

Summary
Recently, bi‐functional molecules that can redirect immune effectors to tumour cells have emerged as potentially robust mediators of tumour regression in clinical trials. Two modalities in particular, bi‐specific antibodies for T‐cell redirection and activation (BiTe) and immune‐mobilizing monoclonal T‐cell receptors against cancer (ImmTAC), are being evaluated in efficacy studies as ‘off‐the‐shelf’ reagents. Optimal therapy will require an understanding and means to address regulatory mechanisms of limiting efficacy. In light of this, we evaluated the impact of induced regulatory T (iTreg) cells on the efficacy of tumour cell killing redirected by ImmTAC and demonstrated down‐regulation of T‐cell proliferation and expression of CD25, CD107a, Granzyme B and Perforin by ImmTAC‐redirected T cells. Significant recovery of ImmTAC potency, however, could be achieved when combined with an anti‐programmed cell death protein 1 monoclonal antibody. Furthermore, we found that among lung cancer patients failing to respond to ImmTAC therapy, there was a significantly higher fraction of Treg cells in the peripheral blood mononuclear cells of lung cancer patients than in healthy donors. These results provide in vitro evidence for an iTreg cell‐mediated immunosuppression of ImmTAC‐redirected T‐cell responses. Whilst immune checkpoint blockade can reverse the Treg cell suppression, it forms a rational basis for a combination of the blockade with ImmTAC in clinical trials.
Keywords: anti‐PD‐1 monoclonal antibody, cancer, ImmTAC, immunotherapy, regulatory T cells
Abbreviations
- anti‐PD‐1mAb
antibody programmed death‐1 monoclonal antibody
- CFSE
carboxyfluorescein succinimdyl ester
- CTLA‐4
cytotoxic T‐lymphocyte‐associated protein 4
- FACS
fluorescence‐activated cell sorting
- FoxP3
forkhead box P3
- GITR
glucocorticoid‐induced tumor necrosis factor receptor
- ‘IC’
induction cocktail
- IL‐10
interleukin‐10
- ImmTAC
Immune‐mobilizing monoclonal T‐cell receptors against cancer
- iTreg cells
induced regulatory T cells
- mAb
monoclonal antibody
- MHC
major histocompatibility complex
- nTreg cells
natural regulatory T cells
- PBMC
peripheral blood mononuclear cells
- PD‐1
programmed cell death protein 1
- PD‐L1
PD‐1 ligand‐1
- TCR
T‐cell receptor
- TGF‐β1
transforming growth factor‐β 1
- TME
tumour microenvironment
- Treg cell
regulatory T cell
Introduction
Immune‐based therapy for cancer is emerging as an important treatment modality for patients for whom conventional therapy has failed. Although remarkable results have been observed for some cancers, tumour immune escape remains a challenge facing researchers and clinicians.1 The contribution of tumour infiltrating lymphocytes, found in the tumour microenvironment (TME), in tumour control2 may be mitigated by the presence of regulatory T (Treg) cells, which preferentially traffic to tumour sites.3, 4 In animal models, selective Treg cell depletion can enhance immunotherapeutic strategies,5 and clinical studies have suggested poorer prognosis in patients with elevated Treg cell levels.6, 7
Two types of Treg cells have been identified: ‘natural’ Treg (nTreg) cells,8 which develop in thymus, and ‘induced’ Treg (iTreg) cells, which can arise in the periphery from conventional T cells in the TME via tumour‐derived signals, including transforming growth factor‐β (TGF‐β).9, 10, 11 Epigenetic differences between these two types of Treg cells have shown that nTreg cells are associated with stable forkhead box P3 (Foxp3) expression and wider demethylation patterns compared with iTreg cells.12 Although, the contribution of nTreg versus iTreg cells in maintaining tolerance as well as tumour escape from the TME is not completely understood,13, 14, 15 circulating Treg cells in healthy donors are largely represented by nTreg cells, whereas iTreg cells seem to play a key role in the TME in suppressing the antitumour immune response.14, 16
With respect to tumour tolerance in the TME, augmenting tumour‐specific cytotoxic T lymphocyte (CTL) response strategies may be negatively affected by the increased numbers of iTreg cells. In addition, tumours will down‐regulate the expression of major histocompatibility complexes (MHCs) to evade immune scrutiny, which will present a huge challenge to the T cells with naturally expressing low‐affinity antigen receptors to complete the immune tasks.17 To overcome the problem of the natural immune‐synapse formed with low‐affinity interactions, affinity‐enhanced T‐cell receptors (TCRs) are designed.18 On the other hand, to harness a patient's own CTLs, an immune‐mobilizing monoclonal TCR against cancer (ImmTAC) was constructed to hold two ends: one with a high‐affinity TCR to engage the tumour antigen‐derived peptide–MHC complex on the target cells and the other, a humanized anti‐CD3 scFv (single‐chain variable fragment) to recruit CTLs. The fusion molecule may form a central molecular complex that establishes an artificial immune‐synapse for activating CD3+ T cells in the proximity of tumour cells in the TME.18, 19, 20 Such a high‐affinity TCR‐made immune‐synapse will allow CTLs to express essential molecules, such as CD25, CD107a, Granzyme B and Perforin to complete the killing process. In general, ImmTAC can mediate very effective killing of tumour cells in vitro and in vivo, but it is not clear whether iTreg cells are involved in suppression of the ImmTAC‐redirected antitumour responses.
The programmed cell death protein 1 (PD‐1) /PD‐1 ligand 1 (PD‐L1) pathway represents an important checkpoint limiting the antitumour immune response.21, 22 PD‐1 is known to be expressed on iTreg cells and appears to play a functional role in the de novo generation of CD4+ Foxp3+ Treg cells from naive CD4+ T cells and activation of the PD‐1/PD‐L1 axis can convert human T helper type 1 cells toward a Foxp3+ Treg lineage.23, 24 Expression of Foxp3 is required for iTreg cell development and appears to control a genetic programme specifying the cell fate.25 Although the PD‐1/PD‐L1 pathway drives the differentiation and maintenance of Foxp3+ Treg cells by blocking the Akt/mammalian target of rapamycin pathway, PD‐1 deficiency impairs the expression of FoxP3 in iTreg cells in vivo.26 These results demonstrated that PD‐1/PD‐L1 signalling has a vital role in iTreg cell development and sustaining iTreg cell function. Blockade of the PD‐1/PD‐L1 pathway using anti‐PD‐1 or anti‐PD‐L1 antibodies reverses the suppressive ability of iTreg cells.27
In this study, we first examined whether iTreg cells posed a significant barrier to the antitumour effect of ImmTAC‐redirected CD8+ T cells and second whether a treatment in combination with anti‐PD‐1 antibody could reverse iTreg cell‐mediated immunosuppression of ImmTAC. We further examined this question using samples of peripheral blood from lung cancer patients. The observation can be used as a rationale for embarking on clinical studies combining ImmTAC and immune checkpoint blockade regimens.
Materials and methods
Cell preparation and culture
We enrolled 29 patients with lung cancer in the First Affiliated Hospital of Guangzhou Medical University and 29 healthy donors. The NCI‐H1299 non‐small cell lung cancer cells (HLA‐A*0201, HLA‐A*2402, NY‐ESO‐1 positive determined with nanostring) were a gift from XiangXue Life Sciences Research Centre (XiangXue Pharmaceutical Co. Ltd, Guangzhou, China), and cultured with RPMI‐1640 medium supplemented with 10% fetal bovine serum. We verified the antigen expression and HLA restriction of the cell strain using Western blotting and fluorescence‐activated cell sorting (FACS) (see Supplementary material, Fig. S1). The T2 cells and the T cells were cultured with Iscove's modified Dulbecco's medium supplemented with 10% fetal bovine serum. These cells were all free from mycoplasma detected by polymerase chain reaction every month.
FACS analysis
The cells were washed once with FACS staining buffer and then stained for 30 min at 4° with antibodies specific for human CD4‐fluorescein isothiocyanate (Clone: RPA‐T4, Cat: 555346, BD Bioscience, San Jose, CA), CD25‐phycoerythrin (Clone: M‐A251, Cat: 555432, BD Bioscience), CD45RA‐phycoerythrin‐Cy7 (Clone: HI100, Cat: 560675, BD Bioscience), CD8‐allophycocyanin (Clone: RPA‐T8, Cat: 301014, Biolegend, San Diego, CA), glucocorticoid‐induced tumour necrosis factor receptor (GITR) ‐allophycocyanin (Clone: DT5D3, Cat: 130098575, Miltenyi Biotec, Bergisch Gladbach, Germany), PD‐1‐phycoerythrin (Clone: EH12, Cat: 560795, BD Bioscience), PD‐L1‐fluorescein isothiocyanate (Clone:MIH1, Cat: 558065, BD Bioscience) and CD107a‐Alexa Fluor 647 (Clone:H4A3, Cat: 328612, Biolegend), respectively. For intracellular staining, the cells were fixed, permeabilized, and stained with antibodies against FoxP3‐Alexa Fluor 647 (Clone: 259D/C7, Cat: 560045, BD Bioscience), cytotoxic T‐lymphocyte‐associated protein 4 (CTLA‐4) ‐phycoerythrin (Clone: BN13, 130097684, Miltenyi Biotec), IL‐10‐allophycocyanin (Clone: JES319F1, Cat: 554707, BD Bioscience), Caspase‐3‐Alexa Fluor 647 (Clone: C92‐605, Cat: 560626, BD Bioscience), Granzyme B‐Alexa Fluor 647 (Clone: GB11, Cat: 561999, BD Bioscience), or Perforin‐Peridinin chlorophyll protein‐Cy5.5 (Clone: ŎG9, Cat: 563762, BD Bioscience), respectively.
Induction of human iTreg cells in vitro
The stimulation of CD4+ CD45RA+ CD25− T cells with human T‐activation anti‐CD3/CD28 beads (Dynabeads, Cat: 11131D; Gibco, ThermoFisher, Waltham, MA) at a cell to bead ratio of 3 : 1 was carried out by culturing in 24‐well plates with AIM medium which was supplemented with IL‐2 (400 U/ml, R&D Systems, Minneapolis, MN), TGF‐β 1 (25 ng/ml, R&D) and rapamycin (400 ng/ml, R&D). This cytokine cocktail was referred to as an ‘induction cocktail’ (IC) and the concentration of the CD4+ CD45RA+ CD25− T cells was 2 million/ml at the beginning of the induction assay. The activated CD4+ T‐cell control culture was set similarly with the same concentration of anti‐CD3/CD28 beads and 100 U/ml IL‐2. At day 5, cells were harvested for FACS analysis to detect the expression of FoxP3, or carried on for further experiments.
Enzyme‐linked immunosorbent assay
The iTreg cells were cultured with AIM medium for 2 days, supernatants were collected to detect TGF‐β 1 level using an enzyme‐linked immunosorbent assay (ELISA) Kit (Cat: DKW12‐1710‐096; Dakewe, Guangdong, China). All protocols were conducted according to the manufacturer's instructions.
In vitro suppression assay
The carboxyfluorescein succinimidyl ester (CFSE; Cat: V12883; Invitrogen, Carlsbad, CA) ‐based suppression assay was performed as described previously.28 Briefly, CFSE‐labelled autologous peripheral blood mononuclear cells (PBMCs) were stimulated with anti‐CD3/CD28 monoclonal antibody (mAb) beads at a cell to bead ratio of 3 : 1 and incubated with iTreg/CD4+ T cells at a ratio of PBMCs to iTreg/CD4+ T‐cells of 4 : 1. For the proliferation assays redirected by ImmTAC‐NYE, the CFSE‐labelled autologous PBMCs were cultured with iTreg/CD4+ T cells at a PBMC to iTreg/CD4+ T‐cell ratio of 2 : 1 in the presence of tumour cells and ImmTAC‐NYE. After 3 days, these cells were harvested and stained with allophycocyanin‐conjugated anti‐CD8 antibodies to analyse the proliferation of CD8+ T cells by FACS.28, 29
Transwell and co‐culture assays
Transwell experiment was performed in 96‐well plates with 0·4‐μm pore sizes in inner wells (Cat: 3381; Corning, Corning, NY) to physically separate iTreg cells and the co‐culture of PBMCs with NCI‐H1299 cells in the presence of ImmTAC‐NYE (1 × 10−9 m). The co‐cultures of PBMCs (1 × 105) with NCI‐H1299 cells (2 × 104) were grown in 175 μl RPMI‐1640 medium containing 10% fetal bovine serum using the outer wells. A total of 1 × 105 iTreg cells was added into the inner wells in 50 μl of the same medium and the plates were incubated for 48 hr. Then the co‐cultures of PBMCs with NCI‐H1299 cells were collected for detection of the expression of CD107a. This co‐culture experiment was performed in 96‐well round plates containing the iTreg cells (1 × 105) cultured together with PBMCs (1 × 105) and NCI‐H1299 cells (2 × 104) for 48 hr in the presence of ImmTAC‐NYE (1 × 10−9 m). These cells from the co‐culture system were collected for detection of the expression of CD107a.
Cytotoxic T lymphocyte assays
The CytoTox96 Non‐Radioactive Cytotoxicity Assay kit (Cat: G1782, Promega, Madison, WI) was used to determine the CTL activity of PBMCs. In brief, triplicate wells containing 1 × 105 cells/well autologous PBMCs at an effector to target (NCI‐H1299, T2) ratio of 5 : 1 were incubated with/without iTreg/CD4+ T cells in the presence of ImmTAC‐NYE (10−9 m) or anti‐PD‐1 mAb (WuXi AppTec, Shanghai, China; 10 μg/ml) for 24 hr, iTreg/CD4+ T cells : PBMC = 1 : 2. T2 cells were loaded with NY‐ESO‐1157–165 peptide (SLLMWITQC; GenScript, Piscataway, NJ) or gp100280–288 (YLEPGPVTV; GenScript) at final concentration of 10−7 m for 30 min at 37°. The detection of lactate dehydrogenase in the supernatant was completed following the manufacturer's instructions, and results were calculated with the formula: Cytotoxicity = (Experimental – Effector Spontaneous – Target Spontaneous)/(Target Maximum – Target Spontaneous) × 100%.
Statistical analysis
All graphics and statistical analyses were performed using graphpad prism 5 (GraphPad, San Diego, CA). Unpaired two‐tailed t‐tests were performed using the Student's t‐test; Significances are indicated as *P < 0·05, **P < 0·01, ***P < 0·001, and NS, P > 0·05. Data are representative of at least three independent experiments and are means ± SEM.
Results
Induction and characterization of human iTreg cells
To examine the role of iTreg cells in vitro, a standard protocol was used to generate iTreg cells from the peripheral blood. A population of CD4+ CD45RA+ CD25− T cells (see Supplementary material, Fig. S2) were cultured with 400 U/ml IL‐2, 25 ng/ml TGF‐β 1 and 400 ng/ml rapamycin, which was referred to as the ‘induction cocktail’ or ‘IC’.10, 11 After 5 days of culture in AIM medium containing IC, the FoxP3 expression of iTreg cells was detected by FACS. Figure 1(a) shows the condition of the induced iTreg cells from naive CD4+ T cells. We verified the induction efficiency on 10 isolated healthy naive CD4+ T‐cell samples. The iTreg populations were detected as 78·7% on average (range 67·3–90·5%) expressing FoxP3 on day 5 with IC, which was significantly higher than the 19·98% (range 14·1–26·7%) for control cells stimulated only by the anti‐CD3/CD28 beads without the IC (Fig. 1b).
Figure 1.
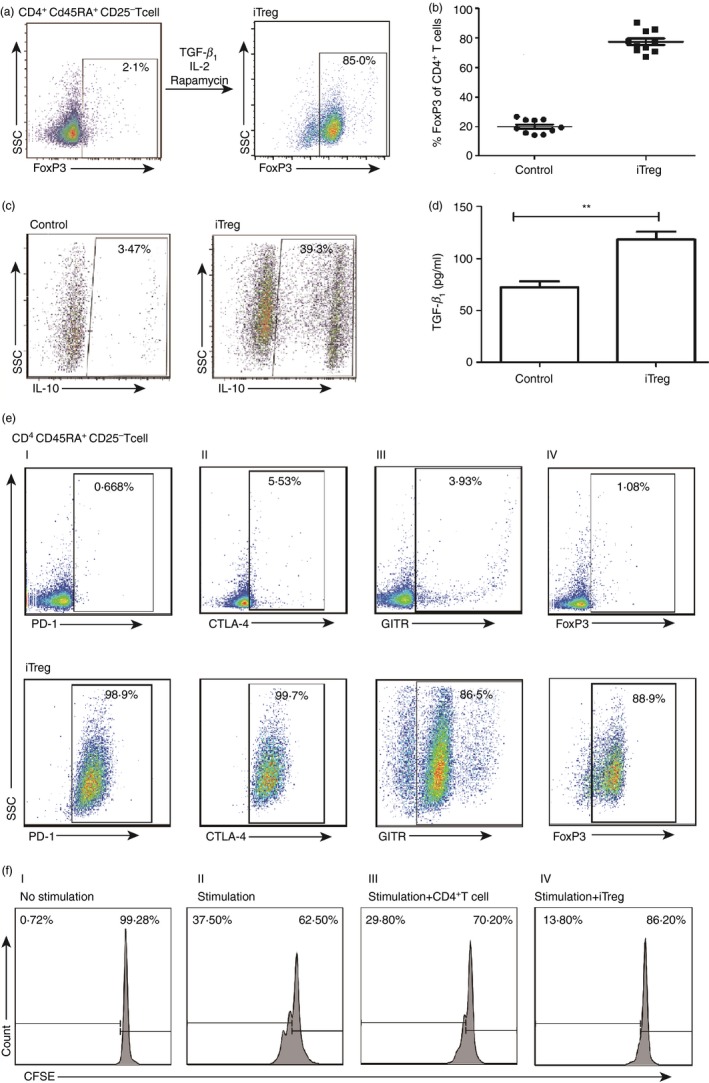
Induction and characterization of human induced regulatory T (iTreg) cells. (a) A representative flow plot to show the induction conditions of the iTreg cells. The CD45RA + CD4+ CD25− T cells, stimulated with anti‐CD3/CD28 monoclonal antibody beads at the cell to bead ratio of 3 : 1, were cultured with the ‘induction cocktail’ (IC). The Foxhead box protein 3 (FoxP3) expression was assessed at day 5 by FACS. (b) FoxP3 expression by the iTreg cells and control cells from 10 different healthy donors. Scattered dot plots depict the percentages of FoxP3 expression in cells treated with IC or without IC (n = 10), each symbol represents one donor. (c) The interleukin‐10 (IL‐10) expression by the iTreg cells. The iTregs and control cells were assessed for secretion of IL‐10 by FACS. (d) The transforming growth factor‐β 1 (TGF‐β 1) secretion by the iTreg cells. The secretion of TGF‐β 1 by the iTreg cells and control cells was detected by ELISA after culturing in AIM medium for 48 hr. Unpaired two‐tailed t‐test, **P < 0·01. (e) Expression of inhibitory markers by the iTreg cells. The expression of inhibitory markers by the iTreg cells was assessed by FACS (lower panel), for programmed cell death protein 1 (PD‐1) (I), cytotoxic T‐lymphocyte associated protein 4 (CTLA‐4) (II), glucocorticoid‐induced tumour necrosis factor receptor (GITR) (III) and FoxP3 (IV), respectively. CD45RA + CD4+ CD25− T cells without induction were used as negative control (upper panel). (f) Suppression of CD8+ T‐cell proliferation by the iTreg cells. The in vitro suppressive activity of the iTreg cells was measured by mixing CFSE‐labelled peripheral blood mononuclear cells (PBMCs) with the iTreg cells, using ratios of PBMC : anti‐CD3/CD28 beads of 3 : 1 and iTreg/CD4+ T cells : PBMC of 1 : 4. After culturing for 3 days, the CFSE dilution was measured by FACS for the determination of CD8+ T‐cell proliferation.
To analyse the suppressive phenotype of the cells, intracellular IL‐10 was measured. Results showed that an average of 39·3% of cells produced IL‐10 in the iTreg cell population, which was over 10‐fold higher than the control cells (Fig. 1c). Similarly, we detected TGF‐β 1 levels in the culture supernatant by ELISA after the cells were cultured with AIM medium for 2 days. The results showed that amounts of TGF‐β 1 secreted from the iTreg population were significantly higher than from the control cells (Fig. 1d). We analysed the expression of Treg cell‐related immunosuppressive cell markers by FACS,30, 31, 32, 33 and found that the iTreg populations expressing GITR, PD‐1, CTLA‐4 and FoxP3 were 86·5%, 98·9%, 99·7% and 88·9%, respectively (Fig. 1e). The negative control was CD4+ CD45RA+ CD25− T cells without IC induction, and results showed significantly lower expression levels for all these markers. Finally, the functional suppressive capacity of the iTreg cells was analysed using an in vitro proliferation assay, in which CFSE‐labelled autologous PBMCs stimulated with anti‐CD3/CD28 mAb beads were cultured with iTreg cells or activated CD4+ T cells without the IC induction at cell to PBMC ratios of 1 : 4. The viability of iTreg cells and control CD4+ T cells were similar as detected by FACS (see Supplementary material, Fig. S3). The fraction of proliferating cells among stimulation of CD8+ T cells was 37·50%, whereas cultures exposed to the iTreg cells yielded a proliferating population of only 13·80%, but in the control CD4+ T cells yielded 29·80% proliferation (Fig. 1f). Hence, the iTreg cells had an obviously superior suppressive capability in comparison to activated CD4+ T cells.
iTreg cells suppressed CTL killing redirected by ImmTAC‐NYE
In this study, we refolded and purified the ImmTAC construct using previously generated high‐affinity TCR (KD = 26 pm) specific for the HLA‐A*0201 restricted epitope of NY‐ESO‐1157–165,18 and named as ImmTAC‐NYE. We demonstrated that the ImmTAC‐NYE could redirect effector cells specifically to release interferon‐γ for the lysis of the target cells including both the antigen peptide loaded T2 (see Supplementary material, Fig. S4A,B) and NCI‐H1299 cells (see Supplementary material, Fig. S4C,D).
To evaluate the suppressive capacity of the iTreg cells, we examined the ability of in vitro induced iTreg cells to suppress ImmTAC‐NYE‐redirected target killing of NCI‐H1299 cells or NY‐ESO‐1 peptide pulsed T2 cells. First, iTreg cells and activated CD4+ T cells without induction were added at the ratios of testing cell : PBMC of 1 : 2 to the co‐culture system containing PBMCs and target cells in the presence of ImmTAC‐NYE. The specific lysis results showed that activated CD4+ T cells without IC induction did not inhibit the lysis of NCI‐H1299 cells and NY‐ESO‐1157–165‐pulsed T2 cells. However, iTreg cells did inhibit the ImmTAC‐NYE‐redirected killing significantly in comparison to that of the activated CD4+ T cells (Fig. 2a,b). The supplement of the activated CD4+ T cells might in fact promote the target cell lysis for both the peptide loaded T2 and NCI‐H1299 cells, although without significance. Further investigation about the inhibition of ImmTAC‐NYE redirected killing was also carried out with a range of different iTreg cells to PBMC ratios from 4 : 1 to 1 : 64. Results showed that even with the iTreg to PBMC ratio set to 1 : 64, suppression of ImmTAC‐NYE‐redirected killing could also be observed (see Supplementary material, Fig. S5).
Figure 2.
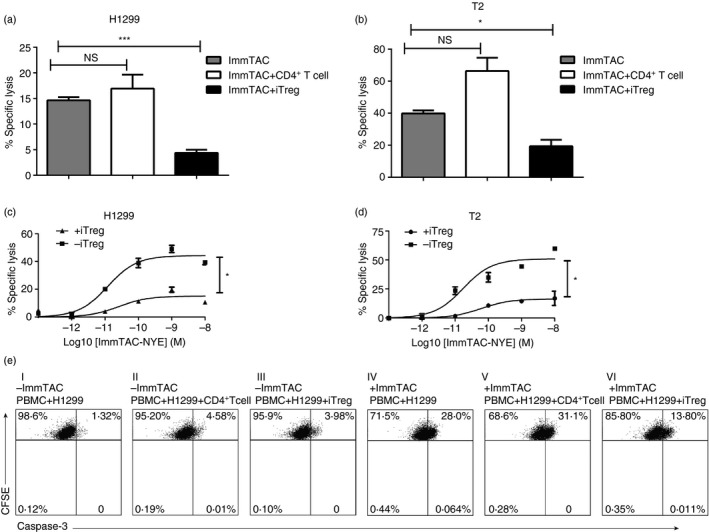
Induced regulatory T (iTreg) cells suppressed cytotoxic T lymphocyte (CTL) killing redirected by ImmTAC‐NYE. (a) iTreg cells suppressed ImmTAC‐NYE‐redirected NCI‐H1299 cell lysis. Activated CD4+ T cells/iTreg cells : peripheral blood mononuclear cells (PBMC) at ratio of 1 : 2. (b) iTreg cells suppressed the ImmTAC‐NYE‐redirected T‐cell killing for T2 cells at a ratio of iTreg/activated CD4+ T cell to PBMC of 1 : 2. (c, d) CTL assays showing the killing activity redirected by titrated concentrations of the ImmTAC‐NYE with or without the presence of the iTreg cells at a PBMC to iTreg cell ratio of 2 : 1. The iTreg cells suppressed the lysis of the NCI‐H1299 cells (HLA‐A*0201, NY‐ESO‐1‐positive) (c) or T2 cells pulsed with NY‐ESO‐1157–165 peptide at a concentration of 10−7 m (d). Unpaired two‐tailed t‐test, *P < 0·05. (e) Representative flow charts showed that the iTreg cells could suppress cleaved caspase‐3 expression of the NCI‐H1299 cells during the CTL assays. The iTreg/CD4+ T cell : PBMC ratio was set as 1 : 2 (I to VI).
Assays of CTLs were performed in the presence or absence of iTreg cells with ImmTAC‐NYE concentrations ranging from 10−8 m to 10−13 m and a 1 : 2 ratio of iTreg cells to PBMCs. Interestingly, the iTreg cells could inhibit ImmTAC‐NYE‐redirected killing of NCI‐H1299 cells and NY‐ESO‐1157–165‐pulsed T2 cells at all concentrations of ImmTAC‐NYE used in these assays (Fig. 2c,d). The values of lysis EC50 were increased in the presence of iTreg cells from 3·951 × 10−11 to 1·953 × 10−10 for the T2 cells, and 1·856 × 10−11 to 1·179 × 10−10 for the NCI‐H1299 cells.
Finally, to further demonstrate the iTreg cell suppressive function against tumour cell killing, we verified cleaved caspase‐3 expression by NCI‐H1299 cells in ImmTAC‐NYE‐redirected CTL assays using an iTreg to PBMC ratio of 1 : 2. Activated CD4+ T cells without the IC‐induction were used as a control. The NCI‐H1299 cells were stained for verifying the true source of cleaved caspase‐3 by gating the CFSE‐positive cells. The result showed that the iTreg cells, but not the activated CD4+ T cells without the IC‐induction were able to inhibit the expression of cleaved caspase‐3 of the NCI‐H1299 cells (Fig. 2e).
Mechanisms by which iTreg cells suppressed ImmTAC‐NYE‐redirected CTL killing
To unveil the mechanism whereby the iTreg cells suppressing the function of ImmTAC‐NYE redirected T cells, the CD8+ T‐cell proliferation was studied. We added the ImmTAC‐NYE to cell mixtures of CFSE‐labelled PBMCs (effector) and NCI‐H1299 cells (target) at a ratio of 5 : 1 and cultured them for 3 days with or without the autologous iTreg cells at a testing cell to PBMC ratio of 1 : 2. We analysed the CFSE dilution by gating CD8+ T cells. A separate control assay was set with the CD4+ T cells activated without the IC induction at the same ratio. The fraction of proliferating cells among ImmTAC‐NYE‐redirected CD8+ T cells was 93·80%, whereas cultures exposed to iTreg cells yielded a proliferating population of only 3·16%, cultures exposed to activated CD4+ T cells yielded a proliferating population of 24·60% (Fig. 3a). These data indicated significant iTreg cell suppressive effects on the ImmTAC‐NYE redirected and activated CTL in the current conditions.
Figure 3.
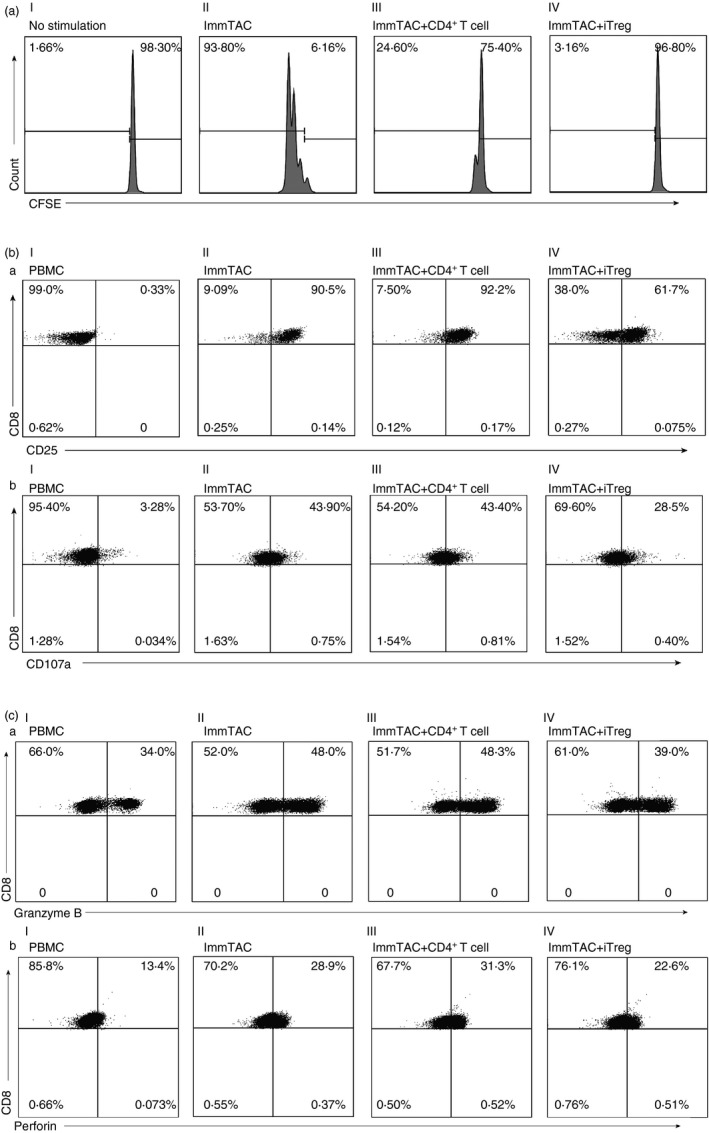
Mechanisms by which induced regulatory T (iTreg) cell suppressed ImmTAC‐NYE‐redirected cytotoxic T lymphocyte (CTL) killing. (a) The iTreg cells suppressed the proliferation of CD8+ T cells redirected by the ImmTAC‐NYE. A ratio of iTreg/activated CD4+ T cell : peripheral blood mononuclear cell (PBMC) of 1 : 2. The CFSE‐labelled PBMC was used as negative control. The CFSE intensity was determined after the cells were gated with anti‐CD8 antibody staining. (b, c) The PBMC was cultured with NCI‐H1299 cells at an effector to target ratio of 5 : 1, with or without the iTreg/activated CD4+ T cell at a 2 : 1 ratio of PBMC to iTreg/activated CD4+ T cell in the presence of ImmTAC‐NYE (10−9 m) over a 24‐hr period. Cultured cells were collected, after gating with anti‐CD8 antibody staining, the expression of CD25, CD107a (b), Granzyme B, Perforin (c) were determined for the CD8+ T cells. ImmTAC group: ImmTAC‐NYE + PBMC + NCI‐H1299; ImmTAC + CD4+ T‐cell group: ImmTAC‐NYE + PBMC + NCI‐H1299 + activated CD4+ T cells without induction; ImmTAC + iTreg group: ImmTAC‐NYE + PBMC + NCI‐H1299 + iTreg cells.
In addition, we investigated the effect of iTreg cells on the expression levels of several T‐cell‐activated and cytolytic markers. ImmTAC‐NYE (10−9 m) ‐redirected CTL assays were carried out by culturing for 24 hr at an effector : target ratio of 5 : 1 with or without the iTreg cells or the activated CD4+ T cells. These co‐culture cells were collected and stained with the corresponding antibodies for analysis of the expression of CD25, CD107a, Granzyme B and Perforin by the CD8+ T effector cells. Results showed that the expression of CD25, CD107a, Granzyme B and Perforin were decreased significantly in the CD8+ T cells when treated with the iTreg cells, but not the activated CD4+ T cells without induction during the CTL assays (Fig. 3b,c). By summarizing the result of three experiments, we could see a significant decrease in the expression of activated/cytolytic markers by CD8+ T cells with the presence of iTreg cells (see Supplementary material, Fig. S6). These results indicated that the iTreg cells suppressed the proliferation, activation and cytolytic functions of the T cells.
Anti‐PD‐1 mAb could reverse the suppressive effect of iTreg cells on ImmTAC‐NYE‐redirected cell killing
To investigate the relation between the iTreg cell function and the PD‐1 axis, we assessed the effect of blocking the PD‐1 pathway on both the iTreg cell conversion and the suppression of the ImmTAC‐NYE‐redirected CTL responses. First, we investigated whether the PD‐1 blockade could reduce the conversion of the iTreg cells during the induction of CD45RA+ CD4+ CD25− T cells with the IC. We analysed the FoxP3 expression of the T cells after a 5‐day induction with 10 μg/ml anti‐PD‐1 mAb added. The result showed that the conversion of the cells to CD4+ Foxp3+ iTreg cells was decreased by the addition of anti‐PD‐1 mAb (Fig. 4a). Second, we analysed the FoxP3 expression in the iTreg cells cultured with the supplement of 10 μg/ml anti‐PD‐1 mAb for a further 2 days after the 5‐day induction with just IC. In this case, we found that the addition of anti‐PD‐1 mAb could reduce the FoxP3 expression in already converted iTreg cells (Fig. 4b), indicating that the stability of the iTreg cells could be reduced by the blockade of the PD‐1 pathway. These results demonstrated that the anti‐PD‐1 mAb might potentially overturn the conversion and stability of iTreg cells in the immune‐suppressive microenvironment.
Figure 4.
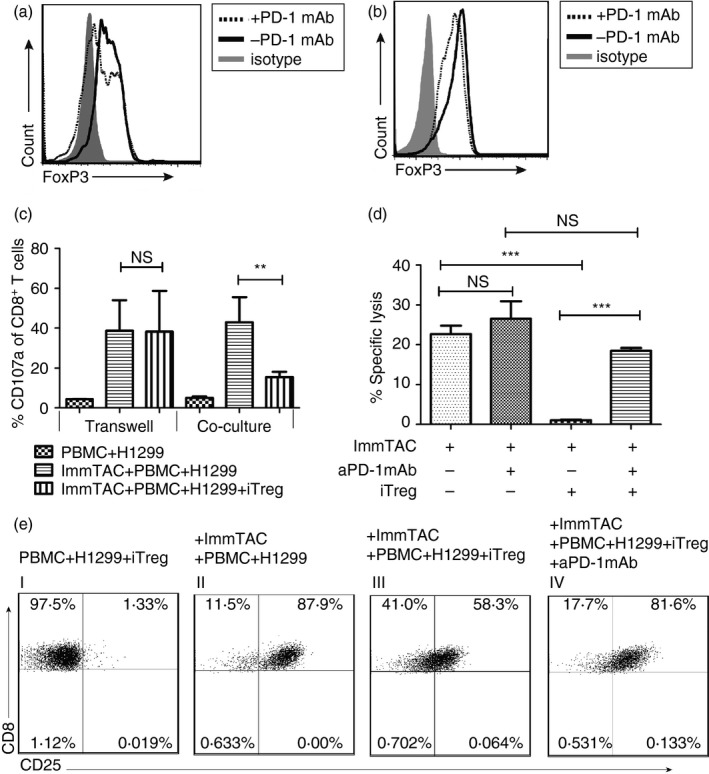
Anti‐programmed cell death protein 1 (PD‐1) monoclonal antibody (mAb) could reverse the suppressive effect of induced regulatory T (iTreg) cells on ImmTAC‐NYE‐redirected cell killing. (a) The Foxhead box protein 3 (FoxP3) expression of the iTreg cells was down‐regulated by an anti‐PD‐1 mAb (10 μg/ml) (black dotted line) during the induction of CD4+ CD45RA + CD25− T cells with the ‘induction cocktail’ (IC), in comparison with the absence of the antibody (black solid line). (b) The anti‐PD1 antibody could destabilize the expression of FoxP3 in the iTreg cells. After a complete induction, the induced iTreg cells was cultured with (black dotted line) or without (black solid line) anti‐PD‐1 mAb (10 μg/ml) for a further 2 days, then the FoxP3 expression was detected by FACS. The grey dashed area was an isotype control for the FoxP3 antibody. (c) Comparison of the iTreg cell inhibition of the CD107a expression by the ImmTAC‐NYE‐stimulated T cells in the transwell and co‐culture system. The cytotoxic T‐lymphocytes (CTL) assays; CTLs in the presence of iTreg cells [ratio of peripheral blood mononuclear cell (PBMC) to iTreg cells = 1 : 1] were cultured in 96‐well transwell or round‐bottom plates (co‐culture), 48 hr later, cells were collected to detect CD107a expression after gating the CD8‐positive cells by FACS. (d) The anti‐PD‐1 antibody reversed the iTreg cell suppressive effects on the ImmTAC‐NYE redirected tumour cell killing. After the anti‐PD‐1 antibody (10 μg/ml) was added to the CTL assays in the presence of iTreg cells (PBMC : iTreg cells = 1 : 1) over a 24‐hr period, lactate dehydrogenase (LDH) release was detected. Unpaired two‐tailed t‐test, ***P < 0·001, NS P > 0·05. (e) The anti‐PD‐1 mAb enhanced CD25 expression of CD8+ T cells suppressed by the iTreg cells. The cultured cells in the CTL assays were collected for the detection of the CD25 expression by CD8+ T cells after gating on CD8+ T cells by staining with anti‐CD8 antibody.
Because cancer cells usually express high levels of PD‐L1, they can efficiently trigger suppressive signals on activated T cells that normally up‐regulate PD‐1 surface expression. It is not surprising that 35·0% of the cells in the lung cancer cell line of NCI‐H1299 constitutively express PD‐L1 (see Supplementary material, Fig. S7A). Additionally, transwell assay showed that the iTreg cells exerted the suppressive ability mainly through cell‐to‐cell contact (Fig. 4c), in comparison with the effector and iTreg cell co‐culture assay, we observed no significant reduction of CD107a expression in the transwell assay, where the iTreg cells could not contact the effector cells. These data indicate that the cell‐to‐cell contact mechanism acting through the PD‐1 pathway might play an important role in the suppressive activities. Therefore, we investigated whether PD‐1 pathway blockade with the anti‐PD‐1 mAb could reverse the iTreg cell suppressive effects on killing NCI‐H1299 cells by ImmTAC‐NYE‐redirected T cells. The result showed that the ImmTAC‐NYE combined with the anti‐PD‐1 mAb could enhance the CTL response to NCI‐H1299 cells. Most importantly, the killing effect observed with the combination of ImmTAC‐NYE and anti‐PD‐1 mAb did not show significant reduction with the supplement of iTreg cells (Fig. 4d). In comparison to ImmTAC‐NYE with only iTreg cells added, the addition of anti‐PD‐1 mAb recovered over 81% of the tumour lysis by the redirected CD8+ T cells (or increased killing 1000‐fold over the test with no PD‐1 blockade). In addition, the CD25 expression of the CD8+ T cells increased significantly from 58·3% to 81·6% detected in the assays with the addition of the PD‐1 blockade in the presence of iTreg cells (Fig. 4e). By summarizing three experiment results, we could see a significant increase in CD25 expression of CD8+ T cells with the presence of anti‐PD‐1 mAb (see Supplementary material, Fig. S7B). The data indicated that the PD‐1 pathway blockade with anti‐PD‐1 mAb could recover the activation phenotype of CD8+ T cells.
Comparison of the killing by ImmTAC‐NYE‐redirected CTLs between lung cancer patients and healthy donors
To determine whether PD1 blockade may have a role in combination with ImmTAC therapy, we collected PBMCs from 29 lung cancer patients and 29 healthy donors and evaluated the effectiveness of cancer cell killing by the ImmTAC‐NYE‐redirected T cells. The result of ImmTAC‐NYE redirected lactate dehydrogenase release showed that the specific cancer cell lysis by the PBMCs was usually lower than that of healthy donors (Fig. 5a), and the percentage of the CD4+ FoxP3+ Treg cells in these lung cancer patients’ PBMCs was significantly higher than that in healthy donors (Fig. 5b). Therefore, we wondered whether Treg cells were one of the dominant mechanisms for suppressing the ImmTAC‐redirected CTL response in lung cancer patients.
Figure 5.
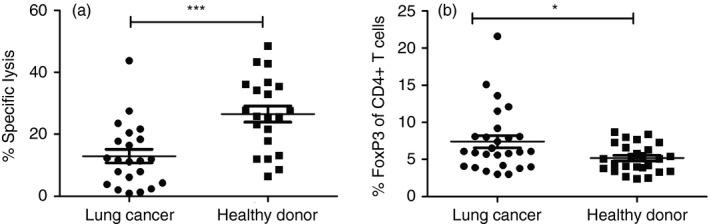
Comparison of the killing by ImmTAC‐NYE redirected cytotoxic T lymphocytes (CTLs) between lung cancer patients and healthy donors. (a) Scattered dot plots depicted the specific lysis between lung cancer patients and healthy donors. CTL assays were performed using effector T cells [peripheral blood mononuclear cell (PBMC) of lung cancer patients or healthy donors, 1 × 105/well] and target cells (lung cancer cell NCI‐H1299, 2 × 104/well) in the presence of ImmTAC‐NYE (10−9 m), lactate dehydrogenase (LDH) release was detected after incubation of the cell mixture for 24 hr, with samples of lung cancer patients (n = 22) or healthy donors (n = 21). (b) Comparison of the CD4+ FoxP3+ Treg populations in the peripheral blood between healthy donors (n = 27) and lung cancer patients (n = 27). Unpaired two‐tailed t‐tests, *P < 0·05, ***P < 0·001.
Combination ImmTAC‐NYE with anti‐PD‐1 mAb produced synergistic tumour cell killing in vitro for lung cancer patients
To better understand the mechanism of anti‐PD‐1 mAb overcoming the suppressive effects of the iTreg cells, we detected PD‐1 expression levels on the PBMCs from lung cancer patients and healthy donors. We found that the PD‐1 expression levels of CD4+ T cells from the patient group were up‐regulated in comparison to the healthy donors (Fig. 6a). When anti‐PD‐1 antibody was added to the CTL assay co‐culture system, the enhancement of NCI‐H1299 cell lysis in the lung cancer patient group was higher than in the healthy donors. Significantly, one patient sample showed that the PD‐1 blockade enhanced the killing nearly eightfold in comparison to almost no change for a representative healthy control (Fig. 6b,c).
Figure 6.
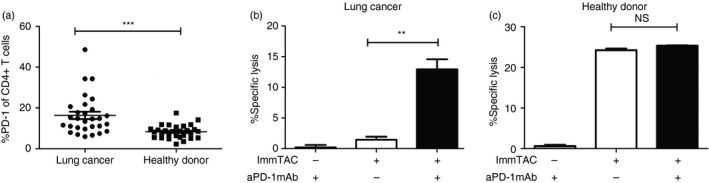
Combination ImmTAC‐NYE with anti‐programmed cell death protein 1 (PD‐1) monoclonal antibody (mAb) produced synergistic tumour cell killing in vitro for lung cancer patients. (a) Comparison of the PD‐1 expression level of CD4+ T cells in the peripheral blood between healthy donors (n = 29) and lung cancer patients (n = 29). P value was calculated using unpaired Student's t‐test. Significance is indicated as *P < 0·05, ***P < 0·001. (b, c) The anti‐PD‐1 mAb enhanced the ImmTAC‐NYE‐redirected cell killing in lung cancer patients and healthy donors. The peripheral blood mononuclear cells (PBMCs) (1 × 105/well) were cultured for 24 hr with NCI‐H1299 (2 × 104/well) cells at a 5 : 1 effector to target ratio in the presence of ImmTAC‐NYE (10−9 m) and anti‐PD‐1 mAb (10 μg/ml) was added or not. The co‐culture system without ImmTAC‐NYE was used as negative control. Lactate dehydrogenase release was detected for lung cancer patients (b) or healthy donors (c). Unpaired Student's t‐test, **P < 0·01, NS P > 0·05.
Discussion
Three fundamental immunotherapeutic strategies are emerging as important anti‐tumour approaches: (i) chimeric antibody receptor or TCR engineered T cells,34, 35 (ii) bi‐functional molecules such as BiTe,36 ImmTAC19 or HATac to redirect T cells to kill tumour cells,37 and (iii) blocking immunosuppressive mechanisms using immune checkpoint reagents such as anti‐PD‐1,38 anti‐PD‐L139 or anti‐CTLA4 antibodies.40 ImmTAC has been shown to redirect T cells to kill target tumour cells in vitro and in vivo with promising results. A previous study demonstrated that ImmTAC‐gp100 could redirect and activate T cells to directly kill melanoma cells even in the presence of Treg cells;41 however, the use of unstimulated nTreg cells in that study may have weak suppressive capacity. Our own studies confirmed that the use of unstimulated nTreg cells from healthy donors failed to demonstrate suppressive capability, but activated nTreg cells could mediate strong suppressive effects (unpublished data). The thymus‐derived nTreg cells are responsible for peripheral tolerance in healthy individuals, and in pathological situations, nTreg cells are largely replaced by iTreg cells, which are the major participants in shaping the immune response in situ.14 Therefore, iTreg cells can play a critical role in tumour immunosuppression. We used established in vitro methods to generate iTreg cells, which suppressed the activation and proliferation of ImmTAC‐NYE‐redirected T cells, and down‐regulated the expression of CD25, CD107a, Granzyme B and Perforin. The results suggested that the strong immunosuppressive function of iTreg cells would become a hurdle for such immunotherapy.
In addition, it has been suggested recently that the expression of PD‐1 on Treg cells contributed to the suppressive capability in vitro.42 Studies have shown that PD‐L1‐immunoglobulin‐coated beads could promote not only the development, but also the suppressive capacity of PD‐1‐expressing iTreg cells in vitro.24 In vitro Treg cell suppression assays have suggested that PD‐1−/− Treg cells showed weakened suppression of the CD8+ T‐cell proliferation and cytokine production.43 Taken together, we postulated that inhibition of this immune checkpoint pathway may override the suppressive effect of iTreg cells on the ImmTAC‐NYE redirected CTL response. The transwell assays in this study indicated that the iTreg cells wielded their suppressive effects through cell‐to‐cell contact, and co‐cultivation of the ImmTAC‐NYE‐redirected CTL and iTreg cells in the presence of PD‐1 blockade was able to reverse iTreg cell‐mediated suppression of the response. As activated conventional T cells are known to express PD‐L1, it may confirm that anti‐PD‐1 mAb blockades the interactions between PD‐1‐expressing iTreg cells and PD‐L1‐expressing conventional T cells, leading to the relief of immune tolerance built by the PD‐1 axis. As the immune checkpoint protein PD‐1 is also expressed on cytotoxic T cells, anti‐PD‐1 mAb may have a dual role in blocking immune checkpoints on both cytotoxic T cells and Treg cells to enhance the ImmTAC‐NYE redirected cancer cell killing. As previous results showed that the function and development of iTreg cells were regulated by the PD‐1/PD‐L1 pathway, binding of anti‐PD‐1 mAb could decrease the expression of FoxP3 during the induction of iTreg cells and accelerate the loss of FoxP3 of iTreg cells. Previous investigations had demonstrated that Akt signalling was essential for naive T‐cell activation and proliferation, furthermore, PD‐1/PD‐L1 interactions down‐regulated the Akt‐mammalian target of rapamycin signalling axis during stimulation of CD4+ T cells, promoting differentiation of CD4+ T cells into iTreg cells.24 Hence, we suspected that blockade of PD‐1 signalling might restore Akt signalling, which assisted activation and proliferation of conventional CD4+ T cells rather than converting them into iTreg cells. In addition, the lack of PD‐1 signalling in stimulated CD4+ T cells led to enhanced signal strength through the TCR, which was previously shown to inhibit iTreg cell generation in a nuclear factor‐κB‐dependent manner even in the presence of TGF‐β and IL‐2.44 This is another explanation for how the blockade of PD‐1 axis with antibodies can enhance the ImmTAC‐NYE‐redirected CTL responses during the presence of iTreg cells. Therefore, blocking of the PD‐1 pathway should down‐regulate FoxP3 expression. This also suggests that PD‐1 signalling may play a key role in TGF‐β‐induced Treg cell conversion, strengthening the evidence for a link between PD‐1 and TGF‐β signalling in iTreg cells.
Finally, we demonstrated that the use of ImmTAC‐NYE‐redirected CTLs from PBMCs of lung cancer patients yielded significantly lower lytic capacity than those from PBMCs from healthy donors. This might be attributable to the higher iTreg cell level in the lung cancer patients compared with the healthy donors. The PBMCs from both the lung cancer patients and the healthy donors demonstrated enhanced ImmTAC‐NYE‐redirected T‐cell activity with the presence of anti‐PD‐1 mAb in comparison without the antibody, but the extent of the enhancement was significantly greater for the lung cancer patients than for the healthy donors. As FoxP3 and PD‐1 were all up‐regulated in lung cancer patients, anti‐PD‐1 mAb was able to reverse the up‐regulated iTreg cell‐mediated suppression by blocking up‐regulated PD‐1 signalling and down‐regulating FoxP3 expression. These results suggested that the combination of ImmTAC‐NYE with anti‐PD‐1 mAb might produce a synergistic anti‐tumour effect in lung cancer patients for breaking iTreg cell‐mediated immunosuppression. Blockade of the PD‐1/PD‐L1 pathway with checkpoint‐specific antibodies in lung cancer patients might enhance anti‐tumour T‐cell responses not only through re‐activation of effector T cells but also by preventing the suppression of iTreg cells. These findings are of particular importance for clinical studies combining the ImmTAC and immune checkpoint blockade regimens.
Disclosures
The authors declare no potential conflicts of interest.
Supporting information
Figure S1. Identification of NCI‐H1299 cell line.
Figure S2. Naive CD4+ T‐cell purity was detected by FACS.
Figure S3. The viability of induced regulatory T cells and activated CD4+ T cells were detected by FACS.
Figure S4. ImmTAC‐NYE redirected and activated T cells to lyse tumour cells.
Figure S5. Induced regulatory T cells suppressed cytotoxic T lymphocyte killing redirected by ImmTAC‐NYE.
Figure S6. Mechanisms by which induced regulatory T cells suppressed ImmTAC‐NYE‐redirected cytotoxic T lymphocyte killing.
Figure S7. Programmed cell death protein ligand 1 (PD‐L1) expression detected by FACS and anti‐PD‐1 monoclonal antibody could reverse the suppressive effect of induced regulatory T cells on ImmTAC‐NYE redirected cell killing.
Acknowledgements
This study was supported by the Guangzhou Science Technology and Innovation Commission Project Grants 201504010016, The National key R&D Programme (2016YFC1303404) and The Science and Technology Programme of Guangzhou (201704020220) and National Natural Science Foundation of China to X.L. (81301798). Conception and design: Yi Li, Huanling Zhang, Mengyong Yan; development of methodology: Yi Li, Huanling Zhang, Yanyan Li, Zhaoduan Liang; contribution of critical reagents: Qiang Liu, Anan Chen, Yifeng Bao, Chengzhi Zhou, Shiyue Li; acquisition of data: Huanling Zhang; analysis and interpretation of data: Yi Li, Huanling Zhang. Writing; and review and/or revision of the manuscript: Yi Li, Huanling Zhang, Xiaoping Liu, Mengyong Yan,Cassian Yee. We thank members of the Guangzhou Xiangxue Life Sciences Research Centre for helpful suggestions.
References
- 1. Nguyen AH, Berim IG, Agrawal DK. Cellular and molecular immunology of lung cancer: therapeutic implications. Expert Rev Clin Immunol 2014; 10:1711–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gooden MJ, de Bock GH, Leffers N, Daemen T, Nijman HW. The prognostic influence of tumour‐infiltrating lymphocytes in cancer: a systematic review with meta‐analysis. Br J Cancer 2011; 105:93–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Curiel TJ. Tregs and rethinking cancer immunotherapy. J Clin Invest 2007; 117:1167–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Oleinika K, Nibbs RJ, Graham GJ, Fraser AR. Suppression, subversion and escape: the role of regulatory T cells in cancer progression. Clin Exp Immunol 2013; 171:36–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mattarollo SR, Steegh K, Li M, Duret H, Foong Ngiow S, Smyth MJ. Transient Foxp3+ regulatory T‐cell depletion enhances therapeutic anticancer vaccination targeting the immune‐stimulatory properties of NKT cells. Immunol Cell Biol 2013; 91:105–14. [DOI] [PubMed] [Google Scholar]
- 6. Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P et al Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med 2004; 10:942–9. [DOI] [PubMed] [Google Scholar]
- 7. Shang B, Liu Y, Jiang SJ, Liu Y. Prognostic value of tumor‐infiltrating FoxP3+ regulatory T cells in cancers: a systematic review and meta‐analysis. Sci Rep 2015; 5:15179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Singh AK, Seavey CN, Horvath KA, Mohiuddin MM. Ex‐vivo expanded baboon CD4+ CD25Hi Treg cells suppress baboon anti‐pig T and B cell immune response. Xenotransplantation 2012; 19:102–11. [DOI] [PubMed] [Google Scholar]
- 9. Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N et al Conversion of peripheral CD4+ CD25– naive T cells to CD4+ CD25+ regulatory T cells by TGF‐β induction of transcription factor Foxp3. J Exp Med 2003; 198:1875–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fu S, Zhang N, Yopp AC, Chen D, Mao M, Chen D et al TGF‐β induces Foxp3+ T‐regulatory cells from CD4+ CD25– precursors. Am J Transplant 2004; 4:1614–27. [DOI] [PubMed] [Google Scholar]
- 11. Hadaschik EN, Enk AH. TGF‐β1‐induced regulatory T cells. Hum Immunol 2015; 76:561–4. [DOI] [PubMed] [Google Scholar]
- 12. Waight JD, Takai S, Marelli B, Qin G, Hance KW, Zhang D et al Cutting edge: epigenetic regulation of Foxp3 defines a stable population of CD4+ regulatory T cells in tumors from mice and humans. J Immunol 2015; 194:878–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Haribhai D, Williams JB, Jia S, Nickerson D, Schmitt EG, Edwards B et al A requisite role for induced regulatory T cells in tolerance based on expanding antigen receptor diversity. Immunity 2011; 35:109–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Whiteside TL. Regulatory T cell subsets in human cancer: are they regulating for or against tumor progression? Cancer Immunol Immunother 2014; 63:67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhou G, Levitsky HI. Natural regulatory T cells and de novo‐induced regulatory T cells contribute independently to tumor‐specific tolerance. J Immunol 2007; 178:2155–62. [DOI] [PubMed] [Google Scholar]
- 16. Whiteside TL, Schuler P, Schilling B. Induced and natural regulatory T cells in human cancer. Expert Opin Biol Ther 2012; 12:1383–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science 2011; 331:1565–70. [DOI] [PubMed] [Google Scholar]
- 18. Li Y, Moysey R, Molloy PE, Vuidepot AL, Mahon T, Baston E et al Directed evolution of human T‐cell receptors with picomolar affinities by phage display. Nat Biotechnol 2005; 23:349–54. [DOI] [PubMed] [Google Scholar]
- 19. Liddy N, Bossi G, Adams KJ, Lissina A, Mahon TM, Hassan NJ et al Monoclonal TCR‐redirected tumor cell killing. Nat Med 2012; 18:980–7. [DOI] [PubMed] [Google Scholar]
- 20. Molloy PE, Sewell AK, Jakobsen BK. Soluble T cell receptors: novel immunotherapies. Curr Opin Pharmacol 2005; 5:438–43. [DOI] [PubMed] [Google Scholar]
- 21. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF et al Safety, activity, and immune correlates of anti‐PD‐1 antibody in cancer. N Engl J Med 2012; 366:2443–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brahmer JR. PD‐1‐targeted immunotherapy: recent clinical findings. Clin Adv Hematol Oncol 2012; 10:674–5. [PubMed] [Google Scholar]
- 23. Amarnath S, Mangus CW, Wang JC, Wei F, He A, Kapoor V et al The PDL1‐PD1 axis converts human TH1 cells into regulatory T cells. Sci Transl Med 2011; 3:111ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK et al PD‐L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med 2009; 206:3015–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li Z, Li D, Tsun A, Li B. FOXP3+ regulatory T cells and their functional regulation. Cell Mol Immunol 2015; 12:558–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Qiao G, Yang L, Li Z, Ying H, Hassen Y, Yin F et al Program death‐1 regulates peripheral T cell tolerance via an anergy‐independent mechanism. Clin Immunol 2012; 143:128–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang W, Lau R, Yu D, Zhu W, Korman A, Weber J. PD1 blockade reverses the suppression of melanoma antigen‐specific CTL by CD4+ CD25Hi regulatory T cells. Int Immunol 2009; 21:1065–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Golovina TN, Mikheeva T, Suhoski MM, Aqui NA, Tai VC, Shan X et al CD28 costimulation is essential for human T regulatory expansion and function. J Immunol 2008; 181:2855–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Plesa G, Zheng L, Medvec A, Wilson CB, Robles‐Oteiza C, Liddy N et al TCR affinity and specificity requirements for human regulatory T‐cell function. Blood 2012; 119:3420–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kitagawa Y, Ohkura N, Sakaguchi S. Epigenetic control of thymic Treg‐cell development. Eur J Immunol 2015; 45:11–6. [DOI] [PubMed] [Google Scholar]
- 31. McHugh RS, Whitters MJ, Piccirillo CA, Young DA, Shevach EM, Collins M et al CD4+CD25+ immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid‐induced TNF receptor. Immunity 2002; 16:311–23. [DOI] [PubMed] [Google Scholar]
- 32. Read S, Malmstrom V, Powrie F. Cytotoxic T lymphocyte‐associated antigen 4 plays an essential role in the function of CD25+CD4+ regulatory cells that control intestinal inflammation. J Exp Med 2000; 192:295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhong A, Pan X, Shi M. Expression of PD‐1 by CD4+CD25+CD127low Treg cells in the peripheral blood of lung cancer patients. Onco Targets Ther 2015; 8:1831–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bobisse S, Rondina M, Merlo A, Tisato V, Mandruzzato S, Amendola M et al Reprogramming T lymphocytes for melanoma adoptive immunotherapy by T‐cell receptor gene transfer with lentiviral vectors. Cancer Res 2009; 69:9385–94. [DOI] [PubMed] [Google Scholar]
- 35. Wang Z, Guo Y, Han W. Current status and perspectives of chimeric antigen receptor modified T cells for cancer treatment. Protein Cell 2017; 8:896–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mack M, Riethmuller G, Kufer P. A small bispecific antibody construct expressed as a functional single‐chain molecule with high tumor cell cytotoxicity. Proc Natl Acad Sci U S A 1995; 92:7021–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang H, Zhang J, Chen L, Weng Z, Tian Y, Zhao H et al Targeting naturally occurring epitope variants of hepatitis C virus with high‐affinity T‐cell receptors. J Gen Virol 2016; 98:374–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rizvi NA, Mazières J, Planchard D, Stinchcombe TE, Dy GK, Antonia SJ et al Activity and safety of nivolumab, an anti‐PD‐1 immune checkpoint inhibitor, for patients with advanced, refractory squamous non‐small‐cell lung cancer (CheckMate 063): a phase 2, single‐arm trial. Lancet Oncol 2015; 16:257–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Powles T, Eder JP, Fine GD, Braiteh FS, Loriot Y, Cruz C et al MPDL3280A (anti‐PD‐L1) treatment leads to clinical activity in metastatic bladder cancer. Nature 2014; 515:558–62. [DOI] [PubMed] [Google Scholar]
- 40. Marabelle A, Kohrt H, Levy R. Intratumoral anti‐CTLA‐4 therapy: enhancing efficacy while avoiding toxicity. Clin Cancer Res 2013; 19:5261–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Boudousquie C, Bossi G, Hurst JM, Rygiel KA, Jakobsen BK, Hassan NJ. Polyfunctional response by ImmTAC (IMCgp100) redirected CD8+ and CD4+ T cells. Immunology 2017; 152:425–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tripathi S, Guleria I. Role of PD1/PDL1 pathway, and TH17 and Treg cells in maternal tolerance to the fetus. Biomed J 2015; 38:25–31. [DOI] [PubMed] [Google Scholar]
- 43. Zhou Q, Munger ME, Highfill SL, Tolar J, Weigel BJ, Riddle M et al Program death‐1 signaling and regulatory T cells collaborate to resist the function of adoptively transferred cytotoxic T lymphocytes in advanced acute myeloid leukemia. Blood 2010; 116:2484–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Molinero LL, Miller ML, Evaristo C, Alegre ML. High TCR stimuli prevent induced regulatory T cell differentiation in a NF‐κB‐dependent manner. J Immunol 2011; 186:4609–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Identification of NCI‐H1299 cell line.
Figure S2. Naive CD4+ T‐cell purity was detected by FACS.
Figure S3. The viability of induced regulatory T cells and activated CD4+ T cells were detected by FACS.
Figure S4. ImmTAC‐NYE redirected and activated T cells to lyse tumour cells.
Figure S5. Induced regulatory T cells suppressed cytotoxic T lymphocyte killing redirected by ImmTAC‐NYE.
Figure S6. Mechanisms by which induced regulatory T cells suppressed ImmTAC‐NYE‐redirected cytotoxic T lymphocyte killing.
Figure S7. Programmed cell death protein ligand 1 (PD‐L1) expression detected by FACS and anti‐PD‐1 monoclonal antibody could reverse the suppressive effect of induced regulatory T cells on ImmTAC‐NYE redirected cell killing.