

 This work is licensed under a
This work is licensed under a Abstract
Mitochondria perform essential roles as crucial organelles for cellular and systemic energy homeostasis, and as signaling hubs, which coordinate nuclear transcriptional responses to the intra- and extra-cellular environment. Complex human diseases, including diabetes, obesity, fatty liver disease and aging-related degenerative diseases are associated with alterations in mitochondrial oxidative phosphorylation (OxPhos) function. However, a recent series of studies in animal models have revealed that an integrated response to tolerable mitochondrial stress appears to render cells less susceptible to subsequent aging processes and metabolic stresses, which is a key feature of mitohormesis. The mitochondrial unfolded protein response (UPRmt) is a central part of the mitohormetic response and is a retrograde signaling pathway, which utilizes the mitochondria-to-nucleus communication network. Our understanding of the UPRmt has contributed to elucidating the role of mitochondria in metabolic adaptation and lifespan regulation. In this review, we discuss and integrate recent data from the literature on the present status of mitochondrial OxPhos function in the development of metabolic diseases, relying on evidence from human and other animal studies, which points to alterations in mitochondrial function as a key factor in the regulation of metabolic diseases and conclude with a discussion on the specific roles of UPRmt and mitohormesis as a novel therapeutic strategy for the treatment of obesity and insulin resistance.
Keywords: mitochondria, oxidative phosphorylation, mitochondrial unfolded protein response, diabetes, insulin resistance
Introduction
Mitochondria are double membrane-bound organelles that resemble α-proteobacteria, from which they are thought to have originated by endocytosis more than 1 billion years ago (Gray et al. 2001). As the powerhouses of the cell, mitochondria generate most of the energy through oxidative phosphorylation (OxPhos), a process in which electrons are passed along a series of OxPhos subunit proteins embedded in the inner mitochondrial membrane (Huttemann et al. 2007). Apart from cellular respiration and energy synthesis, mitochondria are also required for the metabolism of nucleotides as well as biosynthesis of amino acids and lipids.
The discovery in the early 1960s that mitochondria have their own DNA and translation system stimulated an increase in articles related to mitochondria and oxidative phosphorylation from the late 1960s to 1980 (Figs 1 and 2). Mitochondrial ATPase was initially purified from beef heart mitochondria and its action as a coupling factor was also demonstrated (Penefsky et al. 1960, Pullman et al. 1960). Moreover, the positional papers regarding the evidences for electron transport-linked proton pumps and oxidative phosphorylation were published in the 1970s (Figs 1 and 2) (Guerrieri & Nelson 1975, Brand et al. 1976, Wikstrom & Krab 1979). These intensive works on mitochondrial OxPhos complex contributed to launching the golden era of mitochondrial research from the 1960s to late 1970s.
Figure 1.

Scientific interests in mitochondria from 1950 to 2016. The articles were identified in PubMed using the term ‘mitochondria’ for each year from 1950 to 2016. The articles were expressed as the total number of mitochondria articles out of all articles, as well as mitochondria articles as a percentage of all articles in PubMed. The essential milestones and discoveries in the field of mitochondrial research are described by the year of publication.
Figure 2.

Total number and percentage of ‘Mitochondria,’ ‘Oxidative phosphorylation,’ ‘UPRmt’ and ‘Metabolic disease’ articles in PubMed from 1950 to 2016.
Recently, a second renaissance in mitochondrial biology is emerging due to the recognition of the mitochondria as a central regulator in metabolic homeostasis and longevity. In 1998, 46 mitochondrial proteins were reported by 2D gel electrophoresis of purified human placental mitochondria, and the mitochondrial proteins were also identified by peptide mass fingerprinting (Fig. 1) (Rabilloud et al. 1998). Five years later, in 2003, state-of-the art mass spectrometry-based proteomics enabled us to discover more mitochondrial proteins in yeast, mice and humans (Mootha et al. 2003a , Sickmann et al. 2003, Taylor et al. 2003). These data have been consolidated in the MitoCarta, an inventory of mammalian mitochondrial genes and provides integrated datasets of mitochondrial protein localizations in various tissues through mass spectrometry and large-scale GFP tagging/microscopy (Pagliarini et al. 2008). In addition, a series of recent studies revealed the complete structure of the mammalian respiratory chain complex and mitochondrial ribosomes by single-particle cryoelectron microscopy (Brown et al. 2014, Gu et al. 2016, Englmeier et al. 2017). Thus, new technologies for discovery of uncharacterized mitochondrial proteins are promoting the development of mitochondrial biology (Fig. 1) (Kazak et al. 2013, Rhee et al. 2013). To date, the successful efforts to understand the mitochondrial OxPhos complex and to catalog the mitochondrial proteome have established the relevance of mitochondrial dysfunction in a broad spectrum of metabolic diseases.
By far, one of the most significant discoveries during this second renaissance has been the identification of the mitochondrial unfolded protein response (UPRmt), a retrograde stress response induced by mitochondrial proteotoxic stress. The UPRmt was discovered in the late 1990s and is an area of increased scientific interest for research on aging and metabolic diseases (Figs 1 and 2) (Martinus et al. 1996, Ryan et al. 1997). Although initially characterized in mammalian cells, mechanistic insight on the UPRmt was gleaned from studies in worms and flies. Recently, the mechanism of UPRmt regulation was partially revealed in mammals as well as worms (Haynes et al. 2010, Nargund et al. 2015, Chung et al. 2017b ). Thus, many researchers are trying to define the role of UPRmt in mammalian cells and tissues, and its possible role in mitohormesis. The mitochondrial chaperones and proteases predicted to be involved in the regulation of mitochondrial proteostasis may have an essential role in the modulation of metabolic diseases in mammalian cells and mice. While many review articles focused on the role of UPRmt in lower organisms, a review focusing on the effects of UPRmt on metabolic diseases are scarce in mammals.
In this review, we discuss the recent remarkable progress in understanding the role of mitochondrial OxPhos perturbations on systemic energy metabolism that may be relevant in the treatment of obesity and diabetes. We also describe the effects of UPRmt on cellular and systemic physiology during the conditions of mitochondrial dysfunction and proteotoxicity. Lastly, we discuss the UPRmt and mitohormesis as new therapeutic targets for treatment of metabolic diseases.
OxPhos function in a variety of metabolic diseases
OxPhos, as a main platform for energy production in eukaryotic cells, consists of two major components including the phosphorylation of ADP to ATP using energy from the chemiosmotic proton gradient, as well as the oxidation of molecules generated during the breakdown of glucose. Five multimeric complexes embedded in the inner mitochondrial membrane comprise the OxPhos system. Complexes I-IV are multi-subunit enzymes, which work to generate an electrochemical proton gradient across the mitochondrial inner membrane, which is used by the complex V (ATP synthase) to produce ATP (Chaban et al. 2014). Thus, dysfunction of the mitochondrial OxPhos system not only is correlated with mitochondrial genetic disorders but also complex human diseases such as metabolic and neurodegenerative diseases. Therefore, understanding the systems and functions of OxPhos allows us to unravel not only the significance of mitochondrial respiratory chains but also the novel therapeutic options in the etiology and progression of metabolic diseases.
OxPhos function and metabolic diseases
Metabolic diseases including type 2 diabetes mellitus (T2DM) and obesity are closely associated with the alteration of gene expression involved in OxPhos. The skeletal muscle of patients with T2DM showed a decrease in expression of a global set of genes of the OxPhos pathway (Sreekumar et al. 2002, Mootha et al. 2003b ). High-fat diet decreases the expression of the genes necessary for OxPhos, mitochondrial proteins and mitochondrial biogenesis in humans and mice (Sparks et al. 2005). The rate of mitochondrial OxPhos activity in skeletal muscle were 30% lower in insulin-resistant subjects compared to control subjects (Petersen et al. 2004). Additionally, the intramyocellular triglyceride content in the soleus muscle was 80% higher in the subjects with insulin resistance (Petersen et al. 2004). Impairment in OxPhos function induced by deficiency of Crif1, an integral mitoribosomal factor for the insertion of nascent OxPhos polypeptides into the inner mitochondrial membrane, in beta cells triggers progressive failure of insulin secretion and cellular proliferation (Kim et al. 2015). Beta-cell-specific knockout of Tfam, a transcription factor of mtDNA-encoded OxPhos polypeptides, promotes hyperglycemia by impaired secretion of insulin from islets in response to glucose stimulation (Silva et al. 2000). Haploinsufficiency of pentatricopeptide repeat domain 1 protein, known as a regulator of mitochondrial gene expression, reduces mitochondrial respiratory complex biogenesis and function, thus resulting in obesity and insulin resistance (Perks et al. 2017). These data suggest that impaired OxPhos has been associated with the development of insulin resistance, the decrease of insulin secretion from beta cells, and the dysregulation of fatty acid metabolism in mice and humans.
However, multiple lines of evidences indicate that there may not be a causal relation in all circumstances. For example, Asian Indians with insulin resistance have similar skeletal muscle mitochondrial OxPhos capacity as nondiabetic controls. In addition, regardless of diabetic status, Indians have higher OxPhos capacity than Northern European Americans, indicating that mitochondrial OxPhos dysfunction cannot account for all insulin resistance in Asian Indians (Nair et al. 2008). Further, mitochondrial OxPhos capacity can be altered by fuel intake, oxidative load, epigenetics and environmental factors (Patti & Corvera 2010). Therefore, whether impaired mitochondrial oxidative activity causes insulin resistance or results from insulin resistance is not certain and requires further research.
It is well established that reactive oxygen species (ROS) is an important mediator of metabolic dysfunction (Rani et al. 2016). Mitochondria are the primary site of ROS production, most notably due to premature leakage of electrons from complexes I and III of the electron transport chain. Thus, it is unsurprising that reduction of mitochondrial ROS leads to an improved metabolic phenotype. The Mediterranean diet showing health-promoting effects reduces the expression of OxPhos genes, thereby leading to reduction of ROS in peripheral blood mononuclear cells (van Dijk et al. 2012). Mitochondria-derived ROS is also associated with massive macrophage accumulation in adipose tissue, leading to exacerbation of insulin resistance and systemic inflammation (Han 2016). Several studies showed that caloric restriction, which induces protection from aging and metabolic diseases, prevents the decline in mitochondrial respiratory function (Hempenstall et al. 2012, Lanza et al. 2012). However, caloric restriction also decreases the expression of the genes involved in OxPhos and oxidative stress in peripheral blood mononuclear cells and skeletal muscle (Kayo et al. 2001, Crujeiras et al. 2008, Lanza et al. 2012). In addition, intracellular ROS produced by OxPhos pathway is associated with hyperglycemia and is important in the pathogenesis of diabetic nephropathy (Huang et al. 2006). Muscle- or liver-specific deletion of mitochondrial flavoprotein apoptosis inducing factor results in OxPhos deficiency, but leads to the improvement of glucose tolerance and insulin sensitivity in mice (Pospisilik et al. 2007).
Thus, the relationship between mitochondrial OxPhos function and metabolic diseases is highly complicated and is also likely to be context dependent. Although mitochondrial OxPhos capacity has been considered as a key factor underlying aging and metabolic diseases, the attenuation of OxPhos function may be a useful therapeutic strategy for obesity and insulin resistance. More in-depth mechanistic insight can be gained from studies on the OxPhos quality control systems.
The role of mitochondrial chaperone systems in metabolism
As the majority of mitochondrial proteins are encoded by the nuclear genome and imported into the mitochondria in an unfolded state, mitochondrial chaperones play an important role in maintaining normal mitochondrial function. In conditions of increased mitochondrial proteotoxic stress, the mitochondrial unfolded protein response (UPRmt) is activated and upregulates a specific set of genes, of which the mitochondrial chaperone system is involved (Aldridge et al. 2007). In this context, the mitochondrial chaperone systems are required for facilitating protein-folding within the mitochondria (Fig. 3). While HSP60 plays a prominent role in the UPRmt response, other mitochondrial chaperones are yet to be fully explored in this context.
Figure 3.
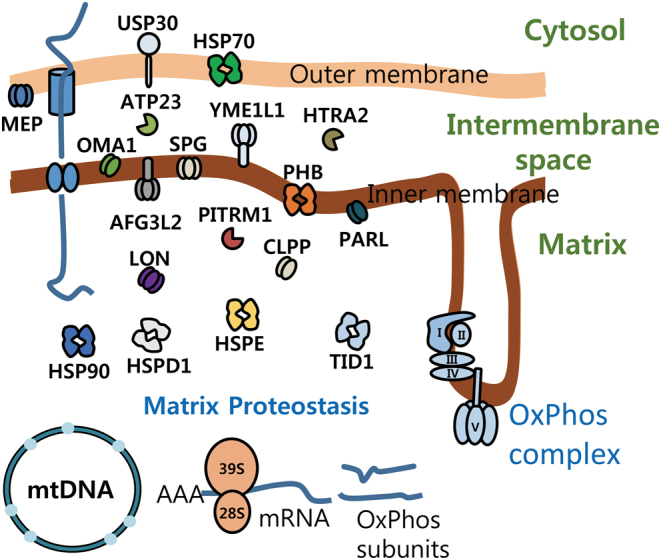
Mitochondrial chaperones and proteases. Schematic cartoon describes the major factors involved in mitochondrial proteostasis. The chaperones and proteases are located in mitochondrial outer and inner membrane, intermembrane space and matrix.
The heat shock protein 60 (HSP60) has been studied extensively and is established as a marker of the UPRmt induction in lower organisms. HSP60 is reduced in the hypothalamus of obese or diabetic mice and humans, which is associated with central insulin resistance and mitochondrial dysfunction. The role of leptin on mitochondrial function and insulin sensitivity in hypothalamus is dependent on mitochondrial HSP60, suggesting that the hypothalamic mitochondrial chaperone system has an important effect on systemic energy homeostasis in obesity and metabolic diseases (Kleinridders et al. 2013). Moreover, heat shock protein family D member 1 (Hspd1) knockdown decreased mitochondrial respiration, levels of respiratory subunits and mtDNA levels in the murine hypothalamic cell line (Kleinridders et al. 2013). Furthermore, myocardial HSPD1 increases in high-fructose-fed rats, which is associated with prevention of hyperglycemia and severe cardiac injury (Chen et al. 2009). siRNA-induced Hspd1 knockdown also increased intracellular protein aggregation and oxidized proteins in renal tubular cells, which may contribute to diabetes-induced renal tubular dysfunction (Aluksanasuwan et al. 2017). Thus, abnormal regulation of HSP60 may decrease the capacity of mitochondrial function and the UPRmt response, which potentially links the UPRmt to metabolic disorders. However, more extensive studies would be required, especially in mammalian systems, to firmly establish a causative role for UPRmt dysregulation in the etiology and progression of diseases.
Several studies have revealed that the mitochondrial HSP90s play a key role in organelle homeostasis such as protein-folding, quality control, redox balance and the regulation of metabolic pathways (Chae et al. 2013). The HSP90 chaperone machinery in association with HSP70 is also involved in protein-folding and activation of newly synthesized proinflammatory signal transducers (Caplan et al. 2007). HSP90 inhibition induced by 17-dimethylaminoethylamino-17-demethoxygeldanamycin attenuates inflammatory signaling pathways including NF-kB, STAT1 and STAT3 in diabetic kidneys, thus resulting in amelioration of diabetic nephropathy and atherosclerosis by the induction of protective HSP70 (Lazaro et al. 2015).
TNFR-associated protein 1 (TRAP1), which belongs to the HSP90 family, is also involved in the regulation of mitochondrial respiration and glycolysis. Loss of TRAP1 leads to a reduction in oxidative damage and cell-cycle defects by global reprogramming of cellular bioenergetics, leading to decreases in aging-related pathologies including obesity and tumor formation (Lisanti et al. 2014).
Prohibitin, one of the evolutionarily conserved mitochondrial chaperones, has been implicated in adipose tissue biology from worms to mice (Merkwirth & Langer 2009). Overexpression of prohibitin in adipocytes leads to an increase in mitochondrial biogenesis and obesity development (Ande et al. 2014). However, the metabolic phenotypes of obesity induced by overexpression of prohibitin in adipocytes are sex specific due to the prohibitin translocation from mitochondria to the nucleus in response to estrogen (Dong et al. 2013). Overexpression of prohibitin in adipocytes impaired glucose homeostasis in male mice, but female mice did not show any changes in the glucose tolerance test (Ande et al. 2014). This suggests that mitochondrial chaperones do not always result in the improvement of metabolic phenotype in mice.
Other studies suggest that HSP72 is closely associated with insulin resistance and type 2 diabetes. Muscle tissues of patients with type 2 diabetes exhibit lower expression of HSP72 compared to those of normal controls (Kurucz et al. 2002). Intramuscular Hsp72 mRNA expression is inversely correlated with insulin-stimulated glucose disposal rate during a hyperinsulinemic–euglycemic clamp in patients with type 2 diabetes (Bruce et al. 2003). Overexpression of Hsp72 in skeletal muscle increases mitochondrial number and OxPhos function, leading to enhanced energy expenditure and insulin sensitivity in mice (Henstridge et al. 2014). In contrast, Hsp72-deficient mice exhibit obesity and insulin resistance, suggesting that HSP72 enhances muscle insulin sensitivity by promoting fatty acid oxidation and reducing fat storage and adiposity in skeletal muscle via the HSP72-Parkin axis (Drew et al. 2014).
Although it is unclear how mitochondrial chaperones contribute to the improvement of obesity and insulin resistance, recent works indicate a role in the recovery of mitochondrial function and biogenesis in mice (Ande et al. 2014, Bottinger et al. 2015). However, it should be noted that while the mitochondrial chaperone-mediated regulation of metabolic homeostasis is established in worms and mice, less is known regarding the regulation of mitochondrial proteostasis in humans. To date, it is unknown as to which key factors regulate the UPRmt in mammals. Additionally, knockout models of specific mitochondrial chaperones and proteases in mammals did not reveal any abnormal mitochondrial proteostasis. Therefore, many studies have just shown the transcript levels of mitochondrial chaperones and proteases as markers of UPRmt induction in vitro and in vivo (Aldridge et al. 2007, Haynes et al. 2010, Fiorese et al. 2016). Recent studies demonstrated that the ATF4 pathway is activated in mammals upon mitochondrial stress and mediates the mitochondrial stress response (Quiros et al. 2017). However, mitochondrial stress caused by LONP1 knockdown or mdivi-1 exposure in mammalian cells does not induce prototypical UPRmt, as defined by the induction of HSP60, HSP10 and CLPP. This suggests that mitochondrial stress-mediated activation of the ATF4 pathway is not always involved in the regulation of mitochondrial chaperones and proteases in mammalian cells.
Role of intrinsic mitochondrial proteases in metabolism
In addition to the chaperone system, mitochondria also contain evolutionarily conserved mitochondrial proteases as crucial elements of mitochondrial quality control (Fig. 3). The inner mitochondrial membrane protease OMA1 has essential roles in mitochondrial quality control and metabolic homeostasis. Deficiency of Oma1 leads to increased body weight and fat contents, as well as impaired thermogenesis after diet-induced obesity in mice. Oma1-deficient mice displayed improved glucose intolerance and increased insulin sensitivity on a normal chow diet. However, Oma1-deficient mice lost these metabolic benefits in the condition of high-fat diet-induced obesity (Quiros et al. 2012).
The LON protease (also LONP1) is an important enzyme in the degradation of oxidized proteins within the mitochondrial matrix, which is highly induced in response to acute stress. A decline in LON levels is associated with aging and chronic stress, thereby promoting the development of aging-related diseases by losing the ability to induce LON during acute stress (Ngo et al. 2011). LON is also involved in the regulation of hepatic insulin resistance and gluconeogenesis. Treatment with LON-specific siRNA induces mitochondrial dysfunction such as reduction in cellular ATP contents and mitochondrial membrane potential. Deficiency of LON protease also causes hepatic gluconeogenesis by induction of glucose-6-phosphatase and PGC-1a in human liver SK-HEP-1 cells. Overexpression of LON protease ameliorates hepatic insulin resistance in the cells containing dysfunctional mitochondria (Lee et al. 2011). These data suggest that mitochondrial proteases contribute to the improvement of systemic energy metabolism via regulation of mitochondrial function and quality control.
CLPP, another matrix protease, plays a crucial role in the UPRmt. Loss of CLPP causes moderate mitochondrial respiratory deficiency by defective mitoribosome assembly and a decrease in mitochondrial translation rates (Szczepanowska et al. 2016). Depletion of Clpp ameliorates cardiomyopathy in heart-specific DARS2-deficient mice via increased mitochondrial OxPhos function (Seiferling et al. 2016). In addition, the expression of CLPP and LON, which are major mitochondrial proteases, are used as an indicator of UPRmt activation in vivo as well as in vitro (Houtkooper et al. 2013, Munch & Harper 2016). ATP-dependent metalloprotease Yme1l deficiency in cardiomyocytes causes dilated cardiomyopathy and heart failure by activation of OPA1-mediated mitochondrial fragmentation. Cardiac function and mitochondrial dynamics were restored by Oma1 ablation in cardiomyocyte-specific Yme1l-deficient mice (Wai et al. 2015).
Mitochondrial protease presenilin-associated rhomboid-like (PARL) also contributes to the regulation of skeletal muscle OxPhos and insulin signaling. PARL mRNA expression in skeletal muscle is reduced in elderly subjects and patients with type 2 diabetes. The expression of PARL in gastrocnemius muscle is correlated with insulin sensitivity as assessed by whole-body glucose disposal during a hyperinsulinemic–euglycemic clamp. Moreover, Leu262Val in PARL is associated with increased plasma insulin levels in human subjects (Walder et al. 2005). Muscle-specific knockdown of PARL results in lower mitochondrial energetics and impaired insulin signaling as well as malformation of mitochondrial cristae and increased oxidative stress in mice (Civitarese et al. 2010). This suggests that PARL is an important candidate gene for the development of insulin resistance and type 2 diabetes. Taken together, a network of conserved proteases distributed across mitochondrial compartments may not only regulate the onset and progression of obesity and insulin resistance, but also play pivotal roles in the maintenance of mitochondrial function.
UPRmt and mitohormesis in organismal models
In contrast to linear models of mitochondrial dysfunction and disease progression, studies in the past decade have established a new field of research with important ramifications for the field of longevity and aging-related diseases, including metabolic diseases. Although mitochondrial perturbation generally leads to metabolic dysfunction, mild mitochondrial perturbation in yeast showed increased replicative lifespan through a retrograde stress response from the mitochondria to the nucleus (Kirchman et al. 1999). This was corroborated in studies of Caenorhabditis elegans and Drosophila, where mild mitochondrial perturbation led to increased lifespan in both models (Dillin et al. 2002, Owusu-Ansah et al. 2013). This concept has come to be defined as mitochondrial hormesis or mitohormesis, where mild mitochondrial perturbation increases fitness and confers a resistance to subsequent stresses (Yun & Finkel 2014). Although various pathways have been reported to be involved in the mitohormetic process, one area of interest is the UPRmt, which will be explored in more detail in the following sections.
The mitochondrial unfolded protein response (UPRmt)
Due to the endosymbiotic origin from α-proteobacteria, mitochondria contain their own genome, the mtDNA, which only encodes ~1% of the total mitochondrial proteome including 13 OxPhos proteins, 22 transfer RNAs and 2 ribosomal RNAs (Calvo & Mootha 2010). Multiple factors such as absence of protective histone molecules and proximity of the mtDNA to the inner mitochondrial membrane, where ROS are generated, contribute to a higher mutation rate in mtDNA, thereby promoting mitochondrial proteotoxicity and a decline in mitochondrial function (Krishnan & Turnbull 2010, Cha et al. 2015). In this context, the concerted efforts of the mitochondrial system of proteases and chaperones are pivotal in maintaining protein homeostasis or proteostasis within the mitochondria.
Altered mitochondrial proteostasis and dysfunction triggers the activation of a retrograde signaling pathway from mitochondria to the nucleus that results in the upregulation of mitochondrial chaperones and proteases to re-establish mitochondrial function. This response is known as the UPRmt. The UPRmt was first characterized in mammalian COS7 cells, where the presence of mutated ornithine transcarbomylase (deltOT) within the mitochondrial matrix resulted in the mitochondrial-specific upregulation of Hsp60 and Clpp, in the absence of endoplasmic reticulum stress (Zhao et al. 2002). Despite its origins in mammalian cells, mechanistic insight was gained from studies in C. elegans. Most notably, the master regulator of the UPRmt in worms was revealed to be Atfs1 and reliant on the co-factors DVE1 and UBL5 (Haynes et al. 2007, 2010). A subsequent study by the same group found that the UPRmt in worms is regulated by two complementary pathways; an Atfs1-dependent pathway and ROS-mediated pathway dependent on the phosphorylation of the GCN2, a eukaryotic initiation factor 2 alpha (eif2α) kinase (Baker et al. 2012). This suggests that the UPRmt may be regulated in a similar fashion to the UPRer, where temporary translation inhibition may be accompanied by upregulation of chaperones and proteases to re-establish organellular homeostasis. Moreover, the reliance on ROS may suggest that there may be an interconnection between the UPRmt and mitohormetic pathways, as low levels of ROS have been implicated in improved cellular fitness and lifespan extension in various animal models (Lapointe & Hekimi 2008, Yang & Hekimi 2010, Owusu-Ansah et al. 2013).
Recent efforts to define this process in mammalian models suggests that the UPRmt response may be differentially regulated in higher organisms, and likely reliant on an integrated stress response (Melber & Haynes 2018). Due to previous studies which have established Hsp60 and Clpp as important factors upregulated during the UPRmt, the current gold standard in mammals is the detection of these markers under conditions of mitochondrial perturbation. However, it should be noted that indiscriminate use of these markers may lead to inaccurate conclusions as the UPRmt may be regulated differently in mammalian models. Nevertheless, the UPRmt is essential for the repair and recovery of the mitochondrial protein network, leading to proper cellular function.
The UPRmt as a mitohormetic response
The concept of UPRmt and mitohormesis has been explored previously in the context of longevity studies in worms and flies. Mitohormesis was first described in a Saccharomyces cerevisiae model, where engineered yeast cells lacking mtDNA activated a diverse set of Rtg-dependent nuclear genes in a retrograde manner, which led to increased replicative lifespan (Kirchman et al. 1999). Later studies in C. elegans established a link between the UPRmt and longevity. Early studies in 2002 found that perturbation of nuclear-encoded mitochondrial oxphos complexes, specifically complexes I, III and IV extended worm lifespan (Dillin et al. 2002). This was later found to be reliant on the activation of a UPRmt machinery, thus suggesting that the mitohormetic benefits of mild mitochondrial perturbation was reliant on the activation of this pathway (Durieux et al. 2011). Moreover, glucose restriction induces formation of ROS, which increases life span through mitohormetic response in worms (Schulz et al. 2007). Physical exercise-induced ROS also improve insulin resistance via mitohormesis in human subjects (Ristow et al. 2009). Further studies in the Drosophila model suggested that the induction of the UPRmt and insulin-like growth factor-binding proteins (IGFBP) co-regulated the longevity of flies (Owusu-Ansah et al. 2013). Thus, the UPRmt-mediated mitohormetic response is conserved from worms to humans.
Due to the longer average lifespan of mouse models, longevity studies are likely less common in mammalian models than in the lower organisms. Despite this limitation, several studies in mice have looked at longevity through the genetic ablation of mclk1 and Surf1 (Liu et al. 2005, Dell’agnello et al. 2007). Although the mechanism was found to be different, both mouse models showed markedly increased lifespan and increased cellular fitness. Heterozygous mclk1 mice showed decreased activity of complex II and ROS production, which contributed to the longevity of the mice, while Surf1 mice showed decreased mitochondrial Ca2+ intake and resistance to neurodegeneration. Despite the mitochondrial involvement in both models and a definite mitohormetic response, neither looked at it from the perspective of the UPRmt, thus necessitating further studies to establish a causal link.
One aspect of increased longevity is improved metabolic performance. Metabolism and mitochondrial integrity have been intricately linked with aging and longevity ((Lopez-Otin et al. 2016). In this regard, one way to study the implications of UPRmt activation and its effects is through the transient modulation of systemic metabolism. A mediator of this response may be signaling factors released from tissues or organs with primary mitochondrial defects. These mediators are known as mitochondrial cytokines or mitokines and are secreted in response to mitochondrial dysfunction to re-establish systemic metabolic homeostasis. Thus, mitokine action and its improvement of metabolic parameters can be considered as a type of transient mitohormetic response and will be discussed in more detail in the following section.
Putative mitokine factors as a regulator of systemic energy metabolism
Previous studies in C. elegans have established a class of secretory proteins defined as mitochondrial cytokines or mitokines (Durieux et al. 2011). In this study, activation of the UPRmt in the brain resulted in the distal activation of the UPRmt in the worm gut and was suggested to be mediated by an unknown factor, which the authors defined as a mitokine. A subsequent study revealed that this mitokine was serotonin (Berendzen et al. 2016). In contrast to worm models, mitokines in the mammalian context may serve a different role. This may be explained by the increased complexity and interorgan communication distance that would be required in higher organisms, although this would require more in-depth studies to prove. Mitokine responses in mammals can be considered as critical cell non-autonomous modifiers in disease severity and progression. Secretion of mitokines can be a critical adaptive response that occurs during a specific period of disease progression (Fig. 4). Individual mitokines may act as disease markers, and utilization of these mitokine factors may serve as potential therapeutics for metabolic diseases.
Figure 4.

Schematic model regarding the importance of mitokine response in progression of metabolic diseases.
Fibroblast growth factor 21 (FGF21) was reported as the first mitokine factor in mice and is induced by Atg7-knockout-mediated autophagy deficiency and mitochondrial dysfunction. Inhibition of mitochondrial function induces ATF4-dependent induction of Fgf21 expression, leading to amelioration of diet-induced obesity and insulin resistance in mice (Kim et al. 2013). Currently, FGF21 is a useful serum biomarker of mitochondrial diseases, although skeletal muscle biopsy is the gold standard for diagnosis of mitochondrial translation and mtDNA maintenance disorders in humans (Suomalainen et al. 2011). FGF21 induced by decreased muscle fat oxidation is dependent on the AMPK and Akt1 signaling, which promotes the increase of glucose uptake in muscle and white adipose tissue browning in mice (Vandanmagsar et al. 2016). Moreover, FGF21 provides a thermoregulatory defense against hypothermia through activation of adipose tissue browning by enhancing adipose PGC1α (Fisher et al. 2012). Additionally, FGF21 attenuates diabetic cardiomyopathy by increasing AMPK activity and paraoxonase 1 signaling in streptozotocin/high-fat diet-induced hyperglycemic mice (Wu et al. 2017).
Emerging studies report the role of UPRmt and mitohormesis on metabolic diseases across multiple species. Administration of nicotinamide riboside, a precursor of NAD+ biosynthesis, prevented hepatic fat accumulation in high-fat high-sucrose diet-fed mice by sirtuin-mediated UPRmt, triggering an adaptive mitohormesis in the liver of mice (Gariani et al. 2016). The UPRmt induced by skeletal muscle-specific OxPhos deficiency is also associated with enhanced lipolysis and fatty acid oxidation by the induction of growth differentiation factor 15 (GDF15), thereby protecting against the adverse effects of high-fat diet-induced obesity in mice (Chung et al. 2017b ). Moreover, serum GDF15 levels predict insulin resistance and glucose intolerance in nondiabetic subjects and patients with type 2 diabetes (Dostalova et al. 2009, Kempf et al. 2012, Hong et al. 2014). Additionally, GDF15 deficiency aggravates chronic alcohol- and carbon tetrachloride-induced liver injury by inducing infiltration of neutrophils, monocytes and activated T cells in the liver (Chung et al. 2017a ). It should be noted that the relationship between the UPRmt and GDF15 induction is merely correlative and requires further confirmation in follow-up studies.
Angiopoietin-like 6 is also induced by OxPhos dysfunction caused by Crif1 knockout or treatment with oligomycin and rotenone in cultured adipocytes. Angiopoietin-like 6 enhances Fgf21 transcription via activation of ERK–MAPK pathway-mediated Pparα in cultured adipocytes and adipose tissue of mice (Kang et al. 2017). Angiopoietin-like 6 also enhances insulin sensitivity and glucose tolerance resulting from increased energy expenditure and reduction of body weight in mice (Oike et al. 2005).
Recently, several groups revealed that adrenomedullin 2 (ADM2), an endogenous bioactive peptide belonging to the calcitonin gene-related peptide family is an inducible gene in response to mitochondrial stress caused by inhibition of mitochondrial respiration chain in human cancer cell lines. ADM2 has a putative ATF4-binding site GTTGCATCA, located at a distance of 30 bp downstream of the translation start codon within the ADM2 gene. Transcription of ADM2 is tightly regulated by ATF4 during the integrated stress response, which is also associated with tumor angiogenesis by inducing vascular endothelial growth factor in the cells (Kovaleva et al. 2016). Moreover, ADM2 levels in plasma are negatively correlated with body weight in Chinese subjects. ADM2 overexpression in adipocytes or treatment with recombinant ADM2 improves metabolic phenotypes by inducing beiging with upregulation of UCP1, as well as M2 macrophage activation in white adipose tissues in high-fat-fed mice (Lv et al. 2016). Moreover, treatment with ADM2 inhibits obesity-induced insulin resistance in mice via suppression of MHCII antigen presentation in adipocytes and reduction of the activation of CD4+ T cells (Zhang et al. 2016). These data suggest that mitokines induced by mitochondrial stress may be a novel target for obesity and diabetes. Table 1 summarizes the effects of UPRmt signaling components including mitochondrial chaperones, proteases and mitokines on metabolic phenotypes in mice and humans.
Table 1.
Effects of UPRmt signaling components on metabolic phenotypes in mammals.
| Function | Gene name | Protein name | Metabolic phenotype | References |
|---|---|---|---|---|
| Chaperone | Hspd1 | HSPD1 | Reduction in hypothalamus of obese and diabetic subjects; Increase of myocardial Hspd1 in high-fructose fed rats; knockdown induces diabetes-mediated renal tubular dysfunction | Kleinridders et al. (2013), Aluksanasuwan et al. (2017) |
| Trap1 | TRAP1 | Decrease of obesity and tumor formation in TRAP1 knockout | Lisanti et al. (2014) | |
| Phb | PHB | Overexpression in adipocytes leads to obesity in both genders, but impairment in glucose homeostasis only in male mice | Dong et al. (2013), Ande et al. (2014) | |
| Hspa1a | HSP72 | Lower muscular Hsp72 expression in patients with T2DM. Overexpression enhances energy expenditure and insulin sensitivity. Knockout induces obesity and insulin resistance | Kurucz et al. (2002), Bruce et al. (2003), Drew et al. (2014), Henstridge et al. (2014) | |
| Protease | Oma1 | OMA1 | Increased body weight and fat content, improved glucose tolerance and insulin sensitivity on NCD, but loss of metabolic benefits on HFD. Restoration of cardiac function in Yme1l1-deficient mice | Quiros et al. (2012) |
| Lonp1 | LON | Increased hepatic gluconeogenesis | Lee et al. (2011), Ngo et al. (2011) | |
| Clpp | CLPP | Amelioration of cardiomyopathy | Seiferling et al. (2016) | |
| Yme1l1 | YME1L1 | Induces dilated cardiomyopathy and heart failure | Wai et al. (2015) | |
| Parl | PARL | Impaired insulin signaling in skeletal muscle | Walder et al. (2005), Civitarese et al. (2010) | |
| Mitokine | Gdf15 | GDF15 | GDF15 levels in serum increase in patients with T2DM. Associated with fatty acid oxidation and lipolysis. Reduces liver injury by modulating immune cell infiltration | Dostalova et al. (2009), Kempf et al. (2012), Hong et al. (2014), Chung et al. (2017a ,b) |
| Adm2 | ADM2 | Negative correlation with body weight. Overexpression in adipocytes induces beiging of white adipose tissue as well as M2 polarization of adipose macrophages. Recombinant ADM2 inhibits insulin resistance by deactivating adipose CD4+ T cells | Lv et al. (2016), Zhang et al. (2016) | |
| Angptl6 | ANGPTL6 | Required for Fgf21 expression in adipocytes. Increases insulin sensitivity, glucose tolerance and energy expenditure | Oike et al. (2005), Kang et al. (2017) | |
| Fgf21 | FGF21 | Increases fat oxidation and mitochondrial function. Enhances glucose uptake in muscle and browning in white adipose tissue. Attenuates diabetic cardiomyopathy | Fisher et al. (2012), Kim et al. (2013), Vandanmagsar et al. (2016), Wu et al. (2017) |
HFD, high-fat diet; NCD, normal chow diet; T2DM, type 2 diabetes mellitus.
Lastly, mitochondrial-derived peptides also have a key role in stress resistance as a mitochondrial autocrine, paracrine and endocrine signal. Humanin, which is translated in the mitochondria (21-amino-acid peptide) or the cytoplasm (24-amino-acid peptide), has been shown to suppress neuronal cell death caused by Alzheimer’s disease and to increase cytoprotective effects against stress and disease models (Lee et al. 2013). MOTS-c is also a mitochondrial-derived peptide that improves obesity and insulin resistance in mice (Lee et al. 2015), but the mechanism regarding translation of MOTS-c is not fully understood and requires further studies for understanding the natural course and tissue specificity of MOTS-c expression, as well as the metabolic effects of MOTS-c in humans.
Induction of UPRmt in physiological and pharmacological models
The UPRmt can be activated in mammals as well as worms by physiological and pharmacological perturbations. Nuclear transcriptional response is induced when the mis-or unfolded proteins within the mitochondrial matrix accumulate by a variety of mitochondrial stresses. In worms, activation of UPRmt requires the activating transcription factor associated with stress-1 (ATFS-1), which is essential for regulating an important transcriptional response to recover mitochondrial function (Nargund et al. 2015). The degraded peptides in matrix by mitochondrial proteases were exported into the cytosol via HAF-1, an ATP-binding cassette transporter protein (Haynes et al. 2010). HAF-1 also modulates ATFS-1-mediated UPRmt activation by inhibiting mitochondrial import of ATFS-1 (Nargund et al. 2012). ATFS-1 was also involved in the maintenance and propagation of deleterious mtDNA in a heteroplasmic C. elegans strain (Lin et al. 2016). In mammals, the UPRmt and mitokine induced by mitochondrial dysfunction requires CHOP-mediated p38 kinase activation in skeletal muscle (Chung et al. 2017b ). This study also revealed putative CHOP-responsive elements in the human promoter of GDF15, a putative mitokine factor (Chung et al. 2017b ). The UPRmt response element was also identified within the promoters of both CHOP and C/EBPβ genes in mammalian cells (Horibe & Hoogenraad 2007). Moreover, activating transcript factor 5 (ATF5), one of the bZIP transcription factors, regulates the activation of UPRmt by a stress-dependent shift in its cellular localization, thereby promoting proliferation and recovery from mitochondrial stress in mammalian cells (Fiorese et al. 2016). These data suggest that UPRmt is an evolutionally conserved mechanism from worms to mammals.
Pharmacological induction of UPRmt is also extensively characterized in mammalian cells. Treatment with doxycycline induces mitonuclear protein imbalance in cultured cells as well as in tissues from mice, thereby triggering the UPRmt to recover the proper mitochondrial function (Moullan et al. 2015). Gamitrinib-triphenylphosphnium (GTPP), a specific inhibitor of the mitochondrial matrix heat shock protein 90 (HSP90), induces the transcription of HSPD1 and HSPE1 as markers of UPRmt in HeLa cells (Munch & Harper 2016). GTPP also promotes the accumulation of misfolded proteins in the mitochondria, thereby activating SIRT3 and its downstream target genes (Papa & Germain 2014). Actinonin generates an aberrant accumulation of de novo mitochondrial proteins in the inner membrane and impairs the turnover of de novo mitochondrial protein synthesis (Richter et al. 2015). MitoBloCK compounds (Mitochondrial protein import Blockers from the Carla Koehler lab), a selective inhibitor of the sulfhydryl oxidase Erv1 activity, regulates the translocation of redox-regulated proteins into mitochondria and oxidation of Tim13 and Cmc1 (Dabir et al. 2013). Moreover, 2-cyano-3,12-dioxo-oleana-1,9(11)-dien-28-oic acid (CDDO), an inhibitor of the LON matrix protease, triggers the robust upregulation of mitochondrial chaperones and proteases in C2C12 myotubes (Chung et al. 2017b ).
Although various UPRmt inducers have been used in experimental studies, these compounds are not free from the issues of mitochondrial specificity or poor property. However, a variety of studies showing the physiological and pharmacological activation of UPRmt signaling demonstrates the cell autonomous footprint of the UPRmt, which leads to beneficial phenotypic responses such as improved metabolic fitness and longevity in worms and flies. Moreover, genetic or pharmacologic modulation of OxPhos system regulates metabolic phenotypes across the animal kingdom through induction of UPRmt and mitohormesis (Durieux et al. 2011, Shao et al. 2016, Chung et al. 2017b ). Thus, the utilization of these pharmacological UPRmt inducers can facilitate further studies on the effects of UPRmt and its physiological relevance, including metabolic effects.
UPRmt and mitohormesis as new therapeutic targets for metabolic diseases
What is the implication of UPRmt and mitohormesis for the development of metabolic diseases? Indeed, a modest or transient mitochondrial stress provides fundamental protection to the host through cell autonomous and cell-non-autonomous signaling, leading to prevention of metabolic diseases and aging-related disorders. The protective function of UPRmt and mitohormesis in multiple organisms, including accumulation of deleterious mitochondrial genomes, pathogen infection, hematopoietic stem cell maintenance and aging has been established (Lin & Haynes 2016, Qureshi et al. 2017). The UPRmt-mediated metabolic adaptation may serve to rewire cellular metabolism to recover or prevent the metabolic deterioration in humans.
Recently, we revealed that OxPhos deficiency in skeletal muscle (Chung et al. 2017a ) activated UPRmt and the induction of mitokines, thereby maintaining systemic energy homeostasis in metabolic deterioration during diet-induced obesity. Additionally, FGF21, a starvation-induced hormone, is a well-known mitokine that elicits metabolic benefits by increasing fat oxidation and improving mitochondrial function in obese subjects (Kim et al. 2013, Wall et al. 2015). In addition to its physiologic actions, the pharmacologic functions of FGF21 play a key role in the maintenance of systemic energy balance through modulating feeding behavior and energy expenditure (Kim & Lee 2014, Laeger et al. 2014). As discussed earlier, the mitokines such as angiopoietin-like 6 and ADM2 may also have the therapeutic value in the treatment of obesity and insulin resistance in humans. Despite these studies, the relevance of the UPRmt and mitohormesis is only beginning to emerge and it is still unknown if the mitonuclear communication network and cell non-autonomous hormonal factors exert beneficial effects in human metabolic diseases.
Conclusions and perspectives
Metabolic diseases, including diabetes, obesity and cardiovascular diseases, which are strongly associated with mitochondrial dysfunction, have become a global epidemic and is a major worldwide health concern. The growing prevalence of metabolic diseases induced by genetic and environmental factors increases annual direct medical costs in the world. Mitochondrial research has been mainly focused on the association between OxPhos and metabolic diseases as well as basic structural principles of OxPhos. Until recently, the molecular and holistic approaches toward mitochondrial proteostasis were merely developed in the physiologic and pathologic contexts from yeast to mammals. In the near future, the UPRmt may be an attractive therapeutic target for the treatment of metabolic diseases that result from mitochondrial dysfunction. Chemical and pharmacologic agents that target mitochondrial oxidative phosphorylation and proteostasis should be developed and investigated in the basic and clinical fields.
We have highlighted the relationship between mitochondrial OxPhos and metabolic diseases in multiple organisms where the UPRmt and mitohormesis have been demonstrated to play a protective role in obesity and diabetes. However, important questions remain unanswered about the UPRmt and mitohormesis in the development and maintenance of metabolic diseases. The most important question would be how UPRmt regulates the susceptibility of metabolic diseases in mammals. It would also be interesting to determine how mitohormesis affects systemic energy metabolism, leading to inhibition of obesity and insulin resistance.
Although the physiologic roles of the UPRmt and mitohormesis in adapting metabolism to recover mitochondrial proteostasis and energy homeostasis are beginning to emerge, the data provide convincing evidences to suggest that UPRmt-mediated cellular or systemic protective effects may be beneficial for the development of therapeutics for diverse metabolic diseases such as diabetes, neurodegenerative diseases and aging-related disorders. However, it should be noted that while the association between UPRmt/mitohormesis and metabolic diseases is well characterized from worms to flies, less is known regarding the mechanism of the UPRmt and the receptors for mitokines in mammals. It is reasonable to predict that mitokines may have autocrine, paracrine and endocrine effects on other cell types within the tissue. Furthermore, mitokine receptors may be present in distant organs, suggesting that mitokines play endocrine roles in interorgan communication involved in systemic homeostasis.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of this review.
Funding
This work was supported by Global Research Laboratory (GRL) Program, National Research Foundation of Korea, Ministry of Science and ICT (2017K1A1A2013124, 2017R1E1A1A01075126), as well as by a grant (H S Y, 2017F-1) from the Korean Diabetes Association. H S Y was also supported by the Basic Science Research Program, National Research Foundation, Ministry of Science and ICT, Future Planning, Korea (2015R1C1A1A01052432, NRF-2018R1C1B6004439). J Y C was supported by the Basic Science Research Program through the NRF funded by the Ministry of Science, ICT, and Future Planning, Korea (NRF-2014M3A9D8034464).
Acknowledgements
The authors apologize in advance for the authors who have been unintentionally left out in this review.
References
- Aldridge JE, Horibe T, Hoogenraad NJ. 2007. Discovery of genes activated by the mitochondrial unfolded protein response (mtUPR) and cognate promoter elements. PLoS ONE 2 e874 ( 10.1371/journal.pone.0000874) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aluksanasuwan S, Sueksakit K, Fong-Ngern K, Thongboonkerd V. 2017. Role of HSP60 (HSPD1) in diabetes-induced renal tubular dysfunction: regulation of intracellular protein aggregation, ATP production, and oxidative stress. FASEB Journal 31 2157–2167. ( 10.1096/fj.201600910RR) [DOI] [PubMed] [Google Scholar]
- Ande SR, Nguyen KH, Padilla-Meier GP, Wahida W, Nyomba BL, Mishra S. 2014. Prohibitin overexpression in adipocytes induces mitochondrial biogenesis, leads to obesity development, and affects glucose homeostasis in a sex-specific manner. Diabetes 63 3734–3741. ( 10.2337/db13-1807) [DOI] [PubMed] [Google Scholar]
- Baker BM, Nargund AM, Sun T, Haynes CM. 2012. Protective coupling of mitochondrial function and protein synthesis via the eIF2alpha kinase GCN-2. PLoS Genetics 8 e1002760 ( 10.1371/journal.pgen.1002760) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendzen KM, Durieux J, Shao LW, Tian Y, Kim HE, Wolff S, Liu Y, Dillin A. 2016. Neuroendocrine coordination of mitochondrial stress signaling and proteostasis. Cell 166 1553.e1510–1563.e1510. ( 10.1016/j.cell.2016.08.042) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottinger L, Oeljeklaus S, Guiard B, Rospert S, Warscheid B, Becker T. 2015. Mitochondrial heat shock protein (Hsp) 70 and Hsp10 cooperate in the formation of Hsp60 complexes. Journal of Biological Chemistry 290 11611–11622. ( 10.1074/jbc.M115.642017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand MD, Reynafarje B, Lehninger AL. 1976. Stoichiometric relationship between energy-dependent proton ejection and electron transport in mitochondria. PNAS 73 437–441. ( 10.1073/pnas.73.2.437) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A, Amunts A, Bai XC, Sugimoto Y, Edwards PC, Murshudov G, Scheres SHW, Ramakrishnan V. 2014. Structure of the large ribosomal subunit from human mitochondria. Science 346 718–722. ( 10.1126/science.1258026) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce CR, Carey AL, Hawley JA, Febbraio MA. 2003. Intramuscular heat shock protein 72 and heme oxygenase-1 mRNA are reduced in patients with type 2 diabetes: evidence that insulin resistance is associated with a disturbed antioxidant defense mechanism. Diabetes 52 2338–2345. ( 10.2337/diabetes.52.9.2338) [DOI] [PubMed] [Google Scholar]
- Calvo SE, Mootha VK. 2010. The mitochondrial proteome and human disease. Annual Review of Genomics and Human Genetics 11 25–44. ( 10.1146/annurev-genom-082509-141720) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caplan AJ, Mandal AK, Theodoraki MA. 2007. Molecular chaperones and protein kinase quality control. Trends in Cell Biology 17 87–92. ( 10.1016/j.tcb.2006.12.002) [DOI] [PubMed] [Google Scholar]
- Cha MY, Kim DK, Mook-Jung I. 2015. The role of mitochondrial DNA mutation on neurodegenerative diseases. Experimental and Molecular Medicine 47 e150 ( 10.1038/emm.2014.122) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaban Y, Boekema EJ, Dudkina NV. 2014. Structures of mitochondrial oxidative phosphorylation supercomplexes and mechanisms for their stabilisation. Biochimica et Biophysica Acta 1837 418–426. ( 10.1016/j.bbabio.2013.10.004) [DOI] [PubMed] [Google Scholar]
- Chae YC, Angelin A, Lisanti S, Kossenkov AV, Speicher KD, Wang H, Powers JF, Tischler AS, Pacak K, Fliedner S, et al 2013. Landscape of the mitochondrial Hsp90 metabolome in tumours. Nature Communications 4 2139 ( 10.1038/ncomms3139) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HS, Wu TE, Juan CC, Lin HD. 2009. Myocardial heat shock protein 60 expression in insulin-resistant and diabetic rats. Journal of Endocrinology 200 151–157. ( 10.1677/JOE-08-0387) [DOI] [PubMed] [Google Scholar]
- Chung HK, Kim JT, Kim HW, Kwon M, Kim SY, Shong M, Kim KS, Yi HS. 2017a. GDF15 deficiency exacerbates chronic alcohol- and carbon tetrachloride-induced liver injury. Scientific Reports 7 17238 ( 10.1038/s41598-017-17574-w) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung HK, Ryu D, Kim KS, Chang JY, Kim YK, Yi HS, Kang SG, Choi MJ, Lee SE, Jung SB, et al 2017b. Growth differentiation factor 15 is a myomitokine governing systemic energy homeostasis. Journal of Cell Biology 216 149–165. ( 10.1083/jcb.201607110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civitarese AE, MacLean PS, Carling S, Kerr-Bayles L, McMillan RP, Pierce A, Becker TC, Moro C, Finlayson J, Lefort N, et al 2010. Regulation of skeletal muscle oxidative capacity and insulin signaling by the mitochondrial rhomboid protease PARL. Cell Metabolism 11 412–426. ( 10.1016/j.cmet.2010.04.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crujeiras AB, Parra D, Milagro FI, Goyenechea E, Larrarte E, Margareto J, Martinez JA. 2008. Differential expression of oxidative stress and inflammation related genes in peripheral blood mononuclear cells in response to a low-calorie diet: a nutrigenomics study. OMICS 12 251–261. ( 10.1089/omi.2008.0001) [DOI] [PubMed] [Google Scholar]
- Dabir DV, Hasson SA, Setoguchi K, Johnson ME, Wongkongkathep P, Douglas CJ, Zimmerman J, Damoiseaux R, Teitell MA, Koehler CM. 2013. A small molecule inhibitor of redox-regulated protein translocation into mitochondria. Developmental Cell 25 81–92. ( 10.1016/j.devcel.2013.03.006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell’agnello C, Leo S, Agostino A, Szabadkai G, Tiveron C, Zulian A, Prelle A, Roubertoux P, Rizzuto R, Zeviani M. 2007. Increased longevity and refractoriness to Ca(2+)-dependent neurodegeneration in Surf1 knockout mice. Human Molecular Genetics 16 431–444. [DOI] [PubMed] [Google Scholar]
- Dillin A, Crawford DK, Kenyon C. 2002. Timing requirements for insulin/IGF-1 signaling in C. elegans. Science 298 830–834. ( 10.1126/science.1074240) [DOI] [PubMed] [Google Scholar]
- Dong P, Jiang L, Liu J, Wu Z, Guo S, Zhang Z, Zhou F, Liu Z. 2013. Induction of paclitaxel resistance by ERalpha mediated prohibitin mitochondrial-nuclear shuttling. PLoS ONE 8 e83519 ( 10.1371/journal.pone.0083519) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dostalova I, Roubicek T, Bartlova M, Mraz M, Lacinova Z, Haluzikova D, Kavalkova P, Matoulek M, Kasalicky M, Haluzik M. 2009. Increased serum concentrations of macrophage inhibitory cytokine-1 in patients with obesity and type 2 diabetes mellitus: the influence of very low calorie diet. European Journal of Endocrinology 161 397–404. ( 10.1530/EJE-09-0417) [DOI] [PubMed] [Google Scholar]
- Drew BG, Ribas V, Le JA, Henstridge DC, Phun J, Zhou Z, Soleymani T, Daraei P, Sitz D, Vergnes L, et al 2014. HSP72 is a mitochondrial stress sensor critical for Parkin action, oxidative metabolism, and insulin sensitivity in skeletal muscle. Diabetes 63 1488–1505. ( 10.2337/db13-0665) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durieux J, Wolff S, Dillin A. 2011. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 144 79–91. ( 10.1016/j.cell.2010.12.016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englmeier R, Pfeffer S, Forster F. 2017. Structure of the human mitochondrial ribosome studied in situ by cryoelectron tomography. Structure 25 1574.e1572–1581.e1572. ( 10.1016/j.str.2017.07.011) [DOI] [PubMed] [Google Scholar]
- Fiorese CJ, Schulz AM, Lin YF, Rosin N, Pellegrino MW, Haynes CM. 2016. The transcription factor ATF5 mediates a mammalian mitochondrial UPR. Current Biology 26 2037–2043. ( 10.1016/j.cub.2016.06.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher FM, Kleiner S, Douris N, Fox EC, Mepani RJ, Verdeguer F, Wu J, Kharitonenkov A, Flier JS, Maratos-Flier E, et al 2012. FGF21 regulates PGC-1alpha and browning of white adipose tissues in adaptive thermogenesis. Genes and Development 26 271–281. ( 10.1101/gad.177857.111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gariani K, Menzies KJ, Ryu D, Wegner CJ, Wang X, Ropelle ER, Moullan N, Zhang H, Perino A, Lemos V, et al 2016. Eliciting the mitochondrial unfolded protein response by nicotinamide adenine dinucleotide repletion reverses fatty liver disease in mice. Hepatology 63 1190–1204. ( 10.1002/hep.28245) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray MW, Burger G, Lang BF. 2001. The origin and early evolution of mitochondria. Genome Biology 2 REVIEWS1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J, Wu M, Guo R, Yan K, Lei J, Gao N, Yang M. 2016. The architecture of the mammalian respirasome. Nature 537 639–643. ( 10.1038/nature19359) [DOI] [PubMed] [Google Scholar]
- Guerrieri F, Nelson BD. 1975. Studies on the characteristics of a proton pump in phospholipid vesicles inlayed with purified complex III from beef heart mitochondria. FEBS Letters 54 339–342. ( 10.1016/0014-5793(75)80935-5) [DOI] [PubMed] [Google Scholar]
- Han CY. 2016. Roles of reactive oxygen species on insulin resistance in adipose tissue. Diabetes and Metabolism: Journal 40 272–279. ( 10.4093/dmj.2016.40.4.272) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes CM, Petrova K, Benedetti C, Yang Y, Ron D. 2007. ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Developmental Cell 13 467–480. ( 10.1016/j.devcel.2007.07.016) [DOI] [PubMed] [Google Scholar]
- Haynes CM, Yang Y, Blais SP, Neubert TA, Ron D. 2010. The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Molecular Cell 37 529–540. ( 10.1016/j.molcel.2010.01.015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hempenstall S, Page MM, Wallen KR, Selman C. 2012. Dietary restriction increases skeletal muscle mitochondrial respiration but not mitochondrial content in C57BL/6 mice. Mechanisms of Ageing and Development 133 37–45. ( 10.1016/j.mad.2011.12.002) [DOI] [PubMed] [Google Scholar]
- Henstridge DC, Bruce CR, Drew BG, Tory K, Kolonics A, Estevez E, Chung J, Watson N, Gardner T, Lee-Young RS, et al 2014. Activating HSP72 in rodent skeletal muscle increases mitochondrial number and oxidative capacity and decreases insulin resistance. Diabetes 63 1881–1894. ( 10.2337/db13-0967) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong JH, Chung HK, Park HY, Joung KH, Lee JH, Jung JG, Kim KS, Kim HJ, Ku BJ, Shong M. 2014. GDF15 is a novel biomarker for impaired fasting glucose. Diabetes and Metabolism: Journal 38 472–479. ( 10.4093/dmj.2014.38.6.472) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horibe T, Hoogenraad NJ. 2007. The chop gene contains an element for the positive regulation of the mitochondrial unfolded protein response. PLoS ONE 2 e835 ( 10.1371/journal.pone.0000835) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, Knott G, Williams RW, Auwerx J. 2013. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 497 451–457. ( 10.1038/nature12188) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Kim Y, Caramori ML, Moore JH, Rich SS, Mychaleckyj JC, Walker PC, Mauer M. 2006. Diabetic nephropathy is associated with gene expression levels of oxidative phosphorylation and related pathways. Diabetes 55 1826–1831. ( 10.2337/db05-1438) [DOI] [PubMed] [Google Scholar]
- Huttemann M, Lee I, Samavati L, Yu H, Doan JW. 2007. Regulation of mitochondrial oxidative phosphorylation through cell signaling. Biochimica et Biophysica Acta 1773 1701–1720. ( 10.1016/j.bbamcr.2007.10.001) [DOI] [PubMed] [Google Scholar]
- Kang SG, Yi HS, Choi MJ, Ryu MJ, Jung S, Chung HK, Chang JY, Kim YK, Lee SE, Kim HW, et al 2017. ANGPTL6 expression is coupled with mitochondrial OXPHOS function to regulate adipose FGF21. Journal of Endocrinology 233 105–118. ( 10.1530/JOE-16-0549) [DOI] [PubMed] [Google Scholar]
- Kayo T, Allison DB, Weindruch R, Prolla TA. 2001. Influences of aging and caloric restriction on the transcriptional profile of skeletal muscle from rhesus monkeys. PNAS 98 5093–5098. ( 10.1073/pnas.081061898) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazak L, Reyes A, Duncan AL, Rorbach J, Wood SR, Brea-Calvo G, Gammage PA, Robinson AJ, Minczuk M, Holt IJ. 2013. Alternative translation initiation augments the human mitochondrial proteome. Nucleic Acids Research 41 2354–2369. ( 10.1093/nar/gks1347) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempf T, Guba-Quint A, Torgerson J, Magnone MC, Haefliger C, Bobadilla M, Wollert KC. 2012. Growth differentiation factor 15 predicts future insulin resistance and impaired glucose control in obese nondiabetic individuals: results from the XENDOS trial. European Journal of Endocrinology 167 671–678. ( 10.1530/EJE-12-0466) [DOI] [PubMed] [Google Scholar]
- Kim KH, Lee MS. 2014. FGF21 as a stress hormone: the roles of FGF21 in stress adaptation and the treatment of metabolic diseases. Diabetes and Metabolism: Journal 38 245–251. ( 10.4093/dmj.2014.38.4.245) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KH, Jeong YT, Oh H, Kim SH, Cho JM, Kim YN, Kim SS, Kim DH, Hur KY, Kim HK, et al 2013. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nature Medicine 19 83–92. ( 10.1038/nm.3014) [DOI] [PubMed] [Google Scholar]
- Kim YK, Joung KH, Ryu MJ, Kim SJ, Kim H, Chung HK, Lee MH, Lee SE, Choi MJ, Chang JY, et al 2015. Disruption of CR6-interacting factor-1 (CRIF1) in mouse islet beta cells leads to mitochondrial diabetes with progressive beta cell failure. Diabetologia 58 771–780. ( 10.1007/s00125-015-3506-y) [DOI] [PubMed] [Google Scholar]
- Kirchman PA, Kim S, Lai CY, Jazwinski SM. 1999. Interorganelle signaling is a determinant of longevity in Saccharomyces cerevisiae. Genetics 152 179–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinridders A, Lauritzen HP, Ussar S, Christensen JH, Mori MA, Bross P, Kahn CR. 2013. Leptin regulation of Hsp60 impacts hypothalamic insulin signaling. Journal of Clinical Investigation 123 4667–4680. ( 10.1172/JCI67615) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovaleva IE, Garaeva AA, Chumakov PM, Evstafieva AG. 2016. Intermedin/adrenomedullin 2 is a stress-inducible gene controlled by activating transcription factor 4. Gene 590 177–185. ( 10.1016/j.gene.2016.06.037) [DOI] [PubMed] [Google Scholar]
- Krishnan KJ, Turnbull DM. 2010. Mitochondrial DNA and genetic disease. Essays in Biochemistry 47 139–151. ( 10.1042/bse0470139) [DOI] [PubMed] [Google Scholar]
- Kurucz I, Morva A, Vaag A, Eriksson KF, Huang X, Groop L, Koranyi L. 2002. Decreased expression of heat shock protein 72 in skeletal muscle of patients with type 2 diabetes correlates with insulin resistance. Diabetes 51 1102–1109. ( 10.2337/diabetes.51.4.1102) [DOI] [PubMed] [Google Scholar]
- Laeger T, Henagan TM, Albarado DC, Redman LM, Bray GA, Noland RC, Munzberg H, Hutson SM, Gettys TW, Schwartz MW, et al 2014. FGF21 is an endocrine signal of protein restriction. Journal of Clinical Investigation 124 3913–3922. ( 10.1172/JCI74915) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanza IR, Zabielski P, Klaus KA, Morse DM, Heppelmann CJ, Bergen HR, 3rd, Dasari S, Walrand S, Short KR, Johnson ML, et al 2012. Chronic caloric restriction preserves mitochondrial function in senescence without increasing mitochondrial biogenesis. Cell Metabolism 16 777–788. ( 10.1016/j.cmet.2012.11.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapointe J, Hekimi S. 2008. Early mitochondrial dysfunction in long-lived Mclk1+/- mice. Journal of Biological Chemistry 283 26217–26227. ( 10.1074/jbc.M803287200) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazaro I, Oguiza A, Recio C, Mallavia B, Madrigal-Matute J, Blanco J, Egido J, Martin-Ventura JL, Gomez-Guerrero C. 2015. Targeting HSP90 ameliorates nephropathy and atherosclerosis through suppression of NF-kappaB and STAT signaling pathways in diabetic mice. Diabetes 64 3600–3613. ( 10.2337/db14-1926) [DOI] [PubMed] [Google Scholar]
- Lee HJ, Chung K, Lee H, Lee K, Lim JH, Song J. 2011. Downregulation of mitochondrial lon protease impairs mitochondrial function and causes hepatic insulin resistance in human liver SK-HEP-1 cells. Diabetologia 54 1437–1446. ( 10.1007/s00125-011-2074-z) [DOI] [PubMed] [Google Scholar]
- Lee C, Yen K, Cohen P. 2013. Humanin: a harbinger of mitochondrial-derived peptides? Trends in Endocrinology and Metabolism 24 222–228. ( 10.1016/j.tem.2013.01.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Zeng J, Drew BG, Sallam T, Martin-Montalvo A, Wan J, Kim SJ, Mehta H, Hevener AL, de Cabo R, et al 2015. The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metabolism 21 443–454. ( 10.1016/j.cmet.2015.02.009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YF, Haynes CM. 2016. Metabolism and the UPR(mt). Molecular Cell 61 677–682. ( 10.1016/j.molcel.2016.02.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YF, Schulz AM, Pellegrino MW, Lu Y, Shaham S, Haynes CM. 2016. Maintenance and propagation of a deleterious mitochondrial genome by the mitochondrial unfolded protein response. Nature 533 416–419. ( 10.1038/nature17989) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisanti S, Tavecchio M, Chae YC, Liu Q, Brice AK, Thakur ML, Languino LR, Altieri DC. 2014. Deletion of the mitochondrial chaperone TRAP-1 uncovers global reprogramming of metabolic networks. Cell Reports 8 671–677. ( 10.1016/j.celrep.2014.06.061) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Jiang N, Hughes B, Bigras E, Shoubridge E, Hekimi S. 2005. Evolutionary conservation of the clk-1-dependent mechanism of longevity: loss of mclk1 increases cellular fitness and lifespan in mice. Genes and Development 19 2424–2434. ( 10.1101/gad.1352905) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Otin C, Galluzzi L, Freije JMP, Madeo F, Kroemer G. 2016. Metabolic control of longevity. Cell 166 802–821. ( 10.1016/j.cell.2016.07.031) [DOI] [PubMed] [Google Scholar]
- Lv Y, Zhang SY, Liang X, Zhang H, Xu Z, Liu B, Xu MJ, Jiang C, Shang J, Wang X. 2016. Adrenomedullin 2 enhances beiging in white adipose tissue directly in an adipocyte-autonomous manner and indirectly through activation of M2 macrophages. Journal of Biological Chemistry 291 23390–23402. ( 10.1074/jbc.M116.735563) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinus RD, Garth GP, Webster TL, Cartwright P, Naylor DJ, Hoj PB, Hoogenraad NJ. 1996. Selective induction of mitochondrial chaperones in response to loss of the mitochondrial genome. European Journal of Biochemistry 240 98–103. ( 10.1111/j.1432-1033.1996.0098h.x) [DOI] [PubMed] [Google Scholar]
- Melber A, Haynes CM. 2018. UPR(mt) regulation and output: a stress response mediated by mitochondrial-nuclear communication. Cell Research 28 281–295. ( 10.1038/cr.2018.16) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkwirth C, Langer T. 2009. Prohibitin function within mitochondria: essential roles for cell proliferation and cristae morphogenesis. Biochimica et Biophysica Acta 1793 27–32. ( 10.1016/j.bbamcr.2008.05.013) [DOI] [PubMed] [Google Scholar]
- Mootha VK, Bunkenborg J, Olsen JV, Hjerrild M, Wisniewski JR, Stahl E, Bolouri MS, Ray HN, Sihag S, Kamal M, et al 2003a. Integrated analysis of protein composition, tissue diversity, and gene regulation in mouse mitochondria. Cell 115 629–640. ( 10.1016/S0092-8674(03)00926-7) [DOI] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, et al 2003b. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nature Genetics 34 267–273. ( 10.1038/ng1180) [DOI] [PubMed] [Google Scholar]
- Moullan N, Mouchiroud L, Wang X, Ryu D, Williams EG, Mottis A, Jovaisaite V, Frochaux MV, Quiros PM, Deplancke B, et al 2015. Tetracyclines disturb mitochondrial function across eukaryotic models: a call for caution in biomedical research. Cell Reports pii: S2211-1247(15)00180-1. ( 10.1016/j.celrep.2015.02.034) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munch C, Harper JW. 2016. Mitochondrial unfolded protein response controls matrix pre-RNA processing and translation. Nature 534 710–713. ( 10.1038/nature18302) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair KS, Bigelow ML, Asmann YW, Chow LS, Coenen-Schimke JM, Klaus KA, Guo ZK, Sreekumar R, Irving BA. 2008. Asian Indians have enhanced skeletal muscle mitochondrial capacity to produce ATP in association with severe insulin resistance. Diabetes 57 1166–1175. ( 10.2337/db07-1556) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nargund AM, Pellegrino MW, Fiorese CJ, Baker BM, Haynes CM. 2012. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 337 587–590. ( 10.1126/science.1223560) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nargund AM, Fiorese CJ, Pellegrino MW, Deng P, Haynes CM. 2015. Mitochondrial and nuclear accumulation of the transcription factor ATFS-1 promotes OXPHOS recovery during the UPR(mt). Molecular Cell 58 123–133. ( 10.1016/j.molcel.2015.02.008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo JK, Pomatto LC, Bota DA, Koop AL, Davies KJ. 2011. Impairment of lon-induced protection against the accumulation of oxidized proteins in senescent wi-38 fibroblasts. Journals of Gerontology: Series A, Biological Sciences and Medical Sciences 66 1178–1185. ( 10.1093/gerona/glr145) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oike Y, Akao M, Yasunaga K, Yamauchi T, Morisada T, Ito Y, Urano T, Kimura Y, Kubota Y, Maekawa H, et al 2005. Angiopoietin-related growth factor antagonizes obesity and insulin resistance. Nature Medicine 11 400–408. ( 10.1038/nm1214) [DOI] [PubMed] [Google Scholar]
- Owusu-Ansah E, Song W, Perrimon N. 2013. Muscle mitohormesis promotes longevity via systemic repression of insulin signaling. Cell 155 699–712. ( 10.1016/j.cell.2013.09.021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, Walford GA, Sugiana C, Boneh A, Chen WK, et al 2008. A mitochondrial protein compendium elucidates complex I disease biology. Cell 134 112–123. ( 10.1016/j.cell.2008.06.016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papa L, Germain D. 2014. SirT3 regulates the mitochondrial unfolded protein response. Molecular and Cellular Biology 34 699–710. ( 10.1128/MCB.01337-13) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patti ME, Corvera S. 2010. The role of mitochondria in the pathogenesis of type 2 diabetes. Endocrine Reviews 31 364–395. ( 10.1210/er.2009-0027) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penefsky HS, Pullman ME, Datta A, Racker E. 1960. Partial resolution of the enzymes catalyzing oxidative phosphorylation. II. Participation of a soluble adenosine tolphosphatase in oxidative phosphorylation. Journal of Biological Chemistry 235 3330–3336. [PubMed] [Google Scholar]
- Perks KL, Ferreira N, Richman TR, Ermer JA, Kuznetsova I, Shearwood AJ, Lee RG, Viola HM, Johnstone VPA, Matthews V, et al 2017. Adult-onset obesity is triggered by impaired mitochondrial gene expression. Science Advances 3 e1700677 ( 10.1126/sciadv.1700677) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. 2004. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. New England Journal of Medicine 350 664–671. ( 10.1056/NEJMoa031314) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pospisilik JA, Knauf C, Joza N, Benit P, Orthofer M, Cani PD, Ebersberger I, Nakashima T, Sarao R, Neely G, et al 2007. Targeted deletion of AIF decreases mitochondrial oxidative phosphorylation and protects from obesity and diabetes. Cell 131 476–491. ( 10.1016/j.cell.2007.08.047) [DOI] [PubMed] [Google Scholar]
- Pullman ME, Penefsky HS, Datta A, Racker E. 1960. Partial resolution of the enzymes catalyzing oxidative phosphorylation. I. Purification and properties of soluble dinitrophenol-stimulated adenosine triphosphatase. Journal of Biological Chemistry 235 3322–3329. [PubMed] [Google Scholar]
- Quiros PM, Ramsay AJ, Sala D, Fernandez-Vizarra E, Rodriguez F, Peinado JR, Fernandez-Garcia MS, Vega JA, Enriquez JA, Zorzano A, et al 2012. Loss of mitochondrial protease OMA1 alters processing of the GTPase OPA1 and causes obesity and defective thermogenesis in mice. EMBO Journal 31 2117–2133. ( 10.1038/emboj.2012.70) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiros PM, Prado MA, Zamboni N, D’Amico D, Williams RW, Finley D, Gygi SP, Auwerx J. 2017. Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. Journal of Cell Biology 216 2027–2045. ( 10.1083/jcb.201702058) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qureshi MA, Haynes CM, Pellegrino MW. 2017. The mitochondrial unfolded protein response: Signaling from the powerhouse. Journal of Biological Chemistry 292 13500–13506. ( 10.1074/jbc.R117.791061) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabilloud T, Kieffer S, Procaccio V, Louwagie M, Courchesne PL, Patterson SD, Martinez P, Garin J, Lunardi J. 1998. Two-dimensional electrophoresis of human placental mitochondria and protein identification by mass spectrometry: toward a human mitochondrial proteome. Electrophoresis 19 1006–1014. ( 10.1002/elps.1150190616) [DOI] [PubMed] [Google Scholar]
- Rani V, Deep G, Singh RK, Palle K, Yadav UC. 2016. Oxidative stress and metabolic disorders: pathogenesis and therapeutic strategies. Life Sciences 148 183–193. ( 10.1016/j.lfs.2016.02.002) [DOI] [PubMed] [Google Scholar]
- Rhee HW, Zou P, Udeshi ND, Martell JD, Mootha VK, Carr SA, Ting AY. 2013. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science 339 1328–1331. ( 10.1126/science.1230593) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter U, Lahtinen T, Marttinen P, Suomi F, Battersby BJ. 2015. Quality control of mitochondrial protein synthesis is required for membrane integrity and cell fitness. Journal of Cell Biology 211 373–389. ( 10.1083/jcb.201504062) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ristow M, Zarse K, Oberbach A, Kloting N, Birringer M, Kiehntopf M, Stumvoll M, Kahn CR, Bluher M. 2009. Antioxidants prevent health-promoting effects of physical exercise in humans. PNAS 106 8665–8670. ( 10.1073/pnas.0903485106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan MT, Herd SM, Sberna G, Samuel MM, Hoogenraad NJ, Hoj PB. 1997. The genes encoding mammalian chaperonin 60 and chaperonin 10 are linked head-to-head and share a bidirectional promoter. Gene 196 9–17. ( 10.1016/S0378-1119(97)00111-X) [DOI] [PubMed] [Google Scholar]
- Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M, Ristow M. 2007. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metabolism 6 280–293. ( 10.1016/j.cmet.2007.08.011) [DOI] [PubMed] [Google Scholar]
- Seiferling D, Szczepanowska K, Becker C, Senft K, Hermans S, Maiti P, Konig T, Kukat A, Trifunovic A. 2016. Loss of CLPP alleviates mitochondrial cardiomyopathy without affecting the mammalian UPRmt. EMBO Reports 17 953–964. ( 10.15252/embr.201642077) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao LW, Niu R, Liu Y. 2016. Neuropeptide signals cell non-autonomous mitochondrial unfolded protein response. Cell Research 26 1182–1196. ( 10.1038/cr.2016.118) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sickmann A, Reinders J, Wagner Y, Joppich C, Zahedi R, Meyer HE, Schonfisch B, Perschil I, Chacinska A, Guiard B, et al 2003. The proteome of Saccharomyces cerevisiae mitochondria. PNAS 100 13207–13212. ( 10.1073/pnas.2135385100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva JP, Kohler M, Graff C, Oldfors A, Magnuson MA, Berggren PO, Larsson NG. 2000. Impaired insulin secretion and beta-cell loss in tissue-specific knockout mice with mitochondrial diabetes. Nature Genetics 26 336–340. ( 10.1038/81649) [DOI] [PubMed] [Google Scholar]
- Sparks LM, Xie H, Koza RA, Mynatt R, Hulver MW, Bray GA, Smith SR. 2005. A high-fat diet coordinately downregulates genes required for mitochondrial oxidative phosphorylation in skeletal muscle. Diabetes 54 1926–1933. ( 10.2337/diabetes.54.7.1926) [DOI] [PubMed] [Google Scholar]
- Sreekumar R, Halvatsiotis P, Schimke JC, Nair KS. 2002. Gene expression profile in skeletal muscle of type 2 diabetes and the effect of insulin treatment. Diabetes 51 1913–1920. ( 10.2337/diabetes.51.6.1913) [DOI] [PubMed] [Google Scholar]
- Suomalainen A, Elo JM, Pietilainen KH, Hakonen AH, Sevastianova K, Korpela M, Isohanni P, Marjavaara SK, Tyni T, Kiuru-Enari S, et al 2011. FGF-21 as a biomarker for muscle-manifesting mitochondrial respiratory chain deficiencies: a diagnostic study. Lancet Neurology 10 806–818. ( 10.1016/S1474-4422(11)70155-7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczepanowska K, Maiti P, Kukat A, Hofsetz E, Nolte H, Senft K, Becker C, Ruzzenente B, Hornig-Do HT, Wibom R, et al 2016. CLPP coordinates mitoribosomal assembly through the regulation of ERAL1 levels. EMBO Journal 35 2566–2583. ( 10.15252/embj.201694253) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SW, Fahy E, Zhang B, Glenn GM, Warnock DE, Wiley S, Murphy AN, Gaucher SP, Capaldi RA, Gibson BW, et al 2003. Characterization of the human heart mitochondrial proteome. Nature Biotechnology 21 281–286. ( 10.1038/nbt793) [DOI] [PubMed] [Google Scholar]
- van Dijk SJ, Feskens EJ, Bos MB, de Groot LC, de Vries JH, Muller M, Afman LA. 2012. Consumption of a high monounsaturated fat diet reduces oxidative phosphorylation gene expression in peripheral blood mononuclear cells of abdominally overweight men and women. Journal of Nutrition 142 1219–1225. ( 10.3945/jn.111.155283) [DOI] [PubMed] [Google Scholar]
- Vandanmagsar B, Warfel JD, Wicks SE, Ghosh S, Salbaum JM, Burk D, Dubuisson OS, Mendoza TM, Zhang J, Noland RC, et al 2016. Impaired mitochondrial fat oxidation induces FGF21 in muscle. Cell Reports 15 1686–1699. ( 10.1016/j.celrep.2016.04.057) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wai T, Garcia-Prieto J, Baker MJ, Merkwirth C, Benit P, Rustin P, Ruperez FJ, Barbas C, Ibanez B, Langer T. 2015. Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science 350 aad0116 ( 10.1126/science.aad0116) [DOI] [PubMed] [Google Scholar]
- Walder K, Kerr-Bayles L, Civitarese A, Jowett J, Curran J, Elliott K, Trevaskis J, Bishara N, Zimmet P, Mandarino L, et al 2005. The mitochondrial rhomboid protease PSARL is a new candidate gene for type 2 diabetes. Diabetologia 48 459–468. ( 10.1007/s00125-005-1675-9) [DOI] [PubMed] [Google Scholar]
- Wall CE, Whyte J, Suh JM, Fan W, Collins B, Liddle C, Yu RT, Atkins AR, Naviaux JC, Li K, et al 2015. High-fat diet and FGF21 cooperatively promote aerobic thermogenesis in mtDNA mutator mice. PNAS 112 8714–8719. ( 10.1073/pnas.1509930112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wikstrom M, Krab K. 1979. Proton-pumping cytochrome c oxidase. Biochimica et Biophysica Acta 549 177–122. ( 10.1016/0304-4173(79)90014-4) [DOI] [PubMed] [Google Scholar]
- Wu F, Wang B, Zhang S, Shi L, Wang Y, Xiong R, Pan X, Gong F, Li X, Lin Z. 2017. FGF21 ameliorates diabetic cardiomyopathy by activating the AMPK-paraoxonase 1 signaling axis in mice. Clinical Science 131 1877–1893. ( 10.1042/CS20170271) [DOI] [PubMed] [Google Scholar]
- Yang W, Hekimi S. 2010. A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PLoS Biology 8 e1000556 ( 10.1371/journal.pbio.1000556) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun J, Finkel T. 2014. Mitohormesis. Cell Metabolism 19 757–766. ( 10.1016/j.cmet.2014.01.011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SY, Lv Y, Zhang H, Gao S, Wang T, Feng J, Wang Y, Liu G, Xu MJ, Wang X, et al 2016. Adrenomedullin 2 improves early obesity-induced adipose insulin resistance by inhibiting the class II MHC in adipocytes. Diabetes 65 2342–2355. ( 10.2337/db15-1626) [DOI] [PubMed] [Google Scholar]
- Zhao Q, Wang J, Levichkin IV, Stasinopoulos S, Ryan MT, Hoogenraad NJ. 2002. A mitochondrial specific stress response in mammalian cells. EMBO Journal 21 4411–4419. ( 10.1093/emboj/cdf445) [DOI] [PMC free article] [PubMed] [Google Scholar]