

Abstract
High body burdens of polybrominated diphenyl ethers (PBDEs) in infants and young children have led to increased concern over their potential impact on human development. PBDE exposure can alter the expression of genes involved in thyroid homeostasis, including those of ATP-binding cassette (ABC) transporters, which mediate cellular xenobiotic efflux. However, little information exists on how PBDEs interact with ABC transporters such as P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP). The purpose of this study was to evaluate the interactions of 2,20 ,4,40 -tetrabromodiphenyl ether (BDE-47) and its hydroxylated metabolite 6-OH-BDE-47 with P-gp and BCRP, using human MDR1- and BCRP-expressing membrane vesicles and stably transfected NIH-3T3-MDR1 and MDCK-BCRP cells. In P-gp membranes, BDE-47 did not affect P-gp activity; however, 6-OH-BDE-47 inhibited P-gp activity at low mM concentrations (IC50¼11.7 mM). In BCRP membranes, BDE-47 inhibited BCRP activity; however, 6-OH-BDE-47 was a stronger inhibitor [IC50¼45.9 mM (BDE-47) vs. IC50¼9.4 mM (6-OH-BDE-47)]. Intracellular concentrations of known P-gp and BCRP substrates [(3 H)-paclitaxel and (3 H)-prazosin, respectively] were significantly higher (indicating less efflux) in NIH-3T3-MDR1 and MDCK-BCRP cells in the presence of 6-OH-BDE-47, but not BDE-47. Collectively, our results indicate that the BDE-47 metabolite 6-OH-BDE-47 is an inhibitor of both P-gp and BCRP efflux activity. These findings suggest that some effects previously attributed to BDE-47 in biological systems may actually be due to 6-OH-BDE-47. Considerations for human exposure are discussed.
Keywords: MDR1, P-gp, BCRP, ABC Transporters, Polybrominated diphenyl ethers (PBDEs), brominated flame retardants
Introduction
Polybrominated diphenyl ethers (PBDEs) are flame retardant chemicals that were used in large amounts in consumer products (electronics, foams, and plastics), leading to widespread environmental contamination. PBDEs were produced in 3 mixtures: PentaBDE (tri-through hexa-brominated congeners); OctaBDE (hexa- and hepta-brominated congeners); and DecaBDE (decabrominated BDE-209). PentaBDE and OctaBDE were withdrawn from the U.S. market in 2004 and DecaBDE was phased-out in 2013. However, human exposures to PBDEs through contaminated food sources and indoor air/dust (Johnson et al., 2010; Schecter et al., 2006) are expected to continue for many years due to their ubiquitous use and environmental persistence.
In humans, the highest body burdens of PBDEs are found in infants and young children, primarily from ingesting breast milk and house dust, respectively, which has led to increased concerns over the potential for these compounds to adversely impact human development (Costa et al., 2008; Johnson-Restrepo and Kannan, 2009; Rose et al., 2010; Toms et al., 2008). In general, animal and in vitro studies suggest that PBDE exposure can lead to adverse neurodevelopmental, reproductive, and thyroid effects (Birnbaum, 2007; Schreiber et al., 2010); however, associations between perinatal exposure to PBDEs and suboptimal neurodevelopmental outcomes in children (Gascon et al., 2012; Herbstman et al., 2010) are not well understood. A further confounding factor is evidence suggesting that cytochrome P450 (CYP) enzymes bio-activate PBDEs in humans to form hydroxylated metabolites (OH-PBDEs) with potentially greater toxicity than parent compounds (Dingemans et al., 2008; Hamers et al., 2008).
2,2′,4,4′-Tetrabromodiphenyl ether (BDE-47) is a major component of PentaBDE and is the predominant congener found in most human samples (serum, breast milk, and adipose tissue) (Marchitti et al., 2013b; Sjodin et al., 2014). CYP-mediated metabolism of BDE-47 (Figure 1) produces various hydroxylated metabolites (OH-PBDEs) that accumulate in human serum at levels similar to or greater than BDE-47 (Feo et al., 2013; Staskal et al., 2006). 6-OH-BDE-47 is one of the most frequently detected congeners in human samples (Athanasiadou et al., 2008; Chen et al., 2016; Qiu et al., 2009) and evidence suggests that it may be the most toxic (Macaulay et al., 2015; Usenko et al., 2012). Like BDE-47, 6-OH-BDE-47 has been detected in maternal serum, umbilical cord blood, and breast milk, leading to concerns over its potential impact to developing infants (Aylward et al., 2014; Chen et al., 2016; Stapleton et al., 2011; Zota et al., 2011).
FIG. 1.

CYP450-mediated metabolism of BDE-47 to produce the hydroxylated metabolite 6-OH-BDE-47. The thyroid hormone, thyroxine (T4) is shown for structural comparison.
OH-PBDEs may contribute to neurodevelopmental disorders through direct neurotoxicity or, indirectly, through thyroid disruption (Dingemans et al., 2011; Hendriks et al., 2010; Kim et al., 2011). Structurally similar to endogenous thyroid hormones (Figure 1), OH-PBDEs may disrupt thyroid homeostasis via interference with transporters, protein binding, or gene expression (Dallaire et al., 2009). OH-PBDEs inhibit thyroid sulfotransferase and deiodinase enzymes (Butt and Stapleton, 2013; Butt et al., 2011) and compete with the natural thyroid hormone thyroxine (T4) for binding to human transthyretin and thyroxine-binding globulin (Szabo et al., 2009; Marchesini et al., 2008). Exposure to BDE-47 and metabolites in adult and developing rodents leads to changes in the expression of genes involved in thyroid homeostasis, including multidrug resistance transporters of the ATP binding cassette (ABC) family such as P-glycoprotein [P-gp (also known as MDR1, ABCB1)] and breast cancer resistance protein [BCRP (also known as ABCG2)] (Richardson et al., 2008; Szabo et al., 2009). P-gp and BCRP are cellular membrane transporters that actively (ie, ATP-dependently) efflux chemical substrates, thus playing critical roles in the cellular protection against potentially toxic compounds and in the absorption and distribution of xenobiotics (Chu et al., 2013). In the placenta, P-gp and BCRP limit the entry of xenobiotics into the fetal circulation (Aye and Keelan, 2013).
Understanding early life exposure is important for accurate exposure and risk assessments in infants and children. Inhibition of ABC transporters or changes in their expression may alter the pharmacokinetics of xenobiotics (Kaddoumi et al., 2007; Mistry et al., 2001). Evaluating how drugs interact with ABC transporters has been critical to understanding drug absorption, chemotherapeutic resistance, and drug–drug interactions. Environmental chemicals, including pesticides and their metabolites, have also been shown to interact with ABC transporters as competitive substrates or non-specific inhibitors, with impacts on intracellular and systemic exposure concentrations of xenobiotics (Mazur et al., 2012, 2014; Oosterhuis et al., 2008). However, limited information exists on the potential for PBDEs to interact with P-gp and BCRP, and no studies have evaluated their hydroxylated metabolites. In this study, we present an in-depth evaluation of BDE-47 and 6-OH-BDE-47 with P-gp and BCRP using in vitro P-gp and BCRP vesicle assay systems, human P-gp (MDR1)-transfected NIH-3T3 cells, and human BCRP-transfected Madin–Darby Canine Kidney Epithelial (MDCK) cells.
Materials and Methods
Chemicals and reagents
BDE-47 (CAS no. 5436-43-1) and 6-OH-BDE-47 (CAS no. 79755-43-4) were purchased from Accustandard (New Haven, Connecticut). Verapamil and erythromycin were obtained from Sigma (St. Louis, Missouri). Omeprazole, Ko134, and PSC833 and all components and chemical reagents used for P-gp and BCRP vesicle transport assays were included in assay kits or purchased separately from Solvo Biotechnology (Szeged, Hungary). All organic solvents were Optima grade and purchased from Fisher Scientific (Pittsburgh, Pennysylvania).
P-gp and BCRP membrane vesicle transport assays
Human P-gp and BCRP-expressed “inside-out” membrane vesicles were used to measure the inhibitory effect of BDE-47 and its hydroxylated metabolite 6-OH-BDE-47 on P-gp- and BCRP-mediated transport of the probe substrates N-methyl quinidine (NMQ) and Lucifer Yellow, respectively. In this system, the inverted membrane conformation allows P-gp or BCRP transport (efflux) of substrates into the vesicle lumen; after membrane solubilization, the amount of transported substrate can be determined. Human P-gp and BCRP membrane vesicles and all chemical reagents were purchased as PREDIVEZ assay kits from Solvo Biotechnology (Szeged, Hungary). All transporter assay kit components and reagents were stored at −80°C until use.
The ATP-dependent efflux of the P-gp substrate NMQ (2 μM final concentration) into human P-gp vesicles was measured using a rapid filtration technique in the absence and the presence of ATP, following the protocol of the manufacturer. Stock solutions of test chemicals were prepared in DMSO with final assay concentrations ranging from 0.02 μM to 200 μM. Briefly, human P-gp expressing mammalian cell membrane suspensions (50 μL) were loaded onto 96-well flat bottom tissue culture plates (50 μg protein), followed by the addition of the chemicals (0.75 μL). Plates were pre-incubated for 15 min at 37°C. Reactions were started by adding 25 μL assay buffer (Solvo kit) with and without ATP, and allowed to proceed for 3 min at 37°C. Reactions were terminated with 200 μL of ice cold “washing mix” (Solvo kit), and the solution was transferred to a glass fiber (Type B) filter plate (Millipore, Billerica, Massachusetts) and washed 5 times with “washing mix” using a Millipore Multiscreen™ rapid filtration vacuum manifold. Vesicles were solubilized in methanol:water (70:30, v/v) at room temperature and vacuum collected. Quinidine (25 ng/mL in DMSO) was included in all experimental samples as the internal standard for LC/MS/MS analysis. Verapamil and erythromycin, known inhibitors of P-gp, were used as controls (Eberl et al., 2007; Heredi-Szabo et al., 2013). The ATP-dependent efflux of the BCRP substrate Lucifer Yellow (2 μM final concentration) into human BCRP vesicles was determined using the same methods, except that after vacuum filtration, filters were dried and a fluorescent detection solution (1× Detector) was added to each well and incubated for 10 min at room temperature. The liquid was transferred under vacuum to white-walled, clear bottom 96-well plates; transported Lucifer Yellow was measured fluorescently on a BioTek Instruments Synergy HT microplate reader (Winooski, Vermont) at Ex: 430, Em: 538 nm. Substrate concentrations measured in experimental wells lacking ATP were treated as negative background controls, and subtracted from measurements taken from wells with ATP (representing active transport). DMSO served as the vehicle control; test chemical interactions with P-gp or BCRP were measured by dividing transported NMQ and Lucifer Yellow concentrations, respectively, by those measured for DMSO to determine % P-gp transport activity. IC50 values were determined for each test chemical by curve-fitting the data using non-linear regression with a 4-parameter logistic fit (Sigma Plot, version 11.0).
To evaluate whether BDE-47 and 6-OH-BDE-47 function as P-gp or BCRP substrates, P-gp and BCRP membrane vesicle assays were conducted using identical protocols as above, except that the intra-vesicular concentrations of BDE-47 and 6-OH-BDE-47 in vesicles was directly measured. Concentrations of BDE-47 were analyzed by GC/MS, whereas those of 6-OH-BDE-47 were analyzed by LC/MS/MS. NMQ and Lucifer Yellow samples with and without ATP served as controls for P-gp and BCRP transport, respectively.
LC/MS/MS analysis
Quantitation of NMQ was performed as described (Mazur et al., 2012) using an Agilent 1200 high-performance liquid chromatograph (HPLC) coupled to a 6420 triple quad mass spectrometer (Agilent, Santa Clara, California). Injections (2 μL) at a 0.65 ml/min flow rate were made onto a Waters Atlantis C18 column (2.1 mm × 100 mm, 5 μm particle diameter; Milford, Massachusetts) maintained at 35°C. Gradient elution with 0.2% formic acid in methanol (solvent A) or water (solvent B) was applied under these conditions: 15% A for 0.1 min, followed by a linear gradient to 45% A at 2.5 min, increasing to 100% A at 2.6 min, and held for a 6 min stop time. The column was then allowed to re-equilibrate under the original conditions for a 3 min post-time. MS/MS detection was conducted using ESI+ in multiple reaction mode under these conditions: NMQ quantifying transition ion m/z 339–172, with qualifying ion transitions m/z 339–160 and 339–96.2 (collision energies 44 V, 34 V, and 40 V, respectively), with the fragmenting voltage set to 165 V. The internal standard quinidine was quantified based on the transition ion m/z 325–81 with the fragmenting voltage, collision energy, and cell accelerator set to 140 V, 40 V, and 4 V, respectively. ESI source parameters were applied according to the following: source gas temperature 350°C, gas flow 11 L/min, nebulizer 50 psi, capillary 4500 V. NMQ standard curves (0.5–70 ng/mL) were prepared in (70:30, MeOH:H2O) using quinidine as an internal standard. Instrument calibration based on a response factor was conducted for each vesicle transport assay and verified during analysis by running a check standard every 10 samples.
Analysis of the BDE-47 metabolite 6-OH-BDE-47 was performed using a previously developed method (Sun et al., 2013) with the same LC/MS/MS system as above. Injections (10 μL) were made onto a Thermo Scientific Acclaim™ RSLC 120 C18 column (2.2 mm × 100 mm, 2.2 μm particle diameter; Waltham, Massachusetts) maintained at 35 °C with a flow rate of 0.38 mL/min. The mobile phase consisted of acetonitrile (solvent A) and water (solvent B) and was used with a gradient elution of A:B from 55:45 to 75:25 in 20 min, followed by a 5 min post re-equilibration time. MS/MS detection of 6-OH-BDE-47 was conducted using ESI source in the negative multiple reaction (MRM) mode. The 6-OH-BDE-47 precursor ion was m/z 500.7 and product ion m/z 80.8. The collision energy, fragmenting voltage, and cell accelerator were set to 8V, 100 V, and 7 V respectively. ESI source parameters were set to the following: source gas temperature 350 °C, gas flow 13 L/min, nebulizer 55 psi, Delta electron multiplier voltage (EMV) was 400 V and capillary 5000 V. Data acquisition and analysis were processed by MassHunter WorkStation software.
GC/MS analysis
Quantification of the parent compound BDE-47 and internal standard [4′-fluoro-2,3′,4,6-tetrabromodiphenyl ether (FBDE)] was performed using a HP 6890 Series gas chromatogram coupled to a HP 5973 Mass Selective Detector (MSD) and equipped with a HP 6890 Series Injector (Agilent Technologies, Santa Clara, California). The following temperature profile was used: initial oven temperature of 90 °C, hold for 1 min, ramp at 20 °C/min to 340 °C, and hold for 2.5 min (total run time of 16 min). Chromatographic separation was achieved with a DB-5ms column (30 m × 0.25 mm, 0.25 μm film thickness, Agilent Technologies, Santa Clara, CA) and helium was used as the carrier gas and held at a constant flow of 1.0 mL/min. Samples were injected (2 μL) in pulsed splitless injection mode (pulse pressure of 15.8 psi at 1.8 min) at a temperature of 280 °C. The MS quad, source, and interface temperatures were 250 °C, 230 °C, and 280 °C, respectively. The MSD was operated in selected ion monitoring mode monitoring 2 ions for each analyte, 485.7 and 325.8 m/Z for BDE-47 and 503.7 and 343.8 for the internal standard.
Cell culture
The parental NIH-3T3 murine fibroblast cell line and its human MDR1 transfected counterpart, the NIH-3T3-G185 cell line (NIH-3T3-MDR1), were kindly provided by Dr. M.M. Gottesman (The National Cancer Institute at the National Institutes of Health, Bethesda, Maryland) (Cardarelli et al., 1995). MDCK-pcDNA3 vector control cells and MDCK cells transfected with human BCRP (MDCK-BCRP) were kindly provided by Dr. Qingcheng Mao (University of Washington, Seattle, Washington). NIH-3T3 cells were maintained in Dulbecco’s modified Eagle medium (DMEM), supplemented with 10% FBS (ATCC, Rockville, Maryland). The drug-resistant NIH-3T3-MDR1 cell line was maintained in medium supplemented with 60 ng/mL of colchicine (Sigma, St. Louis, Missouri). MDCK cells were maintained in DMEM with high glucose and L-glutamine, 10% FBS (ATCC, Rockville, Maryland), and 500 μg/mL of G418 (Life Technologies, Carlsbad, California). All cells were maintained in media supplemented with antibiotics [100 U/mL penicillin; 100 μg/mL streptomycin (Life Technologies)] and 1 μL/mL MycoZap (Lonza, Basel, Switzerland) and a humidified atmosphere with 5% CO2 at 37°C.
Western blot analysis
The expression of human P-gp in parental NIH-3T3 and NIH-3T3-MDR1 cells was determined by Western blot analysis, using the C219 monoclonal antibody (1:200 overnight, 4°C) (Covance, Dedham, Massachusetts) which recognizes all MDR1 isoforms (Georges et al., 1990). The expression of human BCRP in MDCK-Vector and MDCK-BCRP cells was determined by Western blot analysis, using the BXP-21 monoclonal antibody which reacts with an internal epitope of human BCRP (1:500 1 hr, room temperature) (Millipore, Billerica, Massachusetts). Briefly, cells were lysed in RIPA lysis buffer, followed by ultrasonication. Cell lysate protein content was determined using a Pierce BCA protein assay kit according to the guidelines of the manufacturer. Samples (30–50 μg) were separated on 10% SDS-PAGE gels and transferred to PVDF membranes (Hybond-P, GE Healthcare, Piscataway, New Foundland) which were blocked overnight at 4°C in 1× TBST (0.1% Tween) buffer containing 5% non-fat milk. Membranes were probed with primary antibodies, followed with secondary goat anti-mouse HRP antibody (1:1000 1 h, room temperature) (Bethyl, Montgomery, Texas). Blots were washed and visualized using enhanced chemiluminescence and imaged with a Fluorchem SP digital imager (Alpha Innotech, San Leandro, California). Membranes were re-probed for β-actin as a loading control using β-actin primary antibody (1:1000) (Sigma Aldrich, St. Louis, Missouri).
Cytotoxicity assays
The cytotoxicity of known P-gp and BCRP substrates (paclitaxel and mitoxantrone, respectively), and the test chemicals BDE-47 and 6-OH-BDE-47, was determined in parental NIH-3T3-MDR1 and MDCK-BCRP cells compared with control cells. Cells were seeded at a density of 3 × 103 cells/well in 96-well tissue culture treated plates (Corning Costar, Tewksbury, Massachusetts) and were exposed to chemicals (0.01–100 μM in DMSO) for 48 h. Final DMSO concentrations did not exceed 1%. Cell viability was determined with the colorimetric MTS (3-(4,5-dimethyl-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt) assay following the guidelines of the manufacturer (Promega, Madison, Wisconsin). The absorbance was measured with a BioTek Instruments Synergy HT microplate reader (Winooski, Vermont) at 490 nm. This assay is based on reduction of the MTS tetrazolium compound by metabolically viable cells to a colored formazan product that is soluble in tissue culture medium. The cell viability was expressed relative to vehicle-treated (DMSO) control cells; data presented are the mean ± SEM of experiments run in triplicate on at least 2 separate days. In NIH-3T3-MDR1 cells, the specific P-gp inhibitor PSC833 (10 μM) was used as a control.
Cellular efflux assays using radiolabeled substrates
The inhibitory effect of BDE-47 and 6-OH-BDE-47 on P-gp- and BCRP-mediated transport in NIH-3T3-MDR1 and MDCK-BCRP cells was evaluated using radiolabeled P-gp and BCRP substrates [[3H]-paclitaxel and [3H]-prazosin, respectively (American Radiolabeled Chemicals, St. Louis, Missouri)]. Cells were seeded at 40 × 103 cells/well in 24-well plates and allowed to grow to 90–100% confluency. Cells were washed with transport buffer (10 mM HEPES in HBSS), followed by incubation in transport buffer containing test chemicals [BDE-47 or 6-OH-BDE-47 (0.1–100 μM in DMSO)] for 15 min, followed by aspiration and addition of transport buffer containing test chemicals and [3H]-paclitaxel or [3H]-prazosin [1 mCi/mL (1:10 000 dilution)] for 5 min. DMSO content was normalized across treatments to 0.5%. Cells were washed 2 times with 1 mL of ice cold PBS, then lysed, vortexed and spun at 10 000 rpm for 10 min. Protein content analysis was performed using BCA protein assay kits (Pierce) on cell lysates and 100 μL of cell lysis was mixed with 3 mL of Ecolite (+) liquid scintiallation cocktail (MP Biomedicals, Santa Ana, California) and analyzed on a Beckman Coulter LS6500 scintillation counter to measure cellular concentrations (pmol/mg protein) of [3H]-paclitaxel or [3H]-prazosin. Each assay was performed in triplicate on at least 3 separate occasions. To determine if BDE-47 or 6-OH-BDE-47 inhibited P-gp or BCRP cellular efflux activity, intracellular concentrations of paclitaxel or prazosin in the presence of these test chemicals was compared with cells treated with paclitaxel or prazosin alone. Cellular efflux of paclitaxel or prazosin in the presence of the respective inhibitors for P-gp [cyclosporine A (25 μM)] or BCRP [Ko134 (1 μM)] were used as positive control groups.
Statistics
P-gp and BCRP-expressed membrane vesicle IC50 values were determined for each test chemical by curve-fitting the data using non-linear regression with a 4-parameter logistic fit (SigmaPlot, version 13.0). To analyze for statistical differences between groups, Student’s unpaired t-tests and 2-way ANOVA for multiple groups were used (SigmaPlot); P < .05 was considered significant. All values are expressed as means ± SEM.
Results
Influence of BDE-47 and 6-OH-BDE-47 on P-gp Transport in Membrane Vesicles
BDE-47 did not affect P-gp-mediated transport of the probe substrate NMQ in P-gp membrane vesicles (Figure 2 and Table 1). In contrast, 6-OH-BDE-47 inhibited P-gp-mediated transport of NMQ in a concentration-dependent manner with an IC50value of 11.7 μM. 6-OH-BDE-47 demonstrated a greater ability to inhibit P-gp-mediated transport of NMQ than the P-gp inhibitor erythromycin (IC50 value, 145.1 μM), but was a weaker inhibitor of P-gp than verapamil (IC50 value, 1.8 μM). At 40 μM 6-OH-BDE-47, no measurable P-gp-mediated transport of NMQ was detected. In contrast, P-gp-mediated transport was unaffected at 200 μM BDE-47, the highest concentration evaluated. IC50 values for verapamil and erythromycin were consistent with quality assurance reports and assay kit documentation [Solvo (Heredi-Szabo et al. 2013)]. Control membranes lacking P-gp expression displayed no significant efflux of NMQ (data not shown).
FIG. 2.
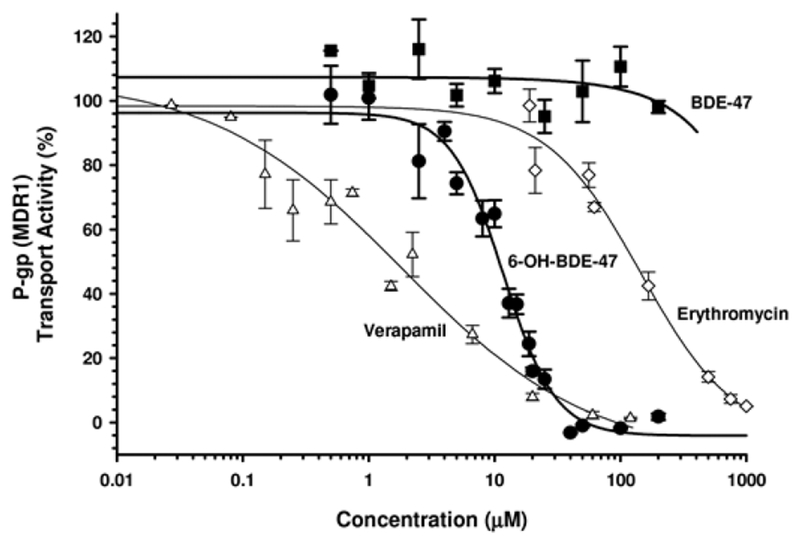
P-gp (MDR1)-mediated transport (of the probe substrate NMQ) in MDR1-expressed membrane vesicles in the presence of varying concentrations of BDE-47 (▪), 6-OH-BDE-47 (•), erythromycin (◊), and verapamil (Δ). Data are expressed relative to vehicle control (DMSO) treated membranes. Each experiment was performed in duplicate and the data represent the mean ± SEM of independent experiments. Results indicate that 6-OH-BDE-47, but not BDE-47, is an inhibitor of P-gp mediated efflux.
TABLE 1.
IC50 Values (μM) for Various Environmental Chemicals and Transporter Inhibitors in P-gp (MDR1) and BCRP
| Membrane vesicle assay | ||
|---|---|---|
| Chemical | MDR1 | BCRP |
| BDE-47 | no inhibition | 45.9 ± 48.1 |
| 6-OH-BDE-47 | 11.7 ± 0.9 | 9.4 ± 1.1 |
| Erythromycin | 145.1 ± 56.2 | n.d. |
| Verapamil | 1.8 ± 1.1 | n.d. |
| Omeprazole | n.d. | 56.9 ± 26.9 |
| Ko134 | n.d. | 0.1 ± 0.01 |
n.d., not determined.
Influence of BDE-47 and 6-OH-BDE-47 on BCRP Transport in Membrane Vesicles
BDE-47 inhibited (IC50 value, 45.9 μM) BCRP-mediated transport of the probe substrate Lucifer Yellow in membrane vesicles in a dose–response manner, although this inhibition was less than that of 6-OH-BDE-47 (IC50 value, 9.4 μM) (Figure 3 and Table 1). Both compounds demonstrated a greater ability to inhibit BCRP-mediated transport than the known BCRP inhibitor omeprazole (IC50 value, 56.9 μM), but were weaker inhibitors than the more specific BCRP inhibitor Ko134 (IC50value, 0.1 μM). BCRP transport activity was essentially zero at 50 μM 6-OH-BDE-47, although approximately 50% activity remained at 50 μM BDE-47. IC50 values for omeprazole and Ko134 were consistent with quality assurance reports and assay kit documentation [Solvo (Heredi-Szabo et al. 2013)]. Control vesicle membranes lacking BCRP expression displayed no significant efflux of Lucifer Yellow (data not shown).
FIG. 3.
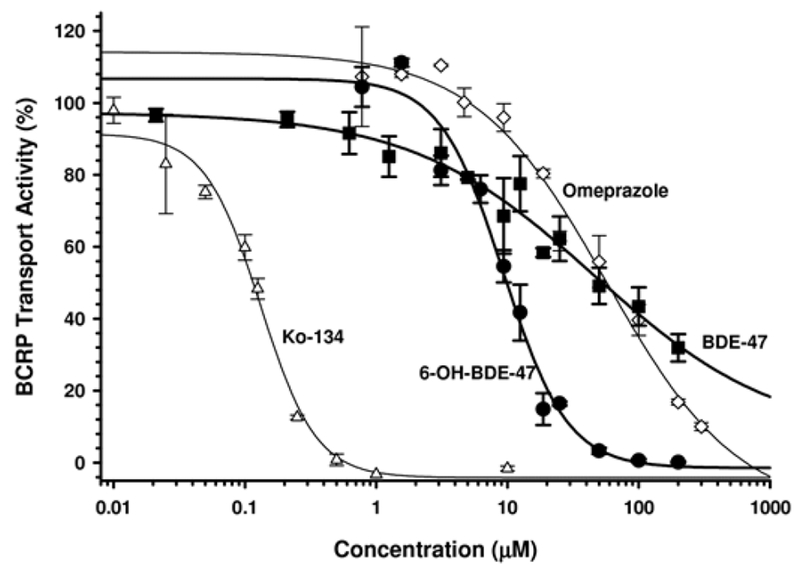
BCRP-mediated transport (of the probe substrate Lucifer Yellow) in BCRP-expressed membrane vesicles in the presence of varying concentrations of BDE-47 (▪), 6-OH-BDE-47 (•), omeprazole (◊), and Ko134 (Δ). Data are expressed relative to vehicle control (DMSO)-treated membranes. Each experiment was performed in duplicate and the data represent the mean ± SEM of multiple independent experiments. Results indicate that 6-OH-BDE-47 is a stronger inhibitor of BCRP efflux activity than the parent BDE-47.
Transport of BDE-47 and 6-OH-BDE-47 in “Inside–Out” P-gp and BCRP Membrane Vesicles
To explore whether BDE-47 or 6-OH-BDE-47 potentially interact with P-gp or BCRP as substrates, the efflux of these compounds using “inside–out” membrane assays were performed with the exception that intra-vesicular concentrations (pmol/mg/min) of BDE-47 and 6-OH-BDE-47 were directly analyzed by GC/MS and LC/MS/MS, respectively. In this system, measurable intra-vesicular concentrations of xenobiotics can indicate either active transport (ATP-dependent), passive diffusion, or non-specific binding to vesicular membranes (non-ATP-dependent). The actively transported P-gp and BCRP substrates NMQ and Lucifer Yellow, respectively, were used as controls. Although internal concentrations of BDE-47 and 6-OH-BDE-47 in P-gp and BCRP membrane vesicles were relatively high, no differences in concentrations were observed in the presence or absence of ATP (Figure 4A and B). These results suggest that BDE-47 and 6-OH-BDE-47 likely bind non-specifically to vesicle membranes or cross vesicle membranes via passive diffusion, suggesting that they may not be strong substrates of P-gp or BCRP. Additional chemical concentrations and experimental reaction times did not affect these results (data not shown); however, if BDE-47 or 6-OH-BDE-47 were actively transported by P-gp or BCRP to a minor extent, it would have been difficult to detect because of the high degree of passive diffusion observed for these compounds.
FIG. 4.
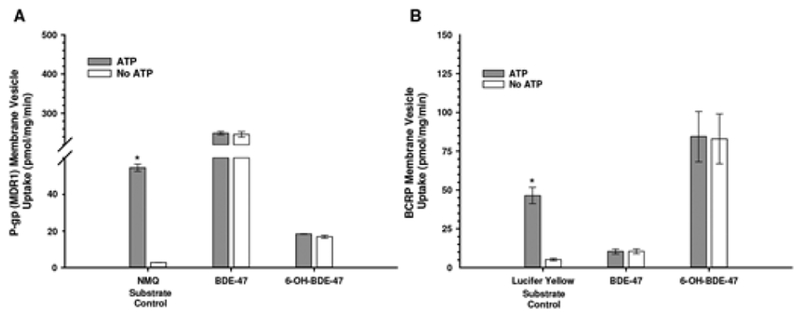
Accumulation of BDE-47 and 6-OH-BDE-47 (1 μM) in (A) MDR1-expressed “inside-out” membrane vesicles and (B) BCRP-expressed “inside–out” membrane vesicles, in the presence or absence of ATP. Probe substrates NMQ and Lucifer Yellow, respectively, served as positive substrate controls. Results indicate that neither BDE-47 nor 6-OH-BDE-47 are actively transported substrates of P-gp or BCRP.
Cytotoxicity Assays
Expression and function of MDR1 and BCRP protein in NIH-3T3-MDR1 and MDCK-BCRP cells was confirmed prior to evaluating test chemicals for cytotoxicity. MDR1 and BCRP expressions were demonstrated by Western blot in NIH-3T3-MDR1 (Figure 5A, lane 2) and MDCK-BCRP cells (Figure 5B, lane 2), respectively; no expression was detectable in parental or vector control cells (Figure 5A and B, lane 1).
FIG. 5.
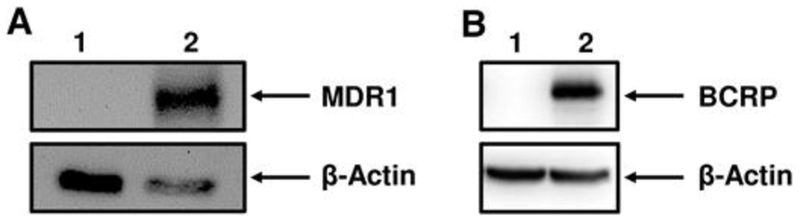
P-gp (MDR1) and BCRP expression in NIH-3T3 and MDCK cell lines, respectively. Cell lysates were processed for Western blot analysis (30 μg total protein), separated by SDS PAGE and transferred to nitrocellulose membranes. A, The MDR1-specific C219 antibody was used to probe expression of MDR1 (170 kDa) in Parental NIH-3T3 cells (lane 1) and transfected NIH-3T3-MDR1 cells (lane 2). B, The BCRP-specific BCRP-21 antibody was used to probe expression of BCRP (72 kDa) in MDCK-vector control cells (lane 1) and transfected MDCK-BCRP cells (lane 2). Expression was visualized by chemiluminescence as described in materials and methods. β-actin was used as a loading control.
Using MTS cytotoxicity assays, a significant protective effect against the control P-gp substrate paclitaxel was demonstrated in NIH-3T3-MDR1 cells compared with parental cells (Figure 6A). The specific P-gp inhibitor PSC-833 had no additional effect on parental cell viability in response to paclitaxel, but decreased cell viability in NIH-3T3-MDR1 cells to levels demonstrated in parental cells (data not shown). Cytotoxicity in response to BDE-47 was evident in NIH-3T3-MDR1 and parental cells only at relatively high (100 μM) concentrations (Figure 6B). No significant differences in viability between NIH-3T3-MDR1 and parental cells in response to BDE-47 were demonstrated. Cytotoxicity in response to 6-OH-BDE-47 was evident at 10 μM with complete cell death at 100 μM (Figure 6C). A modest but significant cytoprotective effect (29% vs 5% cell viability) of P-gp against 6-OH-BDE-47 was seen in NIH-3T3-MDR1 cells compared with parental cells, respectively.
FIG. 6.
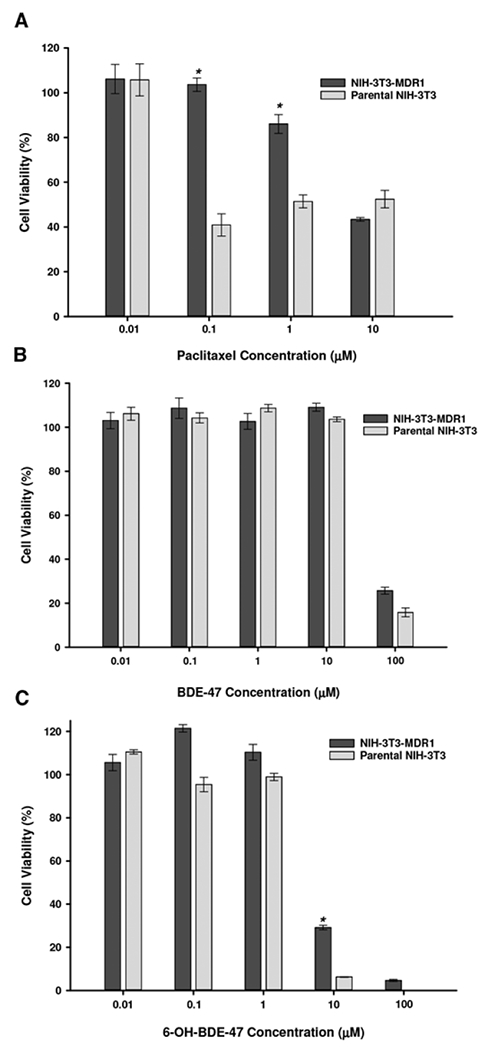
Cell viability of NIH-3T3-MDR1 and parental NIH-3T3 cells in response to varying concentrations of paclitaxel (A), BDE-47 (B), and 6-OH-BDE-47 (C). Data are expressed relative to vehicle control (DMSO)-treated cells. Each experiment was performed in triplicate and data represent mean ± SEM of multiple independent experiments. * Statistically significant difference in the cytotoxic dose–response between NIH-3T3-MDR1 and parental NIH-3T3 cells (P < .05).
In MDCK cells, a significant protective effect was seen in MDCK-BCRP cells in response to the control BCRP substrate mitoxantrone compared with vector control cells (Figure 7A). Similar to NIH-3T3 cells, BDE-47 cytotoxicity in MDCK cells was evident only at 100 μM; however, BDE-47 was less toxic overall to MDCK cells compared with NIH-3T3 cells (Figure 7B). 6-OH-BDE-47 significantly decreased cell viability of MDCK-BCRP and vector control cells by approximately 50% at 10 μM concentrations (compared with 80% in NIH-3T3 cells) (Figure 7C). No significant differences in viability between MDCK-BCRP and vector control cells were demonstrated in response to BDE-47 or 6-OH-BDE-47.
FIG. 7.
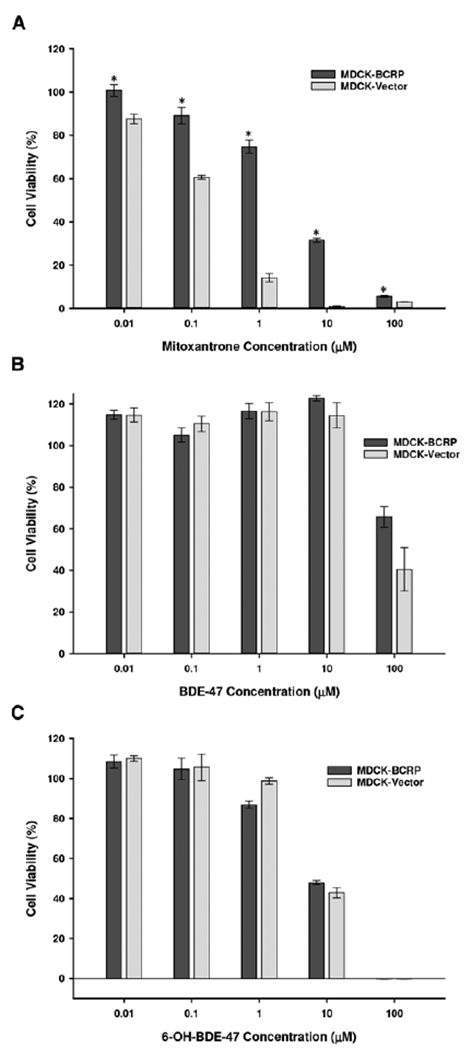
Cell viability of MDCK-BCRP and MDCK-vector control cells in response to varying concentrations of mitoxantrone (A), BDE-47 (B), and 6-OH-BDE-47 (C). Data are expressed relative to vehicle control (DMSO)-treated cells. Each experiment was performed in triplicate and data represent mean ± SEM of multiple independent experiments. * Statistically significant difference in the cytotoxic dose–response between MDCK-BCRP and MDCK-Vector control cells (P < .05).
Cellular Efflux Assays
To determine if exposure to BDE-47 (0.1–100 μM) and 6-OH-BDE-47 (0.1–100 μM) affect transport (efflux) activities of P-gp and BCRP in transfected cells, cellular efflux assays were performed using the known P-gp and BCRP substrates paclitaxel and prazosin, respectively. In this system, an increase in the intracellular concentrations of paclitaxel and prazosin would suggest a decrease in P-gp and BCRP efflux activity, respectively. Although no significant changes in efflux of [3H]-paclitaxel were observed in NIH-3T3-MDR1 or parental cells in response to BDE-47 (Figure 8A), 6-OH-BDE-47 significantly decreased the cellular efflux of [3H]-paclitaxel in NIH-3T3-MDR1 cells in a dose-dependent manner (Figure 8B). At 50 μM and 100 μM 6-OH-BDE-47, intracellular concentrations of [3H]-paclitaxel increased by approximately 250% and 300%, respectively, in NIH-3T3-MDR1 cells, compared with cells exposed to [3H]-paclitaxel alone. This response was comparable with the intracellular increase in [3H]-paclitaxel observed in NIH-3T3-MDR1 cells in response to the known P-gp inhibitor cyclosporin-A (25 μM), which was used as a positive control. Cellular efflux of [3H]-paclitaxel in parental NIH-3T3 cells was primarily unaffected by 6-OH-BDE-47, although at 100 μM 6-OH-BDE-47, a 25% increase in intracellular concentrations of [3H]-paclitaxel was observed, which could indicate non-specific toxic effects.
FIG. 8.
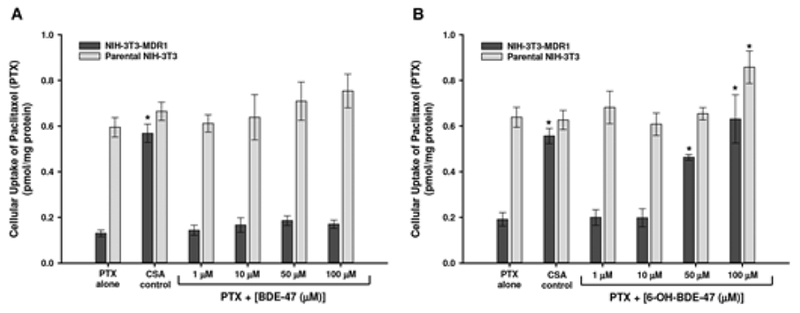
Cellular efflux of [3H]paclitaxel (PTX) in NIH-3T3-MDR1 and parental NIH-3T3 control cells in response to varying concentrations (1–100 μM) of BDE-47 (A) and 6-OH-BDE-47 (B). The P-gp inhibitor cyclosporine-A (CSA; 25 μM) was used as a positive control (A and B). Each experiment was performed in triplicate and data represent mean ± SEM of multiple independent experiments. *Statistically significant increase in intracellular concentrations (decreased efflux) of PTX in response to test chemical exposure compared with PTX alone (p < 0.05).
In MDCK-BCRP and MDCK-vector control cells, BDE-47 (0.1–100 μM) had no effect on the cellular efflux of [3H]-prazosin (Figure 9A). In contrast, 6-OH-BDE-47 significantly decreased the cellular efflux of [3H]-prazosin (leading to intracellular accumulation) in MDCK-BCRP cells in a dose–response manner, beginning at 1 μM (Figure 9B). At 1 and 10 μM 6-OH-BDE-47, this intracellular increase was approximately 200%–220%, whereas 100 μM 6-OH-BDE-47 increased prazosin accumulation by approximately 550% in MDCK-BCRP cells. 6-OH-BDE-47 did not affect [3H]-prazosin efflux in MDCK-vector control cells. Similarly, the control BCRP inhibitor Ko134 increased intracellular concentrations of [3H]-prazosin by approximately 600%, but had no effect on vector control cells (Figure 9).
FIG. 9.
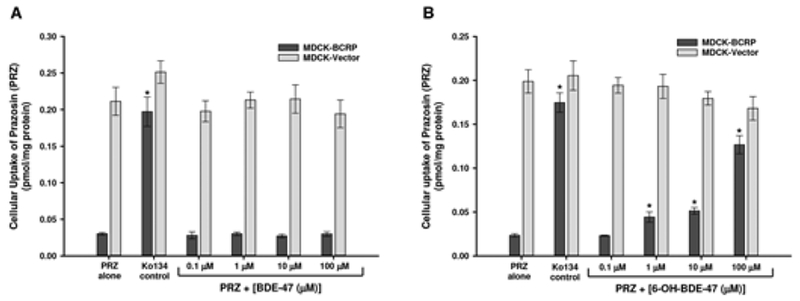
Cellular efflux of [3H]prazosin (PRZ) in MDCK-BCRP and MDCK-Vector control cells in response to varying concentrations (1–100 μM) of BDE-47 (A) and 6-OH-BDE-47 (B). The BCRP inhibitor Ko134 (1 μM) was used as a positive control (A and B). Each experiment was performed in triplicate and data represent mean ± SEM of multiple independent experiments. *Statistically significant increase in intracellular concentrations (decreased efflux) of PRZ in response to test chemical exposure compared with PRZ alone (p < 0.05).
Discussion
Transport proteins are present in virtually all tissues throughout the body where they play critical roles in determining intracellular and systemic concentrations of xenobiotics (Chu et al., 2013; Hillgren et al., 2013). We have previously shown that environmental chemicals and their metabolites, including the agricultural fungicide propiconazole and bisphenol-A (BPA), interact with ABC transporters such as P-gp and BCRP, either as potential substrates or inhibitors (Mazur et al. 2012, 2014). These studies indicated that interactions with ABC transporters can iffer substantially among parent compounds and their metabolites, which may have important pharmacokinetic implications. Common use pesticides, including structurally diverse organochlorines, organophosphates, and chloroacetanilides have also been shown to have specific interactions with ABC transporters, including the potent inhibition of P-gp activity with the potential to modulate drug absorption and cause the chemosensitization of cells (Oosterhuis et al. 2008; Pivcevic and Zaja, 2006). Prior to this study, little was known whether other environmental chemicals, such as PBDEs and their hydroxylated metabolites, interact with ABC transporters and what impact this may have for pharmacokinetic and exposure assessments. In the U.S., concentrations of PBDEs in the serum of pregnant women and in breast milk samples are up to 10-fold higher than in Asia and Europe (Hites, 2004; Marchitti et al., 2013a; Sjodin et al., 2014). Both PBDEs and OH-PBDEs have been shown to enter the fetal circulation and distribute into breast milk, resulting in high body burdens in infants and young children. PBDEs and OH-PBDEs have been shown to produce adverse reproductive and neurodevelopmental effects (Birnbaum, 2007; Gascon et al., 2012; Herbstman et al., 2010; Macaulay et al., 2015; Schreiber et al., 2010), thus, there is increasing concern over their potential to impact fetal and infant neurodevelopment. In this study, we examined the interactions of BDE-47 and one of its most abundant and toxic hydroxylated metabolites, 6-OH-BDE-47, with the ABC transporters P-gp and BCRP.
BDE-47 and 6-OH-BDE-47 varied substantially in their interactions with P-gp and BCRP. In P-gp membrane vesicles, whereas the parent BDE-47 had no effect on P-gp efflux activity, 6-OH-BDE-47 was a strong inhibitor with an IC50 value in the low micromolar range. P-gp inhibition by 6-OH-BDE-47 was similar to the known P-gp inhibitor verapamil. In BCRP membrane vesicles, both BDE-47 and 6-OH-BDE-47 inhibited BCRP transport activity in a dose–response manner; however, the metabolite 6-OH-BDE-47 demonstrated a 5-fold lower IC50 value than did the parent compound. BCRP inhibition by 6-OH-BDE-47 was significantly stronger than that observed for the known BCRP inhibitor omeprazole but appreciably weaker than the specific inhibitor Ko134.
To explore whether BDE-47 or 6-OH-BDE-47 possesses substrate activity with P-gp or BCRP, direct analysis of intravesicular concentrations of BDE-47 and 6-OH-BDE-47 in “inside– out” P-gp and BCRP vesicles was performed. Although these results suggest that neither compound is likely a strong substrate for P-gp or BCRP, further research into the potential substrate properties of BDE-47 and 6-OH-BDE-47 may be warranted. Due to the greater non-specific binding and passive diffusion that lipophilic compounds display, false-negative results can occur in both membrane vesicle assays and ATPase assays when lipophilic compounds are directly quantified as potential substrates (Brouwer et al., 2013, p. 95). However, cytotoxicity assays of NIH-3T3-MDR1 and MDCK-BCRP cells exposed to BDE-47 and 6-OH-BDE-47 indicated no significant protection from BCRP against these compounds, supporting preliminary vesicle findings. Similarly, P-gp did not significantly protect against BDE-47 in NIH-3T3-MDR1 cells; however, modest cytoprotection of P-gp against 6-OH-BDE-47 at 10 mM was observed, suggesting 6-OH-BDE-47 may display minor substrate activity. P-gp has been reported to transport thyroid hormones, which are structurally similar to 6-OH-BDE-47, and shown to bind (but not necessarily transport) some lipophilic pesticides with molecular weights similar to BDE-47 and 6-OH-BDE-47 (Bain and LeBlanc, 1996; Mitchell et al., 2005). A study investigating the distribution and elimination of PBDEs in mice indicated P-gp may play a minor role in the transport of BDE-47 as a secondary urinary elimination pathway, but the binding affinity of P-gp for BDE-47 may be low (Emond et al., 2013). Although the findings of our study clearly demonstrate the inhibitory properties of 6-OH BDE-47, the substrate properties of PBDEs and their active metabolites towards drug transporters remain to be fully clarified.
In the placenta, both P-gp and BCRP are highly expressed and localized to the brush border of syncytiotrophoblasts, the single limiting layer of multinuclear cells that make up the placental barrier (Prouillac and Lecoeur, 2010). This microvillous brush border membrane is in direct contact with maternal blood; efflux transporters localized to the brush border are responsible for protecting the fetus via the direct efflux of potentially toxic xenobiotics from the placenta into the maternal circulation. Compounds that are not substrates for this protective mechanism would enter the fetal circulation unhindered and potentially be a source of fetal exposure. Concentrations of BDE-47 and 6-OH-BDE-47 in the serum of pregnant women have been reported to be approximately 13–43 ng/g lipid (geometric mean) for BDE-47 and 5–25 pg/mL (geometric mean) for 6-OH-BDE-47, depending on the study population (Chen et al., 2013; Stapleton et al., 2011; Zota et al., 2011). Recent studies have shown that BDE-47 and 6-OH-BDE-47 are able to distribute from the maternal serum and cross the placental barrier into the fetal circulation (Aylward et al., 2014; Chen et al., 2016); our results suggest that these compounds are not strong candidates for P-gp or BCRP efflux support these findings. In studies analyzing paired maternal and infant samples, higher concentrations of BDE-47 and 6-OH-BDE-47 have been reported in umbilical cord serum (ie, fetal circulation) compared with maternal serum, with approximate cord:maternal ratios of 1.3 and 1.4, respectively (Aylward et al., 2014; Chen et al., 2013). Similarly, OH-PBDEs appear to preferentially partition into fetal cord blood compared with placenta with concentrations of 6-OH-BDE-47 in fetal cord blood (median, 8 pg/g wet weight) reportedly exceeding those in the placenta by approximately 4-fold. High binding affinity of OH-PBDEs for thyroid hormone transport proteins has been suggested as an underlying mechanism for the maternal-to-fetal transport of OH-PBDEs (Chen et al., 2016). In breast milk, concentrations of OH-PBDEs appear to be correlated with those in placenta (Chen et al., 2016). In the mammary gland, BCRP is expressed in alveolar epithelial cells during pregnancy and lactation, where it actively secretes a variety of substrate drugs and toxins directly into breast milk (van Herwaarden and Schinkel, 2006). This is in apparent contradiction with the fetal protecting role of BCRP expressed in the placenta resulting in possible exposure of breastfeeding infants to xenobiotics. Given our results, we would not expect BCRP to be actively involved in the efflux of BDE-47 or 6-OH-BDE-47 into human milk, but rather, these compounds likely distribute into breast milk through passive diffusion. Although estimated daily intakes of BDE-47 for U.S. infants are believed to exceed 100 ng/kg bw/day (Marchitti et al., 2016), initial evidence suggests infants are exposed to substantially less OH-PBDEs through breast milk compared with parent PBDE compounds. In a Chinese population, the median breast milk concentration of 6-OH-BDE-47 was reported to be 1.8 pg/g (wet weight), corresponding to a mean estimated daily infant intake of approximately 0.3 ng/kg bw/day (Chen et al., 2016).
To further evaluate the interactions of BDE-47 and 6-OHBDE-47 on P-gp and BCRP efflux activity, NIH-3T3-MDR1 and MDCK-BCRP cells were used in experiments with radiolabeled P-gp and BCRP substrates. Results correlated well with those from membrane vesicle assays. Although BDE-47 did not significantly affect intracellular concentrations (or cellular efflux) of the P-gp substrate [3H]paclitaxel, exposure to 6-OH-BDE-47 in NIH-3T3-MDR1 cells increased intracellular concentrations of [3H]paclitaxel by approximately 5-fold, indicating a decrease in P-gp efflux activity. This was similar to the effect demonstrated by the P-gp inhibitor cyclosporin-A. Although some inhibition of BCRP efflux activity was seen in response to BDE-47 in BCRP membrane vesicle assays, BDE-47 exposure in MDCK-BCRP cells had no effect on the cellular efflux of [3H]prazosin. However, 6-OH-BDE-47 decreased the cellular efflux of [3H]prazosin in a dose–response manner beginning with a 2.5-fold increase in intracellular concentrations of [3H]prazosin at 1 mM. Collectively, our vesicle assay and cell results suggest that BDE-47 and its hydroxylated metabolite 6-OH-BDE-47 are not P-gp or BCRP substrates; however, hydroxylation of BDE-47 to 6-OHBDE-47 produces a metabolite capable of inhibiting the efflux activity of both transporters. Results from vesicle- and cellbased systems correlated well, suggesting that in vitro membrane vesicle assays may be useful for rapidly screening chemicals for transporter interactions in a high-throughput manner.
Due to limited data, many chemical exposure and risk assessments cannot fully consider the potential impact of chemical metabolism, including the bioactivation of xenobiotics to chemical species with greater toxicity. However, evaluating the interactions of parent xenobiotics and their metabolites with key transport proteins such as the ABC efflux transporters is important to fully understand xenobiotic pharmacokinetics and make informed risk assessment decisions. The marked difference in transporter inhibition we observed between the parent BDE-47 and one of its most abundant active metabolites should be considered when performing PBDE exposure and risk assessments, particularly in light of the low IC50 values demonstrated for 6-OH-BDE-47 towards P-gp and BCRP transport activity. Although associations between PBDEs and thyroid hormone disruption in humans are not fully understood (Abdelouahab et al., 2013; Stapleton et al., 2011; Zota et al., 2011), it has been suggested that OH-PBDEs may actually be responsible for some of the observed effects; however, metabolites are not frequently measured in epidemiological studies (Macaulay et al., 2015). Prior to our study, limited information existed on the interactions of BDE-47 with ABC transporters, and no studies evaluated its hydroxylated metabolites. We previously have shown that hydroxylation of the systemic fungicide propiconazole results in metabolites that interact significantly less with P-gp compared with the parent compound (Mazur et al., 2014). This is in contrast to what has been observed for some pharmaceutical compounds [eg, tamoxifen, indinavir (Hochman et al. 2000; Teft et al., 2011)] and to our results of BDE-47 and 6-OH-BDE-47 where hydroxylation led to enhanced P-gp interaction, indicating that the functional consequences of metabolite hydroxylation may differ among chemical classes.
Because P-gp and BCRP greatly impact the absorption and excretion of xenobiotics (both drugs and pesticides) entering the systemic circulation, modulation or inhibition of P-gp by parent xenobiotics and/or their metabolites can affect the pharmacokinetics, tissue levels and safety of therapeutic applications (Akhtar et al., 2011; Hartter et al., 2013). During scenarios of coexposure of multiple xenobiotics, systemic concentrations of the therapeutic agent may be altered due to P-gp or BCRP inhibition, resulting in undesirable effects. To our knowledge, these results are the first to provide comprehensive evidence that 6-OH-BDE-47 is a P-gp and BCRP inhibitor. Further research is needed to improve understanding of the consequences of 6-OHBDE-47-mediated transporter inhibition, whether it occurs at environmental concentrations relevant to PBDE exposure, and to what degree this impacts xenobiotic transport and cellular protective mechanisms.
Acknowledgements
This research received funding from the U.S. Environmental Protection Agency (U.S. EPA) and by an appointment to the Postdoctoral Research Program at the Ecosystems Research Division, administered by the Oak Ridge Institute for Science and Education through Interagency Agreement No. (DW8992298301) between the U.S. Department of Energy and the U.S. EPA.
The views expressed in this article are those of the author[s] and do not necessarily represent the views or policies of the U.S. Environmental Protection Agency.
We would like to thank Ramya Kolli for technical assistance, and the National Cancer Institute and the University of Washington for providing cell lines.
REFERENCES
- Abdelouahab N, Langlois MF, Lavoie L, Corbin F, Pasquier JC, Takser L. 2013. Maternal and cord-blood thyroid hormone levels and exposure to polybrominated diphenyl ethers and polychlorinated biphenyls during early pregnancy. American journal of epidemiology 178(5): 701–713. [DOI] [PubMed] [Google Scholar]
- Akhtar N, Ahad A, Khar RK, Jaggi M, Aqil M, Iqbal Z, et al. 2011. The emerging role of P-glycoprotein inhibitors in drug delivery: a patent review. Expert opinion on therapeutic patents 21(4): 561–576. [DOI] [PubMed] [Google Scholar]
- Allen JG, McClean MD, Stapleton HM, Nelson JW, Webster TF. 2007. Personal exposure to polybrominated diphenyl ethers (PBDEs) in residential indoor air. Environ Sci Technol 41(13): 4574–4579. [DOI] [PubMed] [Google Scholar]
- Athanasiadou M, Cuadra SN, Marsh G, Bergman A, Jakobsson K. 2008. Polybrominated diphenyl ethers (PBDEs) and bioaccumulative hydroxylated PBDE metabolites in young humans from Managua, Nicaragua. Environ Health Perspect 116(3): 400–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain LJ, LeBlanc GA. 1996. Interaction of structurally diverse pesticides with the human MDR1 gene product P-glycoprotein. Toxicol Appl Pharmacol 141(1): 288–298. [DOI] [PubMed] [Google Scholar]
- Bardelmeijer HA, Ouwehand M, Beijnen JH, Schellens JH, van Tellingen O. 2004. Efficacy of novel P-glycoprotein inhibitors to increase the oral uptake of paclitaxel in mice. Investigational new drugs 22(3): 219–229. [DOI] [PubMed] [Google Scholar]
- Birnbaum L 2007. Health effects of brominated flame retardants (BFRs). Organohalogen Compounds 69: 670–673. [Google Scholar]
- Birnbaum LS, Cohen Hubal EA. 2006. Polybrominated diphenyl ethers: a case study for using biomonitoring data to address risk assessment questions. Environ Health Perspect 114(11): 1770–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borst P, Elferink RO. 2002. Mammalian ABC transporters in health and disease. Annual review of biochemistry 71: 537–592. [DOI] [PubMed] [Google Scholar]
- Cardarelli CO, Aksentijevich I, Pastan I, Gottesman MM. 1995. Differential effects of Pglycoprotein inhibitors on NIH3T3 cells transfected with wild-type (G185) or mutant (V185) multidrug transporters. Cancer research 55(5): 1086–1091. [PubMed] [Google Scholar]
- Chao HR, Tsou TC, Huang HL, Chang-Chien GP. 2011. Levels of breast milk PBDEs from southern Taiwan and their potential impact on neurodevelopment. Pediatric research 70(6): 596–600. [DOI] [PubMed] [Google Scholar]
- Chen A, Park JS, Linderholm L, Rhee A, Petreas M, DeFranco EA, et al. 2013. Hydroxylated polybrominated diphenyl ethers in paired maternal and cord sera. Environ Sci Technol 47(8): 3902–3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu X, Korzekwa K, Elsby R, Fenner K, Galetin A, Lai Y, et al. 2013. Intracellular drug concentrations and transporters: measurement, modeling, and implications for the liver. Clinical pharmacology and therapeutics 94(1): 126–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa LG, Giordano G. 2007. Developmental neurotoxicity of polybrominated diphenyl ether (PBDE) flame retardants. Neurotoxicology 28(6): 1047–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa LG, Giordano G, Tagliaferri S, Caglieri A, Mutti A. 2008. Polybrominated diphenyl ether (PBDE) flame retardants: environmental contamination, human body burden and potential adverse health effects. Acta bio-medica : Atenei Parmensis 79(3): 172–183. [PubMed] [Google Scholar]
- Dallaire R, Dewailly E, Pereg D, Dery S, Ayotte P. 2009. Thyroid function and plasma concentrations of polyhalogenated compounds in Inuit adults. Environ Health Perspect 117(9): 1380–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnerud PO, Eriksen GS, Johannesson T, Larsen PB, Viluksela M. 2001. Polybrominated diphenyl ethers: occurrence, dietary exposure, and toxicology. Environ Health Perspect 109 Suppl 1: 49–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingemans MM, de Groot A, van Kleef RG, Bergman A, van den Berg M, Vijverberg HP, et al. 2008. Hydroxylation increases the neurotoxic potential of BDE-47 to affect exocytosis and calcium homeostasis in PC12 cells. Environ Health Perspect 116(5): 637–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingemans MM, Heusinkveld HJ, Bergman A, van den Berg M, Westerink RH. 2010a. Bromination pattern of hydroxylated metabolites of BDE-47 affects their potency to release calcium from intracellular stores in PC12 cells. Environ Health Perspect 118(4): 519–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingemans MM, van den Berg M, Bergman A, Westerink RH. 2010b. Calcium-related processes involved in the inhibition of depolarization-evoked calcium increase by hydroxylated PBDEs in PC12 cells. Toxicological sciences : an official journal of the Society of Toxicology 114(2): 302–309. [DOI] [PubMed] [Google Scholar]
- Dingemans MM, van den Berg M, Westerink RH. 2011. Neurotoxicity of brominated flame retardants: (in)direct effects of parent and hydroxylated polybrominated diphenyl ethers on the (developing) nervous system. Environ Health Perspect 119(7): 900–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunnick JK, Nyska A. 2009. Characterization of liver toxicity in F344/N rats and B6C3F1 mice after exposure to a flame retardant containing lower molecular weight polybrominated diphenyl ethers. Experimental and toxicologic pathology : official journal of the Gesellschaft fur Toxikologische Pathologie 61(1): 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberl S, Renner B, Neubert A, Reisig M, Bachmakov I, Konig J, et al. 2007. Role of p-glycoprotein inhibition for drug interactions: evidence from in vitro and pharmacoepidemiological studies. Clinical pharmacokinetics 46(12): 1039–1049. [DOI] [PubMed] [Google Scholar]
- Emond C, Sanders JM, Wikoff D, Birnbaum LS. 2013. Proposed mechanistic description of dose-dependent BDE-47 urinary elimination in mice using a physiologically based pharmacokinetic model. Toxicol Appl Pharmacol 273(2): 335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson P, Jakobsson E, Fredriksson A. 2001. Brominated flame retardants: a novel class of developmental neurotoxicants in our environment? Environ Health Perspect 109(9): 903–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feo ML, Gross MS, McGarrigle BP, Eljarrat E, Barcelo D, Aga DS, et al. 2013. Biotransformation of BDE-47 to potentially toxic metabolites is predominantly mediated by human CYP2B6. Environ Health Perspect 121(4): 440–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer D, Hooper K, Athanasiadou M, Athanassiadis I, Bergman A. 2006. Children show highest levels of polybrominated diphenyl ethers in a California family of four: a case study. Environ Health Perspect 114(10): 1581–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederiksen M, Vorkamp K, Thomsen M, Knudsen LE. 2009. Human internal and external exposure to PBDEs--a review of levels and sources. Int J Hyg Environ Health 212(2): 109–134. [DOI] [PubMed] [Google Scholar]
- Gascon M, Fort M, Martinez D, Carsin AE, Forns J, Grimalt JO, et al. 2012. Polybrominated Diphenyl Ethers (PBDEs) in Breast Milk and Neuropsychological Development in Infants. Environ Health Perspect 120(12): 1760–1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georges E, Bradley G, Gariepy J, Ling V. 1990. Detection of P-glycoprotein isoforms by gene-specific monoclonal antibodies. Proceedings of the National Academy of Sciences of the United States of America 87(1): 152–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamers T, Kamstra JH, Sonneveld E, Murk AJ, Visser TJ, Van Velzen MJ, et al. 2008. Biotransformation of brominated flame retardants into potentially endocrine-disrupting metabolites, with special attention to 2,2’,4,4’-tetrabromodiphenyl ether (BDE-47). Molecular nutrition & food research 52(2): 284–298. [DOI] [PubMed] [Google Scholar]
- Hartter S, Sennewald R, Nehmiz G, Reilly P. 2013. Oral bioavailability of dabigatran etexilate (Pradaxa(®)) after co-medication with verapamil in healthy subjects. Br J Clin Pharmacol 75(4): 1053–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendriks HS, Antunes Fernandes EC, Bergman A, van den Berg M, Westerink RH. 2010. PCB-47, PBDE-47, and 6-OH-PBDE-47 differentially modulate human GABAA and alpha4beta2 nicotinic acetylcholine receptors. Toxicological sciences : an official journal of the Society of Toxicology 118(2): 635–642. [DOI] [PubMed] [Google Scholar]
- Herbstman JB, Sjodin A, Kurzon M, Lederman SA, Jones RS, Rauh V, et al. 2010. Prenatal exposure to PBDEs and neurodevelopment. Environ Health Perspect 118(5): 712–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heredi-Szabo K, Palm JE, Andersson TB, Pal A, Mehn D, Fekete Z, et al. 2013. A P-gp vesicular transport inhibition assay - optimization and validation for drug-drug interaction testing. European journal of pharmaceutical sciences : official journal of the European Federation for Pharmaceutical Sciences 49(4): 773–781. [DOI] [PubMed] [Google Scholar]
- Hillgren KM, Keppler D, Zur AA, Giacomini KM, Stieger B, Cass CE, et al. 2013. Emerging transporters of clinical importance: an update from the International Transporter Consortium. Clinical pharmacology and therapeutics 94(1): 52–63. [DOI] [PubMed] [Google Scholar]
- Hites RA. 2004. Polybrominated diphenyl ethers in the environment and in people: a meta-analysis of concentrations. Environ Sci Technol 38(4): 945–956. [DOI] [PubMed] [Google Scholar]
- Hochman JH, Chiba M, Nishime J, Yamazaki M, Lin JH. 2000. Influence of P-glycoprotein on the transport and metabolism of indinavir in Caco-2 cells expressing cytochrome P-450 3A4. The Journal of pharmacology and experimental therapeutics 292(1): 310–318. [PubMed] [Google Scholar]
- International Transporter C, Giacomini KM, Huang SM, Tweedie DJ, Benet LZ, Brouwer KL, et al. 2010. Membrane transporters in drug development. Nature reviews Drug discovery 9(3): 215–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson-Restrepo B, Kannan K. 2009. An assessment of sources and pathways of human exposure to polybrominated diphenyl ethers in the United States. Chemosphere 76(4): 542–548. [DOI] [PubMed] [Google Scholar]
- Johnson PI, Stapleton HM, Sjodin A, Meeker JD. 2010. Relationships between polybrominated diphenyl ether concentrations in house dust and serum. Environ Sci Technol 44(14): 5627–5632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaddoumi A, Choi SU, Kinman L, Whittington D, Tsai CC, Ho RJ, et al. 2007. Inhibition of Pglycoprotein activity at the primate blood-brain barrier increases the distribution of nelfinavir into the brain but not into the cerebrospinal fluid. Drug metabolism and disposition: the biological fate of chemicals 35(9): 1459–1462. [DOI] [PubMed] [Google Scholar]
- Kiki-Mvouaka S, Menez C, Borin C, Lyazrhi F, Foucaud-Vignault M, Dupuy J, et al. 2010. Role of P-glycoprotein in the disposition of macrocyclic lactones: A comparison between ivermectin, eprinomectin, and moxidectin in mice. Drug metabolism and disposition: the biological fate of chemicals 38(4): 573–580. [DOI] [PubMed] [Google Scholar]
- Kim KH, Bose DD, Ghogha A, Riehl J, Zhang R, Barnhart CD, et al. 2011. Para- and ortho-substitutions are key determinants of polybrominated diphenyl ether activity toward ryanodine receptors and neurotoxicity. Environ Health Perspect 119(4): 519–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macaulay LJ, Chen A, Rock KD, Dishaw LV, Dong W, Hinton DE, et al. 2015. Developmental toxicity of the PBDE metabolite 6-OH-BDE-47 in zebrafish and the potential role of thyroid receptor beta. Aquat Toxicol 168: 38–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchesini GR, Meimaridou A, Haasnoot W, Meulenberg E, Albertus F, Mizuguchi M, et al. 2008. Biosensor discovery of thyroxine transport disrupting chemicals. Toxicol Appl Pharmacol 232(1): 150–160. [DOI] [PubMed] [Google Scholar]
- Marchitti SA, LaKind JS, Naiman DQ, Berlin CM, Kenneke JF. 2013. Improving infant exposure and health risk estimates: using serum data to predict polybrominated diphenyl ether concentrations in breast milk. Environ Sci Technol 47(9): 4787–4795. [DOI] [PubMed] [Google Scholar]
- Marsh G, Athanasiadou M, Athanassiadis I, Sandholm A. 2006. Identification of hydroxylated metabolites in 2,2’,4,4’-tetrabromodiphenyl ether exposed rats. Chemosphere 63(4): 690–697. [DOI] [PubMed] [Google Scholar]
- Mazur CS, Marchitti SA, Dimova M, Kenneke JF, Lumen A, Fisher J. 2012. Human and Rat ABC Transporter Efflux of Bisphenol A and Bisphenol A Glucuronide: Interspecies Comparison and Implications for Pharmacokinetic Assessment. Toxicological sciences : an official journal of the Society of Toxicology 128(2): 317–325. [DOI] [PubMed] [Google Scholar]
- Mazur CS, Marchitti SA, Zastre J. 2014. P-glycoprotein inhibition by the agricultural pesticide propiconazole and its hydroxylated metabolites: Implications for pesticide-drug interactions. Toxicol Lett 232(1): 37–45. [DOI] [PubMed] [Google Scholar]
- Mistry P, Stewart AJ, Dangerfield W, Okiji S, Liddle C, Bootle D, et al. 2001. In vitro and in vivo reversal of P-glycoprotein-mediated multidrug resistance by a novel potent modulator, XR9576. Cancer research 61(2): 749–758. [PubMed] [Google Scholar]
- Mitchell AM, Tom M, Mortimer RH. 2005. Thyroid hormone export from cells: contribution of P-glycoprotein. The Journal of endocrinology 185(1): 93–98. [DOI] [PubMed] [Google Scholar]
- Oosterhuis B, Vukman K, Vagi E, Glavinas H, Jablonkai I, Krajcsi P. 2008. Specific interactions of chloroacetanilide herbicides with human ABC transporter proteins. Toxicology 248(1): 45–51. [DOI] [PubMed] [Google Scholar]
- Pivcevic B, Zaja R. 2006. Pesticides and their binary combinations as P-glycoprotein inhibitors in NIH 3T3/MDR1 cells. Environmental toxicology and pharmacology 22(3): 268–276. [DOI] [PubMed] [Google Scholar]
- Prouillac C, Lecoeur S. 2010. The role of the placenta in fetal exposure to xenobiotics: importance of membrane transporters and human models for transfer studies. Drug metabolism and disposition: the biological fate of chemicals 38(10): 1623–1635. [DOI] [PubMed] [Google Scholar]
- Qiu X, Bigsby RM, Hites RA. 2009. Hydroxylated metabolites of polybrominated diphenyl ethers in human blood samples from the United States. Environ Health Perspect 117(1): 93–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X, Mercado-Feliciano M, Bigsby RM, Hites RA. 2007. Measurement of polybrominated diphenyl ethers and metabolites in mouse plasma after exposure to a commercial pentabromodiphenyl ether mixture. Environ Health Perspect 115(7): 1052–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson VM, Staskal DF, Ross DG, Diliberto JJ, DeVito MJ, Birnbaum LS. 2008. Possible mechanisms of thyroid hormone disruption in mice by BDE 47, a major polybrominated diphenyl ether congener. Toxicol Appl Pharmacol 226(3): 244–250. [DOI] [PubMed] [Google Scholar]
- Rose M, Bennett DH, Bergman A, Fangstrom B, Pessah IN, Hertz-Picciotto I. 2010. PBDEs in 2-5 year-old children from California and associations with diet and indoor environment. Environ Sci Technol 44(7): 2648–2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roze E, Meijer L, Bakker A, Van Braeckel KN, Sauer PJ, Bos AF. 2009. Prenatal exposure to organohalogens, including brominated flame retardants, influences motor, cognitive, and behavioral performance at school age. Environ Health Perspect 117(12): 1953–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders JM, Lebetkin EH, Chen LJ, Burka LT. 2006. Disposition of 2,2’,4,4’,5,5’-hexabromodiphenyl ether (BDE153) and its interaction with other polybrominated diphenyl ethers (PBDEs) in rodents. Xenobiotica 36(9): 824–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schecter A, Papke O, Harris TR, Tung KC, Musumba A, Olson J, et al. 2006. Polybrominated diphenyl ether (PBDE) levels in an expanded market basket survey of US food and estimated PBDE dietary intake by age and sex. Environmental Health Perspectives 114(10): 1515–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber G 2002. The evolutionary and integrative roles of transthyretin in thyroid hormone homeostasis. The Journal of endocrinology 175(1): 61–73. [DOI] [PubMed] [Google Scholar]
- Schreiber T, Gassmann K, Gotz C, Hubenthal U, Moors M, Krause G, et al. 2010. Polybrominated diphenyl ethers induce developmental neurotoxicity in a human in vitro model: evidence for endocrine disruption. Environ Health Perspect 118(4): 572–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schussler GC. 2000. The thyroxine-binding proteins. Thyroid : official journal of the American Thyroid Association 10(2): 141–149. [DOI] [PubMed] [Google Scholar]
- Sjodin A, Jones RS, Caudill SP, Wong LY, Turner WE, Calafat AM. 2014. Polybrominated diphenyl ethers, polychlorinated biphenyls, and persistent pesticides in serum from the national health and nutrition examination survey: 2003-2008. Environ Sci Technol 48(1): 753–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stapleton HM, Eagle S, Anthopolos R, Wolkin A, Miranda ML. 2011. Associations between polybrominated diphenyl ether (PBDE) flame retardants, phenolic metabolites, and thyroid hormones during pregnancy. Environ Health Perspect 119(10): 1454–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staskal DF, Hakk H, Bauer D, Diliberto JJ, Birnbaum LS. 2006. Toxicokinetics of polybrominated diphenyl ether congeners 47, 99, 100, and 153 in mice. Toxicological sciences : an official journal of the Society of Toxicology 94(1): 28–37. [DOI] [PubMed] [Google Scholar]
- Sun J, Liu J, Liu Q, Ruan T, Yu M, Wang Y, et al. 2013. Hydroxylated polybrominated diphenyl ethers (OH-PBDEs) in biosolids from municipal wastewater treatment plants in China. Chemosphere 90(9): 2388–2395. [DOI] [PubMed] [Google Scholar]
- Suvorov A, Girard S, Lachapelle S, Abdelouahab N, Sebire G, Takser L. 2009. Perinatal exposure to low-dose BDE-47, an emergent environmental contaminant, causes hyperactivity in rat offspring. Neonatology 95(3): 203–209. [DOI] [PubMed] [Google Scholar]
- Szabo DT, Richardson VM, Ross DG, Diliberto JJ, Kodavanti PR, Birnbaum LS. 2009. Effects of perinatal PBDE exposure on hepatic phase I, phase II, phase III, and deiodinase 1 gene expression involved in thyroid hormone metabolism in male rat pups. Toxicological sciences : an official journal of the Society of Toxicology 107(1): 27–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teft WA, Mansell SE, Kim RB. 2011. Endoxifen, the active metabolite of tamoxifen, is a substrate of the efflux transporter P-glycoprotein (multidrug resistance 1). Drug metabolism and disposition: the biological fate of chemicals 39(3): 558–562. [DOI] [PubMed] [Google Scholar]
- Toms LM, Harden F, Paepke O, Hobson P, Ryan JJ, Mueller JF. 2008. Higher accumulation of polybrominated diphenyl ethers in infants than in adults. Environ Sci Technol 42(19): 7510–7515. [DOI] [PubMed] [Google Scholar]
- Usenko CY, Hopkins DC, Trumble SJ, Bruce ED. 2012. Hydroxylated PBDEs induce developmental arrest in zebrafish. Toxicol Appl Pharmacol 262(1): 43–51. [DOI] [PubMed] [Google Scholar]
- van Herwaarden AE, Schinkel AH. 2006. The function of breast cancer resistance protein in epithelial barriers, stem cells and milk secretion of drugs and xenotoxins. Trends in pharmacological sciences 27(1): 10–16. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Ruan X, Li Y, Yan M, Qin Z. 2013. Polybrominated diphenyl ethers (PBDEs) in aborted human fetuses and placental transfer during the first trimester of pregnancy. Environ Sci Technol 47(11): 5939–5946. [DOI] [PubMed] [Google Scholar]
- Zota AR, Park JS, Wang Y, Petreas M, Zoeller RT, Woodruff TJ. 2011. Polybrominated diphenyl ethers, hydroxylated polybrominated diphenyl ethers, and measures of thyroid function in second trimester pregnant women in California. Environ Sci Technol 45(18): 7896–7905. [DOI] [PMC free article] [PubMed] [Google Scholar]