

Abstract
Cobamides (Cbas) are synthesized by many archaea, but some aspects of Cba biosynthesis in these microorganisms remain unclear. Here, we demonstrate here that the ORF MM2060 in the archaeum Methanosarcina mazei strain Gö1 encodes a bifunctional enzyme with L-threonine-O-3-phosphate (L-Thr-P) decarboxylase (EC 4.1.1.81) and L-Thr kinase activities (EC 2.7.1.177). In Salmonella enterica, where Cba biosynthesis has been extensively studied, the above-mentioned activities are encoded by separate genes, namely, cobD and pduX, respectively. The activities associated with the MM2060 protein (MmCobD) were validated in vitro and in vivo. In vitro, MmCobD used ATP and L-Thr as substrates and generated ADP, L-Thr-P, and (R)-1-aminopropan-2-ol O-phosphate as products. Notably, MmCobD has a 111–amino acid C-terminal extension of unknown function, which contains a putative metal-binding motif. This C-terminal domain alone did not display either activity in vivo or in vitro. Although the C-terminal MmCobD domain was not required for L-Thr-P decarboxylase or L-Thr kinase activities in vivo, its absence negatively affected both activities. In vitro results suggested that this domain may have a regulatory or substrate-gating role. When purified under anoxic conditions, MmCobD displayed Michaelis-Menten kinetics and had 1000-fold higher affinity for ATP and 1300-fold higher catalytic efficiency than MmCobD purified under oxic conditions. To our knowledge, MmCobD is the first example of a new class of L-Thr-P decarboxylases that also have L-Thr kinase activity. An archaeal protein with L-Thr kinase activity had not been identified prior to this work.
Keywords: Bacterial metabolism, kinase, decarboxylase, coenzyme biosynthesis, zinc finger, metalloprotein, adenosylcobalamin, B12
INTRODUCTION
Corrinoids are tetrapyrroles with two axial ligands coordinated to a central cobalt ion. The upper ligand in the coenzymatic form is a 5’-deoxyadenosine (Ado) group. The lower ligand can be benzimidazoles, phenolics, purines, or analogs and varies depending on the organism which produces it 1-3. Corrinoids that contain a lower ligand are referred to as a complete cobamide (Cba) 4. Cobalamin (Cbl) is the most well-known Cba and contains a purine analog 5,6-dimethylbenzamidazole (DMB) lower ligand base. Salmonella enterica subsp. enterica sv Typhimurium strain LT2 (hereafter S. enterica) is known to produce Cbl as well as pseudoCbl, which has adenine as the lower ligand, and Factor A, which has 2-methyladenine as the base 1-3, 5. Methanogenic archaea produce either pseudoCbl, or a Cba with 5-hydroxybenzimidazolyl as the lower ligand, known as Factor III (Fig. 1 inset).
Figure 1.

Assembly of the nucleotide loop in bacteria and archaea - Non-homologous archaeal enzyme names are boxed in black. The relevant reactions are boxed in red with enzyme names in bold. Highlighted in dark gray are the hydroxyl group of L-threonine (L-Thr) that is phosphorylated by PduX in S. enterica and the MmCobD archaeal kinase, the resulting phosphate of L-threonine-O-3-phosphate (L-Thr-P), and the (R)-1-aminopropan-2-ol O-phosphate (AP-P) linker which is subsequently attached to the corrinoid ring to form the linker between the ring and nucleotide base. The purine analog base 5,6-dimethylbenzimidazole that is particular to cobalamin (Cbl) is highlighted in a light gray oval. Inset: Boxed in light gray are purine and purine analog bases incorporated in cobamides synthesized by S. enterica and M. mazei. AdoCby, adenosylcobyric acid; AdoCbi-P, adenosylcobinamide phosphate; AdoCbi-GDP, adenosylcobinamide-GDP; AP-P, (R)-1-aminopropan-2-ol O-phosphate; L-Thr-P, L-threonine-O-3-phosphate; L-Thr, L-threonine; α-ribazole phosphate, DMB, 5,6-dimethylbenzimidazole; NaMN, nicotinic acid mononucleotide; Nm, Nicotinic Acid; PPi, pyrophosphate; Pi, orthophosphate; CbiB, AdoCbi-P synthase; CobY, AdoCbi-P guanylyltransferase; CobS, AdoCba-5’P synthase; CobD, L-Thr-P decarboxylase; CobT, NaMN:DMB phosphoribosyltransferase; CobU, AdoCbi kinase / AdoCbi-P guanylyltransferase; PduX, L-Thr kinase; CobC, AdoCba-5’-P phosphatase; CobZ; AdoCba-5’-P phosphatase.
In methanogens Cbas play a central role in methanogenesis from CO2 and methylamines 6-9. In this pathway, Cba-dependent enzymes serve as methyl-group carriers, transferring the C1 unit to coenzyme M 6. Similarly, methionine synthase, MetH, also uses Cbas to methylate homocysteine to yield methionine 10,11. Some archaea synthesize these complex molecules de novo, others can only synthesize them from precursors present in the environment, and yet others depend on acquiring complete molecules.
The adenosylcobamide (AdoCba) biosynthetic pathway has been extensively studied in bacteria such as S. enterica 12, however, gaps in our knowledge of how archaea synthesize Cbas remain. In archaea, only about half of the genes have assigned functions, and a fraction of these do not have orthologues in bacteria 13,14. Our work, and that of others, has employed comparative genomics as a tool for the identification of putative archaeal orthologues of bacterial cobamide biosynthetic genes 15-21.
While bioinformatics analysis of AdoCba biosynthesis gene clusters has been reported 18, 22, orthologues of the MM2060 protein have been overlooked despite its frequent association with cob biosynthetic genes. Another enzyme also missing in archaea is the L-threonine (L-Thr) kinase, which is encoded by pduX in S. enterica 23, 24. Many of the AdoCba biosynthetic enzymes in methanogenic archaea and bacteria are homologous, including the L-Thr-P decarboxylase CobD protein. Notably, the CobD proteins encoded in the genomes of many methanogenic archaea have an extended C-terminal domain about 111 amino acids in length, a domain that is not present in CobD from S. enterica or other bacteria (Fig. S1). At present, the function of this protein extension is unclear.
The studies presented herein focused on the identification of the archaeal enzyme responsible for the pyridoxal-5’-phosphate (PLP)-dependent decarboxylation of L-threonine-O-3-phosphate (L-Thr-P) that produces (R)-1-amino-propan-2-ol O-phosphate (a.k.a (R)-1-amino-2-propanol O-2-phosphate, AP-P), which in turn is used as co-substrate in the last step of the de novo corrin ring biosynthetic branch of the pathway (Fig. 1). In S. enterica, CobD (EC 4.1.1.81; hereafter SeCobD) catalyzes the decarboxylation of L-Thr-P 25. Here, we report genetic and biochemical data that support the functional assignment of the MM2060 protein from M. mazei Gö1 as the L-Thr-P decarboxylase of this archaeum. We show that the MM2060 protein also has L-Thr kinase activity, making the MM2060 enzyme unique among L-Thr-P decarboxylases. In addition, we show that although the putative metal-binding C-terminal domain of MM2060 is not required for L-Thr-P decarboxylase or L-Thr kinase activities, in its absence the efficiency of the L-Thr-P decarboxylase and L-Thr kinase activities of the enzyme are impaired. MM2060 has optimal enzymatic activity under anoxic conditions after incubation with iron(II) and sulfide, indicating the potential presence of an O2 sensitive metal center. To our knowledge, this is the first report of a single protein with both L-Thr kinase and L-Thr-P decarboxylase activities in any organism.
MATERIALS AND METHODS
Bacterial strains.
Strains and plasmids used in this work are described in Table S1. S. enterica strains carried a null allele of the metE gene that encodes the Cba-independent methionine synthase (MetE) enzyme 26. In the absence of MetE the cell uses the Cba-dependent methionine synthase (MetH) enzyme 10, 27-29. All S. enterica strains also carry an undefined mutation in the arabinose locus (allele ara-9), which prevents the utilization of arabinose as a carbon and energy source. Gene deletions in S. enterica were constructed using the phage lambda Red recombinase system as described elsewhere 30.
Culture media and growth conditions.
No-carbon essential (NCE) 31 with glycerol (22 mM) as the carbon and energy source was used as minimal growth medium. When added to the medium, the following supplements were at the indicated concentrations: trace minerals 32 (10 mL L−1), MgSO4 (1 mM), 5,6-dimethylbenzimidazole (DMB, 0.15 mM), ampicillin (0.1 mg mL−1), arabinose (0.5 mM). All corrinoids (cobyric acid dicyanide [(CN)2Cby], cobinamide dicyanide [(CN)2Cbi], and cyanocobalamin (CNCbl)) were added at (1 or 10 nM) final concentrations. When ethanolamine (90 mM) was used as a carbon and energy source, Fe(III)-citrate (0.05 mM) was also added to the medium with corrinoids (300 nM). (CN)2Cby was a gift from Paul Renz (Universität-Hohenheim, Stuttgart, Germany). All other chemicals were purchased from Sigma-Aldrich. S. enterica strains were cultured in Nutrient Broth (NB, Difco Laboratories) (0.8% w/v) containing NaCl (85 mM). Lysogeny broth (LB) 33, 34 was used as rich medium to culture Escherichia coli strains unless otherwise indicated.
Plasmid construction.
M. mazei strain Gö1 genomic DNA for PCR-gene amplification was a gift from Gerhard Gottschalk (Georg-August-Universität, Göttingen, Germany). Genomic DNA from S. enterica strain JE7088 (metE2702 ara-9) were extracted by heating cells at 90°C suspended in double distilled H2O for 5 min to release DNA. Cell debris was separated from DNA in the supernatant by centrifugation; this was the source of DNA used as a template for PCR amplification. Oligonucleotide primers were purchased from Integrated DNA Technologies Inc. A list of primer sequences can be found in Table S2. Primers for cloning were designed using the Saccharomyces Genome Database web-based primer design tool available at http://www.yeastgenome.org/cgi-bin/web-primer. Genes were PCR amplified from the appropriate genomic DNA template with PCR Extender Polymerase (5 Prime) and the primer pairs listed in Table S2. PCR products and vectors were treated with restriction endonucleases (Fermentas) indicated in the primer name in Table S2 and purified with the Wizard® SV Gel and PCR Clean-Up kit (Promega). Cloning vectors were treated with FastAP alkaline phosphatase (Fermentas). PCR fragments and vectors were ligated together using Fastlink™ Ligase (Epicentre) and introduced into E. coli DH5α 35, 36 via electroporation 37. Plasmid DNA was purified using the Wizard® Plus SV Miniprep kit (Promega). Plasmid sequence was confirmed by using BigDye® (ABI PRISM) protocols (University of Wisconsin-Madison Biotechnology Center and University of Georgia Genomics Facility). Table S1 lists the resulting plasmids. The start codon for wild-type M. mazei cobD (ORF MM2060) was changed from GTG to ATG. The N-terminus (MmCobD1-385) was cloned with codons encoding amino acids 1-385 with two stop codons TAA TAA added after the last residue. The C-terminal domain (MmCobD386-497) was cloned separately from codons encoding residues 386-497 with the addition of a methionine as the first residue. pBAD24 vector 38 was used for complementation and pTEV5 vector 39 was used for protein overproduction.
Complementation of function.
To determine whether or not a protein of interest was functional in vivo, plasmids were introduced into S. enterica by electroporation 37. S. enterica strains were grown to full density (~2 × 109 cfu mL−1) in nutrient broth (NB, Difco) supplemented with ampicillin (0.1 mg mL−1) for plasmid maintenance. Strains were grown in triplicate in sterile 96-well tissue culture plates (Falcon) where 2 μL of an overnight culture was used to inoculate 198 μL of fresh minimal (NCE) medium supplemented with glycerol, MgSO4, and trace minerals. For growth in minimal medium supplemented with ethanolamine, 10 μL of culture was used to inoculate 190 μL of medium. Corrinoids were added as indicated above. Growth behavior was monitored using Gen5 software (BioTek Instruments) during growth at 37°C in an EL808 Ultra Microplate Reader (BioTek Instruments) with continuous shaking using the slow shake instrument setting. Cell density measurements at 630 nm were acquired every 15 or 30 min for 24 or 60 h, respectively. Data were analyzed using the Prism v6 software package (GraphPad).
Normoxic protein overproduction.
SeCobD was overproduced and purified as described elsewhere 25, 40, 41. N-terminal, TEV-cleavable H6-tagged MmCobD, MmCobD1-385 and MmCobD386-497 proteins were overproduced in E. coli C43 (λDE3) 42 cells (Lucigen) from plasmids pMmCOBD18, pMmCOBD9, and pMmCOBD19, respectively. A sample (20 mL) of an overnight culture carrying the above plasmids was used to inoculate 1.5 L of Terrific Broth (TB) 43, 44 containing ampicillin (0.1 mg mL−1) supplemented with pyridoxine•HCl (1 mM), Fe(III)-citrate (0.05 mM), and δ-aminolevulinic acid (δ-ALA, 0.5 mM) to increase the intracellular concentration of PLP, Fe, and heme, respectively 45. Cultures were grown at 37°C with shaking (200 rpm) to an OD600 ~0.8, followed by a temperature downshift to 25°C and induction of gene expression by the addition of isopropyl-β-D-thiogalactopyranoside (IPTG, 0.3 mM) to the medium. Cultures were incubated overnight at 25°C with shaking (200 rpm). Cells were harvested by centrifugation at 6,000 × g at 4°C for 10 min in an Avanti J-20 XPI Beckman/Coulter refrigerated centrifuge equipped with a JLA 8.1000 rotor; cell pastes were stored at −20°C until used. Cells were resuspended in 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES-NaOH) buffer (50 mM, pH 7.9 at 4°C) containing NaCl (500 mM), imidazole (5 mM), lysozyme (1 mg mL−1), DNaseI (1 mg mL−1), and protease inhibitor phenylmethanesulfonyl fluoride (PMSF, 0.1 mM). Cells were lysed at 1.9×108 kPa using a TS Series (1.1 kW) bench top cell disrupter (Constant Systems Ltd.), equipped with a cooling jacket on the disruptor head to maintain a 6°C temperature using a Neslab ThermoFlex 900 recirculating chiller (Thermo Scientific). Debris was removed by centrifugation at 39,000 × g for 20 min. Proteins were purified at 4°C by Ni-affinity chromatography using a 1.5-mL bed volume of HisPur Ni-NTA resin (Thermo Scientific). The resin was equilibrated with bind buffer [HEPES (50 mM, pH 7.9 at 4°C), NaCl (500 mM), imidazole (5 mM)] before clarified supernatant was applied to the column. After binding to the column, the column was washed with four column volumes of bind buffer before proteins were eluted stepwise by increasing the imidazole concentration from 20 to 100 mM at 20 mM increments. Fractions (4 mL each) were collected for each step. A final wash step was performed with 300 mM imidazole. H6-MmCobD1-385 eluted with 80-100 mM imidazole and H6-MmCobD and H6-MmCobD386-497 eluted with 100-300 mM imidazole. Fractions containing H6-tagged proteins were pooled and the tag was cleaved with rTEV protease (1:100 rTEV:H6-protein ratio) for 3 h at 25°C in bind buffer containing 1,4-dithiothreitol (DTT, 1 mM). The cleaved protein was dialyzed into bind buffer containing ethylenediaminetetracetic acid (EDTA, 1 mM) at 25°C for 20 min, then dialyzed twice more in the same buffer without EDTA. The protein was passed over a Ni(NTA) column again to remove the cleaved H6-tag and H7-tagged rTEV protease using the buffers employed in the first purification step. Untagged proteins eluted in the flow-through fraction were pooled and dialyzed at 25°C for 20 min into desalting buffer 1 [HEPES (10 mM, pH 7.9 at 25°C), NaCl (300 mM), EDTA (1 mM)], followed by desalting buffer 2 [(HEPES (10 mM, pH 7.9 at 25°C), NaCl (200 mM), desalting buffer 3 [HEPES (10 mM, pH 7.9 at 25°C), NaCl (100 mM)], and storage buffer [HEPES (10 mM, pH 7.9 at 25°C), glycerol (10%, v/v)]. Proteins were concentrated using Amicon Ultracel centrifugal filters (Millipore) with a 10-kDa molecular mass cut off, and frozen drop-wise into liquid N2, and stored at −80°C until used. Protein concentrations were determined using a NanoDrop 1000 spectrophotometer (Thermo Scientific), using theoretical molecular weights and A280 molar extinction coefficients for each protein, which were obtained from the ExPASy Protparam database 46, 47. Purified proteins were resolved using a 15% SDS-PAGE gel and protein purity was estimated using band densitometry with a Fotodyne imaging system and Foto/Analyst v.5.00 software (Fotodyne Inc.) for image acquisition and TotalLab v.2005 software for analysis (Nonlinear Dynamics). MmCobD, MmCobD1-385, MmCobD386-497, and SeCobD proteins were purified to 85%, 97%, 65%, and 84% purity, respectively. The MmCobD protein produces two bands (Fig. S7). Both bands were excised and verified to be full-length MmCobD by in-gel trypsin digestion, followed by MALDI mass spectrometry and peptide mass fingerprinting with protein identification via Mascot (Matrix Science) protein identification software using peptide sequence databases performed by the Proteomics and Mass Spectrometry Core Facility at the University of Georgia. Both bands produced nearly identical mass spectrometry fragmentation patterns and identical peptide fragments with coverage of most of the N- and C-terminal resides indicating that bands were the full-length MmCobD and not a proteolyzed or truncated protein product (Fig. S8).
Anoxic overproduction of M. mazei CobD.
Anoxic overproduction and purification of MmCobD and MmCobD386-497 was performed as described above for the normoxic purification with the following exceptions. Cells were broken in an anaerobic chamber (Coy) by sonication (2 min, continuous pulse with stirring, amplitude 80 m) with a Qsonica Q55 sonicator equipped with microtip probe. All handling of cell extracts and resulting proteins were conducted under anoxic conditions in an anaerobic chamber (Coy) at 24-26°C. After collection of untagged protein in the flow through, proteins were concentrated by first precipitating by bring fractions to ~60% (NH4)2SO4 saturation. Precipitated protein was gently pelleted by centrifugation at 2000 × g. The supernatant was discarded and the protein pellet was gently resuspended in 1 mL 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer (10 mM, pH 7.9 at 25°C). Proteins were further concentrated and ammonium sulfate removed by washing samples with HEPES buffer over Amicon Ultracel centrifugal filters (Millipore) with a 10-kDa molecular mass cut off. 50% glycerol was added to protein samples for storage. Protein samples (0.25 mL) were sealed in glass anoxic vials and stored at −20°C until used.
In vitro L-Thr-P decarboxylase activity assay.
Reaction mixtures (25 μL) contained HEPES buffer (50 mM, pH 8.5 at 25 °C), L-Thr-P (5 μM), and normoxically purified protein (72 nM). When required for radiolabeled assays, a mixture of [14C-U]-L-Thr-P and L-Thr-P in a 1:10 ratio was used as substrate. (MP No enzyme reactions with [14C-U]-L-Thr (Biochemicals (9.25 mBq mmol−1 at a concentration of 0.1 mCi mL−1)) and [14C-U]-L-Thr-P (American RadioChemicals (3.7-5.55 mBq mmol−1), at 0.1 mCi mL−1 concentration)) were used as controls. Reactions were incubated at 37°C, and after 1 h, 5-μL samples were spotted onto C-Fertigfolien Polygram CEL 400 0.1 mm cellulose thin layer chromatography (TLC) plates (Macherey-Nagel) for product separation and analysis (see below for details).
Thin-layer chromatography (TLC) analysis.
Products resulting from the decarboxylation of [14C-U]-L-Thr-P were detected by TLC on 10×10 cm cellulose plates with 7 mm lanes and 3 mm spacers developed for 1 h with an ammonium acetate (2.5 M):ethanol (95%; v/v) (30:70 ratio) mobile phase. Use of plates pre-developed with distilled water and allowed to air-dry prior to applying samples provided the best resolution. A Typhoon Trio Variable Mode Imager (GE Healthcare) with ImageQuant v5.2 software was used to visualize the results. Reactions and TLC separation were repeated in three independent experiments, with representative TLC phosphor image presented herein.
ATPase activity assay.
ATPase activity was assessed using the ADP-Glo™ Kinase/ATPase Assay kit (Promega). This two-step endpoint assay was used in the following manner per the manufacturer’s instructions 48. MmCobD reactions containing ATP and L-Thr are incubated for 1 h. Reactions are stopped by the addition of reagents from the ADP-Glo™ kit that uses a proprietary reagent to deplete any remaining unused ATP in the reaction mixtures. The ADP that was generated in the MmCobD reaction is then converted back to ATP by a second proprietary reagent. The ATP is then used in a Luciferase luminescence producing reaction, which is measured at 560 nm with a SpectraMax Plus Gemini EM microplate spectrophotometer (Molecular Devices) equipped with SoftMax Pro v4 software. Reaction mixture consisting of HEPES (50 mM, pH 8.5 at 25°C), MgCl2 (1 mM), ATP (0.1-10 mM), L-Thr (0.3-50 mM) and protein (0.1 μM or 2 μM) were incubated at 37°C for 1 h. When indicated L-Ser, L-Ser-P, and L-Thr-P were used at 10 mM. Nunc 96-well round-bottom black polypropylene microtiter plates (Thermo Fisher) were used to minimize background. No-enzyme controls were subtracted to reduce background. Values were compared to a standard curve of luminescence (relative light units; RLU) vs % ATP to ADP conversion and converted into units of ADP produced (μM or mM) per mg or μM of protein. For ATPase inhibition assay the following inhibitors were used ADP (200 mM), AMP (200 mM), sodium pyrophosphate (PPi, 10 mM), sodium triphosphate (PPPi, 10 mM), adenosine 5’-[γ-thio]triphosphate (ADP-γ-S, 0.1 mM), sodium ortho-vanadate (Na3VO4, 1 mM), and sodium beryllium fluoride (BeF2, 2 mM) in HEPES buffer (50 mM, pH 7.5 at 25°C) with MmCobD (6 μM). Data were graphed using Prism v6 (GraphPad) and standard error, R2, and P values calculated using the data analysis features of the software.
13Carbon nuclear magnetic resonance (13C-NMR) analysis of L-Thr kinase reaction products.
Proton-decoupled 13C-NMR spectra were obtained using a Bruker Avance III HD 400 MHz NMR spectrometer (Chemical Sciences Magnetic Resonance Facility, University of Georgia) with the following parameters: relaxation delay 2 s, spectral width 24.03 kHz, acquisition time 1.36 s, 512 scans. Decarboxylation reaction mixtures consisted of sodium phosphate buffer (5 mM, pH 8.5 at 25 °C), L-Thr-P or O-phospho-L-serine (L-Ser-P) (40 mM), and enzyme (0.68 μM) incubated at 37°C for 1 h. Kinase reaction mixture consisted of sodium phosphate buffer (5 mM, pH 8.5 at 25°C), MgCl2 (5 mM), ATP (40 mM), L-Thr or L-Ser (40 mM), and enzyme (0.68 μM) incubated at 37°C for 1 h. No-enzyme control reactions consisted of sodium phosphate buffer (5 mM, pH 8.5 at 25 °C), and L-Thr, L-Ser, L-Thr-P, L-Ser-P, 1-amino-propan-2-ol (AP), ethanolamine (EA), or ethanolamine phosphate (EA-P) (40 mM). Protein was removed from reaction mixtures by filtration using Amicon Ultracel filters (Millipore) with 10-kDa molecular mass size exclusion. Reaction mixtures (0.5 mL) were brought up to a final volume of 0.6 mL in D2O (17% v/v). Two independent experiments were performed and a representative spectrum presented herein. Spectra were processed with MestReNova software version v11.02 (Mestrelab Research). All chemical structures presented herein were generated with ChemDraw Professional 15.0 (PerkinElmer Informatics) and the 13C-NMR spectra prediction feature was used as a reference for experimentally generated spectra. All figures were created using Illustrator CS6 or CC (Adobe).
31Phosphorous nuclear magnetic resonance (31P-NMR) analysis of L-Thr kinase reaction products.
Proton-decoupled 31P-NMR spectra were obtained using a Varian Unity Inova 500 MHz NMR spectrometer (Chemical Sciences Magnetic Resonance Facility, University of Georgia) with the following parameters: relaxation delay 1 s, excitation pulse 3.88 μs, spectral width 12.11 kHz, acquisition time 0.810 s, 256 scans. The 0.5 mL reaction mixture consisted of HEPES (50 mM, pH 8.5 at 25 °C), MgCl2 (1 mM), ATP (3 mM), L-Thr (3 mM), and enzyme (3 μM) incubated at 37°C for 1 h. Reaction mixtures (0.5 mL) were brought up to a final volume of 0.6 mL in D2O (17% v/v). For no enzyme controls freshly prepared PLP (0.5 mM) and L-Thr-P (3 mM) were used. Independent experiments were performed in quadruplicate. Spectra were processed with MestReNova software version v7.0 (Mestrelab Research).
Bioinformatics and phylogenetic analysis.
Sequences were obtained using BLAST 49 search for homology in the Integrated Microbial Genome (IMG) database 50. The protein sequence for ORFs MM2060 was used as the query sequences. Only finished genomes with bit scores > 50 or e values > 1.0e−7 were used in the analysis. Sequence header files were simplified with the find/replace and grep functions of TextWrangler (Bare Bones Software). Outliers with extreme sequence divergence were not included nor were alleles not associated with Cba biosynthetic genes on the chromosome. FASTA formatted sequences were aligned using the MUSCLE 51 plugin within Geneious R8.1.7 software (Biomatters Ltd.) with default settings. ESPript 3.0 52 was used to generate images of alignments.
Oligomeric state analysis of MmCobD and its truncated variants.
Gel filtration was performed using a HiPrep 26/60 Sephacryl S-100 high resolution column (GE Healthcare) connected to a computer-controlled AKTA fast protein liquid chromatography (FPLC) system. The column was equilibrated with HEPES buffer (50 mM, pH 7.5 at 4°C) containing NaCl (150 mM). MmCobD and truncated proteins (2 mg) was applied to the column, which was developed isocratically at a rate of 2 mL min−1. Molecular mass calibrations were performed using ovalbumin (44 kDa), myoglobin (17 kDa), and vitamin B12 (1.35 kDa) components of the Bio-Rad gel filtration standards along with bovine serum albumin (66.4 kDa, Promega) and DNaseI (31 kDa, Sigma).
Spectrophotometric kinase assay.
MmCobD kinase activity was measured using an NADH-consuming assay 53-56. All substrate stocks were made fresh. Reaction mixtures (0.1 mL) contained HEPES buffer (50 mM, pH 8.5 at 25°C), MgCl2 (5 mM), phosphoenolpyruvate (PEP, 3 mM), NADH (0.1 mM), pyruvate kinase (1 U), and lactate dehydrogenase (1.5 U) incubated at 25°C with measurements taken every 15 s over a 40-min period. For ATPase specific activity L-Thr concentration was held at 10 mM while ATP concentration was varied (0 – 200 mM). Reactions were started by the addition of MmCobD (3 μM). The absorbance at 340 nm was monitored in a 96-well plate using a Spectramax Plus UV-visible spectrophotometer (Molecular Devices) equipped with SoftMax Pro v6.2. The initial velocity of NADH consumption was derived from the slope of the linear range of the curve. Initial velocity values were acquired as milli-units of A340/min and normalized to a path length of 1 cm by the PathCheck Sensor feature of the instrument. Initial velocities were converted to μM s−1 using Beer’s Law (A = εlc), where A was the absorption at 340 nm, εwas the molar extinction coefficient of NADH (6.22 mM−1 cm−1 at 340 nm) 53, l was the path length (1 cm), and c was the concentration. Graphs of initial velocity vs substrate concentration were plotted and pseudo-first-order kinetic parameters were determined using Prism v6 (GraphPad). To determine cooperativity kinetic parameters for ATP, data were fitted to the equation V0 = (Vmax [S]h)/(Kh0.5 + [S]h), where V0 was the apparent initial velocity, Vmax was the apparent maximum velocity, [S] was the substrate concentration, h represented the hill coefficient, and Kh0.5 denotes the substrate concentration for half-maximal velocity. The turnover number, kcat was determine with the equation Vmax = kcat[E], where [E] was the enzyme concentration. To determine Michaelis-Menten kinetic parameters for ATP, data were fitted to the equation V0 = (Vmax ([S]/ (Km + [S]), where Km. represents the substrate concentration for half-maximal velocity. Specific activities were calculated as described elsewhere 55. Specific activity data are presented with standard deviation from duplicate independent experiments each performed in technical triplicates.
RESULTS
Bioinformatics analysis ofM. mazei ORF MM2060 and its homologues in other Methanosarcinales.
Figure 2 shows a protein sequence alignment of M. mazei MM2060 (MmCobD) and the S. enterica CobD (SeCobD) and supporting figure S1 shows a protein sequence alignment of CobD from several methanogenic archaea and S. enterica. As seen in figure 2, the primary sequence of these proteins shows end-to-end ~37% similarity and ~23% identity.
Figure 2.

Protein sequence alignment - Protein sequence alignment of CobD from S. enterica and M. mazei. Conserved residues are highlighted in red, and residues with similar properties are boxed in blue. The pyridoxal-5’-phosphate (PLP)-binding domain is bracketed and the active site lysine is marked with an asterisk. The cysteine-rich, putative metal-binding domain is indicated with brackets, and the cysteinyl and histidinyl residues in this region are boxed in yellow.
The MmCobD protein has an N-terminus that is 13 amino acids longer than the SeCobD protein, which also has two small deletions spanning position 56-62, and 262-266. The most striking difference is the 111-residue extended C-terminus of MmCobD. Information currently available in databases shows that genome sequences of all Methanosarcinales encode CobD proteins with C-terminal extensions. Notably, the C-terminus of MmCobD contains a cysteine-rich putative metal-binding zinc finger-like domain, starting at residue 390. This domain contains 5 His and 9 Cys arranged in 4 clusters (CX4CH, CX2CXCX4C, CX2CHX2H, CX2H) including what appears to be a heme-binding motif (CXXCH). Methanosarcina acetivorans and Methanosarcina barkeri possess this cytochrome c-like heme-binding motif, whereas Methanosarcina burtonii, Methanosarcina psychrophilus, and Methanosarcina hallandica appear to have two slightly different copies of the cobD gene adjacent to each other on the chromosome, that encode proteins with variant CXXCV or CXXCN motifs. CobD from M. psychrophilus, also has a particularly long N-terminus, which is 37 amino acids longer than SeCobD (Fig. S1).
A gene encoding a protein of unknown function with a cysteine-rich putative metal-binding motif is associated with cob genes across many genera of bacteria and archaea.
Further bioinformatics analysis of the putative metal-binding zinc finger-like protein of MmCobD revealed that this protein was found fused to other AdoCba biosynthetic proteins or encoded separately as open reading frames associated with cob genes. Most methanogenic archaea genomes encode proteins with the zinc finger-like protein fused to the C-termini of CobDs, however in Methanocorpusculum labreanm the zinc finger-like protein is fused to the corrinoid amidohydrolase CbiZ enzyme, in Methanothermus fervidus it is fused to the methyltransferase CbiH enzyme, and in the extremophile Methanopyrus kandleri it is not fused to any protein. This zinc finger-like protein can also be found fused to the corrinoid transporter protein BtuC in Butyrivibrio, to cobyrinic acid a,c-diamide synthetase CbiA in Treponema sp, and to the N-terminus of the cobalt-precorrin-5B (C1)-methyltransferase CbiD enzyme in Slackia heliotrinireducens. Genes encoding zinc finger-like proteins as independent ORFs can be found clustered with cob genes. The latter type is found in many Cba-producing bacteria, including representatives of the genera Clostridium, Bacillus, Spirochaetes, and Rhodobacterales, Rhizobiales, Alpha-, Beta-, and Gamma-proteobacteria. Genes encoding zinc finger-like proteins that are clustered with cob genes are found in organisms that utilize both the early-cobalt-insertion (a.k.a. anaerobic or O2-independent) or the late-cobalt-insertion (a.k.a. aerobic or O2-dependent) pathways [reviewed in 12,57]. Notably, this type of zinc finger-like protein is found only in Cba producers that do not encode the bona fide L-Thr kinase PduX enzyme. To date, non-orthologous replacements of PduX or alternative pathways for the production of L-Thr-P have not been found in any Cba producer that lacks a homologue of PduX. The function of these putative zinc finger-like proteins is unknown. For simplicity, we will refer to the fused zinc finger-like protein, spanning residues 386-497, as the C-terminal domain of MmCobD.
In vivo evidence that MmCobD has L-Thr-P decarboxylase activity.
To verify that MmCobD had L-Thr-P decarboxylase activity in vivo we used a S. enterica cobD strain to block the synthesis of AP-P. All S. enterica strains used in this study relied on Cba biosynthesis for the synthesis of methionine via the Cba-dependent methionine synthase (MetH, EC 2.1.1.13). That is, all strains lacked the Cba-independent methionine synthase (MetE, EC 2.1.1.1.14) enzyme.
Cultures were grown under normoxic conditions to block de novo synthesis of the corrin ring 58,59. The medium was supplemented with cobyric acid (CN)2Cby (Fig. 1), which is converted into adenosylcobinamide-phosphate (AdoCbi-P, Fig. 1) by the AdoCbi-P synthase (CbiB, EC 6.3.1.10) enzyme in what is considered to be the last step in the de novo corrin ring biosynthetic pathway 25 (Fig. 1).
We introduced plasmids carrying genes encoding the full-length MmCobD protein, the N-terminal decarboxylase domain, MmCobD1-385, or the C-terminal, putative metal-binding domain only, MmCobD386-497, into a S. enterica cobD strain (Table S1). A S. enterica cobD strain carrying a plasmid encoding SeCobD was used as positive control.
The growth behavior of the strain expressing MmCobD was comparable to that of the wild-type strain, and to that of the strain synthesizing SeCobD from a plasmid (4-h lag, 1.5-h doubling time) (Fig. 3A, triangles vs open squares). We observed a lag before the onset of exponential growth of cells expressing only the N-terminal region, (13-h lag, 4.1-h doubling time; Fig. 3A, inverted triangles), suggesting that although the putative metal-binding domain was not required for L-Thr-P decarboxylase activity, it appeared to be important for efficient MmCobD activity in vivo. The C-terminal putative metal-binding domain alone failed to complement the S. enterica cobD strain (Fig. 3A, solid squares).
Figure 3.

MmCobD has L-Thr-P decarboxylase activity in vivo and in vitro. (A) Cobalamin-dependent growth assessment of S. enterica cobD+ (open squares) and ΔcobD (diamonds) strains with plasmids synthesizing MmCobD (triangles), MmCobD1-385 (inverted triangles), MmCobD386-497 (solid squares), or SeCobD (circles) proteins. Plasmid pBAD24 was introduced into the strains to monitor their growth behavior when the empty cloning vector was present in the cell. Cells were grown aerobically at 37°C in NCE minimal medium containing glycerol (22 mM) as the sole carbon and energy source, supplemented with (CN)2Cby (1 nM), arabinose (0.25 mM), ampicillin (0.1 mg mL−1), and MgSO4 (1 mM). Growth analysis was performed in triplicate and repeated in three independent experiments. Error bars represent the standard error of the mean (SEM). (B) Phosphor image of products and reactants resolved by TLC with retention factors (Rf) indicated. A sample (5 μL) of reactions containing HEPES buffer (50 mM, pH 8.5 at 25 °C), normoxically purified protein (72 nM), and a 1:10 ratio of [14C-U]-L-Thr-P and L-Thr-P (5 μM), was spotted onto a cellulose TLC plate. Plates were developed for 1 h with ammonium acetate (2.5 M):ethanol (95%; v/v) (30:70 ratio) mobile phase. Reactions and TLC separations were repeated in three independent experiments. AP-P, (R)-1-aminopropan-2-ol-O-phosphate; L-Thr-P, L-threonine-O-3-phosphate; L-Thr, L-threonine.
The MmCobD protein decarboxylates L-Thr-P yielding AP-P.
Figure 3B shows a representative set of results from in vitro experiments aimed at detecting L-Thr-P decarboxylase enzymatic activity. Lanes 1 and 2 of the phosphor image show that MmCobD and the N-terminal domain containing only the CobD decarboxylase domain (MmCobD1-385) converted [14C-U]-L-Thr-P to [14C-U]-AP-P, as did the SeCobD positive control (lane 4). Small amounts of [14C-U]-L-Thr can be seen in each lane as a decomposition product. The cysteine-rich C-terminal domain alone (MmCobD386-497, lane 3) failed to generate [14C-U]-AP-P from [14C-U]-L-Thr-P. These data support the in vivo results indicating that the N-terminal domain of MmCobD possesses the L-Thr-P decarboxylase activity, and that the removal of the C-terminal domain does not abolish decarboxylase activity under the conditions tested.
The intracellular concentration of cobyric acid (Cby) is the limiting factor for the complementation of S. enterica by MmCobD.
We performed complementation studies with varying concentrations of Cby. Supporting figure S2 shows the effect of increasing levels of Cby on the ability of the MmCobD1-385 protein (Fig. S2A) and MmCobD protein (Fig. S2B) to restore AdoCba biosynthesis in a S. enterica cobD strain relative to the growth behavior of a S. enterica cobD strain expressing SeCobD (black squares). When genes were placed under the control of an arabinose inducible promoter 38 the concentration of inducer did not influence the level of complementation (data not shown), suggesting that the Cby substrate concentration, not protein level, was the limiting factor for complementation by the MmCobD1-385 protein.
MmCobD restores AdoCba biosynthesis in a S. enterica pduX strain.
M. mazei and other AdoCba producers that encode a version of the cysteine-rich, putative metal-binding domain also lack genes encoding homologues of the S. enterica L-Thr kinase, PduX. This observation led us to investigate the possibility that the C-terminal domain of MmCobD might generate L-Thr-P. Figure 4 shows the growth analysis of S. enterica pduX strains carrying plasmids encoding MmCobD protein, the N- (MmCobD1-385) or the C-terminal (MmCobD386-497) domains. Figure 4A shows the growth behavior on glycerol supplemented with Cby (1 nM), a condition that required only low levels of AdoCba biosynthesis for growth. In contrast, figure 4B shows the growth behavior on ethanolamine as the sole source of carbon and energy supplemented with Cby (300 nM), a condition that demanded a high level of AdoCba production for growth 60-62. Both the MmCobD and MmCobD1-385 proteins supported growth of a pduX strain under the less demanding condition (Fig. 4A, diamonds, solid circles, respectively) although not as well as the SePduX protein (Fig. 4A, solid squares). Under growth conditions where higher levels of AdoCba were required (e.g., ethanolamine catabolism), only the full-length MmCobD protein supported growth (Fig. 4B, diamonds). The strain that synthesized MmCobD1-385 grew as poorly on ethanolamine as the strain synthesizing MmCobD386-497 (Fig. 4B, solid circles vs open triangles). On glycerol (Fig. 4A), the strain that synthesized MmCobD had a 7-h lag time and a doubling time of 6.2-h, while the strain that synthesized MmCobD1-385 had an 8-h lag time and a 7.7-h doubling time (Fig. 4A, diamonds, solid circles, respectively), compared to the 3.3-h lag time and 3-h and 2.1-h doubling time of the wild-type strain and strains expressing SePduX (Fig. 4A, open circles, solid squares, respectively). Cells that synthesized MmCobD386-497 on glycerol had a 16.2-h doubling time and onset of exponential growth was not observed for that strain or the strain carrying the empty vector.
Figure 4.

MmCobD compensates for the absence of PduX in a S. enterica pduX strain in vivo. Cobamide-dependent growth was assessed with S. enterica cells grown aerobically at 37°C in NCE minimal medium with (A) glycerol (22 mM) and (CN)2Cby (1 nM) or (B) ethanolamine (90 mM) and (CN)2Cby (300 nM), supplemented with, DMB (0.15 mM), ampicillin (0.1 mg mL−1), L(+)-arabinose (0.5 mM) and MgSO4 (1 mM). S. enterica pduX+ and ΔpduX strains with plasmids encoding MmCobD, MmCobD1-385, MmCobD386-497, SePduX, or pBAD24 as the empty vector control. Growth analyses were repeated in three independent experiments with the error bars representing the standard error mean of cultures grown in triplicate.
Likewise, when we demanded growth on ethanolamine, cultures of strains that synthesized MmCobD (Fig. 4B, diamonds) displayed a 27-h lag time and 10.7-h doubling time compared to the 18-h lag time and 3.4-h doubling time and 11-h lag time and 2.5-h doubling time for the wild-type strain and the strain synthesizing SePduX (Fig. 4B, open circles, solid squares, respectively).
Collectively, these results suggested that the C-terminal domain of MmCobD contributed to the production of L-Thr-P, but was not required for function, at least under growth conditions that demand low levels of AdoCba. These results also indicated that the N-terminus of MmCobD had both L-Thr kinase and L-Thr-P decarboxylase activities in vivo.
MmCobD has ATPase activity.
To test whether MmCobD had L-Thr kinase activity in vitro we assayed for ATPase activity as described under Materials and Methods.
Data presented in figure 5A supported the conclusion that the N-terminal domain (MmCobD1-385) had ATPase activity comparable to that of SePduX.
Figure 5.
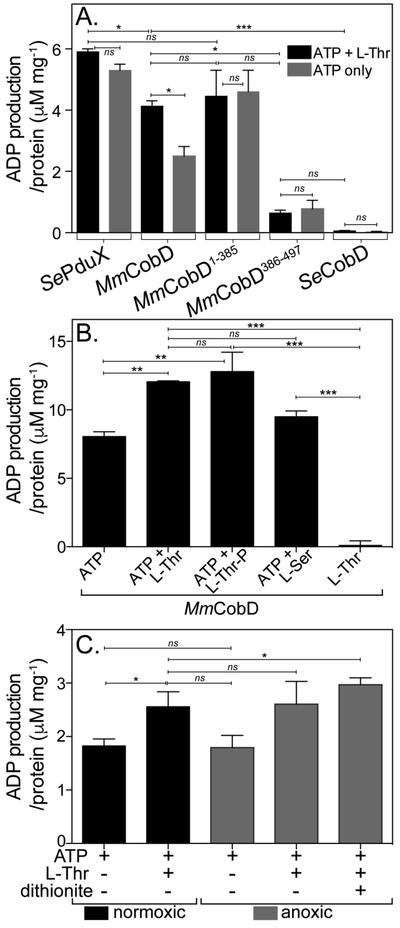
MmCobD has ATPase activity in vitro. ATPase activity assayed with ADP-Glo™ Kit (Promega). See Materials and Methods for a detailed description of the endpoint assay, which indirectly measures the enzymatic conversion of ATP to ADP via luminescence. The y-axis shows the conversion of ATP to ADP (μM) per mg of protein. The reaction mixture contained HEPES buffer (50 mM, pH 7.5 at 25°C), MgCl2 (1 mM), ATP (0.1 mM), L-Thr (0.3 mM), and normoxically purified protein (100 nM) incubated at 25°C for 1 h. (A) ATPase activity assayed in the presence and absence of L-Thr as co-substrate. Comparisons of SePduX, SeCobD, full-length MmCobD, and truncated enzymes in reactions with ATP + Thr (black bars) or ATP only (gray bars). Unpaired t test was used to calculate P values < 0.0003 (***), < 0.03 (*), or not significant (ns), with R2 = 0.96. (B) ATPase activity of MmCobD in the presence of cosubstrates or products. Reaction mixture containing HEPES buffer (50 mM, pH 7 at 25°C), MgCl2 (1 mM), MmCobD (72 nM) and ATP, L-Thr, L-Ser, or L-Thr-P (10 mM) where indicated; incubated at 25°C for 1 h. Unpaired t test was used to calculate P values < 0.0001 (***), <0.004 (**), and not significant (ns), with R2 = 0.99. (C) Reaction mixtures containing normoxically purified MmCobD enzyme were assayed under normoxic (black) and anoxic (gray) conditions. Reaction mixtures contained HEPES buffer (50 mM, pH 7.5 at 25°C), TCEP (2 μM MgCl2 (1 mM), ATP (10 mM), L-Thr (50 mM), protein (72 nM), and dithionite (2 mM) where indicated. Unpaired t test was used to calculate P values < 0.03 and 0.05 (*), and not significant (ns), R2 = 0.8. No-enzyme controls were subtracted to reduce background. Values were compared to a standard curve of luminescence vs ATP to ADP concentration, to generate a percent ATP conversion curve that was then transformed into units of ATP produced (μM) per mg of protein, with the standard error of the mean of triplicate reactions represented by the error bars. Calculations and graphs were generated with Prism v6 (GraphPad).
The full-length MmCobD protein also had ATPase activity comparable to the MmCobD1-385 protein when L-Thr was present as co-substrate but ~40% less than MmCobD1-385 when only ATP was present. S. enterica CobD (SeCobD) did not have any detectable ATPase activity. Surprisingly, the MmCobD386-497 protein had ~11% of the ATPase activity associated with MmCobD1-385. We are cautious to dismiss the slight ATPase activity of MmCobD386-497 as background. The activity may be significant, in particular when compared to the complete absence of activity of SeCobD (Fig. 5A and S6D) and the no-ATP control (Fig. 5B). These results are further discussed below.
Figure 5B shows that MmCobD had ATPase activity in the absence of L-Thr. However, the ATPase activity of MmCobD was enhanced by 33% when either L-Thr, or the product L-Thr-P was present in the reaction mixture. L-serine (L-Ser) also slightly enhanced the ATPase activity of MmCobD (by 15%), raising the possibility that MmCobD may be capable of generating ethanolamine phosphate (EA-P) from L-Ser and ATP. However, it is unlikely that L-Ser or L-Ser-P are the natural substrates of MmCobD as M. mazei and other methanogens do not produce norCbas 63, i.e., Cbas in which the linker tethering the nucleotide loop to the corrin ring is EA-P rather than AP-P 64.
Data presented in figures 5A and5B show that the presence of the co-substrate L-Thr or L-Ser, or the product L-Thr-P enhanced the ATPase activity of the full-length MmCobD enzyme. However, there was no statistically significant difference between the ATPase activity of the MmCobD1-385 enzyme with or without the addition of L-Thr (Fig. 5A). These results suggested that the removal of the C-terminus altered the interaction of L-Thr with the enzyme resulting in higher in vitro ATPase activity of the MmCobD1-385 enzyme, relative to what was observed with the full-length enzyme. However, the enhanced ATPase activity may or may not be the result of enhanced affinity for L-Thr or improved phosphotransfer from ATP to L-Thr to generate L-Thr-P. Further analysis is required to draw additional conclusion as to the effects that binding of each substrate has on the enzymatic activity.
The presence of a putative metal-binding domain from an anaerobe prompted us to consider assaying the enzyme under anoxic conditions in the presence of a reducing agent. Purified protein was yellow-brown, which suggested the protein might contain redox active iron. Figure 5C shows the ATPase activity of MmCobD under normoxic (black bars) and anoxic conditions (gray bars). Consistent with previous results, the addition of L-Thr enhanced ATPase activity under normoxic and anoxic conditions, but there was no substantial difference in activity in the presence or absence of air. The ATPase activity, however, was slightly enhanced by the addition of the sodium dithionite (2 mM), an observation that suggested the presence of a redox sensitive center that somehow affected the ATPase activity of MmCobD. Based on the above findings, we concluded that MmCobD and MmCobD1-385 had ATPase activity in vitro in the presence or absence of the L-Thr co-substrate, and that the C-terminal domain was not required for ATPase activity.
L-Ser inhibits growth of a S. enterica pduX cobD strain that synthesizes M. mazei CobD.
Figure 6 shows growth in minimal glycerol medium with Cby of a ΔpduX ΔcobD S. enterica strain carrying a plasmid that directed the synthesis of full-length MmCobD protein (gray triangles), or the pduX+ cobD+ strain carrying the empty vector (gray squares). In some cases, the medium was supplemented with L-Thr (1 mM, black squares, black triangles), L-Ser (1 mM, open squares, open triangles. In Fig. 6B, L-Thr-P (black symbols) or L-Ser-P was added in lieu of L-Thr or L-Ser (1 mM, open symbols), or no additions (gray symbols).
Figure 6.
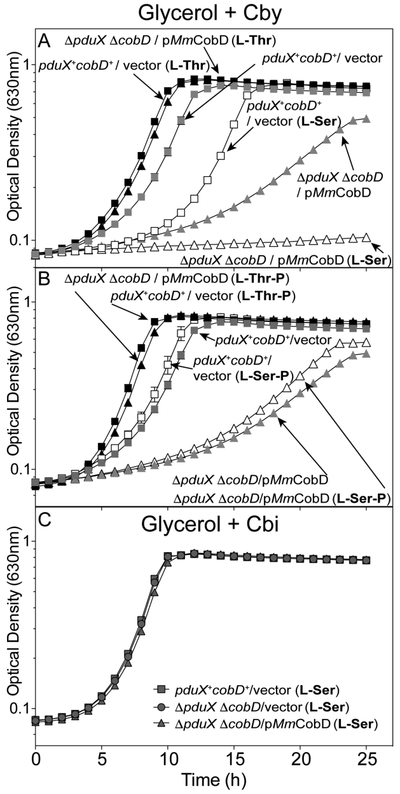
L-Ser blocks growth of a S. enterica pduX cobD strain that synthesizes MmCobD. Cobamide-dependent growth was assessed with S. enterica cells grown aerobically at 37°C in NCE minimal medium with glycerol (22 mM), (CN)2Cby (15 nM), ampicillin (0.1 mg mL−1), L(+)-arabinose (0.5 mM), and MgSO4 (1 mM), with the addition of (A) L-Thr (black symbols) or L-Ser (open symbols) (1 mM each), or no supplementation (gray symbols) as a control, or (B) L-Thr-P (black symbols), L-Ser-P (open symbols), or no supplementation (gray symbols). (C) As a positive control cells were grown in minimal medium supplemented with Cbi (1 nM). Double deletion ΔpduX ΔcobD strains carried plasmids encoding the full-length MmCobD (triangles) or pBAD24 (squares), and wild-type S. enterica pduX+ cobD+ carried pBAD24 (circles) as the empty vector control. Growth analyses were repeated in two independent experiments with the error bars representing the standard error of the mean of cultures grown in triplicate.
Addition of L-Thr to the medium slightly improved growth of the pduX+ cobD+ / vector strain (Fig. 6A, gray squares vs black triangles). In contrast, addition of L-Thr dramatically improved growth of the ΔpduX ΔcobD / pMmCobD strain (gray triangles vs black squares). These results suggested that exogenous L-Thr acted as a substrate for MmCobD with the concomitant increased flux through the late steps of the nucleotide loop assembly pathway, an idea that is not unprecedented 65.
Addition of L-Ser to the medium (open symbols) had different effects on the growth of the above strains. While L-Ser doubled the lag time (3 to 6 h) of the pduX+ cobD+ / vector strain (gray vs open squares), it dramatically increased the doubling time (4 to 23 h) and lag time (17 to 26 h) of the ΔpduX ΔcobD / pMmCobD strain (gray triangles vs open triangles). From these results we concluded that exogenous L-Ser has a negative effect on the growth of S. enterica, which is exacerbated by the expression of MmCobD. These data suggested that MmCobD activity may be inhibited by L-Ser in vivo.
Likewise, the addition of L-Thr-P improved growth of ΔpduX ΔcobD strains expressing either pMmCobD or the empty vector (Fig 6B, black triangles, black squares). While the addition of L-Ser-P had no effect (Fig. 6B, open squares, gray squares), suggesting that MmCobD could not decarboxylate L-Ser-P in vivo to produce EA-P. The AdoCbi-P synthase (CbiB) enzyme of S. enterica is known to use EA-P as a substrate 66. All strains grew in the presence of L-Ser when supplemented with Cbi (Fig. 6C), which bypasses PduX and CobD in the pathway (Fig. 1).
M. mazei CobD produces L-Thr-P and AP-P in vitro.
Using 31P-NMR we showed that MmCobD and MmCobD1-385 synthesize L-Thr-P from ATP and L-Thr (Fig. 7). However, due to the chemical and structural similarity of AP-P and L-Thr-P we could not differentiate between the two using 31P-NMR. Figure 7 shows chemical shifts of L-Thr-P and ATP (Fig. 7A), and PLP (Fig. 7B). In the presence of L-Thr and ATP, the MmCobD (Fig. 7C) and MmCobD1-385 (Fig. 7D) enzymes produced compounds with shifts at 3.6/3.7 ppm that corresponded to L-Thr-P, and 4.1/4.2 ppm that corresponded to PLP. The reactions containing the MmCobD (Fig. 7C, F), MmCobD1-385 (Fig. 7D), and SeCobD (Fig. 7G) enzymes all produced a peak at 4.0, 4.1, or 4.2 ppm. We believe this peak to be PLP because of the close correspondence to the free PLP standard (Fig. 7B), its occurrence only in reactions containing PLP enzymes, and there are no other phosphorylated compounds present in the reactions to account for the presence of the peak. The two peaks at 4.2 and 4.0 ppm in the free PLP standard (Fig. 7B) reflects the protonation state of PLP and the single peak in the reactions containing the proteins reflects the behavior of enzyme bound PLP where the phosphate is fully ionized and bound to a positively charged pocket in the enzyme 67 until the formation of an external aldimine with the substrate L-Thr-P, giving rise to a single peak. The MmCobD386-497 protein did not generate detectable L-Thr-P (Fig. 7E), a result that could be due to its low level of ATPase activity (Fig. 5A). These data agreed with the in vivo data (Fig. 4) that showed MmCobD386-497 did not have L-Thr kinase activity. In Fig. 7, panels F and G show the spectra of the MmCobD and SeCobD L-Thr-P decarboxylation reactions, respectively. The SeCobD spectrum has a peak at 2.9 ppm that is phosphate contamination from the protein purification process. Resonance for L-Thr-P and AP-P were detected at ~3.6 ppm (Fig. 7A, C, F, G). Problems generated by overlapping resonances of AP-P and L-Thr-P were circumvented by 13C-NMR (see below).
Figure 7.

31P-NMR spectra of the reaction catalyzed by MmCobD kinase - Representative 31P-NMR spectra of quadruplicate independent experiments. Spectra were processed with MestReNova (Mestrelab Research). Reaction mixtures containing MgCl2 (1 mM), ATP (0.3 mM), L-Thr (0.3 mM), and protein (3 μM) were incubated for 1 h at 25°C prior to the addition of D2O (17% v/v). Each panel is labeled with the substrate and or protein reaction mixture. Each peak is labeled with the chemical shift value and the substrate that it represents based on the corresponding chemical shifts of the standards in no-enzyme control reactions. (A) ATP standard and L-threonine-O-3-phosphate (Thr-P) standards. (B) PLP standard. (C) Reaction containing ATP, L-Thr, and MmCobD. (D) Reaction containing ATP, L-Thr, and MmCobD1-385. (E) Reaction containing ATP, L-Thr, and MmCobD386-497. (F) Reaction containing L-Thr-P and MmCobD. (G) Reaction containing L-Thr-P and SeCobD.
Figure 8 shows representative 13C-NMR spectra of MmCobD, SeCobD, and control reactions. Panel A shows the sequence of the reactions with each carbon atom labeled with a color-coded letter to differentiate each substrate. Panel B shows the spectrum for the L-Thr-P no-enzyme control reaction. Panels C and D show the conversion of L-Thr-P (peaks A, B, C, D; red) to AP-P (peaks E, F, G; fuchsia circled) by both SeCobD (panel C) and MmCobD (panel D). AP-P is not commercially available and was generated enzymatically using SeCobD (Panel C). The no-enzyme control reactions for the kinase reaction are, L-Thr (panel E), ATP (panel F), and an equimolar mixture of all three substrates (panel G). Panel H shows the conversion of ATP (I-U; blue) and L-Thr (W, X, Y; green) to L-Thr-P by MmCobD (peaks A, B, C, D; red letters), and AP-P (peaks E, F, G; fuchsia circled) as the final product of the reaction(s). These results show that MmCobD uses L-Thr and ATP to generate L-Thr-P, which is then converted to AP-P in vitro.
Figure 8.
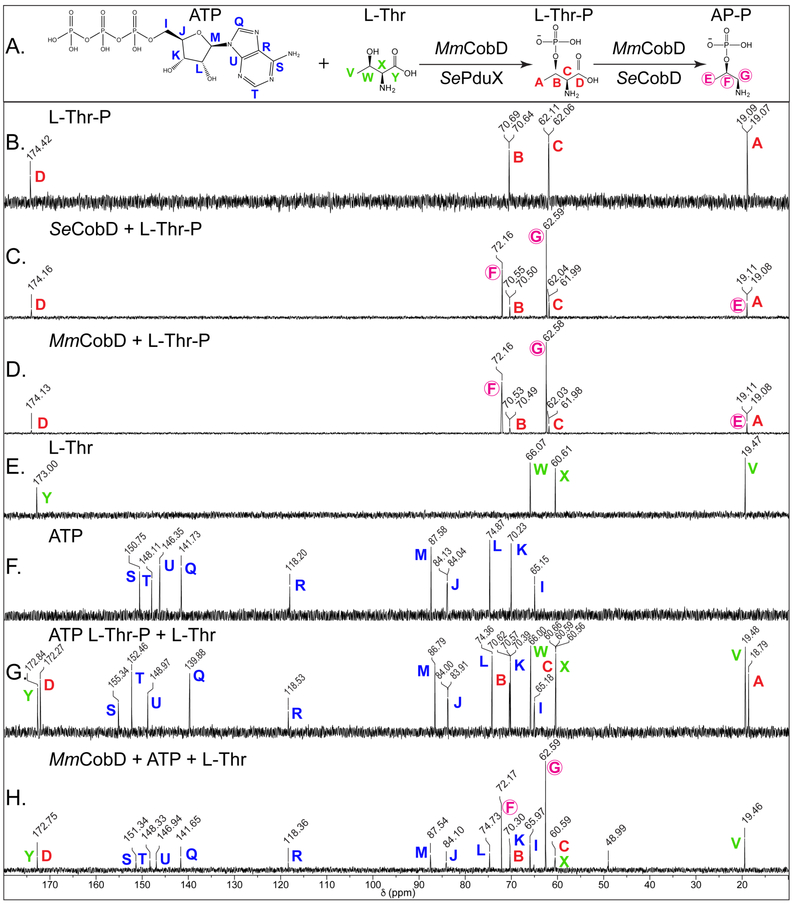
13C-NMR spectra of the reaction catalyzed by MmCobD kinase. Representative 13C-NMR spectra of duplicate independent experiments. Reaction mixtures containing sodium phosphate buffer (5 mM, pH 8.5 at 25°C), MgCl2 (5 mM), ATP (40 mM), L-Thr (40 mM) or L-Thr-P (40 mM), and protein (0.68 μM) were incubated for 1 h at 25°C prior to the addition of D2O (17% v/v). Each panel is labeled with the substrate and or protein reaction mixture. Each peak is labeled with the chemical shift value (ppm) and a color-coded letter corresponding to the carbon atom it represents in the substrates or products, based on the corresponding chemical shifts for the standards in the no-enzyme control reactions. (A) Reactions and chemical structures of substrates with each carbon atom of each reactant or product labeled with a color-coded letter. (B) L-threonine-O-3-phosphate (Thr-P) standard (red). (C) Reaction containing L-Thr-P and Se CobD to generate (R)-1-aminopropan-2-ol O-phosphate (AP-P) standard (circled, fuchsia). (D) Reaction containing L-Thr-P and MmCobD. (E) L-threonine (L-Thr) standard (green). (F) ATP standard (blue). (G) Equimolar mixture of L-Thr, L-Thr-P, and ATP standards. (H) Reaction containing ATP, L-Thr, and MmCobD. Two independent experiments were performed and representative spectra presented. Spectra were processed with MestReNova (Mestrelab Research).
MmCobD does not produce O-phospho-L-serine (L-Ser-P) or ethanolaminephosphate (EA-P).
13C-NMR was performed to assess whether MmCobD could produce EA-P via decarboxylation of L-Ser-P. Figure S3 shows representative 13C-NMR spectra of MmCobD and SeCobD decarboxylation reactions with L-Ser-P as the substrate. Panel A shows the sequence of the reactions with each carbon atom labeled with a color-coded letter to differentiate each substrate. Panels B, C, and D were used as no-enzyme control reactions. Signals at 57.51 and 16.85 ppm were either contaminants or decomposition products from L-Ser-P and EA-P standards; the identity of these compounds was not established. The absence of EA-P signals (boxed purple) with chemical shifts at 61.3 ppm (peak F) and 40.3 ppm (peak G) in both the SeCobD (panel E) and MmCobD (panel F) decarboxylation reactions was consistent with the idea that neither the M. mazei nor S. enterica CobD enzymes could generate EA-P from L-Ser-P.
13C-NMR was performed using L-Ser and ATP as substrates to assess whether MmCobD could produce L-Ser-P (Fig. S4). Panel A of figure S4 shows the sequence of the reactions with each carbon atom labeled with a color-coded letter to differentiate each substrate. The standards used in the no-enzyme control reactions were, L-Ser (Fig. S4B), L-Ser-P (Fig. S4C), ethanolamine phosphate, EA-P (Fig. S4D), ATP (Fig. S4E), and an equimolar mixture of all four (Fig. S4F). Figure S4G shows representative 13C-NMR spectra of MmCobD kinase reactions with L-Ser and ATP as substrates. MmCobD did not use L-Ser and ATP to generate L-Ser-P (red) or EA-P (boxed purple). Carbon atoms with chemical shifts of 62.58 and 72.15 ppm were present in the MmCobD kinase reaction, and in the MmCobD and SeCobD decarboxylation reactions when L-Ser-P was added as substrate. The carbon atoms that these signals correspond to were not identified. These signals may represent a yet-to-be-determined product that is generated by CobD when L-Ser-P is the substrate.
The MmCobD1-385protein has lower specific activity than MmCobD.
Figure 9 shows the specific activity of normoxically purified full-length MmCobD (Fig. 9A) and the MmCobD1-385 (Fig. 9B) proteins as a function of ATP concentration. Both enzymes responded to increasing ATP concentrations. However, the specific activity of MmCobD was ~8-fold higher than that of the truncated protein (Table 1). The full-length enzyme showed saturation at ~50 mM ATP while the truncated protein was not saturated even at twice that concentration (Figs. 9A, 9B). MmCobD activity as a function of ATP concentrations is also shown in supporting figure S5G. These results suggested that the full-length protein likely had a significantly higher affinity for ATP than the truncated protein.
Figure 9.

Specific activities of normoxically purified MmCobD and MmCobD1-385 proteins as a function of substrate concentrations - Indirect measurement of the specific activity of (A) MmCobD and (B) MmCobD1-385 proteins as a function of ATP concentration, and (C), (D) L-Thr concentration expressed as μmol of ATP per min per mg of protein with the standard deviation from the mean (SD) of triplicate reactions represented by the error bars. Activity was measured by a NADH-consuming assay described in the Materials and Methods section. Assays were performed with normoxically purified protein (3 μM), HEPES buffer (50 mM pH 7.5 at 25°C), MgCl2 (5 mM), phosphoenolpyruvate (PEP, 3 mM), NADH (0.1 mM), pyruvate kinase (1 U), lactate dehydrogenase (1.5 U) incubated at 25°C for 20 min under normoxic conditions. For ATPase specific activity L-Thr concentration was held at 50 mM while ATP concentration was varied (5-100 mM). To measure the effect of the co-substrate on ATPase activity, ATP concentration was held at 50 mM while the concentration of L-Thr was varied (0-100 mM).
Table 1. Specific activity measurements.
Specific activity values for normoxically purified full-length MmCobD and MmCobD1-385 enzymes were assayed for ATPase activity in the presence and absence of L-Thr as co-substrates. Values are reported as mean ± standard deviation of three activity measurements. Activity was measured with a coupled NADH consuming assay (see Materials and Methods).
| Protein | ATP (μmol ATP min−1 mg−1) |
L-Thr (μmol ATP min−1 mg−1) |
|---|---|---|
| MmCobD | 10.3 ± 0.30 | 7.6 ± 0.02 |
| MmCobD1-385 | 1.30 ± 0.04 | 3.7 ± 0.11 |
Figures 9C and 9D show the specific activity as a function of L-Thr concentration, where the concentration of ATP was held constant at 50 mM while the concentration of L-Thr was varied. In contrast with the previous in vitro ATPase endpoint assay data (Fig. 5), the addition of L-Thr decreased the specific activity from 10.3 μmol min−1 mg−1 to 7.6 μmol min−1 mg−1 for MmCobD, and conversely increased the specific activity for MmCobD1-385 (1.3 to 3.7 μmol min−1 mg−1) (Table 1). However, there was no significant change in the activity as the concentration of L-Thr was increased (Fig. 9C, Fig. S5F), a result that suggested ATP was limiting. The same observation was made with the MmCobD1-385 enzyme, however, the activity decreased at L-Thr concentrations of 50 mM and above (Fig. 9D) suggesting the possibility of substrate inhibition by L-Thr at high concentrations. The implications of the influence of the C-terminal domain on substrate affinity are discussed below.
Mm CobD displays positive cooperativity in response to ATP under normoxic conditions.
We performed steady state kinetic analysis with normoxically purified MmCobD MmCobD1-385 (Fig. 10A) enzymes. The activity was measured indirectly via a coupled assay that measured the consumption of NADH (see Materials and Methods)53. Neither enzyme displayed Michaelis-Menten behavior when L-Thr was held at a fixed saturating concentration (50 mM) and the ATP concentration was varied. Instead, the kinetics of the reaction for ATP displayed a sigmoidal curve consistent with positive cooperativity 68 (Fig. 10A). This was further confirmed by positive integer Hill coefficients (h) of 5 and 3 (Table 2A) for MmCobD and MmCobD1-385 respectively, and the concavity of the double-reciprocal plots (Fig. 10A inset). For MmCobD, positive cooperative would suggest that the binding of ATP likely increased the affinity of MmCobD for L-Thr as the ATP concentration is increased or vice versa. However, because we were unable to obtain K0.5 values for L-Thr we are unable to definitively make this assertion. The apparent Vmax of the MmCobD enzyme for ATP was 25% higher and the K0.5 1.5-fold lower than that of the MmCobD1-385 enzyme (Table 2A). This along with the 1.5-fold faster turnover (kcat) and 2-fold higher enzymatic efficiency (kcat/K0.5) suggested that the full-length protein was a more efficient enzyme than the truncated protein. The kinetic curves show the MmCobD enzyme reached saturation at ~75 mM ATP while the N-terminus required 200 mM ATP before saturation was reached.
Figure 10.

Kinetic analysis of the ATPase activity. Representative graphs of oxically purified MmCobD (squares) and MmCobD1-385 (triangles) assayed under oxic conditions with the data fit to (A) sigmoidal cooperativity non-linear regression curves. Insets show the double-reciprocal plot. L-Thr concentration was held constant at 50 mM while ATP concentration was varied. Representative graphs of anoxically purified MmCobD (circles) assayed under anoxic conditions with the data fit to (B) Michaelis-Menten non-linear regression curves. Insets show the double-reciprocal plot. L-Thr concentration was held constant at 10 mM while ATP concentration was varied. Assay was performed in duplicate independent experiments with three technical replicates with error bars indicating the standard deviation from the mean. Graphs of initial velocity (μM s−1) vs ATP substrate concentration (mM, for oxic conditions; μM for anoxic conditions) were plotted and pseudo-first-order kinetic parameters were determined using Prism v6 (GraphPad).
Table 2. Kinetic parameters of MmCobD and truncated MmCobD1-385 for ATP.
SeCobD, MmCobD, MmCobD1-385, and MmCobD386-497 enzymes were assayed for ATPase activity. using a NADH consumption assay (see Materials and Methods). Enzymes were purified and assayed oxically or anoxically as indicated. These parameters are apparent kinetic values of the mean ± standard deviation of triplicate independent experiments. For steady-state analysis, the L-Thr concentration was saturating at 10 mM and ATP was gradually increased. ND, not detected.
| A. Cooperativity | |||||||
| ATPa,b,c | |||||||
| Protein | Assay condition |
R2 |
Vmax (μM s−1) |
K0.5 (mM) |
kcat (s−1) |
kcat/K0.5 (M−1 s−1) |
h |
| MmCobD1−385 | oxic | 0.97 | 0.06 ± 0.003 | 72 ± 5 | 0.02 ± 0.001 | 0.27 ± 0.24 | 3 ± 0.4 |
| MmCobD | oxic | 0.91 | 0.08 ± 0.004 | 48 ± 2 | 0.03 ± 0.001 | 0.54 ± 0.53 | 5 ± 1.0 |
| SeCobD | anoxic | - | ND | ND | ND | - | - |
| B.Michaelis-Menten | |||||||
| ATP | |||||||
| Protein | Assay condition |
R2 |
Vmax (μM s−1) |
Km (mM) |
kcat (s−1) |
kcat/Km (M−1 s−1) |
|
| MmCobD | anoxic | 0.97 | 0.08 ± 0.002 | 0.05 ± 0.006 | 0.04 ± 0.0001 | 720 ±150 | |
| MmCobD386−497 | anoxic | - | ND | ND | ND | - | |
These parameters are apparent kinetic values of averages of triplicate independent experiments and standard deviations.
For steady-state analysis, the L-Thr concentration was saturating at 10 mM
ND, not detected
MmCobD displays Michaelis-Menten kinetics and optimal enzymatic activity under anoxic conditions.
Because MmCobD had slightly improved activity in the presence of reducing agents anoxically (Fig. 5C), and purified proteins had a rust brown color, we speculated that an O2 sensitive metal, likely Fe, might be present. We purified MmCobD anoxically and performed steady-state kinetics under anoxic conditions using the NADH consuming assay described in Materials and Methods. In contrast to normoxically purified and assayed protein, the anoxically purified full-length MmCobD displayed Michaelis-Menten kinetics behavior (Fig. 10B). Anoxically purified and assayed MmCobD had a 1000-fold higher affinity for ATP (Km), 1.3 times higher turnover (kcat), and 1300-fold higher enzymatic efficiency (kcat/Km) (Table 2B). Anoxically purified protein also responded to ATP at physiological levels (Fig. 9B). These data indicate that the anoxically purified and assayed MmCobD is a more efficient enzyme.
Factors influencing L-Thr kinase activity of MmCobD.
We optimized the reaction conditions for the kinase reactions with normoxically purified protein (Fig. S5). The optimal pH was 8.5 in HEPES buffer (50 mM; Fig. S5A). Salts were not required for function, however, the presence of KCl (100 mM) increased activity. Reducing agents such as TCEP (1 mM) or sodium dithionite (1 mM) provided slight increases in activity (Fig. S5B). MmCobD activity was optimal with MnCl2 (1 mM); activity was observed with CoCl2 (1 mM) and MgCl2 (1 mM); activity with MgCl2 increased at higher concentrations (10 mM; Fig. S5C). We also measured the ATPase activity of MmCobD and MmCobD1-385 in the presence of L-Ser or L-Thr (0.3 mM; Figs. S5D, S5E), and compared the results of these experiments to those obtained with SePduX and SeCobD. The behavior was the same as previously observed (Fig. 5), with the exception that MmCobD was slightly more active (10%) with a lower concentration of L-Ser (0.3 mM) as co-substrate relative to SePduX or MmCobD1-385. We tested ATPase activity as a function of L-Thr concentration and found no change in the activity of MmCobD with increasing concentrations of L-Thr (Fig. S5F). ATPase activity was assayed as a function of ATP concentration and displayed direct proportionality (Fig. S5G).
ATPase activity in the presence of known ATPase/kinase inhibitors ADP, AMP, PPPi, PPi, ADP-γ-S, Na3VO4, and BeF2 was tested (Fig. S5H). AMP and PPi did not inhibit MmCobD. MmCobD was inhibited in the following order from most inhibitory to least: BeF2>ADP>ADP-γ-S>PPPi>Na3VO4. Additional testing was conducted with ADP-γ-S on MmCobD and MmCobD1-385 with EutP, an acetate kinase from S. enterica (SeEutP) 69 as a positive control. Both MmCobD and SeEutP were inhibited by ADP-γ-S but notably, MmCobD1-385 was not (Fig. S5I). The implication of this surprising result is discussed below.
The C-terminal domain of MmCobD does not affect the multimeric state of the protein.
We investigated whether the absence of the C-terminus of MmCobD affected the multimeric state of the protein. Figure S6 shows the results of a representative gel permeation chromatography analysis. MmCobD (theoretical mass ≈ 55.5 kDa) formed dimers in solution, as did the MmCobD1-385 protein (theoretical mass ≈ 45.8 kDa). The C-terminal domain (MmCobD386-497, theoretical mass ≈ 12.9 kDa) was detected as a monomer. These data suggested that the C-terminal domain was not required for dimerization. Since SeCobD is a dimer 40,70, it was not surprising that MmCobD was also a dimer. If MmCobD were also in the inverted orientation seen in the crystal structure of SeCobD, it is unlikely that the C-terminal domain of each subunit of the MmCobD dimer would be able to interact.
DISCUSSION
A new class of L-Thr-P decarboxylase.
In this work we show that CobD from the methanogenic archaeum M. mazei (MmCobD) is a bifunctional enzyme with L-Thr-P decarboxylase and L-Thr kinase activities. The L-Thr-P decarboxylase activity was expected, as the protein is homologous to the well-characterized CobD enzyme from S. enterica (SeCobD) 25, 40, 70. However, we uncovered a new activity for MmCobD as a L-Thr kinase, an activity that SeCobD does not have (Figs. 5A, S5D). The L-Thr kinase and L-Thr-P decarboxylase activities are both associated with the N-terminal domain (residues 1-385) of MmCobD. This is the first report of these two enzymatic activities being associated with a single polypeptide. The function of the putative ‘zinc finger’ metal-binding C-terminal domain remains unclear. Based on the data reported herein, we propose that M. mazei, and other bacteria and archaea that lack homologues of the known L-Thr kinase (PduX) have evolved a new class of L-Thr-P decarboxylase, one that also has L-Thr kinase activity (Fig. 8). This work fills an important gap in our understanding of AdoCba biosynthesis in methanogenic archaea and other AdoCba producing bacteria.
Bioinformatics analyses suggest that the metal-binding domain is fused to the C-terminus of CobD exclusively in Methanosarcinales (Fig. S1), but it is also found fused to other AdoCba biosynthetic or transport genes such as cbiA, cbiD, cbiH, cbiZ, or btuD as well as an independent ORF in other bacteria and archaea. Our analysis of the genome databases shows that these cob-associated genes encoding putative metal-binding proteins have only been found in genomes lacking pduX homologues. The function of the putative metal-binding protein when fused to CobD is unclear, as MmCobD retains L-Thr decarboxylase activity when the metal-binding domain is removed. L-Thr kinase and L-Thr-P decarboxylase activities associated with a single polypeptide appears to be the ideal placement for shuttling a product of one activity to serve as substrate for the subsequent activity (Fig. 1). We propose that the metal-binding domain somehow optimizes or regulates the L-Thr kinase and L-Thr-P decarboxylase activities of the MmCobD protein. There is at least one precedent for a bifunctional PLP-dependent enzyme with kinase activity in LysA, an aspartate kinase/PLP-diaminopimelate decarboxylase 71, 72. There is also the bifunctional PLP-dependent kynureninase 73 that has hydrolase and transaminase activities as well as a number of other example of other bifunctional PLP-dependent decarboxylases, racemases, hydrolases, and transaminases 74-81.
MmCobD represents a new bifunctional class of CobD enzyme that fills a gap in the archaeal Cba biosynthetic pathway. This new class of CobD might also fill a gap in the Cba biosynthesis pathway of organisms that use the late-cobalt-insertion (a.k.a O2-dependent) pathway 82, 83 [reviewed in 12]. The L-Thr kinase has not been identified in these organisms. The CobD enzymes in organisms that use this pathway may be examples of this new bifunctional class of CobD and represent the missing L-Thr kinase for the late-cobalt-insertion pathway.
L-Ser-P is not a substrate of MmCobD or SeCobD.
S. enterica has not been shown to produce norCbas in nature but will produce norCbas when provided with EA-P exogenously 66, demonstrating that the adenosylcobinamide- phosphate (CbiB) synthase will use either AP-P or EA-P as substrates (Fig. 1). Here we demonstrated that neither MmCobD nor SeCobD could use L-Ser-P to generate EA-P (Fig. S3), further demonstrating that neither organism naturally synthesizes norCbas de novo. In addition, MmCobD did not generate L-Ser-P from ATP and L-Ser in vitro (Fig. S4). In vivo data indicate that L-Ser may inhibit MmCobD or a S. enterica cobD pduX strain more generally (Fig. 6). The ability of the S. enterica PduX enzyme to use L-Ser as substrate has not been established.
MmCobD has optimal enzymatic activity when purified in the absence of oxygen.
The K0.5 for normoxically purified MmCobD is outside the physiological range for the intercellular concentration of ATP for a bacterial cell (~1 mM) 84. The 1000-fold higher enzymatic efficiency and affinity for ATP of the anoxically purified enzyme demonstrates O2 sensitivity that might be attributed to several possible factors. The reason for the apparent poor affinity for ATP in vitro of the normoxically purified protein may be due to the oxidations of thiol groups critical to the function of the putative metal binding C-terminus. The brown color of the purified protein, the high numbers of cysteines and histidines (Fig. 2) capable of coordinating a metal, and the slight enhancement of ATPase activity when reduced with dithionite (Fig. 5B), as well as the strictly anaerobic lifestyle of M. mazei, leads us to speculate that MmCobD may possess a redox sensitive metal such as Fe(II) or one or more Fe-S clusters. It is possible that the disruption of such a metal center may result in conformational changes that are reflected by increased Km for ATP. We are currently investigating what metal(s) maybe present and how the anoxically purified enzyme behaves.
Normoxically purified MmCobD hints at possible mechanism of catalysis.
The positive cooperativity behavior of the normoxically purified MmCobD protein may be artefactual but may reveal something about the mechanism of the enzyme catalysis. However, because we were unable to obtain kinetic parameters for L-Thr or the L-Thr-P decarboxylation reaction we cannot be certain as to the nature of the observed positive cooperativity. The positive cooperativity behavior of MmCobD purified in the presence of air suggests several possibilities. The binding of a substrate to one site could increase the affinity of monomers for the formation of dimers. This explanation is unlikely because MmCobD and MmCobD1-385 form dimers in the absence of substrate (Fig. S6). There is the possibility of two active sites, one for the kinase reaction and another for the decarboxylation reaction. The binding of ATP and phosphorylation of L-Thr in one site might facilitate the binding of L-Thr-P and subsequent decarboxylation in a second site. We think this is unlikely given the involvement of PLP in the reaction and the high sequence homology to SeCobD. Alternatively, the phosphorylation of L-Thr to form L-Thr-P might result in a shift of the product within a single active site to allow decarboxylation. Since ADP inhibits the full-length enzyme (Fig. S5H), the release of ADP from the active site after L-Thr phosphorylation may be required before the L-Thr-P product can be shifted within the active site or shuttled to a second active site for decarboxylation. What is clear is that the absence of the C-terminus reduces the catalytic efficiency and lowers the affinity of the normoxically purified enzyme for ATP by 2-fold (Table 2A). These data support the conclusion that while the C-terminus is not required for activity, its presence improves catalytic efficiency. It is possible that the C-terminus may function as an allosteric regulator for both enzymatic activities, removing products like ADP, or facilitating diffusion of products like L-Thr-P between or within the active site(s).
The role of the C-terminus of MmCobD is complex.
The kinase and decarboxylase activities of MmCobD are localized to the first 385 residues of the protein (Figs. 3 5, S5). However, removal of the last 111 residues has a negative impact on MmCobD function in vivo and in vitro. In vitro the MmCobD1-385 enzyme has ATPase activity comparable to the wild-type enzyme and does not require the presence of L-Thr as co-substrate to enhance the ATPase activity (Fig. 5A). However, kinetic data indicate that MmCobD1-385 is a less efficient enzyme when compared to the full-length enzyme. Suggesting a structural or regulatory role for the C-terminus of the protein. The absence of the C-terminal domain appears to result in better access for substrates or ATP analogs (ADP-γ-S) to the active site. This idea is supported by the 2-fold lower affinity of MmCobD1-385 for ATP (Table 2A), and by the fact that twice as much ATP is needed to saturate the MmCobD1-385 protein than the MmCobD protein (Fig. 9, 10). Along this line of thought, it is worth contrasting the sensitivity of MmCobD and MmCobD1-385 to the ATPase inhibitor, ADP-γ-S. While the ATPase activity of MmCobD and the acetate kinase (SeEutP) control were sensitive to this inhibitor, the ATPase activity of MmCobD1-385 was not (Fig. S5H, 5I). This result would be consistent with the idea that the absence of the C-terminal domain allows unobstructed access to the active site and or lowers the binding affinity for ATP. The 1.5-fold higher K0.5 supports this idea (Table 2A). ADP-γ-S is a non-hydrolyzable ATP analog that inhibits kinases/ATPases by remaining bound to the active site, preventing ATP from entering and locking the enzyme in an inactive state. The ability of MmCobD1-385 to generate ADP in the presence of ADP-γ-S suggest the inhibitor does not remain bound to the active site, or the removal of the C-terminus somehow allows MmCobD1-385 to hydrolyze ADP-γ-S. There is a precedent for enzymes being activated by or using ADP-γ-S as substrate 85. Measurements of the Kd of these proteins for ATP and ADP-γ-S would help clarify these results. There is also a need to identify the ATP-binding site and the binding sites for L-Thr and L-Thr-P. Ongoing efforts to crystallize these proteins in the presence of substrates may provide answers to these questions.
Is L-Thr kinase activity a feature of CobD that is widespread in other Cba producers?
The association of the ATPase activity with the N-terminus of MmCobD raises the question of whether CobD from other organisms also have L-Thr kinase function. If so, how can these bifunctional CobDs be identified and differentiated from CobDs like the one found in S. enterica, which does not have L-Thr kinase activity? On average, CobD homologues are about 28% identical, but CobDs are commonly misannotated as histidinol-phosphate aminotransferases (hisC) due to structural and amino acid sequence similarity between CobD and HisC 40, 70. Given this hurdle, the identification of motifs responsible for L-Thr and ATP binding may be challenging. The protein sequence of MmCobD does not encode canonical ATP-binding motifs. Potential novel ATP or L-Thr binding motifs are the two small insertions spanning position 68-74 and 278-283 (Fig. 2), and the 13-amino acid extension at the N-terminus. The role that these extensions may play in the L-Thr kinase activity is under investigation. Additional work is needed to characterize the mechanism of catalysis.
Supplementary Material
(Multiple sequence alignment of CobD from methanogenic archaea with CobD from S. enterica)
(The intracellular concentration of Cby is the limiting factor for the complementation of S. enterica by MmCobD)
(MmCobD and SeCobD cannot decarboxylate L-Ser-P)
(MmCobD cannot use L-Ser as a substrate)
(Optimization of MmCobD reaction conditions)
(Gel permeation analysis for the determination of the oligomeric state of MmCobD proteins)
(Purification of MmCobD proteins)
(MALDI TOF analysis of MmCobD proteins)
(List of strains and plasmids)
(List of primers).
ACKNOWLEDGEMENTS
This work was supported by NIH grant R37 GM040313 to J.C.E.-S. N.K.T. was supported in part by NIH grant F31 GM095230; C.L.Z. was supported in part by the NIH grant F31 GM64009 and Advanced Opportunity Fellowship awarded by the Graduate School of the University of Wisconsin, Madison. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We thank Paul Renz (Universität Stuttgart, Germany) for his gift of (CN)2Cby, Gerhard Gottschalk (Georg-August-Universität Göttingen, Germany) for his gift of M. mazei genomic DNA. The authors also thank Dongtao Cui at the University of Georgia Chemical Sciences Magnetic Resonance facility for her assistance with NMR. We are grateful to Paula Pappalardo (University of Georgia) for her assistance with phylogenetic analyses.
ABBREVIATIONS:
- AP-P (R)-1
aminopropan-2-ol-O-phosphate
- L-Thr-P
L-threonine-O-3-phosphate
- L-Ser-P
O-phospho-L-serine
- EA-P
ethanolamine phosphate
- Cba
cobamide
- AdoCba
norCba; norCobamide
- Cby
cobyric acid
- ADP-γ-S
adenosylcobamide adenosine 5’-[γ-thio]triphosphate
- Cbi
cobinamide.
Footnotes
CONFLICT OF INTEREST. The authors declare that they have no conflicts of interest with the contents of this article.
REFERENCES
- [1].Renz P (1999) Biosynthesis of the 5,6-dimethylbenzimidazole moiety of cobalamin and of other bases found in natural corrinoids, In Chemistry and Biochemistry of B12. (Banerjee R, Ed.), pp 557–575, John Wiley & Sons, Inc, New York. [Google Scholar]
- [2].Stupperich E, Eisinger HJ, and Kräutler B (1988) Diversity of corrinoids in acetogenic bacteria. p-Cresolylcobamide from Sporomusa ovata, 5-methoxy-6-methylbenzimidazolylcobamide from Clostridium formicoaceticum and vitamin B12 from Acetobacterium woodii, Eur. J. Biochem 172, 459–464. [DOI] [PubMed] [Google Scholar]
- [3].Allen RH, and Stabler SP (2008) Identification and quantitation of cobalamin and cobalamin analogues in human feces, Am. J. Clin. Nutr 87, 1324–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Escalante-Semerena JC (2007) Conversion of cobinamide into adenosylcobamide in bacteria and archaea, J. Bacteriol 189, 4555–4560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Dion HW, Calkins DG, and Pfiffner JJ (1954) 2-Methyladenine, an hydrolysis product of pseudovitamin B12d, J. Amer. Che. Soc 76, 948–949. [Google Scholar]
- [6].Taylor CD, and Wolfe RS (1974) A simplified assay for coenzyme M (HSCH2CH2SO3). Resolution of methylcobalamin-coenzyme M methyltransferase and use of sodium borohydride, J. Biol. Chem 249, 4886–4890. [PubMed] [Google Scholar]
- [7].Thauer RK (2012) The Wolfe cycle comes full circle, Proc. Natl. Acad. Sci. U S A 109, 15084–15085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Krzycki JA (2004) Function of genetically encoded pyrrolysine in corrinoid-dependent methylamine methyltransferases, Curr. Opin. Chem. Biol 8, 484–491. [DOI] [PubMed] [Google Scholar]
- [9].Ferguson T, Soares JA, Lienard T, Gottschalk G, and Krzycki JA (2012) RamA, a protein required for reductive activation of corrinoid-dependent methylamine methyltransferase reactions in methanogenic Archaea (vol 284, pg 2285, 2009), J. Biol. Chem 287, 9328–9328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Taylor RT, and Weissbach H (1973) N5 -methylenetetrahydrofolate-homocysteine methyltransferases, In The Enzymes (Boyer PD, Ed.), pp 121–165, Academic Press, Inc, New York. [Google Scholar]
- [11].Bandarian V, Pattridge KA, Lennon BW, Huddler DP, Matthews RG, and Ludwig ML (2002) Domain alternation switches B(12)-dependent methionine synthase to the activation conformation, Nat. Struct. Biol 9, 53–56. [DOI] [PubMed] [Google Scholar]
- [12].Escalante-Semerena JC, and Warren MJ (2008) Biosynthesis and Use of Cobalamin (B12), In EcoSal - Escherichia coli and Salmonella: cellular and molecular biology (Böck A, Curtiss R III, Kaper JB, Karp PD, Neidhardt FC, Nyström T, Slauch JM, and Squires CL, Eds.), ASM Press, Washington, D. C. [Google Scholar]
- [13].Allers T, and Mevarech M (2005) Archaeal genetics - the third way, Nat. Rev. Genet 6, 58–73. [DOI] [PubMed] [Google Scholar]
- [14].Graham DE, Overbeek R, Olsen GJ, and Woese CR (2000) An archaeal genomic signature, Proc. Natl. Acad. Sci. U. S. A 97, 3304–3308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Woodson JD, Zayas CL, and Escalante-Semerena JC (2003) A new pathway for salvaging the coenzyme B12 precursor cobinamide in archaea requires cobinamide-phosphate synthase (CbiB) enzyme activity, J. Bacteriol 185, 7193–7201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zayas CL, Woodson JD, and Escalante-Semerena JC (2006) The cobZ gene of Methanosarcina mazei Gö1 encodes the nonorthologous replacement of the α-ribazole-5'-phosphate phosphatase (CobC) enzyme of Salmonella enterica, J. Bacteriol 188, 2740–2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Buan NR, Rehfeld K, and Escalante-Semerena JC (2006) Studies of the CobA-type ATP:Co(I)rrinoid adenosyltransferase enzyme of Methanosarcina mazei strain Gö1., J. Bacteriol 188, 3543–3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Rodionov DA, Vitreschak AG, Mironov AA, and Gelfand MS (2003) Comparative genomics of the vitamin B12 metabolism and regulation in prokaryotes, J. Biol. Chem 278, 41148–41159. [DOI] [PubMed] [Google Scholar]
- [19].Woodson JD, Peck RF, Krebs MP, and Escalante-Semerena JC (2003) The cobY gene of the archaeon Halobacterium sp. strain NRC-1 is required for de novo cobamide synthesis, J. Bacteriol 185, 311–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Woodson JD, Reynolds AA, and Escalante-Semerena JC (2005) ABC transporter for corrinoids in Halobacterium sp. strain NRC-1, J. Bacteriol 187, 5901–5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Woodson JD, and Escalante-Semerena JC (2006) The cbiS gene of the archaeon Methanopyrus kandleri AV19 encodes a bifunctional enzyme with adenosylcobinamide amidohydrolase and alpha-ribazole-phosphate phosphatase activities, J. Bacteriol 188, 4227–4235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhang Y, Rodionov DA, Gelfand MS, and Gladyshev VN (2009) Comparative genomic analyses of nickel, cobalt and vitamin B12 utilization, BMC Genomics 10, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Fan C, and Bobik TA (2008) The PDUX enzyme of Salmonella enterica is an L-threonine kinase used for coenzyme B12 synthesis, J. Biol. Chem 283, 11322–11329. [DOI] [PubMed] [Google Scholar]
- [24].Fan C, Fromm HJ, and Bobik TA (2009) Kinetic and functional analysis of L-threonine kinase, the PduX enzyme of Salmonella enterica, J. Biol. Chem 284, 20240–20248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Brushaber KR, O'Toole GA, and Escalante-Semerena JC (1998) CobD, a novel enzyme with L-threonine-O-3-phosphate decarboxylase activity, is responsible for the synthesis of (R)-1-amino-2-propanol O-2-phosphate, a proposed new intermediate in cobalamin biosynthesis in Salmonella typhimurium LT2, J. Biol. Chem 273, 2684–2691. [DOI] [PubMed] [Google Scholar]
- [26].Peariso K, Zhou ZS, Smith AE, Matthews RG, and Penner-Hahn JE (2001) Characterization of the zinc sites in cobalamin-independent and cobalamin-dependent methionine synthase using zinc and selenium X-ray absorption spectroscopy, Biochemistry 40, 987–993. [DOI] [PubMed] [Google Scholar]
- [27].Drennan CL, Huang S, Drummond JT, Matthews RG, and Ludwig ML (1994) How a protein binds B12: A 3.0Å X-ray structure of B12-binding domains of methionine synthase, Science 266, 1669–1674. [DOI] [PubMed] [Google Scholar]
- [28].Hall DA, Vander Kooi CW, Stasik CN, Stevens SY, Zuiderweg ER, and Matthews RG (2001) Mapping the interactions between flavodoxin and its physiological partners flavodoxin reductase and cobalamin-dependent methionine synthase, Proc. Natl. Acad. Sci. USA 98, 9521–9526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Banerjee RV, and Matthews RG (1990) Cobalamin-dependent methionine synthase, FASEB J. 4, 1450–1459. [DOI] [PubMed] [Google Scholar]
- [30].Datsenko KA, and Wanner BL (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products., Proc. Natl. Acad. Sci. USA 97, 6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Berkowitz D, Hushon JM, Whitfield HJ Jr., Roth J, and Ames BN (1968) Procedure for identifying nonsense mutations, J. Bacteriol 96, 215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Balch WE, and Wolfe RS (1976) New approach to the cultivation of methanogenic bacteria: 2-mercaptoethanesulfonic acid (HS-CoM)-dependent growth of Methanobacterium ruminantium in a pressurized atmosphere, Appl. Environ. Microbiol 32, 781–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Bertani G (1951) Studies on lysogenesis. I. The mode of phage liberation by lysogenic Escherichia coli., J. Bacteriol 62, 293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Bertani G (2004) Lysogeny at mid-twentieth century: P1, P2, and other experimental systems., J. Bacteriol 186, 595–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Raleigh EA, Lech K, and Brent R (1989) Selected topics from classical bacterial genetics, In Curr. Protoc. Mol. Biol (Ausubel FA, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, and Struhl K, Eds.), p 1.4, Wiley Interscience, New York. [DOI] [PubMed] [Google Scholar]
- [36].Woodcock DM, Crowther PJ, Doherty J, Jefferson S, De Cruz E, Noyer-Weidner M, Smith SS, Michael MZ, and Graham MW (1989) Quantitative evaluation of Escherichia coli host strains for tolerance to cytosine methylation in plasmid and phage recombinants, Nucl. Acids Res 17, 3469–3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Calvin NM, and Hanawalt PC (1988) High-efficiency transformation of bacterial cells by electroporation, J. Bacteriol 170, 2796–2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Guzman LM, Belin D, Carson MJ, and Beckwith J (1995) Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter, J. Bacteriol 177, 4121–4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Rocco CJ, Dennison KL, Klenchin VA, Rayment I, and Escalante-Semerena JC (2008) Construction and use of new cloning vectors for the rapid isolation of recombinant proteins from Escherichia coli, Plasmid 59, 231–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Cheong CG, Bauer CB, Brushaber KR, Escalante-Semerena JC, and Rayment I (2002) Three-dimensional structure of the L-threonine-O-3-phosphate decarboxylase (CobD) enzyme from Salmonella enterica, Biochemistry 41, 4798–4808. [DOI] [PubMed] [Google Scholar]
- [41].Tavares NK (2016) Biosynthesis of the Aminopropanol Moiety of Cobamides by Bacterial and Archaeal Enzymes, In Dept. of Microbiology, University of Georgia. [Google Scholar]
- [42].Miroux B, and Walker JE (1996) Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels, J. Mol. Biol 260, 289–298. [DOI] [PubMed] [Google Scholar]
- [43].Tartof K, and Hobbs C (1987) Improved media for growing plasmid and cosmid clones, Bethesda Research Laboratories Focus 9, 19. [Google Scholar]
- [44].Sambrook J, Fritsch EF, and Maniatis T (1989) Molecular Cloning: A Laboratory Manual, Second ed., Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y. [Google Scholar]
- [45].Varnado CL, and Goodwin DC (2004) System for the expression of recombinant hemoproteins in Escherichia coli, Protein Expr. Purif 35, 76–83. [DOI] [PubMed] [Google Scholar]
- [46].Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, and Bairoch A (2003) ExPASy: The proteomics server for in-depth protein knowledge and analysis, Nucleic Acids Res. 31, 3784–3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Wilkins MR, Gasteiger E, Bairoch A, Sanchez JC, Williams KL, Appel RD, and Hochstrasser DF (1999) Protein identification and analysis tools in the ExPASy server, Methods Mol. Biol 112, 531–552. [DOI] [PubMed] [Google Scholar]
- [48].Zegzouti H, Zdanovskaia M, Hsiao K, and Goueli SA (2009) ADP-Glo: A Bioluminescent and homogeneous ADP monitoring assay for kinases, Assay Drug Dev. Technol 7, 560–572. [DOI] [PubMed] [Google Scholar]
- [49].Altschul SF, Madden TL, Schaffer AA, Zhang J, Miller W, and Lipmann DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs., Nucl. Acids Res 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Markowitz VM, Korzeniewski F, Palaniappan K, Szeto E, Werner G, Padki A, Zhao X, Dubchak I, Hugenholtz P, Anderson I, Lykidis A, Mavromatis K, Ivanova N, and Kyrpides NC (2006) The integrated microbial genomes (IMG) system, Nucleic Acids Res. 34, D344–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput, Nucleic Acids Res. 32, 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Gouet P, Courcelle E, Stuart DI, and Metoz F (1999) ESPript: multiple sequence alignments in PostScript., Bioinformatics 15, 305–308. [DOI] [PubMed] [Google Scholar]
- [53].Bergmeyer HU, Bergmeyer J, Grassl M, and Berger R (1985) Methods of enzymatic analysis. vol. iv. “enzymes 2: Esterases, glycosidases, lyases, ligases” 3rd Edition. Weinheim; Deerfield Beach, Florida; Basel: Verlag Chemie, 1984. 426 S., 258 DM, Acta Biotechnologica 5, 114–114. [Google Scholar]
- [54].Horswill AR, and Escalante-Semerena JC (2002) Characterization of the propionyl-CoA synthetase (PrpE) enzyme of Salmonella enterica: Residue Lys592 is required for propionyl-AMP synthesis, Biochemistry 41, 2379–2387. [DOI] [PubMed] [Google Scholar]
- [55].Wilson A. C. a. A. B. P. (1962) Regulation of flavin synthesis by Escherichia coli, J. Gen. Microbiol 28, 283–303. [DOI] [PubMed] [Google Scholar]
- [56].Havemann GD, and Bobik TA (2003) Protein content of polyhedral organelles involved in coenzyme B12-dependent degradation of 1,2-propanediol in Salmonella enterica serovar Typhimurium LT2., J. Bacteriol 185, 5086–5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Fang H, Kang J, and Zhang D (2017) Microbial production of vitamin B12: a review and future perspectives, Microb. Cell Fact 16, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Escalante-Semerena JC, and Roth JR (1987) Regulation of cobalamin biosynthetic operons in Salmonella typhimurium, J. Bacteriol 169, 2251–2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Jeter RM, Olivera BM, and Roth JR (1984) Salmonella typhimurium synthesizes cobalamin (vitamin B12) de novo under anaerobic growth conditions, J. Bacteriol 159, 206–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Roof DM, and Roth JR (1988) Ethanolamine utilization in Salmonella typhimurium, J. Bacteriol 170, 3855–3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Stojiljkovic I, Baumler AJ, and Heffron F (1995) Ethanolamine utilization in Salmonella typhimurium: nucleotide sequence, protein expression, and mutational analysis of the cchA cchB eutE eutJ eutG eutH gene cluster, J. Bacteriol 177, 1357–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Kofoid E, Rappleye C, Stojiljkovic I, and Roth J (1999) The 17-gene ethanolamine (eut) operon of Salmonella typhimurium encodes five homologues of carboxysome shell proteins, J. Bacteriol 181, 5317–5329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Stupperich E, and Kräutler B (1988) Pseudo vitamin B12 or 5-hydroxybenzimidazolyl-cobamide are the corrinoids found in methanogenic bacteria, Arch. Microbiol 149, 213–217. [Google Scholar]
- [64].Kräutler B, Fieber W, Osterman S, Fasching M, Ongania K-H, Gruber K, Kratky C, Mikl C, Siebert A, and Diekert G (2003) The cofactor of tetrachloroethene reductive dehalogenase of Dehalospirillum multivorans is Norpseudo-B12, a new type of natural corrinoid., Helv. Chim. Acta 86, 3698–3716. [Google Scholar]
- [65].Zayas CL, and Escalante-Semerena JC (2007) Reassessment of the late steps of coenzyme B12 synthesis in Salmonella enterica: Evidence that dephosphorylation of adenosylcobalamin-5'-phosphate by the CobC phosphatase is the last step of the pathway, J. Bacteriol 189, 2210–2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Zayas CL, Claas K, and Escalante-Semerena JC (2007) The CbiB protein of Salmonella enterica is an integral membrane protein involved in the last step of the de novo corrin ring biosynthetic pathway, J. Bacteriol 189, 7697–7708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Martinez-Carrion M (1975) 31P nuclear-magnetic-resonance studies of pyridoxal and pyridoxamine phosphates. Interaction with cytoplasmic aspartate transaminase, Eur. J. Biochem 54, 39–43. [DOI] [PubMed] [Google Scholar]
- [68].Segel IH (1975) Enzyme Kinetics, John Wiley & Sons, New York. [Google Scholar]
- [69].Moore TC, and Escalante-Semerena JC (2016) The EutQ and EutP proteins are novel acetate kinases involved in ethanolamine catabolism: physiological implications for the function of the ethanolamine metabolosome in Salmonella enterica, Mol. Microbiol 99, 497–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Cheong CG, Escalante-Semerena JC, and Rayment I (2002) Structural studies of the L-threonine-O-3-phosphate decarboxylase (CobD) enzyme from Salmonella enterica: the apo, substrate, and product-aldimine complexes, Biochemistry 41, 9079–9089. [DOI] [PubMed] [Google Scholar]
- [71].Nagradova N (2003) Interdomain communications in bifunctional enzymes: how are different activities coordinated?, IUBMB Life 55, 459–466. [DOI] [PubMed] [Google Scholar]
- [72].Scapin G, and Blanchard JS (1998) Enzymology of bacterial lysine biosynthesis, Adv. Enzymol. Relat. Areas Mol. Biol 72, 279–324. [DOI] [PubMed] [Google Scholar]
- [73].Tanizawa K, and Soda K (1979) Inducible and constitutive kynureninases. Control of the inducible enzyme activity by transamination and inhibition of the constitutive enzyme by 3-hydroxyanthranilate, J Biochem. 86, 499–508. [DOI] [PubMed] [Google Scholar]
- [74].Baker AS, Ciocci MJ, Metcalf WW, Kim J, Babbitt PC, Wanner BL, Martin BM, and Dunaway-Mariano D (1998) Insights into the mechanism of catalysis by the P-C bond-cleaving enzyme phosphonoacetaldehyde hydrolase derived from gene sequence analysis and mutagenesis, Biochemistry 37, 9305–9315. [DOI] [PubMed] [Google Scholar]
- [75].Imoto T, Yamada H, Okazaki K, Ueda T, Kuroki R, and Yasukochi T (1987) Modifications of stability and function of lysozyme, J. Prot. Chem 6, 95–107. [Google Scholar]
- [76].Wang NC, and Lee CY (2006) Molecular cloning of the aspartate 4-decarboxylase gene from Pseudomonas sp. ATCC 19121 and characterization of the bifunctional recombinant enzyme, Appl. Microbiol. Biotechnol 73, 339–348. [DOI] [PubMed] [Google Scholar]
- [77].Stevenson DE, Akhtar M, and Gani D (1990) L-methionine decarboxylase from Dryopteris filix-mas: purification, characterization, substrate specificity, abortive transamination of the coenzyme, and stereochemical courses of substrate decarboxylation and coenzyme transamination, Biochemistry 29, 7631–7647. [DOI] [PubMed] [Google Scholar]
- [78].Ahmed SA, Esaki N, Tanaka H, and Soda K (1985) Mechanism of inactivation of apha-amino-epsilon caprolactam racemase by alpha-amino-delta-valerolactam, Agr. Biolog. Chem 49, 2991–2997. [Google Scholar]
- [79].Kurokawa Y, Watanabe A, Yoshimura T, Esaki N, and Soda K (1998) Transamination as a side-reaction catalyzed by alanine racemase of Bacillus stearothermophilus 1, J. Biochem 124, 1163–1169. [DOI] [PubMed] [Google Scholar]
- [80].Häring D, Lees MR, Banaszak LJ, and Distefano MD (2002) Exploring routes to stabilize a cationic pyridoxamine in an artificial transaminase: site-directed mutagenesis versus synthetic cofactors, Protein Eng. 15, 603–610. [DOI] [PubMed] [Google Scholar]
- [81].Sinz Q, Freiding S, Vogel RF, and Schwab W (2013) A hydrolase from Lactobacillus sakei moonlights as a transaminase, Appl. Environ. Microbiol 79, 2284–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Blanche F, Thibaut D, Debussche L, Hertle R, Zipfel F, and Muller G (1993) Parallels and decisive differences in vitamin-B12 biosyntheses, Angew. Chem. Int. Ed. Engl 32, 1651–1653. [Google Scholar]
- [83].Roessner CA, Santander PJ, and Scott AI (2001) Multiple biosynthetic pathways for vitamin B12: variations on a central theme, Vitam. Horm 61, 267–297. [DOI] [PubMed] [Google Scholar]
- [84].Yaginuma H, Kawai S, Tabata KV, Tomiyama K, Kakizuka A, Komatsuzaki T, Noji H, and Imamura H (2014) Diversity in ATP concentrations in a single bacterial cell population revealed by quantitative single-cell imaging, Sci. Rep 4, 6522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Smith DM, Fraga H, Reis C, Kafri G, and Goldberg AL (2011) ATP binds to proteasomal ATPases in pairs with distinct functional effects, implying an ordered reaction cycle, Cell 144, 526–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(Multiple sequence alignment of CobD from methanogenic archaea with CobD from S. enterica)
(The intracellular concentration of Cby is the limiting factor for the complementation of S. enterica by MmCobD)
(MmCobD and SeCobD cannot decarboxylate L-Ser-P)
(MmCobD cannot use L-Ser as a substrate)
(Optimization of MmCobD reaction conditions)
(Gel permeation analysis for the determination of the oligomeric state of MmCobD proteins)
(Purification of MmCobD proteins)
(MALDI TOF analysis of MmCobD proteins)
(List of strains and plasmids)
(List of primers).