

Abstract
Neutrophils are involved in the pathogenesis of allergy. However, the contribution of the different functionally polarized neutrophils in allergy needs to be clarified. We sought to define the characteristics of interleukin (IL)-33-induced neutrophils and the involvement of this subset of polarized neutrophils in allergic pathogenesis. Freshly isolated neutrophils were treated with different cytokines and the cytokine expression levels were detected by real-time PCR. The gene expression profile of IL-33-induced neutrophils was determined by microarray assay. Adoptive transfer assay was used to investigate the function of IL-33-induced neutrophils in an ovalbumin (OVA)-induced allergic asthma model. IL-33-treated neutrophils selectively produced IL-4, IL-5, IL-9 and IL-13 (referred as to N(IL-33) cells) and displayed a distinctive gene expression profile in sharp contrast to resting and lipopolysaccharide (LPS)-treated neutrophils. IL-33-induced neutrophils expressed high Levels of IL-1R2 on cell surface, whereas resting and LPS-treated neutrophils did not, indicating IL-1R2 might be used as a biomarker for N(IL-33) cells. Importantly, N(IL-33) neutrophils exist in the lungs of OVA-induced allergic asthma mice. Adoptive transfer of N(IL-33) neutrophils significantly promotes the severity of the lung pathogenesis in this model. IL-33 induces neutrophil polarization through c-Jun N-terminal kinase- and nuclear factor-κB-dependent pathways. A previously unappreciated neutrophil polarization driven by IL-33 with unique cell surface markers and cytokine/chemokine-producing gene profile was defined. The newly identified N(IL-33) subpopulation may have significant contribution to IL-33-related pathogenesis.
Keywords: allergy, asthma, IL-9, IL-33, inflammation, neutrophils, polarization
Introduction
Neutrophils (also called polymorphonuclear neutrophil granulocytes) are the most abundant white blood cells in circulation and one of the earliest immune cells recruited to the sites of infection and inflammatory response. Neutrophils that are fully equipped with a variety of granules are essential to host defense against intruding microorganisms and have a critical role in initiating inflammation and innate immunity.1 However, it is highly recognized now that neutrophils also significantly contribute to the various acute and chronic pathological damages in infections, autoimmune diseases and graft rejections in positive or negative ways.2,3,4,5 In fact, beyond cytotoxicity against invading pathogens, neutrophils can switch phenotypes and exert immune-regulatory functions on both innate and adaptive immunity, including dendritic cells, macrophages, natural killer cells, T cells and B cells.6,7 Neutrophils from rheumatoid arthritis patients upregulate major histocompatibility complex class II expression and increase their antigen-presentation ability, leading to T lymphocyte activation.8 Recent studies indicate that neutrophils exhibit considerable plasticity and polarization. They have highly variable transcriptome profiles depending on their tissue location and microenvironment.9 Immunoregulatory agent everolimus can promote the transition of neutrophil from proinflammatory activation to anti-inflammatory model.10 Akin to macrophage activation pathways, neutrophils have distinct subsets, as presented in infections and tumors.11,12,13 A subpopulation of neutrophils present in the spleen produces BAFF, APRIL, CD40L and interleukin-21 (IL-21), activates marginal-zone B cells and promotes immunoglobulin class switching, somatic hypermutation and antibody production.14 Tumor-associated neutrophils were proposed to be polarized between antitumorigenic and protumorigenic neutrophil phenotypes.15 Our recent results also reveal that IL-23-activated neutrophils expresses a unique gene expression profile and participate in the pathological process of colitis.16 The extracellular inducing factors, the intracellualr molecular regulatory networks, the phenotype characteristics and the biological significance of the polarized neutrophils urgently need to be clarified.
IL-33, a member of the IL-1 family, is not only localized in the cell nucleus but also functions as a cell-free cytokine.17 Administration of IL-33 into mice caused massive infiltrations of tissues by eosinophils, epithelial goblet cell hyperplasia and elevations of typical type 2 cytokines such as IL-5, IL-9 and IL-13, contributing to allergy and fibrosis.18,19,20 These responses also occurred in T- and B-cell-deficient Rag knockout recipient mice, indicating that innate cells, including innate lymphoid cells (ILCs), were the important target of IL-33.19,21 Neutrophils can act as target cells for IL-33 in the lungs. IL-33 exacerbates allergic bronchoconstriction in the mice via activation of mast cells.22 There are a large number of neutrophils marginated in lung capillaries. They are key regulatory cells in the immunopathogenesis of asthma.23,24 In the present study, we demonstrate that IL-33-treated neutrophils selectively produced IL-4, IL-5, IL-9 and IL-13 (referred as to N(IL-33) cells) and displayed a distinct gene expression profile in sharp contrast to resting and lipopolysaccharide (LPS)-treated neutrophils (N(LPS)). Importantly, N(IL-33) neutrophils existed in the lung tissue of ovalbumin (OVA)-induced asthma mice. Adoptive transfer of N(IL-33) neutrophils significantly promoted the severity of the lung pathogenesis in this model. IL-33 induced N(IL-33) neutrophil polarization through c-Jun N-terminal kinase (JNK)- and nuclear factor (NF)-κB-dependent pathways. Thus our findings reveal a previously unappreciated neutrophil polarization driven by IL-33 with unique cell surface markers and cytokine/chemokine-producing gene profile. The newly identified N(IL-33) subpopulation may have significant contribution to IL-33-related pathogenesis, such as asthma.
Materials and methods
Mice
Six-to-eight-week old male C57BL/6 (B6) mice were purchased from Beijing University Experimental Animal Center (Beijing, China). IL-33 transgenic mice (C57BL/6J-TgN(CMV-IL33)ZLFILAS, IL-33-TG) were kindly offered by the Key Laboratory of Human Diseases Comparative Medicine, Ministry of Health; Institute of Laboratory Animal Science, CAMS and PUMC.25 All the mice were maintained and bred in specific pathogen-free conditions. All experimental manipulations were undertaken in accordance with the Institutional Guidelines for the Care and Use of Laboratory Animals, Institute of Zoology (Beijing, China).
Reagents
The following cytokines were purchased from PeproTech (Rocky Hill, CT, USA): recombinant mouse IL-1β, IL-2, IL-6, IL-10, IL-12, IL-13, IL-17A, interferon (IFN)-γ and tumor necrosis factor (TNF)-α. The following cytokines were purchased from R&D Systems (Minneapolis, MN, USA): IL-4, IL-21, IL-23, IL-33, transforming growth factor (TGF)-β1 and granulocyte macrophages colony-stimulating factor (GM-CSF). LPS was offered by Sigma-Aldrich (St Louis, MO, USA). G-CSF was bought from Biovision (Milpitas, CA, USA). The concentrations of the recombinant mouse cytokines were used based on our previous studies.26 Except for experiments with treatment of different concentrations of IL-33, IL-33 was used at 100 ng/ml in other experiments.
Phycoerythrin (PE)-cy5-conjugated anti-CD11b (M1/70) and PE-conjugated anti-F4/80 (BM8) were purchased from eBioscience (San Diego, CA, USA). Fluorescein isothiocyanate-conjugated anti-Ly6G (1A8), PE-conjugated anti-IL-1R2 (4E2), PE-conjugated anti-mSiglec F (E50–2440) were purchased from BD Biosciences Pharmingen (San Diego, CA, USA). PE-conjugated anti-mIL-9 (RM9A4) was purchased from Biolegend (San Diego, CA, USA). IL-4 (431104), IL-9 (434804) and IL-5 (431204) Enzyme-linked immunosorbent assay (ELISA) kits were purchased from Biolegend. TNF-α (430904), IL-1β (BMS6002), IL-13 (88–7137), C-C motif chemokine ligand 2 (CCL2; 88–7391) and CCL7 (BMS6006INST) ELISA kits were from eBioscience.
Selective signal transducer and activator of transcription factor 3 (STAT3) inhibitor (NSC74859; 4655/10) was purchased from R&D Systems. P38 inhibitor (SB203580; 559389-1MG) was from The Merck Group (Darmstadt, Germany). STAT1 inhibitor (MTA) and extracellular signal–regulated kinase (ERK) inhibitor (PD98059, 9900) were from Cell Signaling Technology (Danvers, MA, USA). JNK inhibitor (SP600125, S5567) was purchased from Sigma-Aldrich. NF-κB activator inhibitor (481406) was from Calbicohem (Temecula, CA, USA). The OVA and Alum were purchased from Sigma-Aldrich. The primary antibodies against p-JNK (Thr183/Tyr185), JNK, p-p38 (Thr180/Tyr182), p38, p-STAT1 (Ser727), STAT1, p-ERK (Thr202/Tyr204), ERK, p-p65 (Ser536), p65 and H3 were purchased from Cell Signaling Technology. Anti-β-Actin monoclonal antbody (mAb) was purchased from Sigma-Aldrich. The concentrations of these reagents are used as detected in our previous studies.27,28
Isolation and differentiation of neutrophils in vitro
Primary neutrophils (CD11b+Ly6G+F4/80−, referred as to N0 cells) were isolated from bone marrow cells with 52% percoll (GE Healthcare Life Sciences, Piscataway, NJ, USA, 17-0891-01) and sorted by MoFlo XDP (Beckman Coulter, Brea, CA, USA).29 Unless specifically noted in the figure legend, primary neutrophils (2 × 106 cells/ml in 48-well plates) were differentiated under N(LPS) condition (LPS, 1 μg/ml) for 6 h or N(IL-33) condition (IL-33, 100 ng/ml) for 24 h in Dulbecco’s modified Eagle’s medium with 10% (v/v) fetal bovine serum at 37 °C and 5% CO2.
Microarray analysis
Microarray procedure was performed according to the manufacturer’s instructions and previous reports.16 R (version 3.0.2) was used to draw the heatmap of lineage-restricted genes. The fold changes were calculated as the ratio of the expression level in each cell type versus the second highest expression level in all cell types. Differentially expressed genes were defined as genes with at least twofold variance of expression levels in N(LPS) and N(IL-33) neutrophils compared with N0 neutrophils.
Quantitative PCR analysis
Total RNA was isolated with TRIzol (Invitrogen, Carlsbad, CA, USA) and reverse transcription was performed with M-MLV superscript reverse transcriptase as described previously. Real-time PCR was performed using multiple kits (SYBR Premix Ex TaqTM, DRR041A, Takara Bio, Kusatsu, Japan) on CFX96 (Bio-Rad, Hercules, CA, USA). The used primers are summarized in Table 1. The mRNA expression of each gene was normalized to the housekeeping gene hypoxanthinephosphoribosyl transferase (HPRT) as reported previously.27 The data are shown as the ratios of the interested genes to HPRT. For the inhibitor assay, cells were pretreated with various inhibitors for 30 min respectively, and then treated with IL-33 for 24 h.
Table 1.
Primers used for real-time PCR analysis
| Genes | Primer sequence (5′→3′) | |
|---|---|---|
| HPRT | Forward primer | |
| Reverse primer | ||
| IL-1β | Forward primer | |
| Reverse primer | ||
| IL-4 | Forward primer | |
| Reverse primer | ||
| IL-5 | Forward primer | |
| Reverse primer | ||
| IL-9 | Forward primer | |
| Reverse primer | ||
| IL-13 | Forward primer | |
| Reverse primer | ||
| CXCL1 | Forward primer | |
| Reverse primer | ||
| CCL5 | Forward primer | |
| Reverse primer | ||
| CCL2 | Forward primer | |
| Reverse primer | ||
| CX3CL1 | Forward primer | |
| Reverse primer | ||
| CCL7 | Forward primer | |
| Reverse primer | ||
| CCL12 | Forward primer | |
| Reverse primer | ||
| CCR7 | Forward primer | |
| Reverse primer | ||
| IL-21R | Forward primer | |
| Reverse primer | ||
| CXCR5 | Forward primer | |
| Reverse primer | ||
| Tnfrsf8 | Forward primer | |
| Reverse primer | ||
| CCR2 | Forward primer | |
| Reverse primer | ||
| IL-13Ra1 | Forward primer | |
| Reverse primer | ||
| CXCR1 | Forward primer | |
| Reverse primer | ||
| CCR1 | Forward primer | |
| Reverse primer | ||
| IL-1R2 | Forward primer | |
| Reverse primer | ||
| CXCR2 | Forward primer | |
| Reverse primer | ||
| Tnfrsf13c | Forward primer | |
| Reverse primer | ||
| Csf2rb2 | Forward primer | |
| Reverse primer | ||
| Csf2rb | Forward primer | |
| Reverse primer | ||
Abbreviations: CCL, C-C motif chemokine ligand; CXCL, C-X-C motif chemokine ligand; CCR, C-C motif chemokine receptor; CXCR, C-X-C motif chemokine receptor; HPRT, hypoxanthinephosphoribosyl transferase; IL, interleukin; IL-1R, interleukin-1 receptor.
Flow cytometry
For flow cytometric analysis of surface markers, cells were stained with antibodies in phosphate-buffered saline (PBS) containing 0.1% (w/v) bovine serum albumin (BSA) and 0.1% NaN3. For detection of intracellular cytokines, neutrophils were treated with GolgiPlug (BD Biosciences, San Jose, CA, USA) for the last 4–6 h of incubation. Cells were fixed and permeabilized according to the manufacturer’s protocol (BD Biosciences) and stained with anti-IL-9 mAb (1:100). Flow cytometric data were acquired on a FACSCalibur (BD Biosciences) or Epics XL (Beckman Coulter) and analyzed with the CellQuest Version 5.1 software or FlowJo Version 7.6.5 software (TreeStar, San Carlos, CA, USA).
Western blotting
Neutrophils were cultured in 1640 medium with 10% fetal calf serum in six-well plates. Cells were treated with IL-33 (100 ng/ml) for the indicated times. Total or nuclear cell lysates and immunoblot analysis were performed as described previously.30 Protein concentration was determined by bicinchoninic acid assays. Protein bands were visualized by adding horseradish peroxidase substrate (Millipore, Temecula, CA, USA) and then scanned using the Tanon 1600R Gel Image System (Tanon Co., Ltd., Shanghai, China). Anti-Actin and anti-H3 mAbs were used as cytoplasm and nuclear protein loading control, respectively.
ELISA assays
Cells were treated with LPS (1 μg/ml) and IL-33 (100 ng/ml) for the indicated times. Concentrations of cytokines and chemokines in cell culture medium supernatant were measured by ELISA assay kits, according to the manufacturer’s instructions.
Immunofluorescent staining
Cells cultured on coverslips were fixed in 4% paraformaldehyde and permeabilized in 0.2% Triton X-100/PBS for 10 min at 4 °C, respectively.26 Cells were blocked for 1 h in 5% BSA/PBS and incubated with anti-IL-9 (1:100 dilution in blocking buffer) overnight at 4 °C. After the wash step in PBS, cells were stained with Hoechst 33342 (4 μg/ml) for 10 min. Photo micrographs were taken using an LSM510META Laser Scanning Microscope (Zeiss, Jena, Germany).
OVA-induced allergic airway inflammation
The wild-type (WT) and IL-33-TG mice were sensitized by intraperitoneal injections of 10 μg OVA and 1 mg alum at days 0 and 14, whereas control mice were sham sensitized only with PBS. From day 21, the mice were exposed to aerosolized OVA (1% w/v) 30 min per day for 3 consecutive days. Mice were assessed 24 h after the last OVA challenge. Neutrophils from WT mice were stimulated with IL-33 and transferred into OVA-presensitized WT recipients at days 19 and 20. The fresh lungs were isolated and weighed before and after roast at 65 °C for 1 day to determine the ratio of wet/dry lung. Cell numbers in bronchoalveolar lavage (BAL) fluid were counted by a hemocytometer. Cells in BAL fluid were analyzed by flow cytometry with anti-mCD11b, anti-mSiglec F, anti-mLy6G and anti-mF4/80 mAbs. Cells in BAL fluid from control and asthma mice were stained with anti-IL-9 and anti-IL-1r2 mAbs, respectively. RNA was isolated from lung tissue for real-time PCR analysis. Lungs were fixed with formaldehyde, embedded in paraffin and sectioned and then stained with hematoxylin and eosin (H&E) for evaluation of the infiltration of inflammatory cells.
Statistical analysis
All data were presented as the mean±s.d. The two-way analysis of variance was used for comparison among multiple groups with the SPSS 16.0 software (SPSS, Chicago, IL, USA). A Mann–Whitney U-test was used to compare between the two groups. A P-value <0.05 was considered to be statistically significant.
Results
IL-33 selectively induces T helper type 2 (Th2) cytokine expression in neutrophils
Besides their involvement in primary defense against infections, neutrophils produce and release a large variety of cytokines and chemokines either constitutively or upon microenvironmental stimulations, which have a critical role in infections and pathogenesis.31,32 To explore the roles of different cytokines on the expression of Th2-type cytokines such as IL-4, IL-5, IL-9 and IL-13 in neutrophils, we first detected the mRNA expression levels of these genes in freshly isolated neutrophils of mouse bone marrow after different cytokines and LPS stimulation for 24 h by real-time PCR. Among the 15 cytokines and LPS tested in this study, only IL-33 significantly promotes IL-4, IL-5, IL-9 and IL-13 expression levels with the exception of GM-CSF, which also induces IL-4 expression, while the untreated resting neutrophils express almost undetectable levels of these Th2 cytokines (P<0.001, Figure 1a). IL-33 induces the mRNA and protein expression levels of IL-4, IL-5, IL-9 and IL-13 in dose- and time-dependent manners as determined by real-time PCR and ELISA assays (Figures 1b–d). The expression levels of IL-9 in IL-33-treated neutrophils are further confirmed by flow cytometric and confocal microscopic staining (Figures 1e and f). To exclude the potential contamination of other immune cells such as T cells, B cells and ILCs during the differentiation process, we sort the neutrophils of naive mice to obtain highly purified CD11b+Ly6G+ cells with >99% purity and repeat the same induction experiments. Indeed, the sorted CD11b+Ly6G+ cells express high levels of IL-4, IL-5, IL-9 and IL-13 after IL-33 treatment for 24 h (Supplementary Figure 1). IL-33 simulation can also upregulate all these cytokine expression levels in human neutrophils (Supplementary Figure 2). Thus IL-33 has the ability to promote Th2-type cytokines IL-4, IL-5, IL-9 and IL-13 expression levels in resting mouse neutrophils in a very specific manner.
Figure 1.
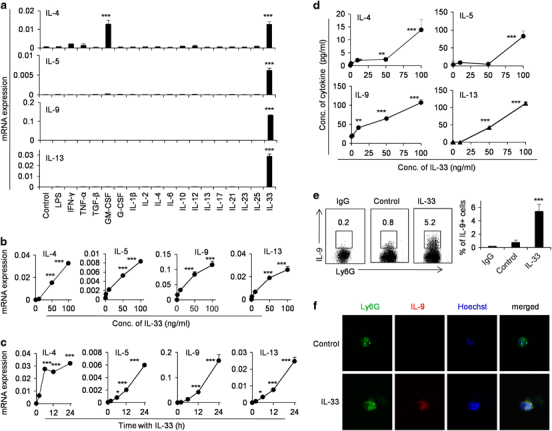
The expression levels of IL-4, IL-5, IL-9 and IL-13 in neutrophils are selectively induced by IL-33. (a) Primary neutrophils freshly isolated from bone marrow of C57BL/6 mice were treated with various cytokines and LPS for 24 h. The IL-4, IL-5, IL-9 and IL-13 mRNA expression levels were determined by real-time PCR. (b) The mRNA expression levels of IL-4, IL-5, IL-9 and IL-13 in primary neutrophils treated with different concentrations of IL-33 for 24 h were determined by real-time PCR. (c) Quantitative PCR analysis of IL-4, IL-5, IL-9 and IL-13 mRNA expression levels in neutrophils treated with IL-33 (100 ng/ml) for different times. (d) The protein levels of IL-4, IL-5, IL-9 and IL-13 in the culture medium of primary neutrophils treated with different concentrations of IL-33 for 24 h were determined by ELISA assays. The protein expression of IL-9 in neutrophils treated with 100 ng/ml IL-33 for 24 h were determined by flow cytometry (e) and confocal microscopy (f). Data are shown as mean±s.d. (n=3), representing one of the three independent experiments. *P<0.05, **P<0.01 and ***P<0.001 compared with control group. Data were analyzed by two-way ANOVA analysis using the SPSS software. ANOVA, analysis of variance; ELISA, enzyme-linked immunosorbent assay; IL, interleukin; LPS, lipopolysaccharide.
IL-33 induces a distinct neutrophil gene expression profile
To investigate whether IL-33 induces a unique neutrophil polarization in contrast to the LPS-induced inflammatory neutrophil subpopulation N(LPS), we thus determined the gene expression profiles of N(LPS) polarized neutrophils and IL-33-induced neutrophils (named as N(IL-33) thereafter) by microarray analysis. Indeed, N(IL-33) neutrophils express a unique panel of genes in sharp contrast to resting neutrophils (N0) and N(LPS) neutrophils (Figure 2a). Specifically, 1294 genes were upregulated and 2486 genes were downregulated in N(IL-33) neutrophils compared with N0 and N(LPS) polarized neutrophils (Figures 2a and b). Interestingly, N(LPS) and IL-33-induced N(IL-33) neutrophils express distinctive gene profiles of cytokines and chemokines, as determined by mRNA microarray and real-time PCR assays (Figures 2c–e, and data not shown). LPS induces TNF-α and IL-1β expression in neutrophils at both mRNA (Figure 2d) and protein levels (Figure 2e) as determined by real-time PCR and ELISA, respectively. However, IL-33 fails to induce the expression levels of TNF-α and IL-1β, as detected by quantitative PCR and ELISA assays (Figures 2d and e). In contrast, IL-33-treated neutrophils specifically express IL-4, IL-5, IL-9 and IL-13 at both mRNA and protein levels (P<0.01, Figures 2d and e). Importantly, LPS and IL-33 induce different chemokine expression patterns (Figure 2f). LPS induces high levels of chemokine (C-X-C motif) ligand-1 (CXCL1) and chemokine (C-C motif) ligand-5 (CCL5) expression levels in neutrophils but IL-33 does not. Both LPS and IL-33 promote CXCL3, CCL7 and CCL12 expression levels at different expression kinetics (Figure 2f). LPS rapidly upregulates CXCL3, CCL7 and CCL12 expression levels in neutrophils and then turn downs these genes quickly too (Figure 2f). However, IL-33 gradually induces CCL7 and CCL12 expression levels in neutrophils in a slower manner than LPS does (Figure 2f). Importantly, IL-33 but not LPS promotes neutrophils to express high level of CCL2 (Figure 2f), an important chemotactic factor for attracting monocytes, memory T lymphocytes and natural killer cells to the site of inflammation through binding to C-C motif chemokine receptor 2 (CCR2) and has been implicated in the pathogeneses of diseases, including psoriasis, rheumatoid arthritis, atherosclerosis, multiple sclerosis and insulin-resistant diabetes.33,34 Altogether, these data promote us to speculate that IL-33 may drive the polarization of resting neutrophils into an undefined state, which results in the production of a distinct panel of cytokines and chemokines compared with LPS-induced inflammatory neutrophils.
Figure 2.
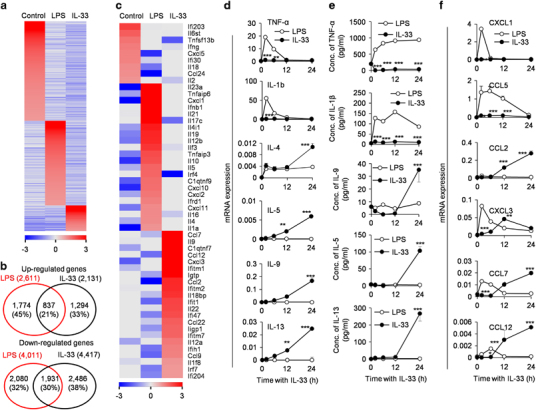
IL-33-treated neutrophils express distinct gene profiles different from the resting N0 or N(LPS) inflammatory subpopulation. Sorted primary neutrophils were cultured with LPS or IL-33 in vitro and microarray analysis of gene expression profiles were performed. (a) Heatmap comparison of gene expression profiles identifies lineage-restricted genes in N0, N(LPS) and N(IL-33) cells. Genes with predominant expression levels in one cell type (at least twofold greater than any other cell types) were organized to draw the plot. (b) Venn diagram showing upregulated genes (n=3 905) and downregulated genes (n=6 497) in N(LPS) and N(IL-33) neutrophils compared with N0 neutrophils. About 33% of upregulated genes and 38% of downregulated genes were specifically differentially expressed in N(IL-33) compared with N0 cells. (c) Expression profiling of lineage-restricted cytokines and chemokines in N0, N(LPS) and N(IL-33) cells. Genes with predominant expression levels in one cell type (at least twofold change compared with other cell types) were organized to draw the plot. Colors represent genes above (red) or below (blue) the second highest expression in three cell types. (d) The levels of cytokines, including TNF-α, IL-1β, IL-4, IL-5, IL-9 and IL-13 expression levels, were determined by real-time PCR after the isolated neutrophils were induced with LPS and IL-33 for the indicated times. (e) The concentrations of TNF-α, IL-1β, IL-9, IL-5 and IL-13 in the culture medium of the isolated neutrophils induced with LPS and IL-33 for indicated times were detected by ELISA assays. (f) The levels of chemokines, including CXCL1, CCL5, CCL2, CXCL3, CCL7 and CCL12, were determined by real-time PCR. Data are expressed as mean±s.d. (n=3–5), showing one representative of the three independent experiments. *P<0.05, **P<0.01 and ***P<0.001 compared with the control or LPS-treated cells. CCL, C-C motif chemokine ligand; CXCL, C-X-C motif chemokine ligand; ELISA, enzyme-linked immunosorbent assay; IL, interleukin; LPS, lipopolysaccharide; TNF, tumor necrosis factor.
IL-1R2 is specifically expressed on IL-33-induced neutrophils
As an effort to identify the specific cell surface markers and the chemotaxis characteristics of IL-33-induced N(IL-33) neutrophils, we determined the chemokine receptors and other cell surface molecule expression levels on N(IL-33) neutrophils. Microarray analysis showed that resting neutrophils, LPS-induced neutrophils and IL-33-induced neutrophils displayed distinct chemokine receptors and membrane protein gene-expressing profiles (Figure 3a). Interestingly, IL-33-induced neutrophils preferentially express high levels of C-X-C motif chemokine receptor 1 (CXCR1), CCR1, IL-1R2 and CXCR2 mRNA compared with LPS-induced neutrophils as indicated by real-time PCR assays, while LPS-induced neutrophils express high levels of CXCR5 (P<0.001, Figure 3b). We further demonstrate that IL-1R2 protein is expressed preferentially on the cell surface of IL-33-induced neutrophils as detected by flow cytometric assays (Figures 3c and d). Therefore, cell surface IL-1R2 expression may be a potential marker for IL-33-induced N(IL-33) neutrophil subpopulation.
Figure 3.
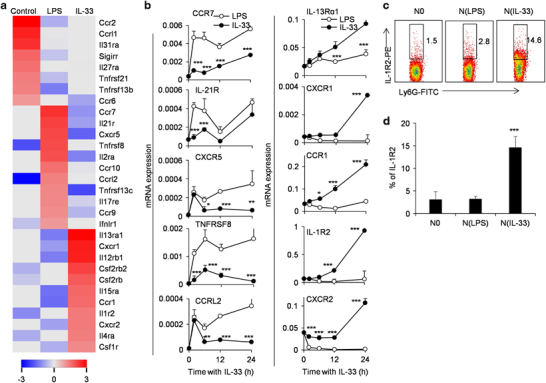
IL-33-induced neutrophils express unique cytokine and chemokine receptors compared with N0- or LPS-treated neutrophils. (a) Expression profiling of lineage-restricted receptors of cytokines and chemokines in N0, N(LPS) and N(IL-33) cells. Genes with predominant expression levels in one cell type (at least twofold change compared with other cell types) were organized to draw the plot. Colors represent genes above (red) or below (blue) the second highest expression in three cell types. (b) Freshly isolated bone marrow neutrophils were stimulated with LPS and IL-33 for the indicated times. The levels of cytokine and chemokine receptors in N(IL-33) cells, including IL-13Rα1, CXCR1, CCR1, IL-1R2 and CXCR2, were determined by real-time PCR. (c) Flow cytometric analysis of IL-1R2 expression on the surface of N0, N(LPS) and N(IL-33) cells. (d) The average percentages of IL-1R2+ cells in different neutrophil subpopulations were summarized. Data were shown as mean±s.d. (n=3). *P<0.05, **P<0.01 and ***P<0.001 compared with control or LPS-treated cells. CCR, C-C motif chemokine receptor; CXCR, C-X-C motif chemokine receptor; IL-1R, interleukin-1 receptor; LPS, lipopolysaccharide.
IL-33 induces Th2 cytokine expression in neutrophils via JNK and NF-κB pathways
IL-33 interaction with its receptor ST2 promotes the activations of Erk1/2, p38, JNK and activator protein 1 (AP-1) and translocation of NF-κB to the nucleus.35 The IL-9 promoter region and two additional regions of conserved non-coding sequences upstream of the promoter region form the cis- and trans-regulatory elements collectively regulating IL-9 gene expression.36,37 Sequence and molecular analysis identified the potential binding sites for STATs, nuclear factor of activated T cells, GATA1, GATA3, Smad (Drosophila mothers against decapentaplegic proteins) and notch as well as NF-κB and AP-1.37,38 It is true that IL-33 significantly induced the activation of P38-STAT1, JNK, AP-1 and NF-κB in neutrophils, as indicated by their protein phosphorylation and re-localization to the nucleus (Figures 4a and b). Inhibition of p38 mitogen-activated protein kinase, STAT1, STAT3 and ERK activation by their specific inhibitors, respectively, failed to significantly decrease the IL-9 mRNA expression in IL-33-induced neutrophils (Supplementary Figure 3). But inhibition of either JNK or NF-κB remarkably impacts IL-33-induced IL-4, IL-5, IL-9, IL-13, CCL2, CCL12 and IL-1R2 expression levels in IL-33-induced neutrophils (P<0.01, Figures 4c and d, Supplementary Figure 4). Meanwhile, the concentrations of IL-9, IL-5, IL-13 and CCL2 in the culture medium of IL-33-treated neutrophils were significantly decreased with the increasing doses of JNK inhibitor (SP600125) and NF-κB inhibitor, respectively (Figures 4e and f). In order to demonstrate that IL-33 acts through ST2 for downstream signaling activation and cytokine production, we used neutrophils isolated from IL-33R(ST2) knockout (KO) mice to perform the similar studies. As expected, ST2 KO neutrophils showed minimal phosphorylation of p65 and JNK after IL-33 treatment (Supplementary Figure 5A). Meanwhile, IL-33 induced poor expression levels of IL-4, IL-5, IL-9 and IL-13 in ST2-deficient neutrophils (Supplementary Figure 5B). These results indicated that the expression levels of Th2-type cytokines, CCL2, CCL12 and IL-1R2 in IL-33-induced neutrophils were dependent on JNK- and NF-κB-mediated pathways.
Figure 4.
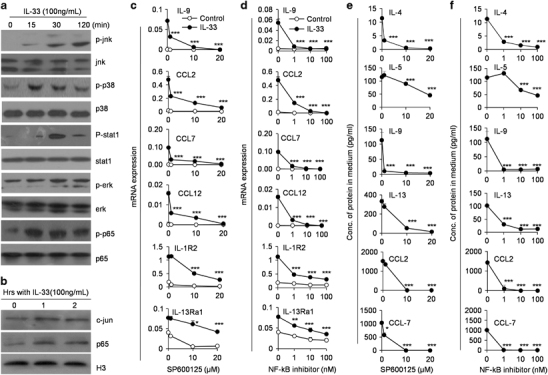
The specific phenotype of IL-33-treated neutrophils is mediated by JNK and NF-κB signaling pathways. (a) Neutrophils freshly isolated from bone marrow were treated with IL-33 for 15, 30 and 120 min, respectively. The activities of JNK, p38, STAT1, Erk and p65 in cytoplasm were detected by western blotting. (b) The c-jun and p65 protein levels in nucleus of neutrophils after IL-33 treatment for 1 and 2 h, respectively, were determined by western blotting. (c and d) Neutrophils were pretreated with the indicated concentrations of JNK inhibitor (SP600125) and NF-κB inhibitor for 30 min and then stimulated with IL-33 for 24 h. The levels of IL-9, CCL2, CCL7, CCL12, IL-1R2 and IL-13Ra1 expression were detected by real-time PCR. (e and f) The concentrations of IL-9, IL-4, IL-5, IL-13, CCL2 and CCL7 in the supernatant of neutrophils pretreated with either SP600125 or NF-κB inhibitor and then cultured with IL-33 for 24 h were determined by ELISA assay kits. Assays were performed more than three times. Data are shown as mean±s.d. (n=3). *P<0.05, **P<0.01 and ***P<0.001 compared with control. CCL, C-C motif chemokine ligand; ELISA, enzyme-linked immunosorbent assay; ERK, extracellular signal–regulated kinase; IL, interleukin; JNK, c-Jun N-terminal kinase; NF, nuclear factor; STAT, signal transducer and activator of transcription factor.
The presence and the roles of N(IL-33) neutrophils in allergic airway inflammatory mice
Th2/Th9 cytokines are critically involved in the pathogenesis of airway allergic inflammation in mice and humans.39,40 We observed the presence of N(IL-33) neutrophils in an OVA-induced airway allergic inflammatory mouse model, in which Th2/Th9 cells and neutrophils have a critical role as previously reported. IL-9+ and IL-1R2+ neutrophils did exist in the BAL fluid of airway allergic inflammatory mice (Figures 5a and b). IL-33-TG mice displayed more severe airway allergic inflammatory pathogenesis in the lung tissues as indicated by H&E staining of lung tissues, the ratio of wet/dry lung weight, BAL fluid cell number and the infiltration by leukocytes and eosinophils (Figures 5b–f, and data not shown). In parallel with the pathological changes, lung tissue of IL-33-TG airway allergic inflammatory mice express significantly higher IL-4, IL-5, IL-9, IL-13, CCL2 and CCL7 molecules than those of WT airway allergic inflammatory mice as determined by real-time PCR (Figure 5g). To identify the role of N(IL-33) cells in the pathogenesis of airway allergic inflammation, we adoptively transferred either induced N(IL-33) or resting N0 neutrophils into OVA-induced airway allergic inflammatory mice in asthma-inducing stage. H&E staining shows that mice receiving N(IL-33) neutrophils suffer more severe pathogenesis of asthma in the lung tissues than mice receiving resting N0 neutrophils (Figure 5h, and data not shown). Adoptive transfer of IL-33-induced N(IL-33) neutrophils enhances BAL fluid cell number and the infiltration of leukocytes and eosinophils (P<0.001, Figures 5i and j). Meanwhile, the lung tissues of mice receiving N(IL-33) neutrophils express significantly more IL-4, IL-5, IL-9, IL-13, CCL2 and CCL7 than those in mice receiving resting neutrophils (Figure 5k). In order to test the role of endogenous neutrophils in the pathogenesis of asthma, we performed loss-of-function experiments by neutrophil depletion during asthma induction. Neutrophil-depleted mice showed significantly reduced allergic inflammation in the lungs compared with control (Supplementary Figure 6). Thus IL-33-induced N(IL-33) neurophils are present in the lung tissues of airway allergic inflammatory mice and have the capability to promote airway allergic inflammatory pathogenesis, at least in mice.
Figure 5.
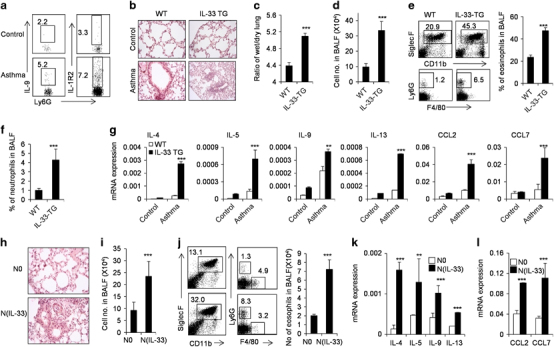
N(IL-33) neutrophils promote OVA-induced allergic airway inflammation in vivo. OVA-presensitized WT and IL-33-TG mice were challenged with aerosolized OVA daily for 3 days as described in the Materials and methods section. (a) IL-9+ and IL-1R2+ neutrophils in BAL fluid of wild-type asthma mice were detected by flow cytometry. (b) H&E staining of lung tissues from control or OVA-induced asthma WT and IL-33-TG mice is presented. (c) Ratio of wet/dry lung weights of WT and IL-33-TG mice after OVA challenge was measured as described in the Materials and methods section. (d) Cell numbers in BAL fluid of WT and IL-33-TG mice after OVA challenge. (e) Flow cytometric analysis of infiltrated cells such as eosinophils in BAL fluid of WT and IL-33-TG mice after OVA challenge. (f) Proportion of neutrophils in BAL fluid of WT and IL-33-TG mice after OVA challenge is presented. (g) The mRNA expression levels of IL-4, IL-5, IL-9, IL-13, CCL2 and CCL7 in lung tissues of WT and IL-33-TG mice after OVA challenge were detected by real-time PCR. (h) Wild-type neutrophils were stimulated with IL-33, and N0 or N(IL-33) cells were transferred into OVA-presensitized syngeneic wild-type recipients before challenged with aerosolized OVA for 3 days as described in the Materials and methods section. H&E staining of lung tissues from mice treated as described. (i) Cell numbers in BAL fluid of OVA-induced asthma mice transferred with N0 and N(IL-33) cells. (j) The infiltrated cells in BAL fluid of asthma mice treated with N0 and N(IL-33) cells were detected by flow cytometry. Eosinophil numbers in BAL fluid were presented. (k and l) Quantitative PCR analysis of IL-4, IL-5, IL-9, IL-13, CCL2 and CCL7 expression levels in lung tissues of asthma mice treated with N0 and N(IL-33) cells. Data are expressed as mean±s.d. (n=3–5) and are one representative of the three independent experiments with similar results. *P<0.05, **P<0.01 and ***P<0.001 compared with control mice. BAL, bronchoalveolar lavage; CCL, C-C motif chemokine ligand; H&E, hematoylin and eosin; IL, interleukin; OVA, ovalbumin; WT, wild type.
Discussion
We herein identified a unique neutrophil subpopulation induced by IL-33 with the following properties: (1) displaying distinctive gene-expressing profiles than resting neutrophils and LPS-activated neutrophils; (2) producing more Th2 type cytokines, including IL-4, IL-5, IL-9 and IL-13 with less TNF-α and IL-1β; (3) expressing more chemokines CCL2 and CCL7; (4) expressing more IL-1R2 on the cell surface; and (5) promoting airway allergic inflammatory pathogenesis. Thus we offered additional evidence for the differentially polarized neutrophils with respect to cytokine and chemokine productions.
IL-33 protein is constitutively present in healthy mice and humans, primarily in the nuclei of non-hematopoietic cells, with particular abundance in specialized populations of epithelial and endothelial cells.41,42 IL-33 contributes to many diseases, such as allergy, fibrosis, multiple sclerosis, rheumatoid arthritis and so on.20,43 Single-nucleotide polymorphism studies showed the association of IL-33–IL-1R-like 1 pathway polymorphisms with asthma in childhood.44 Blockade of IL-33/ST2 ameliorates airway inflammation in a mouse model of allergic asthma.45 Although IL-33 can act on many immune cells to promote the pathogenesis, we found that IL-33, as a cytokine, can directly induce Th2 cytokine productions, including IL-4, IL-5, IL-9 and IL-13, in neutrophils and subsequently promote the process of airway allergic inflammation in a mouse model. IL-9 is secreted by many cell types, including T lymphocytes, eosinophils, mast cells and neutrophils, and is implicated in asthma and in the protection against nematode infections.46,47 Transgenic expression of IL-9 results in allergic inflammation.48 In addition, the IL-33-induced Th2 chemokine CCL2 releasing by neutrophils likely recruit more M2 phenotypic macrophages and modulate the switch of Th1 and Th2 cells.34,49 The involvement of IL-33-induced N(IL-33) neutrophils in airway allergic inflammation is consistent with the previous reports showing the roles of neutrophils in the pathogenesis of airway allergic inflammation,50,51 although using CXCR2 antagonist to reduce neutrophil numbers in the lungs did not reduce the frequency of severe exacerbations in patients with uncontrolled severe asthma.52 This could not exclude the roles of neutrophils in asthma, especially the onset of this disease. Neutrophil extracellular traps can also be generated in asthmatic airways and participate in the disease process.23,53
It is reported that neutrophils can produce IL-1β, TNF-α, IL-12, IL-4 and IL-10 under different stimulations.31 We observed that neutrophils have the ability to release IL-9 and IL-13 after stimulation with IL-33 alone. IL-4 appears to be an important initiator of IL-9 synthesis in CD4+ T cells, as a reduction in IL-9 production has been found after activating CD4+ T cells from IL-4-deficient mice in vitro.54 Although the requirement for IL-4 in enhancing IL-9 production has been suggested in different models,55 a significant amount of IL-9 production is present in IL-4-deficient mice upon protein antigen immunization, suggesting an IL-4-independent pathway for IL-9 production.54 However, in the later case it is unclear whether IL-9 is produced by CD4+ T cells or innate cells such as CD4+ natural killer T cells or CD4+ lymphoid tissue-inducer cells. In the present study, we found IL-33 alone without additional IL-4 supplement can induce IL-9 production in neutrophils, suggesting that IL-4 is not essential for IL-9 expression in resting neutrophils. Molecular studies showed that IL-33 induces Th2 cytokine and chemokine expression levels mainly through JNK and NF-κB-dependent pathways. p38, STAT1, STAT3 and ERK do not significantly participate in IL-33 induction of Th2 cytokine expression levels in neutrophils.
In summary, our present studies demonstrate a Th2/Th9-type-like polarization of neutrophils induced by IL-33. These polarized neutrophils promote the airway allergic inflammatory pathogenesis in a mouse model. Thus we offered additional evidence for the differentially polarized neutrophils with respect to cytokine and chemokine productions. These findings shed light on the presence of neutrophil functional plasticity and polarization, which we previously neglected. The biological significance of the polarized neutrophils in other diseases needs to be investigated, which may provide new insights to the contribution of neutrophils to the pathogenesis caused by inflammatory and immune disorders and may offer novel therapeutic approaches to treat neutrophil-related immune disorders.
Electronic supplementary material
Acknowledgements
We thank Dr Lianjun Zhang and Dr Aqeel Javeed for their kind review of the manuscript. This work was supported by grants from the National Natural Science Foundation of China for General and Key Programs (81530049 and 81130055 to YZ), Knowledge Innovation Program of Chinese Academy of Sciences (XDA04020202-19 to YZ), the CAS/SAFEA International Partnership Program for Creative Research Teams (to YZ) and Beijing Municipal Hospital Authority ‘Yangfan Program’ (ZYLX201408 to XZ).
Author contributions
BS, LZ and YT designed and carried out the experiments, analyzed data and wrote the manuscript; H-XS analyzed microarray data; PW performed real-time PCR assays; YZ performed ELISA assays; YL performed animal models and flow cytometry; XZ and YH analyzed data and revised the manuscript; LZ, NN and YZ designed experiments, analyzed data, wrote the manuscript and provided overall supervision.
Conflict of interest
The authors declare no conflict of interest.
Contributor Information
Lianfeng Zhang, Email: zhanglf@cnilas.org.
Ning Na, Email: nngg20102009@hotmail.com.
Yong Zhao, Email: zhaoy@ioz.ac.cn.
Electronic supplementary material
Supplementary Information for this article can be found on the Cellular & Molecular Immunology website 10.1038/cmi.2017.163
References
- 1.Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13:159–175. doi: 10.1038/nri3399. [DOI] [PubMed] [Google Scholar]
- 2.Xu R, Lin F, Bao C, Huang H, Ji C, Wang S, et al. Complement 5a receptor-mediated neutrophil dysfunction is associated with a poor outcome in sepsis. Cell Mol Immunol. 2016;13:103–109. doi: 10.1038/cmi.2014.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nemeth T, Mocsai A. The role of neutrophils in autoimmune diseases. Immunol Lett. 2012;143:9–19. doi: 10.1016/j.imlet.2012.01.013. [DOI] [PubMed] [Google Scholar]
- 4.Wu T, Sun C, Chen Z, Zhen Y, Peng J, Qi Z, et al. Smad3-deficient CD11b(+)Gr1(+) myeloid-derived suppressor cells prevent allograft rejection via the nitric oxide pathway. J Immunol. 2012;189:4989–5000. doi: 10.4049/jimmunol.1200068. [DOI] [PubMed] [Google Scholar]
- 5.Colgan SP. Neutrophils and inflammatory resolution in the mucosa. Semin Immunol. 2015;27:177–183. doi: 10.1016/j.smim.2015.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jaeger BN, Donadieu J, Cognet C, Bernat C, Ordonez-Rueda D, Barlogis V, et al. Neutrophil depletion impairs natural killer cell maturation, function, and homeostasis. J Exp Med. 2012;209:565–580. doi: 10.1084/jem.20111908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chiewchengchol D, Midgley A, Sodsai P, Deekajorndech T, Hirankarn N, Beresford MW, et al. The protective effect of GM-CSF on serum-induced neutrophil apoptosis in juvenile systemic lupus erythematosus patients. Clin Rheumatol. 2015;34:85–91. doi: 10.1007/s10067-014-2800-2. [DOI] [PubMed] [Google Scholar]
- 8.Cross A, Bucknall RC, Cassatella MA, Edwards SW, Moots RJ. Synovial fluid neutrophils transcribe and express class II major histocompatibility complex molecules in rheumatoid arthritis. Arthritis Rheum. 2003;48:2796–2806. doi: 10.1002/art.11253. [DOI] [PubMed] [Google Scholar]
- 9.Lakschevitz FS, Visser MB, Sun C, Glogauer M. Neutrophil transcriptional profile changes during transit from bone marrow to sites of inflammation. Cell Mol Immunol. 2015;12:53–65. doi: 10.1038/cmi.2014.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vitiello D, Neagoe PE, Sirois MG, White M. Effect of everolimus on the immunomodulation of the human neutrophil inflammatory response and activation. Cell Mol Immunol. 2015;12:40–52. doi: 10.1038/cmi.2014.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scapini P, Nardelli B, Nadali G, Calzetti F, Pizzolo G, Montecucco C, et al. G-CSF-stimulated neutrophils are a prominent source of functional BLyS. J Exp Med. 2003;197:297–302. doi: 10.1084/jem.20021343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mhawech-Fauceglia P, Kaya G, Sauter G, McKee T, Donze O, Schwaller J, et al. The source of APRIL up-regulation in human solid tumor lesions. J Leukoc Biol. 2006;80:697–704. doi: 10.1189/jlb.1105655. [DOI] [PubMed] [Google Scholar]
- 13.Zindl CL, Lai JF, Lee YK, Maynard CL, Harbour SN, Ouyang W, et al. IL-22-producing neutrophils contribute to antimicrobial defense and restitution of colonic epithelial integrity during colitis. Proc Natl Acad Sci USA. 2013;110:12768–12773. doi: 10.1073/pnas.1300318110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Puga I, Cols M, Barra CM, He B, Cassis L, Gentile M, et al. B cell-helper neutrophils stimulate the diversification and production of immunoglobulin in the marginal zone of the spleen. Nat Immunol. 2012;13:170–180. doi: 10.1038/ni.2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: ‘N1’ versus ‘N2’ TAN. Cancer Cell. 2009;16:183–194. doi: 10.1016/j.ccr.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Y, Zhu L, Chu Z, Yang T, Sun HX, Yang F et al. Characterization and biological significance of IL-23-induced neutrophil polarization. Cell Mol Immunol 2017; e-pub ahead of print. [DOI] [PMC free article] [PubMed]
- 17.Molofsky AB, Savage AK, Locksley RM. Interleukin-33 in tissue homeostasis, injury, and inflammation. Immunity. 2015;42:1005–1019. doi: 10.1016/j.immuni.2015.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Humphreys NE, Xu D, Hepworth MR, Liew FY, Grencis RK. IL-33, a potent inducer of adaptive immunity to intestinal nematodes. J Immunol. 2008;180:2443–2449. doi: 10.4049/jimmunol.180.4.2443. [DOI] [PubMed] [Google Scholar]
- 19.Kondo Y, Yoshimoto T, Yasuda K, Futatsugi-Yumikura S, Morimoto M, Hayashi N, et al. Administration of IL-33 induces airway hyperresponsiveness and goblet cell hyperplasia in the lungs in the absence of adaptive immune system. Int Immunol. 2008;20:791–800. doi: 10.1093/intimm/dxn037. [DOI] [PubMed] [Google Scholar]
- 20.von Moltke J, Locksley RM. I-L-C-2 it: type 2 immunity and group 2 innate lymphoid cells in homeostasis. Curr Opin Immunol. 2014;31:58–65. doi: 10.1016/j.coi.2014.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moro K, Yamada T, Tanabe M, Takeuchi T, Ikawa T, Kawamoto H, et al. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature. 2010;463:540–544. doi: 10.1038/nature08636. [DOI] [PubMed] [Google Scholar]
- 22.Sjoberg LC, Gregory JA, Dahlen SE, Nilsson GP, Adner M. Interleukin-33 exacerbates allergic bronchoconstriction in the mice via activation of mast cells. Allergy. 2015;70:514–521. doi: 10.1111/all.12590. [DOI] [PubMed] [Google Scholar]
- 23.Radermecker C, Sabatel C, Toussaint M, Johnston S, Bureau F, Marichal T. Release of neutrophils extracellular traps as a main trigger for asthma onset. Unpublished Conference/Abstract 2017 6th NIF Winter School on Advanced Immunology, January 22 to 26, Singapore, Singapore.
- 24.Foley SC, Hamid Q. Images in allergy and immunology: neutrophils in asthma. J Allergy Clin Immunol. 2007;119:1282–1286. doi: 10.1016/j.jaci.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 25.Gao K, Li X, Zhang L, Bai L, Dong W, Gao K, et al. Transgenic expression of IL-33 activates CD8(+) T cells and NK cells and inhibits tumor growth and metastasis in mice. Cancer Lett. 2013;335:463–471. doi: 10.1016/j.canlet.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 26.Hou Y, Lin H, Zhu L, Liu Z, Hu F, Shi J, et al. The inhibitory effect of IFN-gamma on protease HTRA1 expression in rheumatoid arthritis. J Immunol. 2014;193:130–138. doi: 10.4049/jimmunol.1302700. [DOI] [PubMed] [Google Scholar]
- 27.Zhu L, Yang T, Li L, Sun L, Hou Y, Hu X, et al. TSC1 controls macrophage polarization to prevent inflammatory disease. Nat Commun. 2014;5:4696. doi: 10.1038/ncomms5696. [DOI] [PubMed] [Google Scholar]
- 28.Hou Y, Lin H, Zhu L, Liu Z, Hu F, Shi J, et al. Lipopolysaccharide increases the incidence of collagen-induced arthritis in mice through induction of protease HTRA-1 expression. Arthritis Rheum. 2013;65:2835–2846. doi: 10.1002/art.38124. [DOI] [PubMed] [Google Scholar]
- 29.Sun B, Hu X, Liu G, Ma B, Xu Y, Yang T, et al. Phosphatase Wip1 negatively regulates neutrophil migration and inflammation. J Immunol. 2014;192:1184–1195. doi: 10.4049/jimmunol.1300656. [DOI] [PubMed] [Google Scholar]
- 30.Liu G, Hu X, Sun B, Yang T, Shi J, Zhang L, et al. Phosphatase Wip1 negatively regulates neutrophil development through p38 MAPK-STAT1. Blood. 2013;121:519–529. doi: 10.1182/blood-2012-05-432674. [DOI] [PubMed] [Google Scholar]
- 31.Tecchio C, Micheletti A, Cassatella MA. Neutrophil-derived cytokines: facts beyond expression. Front Immunol. 2014;5:508. doi: 10.3389/fimmu.2014.00508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Taylor PR, Roy S, Leal SM, Jr., Sun Y, Howell SJ, Cobb BA, et al. Activation of neutrophils by autocrine IL-17A-IL-17RC interactions during fungal infection is regulated by IL-6, IL-23, RORgammat and dectin-2. Nat Immunol. 2014;15:143–151. doi: 10.1038/ni.2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bryant VL, Slade CA. Chemokines, their receptors and human disease: the good, the bad and the itchy. Immunol Cell Biol. 2015;93:364–371. doi: 10.1038/icb.2015.23. [DOI] [PubMed] [Google Scholar]
- 34.Lee YG, Jeong JJ, Nyenhuis S, Berdyshev E, Chung S, Ranjan R, et al. Recruited alveolar macrophages, in response to airway epithelial-derived monocyte chemoattractant protein 1/CCl2, regulate airway inflammation and remodeling in allergic asthma. Am J Respir Cell Mol Biol. 2015;52:772–784. doi: 10.1165/rcmb.2014-0255OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Andrade MV, Iwaki S, Ropert C, Gazzinelli RT, Cunha-Melo JR, Beaven MA. Amplification of cytokine production through synergistic activation of NFAT and AP-1 following stimulation of mast cells with antigen and IL-33. Eur J Immunol. 2011;41:760–772. doi: 10.1002/eji.201040718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chang HC, Sehra S, Goswami R, Yao W, Yu Q, Stritesky GL, et al. The transcription factor PU.1 is required for the development of IL-9-producing T cells and allergic inflammation. Nat Immunol. 2010;11:527–534. doi: 10.1038/ni.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perumal NB, Kaplan MH. Regulating Il9 transcription in T helper cells. Trends Immunol. 2011;32:146–150. doi: 10.1016/j.it.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang XO, Zhang H, Kim BS, Niu X, Peng J, Chen Y, et al. The signaling suppressor CIS controls proallergic T cell development and allergic airway inflammation. Nat Immunol. 2013;14:732–740. doi: 10.1038/ni.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chung KF. Targeting the interleukin pathway in the treatment of asthma. Lancet. 2015;386:1086–1096. doi: 10.1016/S0140-6736(15)00157-9. [DOI] [PubMed] [Google Scholar]
- 40.Abreu SC, Antunes MA, Mendonca L, Branco VC, de Melo EB, Olsen PC, et al. Effects of bone marrow mononuclear cells from healthy or ovalbumin-induced lung inflammation donors on recipient allergic asthma mice. Stem Cell Res Ther. 2014;5:108. doi: 10.1186/scrt496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23:479–490. doi: 10.1016/j.immuni.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 42.Pichery M, Mirey E, Mercier P, Lefrancais E, Dujardin A, Ortega N, et al. Endogenous IL-33 is highly expressed in mouse epithelial barrier tissues, lymphoid organs, brain, embryos, and inflamed tissues: in situ analysis using a novel Il-33-LacZ gene trap reporter strain. J Immunol. 2012;188:3488–3495. doi: 10.4049/jimmunol.1101977. [DOI] [PubMed] [Google Scholar]
- 43.Theoharides TC, Petra AI, Taracanova A, Panagiotidou S, Conti P. Targeting IL-33 in autoimmunity and inflammation. J Pharmacol Exp Ther. 2015;354:24–31. doi: 10.1124/jpet.114.222505. [DOI] [PubMed] [Google Scholar]
- 44.Savenije OE, Mahachie John JM, Granell R, Kerkhof M, Dijk FN, de Jongste JC, et al. Association of IL33-IL-1 receptor-like 1 (IL1RL1) pathway polymorphisms with wheezing phenotypes and asthma in childhood. J Allergy Clin Immunol. 2014;134:170–177. doi: 10.1016/j.jaci.2013.12.1080. [DOI] [PubMed] [Google Scholar]
- 45.Lee HY, Rhee CK, Kang JY, Byun JH, Choi JY, Kim SJ, et al. Blockade of IL-33/ST2 ameliorates airway inflammation in a murine model of allergic asthma. Exp Lung Res. 2014;40:66–76. doi: 10.3109/01902148.2013.870261. [DOI] [PubMed] [Google Scholar]
- 46.Morita H, Arae K, Unno H, Miyauchi K, Toyama S, Nambu A, et al. An interleukin-33-mast cell-interleukin-2 axis suppresses papain-induced allergic inflammation by promoting regulatory T cell numbers. Immunity. 2015;43:175–186. doi: 10.1016/j.immuni.2015.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao P, Xiao X, Ghobrial RM, Li XC. IL-9 and Th9 cells: progress and challenges. Int Immunol. 2013;25:547–551. doi: 10.1093/intimm/dxt039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Forbes EE, Groschwitz K, Abonia JP, Brandt EB, Cohen E, Blanchard C, et al. IL-9- and mast cell-mediated intestinal permeability predisposes to oral antigen hypersensitivity. J Exp Med. 2008;205:897–913. doi: 10.1084/jem.20071046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Traynor TR, Herring AC, Dorf ME, Kuziel WA, Toews GB, Huffnagle GB. Differential roles of CC chemokine ligand 2/monocyte chemotactic protein-1 and CCR2 in the development of T1 immunity. J Immunol. 2002;168:4659–4666. doi: 10.4049/jimmunol.168.9.4659. [DOI] [PubMed] [Google Scholar]
- 50.Bruijnzeel PL, Uddin M, Koenderman L. Targeting neutrophilic inflammation in severe neutrophilic asthma: can we target the disease-relevant neutrophil phenotype? J Leukoc Biol. 2015;98:549–556. doi: 10.1189/jlb.3VMR1214-600RR. [DOI] [PubMed] [Google Scholar]
- 51.Makrinioti H, Toussaint M, Jackson DJ, Walton RP, Johnston SL. Role of interleukin 33 in respiratory allergy and asthma. Lancet Respir Med. 2014;2:226–237. doi: 10.1016/S2213-2600(13)70261-3. [DOI] [PubMed] [Google Scholar]
- 52.O'Byrne PM, Metev H, Puu M, Richter K, Keen C, Uddin M, et al. Efficacy and safety of a CXCR2 antagonist, AZD5069, in patients with uncontrolled persistent asthma: a randomised, double-blind, placebo-controlled trial. Lancet Respir Med. 2016;4:797–806. doi: 10.1016/S2213-2600(16)30227-2. [DOI] [PubMed] [Google Scholar]
- 53.Dworski R, Simon HU, Hoskins A, Yousefi S. Eosinophil and neutrophil extracellular DNA traps in human allergic asthmatic airways. J Allergy Clin Immunol. 2011;127:1260–1266. doi: 10.1016/j.jaci.2010.12.1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Monteyne P, Renauld JC, Van Broeck J, Dunne DW, Brombacher F, Coutelier JP. IL-4-independent regulation of in vivo IL-9 expression. J Immunol. 1997;159:2616–2623. [PubMed] [Google Scholar]
- 55.Gessner A, Blum H, Rollinghoff M. Differential regulation of IL-9-expression after infection with Leishmania major in susceptible and resistant mice. Immunobiology. 1993;189:419–435. doi: 10.1016/S0171-2985(11)80414-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.