

ABSTRACT
A number of agents designed for immunotherapy of Acute Myeloid Leukemia (AML) are in preclinical and early clinical development. Most of them target a single antigen on the surface of AML cells. Here we describe the development and key biological properties of a tri-specific agent, the dual-targeting triplebody SPM-2, with binding sites for target antigens CD33 and CD123, and for CD16 to engage NK cells as cytolytic effectors. Primary blasts of nearly all AML patients carry at least one of these target antigens and the pair is particularly promising for the elimination of blasts and leukemia stem cells (LSCs) from a majority of AML patients by dual-targeting agents. The cytolytic activity of NK cells mediated by SPM-2 was analyzed in vitro for primary leukemic cells from 29 patients with a broad range of AML-subtypes. Blasts from all 29 patients, including patients with genomic alterations associated with an unfavorable genetic subtype, were lysed at nanomolar concentrations of SPM-2. Maximum susceptibility was observed for cells with a combined density of CD33 and CD123 above 10,000 copies/cell. Cell populations enriched for AML-LSCs (CD34pos and CD34pos CD38neg cells) from 2 AML patients carried an increased combined antigen density and were lysed at correspondingly lower concentrations of SPM-2 than unsorted blasts. These initial findings raise the expectation that SPM-2 may also be capable of eliminating AML-LSCs and thus of prolonging survival. In the future, patients with a broad range of AML subtypes may benefit from treatment with SPM-2.
Keywords: immunotherapy, AML, CD33-CD16-CD123, dual targeting, single-chain triplebodies, NK cells, redirected lysis, therapeutic antibodies
Introduction
Current chemotherapeutic treatments (CT) induce a complete remission (CR) for 60–80% of AML patients, but more than 50% of the initial responders experience relapse within 5 years after CR.12 Prognosis for patients with relapsed disease is generally poor, and therefore, new treatment options are urgently needed. Recurrence of the disease is thought to be caused by Leukemia Stem Cells (LSCs) and leukemic progenitor cells which survived CT.3–7 New treatment strategies attempting to reach prolonged and deeper remissions therefore need to aim at an improved elimination of AML LSCs. Immuno-therapeutic approaches are promising for this purpose. One possibility is to direct cytolytic effector cells specifically towards AML cells through the identification of suitable target antigens expressed on their surface and the development of antibodies and antibody-derived agents with specificity for these targets. This approach, termed “redirected lysis” (RDL), is pursued by several agents currently under development and in clinical testing.
Suitable target antigens for RDL of both AML blasts and LSCs include CD33 and CD123. Blasts from 85–90% of AML patients as well as normal myeloid progenitors and myelocytes express CD33.8,9 Expression is restricted to normal and malignant hematopoietic cells, including AML-LSCs,10–13 and therefore, CD33 is a promising target for immunotherapies of AML.8,10,13
Gemtuzumab-Ozogamycin (GO, Mylotarg®), a CD33-directed antibody-drug conjugate (ADC), is an approved drug for the treatment of AML.14,15 The agent has clinical efficacy for certain subtypes of AML, but its commercial availability was suspended due to safety concerns.15 However, in spite of these limitations, the agent produced clear clinical benefits, including long-lasting treatment successes in particular after administration in fractionated doses to limit toxicity, probably due to its ability to eliminate some of the relapse-initiating LSCs.16,17 Another CD33-directed antibody-drug conjugate, SGN-CD33A (Vadustuximab Talirine), is in clinical development,18,19 as well as bispecific T cell engagers (BiTEs; AMG 330 and others),20–24 and tetravalent tandabs targeting CD33 and engaging T cells as cytolytic effectors (AMV 564).25 Finally, genetically engineered T cells equipped with transgenic chimeric antigen receptors (CARs) specific for CD33 showed anti-leukemic efficacy in xeno-transplanted mice and were tested in an AML-patient.26–31 Taken together, these developments establish CD33 as a clinically validated target of proven usefulness for the treatment of AML.
To further increase selectivity and efficacy of immunotherapeutic agents, it is desirable to include additional antigens into the spectrum of targets, which can help to discriminate further between normal hematopoietic stem cells (HSCs) and leukemia stem- and progenitor cells. A number of antigens potentially useful towards this goal have been reported, including CLL-1 (also called hMICL), Tim-3, CD96, CD44, CD45RA, CD47, CD32, CD25, CD123 and CD157.32–39 Among these, CD123 has particularly favorable properties for the development of immunotherapeutic agents.
CD123, the α-chain of the interleukin-3 receptor, is expressed on normal myeloid cells and their progenitors, and on blasts and AML-LSCs from 75–89% of AML-patients.8,40–45 In a landmark study, fewer than 1% of CD34pos CD38neg progenitor cells from a normal human bone marrow (BM) expressed CD123, while 99% of the corresponding cells from a patient with CD34-positive AML showed high grade expression. CD34pos CD38neg cells, which are enriched in AML-LSCs for patients with CD34-positive AML, from 16 of 18 primary AML samples showed strong expression of CD123.40 Another prominent study also reported CD123 expressed on the surface of CD34pos CD38neg AML cells, but in this study the antigen was also present on the corresponding cellular subsets from healthy bone marrow (BM) and cord blood (CB), although expression levels on the enriched normal BM cells were at least 10-fold lower than on on the corresponding AML cells.12 CD123 is also expressed on normal HSCs from AML patients in follow-up after induction therapy.46 On aggregate, the published reports agree that CD123 is expressed with far greater frequency and surface densities on AML-LSCs and progenitor cells than on normal HSCs.7 A fraction of normal HSCs may therefore survive therapies targeting CD123.27 Finally, CD123 is also expressed on cells from other hematologic malignancies including Acute Lymphoblastic Leukemia (ALL), Chronic Myeloid Leukemia (CML), Myelo-Dysplastic Syndromes (MDS), Hodgkin Lymphoma (HL), Hairy Cell Leukemia (HCL) and others.44,45
CD123 therefore is an attractive target for the design of immunotherapies against AML. A number of therapeutic agents with similar molecular formats as those described for CD33 have been developed, including immunoglobulins,47–49 a radio-immunoconjugate,50 ADCs and a fusion protein between the cytokine IL-3 and a fragment of diphtheria toxin,51–54 bi-specific T cell-recruiting agents (BiTEs) and Dual-Antigen Recruiting T-cell engagers (DARTs),55–58 dual-targeting triplebodies,59,60 and CAR-transfected T cells.61–64 Expression of CD123 is largely restricted to hematopoietic cells, but expression on endothelial cells has been reported.65 Importantly, CD123 shows low expression on megakaryocytic progenitors.66.
Most of the agents developed to date for the treatment of AML are mono-targeting. However, under treatment of other malignancies with mono-targeting agents, escape variants have often emerged. An example is the CD20 antibody Rituximab. After long-term treatment with this agent, escape variants occurred in almost one third of lymphoma patients.67 Similarly, after treatment of Acute Lymphoblastic Leukemia (ALL) patients with CAR-transfected T-cells specific for CD19, escape variants were frequently observed.
To alleviate this problem of escape variants our team has developed a new class of dual-targeting agents designed for simultaneous binding to one copy each of 2 different targets on the surface of the same cancer cell. We anticipated that this molecular design should permit us to reach 2 main objectives: a) a significant reduction of the probability for emergence of “double-escape variants”, simultaneously unresponsive to agents targeting both antigens; and b) an “enhanced selectivity of lysis”. This term signifies that in a mixed population of normal and malignant cells, as it is typically present in a tumor environment, the agent should preferentially bind and destroy the malignant cells with the help of the cytolytic effectors it engages. We hypothesized that “enhanced selectivity of lysis” can be achieved by dual-targeting triplebodies directed against a suitably chosen pair of target antigens, which are present on the cancer cells in a greater combined surface density than on the corresponding normal cells.68–70 We have searched for a pair of target antigens on AML cells and AML-LSCs fulfilling this condition, and the pair of CD33 and CD123 emerged a promising candidate.
Published studies so far have mostly addressed the expression of CD33 and CD123 individually in patient samples, and the cohorts analyzed in clinical studies were small.8 Larger studies in patients with many different subtypes of the disease are needed to investigate, whether new dual-targeting agents simultaneously addressing both antigens on the same cell 59,60 or combinations of corresponding mono-targeting agents are promising for clinical applications for a broad group of AML patients. In a first large study pursuing this goal, the expression of CD33 and CD123 alone and in combination was investigated on cellular samples from 319 AML patients.8 Samples from 88% of the patients expressed CD33, 9 % expressed CD123 without concomitant expression of CD33, and 69% expressed both antigens simultaneously. Blasts from patients with mutations in the NPM-1 gene showed elevated expression of CD33 and CD123, suggesting that Measurable Residual Disease (MRD)-guided and MRD-directed interventions with immunotherapeutic agents simultaneously targeting CD33 and CD123 may become feasible for these patients.8
Our team has designed the dual-targeting triplebody 123–16-33 with specificity for CD123 and CD33.60 Here we describe the generation of the clinical candidate SPM-2, an optimized variant carrying humanized and disulfide-stabilized single-chain variable fragments (scFvs) as antigen binding sites, plus additional mutations favoring its clinical development. We anticipate that SPM-2 will be able to discriminate at least to a degree between AML-LSCs and remaining normal HSCs of a patient, due to its dual-targeting capacity and the greater combined surface density of CD33 and CD123 on AML-LSCs than on normal HSCs.11,12,24,27,40,46 A preferential elimination of AML-LSCs over normal HSCs would be a distinct advantage, if it could be reached, because the surviving HSCs may be able to reconstitute the patient’s hematopoietic system after the end of therapy at least in part, possibly even without the need for a stem cell transplant. Here we have studied, whether SPM-2 in conjunction with activated human NK cells was capable of eliminating blasts from patients with a very broad range of AML subtypes, as predicted,8 and found the prediction to be fulfilled.
Finally, as a first step towards a test of our hypothesis claiming that SPM-2 should be able to mediate the elimination also of AML-LSCs by NK cells, we asked, whether the subsets of CD34-positive and (CD34-positive, CD38-negative) cells from patients with CD34-positive AML, which are enriched in AML-LSCs, were susceptible to lysis by NK cells plus SPM-2. The initial still incomplete results reported here are consistent with this hypothesis.
Results
Design, production and protein-chemical characterization of SPM-2
Expression vectors for SPM-2 were constructed from pre-existing cDNA-bearing vectors encoding scFv-domains specific for CD33, CD123 and CD16.59 The original scFv domains were of murine origin59 and were humanized, disulfide-stabilized and stability-engineered for incorporation into SPM-2. A final clinical candidate named SPM-2 was identified, and a production process in stably transfected human FreestyleTM 293F-cells, as well as a downstream purification process following industry standard procedures was developed. The protein had a molecular mass of 82.5 kDa and expression yields were in the range of 2 – 5 mg/L of purified protein, sufficient for production in the quantities needed for late preclinical development and clinical use. The final product was highly pure and contained neither abundant breakdown products nor detectable aggregates (Figure 1). A suitable formulation buffer with industry standard composition has been developed, and in long-term stability studies the protein was stable in this buffer at 4°C for 12 months and longer. Only marginal degradation was observed after 12 months of storage (Figure 1B). The catalytic activity after storage for 12 months at 4°C was still in the picomolar range (EC50 values of approx. 50 pM, where EC50 is the concentration, for which half-maximum lysis is obtained) as judged by in vitro RDL assays with the human AML-derived target cell line MOLM-13. This line carries elevated surface densities of CD33 and CD123 and is highly susceptible to lysis by SPM-2 plus NK cells (Figure 2; Supplement Figure 1, Supplement Table 1). The protein had good thermostability, and the monovalent binding affinities (equilibrium dissociation constants; KD) of the individual binding sites for CD33 and CD123 were in the low nanomolar range. Early preclinical development of the agent is advanced, and the agent is ready for late preclinical development and advancement to first-in-human (FIH) clinical studies.
Figure 1.
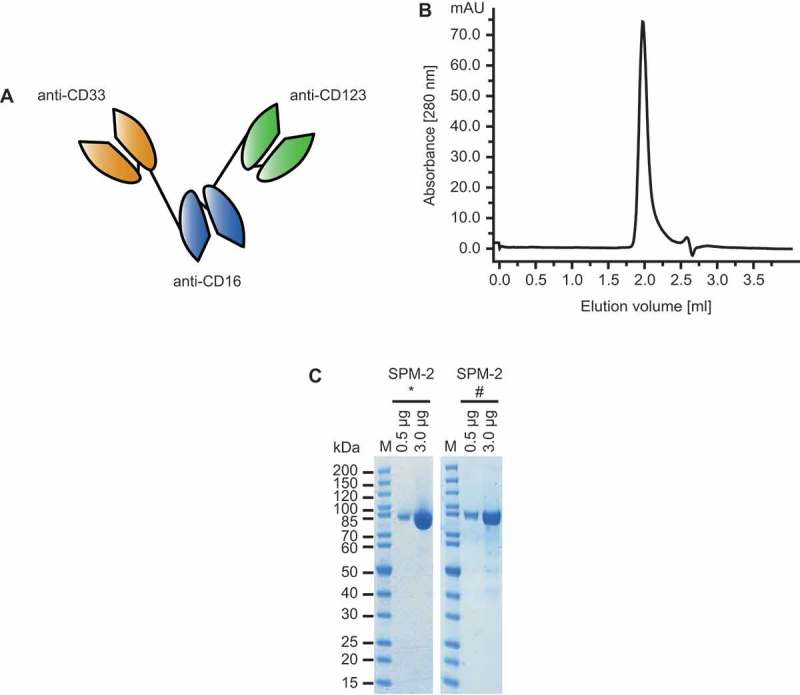
Design and properties of purified SPM-2. A. Molecular structure of triplebody SPM-2 (schematic). Single chain Fragment variable (scFv) domains, consisting of one VH domain joined to one VL domain, are connected into a single polypeptide chain. Black lines: flexible (Gly4Ser) linkers between the domains. One of the two distal scFv domains (orange) is specific for CD33 and the other (green) for CD123. The central scFv domain (blue) binds to CD16 and permits recruitment of various CD16-bearing types of effector cells, including NK and gamma delta T cells. B. After capture with a metal-ion affinity reagent and chromatographic purification the protein appeared as monomeric peak by size exclusion chromatography. C. SMP-2 was analyzed by SDS-PAGE directly after purification (left) and after storage for 12 months at 4°C (right). Only very small quantities of degradation products were observed. *: protein analyzed directly after purification, #: protein analyzed after 12 months of storage at 4°C; M: molecular weight marker in kDa.
Figure 2.
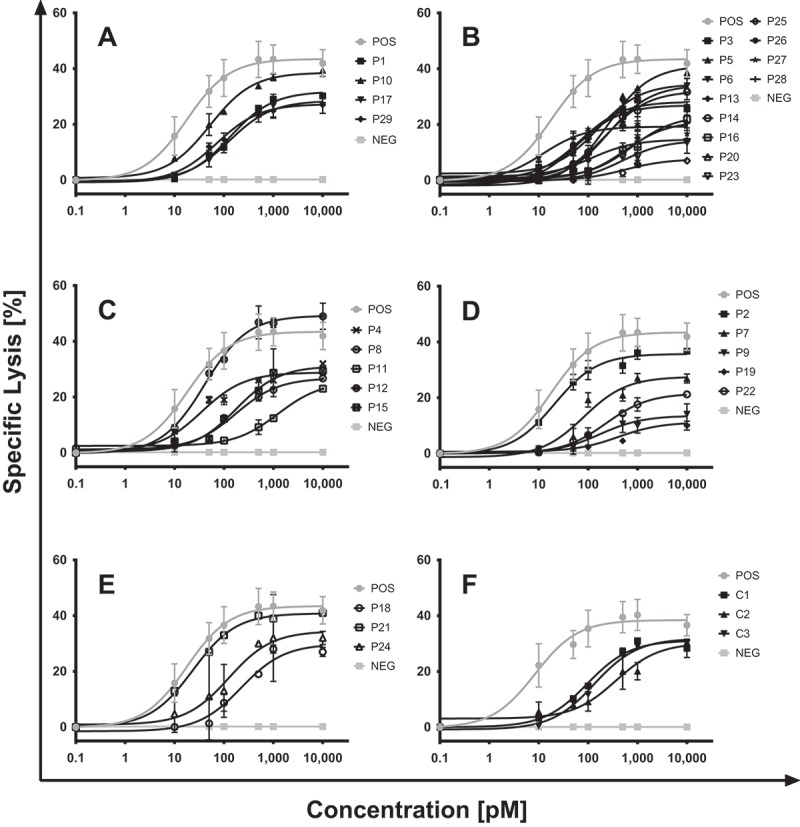
SPM-2-mediated redirected lysis of blasts from patients with different AML subtypes in combination with NK cells from a healthy unrelated donor. Blasts contained in the PBMC or BMMC populations of AML patients were obtained at the disease stages (diagnosis, remission, relapse) specified in Table 1. Blasts were labeled with calcein and used as targets in RDL reactions mediated by SPM-2 or control proteins in combination with ex vivo expanded, IL-2 stimulated NK cells from an unrelated healthy donor. NK cells were part of a population of LAK cells, consisting to 70% of T cells, 25% of NK cells, and 5% of NKT cells, after ex vivo expansion for 20 d in the presence of IL-2 (Material & Methods). The LAK cells were added in a 10: 1 effector to target cell ratio, corresponding to an effective E: T ratio of NK: targets of 2: 1. SPM-2 triplebody was present in the reactions at the concentrations shown in pM. A) Samples from patients with favorable AML subtype according to the ELN (European Leukemia Network) classification2. B) AML with intermediate-I ELN risk subtype. C) Samples from patients with ELN intermediate-II risk subtype. D) samples from patients with adverse ELN risk disease. E) samples from patients with an unclassified disease subtype. F) Myeloid cells from healthy donors (C1, C2), preparatively enriched by immuno-magnetic sorting with CD11b beads show similar susceptibility to SPM-2 mediated lysis as non-enriched blasts from a representative patient sample (C3; patient P1 in Table 1). In all experiments, MOLM-13 cells were carried along as a positive control, and triplebody Her2-16-Her2 as a negative control. Additional controls have previously been performed and reported, showing that target cells devoid of CD33 and/or CD123, such as HEK 293 and CHO cells, failed to bind triplebodies with specificity for CD33 and CD123.58 Specific lysis was computed as outlined in Materials & Methods. Error bars represent the standard error of the mean (SEM) computed for triplicate samples of each measurement point.
Lysis of primary blasts from patients with different subtypes of AML by SPM-2 plus NK cells
To test the prediction that agents capable of bivalent binding to one copy each of CD33 and CD123 on the same AML blast should be able to eliminate blasts from almost all AML patients,8 RDL experiments were performed with primary cells from a panel of 29 patients with a broad range of AML subtypes. The panel included patients with AML belonging to all genetic risk groups according to the ELN (European Leukemia Network) classification,2 (Table 1). For in vitro cytolysis assays the target cells were labeled with calcein.60,68 Peripheral blood mononuclear cells (PBMCs) from an unrelated healthy donor were expanded in culture for 20 d in the presence of IL-2. These cells, called lymphokine-activated killer cells (LAK cells), consisted of approx. 25% NK cells, 70% T cells and a small fraction of NKT cells69,71 and were used at an effector-to-target cell (E: T) ratio of NK cells: targets of 2: 1. After a 4 hr reaction the extent of specific lysis mediated by the agent (beyond the spontaneous lysis achieved by the LAK cells alone, in the absence of added triplebody) was measured by calcein release, and dose-response curves were plotted (Figure 2). From these curves, half-maximum effector concentrations (EC50 values) were computed. Target cells were MNCs enriched by density centrifugation from bone marrow (BM) or peripheral blood (PB) samples (BMMCs; PBMCs), either freshly drawn or stored under liquid nitrogen.
Table 1.
Patient data and characterization of primary cell samples.
| ID | M/F | Age | Diagnosis | Source of material |
Blast Count [%] |
Cytogenetics | NPM1 mut | FLT3-ITD | ELN genetic group |
|---|---|---|---|---|---|---|---|---|---|
| P1 | M | 71 | AML M2 from MDS |
BM | 54 | 46, xy; t(8;21) Runx1-Runx1T1 (AML-ETO) |
ND | ND | favorable |
| P2 | M | 43 | relapsed bi-pheno- typic AL |
BM | 96 | 46, xy; complex aberrant MLL rearranged |
- | - | adverse |
| P3 | M | 61 | AML M4 | PB | 91 | 46, xy MLL-PTD |
+ (Type D) |
+ (Flt3-TKD) |
intermediate-I |
| P4 | M | 24 | AML M4 | PB | ND | 47, xy; +8 | - | + | intermediate-II |
| P5 | M | 74 | AML M1 | BM | 93 | 46, xy | - | - | intermediate-I |
| P6 | F | 22 | AML M5b | BM | 83 | 46, xx | + | + | intermediate-I |
| P7 | M | 77 | AML M6 | BM | 24 | complex aberrant del(5q31); del(ETV6); del(Nup98)(11p15) |
- | ND | adverse |
| P8 | M | 81 | AML M2 | PB | 46 | 46, xy, t(1;21) (p36;q22) |
- | ND | intermediate-II |
| P9 | F | 74 | AML M2 | PB | 54 | complex aberrant RUNX1 amplification |
- | ND | adverse |
| P10 | F | 72 | AML M4 | PB | 93 | 46, xx | + | - | favorable |
| P11 | M | 61 | AML M4 | BM | 92 | del(12)(p12) (partial, 2/8) |
- | - | intermediate-II |
| P12 | F | 55 | AML ND | PB | ND | trisomy 4 | + | + | intermediate-II |
| P13 | F | 75 | AML ND | BM | 75 | 46, xx | ND | + | intermediate-I |
| P14 | M | 20 | AML M1 | BM | 96 | 46, xy | - | - | intermediate-I |
| P15 | M | 72 | AML ND | PB | 93 | 46, xy; t(6;9) (p22;q34) |
- | + | intermediate-II |
| P16 | F | 85 | AML ND | PB | 92 | 46, xx | + | + | intermediate-I |
| P17 | M | 82 | AML ND | PB | 92 | 46, xy | + | - | favorable |
| P18 | M | 85 | AML M1 | BM | ND | ND | + | - | unclassifiable |
| P19 | M | 46 | AML M5 | PB | 87 | 47, xy; +8; t(9;11)(p22;923) |
- | - | adverse |
| P20 | F | 57 | AML ND | PB | 97 | 46, xx | + | + | intermediate-I |
| P21 | M | 23 | AML M3 V | PB | 86 | 46, xy; t(15;17)(q22;q12) |
+ | + | unclassifiable |
| P22 | M | 66 | AML M2 | PB | ND | 44, xy; complex aberrant |
- | - | adverse |
| P23 | F | 64 | AML M1 | PB | 85 | 46, xx | + | + | intermediate-I |
| P24 | F | 69 | AML ND | PB | ND | 46, xx | ND | ND | unclassifiable |
| P25 | M | 76 | AML M5 (from CMML) |
PB | 94 | 46, xy | - | - | intermediate-I |
| P26 | F | 50 | AML M1 | BM | ND | 46, xx | - | - | intermediate-I |
| P27 | M | 69 | AML M4 | PB | 91 | 46, xy | + | + | intermediate-I |
| P28 | M | 42 | AML M1 (refractory) |
PB | 86 | 46, xy | + | + | intermediate-I |
| P29 | M | 61 | AML M4 | BM | 97 | 46, xy | + | - | favorable |
ND: not determined; BM: bone marrow; PB: peripheral blood; genetic risk groups assigned according to European Leukemia Net (ELN) classification [3, 4]; M,F: male, female; TKD = Tyrosine Kinase Domain; blast counts were determined after Ficoll density enrichment of BM MNCs (mononuclear cells) by cytofluorimetry; gates were set in an SSC vs. CD45 plot as illustrated in Supplementary Figure 3
Cells from all 29 patients responded to lysis by SPM-2 plus NK (LAK) cells (Figure 2A-E). First cytolytic effects were seen at 10 pM concentrations of SPM-2 for many samples, and all samples responded at 100 pM and above. For some samples, such as the sample from patient #25, the extent of specific lysis was small, but was still clearly measurable above background within the sensitivity of the assay. MOLM-13 cells were carried along as a positive control (standard of susceptibility), and the triplebody Her2-16-Her2 was used as a negative control. This is a triplebody in the same molecular format as SPM-2, but with binding sites for Her2, a tumor antigen expressed on human breast cancer cells and other carcinoma cells, but not on AML blasts. The Her2-directed triplebody was biologically active in separate control experiments with Her2-positive target cells. In another control, carried along in all experiments, the LAK cells were omitted but the triplebody was present. This control permitted us to evaluate any potential contribution of autologous CD16-bearing effector cells (NK cells, gamma-delta T cells, etc.) contained in the MNC target cell population to the lytic effect. These controls revealed no detectable contributions of autologous effector cells contained in the MNC population. A final control were target cells lacking the expression of CD33 and CD123. For this control (Suppl. Figure1) MNCs prepared from peripheral blood of a healthy donor (MNCs HD) were used as targets, and no measurable specific lysis was observed, although the sample contained approx. 10% of myeloid cells expressing CD33 and CD123. However, averaged over the entire MNC population the densities amounted to only a few hundred copies per cell (Suppl. Table 1), and specific lysis of this entire population remained below the detection limit.
Only one sample (patient 12) manifested a greater extent of lysis than the MOLM-13 control (Figure 2C). Cells from patients # 5, 14, 18, 23, 26 and 28 with AML subtype FAB M1 (Table 2) responded with EC50 values of 242, 229, 221, 475, 58 and 560 pM, respectively (Figure 2B, E). Blasts from patients with a FAB M1 subtype often display a very immature maturation state and express only low levels of CD33, or are CD33 negative, and are often difficult to treat with agents mono-specific for CD33.60 Cells from patients 2 to 9, 11 to 16, 19, 20, 22, 23 and 25 to 28 with intermediate and adverse ELN genetic subtypes (Table 2) were also lysed efficiently by SPM-2 with EC50 values ranging from 10.3 – 1078 pM (Figure 2B-D). These data strongly support the prediction8 that a dual-targeting agent with specificity for CD33 and CD123 acting together with NK cells should be able to eliminate blasts from almost all AML patients with a very broad range of disease subtypes.
Table 2.
Target antigen densities and susceptibility to SPM-2-mediated cytolysis of cell samples.
| ID | % CD33+ | % CD123+ | # 33 | # 123 | Σ # (33 + 123) |
EC50 (pM) | ELN genetic group |
|---|---|---|---|---|---|---|---|
| P1 | 73 | 30 | 2,781 | 5,349 | 8,130 | 131 | favorable |
| P2 | 100 | 74 | 9,765 | 1,388 | 11,153 | 20 | adverse |
| P3 | 100 | 100 | 12,974 | 25,812 | 38,786 | 67 | intermediate-I |
| P4 | 100 | 97 | 9,045 | 25,424 | 34,469 | 32 | intermediate-II |
| P5 | 85 | 99 | 1,459 | 5,852 | 7,311 | 242 | intermediate-I |
| P6 | 99 | 80 | 9,489 | 3,769 | 13,258 | 155 | intermediate-I |
| P7 | 70 | 36 | 6,405 | 2,668 | 9,073 | 79 | adverse |
| P8 | 57 | 30 | 1,197 | 6,411 | 7,608 | 133 | intermediate-II |
| P9 | 94 | 80 | 5,214 | 2,138 | 7,352 | 166 | adverse |
| P10 | 84 | ND | 15,414 | 8,122 | 22,536 | 51 | favorable |
| P11 | 47 | ND | 1,192 | 2,391 | 3,583 | 1,078 | intermediate-II |
| P12 | 84 | ND | 17,635 | 13,101 | 30,736 | 38 | intermediate-II |
| P13 | 65 | ND | 4,987 | 4,994 | 9,481 | 245 | intermediate-I |
| P14 | 52 | ND | 1,010 | 7,123 | 8,133 | 229 | intermediate-I |
| P15 | 95 | ND | 4,797 | 8,206 | 13,003 | 177 | intermediate-II |
| P16 | 90 | ND | 1,877 | 8,257 | 10,134 | 758 | intermediate-I |
| P17 | 95 | ND | 6,463 | 7,448 | 13,911 | 125 | favorable |
| P18 | 79 | ND | 4,544 | 6,408 | 10,952 | 221 | unclassifiable |
| P19 | 82 | ND | 2,876 | 766 | 3,642 | 406 | adverse |
| P20 | 96 | ND | 16,377 | 9,072 | 25,449 | 251 | intermediate-I |
| P21 | 87 | ND | 11,891 | 7,657 | 19,548 | 22 | unclassifiable |
| P22 | 80 | ND | 1,250 | 1,892 | 3,142 | 225 | adverse |
| P23 | 75 | ND | 1,245 | 6,479 | 7,724 | 476 | intermediate-I |
| P24 | 70 | ND | 6,790 | 3,639 | 10,429 | 127 | unclassifiable |
| P25 | 75 | ND | 5,193 | 2,051 | 7,244 | 541 | intermediate-I |
| P26 | 92 | ND | 3,71 | 2,389 | 5,560 | 58 | intermediate-I |
| P27 | 80 | ND | 5,458 | 6,892 | 12,350 | 10 | intermediate-I |
| P28 | 84 | ND | 7,399 | 5,237 | 12,636 | 560 | intermediate-I |
| P29 | 88 | ND | 4,896 | 7,438 | 12,334 | 70 | favorable |
ND: not determined, genetic risk groups assigned according to European Leukemia Net (ELN) classification [3, 4], % CD33+: fraction of MNCs scoring positive for CD33; # 33: antigen copies per cell determined by calibrated cytofluorimetry.
For comparison, the susceptibility of myeloid cells from healthy donors to RDL by SPM-2 or the control triplebody Her2-16-Her2 plus NK (LAK) cells was also examined (Figure 2F). When the total population of PBMCs from healthy donors was used as targets, only marginal lysis was observed (≤ 5% specific lysis; Suppl. Figure 2). This was to be expected, because typically only a small fraction of normal PBMCs (15 – 20%) are myeloid cells. To investigate the effect of SPM-2 plus NK cells on healthy myeloid cells, PBMCs of healthy donors were enriched for CD11b-bearing cells by preparative immuno-magnetic sorting with commercially available beads coated with a CD11b-specific antibody. This resulted in an enrichment of CD11b-bearing myeloid cells to ≥ 90%. These cells were then tested as targets in RDL experiments (Suppl. Table 3). Myeloid cells from normal donors enriched in this manner now demonstrated similar antigen densities and similar susceptibility to lysis by SPM-2 plus NK (LAK) cells as those from the non-enriched blasts of patient 1 (Figure 2F). They showed no greater susceptibility than the malignant cells from AML patients.
Finally, BMMCs from patients with non-leukemic disorders showing no alterations in their hematopoietic system (donors with non-AML disorders; Suppl. Figure 2 and Suppl. Table 2) were tested by RDL, and only low levels of lysis were found. These cells were not enriched for myeloid cells by preparative sorting with CD11b immuno-magnetic beads and therefore showed similar susceptibility as non-enriched PBMCs from unrelated healthy donors. This finding suggests that the hematotoxic damage in the bone marrow that has to be expected for the normal hematopoietic cells of patients with AML under treatment with SPM-2 will be limited relative to the therapeutic effect on the malignant cells.
Surface antigen densities of CD33 and CD123 on primary cells from patients with different subtypes of AML and from patients with non-hematologic disorders
A loose correlation between cytolytic and cytotoxic activity and target antigen density has been reported for the CD33-specific agents AMG330 9 and GO.72 Therefore, we studied here, whether a similar correlation also existed for SPM-2 plus NK cells. To this effect, mean antigen densities of CD33 and CD123 on the patient-derived cells and on myeloid cells from healthy donors were determined with precision by calibrated cytofluorimetry and expressed in copy numbers per cell (Table 2; Suppl. Table 1–3). All samples showed measurable expression of CD33 and CD123, but with considerable variability between samples. The sample from patient 12 had the highest density of CD33 with approx. 17,600 copies/cell, and the sample from patient 3 had the highest density of CD123 with approx. 25,800 copies/cell. The sample from patient 3 also had the highest combined antigen density of CD33 plus CD123 cell with approx. 38,780 copies/cell (Table 2). This combined density is roughly comparable with the value for MOLM-13 cells, which was approximately 46,000 copies/cell (Suppl. Table 1). A weak correlation between the EC50 values and the combined antigen densities of CD33 plus CD123 was detected (Figure 3). A threshold for lysis by SPM-2 appeared to exist at a combined density of approximately 10,000 copies/cell of (CD33 plus CD123). However, this apparent correlation must be interpreted with caution, because the number of samples analyzed is still small and statistical significance has not yet been reached.
Figure 3.
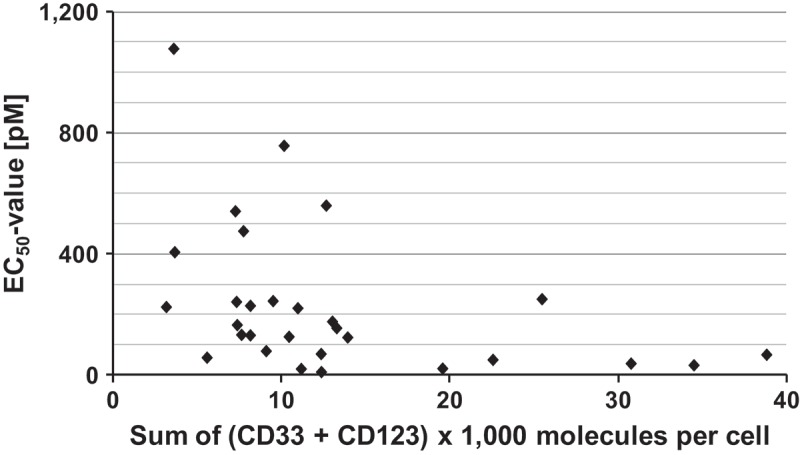
Correlation between susceptibility to lysis by SPM-2 plus NK cells (EC50 values) and combined surface antigen density of (CD33 + CD123) on patient blasts. Mean surface densities of CD33 and CD123 were determined for each patient sample. EC50 values were computed for each sample from the dose response curves shown in Figure2. Numerical values were the same as those shown in Table 3.
Subsets of patient blasts enriched for CD34-bearing cells show increased expression of CD123 and increased susceptibility to lysis by SPM-2 plus NK cells
Our hypothesis is that AML-LSCs should be susceptible to lysis by SPM-2 plus NK cells, because they have been reported to display high surface densities of CD33 and CD123.7,10–12,40,46 We are not aware of any published reports indicating that they should differ from AML blasts in their intrinsic susceptibility to RDL by antibodies or antibody-derived agents in combination with NK cells. For most patients with CD34-positive AML, the relapse-relevant AML-LSCs are contained predominantly in the compartments of CD34-positive and (CD34-positive CD38-negative) cells.12,40,43–46,73 Therefore, we have first enriched CD34-bearing cells from 2 patients with CD34-positive AML, for whom sufficient cell numbers were available (patients 9 & 11; Table 3), by immuno-magnetic sorting with a commercial kit. Both samples carried intermediate combined densities of CD33 plus CD123 on their blasts (MNCs; approx. 8,000 and 3,600 copies/cell, respectively; Tables 2 and 3). Both blast (MNC) samples were lysed by (SPM-2 plus NK cells) in RDL experiments with EC50 values of 166 and 1,078 pM, respectively, prior to enrichment for CD34-positive cells (Tables 2, 3). After the enrichment, CD33 and CD123 densities and susceptibility to RDL were determined again. The CD33 density was now reduced for cells from patient 11 to 500 copies/cell, and the CD123 density was increased to 14,500 copies/cell (Table 3). The EC50 value fell to 681 pM, indicating an approximately 2-fold increased susceptibility for lysis relative to the value for non-enriched cells. For patient 9 the CD33 density on the enriched cells was 13,000 copies/cell, the density of CD123 was 9,100 copies/cell, and the combined density was 22,100 copies/cell. The EC50 value for the enriched cells of this patient was reduced by about 4-fold to 43 pM. The increase in the combined density of CD33 plus CD123 and the decrease in EC50 values in both cases are consistent with our hypothesis positing that cellular compartments encompassing the AML-LSCs more closely may indeed have greater susceptibility to lysis by SPM-2 than bulk AML blasts (MNCs). Similar initial data were also obtained for SPM-2-mediated RDL of cells from the (CD34-positive, CD38-negative) subset of 2 AML patients. From the small number of cases tested we cannot yet draw statistically valid conclusions, but the initial results obtained so far are consistent with our hypothesis, although the data set is still very small.
Table 3.
Antigen expression and susceptibility to redirected lysis of patient blasts and analysis after preparative sorting for CD34- bearing cells.
| ID | Cellular Subset | Antigen Density (# molecules/cell) |
EC50 (pM) | ||
|---|---|---|---|---|---|
| CD33 | CD123 | CD33 + CD123 | |||
| P9 (AML-M2) | Bulk (CD45dim SSClow) | 5,500 | 2,500 | 8,500 | 166 |
| CD34-enriched MNCs | 13,000 | 9,100 | 22,100 | 43 | |
| P11 (AML-M4) | Bulk (CD45dim SSClow) | 1,200 | 2,400 | 3,600 | 1,078 |
| CD34-enriched MNCs | 500 | 14,500 | 15,000 | 681 | |
| LSC-enriched fraction (CD34+ CD38− CD123+) |
300 | 18,000 | 18,300 | ND | |
ND: not determined
Discussion
The main result of this study is that primary blasts from AML patients with a very broad range of disease subtypes responded strongly to treatment with SPM-2 plus NK cells in RDL assays in vitro. Cells from all 29 patients showed a clear response, although the response varied for different patients and was weak for some patients. In addition, we have studied a number of AML-derived cell lines (Suppl. Table 1, Suppl. Figure 1 and others), and all lines expressing either CD123 or CD33 or both on their surface showed a clear RDL response to SPM-2 plus NK cells. These data support the recent prediction that malignant cells from almost all AML patients should be lysed by agents jointly binding CD33 and CD123.8
We expect that this prediction will be further confirmed, when lysis of AML cells by the patients’ autologous NK cells, mediated by SPM-2, will be studied. Such studies are planned for the future, but have not yet been performed so far. They will likely be informative for the question, whether the combination of the new agent with the patients autologous cells is beneficial for patients with a broad range of AML subtypes. With the present study we tried to answer a different question. We tried to isolate the AML subtype as the sole variable and wanted to determine, whether the same molecular agent was effective for samples from patients with different disease subtypes in combination with a constant source of human NK cells. The results obtained here are remarkable, because for other antibody therapeutics in clinical use against AML, such as GO, typically only less than half of the patients with antigen-positive disease responded to the treatment.10,15,72
Correlation between combined surface antigen density of CD33 and CD123 and susceptibility to RDL lysis by SPM-2 plus NK cells
A loose correlation between these two properties was observed for bulk AML cells (Figure 3), and similar correlations have been reported for the CD33-specific BiTE AMG330,9 the CD123 antibody Talacotuzumab (CSL 362),48,49 and the CD3 x CD123 DART agent MGD006.56 While the target antigen density likely dictates the strength of binding of these agents to the target cells and the subsequent engagement of killer cells, it is unlikely that binding strength alone will translate 1:1 into an enhanced lytic activity. Other relevant processes independent of target antigen density will influence the cytolytic outcome. Therefore, it would be unrealistic to expect a very tight correlation between target antigen density and the susceptibility of the patients blasts to lysis by SPM-2 plus NK-cells (LAK cells) in our cell culture RDL assays.
Our findings are consistent with a recent study of the CD33-directed agent AMG330.9 In this study surface densities of CD33 were determined for samples from 621 patients and CD33 was strongly expressed on the blasts from 99% of the patients. However, the surface expression level of CD33 was not the sole determinant of lytic activity, although the degree of lysis differed between strongly and weakly expressing cells. Interestingly, monocytes and dendritic cells (DCs) showed strong expression of CD33, but no lysis of DCs by AMG 330 plus T cells was observed in RDL assays.9 Data about lysis of monocytes and DCs by SPM-2 plus NK cells are not yet available.
Effect of SPM-2 on cellular subfractions enriched in AML LSCs
Our hypothesis is that in concert with NK cells SPM-2 will mediate the elimination not only of bulk AML cells, but also of AML-LSCs. As a first approximation to a test of this hypothesis, cellular subsets enriched for CD34-bearing cells by immuno-magnetic sorting were prepared from primary cells of 2 AML patients with CD34-positive AML. These populations displayed increased surface antigen densities of (CD33 plus CD123) and correspondingly an enhanced susceptibility to lysis by SPM-2 plus NK cells (Table 3). The data are consistent with the hypothesis but do not yet provide direct evidence in its favor. Even though the AML-LSCs are enriched in the populations of CD34-bearing and (CD34-positive, CD38-negative) cells, the LSCs probably still account for only a small fraction of cells in these populations. Therefore, the data presented in Table 3, which represent properties averaged over all cells of the enriched populations, probably do not yet reflect the properties of the far narrower subset of true LSCs.
To test our hypothesis, clinical studies are needed. A suitable flow cytometric method permitting to monitor the effect of a treatment with SPM-2 plus NK-cells on AML LSCs has recently been published. This assay was tested and validated for the detection of LSCs from a majority of patients with CD34-positive AML, which account for approximately 80% of all AML cases.46 We intend to use this and other methods (quantitative polymerase chain reaction, qPCR, where applicable) in the future, to monitor the effect of SPM-2 plus autologous NK cells on the subset of AML-LSCs.
Planned studies of antileukemic efficacy of SPM-2 in xenotransplanted animals and in comparison with corresponding mono-targeting agents in cell culture studies
Efficacy studies with SPM-2 in mice xenotransplanted with human AML cells have so far not been performed but are planned for the near future. These studies were postponed until SPM-2 protein in GMP or GMP-like purity grade became available, so that the data are admissible by the regulatory authorities. Also, the scFv binding domains carried in SPM-2 are uniquely specific for the human target antigens and do not cross-react with their murine homologues. Therefore, both the human target cells and human NK cells need to be transplanted into the mice, and these experiments are difficult and expensive. However, they will be performed with high priority as soon as the first batch of SPM-2 protein in GLP purity grade becomes available. An alternative is to use a mouse model with a xenotransplanted fully human haematopoietic system, and then to transplant only the human AML target cells. This model is available on a collaborative basis, but is very labor-intensive and expensive, and therefore, we need to wait for a first batch of SPM-2 protein in GLP purity grade to become available. Another set of important studies needed to be postponed for the same reason. This was the investigation, whether the final clinical candidate SPM-2 mediates not only enhanced selectivity of lysis in a mixture of two target cell populations, one highly double-positive and the other lowly double-positive for CD33 and CD123 as predicted. We also urgently wish to determine, whether the dual-targeting triplebody mediates more potent lysis at lower EC50 doses in RDL experiments than the mono-targeting related proteins CD33-16 and CD123–16, either individually or in combination, similar to the studies performed earlier with the triplebodies 123-16-33, HLAII-16–19 and 33–3-19.59,68–70
NK cells vs. T cells as cytolytic effectors
When SPM-2 was designed, several studies had reported that T cells engaged by immuno-therapeutic agents caused serious CRS (Cytokine Release Syndrome). If NK cells should turn out to be sufficiently strong killers when engaged by our agent, then we would prefer to promote an agent engaging NK cells into clinical development, because it would likely produce less severe CRS and may be better tolerated by the patients.
Our earlier study 60 and others quoted therein reported that NK cells collected at the time of diagnosis were greatly reduced in numbers and specific cytolytic activity in the BM of AML patients, but that they frequently recovered in both regards by the end of induction CT, when the leukemic burden was greatly reduced. Therefore, SPM-2 was designed for use in a post-induction setting with the objective to improve survival by eliminating the relapse-inducing LSCs, and for this purpose NK cells appeared to be suited in the single case studied so far.60
In a recent publication of data obtained for the CD157-specific antibody MEN1112,39 the authors reported, that autologous NK cells from a sizable cohort of AML patients showed only marginal cytolytic RDL activity in assays with this antibody. However, in this study, NK cells obtained from the patients at the time of diagnosis were used, a time point, when NK cells are typically reduced in numbers and cytolytic activity. We have not yet tested the effects of SPM-2 in combination with a similar set of autologous NK cells from a large cohort of AML patients. We only tested NK cells from a single AML patient so far, prepared from a BM sample obtained in a first remission after standard CT. These NK cells were highly active for lysis of the autologous AML blasts.60 In the study of MEN1112,39 the RDL reaction also was mediated by NK cells binding with their CD16 receptor to the Fc domain of the CD157 antibody, and thereby triggering RDL. In our case, the NK cells were actively engaged by an scFv binding domain with high affinity for CD16 contained in the triplebody. This is an important difference, and therefore, the strong binding of our agent to CD16 on NK cells may conceivably cause a stronger activation of the NK cells than binding of CD16 on NK cells to the Fc-domain of the CD157 antibody. NK cells from the patients studied by Krupka and colleagues39 might still have shown a sufficiently strong response towards autologous AML blasts in concert with SPM-2, if they had been drawn in remission.
Finally, a number of methods have recently been developed, which appear suited to optimize NK cell function in human AML patients either by pharmaceutical agents 74–77 or by adoptive transfer of ex vivo expanded NK cells.71,77 The combination of SPM-2 with some of these approaches may contribute to the elimination of MRD and thus lead to favorable therapeutic outcomes in the future.
The results reported here for SPM-2 are very encouraging, and this agent may therefore become useful for the therapy of AML in a post-induction, MRD-directed setting, even without an adoptive transfer of NK cells. This agent clearly deserves to be advanced into clinical development.
Materials and methods
Patients, healthy donors, and donors with non-aml disorders
Written informed consent was obtained from patients in accordance with the Declaration of Helsinki. After approval by the Institutional Review Boards (IRBs) of the Ludwig-Maximilians-Universität (LMU) Munich (patients 1–9; Table 1) and the Friedrich Alexander Universität (FAU) Erlangen (patients 10–29; Table 1) was obtained, PB and BM samples were collected from healthy donors (HDs) and patients with AML at diagnosis or relapse. HDs were adults free of known disease, and “donors with non-AML disorders” were patients with disorders other than acute leukemias and without symptoms of their hematopoietic system, which were treated at the LMU Medical Center in the context of other clinical studies. Their cells were made available after their use for the present study was approved by the LMU’s IRB.
Expression in mammalian cells, purification and protein-chemical characterization of triplebody SPM-2
The DNA construct coding for SPM-2 was synthesized by a commercial provider (Eurofins/MWG-Operon). To create this construct, the scFv subunits were disulfide-stabilized78-80 humanized81-83 and stability-engineered84 by standard procedures. The TransIT®-LT1 transfection reagent (Mirus Bio LLC, catalog # MIR 2300) was used for transfection according to the manufacturer´s protocol to generate a stable cell pool of FreestyleTM 293F cells (ThermoFisher Scientific, cat. # R79007) for protein expression. Cells were then cultured under continuous selection with hygromycin C. SPM-2 was captured from cell culture supernatants via its C-terminal hexahistidine tag by immobilized zinc ion affinity chromatography followed by anion exchange chromatography using a 1 ml (bed volume) HiTrap Q Sepharose HP column (GE Healthcare). The third purification step was a cation exchange (CEX) chromatography. Here, a 1 ml HiTrap SP Sepharose HP column (GE Healthcare) was used and connected to an Äkta liquid chromatography system (GE Healthcare). SPM-2 preparations were analyzed by size exclusion chromatography (Superdex S200 5/150 GL column, GE Healthcare) for quality control and by SDS polyacrylamide gel electrophoresis (SDS-PAGE) after the final purification step. The protein concentration was determined by UV absorption at 280 nm using a NanoDrop spectrometer (PeqLab). The theoretical extinction coefficient was calculated from the primary amino acid sequence with the computer program ProtParam (www.expasy.ch). A final yield after purification of approx. 2.5 to 5 mg SPM-2 per L of culture medium was achieved in several independent experiments.
Preparation of primary cells from blood and bone marrow of human donors
PB and BM samples were drawn from subjects into either EDTA or heparin solution. Leukemia-derived cells and Mononuclear Cells (MNCs) from healthy donors were enriched by density centrifugation using the Lymphoflot reagent (Bio-Rad, cat. # 824012) according to manufacturer’s instructions. Peripheral Blood Mononuclear Cells (PBMCs) were then either suspended in RPMI medium 1640 with GlutaMAXTM Supplement (ThermoFisher Scientific, cat. # 61870–044) containing 10% GibcoTM fetal bovine serum (FBS; ThermoFisher Scientific, cat. # 10082–147) with GibcoTM penicillin-streptomycin (PS; ThermoFisher Scientific, cat. # 15140–122) at 100 units/ml and 100 µg/ml, respectively, for immediate use, or stored frozen in a solution containing 90% FBS and 10% DMSO. Cell viability was assessed by Trypan blue exclusion before use.
Ex vivo expansion of MNCs from a healthy donor in the presence of IL-2 (LAK-cells)
PBMCs from an adult healthy donor were expanded ex vivo in RPMI medium containing Interleukin-2 (IL-2) (ThermoFisher Scientific, cat. # PHC0023) plus 5% human serum (ThermoFisher Scientific, cat. # 34005–100, discontinued) for 20 days as described.68,71 At the end of this expansion period, the LAK (Lymphokine Activated Killer) cell population consisted of approx. 25% NK cells, 70% T cells and 5% NKT cells. LAK cells were then frozen in aliquots for subsequent use. Prior to use in cytolysis experiments, the cells were thawed and cultured overnight in RPMI medium containing 5% human serum (Invitrogen) plus 50 units/ml and 50 µg/ml PS, respectively, but no additional IL-2.
Enrichment of human CD34-positive cells by preparative sorting with immuno-magnetic beads
CD34-positive cells were enriched by positive selection using the commercial human CD34+ Multisort Kit (Miltenyi Biotec MACS sorting kit, cat. #130–058-701) according to manufacturer’s instructions. Starting material were purified PBMCs from PB or BMMCs from donor BM samples. The enriched CD34-positive cells were then either suspended in RPMI medium containing 10% FBS with PS at 100 units/ml and 100 µg/ml, respectively, for use in RDL assays, or placed in Dulbecco’s phosphate buffered saline (PBS) solution (ThermoFisher Scientific, cat. # 14190–250) containing 1% bovine serum albumin (BSA) (Sigma-Aldrich Co., LLC, cat. # A9418) for flow cytometric analysis.
Flow cytometry
Flow cytometric analysis was performed with an Accuri C6 flow cytometer (BD Biosciences). A typical staining reaction used 300,000 cells per FACS tube containing the specific monoclonal antibody (mAb) and proceeded for 20 min on ice. Wash steps were performed with a PBA solution (PBS; 0.1% Bovine Serum Albumin and 0.02% Sodium Azide). AML blasts were defined as the population of CD45DIM x SSCLOW cells detected among the patient’s PBMCs or BMMCs with a Fluorescein-isothiocyanate (FITC)-conjugated CD45-specific mAb (Beckman Coulter, Immunotest, cat. # IM0782U). To determine the content of CD34-positive cells after enrichment by immuno-magnetic (MACS) beads, staining was performed with a phycoerythrin (PE)-labelled CD34-specific mAb and a FITC-labelled CD45-specific mAb.
Flow cytometric measurement of CD33 and CD123 surface antigen densities
Initial analysis of patient samples was performed in the laboratories of the participating hospitals. The fractions of CD33- and for some samples of CD123-expressing leukocytes were determined by FACS analysis (FACS Navios, Beckman Coulter). Cell surface densities of CD33 and CD123 were measured by using a calibrated cytofluorimetric procedure as described.60,68,85 For this purpose, a kit of fluorescent beads with known numbers of fluorescent chromophores per bead (QIFIKIT®; Agilent Technologies, cat. # K0078) was used for calibration purposes. The procedure allows the investigator to express the measured fluorescence intensity of antibodies bound to a cell surface in terms of average number of antigen copies per cell.60,68,85 PE-labeled mAbs specific for CD33 and CD123, calibrated for QIFI kit measurements of antigen densities on MOLM-13 AML cells, were used as a substitute for the less economic complete QIFI kits. MOLM-13 cells were used as standards because surface densities of CD33 and CD123 on these cells remained sufficiently constant over time to make these cells useful for calibration purposes. To obtain absolute numbers for the calibration-batch of MOLM-13 cells, CD33 and CD123 densities were first measured with the QIFI kit, and then this batch of cells was used as a standard for secondary calibration for the next 20–30 generations of MOLM-13 cells. Antigen densities on patient- or donor-derived “test cells” were then measured by calibrating them against this standard batch of MOLM-13 cells.
PE-labeled CD33 and CD123 mAbs (CD33; Beckmann Coulter Immunotest cat # A07775 and CD123; ebioscience cat # 12–1239-42) were used to stain the standard batch of MOLM-13 cells and the test samples of patient- and donor-derived cells. Mean Fluorescence Intensities (MFI) were measured with an Accuri C6 flow cytometer (BD Biosciences). In the Accuri C6 instrument the laser- and optical alignments are pre-set and locked, and detector voltages are not adjustable, as opposed to other cytometers. This feature permitted reproducible measurements of Mean Fluorescence Intensity (MFI) values for CD33 and CD123 on MOLM-13 cells. MFI data recorded in this manner gave rise to numerically similar values expressed in MFI units as the number of antigen copies per cell (SABC values) measured with the QIFI kit. The close similarity of numerical values obtained with both methods allowed us to use the direct staining protocol as a surrogate for the QIFI kit for routine measurements of antigen copy numbers per cell (T. Braciak; unpublished data).
Re-directed lysis (RDL) assays using calcein release
Cytolysis assays based on the release of calcein from target cells prelabeled with calcein AM (ThermoFisher Scientific, cat. # C3100MP) were performed as described.60,68 In the final cytolytic reaction, no calf or human serum was present. The cytolytic activity of SPM-2 was measured in RDL reactions using NK cells expanded for 20 d in culture in the presence of IL-2 (LAK cells) as mediators of lysis with AML patient’s blasts as targets. The reactions with cells from all 29 patients, AML cell lines and controls (Figure 2; Suppl. Figure1) were performed with LAK cells from the same large standard batch of ex vivo expanded cells that were frozen in aliquots stored under liquid nitrogen. Similarly, all reactions were performed with SPM-2 from the same batch of protein, also frozen in aliquots, to assure that all experimental variables were kept constant and that the sole variable, for which we wanted to test, was the origin of the target cells. The reproducibility of the assay was assessed by using MOLM-13 cells from the same standard batch as calibration standards together with the standard batch of LAK cells. The maximum degree of specific lysis of the MOLM-13 cells varied by less than 10% in different experiments using separate frozen and thawed aliquots of the standard batch of LAK cells, and the EC50 values varied by less than a factor 2. This amounts to a high degree of reproducibility of the assay, given the fact, that the EC50 values were derived from plots, in which the dose of the agent was plotted on an exponential scale. Therefore, the differences seen for samples from different patients truly reflect the contribution of the cellular origin as the main variable. Control reactions using the Her2-specific triplebody Her2-16-Her2 as a negative control were run side by side in each experiment. The Her2-specific triplebody contained 2 scFv (antibody-derived single chain Fragment variable) binding domains for EGFR-2 and the same CD16-specific trigger binding domain as SPM-2. In addition, reactions with MOLM-13 cells as targets were included in each experiment as a positive control. Specific lysis was measured by quantitating the release of calcein from target cells using a fluorimeter/ELISA plate reader and expressed in Relative Light Units (RLU) at 485/535 nm. Calcein release was measured at the 4 hr time point. “Specific lysis” was defined as overall lysis minus the background of spontaneous lysis mediated by the NK cells alone, in the absence of added antibody-reagents, expressed as a fraction of net maximum release. “Specific lysis” was therefore computed by the formula:
% specific lysis = 100 x (Experimental RLU-Background RLU)/(Maximal RLU-Background RLU)
EC50 values and estimates of MABEL doses
EC50 values (concentration of triplebody protein producing 50% of maximum specific lysis) were calculated by using a nonlinear regression curve fit (variable slope) analysis using Graph Pad Prism Software (Graph Pad Software Inc.). MABEL (Minimum Anticipated Biologically Effective Levels) concentrations were deduced from (Figure 2), panels A-E, by visual inspection as the levels, for which a clear cytolytic effect beyond background was detected for more than 90% of the analyzed primary cell samples.
Funding Statement
This research was supported by academic research grants from the DFG (Deutsche Forschungsgemeinschaft; German Research Community, SFB 643, project C3), grant No. 2007.049.1 from the Wilhelm Sander Foundation, Neustadt, Germany; grant No. 109 063 from the Stiftung Deutsche Krebshilfe (German Cancer Foundation), and the Association Kaminkehrer helfen krebskranken Kindern (Chimney Sweeps support children with cancer) to GHF; by a grant from the Bavarian State (Ministry for Commerce and Economy) awarded in the M4 excellence competition to KPH, GHF and FO; by funding from the DFG CRC 1243 to KPH and MS; and by a student research fellowship awarded to CCR by the German José Carreras-Leukemia Foundation (www.carreras-stiftung.de, Scholarship No. DJCLS F 13/05). The collection of cell samples from AML patients maintained at the Department of Internal Medicine 5 of the University of Erlangen received financial support from the University’s Comprehensive Cancer Center (CCC).
Acknowledgments
Technical assistance of D Saul, S Nowecki, K Lämmermann and A Schele is gratefully acknowledged. We thank S Schneider, K Pfannes and K Förster-Poller for help with patient cells and patient data. We are grateful to F Nimmerjahn and J Seissler for permission to use their equipment. Our thanks go to D Wehr and K Heckel for administrative support; to C Stein, C Behrens, J Bruenke, J E Dick, GJ Schuurhuis, W Zeijlemaker and BM Peterlin for valuable scientific advice; to H Domdey, P Burgstaller and C Enke-Stolle from BioM for organizational support and to K Fischer for help with the presentation of data. We are grateful to the volunteer donors and patients, who provided cell samples for this study.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Dombret H, Gardin C.. An update of current treatments for adult acute myeloid leukemia. Blood. 2016;127(1):53–61. doi: 10.1182/blood-2015-08-604520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Döhner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Büchner T, Dombret H, Ebert BL, Fenaux P, Larson RA et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424–447. doi: 10.1182/blood-2016-08-733196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shlush LI, Mitchell A, Heisler L, Abelson S, Ng SWK, Trotman-Grant A, Medeiros JJF, Rao-Bhatia A, Jaciw-Zurakowsky I, Marke R et al. Tracing the origins of relapse in acute myeloid leukaemia to stem cells. Nature. 2017;547(7661):104–108. doi: 10.1038/nature22993. [DOI] [PubMed] [Google Scholar]
- 4.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3(7):730–737. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 5.Dick JE. Stem cell concepts renew cancer research. Blood. 2008;112(13):4793–4807. doi: 10.1182/blood-2008-08-077941. [DOI] [PubMed] [Google Scholar]
- 6.Thomas D, Majeti R. Biology and relevance of human acute myeloid leukemia stem cells. Blood. 2017. doi: 10.1182/blood-2016-10-696054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Horton SJ, Huntly BJP. Recent advances in acute myeloid leukemia stem cell biology. Haematologica. 2012;97(7):966–974. doi: 10.3324/haematol.2011.054734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ehninger A, Kramer M, Rollig C, Thiede C, Bornhauser M, Von Bonin M, Wermke M, Feldmann A, Bachmann M, Ehninger G et al. Distribution and levels of cell surface expression of CD33 and CD123 in acute myeloid leukemia. Blood Cancer J. 2014;4:e218. doi: 10.1038/bcj.2014.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krupka C, Kufer P, Kischel R, Zugmaier G, Bögeholz J, Köhnke T, Lichtenegger FS, Schneider S, Metzeler KH, Fiegl M et al. CD33 target validation and sustained depletion of AML blasts in long-term cultures by the bispecific T-cell–engaging antibody AMG 330. Blood. 2014;123(3):356–365. doi: 10.1182/blood-2013-08-523548. [DOI] [PubMed] [Google Scholar]
- 10.Walter RB, Appelbaum FR, Estey EH, Bernstein ID. Acute myeloid leukemia stem cells and CD33-targeted immunotherapy. Blood. 2012;119(26):6198–6208. doi: 10.1182/blood-2011-11-325050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hauswirth AW, Florian S, Printz D, Sotlar K, Krauth MT, Fritsch G, Schernthaner GH, Wacheck V, Selzer E, Sperr WR et al. Expression of the target receptor CD33 in CD34+/CD38−/CD123+ AML stem cells. Eur J Clin Invest. 2007;37(1):73–82. doi: 10.1111/j.1365-2362.2007.01746.x. [DOI] [PubMed] [Google Scholar]
- 12.Taussig DC, Pearce DJ, Simpson C, Rohatiner AZ, Lister TA, Kelly G, Luongo JL, G-aH D-D, Bonnet D. Hematopoietic stem cells express multiple myeloid markers: implications for the origin and targeted therapy of acute myeloid leukemia. Blood. 2005;106(13):4086–4092. doi: 10.1182/blood-2005-03-1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Laszlo GS, Estey EH, Walter RB. The past and future of CD33 as therapeutic target in acute myeloid leukemia. Blood Rev. 2014;28(4):143–153. doi: 10.1016/j.blre.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 14.Sievers EL, Larson RA, Stadtmauer EA, Estey E, Löwenberg B, Dombret H, Karanes C, Theobald M, Bennett JM, Sherman ML et al. Efficacy and safety of gemtuzumab ozogamicin in patients with CD33-positive acute myeloid leukemia in first relapse. J Clin Oncol. 2001;19(13):3244–3254. doi: 10.1200/jco.2001.19.13.3244. [DOI] [PubMed] [Google Scholar]
- 15.Rowe JM, Löwenberg B. Gemtuzumab ozogamicin in acute myeloid leukemia: a remarkable saga about an active drug. Blood. 2013;121(24):4838–4841. doi: 10.1182/blood-2013-03-490482. [DOI] [PubMed] [Google Scholar]
- 16.Castaigne S, Pautas C, Terré C, Raffoux E, Bordessoule D, Bastie JN, Legrand O, Thomas X, Turlure P, Oumedaly R et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): a randomised, open-label, phase 3 study. Lancet. 2012;379(9825):1508–1516. doi: 10.1016/S0140-6736(12)60485-1. [DOI] [PubMed] [Google Scholar]
- 17.O’Hear C, Inaba H, Pounds S, Shi L, Dahl G, Bowman WP, Taub JW, Pui C-H, Ribeiro RC, Coustan-Smith E et al. Gemtuzumab ozogamicin can reduce minimal residual disease in patients with childhood acute myeloid leukemia. Cancer. 2013;119(22):4036–4043. doi: 10.1002/cncr.28334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kung Sutherland MS, Walter RB, Jeffrey SC, Burke PJ, Yu C, Kostner H, Stone I, Ryan MC, Sussman D, Lyon RP et al. SGN-CD33A: a novel CD33-targeting antibody–drug conjugate using a pyrrolobenzodiazepine dimer is active in models of drug-resistant AML. Blood. 2013;122(8):1455–1463. doi: 10.1182/blood-2013-03-491506. [DOI] [PubMed] [Google Scholar]
- 19.Vadustuxima-b-Talirine (SGN-CD33A) is under clinical investigation: ClinicalTrials.gov identifiers NCT01902329; NCT02785900; NCT02706899 and 2 others All trials have been halted in 2017 due to toxicities; https://www.businesswire.com/news/home/20170619005466/en/Seattle-GeneticsDiscontinues-Phase-3-CASCADE-Trial
- 20.Aigner M, Feulner J, Schaffer S, Kischel R, Kufer P, Schneider K, Henn A, Rattel B, Friedrich M, Baeuerle PA et al. T lymphocytes can be effectively recruited for ex vivo and in vivo lysis of AML blasts by a novel CD33/CD3-bispecific BiTE antibody construct. Leukemia. 2013;27(5):1107–1115. doi: 10.1038/leu.2012.341. [DOI] [PubMed] [Google Scholar]
- 21.Krupka C, Kufer P, Kischel R, Zugmaier G, Lichtenegger FS, Kohnke T, Vick B, Jeremias I, Metzeler KH, Altmann T et al. Blockade of the PD-1/PD-L1 axis augments lysis of AML cells by the CD33/CD3 BiTE antibody construct AMG 330: reversing a T-cell-induced immune escape mechanism. Leukemia. 2016;30(2):484–491. doi: 10.1038/leu.2015.214. [DOI] [PubMed] [Google Scholar]
- 22.Friedrich M, Henn A, Raum T, Bajtus M, Matthes K, Hendrich L, Wahl J, Hoffmann P, Kischel R, Kvesic M et al. Preclinical characterization of AMG 330, a CD3/CD33-bispecific T-cell–engaging antibody with potential for treatment of acute myelogenous leukemia. Mol Cancer Ther. 2014;13(6):1549–1557. doi: 10.1158/1535-7163.mct-13-0956. [DOI] [PubMed] [Google Scholar]
- 23.AMG 330 is under clinical investigation : ClinicalTrials.gov identifier NCT02520427.
- 24.Arndt C, Von Bonin M, Cartellieri M, Feldmann A, Koristka S, Michalk I, Stamova S, Bornhauser M, Schmitz M, Ehninger G et al. Redirection of T cells with a first fully humanized bispecific CD33-CD3 antibody efficiently eliminates AML blasts without harming hematopoietic stem cells. Leukemia. 2013;27(4):964–967. doi: 10.1038/leu.2013.18. [DOI] [PubMed] [Google Scholar]
- 25.AMV-564, a tetravalent bispecific tandab with specificities for CD3 and CD33, is under clinical investigation: clinicalTrials.gov identifier NCT03144245. [Google Scholar]
- 26.Marin V, Pizzitola I, Agostoni V, Attianese GMPG, Finney H, Lawson A, Pule M, Rousseau R, Biondi A, Biagi E. Cytokine-induced killer cells for cell therapy of acute myeloid leukemia: improvement of their immune activity by expression of CD33-specific chimeric receptors. Haematologica. 2010;95(12):2144–2152. doi: 10.3324/haematol.2010.026310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pizzitola I, Anjos-Afonso F, Rouault-Pierre K, Lassailly F, Tettamanti S, Spinelli O, Biondi A, Biagi E, Bonnet D. Chimeric antigen receptors against CD33/CD123 antigens efficiently target primary acute myeloid leukemia cells in vivo. Leukemia. 2014;28(8):1596–1605. doi: 10.1038/leu.2014.62. [DOI] [PubMed] [Google Scholar]
- 28.O’Hear C, Heiber JF, Schubert I, Fey G, Geiger TL. Anti-CD33 chimeric antigen receptor targeting of acute myeloid leukemia. Haematologica. 2015;100(3):336–344. doi: 10.3324/haematol.2014.112748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kenderian SS, Ruella M, Shestova O, Klichinsky M, Aikawa V, Morrissette JJD, Scholler J, Song D, Porter DL, Carroll M et al. CD33-specific chimeric antigen receptor T cells exhibit potent preclinical activity against human acute myeloid leukemia. Leukemia. 2015;29(8):1637–1647. doi: 10.1038/leu.2015.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Q-S, Wang Y, Lv H-Y, Han Q-W, Fan H, Guo B, Wang LL, Han W-D. Treatment of CD33-directed chimeric antigen receptor-modified T cells in one patient with relapsed and refractory acute myeloid leukemia. Mol Ther. 2015;23(1):184–191. doi: 10.1038/mt.2014.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.CD33-specific CAR-T cells are under clinical investigation at the PLA general hospital, Beijing, China: clinicalTrials.gov identifier NCT01864902 and at the MD Anderson Cancer Center, Houston, Texas: clinicalTrials.gov identifier NCT03126864. [Google Scholar]
- 32.Van Rhenen A, Gams VD, Kelder A, Rombouts EJ, Feller N, Moshaver B, Walsum MS-V, Zweegman S, Ossenkoppele G, Jan SG. The novel AML stem cell–associated antigen CLL-1 aids in discrimination between normal and leukemic stem cells. Blood. 2007;110(7):2659–2666. doi: 10.1182/blood-2007-03-083048. [DOI] [PubMed] [Google Scholar]
- 33.Kikushige Y, Shima T, Takayanagi S-I, Urata S, Miyamoto T, Iwasaki H, Takenaka K, Teshima T, Tanaka T, Inagaki Y et al. TIM-3 Is a promising target to selectively kill acute myeloid leukemia stem cells. Cell Stem Cell. 2010;7(6):708–717. doi: 10.1016/j.stem.2010.11.014. [DOI] [PubMed] [Google Scholar]
- 34.Jan M, Chao MP, Cha AC, Alizadeh AA, Gentles AJ, Weissman IL, Majeti R. Prospective separation of normal and leukemic stem cells based on differential expression of TIM3, a human acute myeloid leukemia stem cell marker. Proc Natl Acad Sci. 2011;108(12):5009–5014. doi: 10.1073/pnas.1100551108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hosen N, Park CY, Tatsumi N, Oji Y, Sugiyama H, Gramatzki M, Krensky AM, Weissman IL. CD96 is a leukemic stem cell-specific marker in human acute myeloid leukemia. Proc Natl Acad Sci. 2007;104(26):11008–11013. doi: 10.1073/pnas.0704271104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med. 2006;12(10):1167–1174. http://www.nature.com/nm/journal/v12/n10/suppinfo/nm1483_S1.html. doi: 10.1038/nm1483. [DOI] [PubMed] [Google Scholar]
- 37.Goardon N, Marchi E, Atzberger A, Quek L, Schuh A, Soneji S, Woll P, Mead A, Alford KA, Rout R et al. Coexistence of LMPP-like and GMP-like leukemia stem cells in acute myeloid leukemia. Cancer Cell. 2011;19(1):138–152. doi: 10.1016/j.ccr.2010.12.012. [DOI] [PubMed] [Google Scholar]
- 38.Saito Y, Kitamura H, Hijikata A, Tomizawa-Murasawa M, Tanaka S, Takagi S, Uchida N, Suzuki N, Sone A, Najima Y et al. Identification of Therapeutic Targets for Quiescent, Chemotherapy-Resistant Human Leukemia Stem Cells. Sci Transl Med. 2010;2(17):17ra19–17ra19. doi: 10.1126/scitranslmed.3000349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krupka C, Lichtenegger FS, Kohnke T, Bogeholz J, Bucklein V, Roiss M, Altmann T, Do TU, Dusek R, Wilson K et al. Targeting CD157 in AML using a novel, Fc-engineered antibody construct. Oncotarget. 2017;8(22):35707–35717. The CD157 antibody MEN1112 is currently investigated in a clinical study: ClinicalTrials.gov identifier NCT02353143. doi: 10.18632/oncotarget.16060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jordan CT, Upchurch D, Szilvassy SJ, Guzman ML, Howard DS, Pettigrew AL, Meyerrose T, Rossi R, Grimes B, Rizzieri DA et al. The interleukin-3 receptor alpha chain is a unique marker for human acute myelogenous leukemia stem cells. Leukemia. 2000;14(10):1777–1784. doi: 10.1038/sj.leu.2401903. [DOI] [PubMed] [Google Scholar]
- 41.Hwang K, Park C-J, Jang S, Chi H-S, Kim D-Y, Lee J-H, Lee JH, Lee KH, Im H-J, Seo -J-J. Flow cytometric quantification and immunophenotyping of leukemic stem cells in acute myeloid leukemia. Ann Hematol. 2012;91(10):1541–1546. doi: 10.1007/s00277-012-1501-7. [DOI] [PubMed] [Google Scholar]
- 42.Munoz L, Nomdedeu J, Lopez O, Carnicer M, Bellido M, Aventin A, Brunet S, Sierra J. Interleukin-3 receptor alpha chain (CD123) is widely expressed in hematologic malignancies. Haematologica. 2001;86(12):1261–1269. [PubMed] [Google Scholar]
- 43.Testa U, Riccioni R, Militi S, Coccia E, Stellacci E, Samoggia P, Latagliata R, Mariani G, Rossini A, Battistini A et al. Elevated expression of IL-3Rα in acute myelogenous leukemia is associated with enhanced blast proliferation, increased cellularity, and poor prognosis. Blood. 2002;100(8):2980–2988. doi: 10.1182/blood-2002-03-0852. [DOI] [PubMed] [Google Scholar]
- 44.Testa U, Pelosi E, Frankel A. CD 123 is a membrane biomarker and a therapeutic target in hematologic malignancies. Biomarker Res. 2014;2(1):4. doi: 10.1186/2050-7771-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Florian S, Sonneck K, Hauswirth AW, Krauth M-T, Schernthaner G-H, Sperr WR, Valent P. Detection of molecular targets on the surface of CD34+/CD38− stem cells in various myeloid malignancies. Leuk Lymphoma. 2006;47(2):207–222. doi: 10.1080/10428190500272507. [DOI] [PubMed] [Google Scholar]
- 46.Zeijlemaker W, Kelder A, Oussoren-Brockhoff YJ, Scholten WJ, Snel AN, Veldhuizen D, Cloos J, Ossenkoppele GJ, Schuurhuis GJ. A simple one-tube assay for immunophenotypical quantification of leukemic stem cells in acute myeloid leukemia. Leukemia. 2016;30(2):439–446. doi: 10.1038/leu.2015.252. [DOI] [PubMed] [Google Scholar]
- 47.Jin L, Lee EM, Ramshaw HS, Busfield SJ, Peoppl AG, Wilkinson L, Guthridge MA, Thomas D, Barry EF, Boyd A et al. Monoclonal antibody-mediated targeting of CD123, IL-3 receptor α chain, eliminates human acute myeloid leukemic stem cells. Cell Stem Cell. 2009;5(1):31–42. doi: 10.1016/j.stem.2009.04.018. [DOI] [PubMed] [Google Scholar]
- 48.Busfield SJ, Biondo M, Wong M, Ramshaw HS, Lee EM, Ghosh S, Braley H, Panousis C, Roberts AW, He SZ et al. Targeting of acute myeloid leukemia in vitro and in vivo with an anti-CD123 mAb engineered for optimal ADCC. Leukemia. 2014;28(11):2213–2221. doi: 10.1038/leu.2014.128. [DOI] [PubMed] [Google Scholar]
- 49.CSL362 (Talacotuzumab) is under clinical investigation: clinicalTrials.gov identifiers NCT01632852, NCT02472145, and NCT02992860. [Google Scholar]
- 50.Leyton JV, Hu M, Gao C, Turner PV, Dick JE, Minden M, Reilly RM. Auger electron radioimmunotherapeutic agent specific for the CD123+/CD131− phenotype of the leukemia stem cell population. J Nucl Med. 2011;52(9):1465–1473. doi: 10.2967/jnumed.111.087668. [DOI] [PubMed] [Google Scholar]
- 51.Frankel AE, Woo JH, Ahn C, Pemmaraju N, Medeiros BC, Carraway HE, Frankfurt O, Forman SJ, Yang XA, Konopleva M et al. Activity of SL-401, a targeted therapy directed to the interleukin-3 receptor, in patients with blastic plasmacytoid dendritic cell neoplasm. Blood. 2014. doi: 10.1182/blood-2014-04-566737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Du X, Ho M, Pastan I. New immunotoxins targeting CD123, a stem cell antigen on acute myeloid leukemia cells. J Immunotherapy. 2007;30(6):607–613. doi: 10.1097/CJI.0b013e318053ed8e. [DOI] [PubMed] [Google Scholar]
- 53.Stein C, Kellner C, Kügler M, Reiff N, Mentz K, Schwenkert M, Stockmeyer B, Mackensen A, Fey GH. Novel conjugates of single-chain Fv antibody fragments specific for stem cell antigen CD123 mediate potent death of acute myeloid leukaemia cells. Br J Haematol. 2010;148(6):879–889. doi: 10.1111/j.1365-2141.2009.08033.x. [DOI] [PubMed] [Google Scholar]
- 54.The antibody-drug conjugate SGN CD123A is under clinical investigation: clinicalTrials.gov identifier NCT02848248. [Google Scholar]
- 55.Kuo S-R, Wong L, Liu J-S. Engineering a CD123xCD3 bispecific scFv immunofusion for the treatment of leukemia and elimination of leukemia stem cells. Protein Engg Design Selection. 2012;25(10):561–570. doi: 10.1093/protein/gzs040. [DOI] [PubMed] [Google Scholar]
- 56.Chichili GR, Huang L, Li H, Burke S, He L, Tang Q, Jin L, Gorlatov S, Ciccarone V, Chen F et al. A CD3xCD123 bispecific DART for redirecting host T cells to myelogenous leukemia: preclinical activity and safety in nonhuman primates. Sci Transl Med. 2015;7(289):289ra282–289ra282. doi: 10.1126/scitranslmed.aaa5693. [DOI] [PubMed] [Google Scholar]
- 57.Al-Hussaini M, Rettig MP, Ritchey JK, Karpova D, Uy GL, Eissenberg LG, Gao F, Eades WC, Bonvini E, Chichili GR et al. Targeting CD123 in acute myeloid leukemia using a T-cell–directed dual-affinity retargeting platform. Blood. 2016;127(1):122–131. doi: 10.1182/blood-2014-05-575704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.MGD 006 is under clinical investigation: clinicalTrials.gov identifier NCT02152956. [Google Scholar]
- 59.Kügler M, Stein C, Kellner C, Mentz K, Saul D, Schwenkert M, Schubert I, Singer H, Oduncu F, Stockmeyer B et al. A recombinant trispecific single-chain Fv derivative directed against CD123 and CD33 mediates effective elimination of acute myeloid leukaemia cells by dual targeting. Br J Haematol. 2010;150(5):574–586. doi: 10.1111/j.1365-2141.2010.08300.x. [DOI] [PubMed] [Google Scholar]
- 60.Braciak TA, Wildenhain S, Roskopf CC, Schubert IA, Fey GH, Jacob U, Hopfner K-P, Oduncu FS. NK cells from an AML patient have recovered in remission and reached comparable cytolytic activity to that of a healthy monozygotic twin mediated by the single-chain triplebody SPM-2. J Transl Med. 2013;11(1):289. doi: 10.1186/1479-5876-11-289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tettamanti S, Marin V, Pizzitola I, Magnani CF, Giordano Attianese GMP, Cribioli E, Maltese F, Galimberti S, Af L, Biondi A et al. Targeting of acute myeloid leukaemia by cytokine-induced killer cells redirected with a novel CD123-specific chimeric antigen receptor. Br J Haematol. 2013;161(3):389–401. doi: 10.1111/bjh.12282. [DOI] [PubMed] [Google Scholar]
- 62.Mardiros A, Dos Santos C, McDonald T, Brown CE, Wang X, Budde LE, Hoffman L, Aguilar B, Chang W-C, Bretzlaff W et al. T cells expressing CD123-specific chimeric antigen receptors exhibit specific cytolytic effector functions and antitumor effects against human acute myeloid leukemia. Blood. 2013;122(18):3138–3148. doi: 10.1182/blood-2012-12-474056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gill S, Tasian SK, Ruella M, Shestova O, Li Y, Porter DL, Carroll M, Danet-Desnoyers G, Scholler J, Grupp SA et al. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor–modified T cells. Blood. 2014;123(15):2343–2354. doi: 10.1182/blood-2013-09-529537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.CD123-specific CAR-T cells are under clinical investigation at the affiliated hospital to the academy of military medical sciences, Beijing, China: clinicalTrials.gov identifier NCT03114670; at the university of pennsylvania medical center: identifier NCT02623582; at the city of hope medical center: identifier NCT02159495; and at the weill cornell medical college, NY: identifier NCT03190278. [Google Scholar]
- 65.Brizzi MF, Garbarino G, Rossi PR, Pagliardi GL, Arduino C, Avanzi GC, Pegoraro L. Interleukin 3 stimulates proliferation and triggers endothelial-leukocyte adhesion molecule 1 gene activation of human endothelial cells. J Clin Invest. 1993;91(6):2887–2892. doi: 10.1172/JCI116534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Manz MG, Miyamoto T, Akashi K, Weissman IL. Prospective isolation of human clonogenic common myeloid progenitors. Proc Natl Acad Sci. 2002;99(18):11872–11877. doi: 10.1073/pnas.172384399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rezvani AR, Maloney DG. Rituximab resistance. Best Pract Res Clin Haematol. 2011;24(2):203–216. doi: 10.1016/j.beha.2011.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schubert I, Saul D, Nowecki S, Mackensen A, Fey GH, Oduncu FS. A dual-targeting triplebody mediates preferential redirected lysis of antigen double-positive over single-positive leukemic cells. mAbs. 2014;6(1):286–296. doi: 10.4161/mabs.26768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Roskopf C, Braciak TA, Fenn NC, Kobold S, Fey GH, Hopfner KP, Oduncu FS. Dual- targeting triplebody 33- 3-19mediates selective lysis of biphenotypic CD19+ CD33+ leukemia cells. Oncotarget. 2016;7(16):22579–22589. doi: 10.18632/oncotarget.8022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schubert I, Kellner C, Stein C, Kügler M, Schwenkert M, Saul D, Stockmeyer B, Berens C, Oduncu FS, Mackensen A et al. A recombinant triplebody with specificity for CD19 and HLA-DR mediates preferential binding to antigen double- positive cells by dual-targeting. MAbs. 2012;4(1):45–56. doi: 10.4161/mabs.4.1.18498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Alici E, Sutlu T, Björkstrand B, Gilljam M, Stellan B, Nahi H, Quezada HC, Gahrton G, Ljunggren H-G, Dilber MS. Autologous antitumor activity by NK cells expanded from myeloma patients using GMP-compliant components. Blood. 2008;111(6):3155–3162. doi: 10.1182/blood-2007-09-110312. [DOI] [PubMed] [Google Scholar]
- 72.Pollard JA, Loken M, Gerbing RB, Raimondi SC, Hirsch BA, Aplenc R, Bernstein ID, Gamis AS, Alonzo TA, Meshinchi S. CD33 Expression and its association with gemtuzumab ozogamicin response: results from the randomized phase III children’s oncology group trial AAML0531. J Clin Oncol. 2016;34(7):747–755. doi: 10.1200/jco.2015.62.6846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vergez F, Green AS, Tamburini J, Sarry JE, Gaillard B, Cornillet-Lefebvre P, Pannetier M, Neyret A, Chapuis N, Ifrah N et al. High levels of CD34+CD38low/-CD123+ blasts are predictive of an adverse outcome in acute myeloid leukemia: a groupe ouest-est des leucemies aigues et maladies du Sang (GOELAMS) study. Haematologica. 2011;96(12):1792–1798. doi: 10.3324/haematol.2011.047894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Denman CJ, Senyukov VV, Somanchi SS, Phatarpekar PV, Kopp LM, Johnson JL, Singh H, Hurton L, Maiti SN, Huls MH et al. Membrane-bound IL-21 promotes sustained Ex Vivo proliferation of human natural killer cells. PloS One. 2012;7(1):e30264. doi: 10.1371/journal.pone.0030264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Arriga R, Caratelli S, Coppola A, Spagnoli GC, Venditti A, Amadori S, Lanzilli G, Lauro D, Palomba P, Sconocchia T et al. Enhancement of anti-leukemia activity of NK cells in vitro and in vivo by inhibition of leukemia cell-induced NK cell damage. Oncotarget. 2016;7(2):2070–2079. doi: 10.18632/oncotarget.6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Basher F, Jeng EK, Wong H, Wu J. Cooperative therapeutic anti-tumor effect of IL-15 agonist ALT-803 and co-targeting soluble NKG2D ligand sMIC. Oncotarget. 2016;7(1):814–830. doi: 10.18632/oncotarget.6416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Romee R, Rosario M, Berrien-Elliott MM, Wagner JA, Jewell BA, Schappe T, Leong JW, Abdel-Latif S, Schneider SE, Willey S et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Transl Med. 2016;8(357):357ra123–357ra123. Cytokine-induced memory-like NK cells are under clinical investigation: ClinicalTrials.gov identifier NCT0189793. doi: 10.1126/scitranslmed.aaf2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Reiter Y, Brinkmann U, Lee B, Pastan I. Engineering antibody Fv fragments for cancer detection and therapy: bisulfide-stabilized Fv fragments. Nat Biotech. 1996;14(10):1239–1245. doi: 10.1038/nbt1096-1239. [DOI] [PubMed] [Google Scholar]
- 79.Luo D, Mah N, Krantz M, Wilde K, Wishart D, Zhang Y, Jacobs F, Martin L. Vl-Linker-Vh orientation-dependent expression of single chain fv containing an engineered disulfide-stabilized bond in the framework regions. J Biochemistry. 1995;118(4):825–831. doi: 10.1093/oxfordjournals.jbchem.a124986. [DOI] [PubMed] [Google Scholar]
- 80.Bruenke J, Barbin K, Kunert S, Lang P, Pfeiffer M, Stieglmaier K, Niethammer D, Stockmeyer B, Peipp M, Repp R et al. Effective lysis of lymphoma cells with a stabilised bispecific single-chain Fv antibody against CD19 and FcγRIII (CD16). Br J Haematol. 2005;130(2):218–228. doi: 10.1111/j.1365-2141.2005.05414.x. [DOI] [PubMed] [Google Scholar]
- 81.Kettleborough CA, Saldanha J, Heath VJ, Morrison CJ, Bendig MM. Humanization of a mouse monoclonal antibody by CDR-grafting: the importance of framework residues on loop conformation. Protein Eng. 1991;4(7):773–783. doi: 10.1093/protein/4.7.773. [DOI] [PubMed] [Google Scholar]
- 82.Carter P, Presta L, Gorman CM, Ridgway JB, Henner D, Wong WL, Rowland AM, Kotts C, Carver ME, Shepard HM. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc Natl Acad Sci U S A. 1992;89(10):4285–4289. doi: 10.1073/pnas.89.10.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rothdiener M, Müller D, Castro PG, Scholz A, Schwemmlein M, Fey G, Heidenreich O, Kontermann RE. Targeted delivery of SiRNA to CD33-positive tumor cells with liposomal carrier systems. J of Controlled Release. 2010;144(2):251–258. 10.1016/j.jconrel.2010.02.020. [DOI] [PubMed] [Google Scholar]
- 84.Ewert S, Honegger A, Plückthun A. Stability improvement of antibodies for extracellular and intracellular applications: CDR grafting to stable frameworks and structure-based framework engineering. Methods. 2004;34(2):184–199. 10.1016/j.ymeth.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 85.Olejniczak SH, Stewart CC, Donohue K, Czuczman MS, Quantitative A. Exploration of surface antigen expression in common B-cell malignancies using flow cytometry. Immunol Invest. 2006;35(1):93–114. doi: 10.1080/08820130500496878. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.