

Abstract
We have previously reported that miR-17~92 is critically involved in the pathogenesis of pulmonary hypertension (PH). We also identified two novel mR-17/20a direct targets, PDZ and LIM domain protein 5 (PDLIM5) and prolyl hydroxylase 2 (PHD2), and elucidated the signaling pathways by which PDLIM5 and PHD2 regulate functions of pulmonary artery smooth muscle cells (PASMCs). In addition, we have shown that plasminogen activator inhibitor-1 (PAI-1) is also downregulated in PASMCs that overexpress miR-17~92. However, it is unclear whether PAI-1 is a direct target of miR-17~92 and whether it plays a role in regulating the PASMC phenotype. In this study, we have identified PAI-1 as a novel target of miR-19a/b, two members of the miR-17~92 cluster. We found that the 3′-untranslated region (UTR) of PAI-1 contains a miR-19a/b binding site and that miR-19a/b can target this site to suppress PAI-1 protein expression. MiR-17/20a, two other members of miR-17~92, may also indirectly suppress PAI-1 expression through PDLIM5. PAI-1 is a negative regulator of miR-17~92-mediated PASMC proliferation. Silencing of PAI-1 induces Smad2/calponin signaling in PASMCs, suggesting that PAI-1 is a negative regulator of the PASMC contractile phenotype. We also found that PAI-1 is essential for the metabolic gene expression in PASMCs. Furthermore, although there is no significant change in PAI-1 levels in PASMCs isolated from idiopathic pulmonary arterial hypertension and associated pulmonary arterial hypertension patients, PAI-1 is downregulated in hypoxia/Sugen-induced hypertensive rat lungs. These results suggest that miR-17~92 regulates the PASMC contractile phenotype and proliferation coordinately and synergistically by direct and indirect targeting of PAI-1.
Keywords: miR-19a/b, PAI-1, PASMC
INTRODUCTION
Pulmonary arterial hypertension (PAH) is a debilitating and incurable disease characterized by pulmonary vasoconstriction and vascular remodeling, causing irreversible increases in pulmonary vascular resistance and pulmonary arterial pressure and eventually right ventricular hypertrophy and failure (32). Although advances in treatment have led to significant relief of symptoms and improved length of survival, most patients become resistant to therapies, eventually succumbing to the disease (40). The vascular remodeling leading to PAH can largely be attributed to pulmonary arterial smooth muscle cell (PASMC) dysfunction. MicroRNAs (miRNAs), including miR-130/301 (4, 5), miR-140 (46), miR-143/145 (7, 15, 16), miR-204 (18), miR-21 (48), miR-210 (24, 53), miR-221 (19), and miR-223 (38), are known to regulate PASMC phenotype and thus are implicated in the pathogenesis of PAH (57). We (13) and others (45) have shown that the miR-17~92 cluster is a key regulator of pulmonary hypertension (PH) in vivo and in vitro. We have identified two novel targets of miR-17/20a, two members of the miR-17~92 cluster, and have shown their mechanisms of action, namely 1): direct targeting and suppression of PDZ and LIM domain protein 5 (PDLIM5) to induce the transformng growth factor-β (TGF-β)/Smad signaling pathway and contractile protein expression in PASMCs (13); and 2) direct targeting and suppression of prolyl hydroxylase 2 (PHD2) to induce hypoxia-inducible factor 1α (HIF1α) levels and PASMC proliferation (14). Since the miR-17~92 cluster consists of six mature miRNA members (miR-17, miR-20a, miR-18a, miR-19a, miR-19b, and miR-92a) (31, 43), targets of other members of the miR-17~92 cluster need to be identified to better understand the role of miR-17~92 in the phenotype change of PASMCs and pathogenesis of PH. Using a quantitative proteomics approach, we (13) reported that overexpression of miR-17~92 suppressed 14 proteins in PASMCs, one of which is PDLIM5 and has been studied. It is important to study how the other proteins are regulated by miR-17~92 and whether these proteins regulate PASMC phenotype.
In this study, we sought to characterize how miR-17~92 regulates plasminogen activator inhibitor-1 (PAI-1), another protein that is also suppressed by miR-17~92 in PASMCs. PAI-1 is of particular interest for the following reasons: PAI-1 is produced and secreted by endothelial cells, PASMCs, and macrophages, among other vascular cells (20). Active PAI-1 is the principle inhibitor of both tissue type and urokinase type plasminogen activator (also known as t-PA and u-PA, respectively), both of which actively convert plasminogen to plasmin, a serine protease that degrades fibrin clots and other blood plasma proteins. Additionally, PAI-1 interacts with the extracellular matrix protein vitronectin, modulating the ability of cells to adhere to the matrix by decreasing integrin binding affinity. How PAI-1 participates in PAH appears to be controversial. In one study, lungs of patients with idiopathic PAH (IPAH) and hypoxia-induced pulmonary hypertension (PH) rodents secrete markedly increased levels of PAI-1 (55); in another study, PAI-1 is downregulated in PASMCs in IPAH and negatively regulates PASMC proliferation (35). We found that the 3′-untranslated region (UTR) of PAI-1 mRNA contains a putative binding site for miR-19a and miR-19b. Accordingly, overexpression of the miR-17~92 cluster or miR-19a/b inhibited expression of PAI-1, whereas miR-17~92 or miR-19a/b inhibitors induced expression of PAI-1. Although PAI-1 itself had little effect on PASMC proliferation and death, silencing of PAI-1 restored PASMC proliferation in the presence of the miR-17~92 inhibitors, whereas overexpression of PAI-1 inhibited miR-17~92-induced PASMC proliferation. We found that suppression of PAI-1 increased expression of smooth muscle cell (SMC) markers such as calponin and Smad2, suggesting that miR-19a/b may suppress PAI-1 to induce Smad2 signaling and SMC marker expression. Furthermore, we have found that PAI-1 was essential for the metabolic gene expression in PASMCs. Although we did not find any change of PAI-1 levels between PASMCs isolated from PAH patients and normal donors, we did observe the downregulation of PAI-1 in hypoxia/Sugen-induced pulmonary hypertension rats.
In summary, we provide the first line of evidence that miR-17~92 can directly or indirectly suppress PAI-1 to regulate PASMC proliferation, upregulate Smad2 and PASMC contractile protein expression, and metabolic gene expression, suggesting that PAI-1 may be a key component in miR-17~92-mediated pathogenesis of PH. Future investigations into how PAI-1 contributes to PAH are warranted.
MATERIAL AND METHODS
Cell culture.
Normal human PASMCs were purchased from Lonza (Walkersville, MD). Human PASMCs from explanted lungs of normal donors and patients with idiopathic pulmonary arterial hypertension (IPAH) and PAH patients associated with other diseases (APAH) were provided by the Pulmonary Hypertension Breakthrough Initiative (PHBI) and by Drs. Suzy A. A. Comhair and Serpil C. Erzurum, Department of Pathobiology, Respiratory Institute, Cleveland Clinic (13). APAH subjects include patients with collagen vascular disease/connective tissue disease and congenital systemic-to-pulmonary shunts. Use of these cells was approved by the University of Illinois at Chicago Institutional Review Board. Human PASMCs were maintained in SmGM-2 medium (Lonza) containing 5% fetal bovine serum (FBS), growth factors, and 1% penicillin-streptomycin. Human PASMCs at passages 5–7 were used for most of the experiments. For stably transfected human PASMCs overexpressing miR-17~92 or a control construct, we used cell passages up to 10 due to the extra passages needed for screening. In this case, we used passage-matched human PASMCs overexpressing miR-17~92 or a control construct to minimize the variations between passages. Cell line authentication was carried out by morphology observation, immunostaining of α-smooth muscle actin, and Western blot analysis of a panel of SMC markers. For hypoxia exposure, we incubated cells in an INVIVO2300 hypoxia chamber (Ruskinn, Sanford, ME). All cells were maintained in a humidified incubator with a constant supply of 5% CO2 at 37°C.
Lentivirus-based miR-17~92 overexpression in human PASMCs.
For miRNA overexpression experiments, we inserted the miR-17~92 cluster into pLVX-Puro vector (pLVX-CTRL; Clontech, Mountain View, CA) at XhoI and EcoRI restriction endonuclease sites to generate lentiviral vectors overexpressing miR-17~92 (pLVX-miR-17~92). pLVX-Puro vector without insertion of miRNAs was used as control. High-titer lentivirus was prepared using a Lenti-X HT Packaging System (Clontech) in 293T cells following the user manual. Human PASMCs were infected with lentivirus supernatant and incubated for 2 days, followed by selection with 1.5 µg/ml puromycin (Sigma-Aldrich) for 3–4 days. Overexpression of miRNAs was confirmed by qRT-PCR analysis (13).
Quantitative mass spectrometry analysis to identify miR-17~92 target protein in human PASMCs.
Quantitative mass spectrometry proteomic analysis was performed as described previously (13). Briefly, we cultured human PASMCs stably infected with lentiviral vectors or pLVX-miR-17~92 lentiviruses and extracted proteins, followed by the protein assay to determine protein concentration. We then precipitated 100 µg of proteins from each sample by a cold acetone-TCA method. Proteins were resuspended and reduced, digested with trypsin, and desalted, followed with labeling with differential Tandem Mass Tag labeling reagents (Thermo Fisher Scientific, Rockford, IL) and were subjected to LC/MS/MS analysis. We repeated this experiment twice, and each experiment was conducted in triplicate. We identified 506 proteins and 355 proteins whose expression was altered in the two experiments, respectively. We screened for proteins that were downregulated at least 1.2-fold in both experiments and found 14 hits (13).
miRNA inhibitors and mimics.
miRVana miRNA inhibitors and miRNA mimics were purchased from Ambion (Austin, TX). miRNA inhibitors negative control no. 1 was used as miRNA inhibitor control and miRNA mimics negative control no. 1 was used as control for miRNA mimics. Negative controls are random-sequence miRNA mimic/inhibitor molecules that have been extensively tested in human cell lines according to the manufacturer’s protocol. For miR-17~92 mimics or inhibitors, a mixture of the same amount of miR-17/20a, miR-18a, miR-19a/19b, and miR-92a mimics or inhibitors was used, respectively.
PAI-1 3′-UTR luciferase reporter assay.
To construct the luciferase reporter plasmid with a 0.6-kb PAI-1 3′-untranslated region (UTR), we amplified the 3′-UTR of human PAI-1 gene containing the predicted miR-19a/19b binding site from human genomic DNA and inserted it downstream of the luciferase reporter gene in the pGL3-Promoter vector (Promega) through the XbaI endonuclease restriction site. The construct was sequenced to confirm the DNA sequences.
Human PASMCs stably infected with pLVX-CTRL or pLVX-miR-17~92 were plated in six-well plates and transfected with 2 μg of luciferase reporter plasmid and 1 μg of Renilla reporter plasmid using Lipofectamine 2000 reagent (Invitrogen, Grand Island, NY). Normal human PASMCs were plated in six-well plates and cotransfected with the reporter plasmid Renilla reporter plasmid, and 50 pmol of miRNA mimics or inhibitors (Ambion). After 48-h incubation, cells were lysed, and the luciferase activity was measured and calculated by comparing the firefly/Renilla luciferase ratio as we described previously (13, 14).
Small interfering RNA suppression of PAI-1 and PDZ and LIM domain protein 5.
We plated human PASMCs in 60-mm dishes at ~60–70% confluence and transfected them with a mixture of small interfering (si)RNAs (Ambion) and Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. Negative control siRNA (siRNA NC, Ambion) was used as negative control. Cells were incubated with Complete medium for 2 days before harvest.
Overexpression of PAI-1.
PAI-1 overexpression (PAI-OE) plasmids (PAI1-Bio-His, or Serpine1-bio-His) were purchased from Addgene (Cambridge, MA) and transfected into human PASMCs using Lipofectamine 2000. Vehicle plasmids were transfected as controls. Two days after transfection, cells were harvested for bromodeoxyuridine (BrdU) incorporation assay, CellTiter 96 Aqueous One Solution Cell Proliferation Assay, and lactate dehydrogenase (LDH) assay.
Western blot analysis.
Cells were washed with ice-cold PBS and lysed in mRIPA buffer (50 mM Tris, pH 7.4, 1% NP-40, 0.25% deoxycholate, 150 mM NaCl, and protease inhibitors), followed by centrifugation at 13,000 g for 10 min at 4°C. The supernatants were subjected to the Bio-Rad protein assay (Bio-Rad, Hercules, CA) to determine protein concentrations. Proteins were then separated by SDS-polyacrylamide gel electrophoresis and transferred to BA85 nitrocellulose membrane (PROTRAN; Whatman, Dassel, Germany) and immunoblotted with primary antibodies followed by detection with SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific). The following primary antibodies were used in this study: α-tubulin, α-smooth muscle actin (α-SMA) (52), calponin (Santa Cruz Biotechnology, Santa Cruz, CA, https://www.scbt.com/scbt/product/calponin-1-antibody-calp), smooth muscle protein 22α (SM22α; Abcam, Cambridge, MA) (51), myocardin (R&D Systems) (1), serum response factor (SRF; Cell Signaling Technology) (1), Smad2/3 (Cell Signaling Technology) (13), PAI-1, PDLIM5 (Sigma-Aldrich) (13), proliferating cell nuclear antigen (PCNA; Proteintech Group, Rosemont, IL, https://www.ptglab.com/products/PCNA-Antibody-10205-2-AP.htm), Receptor for advanced glycation end products (RAGE, Abcam) (28), poly(ADP-ribose) polymerase-1 (PARP-1, Proteintech Group) (54), His (Qiagen, Valencia, CA), and anti-mouse, anti-rabbit, and anti-goat IgG-horseradish peroxidase conjugates (Bio-Rad). We used ImageJ software to quantify the gray density of protein bands.
BrdU cell proliferation assay, CellTiter 96 Aqueous One Solution Cell Proliferation Assay, and LDH assay.
Cell proliferation assay was performed as described previously (13, 14). Briefly, human PASMCs were plated into a 96-well plate at a density of 3,000 cells/100 µl per well and incubated overnight. Cells were transfected with siRNAs or plasmids using Lipofectamine 2000 (Life Technology) on the next morning. Cell medium was refreshed 24 h after transfection.
For BrdU assay, BrdU label was added to the culture medium with a dilution of 1:10,000 on the next day after transfection. Cells were cultured for another 16 h, and cell proliferation activities were then measured using a BrdU cell proliferation assay kit (Calbiochem, Gibbstown, NJ) on a GloMax-96 Microplate luminometer (Promega, Madison, WI) at the wavelength of 450 nm.
For CellTiter 96 Aqueous One Solution Cell Proliferation Assay (Promega), 48 h after transfection, cell medium were removed for the following LDH assay, and cells were incubated with a mixture of cell culture medium/cell proliferation assay solution for 1–2 h. Then cell proliferation activity was measured by reading the absorbance at 450 nm on a GloMax-96 Microplate luminometer (Promega).
Cell death was determined using the Cytotoxicity Detection Kit (LDH) (Roche, Indianapolis, IN), according to the manufacturer’s instructions. Briefly, the above-collected cell culture media were incubated with LDH assay buffer for 30 min and LDH activity was measured by reading the absorbance at 450 nm on the GloMax-96 Microplate luminometer (Promega).
Quantitative real-time reverse transcription PCR (qRT-PCR).
Total RNA was isolated using a miRNeasy Mini Kit (Qiagen, Valencia, CA) and was treated with an RNase-free DNase set (Qiagen). For mRNA measurement, total RNA was reverse-transcribed using a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA). Quantitative real-time reverse transcription-PCR (qRT-PCR) was then performed using the SYBR Green PCR Master Mix (Applied Biosystems) on StepOnePlus or ViiA 7 Real-Time PCR System (Applied Biosystems). Ribosomal protein L19 (RPL19) was used as internal control. For the measurement of miRNA levels, a poly(A) tail was first added to the 3′ end of miRNAs using a Poly(A) Polymerase Tailing Kit (Epicenter Biotechnologies, Madison, WI). Poly(A)-tailed-miRNAs were then reverse-transcribed using M-MLV reverse transcriptase (Invitrogen, Grand Island, NY) with a poly(T) adaptor, which consists of a poly(T) sequence and a sequence complementary to the universal primer used in following qRT-PCR analysis. SNORD44, SNORD47, and SNORD48 were used as internal controls. Primer sequences are provided in Table 1.
Table 1.
Primer sequences
| Gene | Forward Primer (5′ to 3′) | Reverse Primer (5′ to 3′) | Application |
|---|---|---|---|
| miR-17 | caaagtgcttacagtgcaggtag | gcgagcacagaattaatacgactcac | qPCR |
| miR-18a | taaggtgcatctagtgcagatag | gcgagcacagaattaatacgactcac | qPCR |
| miR-19a | tgtgcaaatctatgcaaaactga | gcgagcacagaattaatacgactcac | qPCR |
| miR-19b | tgtgcaaatccatgcaaaactga | gcgagcacagaattaatacgactcac | qPCR |
| miR-20a | taaagtgcttatagtgcaggtag | gcgagcacagaattaatacgactcac | qPCR |
| miR-92a | tattgcacttgtcccggcctgt | gcgagcacagaattaatacgactcac | qPCR |
| SNORD44 | ggtcttaattagctctaactgac | gcgagcacagaattaatacgactcac | qPCR |
| SNORD47 | ccgttccattttgattctgag | gcgagcacagaattaatacgactcac | qPCR |
| SNORD48 | taactctgagtgtgtcgctga | gcgagcacagaattaatacgactcac | qPCR |
| PAI-1 | ccgccgcctcttcca | gccatcatgggcacagaga | qPCR |
| UCP2 | cctctcccaatgttgctcgt | ggcaagggaggtcatctgtc | qPCR |
| PDK1 | tatggattgcccatatcacg | catctgtcccgtaaccctct | qPCR |
| VEGF | ggaggcgcagcggttag | aacccggatcaatgaatatcaaa | qPCR |
| Glut1 | tgctcatgggcttctcgaa | aagcggcccaggatcag | qPCR |
| HK2 | gagccatcctgcaacacttagg | cagtgcacacctccttaacaatg | qPCR |
| PGK1 | gggcaaggatgttctgttct | tctccagcaggatgacagac | qPCR |
| ENO1 | atctcacagtgaccaaccca | tggttgactttgagcaggag | qPCR |
| hRPL19 | atcatccgcaagcctgtg | tgaccttctctggcattcg | qPCR |
| PAI-1 3′-UTR | gctgacgtcaccgtccagttacacatcaaaaaagaaagaaagaaaaaccccaaagaaaga | tctttctttggggtttttctttctttcttttttgatgtgtaactggacggtgacgtcagc | Luciferase Reporter Plasmid Cloning |
Hypoxia/Sugen-model of pulmonary hypertension in rats.
We obtained rat lungs of a hypoxia/Sugen-induced PH model as we described in our previous report (12). The experimental protocols were approved by our Institutional Animal Care and Use Committee. All animals were purchased from Jackson Laboratory (Bar Harbor, ME) and were handled according to the National Institutes of Health (NIH) guidelines. Briefly, male Sprague-Dawley (SD) rats (Charles River, 190–200 g, 6 wk old) were administered a dose of Sugen 5416 (20 mg/kg) subcutaneously on the first day of hypoxia exposure (10% O2). After a 3-wk chronic hypoxia exposure, the rats were placed back into room air for 2 wk of reoxygenation. The rat lungs were then collected for the analysis of PAI-1 expression.
RESULTS
MiR-17~92 negatively regulates PAI-1 expression in PASMCs.
To identify novel targets of miR-17~92 in human PASMCs, we conducted a quantitative mass spectrometry analysis, as previously described (13). We repeated the experiment and found that 14 proteins were downregulated at least 1.2-fold by the overexpression of miR-17~92 in both experiments. Of these proteins, peptides of PAI-1 were identified, and a representative peptide and its mass are shown in Fig. 1A. The downregulation of PAI-1 in both experiments is shown in Fig. 1B. We confirmed this downregulation by measuring PAI-1 protein levels in control PASMCs (pLVX-CTRL) or PASMCs with miR-17~92 overexpression (pLVX-miR-17~92) by Western blotting (Fig. 1C). In addition, miR-17~92 mimics suppressed the expression of PAI-1 (Fig. 1D), whereas miR-17~92 inhibitors induced the expression of PAI-1 (Fig. 1E). PAI-1 can be regulated by the receptor for advanced glycation end products (RAGE) and poly(ADP-ribose) polymerase-1 (PARP-1) (47, 58), both of which are implicated in PH (37, 49) (38, 39). Thus, we examined the RAGE and PARP-1 levels after miR-17~92 treatment. We found that miR-17~92 did not alter PARP-1 levels, whereas miR-17~92 mimics decreased RAGE levels but miR-17~92 inhibitor had little effect on RAGE levels (Fig. 1, F–I). Taken together, these results suggest that miR-17~92 negatively regulates PAI-1 expression in PASMCs.
Fig. 1.

MicroRNA (MiR)-17~92 negatively regulates plasminogen activator inhibitor-1 (PAI-1) expression in pulmonary artery smooth muscle cells (PASMCs). A: representative peptide and mass of PAI-1 identified by quantitative proteomics. B: quantification of the quantitative mass spectrometry results of PAI-1 from two experiments. C: human PASMCs were infected with control (pLVX-CTRL) or miR-17~92 overexpressing (pLVX-miR-17~92) lentivirus and then collected for protein extraction and Western blotting. Representative blots and quantification graph are presented. D: human PASMCs were transfected with negative control (NC) miRNA mimic or a mixture of miR-17~92 mimics and collected 48 h later for protein extraction and Western blotting. E: human PASMCs were transfected with NC miRNA inhibitors or a mixture of miR-17~92 inhibitors and collected 48 h later for protein extraction and Western blotting. F–I: human PASMCs were transfected with NC miRNA inhibitors or miR-19a/b mimics or inhibitors and collected 48 h later for protein extraction and Western blotting for poly(ADP-ribose) polymerase-1 (PARP-1; F and G) and receptor for advanced glycation end products (RAGE; H and I). Data are presented as means ± SE. *P ≤ 0.05; **P ≤ 0.01.
MiR-19a/b directly targets PAI-1 in PASMCs.
To address whether miR-17~92 directly targets and suppresses PAI-1 expression, we searched for potential miR-17~92 binding sites on the 3′-UTR of PAI-1 mRNA using the TargetScan program (http://www.targetscan.org/vert_71/) and found that the 3′-UTR of PAI-1 contains a potential binding site for miR-19a and miR-19b of the miR-17~92 cluster (Fig. 2A). To verify whether miR-19a/b binds to this putative binding site, we constructed a luciferase reporter plasmid containing a 0.6-kb PAI-1 3′-UTR region, which contains the putative binding site of miR-19a/b. We transfected this reporter plasmid into PASMC-pLVX-CTRL and PASMC-pLVX-miR-17~92 and found that overexpression of miR-17~92 suppressed the luciferase activity of the reporter plasmid in PASMCs (Fig. 2B). In contrast, a mixture of miR-17~92 inhibitors induced the luciferase activity of the reporter plasmid (Fig. 2C). These results suggest that PAI-1 is a direct target of miR-17~92 in PASMCs. To investigate whether miR-19a/b of the miR-17~92 cluster specifically targets PAI-1, we utilized the miR-19a/b mimics and inhibitors to overexpress or inhibit miR-19a/b in PASMCs. We found that miR-19a/b mimics and inhibitors specifically overexpressed or inhibited miR-19a/b without changes in the expression levels of other miRNAs in this cluster (Fig. 2, D and E). We then cotransfected PASMCs with the reporter plasmid and miR-19a/19b mimics or inhibitors. We found that both miR-19a and miR-19b mimics suppressed the luciferase activity (Fig. 2F), whereas inhibition of miR-19a, but not miR-19b, induced the luciferase activity (Fig. 2G). To further confirm whether miR-19a/b can regulate the expression of PAI-1, we transfected PASMCs with miR-19a/b mimics or inhibitors and then measured the mRNA and protein levels of PAI-1. Our results showed that miR-19a/b mimics suppressed both the mRNA and protein levels of PAI-1 (Fig. 2, H and J). Inhibition of miR-19a/b did not alter PAI-1 mRNA levels (Fig. 2I) but induced PAI-1 protein levels (Fig. 2K). These results suggest that miR-19a/b of the miR-17~92 cluster, especially miR-19a, directly targets and inhibits PAI-1.
Fig. 2.

MiR-19a/b directly targets and suppresses PAI-1 in PASMCs. A: the putative miR-19a/b binding site on the 3′-untranslated region (UTR) of PAI-1. B: the PAI-1 3′-UTR luciferase reporter plasmid containing the putative miR-19a/b binding site was transfected into human PASMCs infected with control (pLVX-CTRL) or miR-17~92-overexpressing lentivirus (pLVX-miR-17~92). Luciferase activity in PASMC-pLVX-CTRL was set as 1 and was used for normalization for that of PASMC-pLVX-miR-17~92. C: the PAI-1 3′-UTR luciferase reporter plasmid was cotransfected with NC or a mixture of miR-17~92 (17~92) inhibitors into PASMCs. Luciferase activity of cells transfected with NC inhibitors was set as 1 and used for normalization for that of PASMCs transfected with miR-17~92 inhibitors. D and E: human PASMCs were transfected with NC miRNA inhibitors or miR-19a/b mimics (D) or inhibitors (E) and the relative amount of the individual miRNAs were measured. F and G: the PAI-1 3′-UTR luciferase reporter plasmid was cotransfected with NC or miR-19a or miR-19b mimics (F) or inhibitors (G) into PASMCs. Luciferase activity of cells transfected with NC was set as 1 and used for normalization for that of PASMCs transfected with miR-19a or miR-19b mimics or inhibitors. H–K: PASMCs were transfected with NC or miR-19a or miR-19b mimics (H and J), or inhibitors (I and K); 48 h later, cells were collected to measure mRNA (H and I) and protein (J and K) levels of PAI-1. Representative blots and quantification are presented. Data are presented as means ± SE. *P ≤ 0.05, **P ≤ 0.01; n = 5.
PDLIM5 is required for miR-17/20a-mediated suppression of PAI-1.
Multiple miRNAs, particularly miRNAs from the same miRNA cluster or family, are known to coordinately regulate one or more targets (3, 8, 22, 36). Thus, we investigated whether other members of the miR-17~92 cluster might also regulate PAI-1 expression in PASMCs. We had previously reported that miR-17/20a could target PDLIM5 and PHD2 to regulate SMC marker expression and proliferation in PASMCs (13, 14); however, there is no potential miR-17/20a binding site in the 3′-UTR of PAI-1. Interestingly, we found that miR-17/20a mimics suppressed the expression of PAI-1 (Fig. 3A), whereas miR-17/20a inhibitors induced the expression of PAI-1 (Fig. 3B), suggesting that miR-17/20a may indirectly suppress PAI-1 expression in PASMCs. Since PDLIM5 is a direct target of miR-17/20a in PASMCs (13), we sought to explore whether PDLIM5 might regulate PAI-1 expression. We found that knockdown of PDLIM5 with siRNA suppressed the expression of PAI-1 in PASMCs (Fig. 3C). On the other hand, knockdown of PAI-1 did not alter the expression of PDLIM5 (Fig. 3D). In addition, we overexpressed PDLIM5 by transfection of a plasmid encoding PDLIM5-V5-His and measured the levels of PAI-1. We found that overexpression of PDLIM5 did not alter the levels of PAI-1 (Fig. 3E). These results suggest that PDLIM5 is necessary but not sufficient to regulate the levels of PAI-1 and that miR-17~92 may regulate PAI-1 expression via two mechanisms: 1) a direct suppression by miR-19a/b and 2) an indirect suppression by miR-17/20a/PDLIM5.
Fig. 3.

PDZ and LIM domain protein 5 (PDLIM5) is required for miR-17/20a-mediated suppression of PAI-1. A and B: human PASMCs were transfected with NC or miR-17/20a mimics (A) or inhibitors (B); 48 h later, cells were collected for measuring the expression of PAI-1. C and D: human PASMCs were transfected with NC or PDLIM5 siRNA (C) or PAI-1 siRNA (D); 48 h later, cells were collected for measuring PAI-1 and PDLIM5 expression levels. Representative blots and quantification are presented. E: human PASMCs were transfected with a control plasmid (pcDNA3) or a plasmid encoding PDLIM5-V5-His; 48 h later, cells were collected for measuring the expression of PAI-1. Data are presented as means ± SE. *P ≤ 0.05, **P ≤ 0.01; n = 5.
PAI-1 is a negative regulator of miR-17~92-mediated PASMC proliferation.
We have previously shown that miR-17~92 promotes PASMC proliferation (13). To address whether PAI-1 plays a role in miR-17~92-mediated PASMC proliferation, we cotransfected siPAI-1 and miR-17~92 inhibitors and measured the levels of PCNA, a biomarker of cell proliferation, BrdU incorporation, cell proliferation, and LDH release (as an indicator of cell death). We found that silencing of PAI-1 itself had little effect on PASMC proliferating cell nuclear antigen (PCNA) levels, BrdU incorporation, proliferation, or death (Fig. 4, A–D). However, miR-17~92 inhibitors decreased PASMC PCNA levels, BrdU incorporation, and proliferation without affecting PASMC death (Fig. 4, A–D). More importantly, in the presence of miR-17~92 inhibitors, silencing of PAI-1 restored PCNA levels, BrdU incorporation, and proliferation (Fig. 4, A, C, and D) and decreased PASMC death (Fig. 4B). These results suggest that PAI-1 is required for miR-17~92-regulated PASMC proliferation. To investigate whether overexpression of PAI-1 could suppress miR-17~92-induced PASMC proliferation, we cotransfected PASMCs with PAI-1-Bio-His plasmid and miR-17~92 mimics and measured cell proliferation and BrdU incorporation. We confirmed the overexpression of PAI-1 using PAI-1 and His tag antibody (Fig. 4E). We showed that overexpression of PAI-1 significantly inhibited miR-17~92-induced PASMC proliferation and BrdU incorporation (Fig. 4, F and G). These results suggest that PAI-1 is necessary and sufficient to regulate miR-17~92-mediated PASMC proliferation.
Fig. 4.

PAI-1 is a negative regulator of miR-17~92-mediated PASMC proliferation. A–D: human PASMCs were transfected with NC (anti-Neg) or a mixture of miR-17~92 inhibitors (anti-17~92) along with PAI-1 siRNA; 48 h later, cells were collected for measuring the expression of (PCNA) or tubulin as internal control (A). Media were collected for measurement of lactate dehydrogenase (LDH) activity (B), and cells were subjected to bromodeoxyuridine (BrdU) incorporation assay (C) and cell proliferation assay (D). E–G: human PASMCs were transfected with NC (Neg mimic) or a mixture of miR-17~92 mimics along with PAI-1 overexpression plasmid (PAI-OE) or vehicle control (vehicle); 48 h later, cells were collected for measuring the expression of exogenous PAI-1 (His) and endogenous PAI-1 (PAI-1) or tubulin as internal control (E). These cells were subjected to cell proliferation assay (F) and BrdU incorporation assay (G). Representative blots are presented and data are presented as means ± SE. *P ≤ 0.05, **P ≤ 0.01; n = 5.
Suppression of PAI-1 induces Smad2/calponin signaling in PASMCs.
Since PDLIM5 downregulates TGF-β/Smad signaling to inhibit SMC marker expression in PASMCs (13), we then investigated the role of PAI-1 in SMC marker expression. We silenced PAI-1 with siRNA and found that suppression of PAI-1 increased the expression of calponin without altering the expression of myocardin, SRF, α-SMA, and SM22α (Fig. 4). Since TGF-β/Smad signaling is known to regulate the expression of SMC marker proteins (2, 6, 27, 29), we also measured the expression of Smad2 and Smad3. Our results showed that suppression of PAI-1 induced the expression of Smad2 but decreased the expression of Smad3 (Fig. 5). These results suggest that PAI-1 specifically inhibits the TGF-β/Smad2/calponin signaling in PASMCs.
Fig. 5.

Suppression of PAI-1 induces Smad2/calponin signaling in PASMCs. Human PASMCw were transfected with NC (siNC) or PAI-1 siRNAs (siPAI-1); 48 h later, cells were collected for measuring protein expression by Western blotting. Representative blots (A) and quantification (B–H) are presented. Data are presented as means ± SE. *P ≤ 0.05, **P ≤ 0.01; n = 5.
PAI-1 is essential for the metabolic gene expression in PASMCs.
Recent studies suggest a metabolic reprogramming in PASMCs during PH (10, 50) and PASMC from animal models of PH and human tissues of PAH display a consistent pattern of reprogrammed cellular metabolism (44). A recent report also shows that miR-17~92, particularly, miR-17, regulates tumor cell metabolism by direct targeting LKB1 and downstream mammalian targt of rapamycin (mTOR) signaling (33). Thus, we examined the role of the miR-17-92/PAI-1 axis on metabolism. We have examined the effects of PAI-1 on the mRNA levels of uncoupling protein-2 (UCP2), pyruvate dehydrogenase kinase-1 (PDK1), vascular endothelial growth factor (VEGF), glucose transporter 1(GLUT1), hexokinase-2 (HK2), phosphoglycerate kinase-1 (PGK1), and (ENO1) and found that silencing of PAI-1 significantly induced UCP2, VEGF, GLUT1, PGK1, and ENO1, particularly VEGF, PGK1, and enolase 1 ENO1 (Fig. 5). We found that inhibition of miR-17~92 decreased levels of UCP2, but increased levels of GLUT1, HK2, VEGF, ENO1, and PGK1. We also measured the mRNA levels of these genes in PASMCs with inhibition of miR-17~92 and silencing of PAI-1, silencing of PAI-1 inhibited induction of HK2, ENO1, and GLUT1 in the presence of the miR-17~92 agonist (Fig. 6). These results suggest multiple functions of PAI-1 in PASMC phenotype (Fig. 6).
Fig. 6.

PAI-1 is essential for metabolic gene expression in PASMCs. A: human PASMCs were transfected with NC (siNeg) or PAI-1 siRNA; 48 h later, cells were collected for measuring mRNA levels of indicated genes by real-time qRT-PCR. B: human PASMCs were transfected with NC or a mixture of miR-17~92 inhibitors (anti-17~92) along with PAI-1 siRNA; 48 h later, cells were collected for measuring mRNA levels of indicated genes by real-time qRT-PCR. Levels of RPL19 were used as loading control. Data are presented as means ± SE. * or **, compared with siNeg/anti-Neg group; #, compared with siPAI-1/anti-Neg group. * or #, P ≤ 0.05; **, P ≤ 0.01; n = 5.
Expression levels of PAI-1 in PAH patients and hypoxia/Sugen-induced PH rats.
We have shown that miR-17~92 contributes to the pathogenesis of PH through the miR-17/20a/PDLIM5/TGF-β/Smad and miR-17/20a/PHD2/HIF1 signaling pathways (13, 14). Since miR-19a/b and miR-17/20a/PDLIM5 directly or indirectly regulate PAI-1 in PASMCs (Figs. 1–3), we set out to investigate whether PAI-1 is also dysregulated in PAH. The general characteristics of IPAH and APAH were similar, as shown in Table 2. First, we compared PAI-1 expression levels in human PASMCs isolated from normal donors or APAH and IPAH patients. We found that APAH PASMCs expressed a similar level of PAI-1 compared with that of normal PASMCs (Fig. 7, A and B). In IPAH PASMCs, there was a trend of decreased PAI-1 levels; however, the change was not significant (Fig. 7, C and D). In a severe hypoxia/Sugen-induced rat PH model, we found that hypertensive lungs expressed decreased PAI-1 (Fig. 7, E and F). These results suggest a dysregulation of of PAI-1 in PH.
Table 2.
General characteristics of PAH patients whose samples were obtained from PHBI
| Control Subjects | IPAH | APAH | |
|---|---|---|---|
| n* | 8 | 9 | 8 |
| Age, yr | NA | 40.6 (15.0) | 36.5 (20.4) |
| Female/male | 3/3 | 3/2 | 4/0 |
| mPAP, mmHg | NA | 60.1 (12.9) | 55.6 (26.9) |
| PVR, wood units | NA | 14.3 (4.9) | 9.5 (2.6) |
| 6MWD, m | NA | 394.6 (126.9) | 327.4 (177.8) |
| Race | 5W/1B | 4W/1A | 2W/1A/1U |
| Weight, lb | NA | 168.7 (28.4) | 109.9 (28.2) |
| Height, cm | NA | 168.8 (6.5) | 158.2 (9.3) |
Data are means (SD). W, White; B. Black or African American; A, Asian; U, unknown.
Information of 2 patients in control subjects, 4 idiopathic pulmonary arterial hypertension (IPAH) patients, and 4 associated PAH (APAH) patients are unavailable.
Fig. 7.

Expression levels of PAI-1 in pulmonary arterial hypertension (PAH) patients or when exposed to hypoxia or hypoxia mimics. A and B: PAI-1 protein levels in human PASMCs of associated (A)PAH patients and normal donors were determined and compared. One normal donor’s PAI-1 level was set as 1 and was used for normalization for that of other normal donors and APAH patients; 8 normal donors and 8 APAH samples. C and D: PAI-1 protein levels in human PASMC of idiopathic (I)PAH patients and normal donors were determined and compared. One normal donor’s PAI-1 level was set as 1 and was used for normalization for that of other normal donors and IPAH patients; 8 normal donors and 9 IPAH samples. E and F: PAI-1 protein levels in lungs of control rats and hypoxia/Sugen-treated rats (HPX/Sugen) were determined and compared. One control rat lung’s PAI-1 level was set as 1 and was used for normalization for that of other control and HPX/Sugen rat lungs; 8 control rats and 9 HPX/Sugen-treated rats. Representative blots and quantification are presented in B, D, and F. Data are presented as means ± SE. *P ≤ 0.05, **P ≤ 0.01.
DISCUSSION
Although we (13) and others (45) have demonstrated the significance of miR-17~92 in the pathogenesis of PH utilizing experiments in cell culture, animal models of PH, and clinical samples from PAH patients, the detailed molecular mechanisms underlying the action of miR-17~92 in PASMCs and PH are still not completely elucidated, as there are multiple members in the miR-17~92 cluster and multiple targets of each member. We addressed this issue by adopting a global protein expression profiling in PASMCs overexpressing miR-17~92 with a quantitative proteomics analysis to identify novel miR-17~92 targets. We discovered that 14 proteins are suppressed by overexpression of miR-17~92 and had previously studied the role of PDLIM5 in regulation of PASMC phenotype and PH (13). The involvement of the other proteins in the PASMC phenotype and PH remained to be explored. In this study, we chose to investigate another protein that is suppressed by miR-17~92, PAI-1. We have found that PAI-1 is a direct target of miR-19a/b (Figs. 1 and 2). Interestingly, miR-17/20a can also indirectly suppress PAI-1 via PDLIM5 (Fig. 3). In addition, we have shown that PAI-1 is a negative regulator of miR-17~92-mediated PASMC proliferation and that silencing of PAI-1 induces Smad2 and SMC marker expression (Figs. 4 and 5). These results suggest that miR-17~92 can regulate the PASMC phenotype through multiple members and multiple targets.
Of six members of the miR-17~92 cluster, miR-17 and miR-20a are the two members that are studied the most. In endothelial cells, miR-17/20a directly targets bone morphogenetic protein receptor 2 (BMPR2), the best known genetic mutation associated with PAH (9). In our previous studies, we also show that miR-17/20a are two main members in regulating PASMC phenotype and the development of PH: the miR-17/20a/PDLIM5/TGF-β/Smad signaling is involved in regulating PASMC contractile phenotype (13) and the miR-17/20a/PHD2/HIF pathway in regulating PASMC proliferation (14). However, miR-19a/b are dysregulated in PAH as miR-17/20a are, and discovery of miR-19a/b targets will likely provide a better and more complete understanding of the role of miR-17~92 in PH. Our finding that miR-19a/b can directly target and suppress PAI-1 in PASMCs (Figs. 1 and 2) provides such a link between its dysregulation and pathogenesis of PH, suggesting that miR-19a/b may also contribute to PH via its target PAI-1. Previous studies shown that RAGE and PARP-1 are positive regulators of PAI-1 (47, 58). We have found that miR-17~92 inhibits PAI-1 and RAGE, but not PARP-1 (Fig. 1, F–I), Thus we cannot rule out that there is an additional mechanism that miR-17~92 may inhibit PAI-1 via inhibition of RAGE. In addition, miR-19 is also known to directly target phosphatase and tensin homolog (PTEN) to induce cancer cell and cardiomyocyte proliferation (11, 42). SMC-specific depletion of PTEN induces histopathological changes that are consistent with PH, including medial SMC hyperplasia and vascular remodeling, and more severe PH in hypoxic mice (30, 41). Thus, miR-19a/b are likely key players in the development of PH.
In addition to direct suppression by miR-19a/b, PAI-1 is also indirectly suppressed by miR-17/20a via an indirect route through PDLIM5 (Fig. 3). Thus, at least four members of the miR-17~92 cluster can suppress PAI-1 via different pathways. Given the generally modest regulation of target proteins by miRNAs, this direct and indirect suppression of PAI-1 by different members of the miR-17~92 cluster suggests a functional synergy between individual miRNAs within the cluster. Another evidence of synergy within the cluster is that both PAI-1 and PDLIM5 (a miR-17/20a target) are negative regulators of Smad2 and SMC markers (Fig. 4) (13). Thus, we conclude that miR-19a/b and miR-17/20a can synergistically regulate PASMC contractile protein expression via PAI-1 and PDLIM5.
PAI-1 is a serine protease inhibitor (serpin) targeting the two plasminogen activators tissue-type plasminogen activator (t-PA) and urokinase-type plasminogen activator (u-PA), which play a key role in hemostasis, thrombosis, and other biological processes by controlling the formation and activity of plasmin (23). However, how PAI-1 regulates vascular remodeling is unclear, and some opposing results suggest that PAI-1 can either promote or prevent vascular remodeling, a phenomenon often referred to as the “PAI-1 paradox” (20). Although in a study PAI-1 is downregulated in PASMCs in IPAH and negatively regulates PASMC proliferation (35), another study by Yung et al. (55) showed that PAI-1 expression levels were induced in the lungs of monocrotaline (MCT) rat model and a SU5416/hypoxia mouse model of PH, suggesting an association between PAI-1 induction and the pathogenesis of PH. However, we show that PAI-1 levels are similar in APAH and IPAH PASMCs compared with normal PASMCs (Fig. 7, A–D). We also show that in a hypoxia/Sugen-induced rat PH model, PAI-1 is downregulated. (Fig. 7, E and F). This seeming discrepancy between our study and Kouri’s findings (35) is likely due to 1) relative small sample size, 2) different sampling cells/tissue (whole lung in Kouri’s study vs. PASMCs in our study). It is also possible that PAI-1 is differently regulated in different types of PAH. For example, in our study, we found that PAI-1 itself had little effect on PASMC proliferation or death, instead, silencing of PAI-1 restored PASMC proliferation in presence of miR-17~92 inhibitors (Fig. 4), suggesting a negative effect of PAI-1 in PASMC proliferation in a different context. Previously we observed a biphasic regulation of miR-17~92 in PH, early induction and later reduction, and have postulated that early induction of miR-17~92 promotes PH, whereas the reduction of miR-17~92 in the later stage is an endogenous feedback mechanism to prevent the further development of PH (13). Thus, a combined reduction of miR-19a/b and induction of PAI-1 may also be a part of this feedback system to serve as an internal brake for the development of PH, which may in part resolve the PAI-1 paradox.
There is a postulation that metabolic reprogramming is critical in the pathogenesis of PH according to recent studies (10, 44, 50). MiR-17 is also known to regulate tumor cell metabolism by direct targeting LKB1 and downstream mTOR signaling (33). Thus, we speculate that PAI-1 may also regulate PASMC metabolism. Indeed, we have found that PAI-1 is essential for the metabolic gene expression in PASMCs, ranging from UCP2 to PDK1, VEGF, GLUT, HK2, PGK1, and ENO1 (Fig. 5). Thus, PAI-1 may also regulate the PASMC metabolic reprogramming to contribute to PH and further investigations on this topic are warranted.
PAI-1 has been identified as a target gene of the hypoxia-inducible factor 1 (HIF1) in response to hypoxia (34). In this study, we also find that hypoxia exposure (3% O2) as well as CoCl2 treatment, which is known to stabilize HIF levels under normoxic conditions, induce PAI-1 levels in PASMCs (data not shown), suggesting a hypoxia-HIF1-PAI-1 pathway in PASMCs. Interestingly, after 6-h incubation in hypoxia, we found the induction of both miR-17~92 and PAI-1 (13) (data not shown), which is seemingly inconsistent with our finding that miR-19a/b and miR-17/20a suppress PAI-1 (Figs. 1–3). We also found that miR-17/20 can suppress PHD2 and activate HIF1 (14), which might suggest that miR-17/20a induces PAI-1, which is again inconsistent with our finding that miR-17/20a suppress PAI-1 (Fig. 3). These seemingly discrepant findings suggest complex and multilayered pathways in the regulation of PAI-1 expression. Another key factor is the timing of the experiment. As we discussed above, there is a biphasic regulation of miR-17~92 in PH (13). The molecular action of miR-17~92 in PASMCs may be complex, and interpretation of the change of miR-17~92 and PAI-1 levels in PH should be put in context with the tissue studied, the stage of PH development, and the contributions from multiple pathways.
Although miR-17~92 suppress PAI-1 directly or indirectly via PDLIM5, both PAI-1 and PDLIM5 inhibit Smad signaling and SMC marker protein expression, however, in an isoform-dependent manner (PAI-1 on Smad2, but PDLIM5 on Smad3) (Fig. 5 and Ref. 13). The inhibitory effects of PAI-1 and PDLIM5 on different SMC markers again suggest a complementary role of multiple pathways in contributing to miR-17~92-mediated SMC marker expression (13). Previously, we showed that miR-17~92 induces both Smad2 and Smad3; however, miR-17/20a-mediated PDLIM5 suppression affects only Smad3 (13). It is unknown how miR-17~92 induces Smad2. In this study, we show that PAI-1 affects SMC marker expression via Smad2 (Fig. 7). Thus, the miR-19a/b/PAI-1/Smad2 signaling pathway supplement the miR-17/20a/PDLIM5/Smad3 signaling to achieve the complete function of miR-17~92 in regulating Smad2/3 signaling and SMC marker expression. Our findings are consistent with the reports in the literature on the impaired TGF-β/Smad signaling in PAH (25, 56), particularly two recent publications on the specific role of Smad3 in Schistosoma-induced PH (26) and that of Smad2 in monocrotaline- and hypoxia/SU5416-induced PH (55).
In summary, we have identified PAI-1 as a novel target of miR-19a/b and as an indirect target of miR-17/20a. We have also provided evidence that PAI-1 is a negative regulator for the miR-17~92-mediated PASMC proliferation and that PAI-1 regulates SMC marker protein expression via Smad2. According to these findings and our previous reports on miR-17/20a-mediated regulation of PDLIM5 and PHD2, we propose that members of the miR-17~92 cluster coordinately regulate the PASMC phenotype in a complex manner (Fig. 8): miR-19a/b directly target and suppress PAI-1 to induce Smad2 and calponin; miR-17/20a can directly suppress PDLIM5 and PHD2 to induce Smad2/3 and HIF, leading to SMC marker expression and PASMC proliferation; miR-17/20a can also regulate PAI-1 expression indirectly via PDLIM5, thus regulating Smad2 and calponin. In this complex network, some functions are redundant (e.g., Smad2 is under control of PAI-1 and PDLIM5), whereas others are rather unique (e.g., Smad3 is controlled only by PDLIM5, and HIF1α is controlled only by miR-17/20a/PHD2). This suggests a complex role of the miR-17~92 signaling in the development of PH, as we have previously hypothesized (13, 14). Further studies are warranted to investigate the function of PAI-1 in PAH and how it works together with other molecules to regulate the pathogenesis of PAH.
Fig. 8.
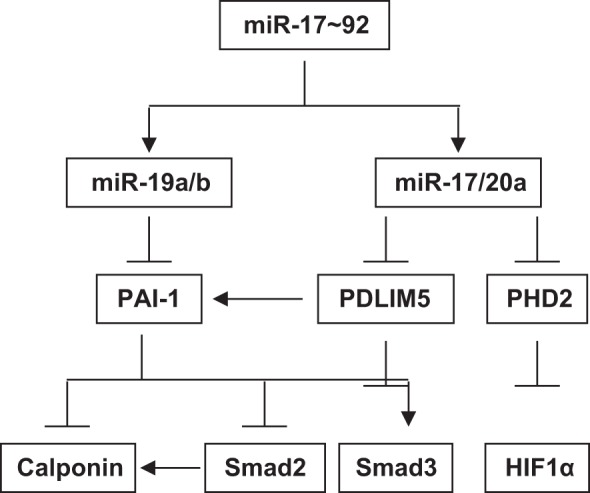
Schematic diagram of miR-17~92-mediated signaling in PASMCs. Members of the miR-17~92 cluster coordinately regulate PASMC phenotype in a complex manner: miR-19a/b directly target and suppress PAI-1 to induce Smad2 and calponin; miR-17/20a can directly suppress PDLIM5 and prolyl hydroxylase 2 (PHD2) to induce Smad2/3 and hypoxia-inducible factor (HIF), leading to smooth muscle cell (SMC) marker expression and PASMC proliferation. miR-17/20a can also regulate PAI-1 expression indirectly via PDLIM5, thus regulating Smad2 and calponin.
GRANTS
This work was supported in part by National Heart, Lung, Blood Institute Grant R01-HL-123804 (J. U. Raj and G. Zhou), National Natural Science Foundation of China Grant 81770050 (G. Zhou), American Lung Association Biomedical Research Grant RG-416135 (T. Chen), Gilead Sciences Research Scholars Program in Pulmonary Arterial Hypertension (T. Chen), and 2014 Alpha Omega Alpha Carolyn L. Kuckein Student Research Fellowship (J. B. Huang).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
T.C. and G.Z. conceived and designed research; T.C., J.B.H., J.D., Q.Z., and G.Z. performed experiments; T.C., J.B.H., J.D., Q.Z., and G.Z. analyzed data; T.C., J.B.H., J.U.R., and G.Z. interpreted results of experiments; T.C. and G.Z. prepared figures; G.Z. drafted manuscript; T.C., J.U.R., and G.Z. edited and revised manuscript; T.C., J.B.H., J.D., Q.Z., J.U.R., and G.Z. approved final version of manuscript..
ACKNOWLEDGMENTS
Normal and APAH human PASMCs were kindly provided by Drs. Suzy A. A. Comhair and Serpil C. Erzurum at the Department of Pathobiology, Respiratory Institute, Cleveland Clinic. We thank Miranda Sun for critical reading of our manuscript.
REFERENCES
- 1.Abdalla M, Goc A, Segar L, Somanath PR. Akt1 mediates α-smooth muscle actin expression and myofibroblast differentiation via myocardin and serum response factor. J Biol Chem 288: 33483–33493, 2013. doi: 10.1074/jbc.M113.504290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adam PJ, Regan CP, Hautmann MB, Owens GK. Positive- and negative-acting Kruppel-like transcription factors bind a transforming growth factor beta control element required for expression of the smooth muscle cell differentiation marker SM22alpha in vivo. J Biol Chem 275: 37798–37806, 2000. doi: 10.1074/jbc.M006323200. [DOI] [PubMed] [Google Scholar]
- 3.Austin ED, Loyd JE. The genetics of pulmonary arterial hypertension. Circ Res 115: 189–202, 2014. doi: 10.1161/CIRCRESAHA.115.303404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bertero T, Cottrill K, Krauszman A, Lu Y, Annis S, Hale A, Bhat B, Waxman AB, Chau BN, Kuebler WM, Chan SY. The microRNA-130/301 family controls vasoconstriction in pulmonary hypertension. J Biol Chem 290: 2069–2085, 2015. doi: 10.1074/jbc.M114.617845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bertero T, Lu Y, Annis S, Hale A, Bhat B, Saggar R, Saggar R, Wallace WD, Ross DJ, Vargas SO, Graham BB, Kumar R, Black SM, Fratz S, Fineman JR, West JD, Haley KJ, Waxman AB, Chau BN, Cottrill KA, Chan SY. Systems-level regulation of microRNA networks by miR-130/301 promotes pulmonary hypertension. J Clin Invest 124: 3514–3528, 2014. doi: 10.1172/JCI74773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Björkerud S. Effects of transforming growth factor-beta 1 on human arterial smooth muscle cells in vitro. Arterioscler Thromb 11: 892–902, 1991. doi: 10.1161/01.ATV.11.4.892. [DOI] [PubMed] [Google Scholar]
- 7.Boettger T, Beetz N, Kostin S, Schneider J, Krüger M, Hein L, Braun T. Acquisition of the contractile phenotype by murine arterial smooth muscle cells depends on the Mir143/145 gene cluster. J Clin Invest 119: 2634–2647, 2009. doi: 10.1172/JCI38864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boross G, Orosz K, Farkas IJ. Human microRNAs co-silence in well-separated groups and have different predicted essentialities. Bioinformatics 25: 1063–1069, 2009. doi: 10.1093/bioinformatics/btp018. [DOI] [PubMed] [Google Scholar]
- 9.Brock M, Trenkmann M, Gay RE, Michel BA, Gay S, Fischler M, Ulrich S, Speich R, Huber LC. Interleukin-6 modulates the expression of the bone morphogenic protein receptor type II through a novel STAT3-microRNA cluster 17/92 pathway. Circ Res 104: 1184–1191, 2009. doi: 10.1161/CIRCRESAHA.109.197491. [DOI] [PubMed] [Google Scholar]
- 10.Calvier L, Chouvarine P, Legchenko E, Hoffmann N, Geldner J, Borchert P, Jonigk D, Mozes MM, Hansmann G. PPARgamma links BMP2 and TGFbeta1 pathways in vascular smooth muscle cells, regulating cell proliferation and glucose metabolism. Cell Metab 25: 1118–1134, 2017. doi: 10.1016/j.cmet.2017.03.011. [DOI] [PubMed] [Google Scholar]
- 11.Chen J, Huang Z-P, Seok HY, Ding J, Kataoka M, Zhang Z, Hu X, Wang G, Lin Z, Wang S, Pu WT, Liao R, Wang D-Z. mir-17-92 cluster is required for and sufficient to induce cardiomyocyte proliferation in postnatal and adult hearts. Circ Res 112: 1557–1566, 2013. doi: 10.1161/CIRCRESAHA.112.300658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen J, Tang H, Sysol JR, Moreno-Vinasco L, Shioura KM, Chen T, Gorshkova I, Wang L, Huang LS, Usatyuk PV, Sammani S, Zhou G, Raj JU, Garcia JGN, Berdyshev E, Yuan JXJ, Natarajan V, Machado RF. The sphingosine kinase 1/sphingosine-1-phosphate pathway in pulmonary arterial hypertension. Am J Respir Crit Care Med 190: 1032–1043, 2014. doi: 10.1164/rccm.201401-0121OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen T, Zhou G, Zhou Q, Tang H, Ibe JCF, Cheng H, Gou D, Chen J, Yuan JXJ, Raj JU. Loss of microRNA-17∼92 in smooth muscle cells attenuates experimental pulmonary hypertension via induction of PDZ and LIM domain 5. Am J Respir Crit Care Med 191: 678–692, 2015. doi: 10.1164/rccm.201405-0941OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen T, Zhou Q, Tang H, Bozkanat M, Yuan JXJ, Raj JU, Zhou G. miR-17/20 controls prolyl hydroxylase 2 (PHD2)/hypoxia-inducible factor 1 (HIF1) to regulate pulmonary artery smooth muscle cell proliferation. J Am Heart Assoc 5: e004510, 2016. doi: 10.1161/JAHA.116.004510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng Y, Liu X, Yang J, Lin Y, Xu DZ, Lu Q, Deitch EA, Huo Y, Delphin ES, Zhang C. MicroRNA-145, a novel smooth muscle cell phenotypic marker and modulator, controls vascular neointimal lesion formation. Circ Res 105: 158–166, 2009. doi: 10.1161/CIRCRESAHA.109.197517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cordes KR, Sheehy NT, White MP, Berry EC, Morton SU, Muth AN, Lee TH, Miano JM, Ivey KN, Srivastava D. miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature 460: 705–710, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Costa RH. FoxM1 dances with mitosis [comment]. Nat Cell Biol 7: 108–110, 2005. doi: 10.1038/ncb0205-108. [DOI] [PubMed] [Google Scholar]
- 18.Courboulin A, Paulin R, Giguère NJ, Saksouk N, Perreault T, Meloche J, Paquet ER, Biardel S, Provencher S, Côté J, Simard MJ, Bonnet S. Role for miR-204 in human pulmonary arterial hypertension. J Exp Med 208: 535–548, 2011. doi: 10.1084/jem.20101812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davis BN, Hata A. Regulation of microRNA biogenesis: a miRiad of mechanisms. Cell Commun Signal 7: 18, 2009. doi: 10.1186/1478-811X-7-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Diebold I, Kraicun D, Bonello S, Görlach A. The ‘PAI-1 paradox’ in vascular remodeling. Thromb Haemost 100: 984–991, 2008. doi: 10.1160/TH08-08-0524. [DOI] [PubMed] [Google Scholar]
- 21.Doebele C, Bonauer A, Fischer A, Scholz A, Reiss Y, Urbich C, Hofmann WK, Zeiher AM, Dimmeler S. Members of the microRNA-17-92 cluster exhibit a cell-intrinsic antiangiogenic function in endothelial cells. Blood 115: 4944–4950, 2010. doi: 10.1182/blood-2010-01-264812. [DOI] [PubMed] [Google Scholar]
- 22.Farha S, Asosingh K, Xu W, Sharp J, George D, Comhair S, Park M, Tang WH, Loyd JE, Theil K, Tubbs R, Hsi E, Lichtin A, Erzurum SC. Hypoxia-inducible factors in human pulmonary arterial hypertension: a link to the intrinsic myeloid abnormalities. Blood 117: 3485–3493, 2011. doi: 10.1182/blood-2010-09-306357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fay WP, Garg N, Sunkar M. Vascular functions of the plasminogen activation system. Arterioscler Thromb Vasc Biol 27: 1231–1237, 2007. doi: 10.1161/ATVBAHA.107.140046. [DOI] [PubMed] [Google Scholar]
- 24.Gou D, Ramchandran R, Peng X, Yao L, Kang K, Sarkar J, Wang Z, Zhou G, Raj JU. miR-210 has an antiapoptotic effect in pulmonary artery smooth muscle cells during hypoxia. Am J Physiol Lung Cell Mol Physiol 303: L682–L691, 2012. doi: 10.1152/ajplung.00344.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goumans M-J, Liu Z, ten Dijke P. TGF-beta signaling in vascular biology and dysfunction. Cell Res 19: 116–127, 2009. doi: 10.1038/cr.2008.326. [DOI] [PubMed] [Google Scholar]
- 26.Graham BB, Chabon J, Gebreab L, Poole J, Debella E, Davis L, Tanaka T, Sanders L, Dropcho N, Bandeira A, Vandivier RW, Champion HC, Butrous G, Wang X-J, Wynn TA, Tuder RM. Transforming growth factor-β signaling promotes pulmonary hypertension caused by Schistosoma mansoni. Circulation 128: 1354–1364, 2013. doi: 10.1161/CIRCULATIONAHA.113.003072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grainger DJ, Metcalfe JC, Grace AA, Mosedale DE. Transforming growth factor-beta dynamically regulates vascular smooth muscle differentiation in vivo. J Cell Sci 111: 2977–2988, 1998. [DOI] [PubMed] [Google Scholar]
- 28.Grimm S, Ott C, Hörlacher M, Weber D, Höhn A, Grune T. Advanced-glycation-end-product-induced formation of immunoproteasomes: involvement of RAGE and Jak2/STAT1. Biochem J 448: 127–139, 2012. doi: 10.1042/BJ20120298. [DOI] [PubMed] [Google Scholar]
- 29.Hautmann MB, Madsen CS, Owens GK. A transforming growth factor beta (TGFbeta) control element drives TGFbeta-induced stimulation of smooth muscle alpha-actin gene expression in concert with two CArG elements. J Biol Chem 272: 10948–10956, 1997. doi: 10.1074/jbc.272.16.10948. [DOI] [PubMed] [Google Scholar]
- 30.Horita H, Furgeson SB, Ostriker A, Olszewski KA, Sullivan T, Villegas LR, Levine M, Parr JE, Cool CD, Nemenoff RA, Weiser-Evans MC. Selective inactivation of PTEN in smooth muscle cells synergizes with hypoxia to induce severe pulmonary hypertension. J Am Heart Assoc 2: e000188, 2013. doi: 10.1161/JAHA.113.000188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, Lang IM, Christman BW, Weir EK, Eickelberg O, Voelkel NF, Rabinovitch M. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol 43, Suppl S: 13S–24S, 2004. doi: 10.1016/j.jacc.2004.02.029. [DOI] [PubMed] [Google Scholar]
- 32.Humbert M, Sitbon O, Simonneau G. Treatment of pulmonary arterial hypertension [see comment]. N Engl J Med 351: 1425–1436, 2004. doi: 10.1056/NEJMra040291. [DOI] [PubMed] [Google Scholar]
- 33.Izreig S, Samborska B, Johnson RM, Sergushichev A, Ma EH, Lussier C, Loginicheva E, Donayo AO, Poffenberger MC, Sagan SM, Vincent EE, Artyomov MN, Duchaine TF, Jones RG. The miR-17∼92 microRNA cluster is a global regulator of tumor metabolism. Cell Reports 16: 1915–1928, 2016. doi: 10.1016/j.celrep.2016.07.036. [DOI] [PubMed] [Google Scholar]
- 34.Kietzmann T, Roth U, Jungermann K. Induction of the plasminogen activator inhibitor-1 gene expression by mild hypoxia via a hypoxia response element binding the hypoxia-inducible factor-1 in rat hepatocytes. Blood 94: 4177–4185, 1999. [PubMed] [Google Scholar]
- 35.Kouri FM, Queisser MA, Königshoff M, Chrobak I, Preissner KT, Seeger W, Eickelberg O. Plasminogen activator inhibitor type 1 inhibits smooth muscle cell proliferation in pulmonary arterial hypertension. Int J Biochem Cell Biol 40: 1872–1882, 2008. doi: 10.1016/j.biocel.2008.01.028. [DOI] [PubMed] [Google Scholar]
- 36.Krek A, Grün D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M, Rajewsky N. Combinatorial microRNA target predictions. Nat Genet 37: 495–500, 2005. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- 37.Meloche J, Courchesne A, Barrier M, Carter S, Bisserier M, Paulin R, Lauzon-Joset J-F, Breuils-Bonnet S, Tremblay É, Biardel S, Racine C, Courture C, Bonnet P, Majka SM, Deshaies Y, Picard F, Provencher S, Bonnet S. Critical role for the advanced glycation end-products receptor in pulmonary arterial hypertension etiology. J Am Heart Assoc 2: e005157, 2013. doi: 10.1161/JAHA.112.005157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meloche J, Le Guen M, Potus F, Vinck J, Ranchoux B, Johnson I, Antigny F, Tremblay E, Breuils-Bonnet S, Perros F, Provencher S, Bonnet S. miR-223 reverses experimental pulmonary arterial hypertension. Am J Physiol Cell Physiol 309: C363–C372, 2015. doi: 10.1152/ajpcell.00149.2015. [DOI] [PubMed] [Google Scholar]
- 39.Meloche J, Pflieger A, Vaillancourt M, Paulin R, Potus F, Zervopoulos S, Graydon C, Courboulin A, Breuils-Bonnet S, Tremblay E, Couture C, Michelakis ED, Provencher S, Bonnet S. Role for DNA damage signaling in pulmonary arterial hypertension. Circulation 129: 786–797, 2014. doi: 10.1161/CIRCULATIONAHA.113.006167. [DOI] [PubMed] [Google Scholar]
- 40.Morrell NW, Adnot S, Archer SL, Dupuis J, Jones PL, MacLean MR, McMurtry IF, Stenmark KR, Thistlethwaite PA, Weissmann N, Yuan JX, Weir EK. Cellular and molecular basis of pulmonary arterial hypertension. J Am Coll Cardiol 54, Suppl: S20–S31, 2009. doi: 10.1016/j.jacc.2009.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nemenoff RA, Simpson PA, Furgeson SB, Kaplan-Albuquerque N, Crossno J, Garl PJ, Cooper J, Weiser-Evans MC. Targeted deletion of PTEN in smooth muscle cells results in vascular remodeling and recruitment of progenitor cells through induction of stromal cell-derived factor-1alpha. Circ Res 102: 1036–1045, 2008. doi: 10.1161/CIRCRESAHA.107.169896. [DOI] [PubMed] [Google Scholar]
- 42.Olive V, Bennett MJ, Walker JC, Ma C, Jiang I, Cordon-Cardo C, Li QJ, Lowe SW, Hannon GJ, He L. miR-19 is a key oncogenic component of mir-17-92. Genes Dev 23: 2839–2849, 2009. doi: 10.1101/gad.1861409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Olive V, Jiang I, He L. mir-17-92, a cluster of miRNAs in the midst of the cancer network. Int J Biochem Cell Biol 42: 1348–1354, 2010. doi: 10.1016/j.biocel.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Paulin R, Michelakis ED. The metabolic theory of pulmonary arterial hypertension. Circ Res 115: 148–164, 2014. doi: 10.1161/CIRCRESAHA.115.301130. [DOI] [PubMed] [Google Scholar]
- 45.Pullamsetti SS, Doebele C, Fischer A, Savai R, Kojonazarov B, Dahal BK, Ghofrani HA, Weissmann N, Grimminger F, Bonauer A, Seeger W, Zeiher AM, Dimmeler S, Schermuly RT. Inhibition of microRNA-17 improves lung and heart function in experimental pulmonary hypertension. Am J Respir Crit Care Med 185: 409–419, 2012. doi: 10.1164/rccm.201106-1093OC. [DOI] [PubMed] [Google Scholar]
- 46.Rothman AM, Arnold ND, Pickworth JA, Iremonger J, Ciuclan L, Allen RM, Guth-Gundel S, Southwood M, Morrell NW, Thomas M, Francis SE, Rowlands DJ, Lawrie A. MicroRNA-140-5p and SMURF1 regulate pulmonary arterial hypertension. J Clin Invest 126: 2495–2508, 2016. doi: 10.1172/JCI83361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sangle GV, Zhao R, Mizuno TM, Shen GX. Involvement of RAGE, NADPH oxidase, and Ras/Raf-1 pathway in glycated LDL-induced expression of heat shock factor-1 and plasminogen activator inhibitor-1 in vascular endothelial cells. Endocrinology 151: 4455–4466, 2010. doi: 10.1210/en.2010-0323. [DOI] [PubMed] [Google Scholar]
- 48.Sarkar J, Gou D, Turaka P, Viktorova E, Ramchandran R, Raj JU. MicroRNA-21 plays a role in hypoxia-mediated pulmonary artery smooth muscle cell proliferation and migration. Am J Physiol Lung Cell Mol Physiol 299: L861–L871, 2010. doi: 10.1152/ajplung.00201.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Spiekerkoetter E, Guignabert C, de Jesus Perez V, Alastalo T-P, Powers JM, Wang L, Lawrie A, Ambartsumian N, Schmidt AM, Berryman M, Ashley RH, Rabinovitch M. S100A4 and bone morphogenetic protein-2 codependently induce vascular smooth muscle cell migration via phospho-extracellular signal-regulated kinase and chloride intracellular channel 4. Circ Res 105: 639–647, 2009. doi: 10.1161/CIRCRESAHA.109.205120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sutendra G, Bonnet S, Rochefort G, Haromy A, Folmes KD, Lopaschuk GD, Dyck JR, Michelakis ED. Fatty acid oxidation and malonyl-CoA decarboxylase in the vascular remodeling of pulmonary hypertension. Sci Transl Med 2: 44ra58–44ra58, 2010. doi: 10.1126/scitranslmed.3001327. [DOI] [PubMed] [Google Scholar]
- 51.Thompson O, Moghraby JS, Ayscough KR, Winder SJ. Depletion of the actin bundling protein SM22/transgelin increases actin dynamics and enhances the tumourigenic phenotypes of cells. BMC Cell Biol 13: 1, 2012. doi: 10.1186/1471-2121-13-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weymouth N, Shi Z, Rockey DC. Smooth muscle α actin is specifically required for the maintenance of lactation. Dev Biol 363: 1–14, 2012. doi: 10.1016/j.ydbio.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.White K, Lu Y, Annis S, Hale AE, Chau BN, Dahlman JE, Hemann C, Opotowsky AR, Vargas SO, Rosas I, Perrella MA, Osorio JC, Haley KJ, Graham BB, Kumar R, Saggar R, Saggar R, Wallace WD, Ross DJ, Khan OF, Bader A, Gochuico BR, Matar M, Polach K, Johannessen NM, Prosser HM, Anderson DG, Langer R, Zweier JL, Bindoff LA, Systrom D, Waxman AB, Jin RC, Chan SY. Genetic and hypoxic alterations of the microRNA-210-ISCU1/2 axis promote iron-sulfur deficiency and pulmonary hypertension. EMBO Mol Med 7: 695–713, 2015. doi: 10.15252/emmm.201404511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yan S, Liu L, Ren F, Gao Q, Xu S, Hou B, Wang Y, Jiang X, Che Y. Sunitinib induces genomic instability of renal carcinoma cells through affecting the interaction of LC3-II and PARP-1. Cell Death Dis 8: e2988, 2017. doi: 10.1038/cddis.2017.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yung L-M, Nikolic I, Paskin-Flerlage SD, Pearsall RS, Kumar R, Yu PB. A selective transforming growth factor-β ligand trap attenuates pulmonary hypertension. Am J Respir Crit Care Med 194: 1140–1151, 2016. doi: 10.1164/rccm.201510-1955OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zakrzewicz A, Kouri FM, Nejman B, Kwapiszewska G, Hecker M, Sandu R, Dony E, Seeger W, Schermuly RT, Eickelberg O, Morty RE. The transforming growth factor-beta/Smad2,3 signalling axis is impaired in experimental pulmonary hypertension. Eur Respir J 29: 1094–1104, 2007. doi: 10.1183/09031936.00138206. [DOI] [PubMed] [Google Scholar]
- 57.Zhou G, Chen T, Raj JU. MicroRNAs in pulmonary arterial hypertension. Am J Respir Cell Mol Biol 52: 139–151, 2015. doi: 10.1165/rcmb.2014-0166TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhu H, Jiang Z, Lei P, Huang W, Yu X. Poly(ADP-ribose) polymerase-1 mediates angiotensin II-induced expression of plasminogen activator inhibitor-1 and fibronectin in rat mesangial cells. Kidney Blood Press Res 34: 320–327, 2011. doi: 10.1159/000327344. [DOI] [PubMed] [Google Scholar]