville, Ontario, Canada; 23 phosphate residues: SPER Chemicals, Clearwater, FL, USA; 14, 60, and 130 phosphate residues: 0.5M RegeneTiss Incorporated, Tokyo, Japan). PolyP chain lengths 23 and 45 stock solutions were made by calculating g/mol of a particular chain length [(molecular mass from molecular formula Nan + 2PnO3n + 1, where n = number of phosphorus atoms)/number of phosphate units in a particular chain length)] and dissolving it in double distilled water (ddH2O) shaking overnight at 4°C. All stock polyP standard solutions were kept at ~30°C at a concentration of 5 mM in H2O until use. PolyP amounts and concentrations are expressed in equivalent phosphate units. Unless otherwise stated, experiments were performed with polyP chain length 45 (polyP-45).
Silica Spin Column Isolation and Fluorometric Quantification of PolyP
One hundred forty microliters of sample was mixed with 280 µL of binding buffer and incubated for 10 minutes at room temperature. Binding buffer consisted of 5.0 M guanidine thiocyanate (GuSCN), 0.9 M sodium acetate, 25 mM ethylenediaminetetraacetic acid (EDTA), 1% β-mercaptoethanol, and 50 mM Tris pH 6.8 (all Sigma-Aldrich). Then, 280 µL of 100% ethanol at room temperature was added to the samples and incubated for 3 minutes at room temperature. To extract polyP, the sample was then loaded on a silica column. Initially columns were prepared manually by packing column shells with silica sheets cut with a skin biopsy punch, but the polyP extraction was not consistent. Then commercially available silica spin columns from different vendors (Qiagen, EconoSpin), were tested. All these columns yielded similar results that were reproducible. EconoSpin (Epoch Life Science, Sugar Land, TX, USA) columns were the most economical and hence used in all remaining experiments. Samples were loaded on the columns and centrifuged (Eppendorf, Mississauga, Ontario, Canada) for 30 seconds at 10,000 × g. Following this step, 550 µL of wash buffer I (1.0 M GuSCN in 80% ethanol) was loaded onto the column and centrifuged for 1 minute at 12,000 × g. Then, 550 µL of wash buffer II (150 mM sodium chloride and 10 mM Tris in 80% ethanol, pH 7.5) was loaded and centrifuged for 1 minute at 12,000 × g. Another 300 µL of wash buffer II was added onto the column and centrifuged for 2 minutes at 12,000 × g. The column was then removed and placed in a fresh recovery tube and 60 µL elution buffer (10 mM Tris, pH 7.2) was added gently to the center of the spin column and incubated at room temperature for 3 minutes followed by centrifugation at 12,000 × g for 1 minute. This elution step was performed 2 more times resulting in a final volume of 180 µL in the recovery tube.
PolyP in the eluent was quantified fluorometrically as previously described17 with the following modifications. Aliquots were dispensed into wells of a black 96-well microplate (Corning, Corning, NY, USA) in triplicates. Then, 50 µL of DAPI solution (50 µg/mL in 10mM Tris pH 7.5 buffer; Invitrogen, Oakville, Ontario, Canada) was added to each well and incubated for 5 minutes at room temperature to achieve steady-state fluorescence signal for all polyP chain lengths examined.18 Fluorescence was measured at an excitation wavelength of 415 nm and an emission wavelength of 558 nm using Fluoroskan Ascent FL microplate fluorometer (Thermo Scientific, Waltham, MA, USA). A standard curve was generated using polyP-45 solution (1-60 µM) and was used to calculate polyP concentration.
Determination of the PolyP Recovery Ratios from Silica Spin Columns
The capacity of the silica spin column to bind and release polyP was determined by loading known amounts of polyP-45 (140 µL/sample). The 415/558 nm DAPI fluorescence was measured in the solution prior to loading and in the eluents from the silica spin columns. The polyP recovery ratio was calculated as the ratio of recovered polyP versus the initially loaded polyP. PolyP quantities, 15.5, 46.6, and 77.6 nmol were loaded to compare the recovery ratios of the current method to previous reported assays.19,20 The lowest amount of polyP that can be reliably recovered using the current protocol was determined by measuring the polyP recovery ratios of 0.25 to 8 nmol (1.79-57.1 µM) polyP. Chain length–dependent recovery ratios were determined using 7 nmol (50 µM) polyP of different chain lengths (14-130 residues).
Determination of Potential Sources of Interference for the PolyP Quantification Assay
To evaluate the effect of possible molecules known to contribute to DAPI fluorescence, solutions of calf thymus DNA, S. cerevisiae RNA, adenosine triphosphate (ATP), sodium pyrophosphate, chondroitin sulfate (the predominant GAG present in cartilage18) and bovine serum albumin (BSA; all Sigma-Aldrich) were prepared at concentrations listed in Table 1 . Fetal bovine serum (FBS) was used to study protein interference as FBS contains 35 mg/mL protein (manufacturer’s analysis). Seven nanomoles (50 µM) polyP were then added to 140 µL of these different macromolecular solutions and the silica spin column isolation step was performed as described in section “Silica Spin Column Isolation and Fluorometric Quantification of PolyP.”
Table 1.
Potential Sources of Extraneous 415 nm/558 nm DAPI Fluorescence Present in Mammalian Tissues and Concentrations Tested.
| Species | Concentration Used in Study | Amount in Cells/Tissue |
|---|---|---|
| DNA | 55 µg/mL | 7.7 pg/bovine articular chondrocyte22 |
| RNA | 180 µg/mL | 20–30 pg/mammalian cell23 |
| ATP | 6.0 µM | 0.5–10 mM/mammalian cell24 |
| Pyrophosphate | 5.0 µM | 655 ± 45 pmol /106 chondrocytes25 |
| Chondroitin sulfate | 6.1 mg/mL | 112 ± 14 µg GAG/µg DNA in bovine cartilage26 |
| Protein | 7.3 mg/mL | 2.2 ± 0.17 mg protein/g of articular cartilage27 (1-1.15 × 106 cells/g of articular cartilage28) |
DAPI = 4′,6-diamidino-2-phenylindole; GAG = glycosaminoglycan.
Elimination of DAPI Fluorescence from DNA and RNA
DNA and RNA were added to a final concentration of 55 and 180 µg/mL, respectively, in 180 µL of 7 nmol polyP (final polyP concentration: 38.9µM) and co-treated with 10 U DNase I and 2.5 U RNase A (Roche Diagnostics, Indianapolis, IN, USA) in a buffer containing 100 µM calcium chloride, 1 mM magnesium chloride, 50 µg/mL bovine serum albumin, and 10 mM Tris pH 7.5 and incubated for 60 minutes at 37°C. Concentrations of nucleic acids were chosen based on the average cellular nucleic acid content ( Table 1 ). After incubation, 6 µL of 0.5 M EDTA was added to the samples to inactivate the enzymes. DAPI fluorescence from enzyme-treated and untreated samples was measured. In selected samples, Tris solution (pH 7.5) of various concentrations up to 500 mM was added to the samples and standards in order to determine if this would decrease fluorescence.
Elimination of DAPI Fluorescence Contributed by Chondroitin Sulfate
PolyP-45 (7 nmol in 140 µL ddH2O) was mixed with either chondroitin sulfate (6.1 mg/mL in ddH2O) or carrier (ddH2O) and loaded onto silica spin column. Extraction was performed and GAG content of the eluents was quantified using dimethylmethylene blue (DMMB) dye binding assay and spectrophotometry (absorbance at 525 nm).29 The 415/558 nm DAPI fluorescence of eluents was measured. In selected samples, Tris solution (3 M, pH 7.5) was added to increase the Tris concentration of the samples to 500 mM. A standard curve was generated by adding DAPI to serial dilutions of chondroitin sulfate made in either 10 mM or 500 mM Tris pH 7.5 and fluorescence measured.
Proteinase K Pretreatment of Samples
To determine if proteinase K is necessary to extract polyP from protein containing tissues/solutions, 7 nmol polyP-45 (50 µM) was mixed with 20% or 50% (v/v) FBS and incubated in the presence or absence of proteinase K (1 mg/mL, Life Technologies, Burlington, Ontario, Canada) in 10 mM Tris, pH 8.0, for 4 hours at 56°C with periodic agitation. Ten millimolar EDTA was also added to selected samples. Silica spin column isolation was performed on proteinase K–treated samples and aliquots of Tris HCl, pH 7.5 solution was added to the eluents (final Tris concentration of 500 mM) before fluorescence was measured. The recovery ratio was calculated by dividing the DAPI fluorescence of eluted samples by the non–enzyme-treated control (50 µM polyP-45 in 10 mM Tris-EDTA, pH 8.0).
PolyP Quantification in the In Vitro Formed and Native Cartilage Samples
Full-thickness and superficial zone (approximately top 15% of tissue) articular cartilage were harvested from calf knee joints (9 to 12 months old). Bovine muscle and adipose tissue were also harvested. To generate in vitro–formed cartilage tissue, articular chondrocytes were isolated from the full-thickness cartilage and cultured as previously described.9 Murine brain, lung, liver, kidney, and heart were harvested from wild-type C3H/HeJ male mice. The wet weight of the tissues was determined and stored at −80°C until analysis. The murine tissue was thawed on the day of analysis and diced into small pieces with a blade. Proteinase K solution (1 mg/mL in 10 mM EDTA and 10 mM Tris pH 8.0) was added to native full thickness cartilage and native murine tissues (1 mL per 100 mg tissue) or 280 µL to in vitro–grown cartilage tissue (weight range 8-15 mg), and incubated for 2 hours at 56°C with periodic agitation. It was determined that 100 µL of 1 mg/mL proteinase K solution per 10 mg wet weight native tissue and 60 µL per 10 mg wet weight of in vitro–formed cartilage was sufficient to achieve complete or near complete digestion of the tissues. Each sample was divided in 2 aliquots of equal volume, and 7 nmol polyP was added to 1 aliquot (50 µM, considered “spiked”). Samples were then incubated for 2 hours at 56°C. As a positive control, 50 µM polyP-45 was incubated in proteinase K solution, in the absence of tissues under identical conditions. Following incubation, the full polyP quantification assay was performed ( Figure 7 ). To confirm that the polyP signals were specific to polyP, 87U of alkaline phosphatase (Thermo Scientific, 87 U/µL, 21.3 mg protein/mL) was added during the DNase and RNase digestion step to selected samples and incubated for 1 hour at 37°C. For each sample, the recovered amount of exogenously added polyP-45 was determined by calculating the difference in DAPI fluorescence between the spiked and nonspiked aliquots. The recovery rate of exogenously added polyP-45 was calculated by dividing the recovered amount of exogenously added polyP-45 by the measured polyP content of the positive control. Then, the polyP content measured in each nonspiked aliquot was divided by the recovery rate to compensate for the loss of polyP during the assay and normalized to the corresponding wet weight to derive the final polyP content of tissues. The GAG content of tissue samples was quantified from nonspiked aliquots using the DMMB assay and normalized to wet weight.
Figure 7.

Line diagram of the complete polyphosphate (polyP) quantification method.
Statistical Analysis
All experiments were repeated at least 3 times. One-way analysis of variance and Tukey’s pairwise post hoc tests were used to compare the recovery ratios of polyP standards in the absence of tissue, fluorescence of potentially interfering molecules in the presence of polyP, fluorescence of nuclease-treated samples in increasing Tris concentration, and the quantified polyP and GAG data from tissues. Two-way analysis of variance and Bonferroni post hoc tests were used to compare the fluorescence and recovery ratios of either untreated/treated or pre-/postisolation samples, as well as for the FBS-supplemented samples. Statistical significance was assigned at P < 0.05.
Safety Considerations
GuSCN can produce a toxic gas, hydrogen cyanide, on contact with acids. As a precaution, it is recommended that GuSCN-containing buffers be prepared and handled in a fume hood. GuSCN-containing waste should be collected into a concentrated sodium hydroxide solution, such that the final concentration of sodium hydroxide solution is no less than 0.5 N when discarded.
Results
Binding and Recovery of polyP from Silica Spin Columns
The recovery ratios of polyP dissolved in water were comparable for quantities of polyP ranging between 15.5 and 77.6 nmol (P = 0.2714, data not shown). These ratios were significantly higher than previously reported recovery ratios at the same polyP quantities. When less than 8 nmol polyP was loaded, a statistically significant decrease in the recovery ratio was observed at 2 nmol or lower ( Fig. 1A ), consistent with previous observations that the recovery ratio decreases with smaller quantities of polyP.21 However, the recovery ratio was consistently over 80% when 0.25 to 2 nmol polyP was loaded. The effect of polyP chain length on the recovery ratio was evaluated by loading equivalent quantities of polyP by phosphate units of varying chain lengths, which revealed an increasing trend of the recovery ratio with chain length ( Fig. 1B ). Compared to the recovery ratio of polyP-45, recovery ratios of polyP chain length 14 and 23 were significantly lower (P < 0.001), while recovery ratios of polyP chain length 60 and 130 were significantly higher (P < 0.01). Although statistically significant, differences in recovery ratios of polyP-45 versus 60 and 130 were minor (11% and 15%). No statistical differences between the recovered and standard polyP were observed in polyP with chain lengths 60 and 130. Therefore, despite the lower recovery ratios of shorter polyP chains, polyP chain lengths of interest (between 45 and 130) could be efficiently recovered from silica spin columns.
Figure 1.

Quantity and chain length affected the polyphosphate (polyP) recovery ratios of the silica spin column at nanomole and subnanomole quantities. (A) Silica spin columns bound and released nanomoles of polyP chain length 45 at a recovery ratio that ranged between 80% and 100%. Results are represented as mean ± SD, n = 3. *P < 0.05, **P < 0.01 compared with either 4 nmol or 8 nmol respectively. (B) The recovery ratio of polyP (7 nmol) increased with increasing chain length. *P < 0.01 compared with polyP chain length 45. Results are represented as mean ± SD, n = 3. P < 0.01 between any 2 values, except the pair marked ns (nonsignificant).
Elimination of DNA and RNA from Samples to Prevent Extraneous DAPI Fluorescence Signals
Elimination of extraneous fluorescence improves the specificity and sensitivity of polyP quantification. DNA, RNA, ATP, pyrophosphate, GAG and protein were identified as potential molecules in mammalian tissues that may also produce 415/558 nm DAPI fluorescence signals and interfere with the fluorometric quantification of polyP.23. To verify this, polyP-45 was mixed with each of these molecules (see Table 1 ) and their subsequent DAPI fluorescence without silica column extraction was measured and compared with polyP samples without these molecules. DAPI fluorescence of polyP samples containing DNA, chondroitin sulfate, RNA or BSA were significantly higher than that of polyP alone ( Fig. 2A ). The mixed samples were loaded on the columns to test whether the silica spin column method could isolate polyP from the mixture. Eluents from samples with DNA and chondroitin sulfate had significantly higher DAPI fluorescence compared with the polyP-only control ( Fig. 2B ). DAPI fluorescence of eluents from samples with RNA was comparable with polyP-only control, which could indicate that spontaneous RNA degradation may be sufficient to prevent its adsorption on silica columns under these conditions. Nuclease treatment (DNase and RNase) of samples supplemented with 55 µg/mL DNA and 180 µg/mL RNA (concentrations chosen based on cellular nucleic acid content; see Table 1 ) failed to eliminate the DAPI fluorescence (Fig. 3A). However, when concentrated Tris buffer was added to nuclease-treated DNA and RNA, the DAPI fluorescence decreased in a concentration-dependent manner, eliminating 96% and 98% of the signals from degraded DNA and RNA, respectively, at a Tris concentration of 500 mM ( Fig. 3B and C ). On the other hand, reduction of DAPI fluorescence from polyP was statistically significant ( Fig. 3D ). Increasing the Tris concentration in the absence of nuclease digestion did not eliminate DAPI fluorescence of intact nucleic acids ( Fig. 3E ), showing that both nuclease treatment and the subsequent addition of Tris were both required to eliminate the signals produced by nucleic acids. Nuclease-treated DNA and RNA supplemented polyP samples produced similar DAPI fluorescence to samples containing polyP alone ( Fig. 3F ).
Figure 2.

DNA, chondroitin sulfate, and fetal bovine serum (FBS) affected the DAPI fluorescence signal of polyP isolated by silica spin columns. Seven nanomoles polyP were added to 140 µL samples of potentially interfering molecules (amount listed in Table 1). DAPI fluorescence signal was measured (A) before and (B) after the silica spin column isolation step and compared with the polyP alone. Results are expressed as mean ± SD, n = 3. *P < 0.01 compared with polyP alone. CS = chondroitin sulfate; BSA = bovine serum albumin; pyroP = sodium pyrophosphate.
Figure 3.

Both nuclease treatment and Tris were required to eliminate DAPI fluorescence signals produced by DNA and RNA following elution from the column. (A) Samples were either treated with nucleases or untreated and DAPI fluorescence was measured as described in the Materials and Methods section. *P < 0.001 compared with untreated. (B-D) Effect of various Tris concentrations on nuclease-treated DNA (B), RNA (C) and polyP (D) sample fluorescence, *P < 0.001 compared with 500 mM. (E) 500 mM Tris concentration by itself did not eliminate DAPI fluorescence of untreated DNA and RNA. *P < 0.001 compared with nucleases untreated samples. (F) DNA and RNA were mixed with polyP and were either untreated or underwent nuclease treatment and DAPI fluorescence was measured in the presence of 500 mM Tris. Results are expressed as mean ± SD, n = 3.
Elimination of GAG from Samples Generating Extraneous DAPI Fluorescence Signals
The dimethylmethylene blue (DMMB) assay confirmed that the silica spin column isolation step eliminated 99% of chondroitin sulfate in the samples, regardless of whether polyP was present in the initially loaded sample ( Fig. 4A ). A standard curve of DAPI fluorescence and chondroitin sulfate revealed a nonlinear relationship with a peak at 100 µg/mL in the presence of 10 mM Tris HCl ( Fig. 4B ). This was consistent with the previous observation that DAPI fluorescence of the eluent from samples containing polyP and chondroitin sulfate was higher ( Fig. 2 ). In the presence of 500 mM Tris, a large reduction of signal was observed at all chondroitin sulfate concentrations examined (50-1000 µg/mL), eliminating 99.7% of signal ( Fig. 4B ). PolyP samples with added chondroitin sulfate produced significantly higher DAPI fluorescence than polyP-only control in the presence of 10 mM Tris HCl, pH 7.5 (P < 0.01, Fig. 4C ).
Figure 4.

DAPI fluorescence signal produced by residual chondroitin sulfate (CS) in postisolation samples were reduced by the addition of Tris. (A) PolyP (7 nmol in 140 µL ddH2O) was mixed with either chondroitin sulfate (6.1 mg/mL in ddH2O) or carrier (ddH2O) and loaded onto silica spin column. After the silica spin column isolation step was performed, chondroitin sulfate in the eluent was quantified with the dimethylmethylene blue dye assay and compared to the original solution. Addition of polyP in samples did not affect the results of GAG quantification. GAG = glycosaminoglycans; ns = not significant. (B) Semilog plot of DAPI fluorescence in the presence of CS. In the presence of 10 mM Tris, DAPI fluorescence peaked at 100 µg/mL and decreased at higher concentrations. In 500 mM Tris, DAPI fluorescence increased linearly (r2 = 0.9938) with CS concentrations >1000 µg/mL. (C) DAPI fluorescence of CS and polyP samples were measured before and after the silica spin column isolation step and compared with the polyP standard alone. Results are expressed as mean ± SD, n = 3. For denoted pairs of data: *P < 0.001.
Recovery of PolyP in Presence of Serum Proteins
The recovery ratios of FBS-containing polyP samples following proteinase K digestion prior to silica spin column isolation were comparable to polyP-only control ( Fig. 5 ). Without EDTA, samples treated with proteinase K prior to isolation produced significantly less DAPI fluorescence, indicating that EDTA was necessary for the proteinase K treatment step to prevent the loss of polyP signal.
Figure 5.
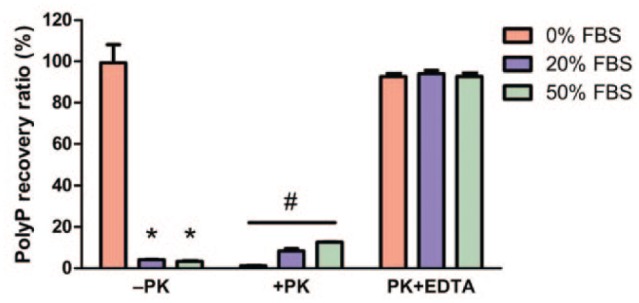
Proteinase K pretreatment enhances polyP recovery. Seven nanomoles polyP samples mixed with various concentrations of fetal bovine serum (FBS) were incubated in the presence (+PK) or absence (–PK) of proteinase K (1 mg/mL), alone or together with ethylene diamine tetraacetic acid (EDTA; 10mM), and incubated for 4 hours in 56°C. 4′,6-Diamidino-2-phenylindole (DAPI) fluorescence was measured after the silica spin column isolation step and compared to similarly incubated polyP standard (no proteinase K treatment) to calculate the recovery ratio. Loss of polyP signal was observed in samples without EDTA during proteinase K pretreatment (+PK). Results are expressed as mean ± SD, n = 3. *P < 0.01 compared with –PK without FBS, #P < 0.01 between one another as well as compared with –PK without FBS.
Quantification of PolyP Content in Native Bovine and Murine Tissues and In Vitro–Formed Cartilage Tissues
Tissues rich in proteoglycans, DNA and RNA that could potentially interfere with the polyP assay were then analyzed using the optimized polyP quantification assay. Tissue samples are first digested with proteinase K in the presence of EDTA before performing the silica spin column polyP extraction. The eluted samples are treated with nucleases for 60 minutes at 37°C, and then the digestion was stopped with the addition of 6 µL of 0.5 M EDTA pH 8.0 (to a final concentration 0.01 M EDTA). Tris and DAPI are then sequentially added before measuring the fluorescence. The polyP recovery rate of each tissue type was measured by quantifying a matching sample that was spiked with a known amount of polyP-45 ( Fig. 6 ). A significantly lower (P < 0.05) recovery rate was measured from both in vitro–grown cartilage tissue samples and full thickness native articular cartilage samples compared with polyP alone (no tissue, 50µM). The optimized polyP quantification assay is summarized in Figure 7 . We measured the polyP content in various murine and bovine tissues using the same methodology ( Table 2 ). The measured DAPI fluorescence signal was specific to polyP as alkaline phosphatase treated samples showed complete elimination of the signal compared to their untreated controls (data not shown). PolyP was detected in all tested murine tissues; however, particulates were observed in samples containing murine brain, kidney, and hearts after 4 hours of proteinase K digestion, indicating incomplete digestion and hence possible under-reporting of the total polyP content in these tissues. PolyP was detected in bovine muscle and adipose tissues, which accumulated similar amounts as the superficial zone of cartilage. Of note, a supernatant lipid layer formed in proteinase K-digested adipose tissue samples. PolyP accumulation was assessed exclusively from the aqueous phase for these tissues, as polyPs are highly polar and therefore unlikely to be found in the lipid layer.
Figure 6.

Quantification of polyP in the in vitro–formed cartilage and native cartilage tissues with correction for recovery rate. Cartilage tissues were grown in vitro on calcium polyphosphate (CPP) substrates. Native full-thickness (FT) or superficial zone (SZ) cartilage tissues were also harvested for polyP quantification. (A) Recovered amounts of exogenously added polyP from each tissue sample were normalized to the recovered polyP from the positive control (tissue digest spiked with 50 µM polyP). Results are expressed as mean ± SD, n = 6. *P < 0.05; **P < 0.01 compared with no tissue control. (B) Measured polyP content of tissues were corrected with the recovery rate and normalized to tissue wet weight. (C) Glycosaminoglycan (GAG) content of tissues was normalized to tissue wet weight. Data points are color-coded for each donor animal, and each point represents the mean of an experiment done in triplicate. Results are expressed as mean ± SD, n = 6. *P < 0.05; **P < 0.01; ***P < 0.001 compared with all others.
Table 2.
Quantified PolyP Contents in Mouse and Bovine Tissues.
| Organism | Tissue | Recovery Rate (%), Mean ± SD | PolyP/Tissue Wet Weight (pmol Pi/mg Tissue), Mean ± SD | n |
|---|---|---|---|---|
| Mouse | Brain | 72.5 ± 2.5 | 48.1 ± 9.1 | 4 |
| Lung | 78.5 ± 1.3 | 56.9 ± 9.4 | 4 | |
| Liver | 76.3 ± 4.8 | 44.2 ± 5.2 | 4 | |
| Kidney | 62.3 ± 6.4 | 90.5 ± 3.3 | 4 | |
| Heart | 72.7 ± 3.0 | 60.4 ± 17.6 | 4 | |
| Bovine | Muscle | 77.7 ± 1.6 | 40.4 ± 6.0 | 3 |
| Adipose | 81.3 ± 0.7 | 31.7 ± 2.9 | 3 | |
| Nucleus pulposus | 80.3 ± 2.7 | 20.0 ± 0.5 | 3 | |
| Inner annulus fibrosus | 79.9 ± 3.6 | 13.9 ± 0.3 | 3 | |
| Outer annulus fibrosus | 78.2 ± 1.9 | 10.1 ± 0.8 | 3 |
Discussion
In this study, we report the development of a simple and sensitive method to isolate and quantify small quantities of polyP ranging from 14 to 130 phosphate residues from mammalian tissues using DAPI-based fluorometric quantification. An effective polyP isolation method from mammalian tissues was required to be able to use this fluorometric quantification technique. We found that Proteinase K digestion in the presence of EDTA was critical. Similarly, elimination of extraneous fluorescence signals was required, which was accomplished by addition of nuclease digestion and Tris addition in post elution steps.
Based on the structural similarities of polyP and nucleic acids, several other articles have reported the use of silica column or glass fiber filters for polyP isolation.17,20 We demonstrate a significant improvement of the silica spin column–based method in both the recovery ratio and the lowest recoverable amounts of polyP using our methodology compared with that published in the literature. The recovery ratios of polyP dissolved in water ranging from 15.5 to 77.6 nmol showed recovery ratios ranging from 99% ± 3% to 97% ± 2%, in contrast to previous studies which reported recovery ratios of 42% to 77%19 and 48% to 77%23 for the same polyP concentrations. Recovery ratios only fell significantly at 2nmol or less using the currently developed method. A recent study by Schlagenhauf et al.30 achieved ≥95% recovery with a silica spin column–based polyP quantification assay as demonstrated by the recovery ratio between extracted and nonextracted polyP samples. Though the authors mention the “matrix effect” as a possible hindrance to polyP quantification and demonstrated that the elution buffer they used was enough to get rid of the interfering matrix molecules, the quantification and effect of those interfering molecules was not shown. Furthermore, this analysis was on cells not tissue as done in the current study, which becomes relevant when quantifying polyP in matrix laden tissues, such as cartilage, disc tissue and brain. It is possible that our better recovery ratios were achieved by removal of the interfering molecules, such as GAGs, and by utilizing an optimized Tris buffer that frees up binding spots on the silica membrane for the polyP to adhere. Investigations of DNA adsorption on silica have shown that shielding intermolecular electrostatic forces, dehydration of the DNA and silica surfaces, and intermolecular hydrogen bond formation in the DNA-silica contact layer are main factors influencing adsorption,31 which can be manipulated by the buffer used to load samples onto silica spin columns. Through the process of optimizing the loading buffer we found that the choice and concentration of the chaotropic reagent, total ionic strength, monovalent cations in the buffer, pH and concentration of ethanol all affected polyP binding as indirectly determined by the recovery ratio in addition to quantifying interference by extracellular matrix. We found that the optimization of wash buffers minimized GAG contamination and increased polyP recovery.
Another major molecule interfering in polyP quantification is protein, which is abundant in tissue and serum samples. These protein molecules cover the silica membrane surface and prevent the binding of polyP to the binding sites. To eliminate interference from proteins, the tissue samples were digested with proteinase K supplemented with 10 mM EDTA. Previously, proteinase K pre-treatment in the presence of 10mM EDTA was demonstrated to be the most effective treatment for maximizing polyP signal in environmental samples.20 Since divalent cations have been shown to cause polyP degradation,32 the reduced DAPI fluorescence without EDTA supplementation during tissue digestion may be due to the divalent cations, not protein, present in FBS. This step makes the assay applicable for quantification of polyP in liquid samples such as blood, serum, and culture media.
In order to develop a polyP-specific quantification method, additional steps to reduce the interfering effects of DNA and RNA were required as DNA and RNA give false positive signals with DAPI at the excitation/emission spectra used to quantify polyP. A previous study reported that enzymatic degradation of DNA and RNA reduced the intensity of shifted DAPI fluorescence to negligible levels,20 but nucleic acids used in that study were at a lower concentration (3 µg/mL). In the current study, concentrations of 101 to 102 µg/mL, which is equivalent to the DNA/RNA content of 1 million cells and mimics the amounts that might be encountered while quantifying polyP in native and engineered tissues was used. Interfering signals were not completely eliminated with nuclease digestion alone. Addition of Tris HCl eliminated the residual interfering signals from DNA and RNA post nuclease digestion. Thus addition of both nucleases and Tris were required.
Interestingly, the polyP content was higher in in vitro–grown tissues compared with native tissue ( Fig. 6B ). There are several possible explanations for this. It may be due to the saturation of the silica membranes of the spin columns by the higher levels of GAGs in native tissue preventing the binding of polyP to the column thus resulting in lower levels. Alternatively, the levels are truly higher perhaps due to sequestration of polyP released from the degrading CPP scaffold or because more polyP is being synthesized by the chondrocytes in the newly formed tissue. It is not clear why the polyP recovery ratio was lower in in vitro–grown tissue. It may be that the abundance of tissue-derived polyP may be preventing binding of the spiked polyP, thereby causing an apparent decrease in the recovery rate compared to no tissue controls and native cartilage.
In conclusion, this method can be easily adapted for use in other laboratories as this protocol closely resembles nucleic acid purification methods, and the enzymes utilized (proteinase K, RNase A, and DNase I) are readily available. Since proteinase K pre-treatment can effectively degrade virtually all proteins, this method can be readily adapted to evaluate most biological tissues/solutions. Since the recovery ratios for polyP chain lengths of less than 14 or greater than 130 Pi residues were not measured, this method may not be suitable for binding very small or very large polyP polymers. In systems that require the quantification of shorter polyP chains, short chain length-specific quantification methods such as high performance liquid chromatography33 may be the better method but further study is required. This new method provides an easy-to-use research tool for future polyP investigation in mammalian biology.
Footnotes
Acknowledgments and Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the Canadian Institutes for Health Research to R.A.K. (CIHR-MOP 12611). R.G. was supported by fellowships from Toronto Musculoskeletal Center (TMC); University of Toronto, The Arthritis Society (TAS) of Canada and National Science and Educational Research Council of Canada (NSERC).
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval: Ethical approval was not sought for the present study because only waste tissues were used.
Animal welfare: Guidelines for humane animal treatment did not apply to the present study because only waste tissue was utilized.
References
- 1. Ruiz FA, Lea CR, Oldfield E, Docampo R. Human platelet dense granules contain polyphosphate and are similar to acidocalcisomes of bacteria and unicellular eukaryotes. J Biol Chem. 2004;279(43):44250-7. [DOI] [PubMed] [Google Scholar]
- 2. Smith SA, Mutch NJ, Baskar D, Rohloff P, Docampo R, Morrissey JH. Polyphosphate modulates blood coagulation and fibrinolysis. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(4):903-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang L, Fraley CD, Faridi J, Kornberg A, Roth RA. Inorganic polyphosphate stimulates mammalian TOR, a kinase involved in the proliferation of mammary cancer cells. Proc Natl Acad Sci U S A. 2003;100(20):11249-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kawazoe Y, Katoh S, Onodera Y, Kohgo T, Shindoh M, Shiba T. Activation of the FGF signaling pathway and subsequent induction of mesenchymal stem cell differentiation by inorganic polyphosphate. Int J Biol Sci. 2008;4(1):37-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zakharian E, Thyagarajan B, French RJ, Pavlov E, Rohacs T. Inorganic polyphosphate modulates TRPM8 channels. PLoS One. 2009;4(4):e5404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Omelon S, Georgiou J, Henneman ZJ, Wise LM, Sukhu B, Hunt T, et al. Control of vertebrate skeletal mineralization by polyphosphates. PLoS One. 2009;4(5):e5634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. St-Pierre JP, Pilliar RM, Grynpas MD, Kandel RA. Calcification of cartilage formed in vitro on calcium polyphosphate bone substitutes is regulated by inorganic polyphosphate. Acta Biomater, 2010;6(8):3302-9. [DOI] [PubMed] [Google Scholar]
- 8. Holmström KM, Marina N, Baev AY, Wood NW, Gourine AV, Abramov AY. Signalling properties of inorganic polyphosphate in the mammalian brain. Nat Commun. 2013;4:1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. St-Pierre JP, Wang Q, Li SQ, Pilliar RM, Kandel RA. Inorganic polyphosphate stimulates cartilage tissue formation. Tissue Eng Part A. 2012:18(11-12):1282-92. [DOI] [PubMed] [Google Scholar]
- 10. Gawri R, Shiba T, Pilliar R, Kandel R. Inorganic polyphosphates enhances nucleus pulposus tissue formation in vitro. J Orthop Res. doi: 10.1002/jor.23288. Epub 2016. May 10. [DOI] [PubMed] [Google Scholar]
- 11. Rao NN, Gomez-Garcia MR, Kornberg A. Inorganic polyphosphate: essential for growth and survival. Annu Rev Biochem. 2009;78:605-47. [DOI] [PubMed] [Google Scholar]
- 12. Kumble KD, Kornberg A. Inorganic polyphosphate in mammalian cells and tissues. J Biol Chem. 1995;270(11):5818-22. [DOI] [PubMed] [Google Scholar]
- 13. Ohtomo R, Sekiguchi Y, Mimura T, Saito M, Ezawa T. Quantification of polyphosphate: different sensitivities to short-chain polyphosphate using enzymatic and colorimetric methods as revealed by ion chromatography. Anal Biochem. 2004;328(2):139-46. [DOI] [PubMed] [Google Scholar]
- 14. Gomez Garcia MR. Extraction and quantification of Poly P. Poly P analysis by urea-PAGE. Bio Protoc. 2014;4(9):e1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Greenfield NJ, Hussain M, Lenard J. Effects of growth state and amines on cytoplasmic and vacuolar pH, phosphate and polyphosphate levels in Saccharomyces cerevisiae: a 31P-nuclear magnetic resonance study. Biochim Biophys Acta. 1987;926(3):205-14. [DOI] [PubMed] [Google Scholar]
- 16. Ault-Riché D, Fraley CD, Tzeng CM, Kornberg A. Novel assay reveals multiple pathways regulating stress-induced accumulations of inorganic polyphosphate in Escherichia coli. J Bacteriol. 1998;180(7):1841-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Werner TP, Amrhein N, Freimoser FM. Novel method for the quantification of inorganic polyphosphate (iPoP) in Saccharomyces cerevisiae shows dependence of iPoP content on the growth phase. Arch Microbiol. 2005;184(2):129-36. [DOI] [PubMed] [Google Scholar]
- 18. Aschar-Sobbi R, Abramov AY, Diao C, Kargacin ME, Kargacin GJ, French RJ, et al. High sensitivity, quantitative measurements of polyphosphate using a new DAPI-based approach. J Fluoresc. 2008;18(5):859-66. [DOI] [PubMed] [Google Scholar]
- 19. Kulakova AN, Hobbs D, Smithen M, Pavlov E, Gilbert JA, Quinn JP. et al. Direct quantification of inorganic polyphosphate in microbial cells using 4′-6-diamidino-2-phenylindole (DAPI). Environ Sci Technol. 2011;45(18): 7799-803. [DOI] [PubMed] [Google Scholar]
- 20. Martin P, Van Mooy BA. Fluorometric quantification of polyphosphate in environmental plankton samples: extraction protocols, matrix effects, and nucleic acid interference. Appl Environ Microbiol. 2013;79(1):273-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sophia Fox AJ, Bedi A, Rodeo SA. The basic science of articular cartilage: structure, composition, and function. Sports Health. 2009;1(6):461-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kim YJ, Sah RL, Doong JY, Grodzinsky AJ. Fluorometric assay of DNA in cartilage explants using Hoechst 33258. Anal Biochem. 1988;174(1):168-76. [DOI] [PubMed] [Google Scholar]
- 23. Alberts B. Molecular biology of the cell. Vol. 1 2nd ed. New York, NY: Garland; 1989. [Google Scholar]
- 24. Beis I, Newsholme EA. The contents of adenine nucleotides, phosphagens and some glycolytic intermediates in resting muscles from vertebrates and invertebrates. Biochem J. 1975;152(1):23-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lust G, Seegmiller JE. A rapid, enzymatic assay for measurement of inorganic pyrophosphate in biological samples. Clin Chim Acta. 1976;66(2):241-9. [DOI] [PubMed] [Google Scholar]
- 26. Waldman SD, Grynpas MD, Pilliar RM, Kandel RA. Characterization of cartilagenous tissue formed on calcium polyphosphate substrates in vitro. J Biomed Mater Res. 2002;62(3):323-30. [DOI] [PubMed] [Google Scholar]
- 27. Bollet AJ, Handy JR, Sturgill BC. Chondroitin sulfate concentration and protein-polysaccharide composition of articular cartilage in osteoarthritis. J Clin Invest. 1963;42:853-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Oseni AO, Butler PE, Seifalian AM. Optimization of chondrocyte isolation and characterization for large-scale cartilage tissue engineering. J Surg Res. 2013;181(1):41-8. [DOI] [PubMed] [Google Scholar]
- 29. Lee WD, Hurtig MB, Kandel RA, Stanford WL. Membrane culture of bone marrow stromal cells yields better tissue than pellet culture for engineering cartilage-bone substitute biphasic constructs in a two-step process. Tissue Eng Part C Methods. 2011;17(9):939-48. [DOI] [PubMed] [Google Scholar]
- 30. Schlagenhauf A, Pohl S, Haidl H, Leschnik B, Gallistl S, Muntean W. Non-enzymatic quantification of polyphosphate levels in platelet lysates and releasates. J Pharm Biomed Anal. 2016;131:1-5. [DOI] [PubMed] [Google Scholar]
- 31. Melzak KA, Sherwood CS, Turner RFB, Haynes CA. Driving forces for DNA adsorption to silica in perchlorate solutions. J Colloid Interf Sci. 1996;181(2):635-44. [Google Scholar]
- 32. Momeni A, Filiaggi MJ. Comprehensive study of the chelation and coacervation of alkaline earth metals in the presence of sodium polyphosphate solution. Langmuir. 2014;30(18): 5256-66. [DOI] [PubMed] [Google Scholar]
- 33. Sekiguchi Y, Matsunaga A, Yamamoto A, Inoue Y. Analysis of condensed phosphates in food products by ion chromatography with an on-line hydroxide eluent generator. J Chromatogr A. 2000;881(1-2):639-44. [DOI] [PubMed] [Google Scholar]