


Abstract
The recent identification and development of RNA-guided enzymes for programmable cleavage of target nucleic acids offers exciting possibilities for both therapeutic and biotechnological applications. However, critical challenges such as expensive guide RNAs and inability to predict the efficiency of target recognition, especially for highly-structured RNAs, remain to be addressed. Here, we introduce a programmable RNA restriction enzyme, based on a budding yeast Argonaute (AGO), programmed with cost-effective 23-nucleotide (nt) single-stranded DNAs as guides. DNA guides offer the advantage that diverse sequences can be easily designed and purchased, enabling high-throughput screening to identify optimal recognition sites in the target RNA. Using this DNA-induced slicing complex (DISC) programmed with 11 different guide DNAs designed to span the sequence, sites of cleavage were identified in the 352-nt human immunodeficiency virus type 1 5′-untranslated region. This assay, coupled with primer extension and capillary electrophoresis, allows detection and relative quantification of all DISC-cleavage sites simultaneously in a single reaction. Comparison between DISC cleavage and RNase H cleavage reveals that DISC not only cleaves solvent-exposed sites, but also sites that become more accessible upon DISC binding. This study demonstrates the advantages of the DISC system for programmable cleavage of highly-structured, functional RNAs.
INTRODUCTION
Argonaute (AGO) proteins are the central enzymes of RNA-induced silencing complex (RISC) pathways in eukaryotes, and homologues are found in the other domains of life (1–4). Along with clustered regularly interspaced short palindromic repeat (CRISPR)/Cas-based systems, AGOs are being exploited for use in biotechnological applications. Recent examples include a programmable artificial DNA restriction enzyme, based on the Pyrococcus furiosus AGO, capable of producing cleaved DNA products with specifically-engineered sticky ends (5), and two programmable RNA-dependent endoribonucleases, established from bacterial CRISPR systems, that are specific for single-stranded (ss) RNA with potential in vivo applications (6,7). Despite these advances, the ability to site-specifically cleave or manipulate highly-structured RNAs in vitro in a time- and cost-effective manner remains to be demonstrated.
Programmable RNA-targeting endoribonucleases for in vitro use have been highly sought after, with applications in RNA structure-function studies, RNA nanotechnology, and therapeutics. In contrast to B-form DNA, which has a uniform and predictable structure, RNA folds into many complex secondary and tertiary structures in an unpredictable manner. Previous approaches using hammerhead (HH) ribozymes (8), catalytic DNAzymes (9), and artificial site-specific RNA endonucleases (ASREs) (10) all suffer limitations such as the need for extensive re-engineering (HH ribozyme/ASREs) or additional selective evolution (DNAzymes) for each new target. Furthermore, CRISPR/Cas-based endoribonucleases require RNA guides that must be either transcribed and purified in vitro or purchased at a significant cost. Moreover, this method has not been shown to recognize structured RNA elements (6) that tend to be hallmarks of many functional RNAs (11,12). An ideal programmable endoribonuclease would exhibit four critical features: efficient site-specific cleavage of RNA without off-target activity, a convenient and inexpensive means of adapting the programmable recognition element, no target site sequence limitations, and activity toward both unstructured and highly structured RNAs. To our knowledge, no existing system satisfies all of these criteria.
Here, we describe a yeast AGO capable of efficient and programmable DNA-dependent endoribonuclease activity in vitro. Once programmed with short, inexpensive and easily interchangeable guide DNAs (gDNAs), this DNA-induced slicing complex (DISC) demonstrates high specificity with extremely low off-target cleavage. DISC was capable of targeting multiple sites spanning the highly-structured human immunodeficiency virus type 1 (HIV-1) 5′-untranslated region (5′UTR) RNA. Additionally, we have adapted a primer extension/capillary electrophoresis method typically used for RNA structure probing to identify optimal sites of target cleavage within the 5′UTR in a high-throughput manner.
MATERIALS AND METHODS
Expression and purification of K. polysporus AGO207
A recombinant protein encompassing Thr207-Ile1251 of Kluyveromyces polysporus AGO (AGO207) was expressed and purified as previously described (13). The concentration of purified AGO207 was determined by Bradford assay (Bio-Rad) and stock aliquots were stored at -80°C in storage buffer (10 mM Tris–HCl pH 7.5, 200 mM NaCl, 5 mM DTT).
Preparation of miR-20a-derived substrates
A list of RNA and DNA oligonucleotides used in this study is provided (Supplementary Tables S1–S4). miR-20a-derived 5′ phosphorylated guide RNA (gRNA) was chemically synthesized (Dharmacon), deprotected, and gel-purified. 5′ phosphorylated gDNAs were chemically synthesized (Sigma Aldrich). The sequences encoding target RNAs were cloned into a pUC19 vector and transcribed in vitro using T7 RNA polymerase. DNase I-treated transcripts were gel purified (10% polyacrylamide, 8 M urea, 1× TBE), capped using ScriptCap m7G Capping System (CellScript) and GTP or [α-32P]-GTP (3000 Ci mmol−1) and gel purified again. DNA targets were chemically synthesized (Sigma Aldrich), 5′ end-labeled with T4 PNK (ThermoFisher) and [γ32P]-ATP (3000 Ci mmol−1) before gel purification. Unlabeled nucleic acid concentrations were quantified by spectrophotometry at 260 nm and calculated using the molar extinction coefficient. All extinction coefficients for substrates synthesized by commercial vendors were calculated or provided by the manufacturer. The extinction coefficient used for capped miR-20a RNA targets is 587 900 l/mol•cm.
miR-20a-mediated cleavage assays
Stock AGO207 was diluted using storage buffer supplemented with 0.5 mg/ml Ultrapure BSA (Ambion) and stored at –80°C. All assays were performed in DISC reaction buffer (20 mM Tris–HCl pH 7.5, 150 mM NaCl, 1 mM MgCl2, 1 mM DTT, 5% glycerol), 0.05 mg/ml BSA, and 0.4 U/μl RiboLock RNase inhibitor (ThermoFisher). AGO207 (1 μM) was mixed with 50 nM gRNA or gDNA and incubated at 25°C for 30 min to form the RISC or DISC, respectively. Cleavage was initiated by adding 1 μl 5′-capped target RNA (final concentration, 25 nM) and trace amounts of 32P-cap-labeled target in a 10 μl reaction and incubating at 30°C for 20 min. For cleavage assays with DNA targets, 32P-end-labeled targets were added to a final concentration of 25 nM. Reactions were quenched with 10 μl formamide loading buffer (95% formamide, 18 mM EDTA, 0.025% SDS, 0.025% bromophenol blue, 0.025% xylene cyanol). Products were resolved on 16% denaturing gels (16% polyacrylamide, 8 M urea, 1× TBE) and visualized on a Typhoon (GE Healthcare) phosphorimager. Bands were quantified by ImageQuant (GE Healthcare). All cleavage percentages were calculated using Equation (1), plotted using Equations (1) or (2), and averaged over three independent experiments. Cleavage of nucleic acid target by either RISC or DISC given as percent:
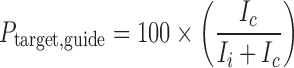 |
(1) |
where P is the percent cleavage of either target RNA or target DNA by RISC or DISC, and Ic and Ii indicate the band intensities of the 5′ cleavage product and intact substrate, respectively.
Relative percent, Prel, target cleavage of a given guide:target pair relative to the gRNA-dependent RNA cleavage:
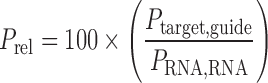 |
(2) |
Identification of appropriate guide: AGO207 ratio
Since AGO207 co-purifies with bound endogenous Escherichia coli RNA (13,14), guide:protein ratios were varied to identify an appropriate amount of gDNA to mix with AGO207 for biochemical assays. AGO207 (500 nM) was pre-incubated with increasing amounts of gDNA (0–100 nM) for 30 min at 25°C followed by addition of cap-labeled miR-20a-derived target (1 nM) and incubation at 30°C for 20 min. Reactions were quenched with formamide loading buffer and resolved by 16% denaturing PAGE (8 M urea, 1× TBE). Gels were visualized on a Typhoon phosphorimager and quantified by ImageQuant. All cleavage percentages were calculated using Equation (1) and averaged over three independent experiments. Cleavage by 500 nM AGO207 saturated at 70 ± 3% target cleaved when guided by 10–25 nM gDNA (Supplementary Figure S1).
Validation of DISC-specificity on unstructured targets
Specificity and fidelity of DISC was determined by performing in vitro cleavage assays using 10 nM gDNA or gRNA and 1 μM AGO207. AGO207 was pre-incubated with gRNA or gDNA followed by addition of either perfectly matched cap-labeled RNA target (25 nM) or the same target but with a dinucleotide mismatch at the cleavage site. For systematic single- and dinucleotide mismatch assays, mismatches were introduced to the guide strands and all reactions were performed with the same target. Products were resolved on 16% denaturing PAGE and gels were visualized on a Typhoon phosphorimager.
Preparation of HIV-1 ΔDIS 5′UTR transcript
The HIV-1 5′UTR variant used in this study contained a stable GAGA tetraloop sequence in place of the dimerization initiation site (DIS) loop (HIV-1 ΔDIS 5′UTR) (see Supplementary Figure S3 and Supplementary Table S2) to eliminate heterogeneity. The HIV-1 ΔDIS 5′UTR was in vitro transcribed from a FokI-digested pUC18 vector with an upstream HH ribozyme using T7 RNA polymerase. DNase I-treated transcripts were purified on denaturing gels (7% polyacrylamide, 8 M urea, 1× TBE) and visualized by UV shadowing. The RNA was eluted from the gel in elution buffer (500 mM ammonium acetate, 1 mM EDTA, 0.1% (w/v) SDS), ethanol precipitated, resuspended in MilliQ water, and quantified by UV absorbance at 260 nm using an extinction coefficient of 3 243 098 l/mol•cm. The RNA was 5′ phosphorylated with T4 PNK4 (ThermoFisher) and ATP or [γ32P]-ATP (3000 Ci mmol−1). In the case of the body-labeled RNA, [α32P]-GTP (3000 Ci mmol−1) was added to the in vitro transcription reaction.
Design of gDNAs for HIV-1 5′UTR cleavage
Guide DNAs for experiments targeting the HIV-1 ΔDIS 5′UTR sequence (Supplementary Tables S3 and S4) were generated by following the workflow outlined in Supplementary Figure S4. To facilitate gDNA design, we developed a Python-based script that will allow users to input an RNA sequence of interest and generate an output list of gDNAs that target the input RNA. Users can select any length of gDNA that they want to generate, as well as specify the interval between cleavage sites. The script commands are based on the workflow outlined in Supplementary Figure S4 and can be downloaded from https://research.cbc.osu.edu/musier-forsyth.1/tools/.
HIV-1 ΔDIS 5′UTR cleavage assays with individual gDNA
All cleavage assays were performed in DISC reaction buffer. HIV-1 ΔDIS 5′UTR substrate was folded by mixing unlabeled RNA substrate (10 nM) and trace amounts of 32P end-labeled RNA in 50 mM HEPES (pH 7.5). Samples were heated at 80°C for two min followed by incubation at 60°C for four min. MgCl2 was added to a final concentration of 10 mM and the sample was transferred to 37°C for 6 min followed by incubation on ice for at least 30 min. Sample homogeneity was evaluated by native PAGE (6% polyacrylamide, 1× TB, 1 mM MgCl2) at 4°C (Supplementary Figure S3). For DISC formation and HIV-1 ΔDIS 5′UTR cleavage, AGO207 (500 nM) was pre-mixed with gDNA (10 nM) for 30 min at 25°C in a 9 μl reaction followed by addition of 1 μl of HIV-1 ΔDIS 5′UTR substrate (final concentration 1 nM) and incubation at 30°C for 20 min. Reactions were quenched with formamide dye and products were resolved on denaturing PAGE (8% polyacrylamide, 8 M urea, 1× TBE). All gels were visualized on a Typhoon phosphorimager and quantified by ImageQuant using Equation (1).
Selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE)
SHAPE probing experiments of HIV-1 ΔDIS 5′UTR were performed as described previously (15). Briefly, in vitro transcribed and gel-purified RNA was refolded as described above. The refolded RNA was then reacted with N-methylisotoic anhydride (NMIA) for 45 min and ethanol precipitated. SHAPE reactivities were determined via primer extension and capillary electrophoresis using a NED™ (Thermo-Fisher)-labeled DNA primer and SuperScriptIII (Invitrogen). SHAPE reactivities were determined using the RiboCAT data analysis tool (15). Results are the average of four replicates.
Cleavage assays using RNase H
All RNase H-mediated RNA cleavage assays were carried out under similar conditions as DISC-mediated cleavage assays except that the order of mixing gDNA, target RNA, and enzyme were modified. To determine the approximate amount of RNase H that results in similar levels of cleavage of the unstructured miR-20a target as DISC, 1 nM of miR-20 cap-labeled target RNA and 10 nM miR-20a gDNA were pre-incubated at 25°C for 30 min to allow heteroduplex formation. RNase H (0.25–5 units, New England Biolabs) was added to the hybrid duplex to a final volume of 10 μl and the reaction was incubated at 30°C for 20 min. Reactions were quenched with formamide loading dye and substrates were resolved from products by denaturing 16% PAGE. Products were quantified using Equation (1). Based on these results, 1.5 units of RNase H was used in each 10 μl reaction for subsequent experiments.
Comparing activity of DISC and RNase H on unstructured or structured targets
DISC reactions were carried out as described above for unstructured and structured targets using 500 nM AGO207, 10 nM gDNA and 1 nM target RNA. AGO207 and gDNAs were mixed to assemble DISCs, followed by addition of the target RNA. RNase H assays were carried out as described above by first incubating 10 nM gDNA with 1 nM target RNA, followed by addition of 1.5 units of RNase H. Cleavage events of the miR-20a gDNA and HIV-1 ΔDIS 5′UTR targets were quantified using Equation (1).
HIV-1 ΔDIS 5′UTR cleavage assays with mixture of gDNAs
Cleavage assays using a mixture of gDNAs were performed similarly to the individually guided cleavage assays except that equimolar amounts of each of the selected gDNAs (20 nM total) were pre-mixed together with AGO207 (2 μM) before adding to the reaction mixture (guide and AGO207 concentrations are indicated when varied for experiments that evaluate assay conditions). The mixture was pre-incubated at 25°C for 30 min to form a mixture of DISCs that would recognize different regions of the HIV-1 ΔDIS 5′UTR substrate. After DISC-formation, 25 nM 32P body-labeled HIV-1 ΔDIS 5′UTR was added and 3-μl aliquots were removed at indicated time points. Reactions were quenched with formamide dye. Products were resolved on denaturing PAGE (8% polyacrylamide, 8 M urea, 1× TBE).
High-throughput analysis of DISC-mediated cleavage of HIV-1 ΔDIS 5′UTR
Cleavage reactions using unlabeled HIV-1 ΔDIS 5′UTR substrate were performed to generate DISC-mediated products for analysis by a reverse transcription/primer extension reaction primed by 5′-NED™ fluorophore-labeled oligonucleotide primer. Eleven gDNAs targeting the HIV-1 ΔDIS 5′UTR at 23 nucleotide (nt) increments between positions 24–276 (gDNA2–12, see Supplementary Table S3) were pre-mixed at equimolar concentrations (20 nM total). AGO207 (500 nM) and the gDNA mixture were pre-incubated at 25°C for 30 min in reaction buffer. Following DISC-formation, folded unlabeled HIV-1 ΔDIS 5′UTR (25 nM) was added and cleavage was performed at 30°C for 60 min. Reactions were quenched and RNA was phenol-chloroform extracted, ethanol precipitated in the presence of glycogen (2 μg), and stored as a pellet at –20°C. Control reactions were performed similarly except gDNAs were excluded for background subtraction.
RNA pellets were resuspended in 9 μl MilliQ water, annealed with 2 μl of 5 μM NED™-labeled primer and extended using Superscript III reverse transcriptase following the manufacturer's protocol (Invitrogen) in a total reaction volume of 20 μl. Remaining RNA was digested by adding 1 μl of 4 M NaOH and heating to 95°C for 3 min. The reactions were then neutralized with 2 μl of 2 M HCl. For each sample, 3 μl of neutralized reaction was added to 17 μl of MilliQ water and ethanol precipitated with 10 μg of glycogen. To allow for sequence alignment, Sanger-style sequencing reactions were also performed using the same NED™-labeled primer with the transcription template plasmid using the Thermo Sequenase Cycle Sequencing Kit (ThermoFisher) per the manufacturer's protocol. All reaction and sequencing pellets were resuspended in formamide supplemented with GeneScan™ 600 LIZ® Size Standard (Applied Biosystems) and resolved using a 3730 DNA Analyzer (Applied Biosystems) at the Plant Microbe Genomics Facility at The Ohio State University. The resulting electropherograms were analyzed using RiboCAT (15). For each dataset, the normalized reactivity values were as described (15) using a sample devoid of gDNA, gDNA(–), as the background control. Reported reactivity values represent the mean of three independent experiments. Raw data will be made available upon request.
RESULTS
DNA-guided RNA cleavage by budding yeast AGO
Recently, human AGO2 was reported to be a catalytically active RNase when programmed with a gDNA (16). Here, we tested the capability of a previously characterized N-terminal truncation variant of the budding yeast K. polysporus AGO, AGO207 (Supplementary Figure S1A), to function as the catalytic part of a DISC (13). The truncation mutant was chosen because it has been well-characterized previously and retains core wild-type function in terms of guide and target recognition and target cleavage (13,14). Furthermore, the N-terminal region varies across different eukaryotic species and is absent in prokaryotes, suggesting that this sequence may have species-specific functionality. K. polysporus AGO was also chosen based on complications associated with other, well-characterized AGO proteins. Pyrococcus furiosus and Thermus thermophilus are both thermophiles that function optimally at high temperature (17,18) while human AGOs are not expressed well in bacteria and would require more complicated expression strategies (19–21).
Previous studies showed that AGO207 uses a 5′ monophosphorylated 23-nt gRNA to cleave complementary RNA targets. To test if gDNA could activate AGO207 to cleave RNA or DNA targets, the purified recombinant protein was loaded with either a miR-20a-derived gRNA or gDNA, followed by addition of either a 5′ labeled 60-nt unstructured complementary RNA or DNA target (Figure 1A and B). Due to the well-documented difficulty of removing cellular RNAs from eukaryotic AGO proteins (13,14,19–21) most of the purified AGO207 molecules are bound to endogenous E. coli RNAs (>63,000 unique sequences) and only the fraction in the RNA-free form is available to be programmed for specific cleavage (13). AGO207 programmed with the gDNA retained ∼80% target RNA cleavage relative to gRNA (Figure 1C and Supplementary Figure S1B and C), demonstrating that gDNA can activate yeast AGO as a functional DISC against unstructured RNAs. In addition, no off-target cleavage was observed indicating that the AGO207 molecules bound to endogenous RNAs do not affect cleavage by DISC loaded with gDNA. Furthermore, neither RISC nor DISC showed appreciable cleavage of the DNA target (Figure 1C). To examine whether RISC and DISC cleave 5′-radiolabeled target RNAs at the same position, their reaction products were resolved on a 16% denaturing polyacrylamide sequencing gel. Both products migrated to the same position (Figure 1D), suggesting that RISC and DISC recognize RNA targets and cleave them in the same manner.
Figure 1.

DNA-guided RNA cleavage activity. (A) Schematic of cleavage assay using ss gRNA or gDNA. RNA-free AGO207 is shown with AGO207 bound to endogenous E. coli RNA that co-purifies with the protein (black and gray strands). Either gRNA or gDNA is loaded into the RNA-free population of AGO207. Following complex formation, a 5′ labeled 60-nt unstructured RNA or DNA target whose sequence perfectly matches the guide is added to the reaction. Yellow circle indicates 32P radiolabel. (B) Schematic of guide and target pairs used in the cleavage assay described in (A) showing combinations of gRNA or gDNA bound to a complementary cap-labeled RNA target or a 5′ end-labeled DNA target. Yellow circle indicates radiolabel. Black arrowhead indicates cleavage site. (C) Cleavage assay using guide and target pairs shown in (B). Black bars indicate average of three independent replicates and dots indicate cleavage percentage of each individual replicate relative to the canonical gRNA targeting the RNA substrate. Inset shows low-level cleavage of DNA targets by either RISC or DISC. (D) Analysis of cleavage site by RISC and DISC. AGO207 was programmed with either a 23-nt gRNA or gDNA followed by addition of a perfectly matched cap-labeled target RNA. Substrates and products were resolved on 16% denaturing PAGE alongside base-hydrolyzed polyuridine RNA. Inset shows expanded view of cleavage products, demonstrating that RISC and DISC both cleave the RNA substrate at the same position.
Optimization of gDNAs
Crystal structures of eukaryotic RISCs indicate that the nt at position 1 (g1) of the gRNA is flipped out and does not participate in target pairing (13,14,19–23). AGO proteins are also known to have different affinity for the 5′-nt of small RNAs (24,25). To test whether the identity of the 5′ g1 nt biases the slicing activity of DISC, we cleaved the 5′ capped miR-20a RNA target using gDNAs that were identical to one another with the exception of the g1 nt. Guides beginning with T, A or C cleaved the target comparably (∼60%) whereas gDNAs with a 5′ G displayed only ∼40% cleavage (Figure 2A). Next, we next tested cleavage of the miR-20a target using gDNAs varying in length from 15 to 25 nt with truncations at the 3′ of the gDNA to determine the minimum length that promotes target cleavage without sacrificing catalytic efficiency (Figure 2B, left). Guide DNAs 23- to 25-nt in length cleaved the RNA target at similar and consistent levels, whereas gDNAs 22-nt and shorter displayed irregular cleavage patterns across replicates (Figure 2B right). Although 24- and 25-nt gDNAs promoted a slight increase in cleavage of the unstructured target compared to the 23-nt gDNA, we found that longer gDNAs had an adverse effect on cleavage activity in some cases, which will be discussed in more detail in the next section. These results indicated that 23-nt gDNAs with a 5′ T display the properties of a suitable gDNA for DISC that retains target specificity.
Figure 2.

Optimization of gDNAs. (A) Evaluating the effect of the 5′ nt on cleavage activity. AGO207 was programmed with gDNAs whose 5′ nt was T, A, G or C followed by addition of a cap-labeled RNA target that perfectly matches all guides from positions 2–23. Black bars indicate average cleavage of three independent replicates and dots indicate cleavage percentage of each individual replicate. (B) Optimizing gDNA length. (Left) miR-20a-derived gDNA sequences with 5′ T were varied in length from 15 – 25 nt. Yellow circle indicates cap-label. Black arrowheads indicate cleavage site. (Right) Quantification of cleavage products using gDNAs of different length. Black bars indicate average of three independent replicates and dots indicate cleavage percentage of each individual replicate. (C) Evaluating sequence-specificity of DISC. (Top) Sequences of gRNA or gDNA paired with a cap-labeled RNA target that contains a dinucleotide mismatch at a non-permissive site known to abrogate cleavage by yeast AGO. Yellow circle indicates cap-label on target strand. Black arrowheads indicate the cleavage site at t10 and t11 (positions 10 and 11 counting from the 5′ end of the guide). (Bottom) AGO207 was pre-incubated with either a gRNA or gDNA followed by addition of either a perfectly matched target or a mismatched target. Substrates and products were resolved by 16% denaturing PAGE. Black arrowhead indicates migration of cleavage product. (D) Single- and dinucleotide mismatch assay. (Left) Schematic of gDNAs (bottom) and target RNA (top) with mismatches in the guide shown in red and bold. Dotted line flanked by black arrowhead indicates cleavage site. Yellow circle indicates cap-label on target strand. (Right) Black bars on plot represent average cleavage by DISC of three independent replicates and gray dots represent individual replicates.
Using 23-nt gDNAs beginning with T, we validated the accuracy of DNA-guided RNA cleavage by introducing a dinucleotide mismatch at the cleavage site in the miR-20a target strand which is known to inhibit cleavage by yeast RISC (13,14). Cleavage of the mismatched target by DISC was not detected, whereas RISC showed minor but detectable cleavage (Figure 2C). To further evaluate the effect of mismatches between guide and target strands, we systematically introduced single- and dinucleotide mismatches to the gDNA spanning an 8-nt window flanking the cleavage site (Figure 2D). Single-nt mismatches to g7, g8 and g14 dramatically reduced target cleavage while single-nt mismatches to any positon between g9–g13 nearly abolished activity. Similarly, all dinucleotide mismatches tested displayed nearly undetectable cleavage of the target RNA (Figure 2D and Supplementary Figure S2).
DISC cleaves highly-structured RNAs
Although a recent study reported that human AGO2-based DISC cleaves 21-nt unstructured RNAs (16), cleavage of structured RNAs remains to be tested. We next examined whether AGO207-based DISC can identify and cleave targeted sequences in a highly-structured RNA that contains a diverse set of conformational features such as varied lengths of helices, bulges, hairpin loops, and single-stranded regions. For this purpose, the HIV-1 5′UTR was selected as a model substrate. This 352-nt RNA is composed of several structured sub-domains; the transactivation response (TAR; nt 1–57) element, polyadenylation signal (poly(A); nt 58–104), primer-binding site (PBS; nt 125–223), and genomic RNA packaging domain (Psi, nt 228–334) (Supplementary Figure S3A) (26). We chose to use the structurally characterized monomeric DIS mutant (HIV-1 ΔDIS 5′UTR, Supplementary Figure S3B) to reduce technical complications associated with RNA dimerization that is typical of the wild-type sequence (27). Native PAGE of the refolded transcript showed a single band, indicating that HIV-1 ΔDIS 5′UTR folds into a single, homogeneous structure (Supplementary Figure S3C).
We systematically designed 14 gDNAs (gDNA1 to gDNA14) end-to-end that span the HIV-1 ΔDIS 5′UTR and recognize 14 target regions (TR1-TR14) (Figure 3A and Supplementary Figure S4). Each of the different cleavage sites was individually targeted using different gDNA sequences in separate reactions. Cleavage products were detected at the expected position for all sites, albeit to different extents (Figure 3B), demonstrating that DISC recognizes the target sequences even in highly-structured RNAs. Since 60-nt unstructured miR-20a targets are cleaved best when gDNAs are 23-nt long or longer (Figure 2B), we tested if the same is true for the structured HIV-1 ΔDIS 5′UTR. Cleavage of target regions 6 and 8 (TR6 and TR8) was tested by DISCs loaded with gDNAs of different length (20–25 nt) (Supplementary Figure S5A and B). TR6 was cleaved most efficiently when gDNA6 was 23- or 24-nt long (Supplementary Figure S5C and D), whereas TR8 cleavage did not depend on guide length (Supplementary Figure S5C and E). This effect may be the result of potential homodimerization properties of each gDNA sequence. Specifically, TR8 cleavage may be independent of length because increasing the length of gDNA8 does not form additional hydrogen bonds in a potential homodimer compared to the additional hydrogen bonds formed between the gDNA6 homodimer as it is lengthened (Supplementary Figure S5F and G). These results suggest a design strategy wherein gDNA length can be extended until it forms a stable homodimer.
Figure 3.

Cleavage of highly structured HIV-1 ΔDIS 5′UTR RNA by DISC. (A) Schematic of DISC-mediated cleavage assay. The target RNA sequence was divided into 23-nt segments (shown in different colors) spanning the entire sequence, with each segment targeted by a different gDNA. AGO207 molecules that are bound to endogenous E. coli RNA are not shown for clarity. Following DISC-assembly, a 5′ end-labeled HIV-1 ΔDIS 5′ UTR transcript was added to the mixture to initiate cleavage by DISC. (B) Substrates and products generated by the assay described in (A) were resolved by denaturing PAGE (8%) revealing cleavability of the viral RNA by DISC. (C) Secondary structure of HIV-1 ΔDIS 5′UTR predicted by SHAPE. Colored circles at nucleotide positions indicate SHAPE reactivity. Guide DNAs targeting the different TRs of the HIV transcript are indicated by light blue bars and arrows (TR1 – TR14). Arrowheads indicate cleavage sites by DISC. The color of each arrowhead reflects the distribution of cleavage percentages by DISC shown in (B) and (D) (white, 0–12.5%; yellow, 12.5–25%; orange, 25–37.5%; red, 37.5–50%). (D) Quantification of DISC-mediated cleavage of each TR. Purple circles indicate nucleotides that are unpaired and green circles indicate nucleotides involved in G•U base pairs. Dotted line flanked by black arrowheads indicates cleavage site. Black bars on plot represent average cleavage by DISC of three independent replicates and gray dots represent individual replicates. Asterisk for set using gDNA13 indicates average of two replicates.
DISC unwinds local RNA structure
DISC cleaved HIV-1 ΔDIS 5′UTR, but the degree of DISC-mediated cleavage varied across the RNA (Figure 3B). We hypothesized that the cleavage efficiency of each site may depend on the local structure of the RNA; however, there was no observed correlation between the DISC cleavage patterns and SHAPE reactivities (Figure 3C). The SHAPE-based secondary structure, which closely matches those previously determined for similar constructs (15,28,29) showed that each TR is involved in unique intramolecular base pairings (Figure 3C and D). For instance, TR4, TR6, TR8 and TR11 have several nt, including those at the cleavage site (i.e., target nt 10 (t10), t11 or both) in base-paired regions. As shown earlier, base pairing of gDNA to t10 and t11 is critical for DISC to cleave unstructured RNA (Figure 2C). Therefore, to test whether gDNA base pairing to t10 and t11 is in fact required for DISC-mediated cleavage of target sequences in highly-structured RNAs, TR4, TR6, TR8 and TR11 of the HIV-1 ΔDIS 5′UTR were targeted by DISC with gDNAs containing 2-nt mismatches at t10 and t11 (Figure 4A). All four TRs were very weakly cleaved or completely resistant to cleavage when programmed with the mismatched gDNA regardless of their reactivity when programmed with the matched gDNAs (Figure 4B and Supplementary Figure S6). Thus, successive, extensive base pairing between the gDNA and target sequence is indispensable for cleaving target sites on highly-structured RNAs. Given that the abovementioned TRs are intramolecularly base-paired (Figure 3C), cleavage at those sites demonstrates that DISC retains an RNA-unwinding activity; i.e. DISC is able to efficiently compete with intramolecular base-pairs in the target sequence allowing for proper annealing and efficient cleavage activity.
Figure 4.

Comparing cleavage activity of DISC and RNase H. (A) Schematic of matched and mismatched guide and target pairs used to target four TRs across the HIV-1 ΔDIS 5′UTR RNA. For each pair, the HIV-1 ΔDIS 5′UTR sequence is shown on top and the perfectly matched gDNA strand is shown on the bottom. Circle indicates target position complementary to the first position of the guide that does not pair due to structural restrains by the protein. Black arrowheads indicate cleavage site. Mismatches between the guide and target strands are indicated by a black box around the bases of the guide that are mutated to the bases shown below the box. (B) Quantified cleavage products from the assay using matched and mismatched guide and target pairs described in (A) are plotted with solid bars representing the average of three replicates and circles representing individual replicates. Cleavage that was not detectable by the assay is indicated by ‘nd’. (C and D) Comparing DISC (circles) and RNase H (triangles) cleavage of the unstructured 60-nt target (C) or of a structured 352-nt RNA target (D). Bars indicate average cleavage of three replicates.
To directly test the potential ability of DISC to compete with target secondary structure, the cleavage activity of DISC was compared with that of E. coli RNase H, an endoribonuclease that is incapable of cleaving dsRNA (30), but is instead targeted to ∼8-nt long single-stranded RNA using a complementary short DNA probe (31). Thus, detection of RNA cleavage by RNase H provides evidence that the target site is solvent-exposed and largely single-stranded. Approximately 1–2 units of commercially available RNase H showed similar cleavage activity as DISC against the unstructured miR-20a target under similar conditions (Figure 4C and Supplementary Figure S7A–C). Therefore, this amount was used for experiments comparing DISC and RNase H cleavage of the structured ΔDIS 5′UTR target. We pre-incubated HIV1 ΔDIS 5′UTR with a subset of the same 23-nt gDNAs used for DISC-mediated cleavage and added RNase H to the mixture. We chose gDNAs that target TR4, which folds into hairpin structure with a predicted 15-nt poly(A) loop (Figure 3C); TR6, which forms a junction upstream of the PBS with a predicted 9-nt unpaired region; TR8 in the hairpin preceding the PBS; and TR11 in Psi, which includes many paired nt and no single-stranded region longer than 4 nt. Relative to cleavage of the unstructured target, RNase H only weakly cleaved all four sites, while DISC demonstrated more robust cleavage of TR6, TR8 and TR11 (Figure 4D and Supplementary Figure S7D). As expected, RNase H cannot cleave strongly base-paired regions to which the gDNA will not spontaneously hybridize. Conversely, DISC efficiently cleaved TRs that were RNase H-resistant, supporting an RNA unwinding activity of DISC.
Assessing DISC-accessible cleavage in a high-throughput manner
To comprehensively identify sites of high DISC cleavage activity in long RNAs, a high-throughput approach is desirable wherein multiple gDNAs can be used in a single reaction. We next tested whether multiple DISC-mediated cleavage events can be performed simultaneously in a one-pot reaction without losing cleavage activity or target site specificity. Equimolar amounts of gDNA4, -5, -6, -7 and -8 were mixed and pre-incubated with AGO207 to assemble five DISCs targeting the ΔDIS 5′UTR (Figure 5A and B). A 32P body-labeled RNA substrate was then added to the assembly, followed by a 60-min time course incubation. The combined DISCs generated cleavage products targeted by all five gDNAs, demonstrating that multiple DISC-mediated cleavage events can be detected from a single reaction (Figure 5C). Moreover, these data show that cleavage observed in the one-pot reaction mimics that observed in the individual reactions. TR7 is consistently poorly cleaved in our experiments, both in the individual reactions and in the one-pot reaction, yielding the two expected cleavage products in each case. We did not detect any appreciable cleavage products at unexpected sites that were not targeted by a gDNA, implying that our results reflect single-hit kinetics. To offer an explanation for why each target was only sliced one time, the effective concentration of DISC was increased relative to the target RNA to see if this promoted multiple slicing events. To do this, the concentration of AGO and HIV-1 ΔDIS 5′UTR were fixed and the amount of gDNA6 and -10 was titrated resulting in a corresponding increase in the level of double-cut 92-nt product, demonstrating that single-hit kinetics are dependent on a low enzyme to substrate ratio (Supplementary Figure S8).
Figure 5.

Detection of multiple cleavage events in a single reaction. (A) Secondary structure of HIV-1 ΔDIS 5′UTR with TRs colored for clarity. Colored arrowheads indicate cleavage sites on each TR. (B) Schematic of cleavage assay using multiple gDNAs targeting different TRs of HIV-1 ΔDIS 5′UTR. For clarity, AGO207 molecules bound to endogenous E. coli RNA are not shown. AGO207 was loaded with five gDNAs (gDNA4–gDNA8). Following assembly of the DISCs, a 32P body-labeled HIV-1 ΔDIS 5′UTR RNA was added to the mixture to initiate cleavage. Cartoon secondary structure showing cleavage products with the 5′ products colored black and 3′ products colored gray. Colored gDNAs show complementary regions between guide and target strands. (C) Multiple DISC-mediated cleavage. Substrates and products formed at the indicated time points were separated by denaturing PAGE (8%). Reactions using only one guide are shown in lanes 3–7 as reference for product migration. The one-pot reaction time course is shown in lanes 8–14.
Another requirement for mapping sites highly-susceptible to DISC cleavage in a high-throughput manner is accurate read-out of the multiple cleavage sites generated by assorted DISCs. To this end, we employed a reverse transcription/primer extension analysis (RT/PE) (Figure 6A). DISCs were assembled with 11 of the 14 gDNAs used in Figure 3 spanning nucleotides 24–276 (TR2–TR12) (noise associated with large peaks corresponding to the primer and full-length RT/PE product limit the applicability of this technique at the 5′ and 3′ termini). An unlabeled HIV-1 ΔDIS 5′UTR substrate was added to the mixture to initiate cleavage. Following a 60-min incubation, reactions were quenched and worked up as described in the Methods. The total RNA pool containing all cleavage products was used to template RT reactions with a fluorophore-labeled primer that is annealed at the 3′ end of the HIV-1 ΔDIS 5′UTR. The extended primers were subjected to capillary electrophoresis and analyzed by RiboCAT software (15) to assign peaks and identify DISC-mediated cleavage sites. The output of the analysis provided a trace of peak intensities corresponding to programmed cleavage sites. In a single experiment, the RT/PE assay allowed us to detect DISC-generated cleavage products across the HIV-1 ΔDIS 5′UTR substrate in 23-nt increments (Figure 6B and Supplementary Figure S9A).
Figure 6.

High-throughput detection of DISC-mediated cleavage events. (A) Schematic of high-throughput assay to detect accessibility of structured RNAs. Guide DNAs spanning the target sequence were mixed together to assemble a mixed population of assorted DISCs. For clarity, AGO207 molecules bound to endogenous E. coli RNA are not shown. An unlabeled HIV-1 ΔDIS 5′UTR was added to the mixture to initiate cleavage. Cleaved RNA products were used as templates to prime reverse transcription using a fluorophore-labeled DNA primer. The cDNA products were detected by capillary electrophoresis yielding an electropherogram of peaks whose positions reflect DISC cleavage sites. (B) Electropherogram of DISC-cleavage sites programmed using 11 gDNAs with 23-nt increments. The average of three individual replicates was plotted as a single trace. Cleavage events were detectable at different sites with varying sensitivity.
Given that detection of cleavage sites in the PAGE and RT/PE analyses relies on the stability of the cleavage products retaining the 5′ and 3′ ends, respectively (Supplementary Figure S9B–E), consistency between the two methods also suggests that our cleavage conditions support near ‘single-hit’ kinetics. The patterns of DISC reactivity were quite similar between the RT/PE and PAGE analyses although TR8 and TR11 showed slightly different reactivities (Supplementary Figure S9F). Thus, our high-throughput method using RT/PE analysis enables a single experiment to map DISC-cleavage sites of a 352-nt region within a structured RNA at 23-nt resolution. In theory, this method should be applicable to the use of many more gDNAs or much longer RNA targets with only minor adjustments.
DISCUSSION
In this study, we showed that AGO207-based DISC is a programmable guide-dependent endoribonuclease with several advantages over existing methods. Our method is cost-effective, as it can be catalytically activated by inexpensive 23-nt synthetic DNA oligonucleotides as guides. In addition to its high specificity, DISC has no target sequence limitations because the target site specificity is determined by the guide sequence alone. This property allows for systematic design of gDNAs against any target RNA (Figure 3A and Supplementary Figure S4). Furthermore, DISC can unwind some RNA secondary structures and cleave target sites known to be in base-paired regions (Figure 4D); however, the specific limitations of this activity require further investigation.
A Cas9-based programmable RNA recognition and cleavage system was recently described (RCas9) (6,7). RCas9 requires the use of ∼120-nt single guide RNAs (sgRNAs) to recognize and cleave complementary sequences on target ssRNAs, in addition to a sequence-specific DNA oligonucleotide protospacer adjacent motif (PAMmer). These requirements limit the practical use of RCas9 for the purpose of mapping RNA susceptibility to cleavage in a high-throughput manner. Although the specificity for PAM can be modified by protein mutagenesis (32), different Cas9 variants need to be prepared in each case. Very recently, Staphylococcus aureus Cas9 was shown to display RNA-dependent RNA targeting activity in a PAM-independent manner (33). However, this variant cleaves ssDNA and dsDNA at higher efficiency than RNA, posing potential complications during in vivo applications (33). In addition, this PAM-independent Cas-based RNase can only cleave unstructured, solvent-exposed regions on the target RNA. Another PAM-independent Cas-based programmable endoribonuclease, Cas13a, requires only ∼56-nt CRISPR RNA (crRNA) for target binding and cleavage (7). However, this Cas protein remains in an enzymatically active state and cleaves any solvent exposed ssRNAs non-specifically once it has recognized and cleaved its target sequence (7). Thus, both RCas9 and Cas13a have target limitations, low target specificity, and/or lack RNA unwinding capability. In contrast, DISC overcomes these limitations and can be used to comprehensively investigate cleavable sites in long RNAs in vitro, even in the absence of any prior knowledge about 3-dimensional structure or target-site accessibility.
DISC was capable of cleaving the majority of structured TRs in the HIV-1 ΔDIS 5′UTR, although a few structured TRs were very poorly cleaved. TR1, 2 and 3 located within the highly-stable stems of TAR and poly(A) were not cleaved (Figure 3C–D) suggesting that there are limitations to the RNA unwinding ability of DISC. In contrast, partially paired TR6, 9 and 10, each having single-stranded stretches 9 nt or longer showed high DISC reactivities. TR5, 11 and 12, each containing successive unpaired regions of no longer than 4 nt, were also sensitive to DISC-mediated cleavage. Thus, DISC reactivity does not seem to correlate with the number of unpaired nt in a predictable manner (Figure 3D).
An immediate application of the in vitro DISC-mediated cleavage method described here includes high-throughput scanning of highly structured RNAs including viral RNAs, long non-coding RNAs, and regulatory regions of mRNAs for identification of sites that may be efficiently targeted by siRNAs or shRNAs in cells. Future therapeutic applications for cleavage of functionally relevant RNAs can also be envisioned but requires additional studies to make the DISC system applicable within biological systems. This technology also has the potential for applications such as targeted adenosine to inosine editing (34,35) and chemical modification (36). These applications would involve engineering of a catalytically-inactive DISC fused to other enzymatically-active proteins such that the DISC would be used to site-specifically localize the fused enzyme partner to the site of the desired reaction, potentially paving the way for more targeted manipulation of large RNAs.
Nature gave rise to many DNA restriction enzymes that recognize different target sequences such as palindromic sequences in dsDNA (37,38). In a biological context, DNA is typically double stranded; thus, target sequences are generally accessible in a highly-conserved canonical conformation. Unlike DNAs, RNAs fold into complex architectures, and therefore potential target sequences are often not solvent exposed. This feature could explain why sequence-specific RNA restriction enzymes are not found in nature. Indeed, while our results indicate a role for solvent accessibility and stability of target RNA structure in determining DISC reactivity, the lack of consistent correlation between structure and reactivity does not support use of DISC for RNA structure probing. Instead, our data indicate that DISC (i) can be readily programmed to target both structured and unstructured RNA sequences without modifying the proteinaceous catalytic machinery, (ii) retains high specificity towards its intended target sites, (iii) possesses no target site sequence limitations, and (iv) can unwind local RNA structure to access base-paired target sites inaccessible to other ribonucleases. Furthermore, we show that target site accessibility by DISC can be determined in a high-throughput manner for relatively long, highly structured RNAs. Future studies will be aimed at extending DISC capability to cleave long, highly structured RNAs in cells and on determining the impact of RNA binding proteins on DISC cleavage.
DATA AVAILABILITY
Data that was generated for the determination of HIV-1 ΔDIS 5′UTR secondary structure by SHAPE will be made available upon request.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Elaina Boyle and Di Ma for their help in the preparation and handling of materials that were used in this study.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
OSU Center for RNA Biology Graduate Student Fellowship (to D.M.D.); Pelotonia Graduate Student Cancer Research Fellowship (to D.M.D.); Pelotonia Postdoctoral Cancer Research Fellowship (to W.A.C.); NIH T32 GM118291 (to J.K.); OSU Center for RNA Biology Seed Grant Program (to K.M.-F. and K.N.); The Ohio State University Start-up Fund (to K.N.); NIH [S10OD023582]. Funding for open access charge: The Ohio State University Center for RNA Biology Seed Grant Program.
Conflict of interest statement. None declared.
REFERENCES
- 1. Yigit E., Batista P.J., Bei Y., Pang K.M., Chen C.C., Tolia N.H., Joshua-Tor L., Mitani S., Simard M.J., Mello C.C.. Analysis of the C. elegans Argonaute family reveals that distinct Argonautes act sequentially during RNAi. Cell. 2006; 127:747–757. [DOI] [PubMed] [Google Scholar]
- 2. Swarts D.C., Jore M.M., Westra E.R., Zhu Y., Janssen J.H., Snijders A.P., Wang Y., Patel D.J., Berenguer J., Brouns S.J. et al. DNA-guided DNA interference by a prokaryotic Argonaute. Nature. 2014; 507:258–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Olovnikov I., Chan K., Sachidanandam R., Newman D.K., Aravin A.A.. Bacterial argonaute samples the transcriptome to identify foreign DNA. Mol. Cell. 2013; 51:594–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nakanishi K. Anatomy of RISC: how do small RNAs and chaperones activate Argonaute proteins. Wiley Interdiscip. Rev. RNA. 2016; 7:637–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Enghiad B., Zhao H.. Programmable DNA-guided artificial restriction enzymes. ACS Synth. Biol. 2017; 6:752–757. [DOI] [PubMed] [Google Scholar]
- 6. O’Connell M.R., Oakes B.L., Sternberg S.H., East-Seletsky A., Kaplan M., Doudna J.A.. Programmable RNA recognition and cleavage by CRISPR/Cas9. Nature. 2014; 516:263–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Abudayyeh O.O., Gootenberg J.S., Konermann S., Joung J., Slaymaker I.M., Cox D.B., Shmakov S., Makarova K.S., Semenova E., Minakhin L. et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science. 2016; 353:aaf5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kharma N., Varin L., Abu-Baker A., Ouellet J., Najeh S., Ehdaeivand M.R., Belmonte G., Ambri A., Rouleau G., Perreault J.. Automated design of hammerhead ribozymes and validation by targeting the PABPN1 gene transcript. Nucleic Acids Res. 2016; 44:e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Silverman S.K. In vitro selection, characterization, and application of deoxyribozymes that cleave RNA. Nucleic Acids Res. 2005; 33:6151–6163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Choudhury R., Tsai Y.S., Dominguez D., Wang Y., Wang Z.. Engineering RNA endonucleases with customized sequence specificities. Nat. Commun. 2012; 3:1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sharp P.A. The centrality of RNA. Cell. 2009; 136:577–580. [DOI] [PubMed] [Google Scholar]
- 12. Dethoff E.A., Chugh J., Mustoe A.M., Al-Hashimi H.M.. Functional complexity and regulation through RNA dynamics. Nature. 2012; 482:322–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nakanishi K., Weinberg D.E., Bartel D.P., Patel D.J.. Structure of yeast Argonaute with guide RNA. Nature. 2012; 486:368–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dayeh D.M., Kruithoff B.C., Nakanishi K.. Structural and functional analyses reveal the contributions of the C- and N-lobes of Argonaute protein to selectivity of RNA target cleavage. J. Bio. Chem. 2018; 203:6308–6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cantara W.A., Hatterschide J., Wu W., Musier-Forsyth K.. RiboCAT: a new capillary electrophoresis data analysis tool for nucleic acid probing. RNA. 2017; 23:240–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Willkomm S., Zander A., Grohmann D., Restle T.. Mechanistic Insights into archaeal and human argonaute substrate binding and cleavage properties. PLoS One. 2016; 11:e0164695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Swarts D.C., Hegge J.W., Hinojo I., Shiimori M., Ellis M.A., Dumrongkulraksa J., Terns R.M., Terns M.P., van der Oost J.. Argonaute of the archaeon Pyrococcus furiosus is a DNA-guided nuclease that targets cognate DNA. Nucleic Acids Res. 2015; 43:5120–5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang Y., Sheng G., Juranek S., Tuschl T., Patel D.J.. Structure of the guide-strand-containing argonaute silencing complex. Nature. 2008; 456:209–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nakanishi K., Ascano M., Gogakos T., Ishibe-Murakami S., Serganov A.A., Briskin D., Morozov P., Tuschl T., Patel D.J.. Eukaryote-specific insertion elements control human ARGONAUTE slicer activity. Cell Rep. 2013; 3:1893–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schirle N.T., MacRae I.J.. The crystal structure of human Argonaute2. Science. 2012; 336:1037–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Park M.S., Phan H.D., Busch F., Hinckley S.H., Brackbill J.A., Wysocki V.H., Nakanishi K.. Human Argonaute3 has slicer activity. Nucleic Acids Res. 2017; 45:11867–11877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Elkayam E., Kuhn C.D., Tocilj A., Haase A.D., Greene E.M., Hannon G.J., Joshua-Tor L.. The structure of human argonaute-2 in complex with miR-20a. Cell. 2012; 150:100–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Faehnle C.R., Elkayam E., Haase A.D., Hannon G.J., Joshua-Tor L.. The making of a slicer: activation of human Argonaute-1. Cell Rep. 2013; 3:1901–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Frank F., Sonenberg N., Nagar B.. Structural basis for 5′-nucleotide base-specific recognition of guide RNA by human AGO2. Nature. 2010; 465:818–822. [DOI] [PubMed] [Google Scholar]
- 25. Frank F., Hauver J., Sonenberg N., Nagar B.. Arabidopsis Argonaute MID domains use their nucleotide specificity loop to sort small RNAs. EMBO J. 2012; 31:3588–3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jones C.P., Cantara W.A., Olson E.D., Musier-Forsyth K.. Small-angle X-ray scattering-derived structure of the HIV-1 5′ UTR reveals 3D tRNA mimicry. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:3395–3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Balakrishnan M., Fay P.J., Bambara R.A.. The kissing hairpin sequence promotes recombination within the HIV-I 5′ leader region. J. Biol. Chem. 2001; 276:36482–36492. [DOI] [PubMed] [Google Scholar]
- 28. Wilkinson K.A., Gorelick R.J., Vasa S.M., Guex N., Rein A., Mathews D.H., Giddings M.C., Weeks K.M.. High-throughput SHAPE analysis reveals structures in HIV-1 genomic RNA strongly conserved across distinct biological states. PLoS Biol. 2008; 6:e96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Watts J.M., Dang K.K., Gorelick R.J., Leonard C.W., Bess J.W. Jr, Swanstrom R., Burch C.L., Weeks K.M.. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature. 2009; 460:711–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kauffmann A.D., Campagna R.J., Bartels C.B., Childs-Disney J.L.. Improvement of RNA secondary structure prediction using RNase H cleavage and randomized oligonucleotides. Nucleic Acids Res. 2009; 37:e121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schultz S.J., Champoux J.J.. RNase H activity: structure, specificity, and function in reverse transcription. Virus Res. 2008; 134:86–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nishimasu H., Yamano T., Gao L., Zhang F., Ishitani R., Nureki O.. Structural basis for the Altered PAM recognition by engineered CRISPR-Cpf1. Mol. Cell. 2017; 67:139–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Strutt S.C., Torrez R.M., Kaya E., Negrete O.A., Doudna J.A.. RNA-dependent RNA targeting by CRISPR-Cas9. Elife. 2018; 7:e32724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nishikura K. A-to-I editing of coding and non-coding RNAs by ADARs. Nat. Rev. Mol. Cell. Biol. 2016; 17:83–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cox D.B.T., Gootenberg J.S., Abudayyeh O.O., Franklin B., Kellner M.J., Joung J., Zhang F.. RNA editing with CRISPR-Cas13. Science. 2017; 358:1019–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Roundtree I.A., Evans M.E., Pan T., He C.. Dynamic RNA modifications in gene expression regulation. Cell. 2017; 169:1187–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pingoud A., Jeltsch A.. Structure and function of type II restriction endonucleases. Nucleic Acids Res. 2001; 29:3705–3727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bath A.J., Milsom S.E., Gormley N.A., Halford S.E.. Many type IIs restriction endonucleases interact with two recognition sites before cleaving DNA. J. Biol. Chem. 2002; 277:4024–4033. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data that was generated for the determination of HIV-1 ΔDIS 5′UTR secondary structure by SHAPE will be made available upon request.