

Abstract
Most older individuals develop inflammageing, a condition characterized by elevated levels of blood inflammatory markers that carries high susceptibility to chronic morbidity, disability, frailty, and premature death. Potential mechanisms of inflammageing include genetic susceptibility, central obesity, increased gut permeability, changes to microbiota composition, cellular senescence, NLRP3 inflammasome activation, oxidative stress caused by dysfunctional mitochondria, immune cell dysregulation, and chronic infections. Inflammageing is a risk factor for cardiovascular diseases (CVDs), and clinical trials suggest that this association is causal. Inflammageing is also a risk factor for chronic kidney disease, diabetes mellitus, cancer, depression, dementia, and sarcopenia, but whether modulating inflammation beneficially affects the clinical course of non-CVD health problems is controversial. This uncertainty is an important issue to address because older patients with CVD are often affected by multimorbidity and frailty — which affect clinical manifestations, prognosis, and response to treatment — and are associated with inflammation by mechanisms similar to those in CVD. The hypothesis that inflammation affects CVD, multimorbidity, and frailty by inhibiting growth factors, increasing catabolism, and interfering with homeostatic signalling is supported by mechanistic studies but requires confirmation in humans. Whether early modulation of inflammageing prevents or delays the onset of cardiovascular frailty should be tested in clinical trials.
With the extension of life expectancy and the rising percentage of older individuals in the general population, understanding why ageing results in progressively higher susceptibility to chronic morbidity, disability, and frailty has become a public health priority1. An interesting hypothesis stems from the observation that older organisms tend to develop a pro-inflammatory status that is characterized by high levels of pro-inflammatory markers in cells and tissues, a condition often named inflammageing, a term first coined in 2000 by Claudio Franceschi2. Strong evidence indicates that inflammageing is a risk factor for cardiovascular disease (CVD), in addition to many age-associated chronic diseases and other adverse health outcomes. Whether inflammageing contributes causally to CVD and other comorbid conditions or is instead a noncausal marker of some other underlying mechanisms is still debated. Other than calorie restriction and physical activity, treatment options for inflammageing rely on small molecules or antibodies that interfere with inflammatory mediators or their biological targets rather than targeting the underlying causes, resulting in highly heterogeneous efficacy.
In this Review, we summarize the current understanding of inflammageing. We explore risk factors and speculate on potential causes, and we look at possible roles of inflammageing in CVD and other conditions that are highly prevalent and often coexist with CVDs in older individuals. We report on findings from intervention studies aimed at modulating inflammation in different diseases, and in particular whether these interventions prevent or attenuate the clinical course of CVD. We continue by examining the role of inflammation in conditions that are typical of ageing and often comorbid with CVD, such as multimorbidity, sarcopenia, and frailty. Finally, we identify gaps in our knowledge and suggest priorities for future research.
Risk factors and causes of inflammageing
Ageing is associated with immune dysregulation, of which the most evident characteristics are high blood levels of pro-inflammatory immunogenic stimulations3,4. The pro-inflammatory state is characterized by high circulating levels of pro-inflammatory markers, including IL-1, IL-1 receptor antagonist protein (IL-1RN), IL-6, IL-8, IL-13, IL-18, C-reactive protein (CRP), IFNα and IFNβ, transforming growth factor-β (TGFβ), tumour necrosis factor (TNF) and its soluble receptors (TNF receptor superfamily members 1A and 1B), and serum amyloid A. At this time, a comprehensive list of pro-inflammatory markers that are associated with ageing has not been compiled owing to the difficulty of applying high-sensitivity discovery proteomics in plasma and serum. High levels of age-associated pro-inflammatory markers are detected in the majority of older individuals, even in the absence of risk factors and clinically active diseases3,5–7. Despite its fundamental physiological role as a defence mechanism against infections or extraneous molecules, when inflammation becomes sustained and prolonged it becomes detrimental to health. According to the antagonistic pleiotropy theory of ageing, inflammation might have been evolutionarily selected because of beneficial effects early in life and in adulthood, although it becomes detrimental in old age when the effect of natural selection is no longer active8. Epidemiological studies have found that inflammageing is a risk factor for CVD, cancer, chronic kidney disease, dementia, and depression as well for global indicators of poor health status, such as multimorbidity, mobility disability and disability in activities of daily living, sarcopenia, frailty, and premature death9–19. On the basis of these findings, many researchers have proposed that inflammageing is a marker of accelerated ageing and should be considered to be one of the pillars of the biology of ageing20. The root causes of inflammageing are poorly understood, as are the mechanisms that connect inflammageing with CVD and with many other health outcomes. A critical question is whether inflammation directly causes the associated pathology or is instead a biomarker for the rate of biological ageing. The answer to this question might depend on the age of the patients and whether we consider CVD by itself or CVD in the context of associated multimorbidity, impairments, and disabilities.
Genetic susceptibility.
Studies in large populations have identified a multitude of genetic variants that affect blood levels of inflammatory mediators21. We focus on associations that have been confirmed by multiple studies and are functionally relevant21–25. Examining common variants of the IL1RN gene revealed that the rs4251961 minor allele is associated with a lowered serum level of IL-1RN and that the rs579543 single nucleotide polymorphism (SNP) is also independently associated with IL-1RN levels, whereas the IL1RN 1018 haplotype correlates with higher concentrations of IL-1β and IFNγ22. These findings have been confirmed in three independent cohorts26, and further research has demonstrated that these factors affect the pathophysiology of human infections27 and the risk of developing insulin resistance28 and knee osteoarthritis29.
A SNP in the promoter region of IL6 at position−174G > C magnifies IL-6 production in response to inflammatory stimuli, but this SNP has been associated inconsistently with baseline IL-6 levels. Carriers of the−174G > C mutation have an increased risk of developing various major diseases, including Alzheimer disease30, CVD31, non-insulin-dependent diabetes mellitus32, bone fragility33, and systemic-onset juvenile chronic arthritis34. A genome-wide association study comparing >2,000 Chinese centenarians to middle-aged controls found that the SNP rs2069837 in IL6 was significantly associated with extreme longevity, confirming the role of IL-6 in conditioning morbidity and mortality, especially in old age35. Confirming the role of IL-6 in health, in a large Mendelian randomization analysis, the IL6R SNP rs7529229, marking a non-synonymous IL6R variant (rs8192284; p.Asp358Ala), was associated with increased circulating IL-6 levels24. Variants in the IL6R gene have been found to be associated with increased risk of coronary artery disease24, rheumatoid arthritis, atrial fibrillation, and abdominal aortic aneurysm, and with increased susceptibility to asthma, type 1 diabetes, and depression36,37. Multiple SNPs in the CRP gene are associated with higher CRP levels and increased risk of myocardial infarction and CVD-related death38.
These data indicate that genetic variability affects the plasma levels of several inflammatory markers and, through this mechanism, increases the risk of many apparently uncorrelated diseases. Therefore, the cumulative effect of these genetic polymorphisms might be a risk factor for multimorbidity and frailty, although this hypothesis has never been fully tested. A gene-expression study conducted on whole-blood RNA samples from a large population cohort in Europe and the USA revealed that immune response and inflammation were the most highly upregulated pathways in association with ageing39. A few gene transcripts mediate the age–IL-6 association, among which the largest affected transcript, SLC4A10 mRNA (encoding the sodium-driven chloride bicarbonate exchanger), explains as much as 19% of this association40. Interestingly, this study did not detect an age-related increase in IL6 mRNA transcript levels, suggesting that the overproduction of circulating proteins occurs in peripheral tissues rather than in blood cells.
Accumulating evidence shows that cellular changes that contribute to inflammageing are mediated by microRNAs (miRNAs), which are non-coding, single-stranded RNAs spanning 17–25 nucleotides that generally modulate protein-expression programmes by interacting with mRNAs that share partial complementarity, thereby reducing mRNA stability and/or translation41. Studies have shown age-related differences in the abundance of specific miRNAs in circulating cells, plasma, and whole blood from older compared with younger individuals42–48. Findings from these studies are inconsistent, possibly owing to differences in sample size, age composition, and health status of the examined individuals, and because miRNA detection methods vary widely in specificity, accuracy, and sensitivity. In addition, given that miRNAs mostly function as intra-cellular modulators of mRNAs, their concentration in whole blood might be a poor indicator of their physiological effects. Nonetheless, miR-25–3p, miR-92a-3p, miR-93–5p, miR-101–3p, miR-106b-5p, miR-142–5p, miR-151a-3p, and miR-181a-5p tend to be under-represented, whereas miR-21–5p and miR-126–3p are over-represented at older ages47,49,50. Age-related changes in miRNAs have been suggested to contribute to inflammageing. For example, miR-126–3p inhibits endothelial inflammation, and low levels of miR-126–3p were found in patients with CVD and diabetes50, whereas miR-21–5p levels are correlated negatively with CRP and fibrinogen levels, and miR-21–5p levels are higher in patients with CVD than in age-matched controls44. Other miRNAs, such as miR-146 and miR-155, might also have a role in inflammageing by affecting cellular senescence or modulating immune responses, although these activities might not be reflected by changes in miRNA concentration or these miRNAs might be detected only in exosomes or other structures carrying miRNAs51. Overall, the contribution of miRNAs to inflammageing is an active area of investigation with high translational potential.
Visceral obesity.
Epidemiological studies provide some information about the origin of inflammageing. Obesity — particularly central obesity — is strongly associated with a pro-inflammatory state52–54. Adipocytes in abdominal, intramuscular, liver, and pericardial fat can produce pro-inflammatory and chemotactic compounds, such as IL-6, IL-1β, TNF, and C-C motif chemokine 2 (CCL2), as well as hormones that modulate inflammation, such as adiponectin and leptin54. The visceral fat tissue of obese individuals is infiltrated by T lymphocytes, macrophages, and monocytes. T lym-phocytes secrete IFNγ, which stimulates the production of several chemokines from adipocytes, including CCL2, CCL5, C-X-C motif chemokine 9 (CXCL9), and CXCL10, which further amplify tissue T cell migration. The number of B lymphocytes and macrophages in visceral adipose tissue from obese individuals is also increased and is correlated with BMI55. Studies with animal models suggest that a specific subset of B cells expressing the TNF ligand superfamily member 9 and producing TNF, IFNγ, and granzyme B is increased in the peritoneal cavity during ageing56. Cytokines released by B cells contribute to the phenotypic switch of adipocytes in the visceral cavity, causing them to release adipokines, other pro-inflammatory markers, and cell debris52. Activated monocytes that give rise to M1 and M2 macrophages produce even more inflammatory compounds that probably appear in the circulation57. Weight loss through reduced dietary intake and possibly bariatric surgery is associated with reductions in primary pro-inflammatory markers, in part owing to normalized expression of inflammation-related genes in white adipose tissue and to downregulation of the NLRP3 inflammasome58–61. In addition, calorie restriction in humans is associated with a substantial reduction in pro-inflammatory markers in the blood62. Weight loss combined with exercise improves functional status and reduces some of the features of frailty in obese older individuals, improves the cardiovascular risk profile, and reduces the risk of CVD, although whether these beneficial effects are caused by reduced inflammation remains unclear63–65.
Microbiota and gut permeability.
A new hypothesis on the origin of inflammation highlights changes that occur in the gut microbiota with ageing as well as age- associated changes in gut permeability. Despite large variability in the gut microbiota found in different populations, geographic regions, and settings, evidence suggests that ageing is associated with a reduction in beneficial commensal microorganisms — such as Coprococcus, Faecalibacterium, and Lactobacillus — as well as a decrease in the Firmicutes: Bacteroidetes ratio66–68. The disappearance of these microorganisms is important because they normally counteract the expansion of pathogenic microbial communities, while also maintaining intestinal barrier integrity by fermenting starches and dietary fibres and producing mucus and lipid metabolites, such as short-chain fatty acids (primarily acetate, propionate, and butyrate)66–68. As beneficial intestinal bacteria decrease in abundance with ageing, other bacteria increase in relative abundance, including symbiotic bacteria that can become pathogenic under inflamed conditions, often termed pathobionts69. This category of microorganisms is enriched in the gut of older adults and is primarily dominated by facultative anaerobes — such as Fusobacterium and Staphylococcus — a state that has been associated with increased levels of inflammatory cytokines in plasma8,70.
Increased gut dysbiosis has been postulated to increase mucosal barrier permeability, thereby allowing bacteria and their products — including pathogen-associated molecular patterns (PAMPs), damage-associated molecular patterns (DAMPs), and microbial-associated molecular patterns (MAMPs)— into the circulatory system. Together, these factors contribute to a chronic pro-inflammatory state71. This theory is supported by studies in animal models, but no definitive evidence exists of increased gut permeability and leakage of pro-inflammatory products in older individuals who are free from overt inflammatory disease72. Dysbiosis seems to be more severe in conditions in which prevalence increases with ageing, such as obesity and type 2 diabetes73. Of note, changes in gut microbiota composition have been shown to be associated with increased frailty74–76, which could be owing to gut dysbiosis-induced inflammation. Centenarians, who can be considered extreme examples of healthy ageing, have an enrichment of Akkermansia, Bifidobacterium, and Christensenellaceae in their intestinal flora67, which promote positive immune function, have anti-inflammatory activity, diminish the effects of obesity, and contribute to metabolic homeostasis77–79.
Consistent with the notion that changes in the gut microbiota composition can affect healthy ageing, calorie restriction — the most powerful strategy to increase longevity in animal models — causes changes in microbiota composition, decreases inflammation, and improves gut barrier integrity80. A healthy intestinal tract flora can theoretically be promoted by the administration of probiotics, prebiotics, or a combination of the two81–83. Some studies have shown that this strategy can reduce systemic inflammation and progression of central obesity, but more research in this area is needed to substantiate this initial evidence and to assess whether the reduction of inflammation owing to microbiota changes has beneficial effects on health84,85.
Cellular senescence.
A number of biological mechanisms that have been identified as hallmarks or pillars of biological ageing might account for inflammageing86,87. Paramount among them is the accumulation of senescent cells in multiple tissues. Cellular senescence is generally considered to be a pre-encoded cancer suppressor mechanism characterized by cell cycle arrest, loss of proliferation capacity, global cell enlargement, characteristic misshaped nuclei, presence of chromatin foci with persistent DNA damage response, increased nuclear factor-κB (NF-κB) signalling, and resistance to apoptosis88,89. Senescent cells are recognized by specific markers, including cyclin-dependent kinase inhibitor 2A (commonly known as p16INK4A) and increased lysosome β-galactosidase activity, although these markers are neither fully sensitive nor specific and, despite intense research, no gold-standard biomarker of cellular senescence has been established90. Jeck and colleagues hypothesized that genetic variants associated with general susceptibility to multiple diseases are enriched in specific areas of the genome. Interestingly, SNPs located near regulators of senescence and inflammation are particularly associated with diseases of ageing, such as cancer, CVD, and type 2 diabetes, and the strongest association was found with a variant in the CDKN2A gene, which encodes the p16INK4A protein that is over-expressed in many forms of senescence. These findings were replicated in a meta-analysis that included 410 genome-wide association studies91,92. In addition, the variant rs2811712 that is close to the CDKN2A gene was associated with poor physical function in two different cohorts93. Therefore, senescence seems to be associated with ageing, inflammation, CVD, and impaired physical function in older individuals, making cell senescence a strong candidate as a mechanism for inflammageing.
Cell senescence can be triggered by many stimuli, including critical telomere shortening, persistent DNA damage, oncogene activation or inactivation, epigenetic alterations, mitochondrial dysfunction, and exposure to DAMPs that are released by stressed cells, with some evidence that the phenotypic manifestations induced by different triggers are heterogeneous88,89. At the core of the replication arrest is increasing levels of cyclin-dependent kinase inhibitors that block the phosphorylation of the retinoblastoma-associated protein and initiate cell cycle arrest. In adulthood, activation of retinoblastoma-associated protein can occur either through the cellular tumour antigen p53 that activates cyclin-dependent kinase inhibitor 1 (commonly known as p21), or directly through the activation of p16INK4A. Theories suggest that senescence is not an acute switch but instead evolves in stages, from a temporary or reversible status to a chronic irreversible condition94.
Relevant to inflammageing, senescent cells acquire a senescence-associated secretory phenotype (SASP) that involves the secretion of a wide range of soluble molecules. The list of these molecules is not comprehensive, and the molecules can vary on the basis of cell type and triggering factors but usually include interleukins (IL-1α, IL-1β, and IL-6), chemokines (IL-8 and growth-regulated-α protein), growth factors (fibroblast growth factor 2 and hepatocyte growth factor), metalloproteinases (interstitial collagenase (also known as MMP1), stromelysin 1 (also known as MMP3), and collagenase 3 (also known as MMP13)), and other insoluble proteins and extracellular matrix components95. These secretory molecules mainly function in a paracrine fashion and can facilitate the development of cellular senescence in neighbouring cells, but some of the soluble mediators are released into the circulation and are likely to contribute to inflammageing96.
Studies have shown that senescent cells accumulate exponentially with ageing in different organs and tissues, both in model organisms and in humans97–100. In humans, the accumulation of senescent markers has been demonstrated in the skin, T lymphocytes, atherosclerotic lesions, insulin-producing β cells, kidney, endothelium, visceral fat, joint cartilage, cardiac muscle, liver, and many others tissues98,101–107. Some tissues are likely to have a greater propensity to developing cellular senescence than others, but research in this area is scarce. Of note, senescent T cell accumulation has been demonstrated in patients with chronic infections such as Cytomegalovirus (CMV) or human immunodeficiency virus (HIV) infection, which might explain why patients with CMV or HIV infection have chronically elevated levels of pro-inflammatory markers and reduced vaccine efficacy108–110. In mice, the clearance of p16INK4A-positive cells extends lifespan and slows the emergence of ageing phenotypes and age-related functional deterioration of organs and tissues111,112. The extent to which the burden of senescent-cell accumulation in humans is associated with inflammageing and organ damage, and whether a plasma protein signature can be developed that correlates with cell senescence burden, are important areas of investigation.
Impaired recycling and elimination of degraded cellular material.
Despite the apparent stability of the human body, a massive turnover of molecules, microorganelles, cells, and cellular components occurs constantly throughout life. A complex and well-regulated molecular machinery constantly surveys cellular components and handles the repair or elimination of biological debris as well as broken or misplaced fragments. Within cells, worn-out macromolecules and organelles are physiologically recycled by proteasome degradation or autophagy. Extracellular debris is recognized by the immune system through different receptors, including pattern recognition receptors, and is then degraded by engulfment in phagocytic vesicles113. Under pathological conditions, molecules are released by stressed cells undergoing necrosis (such as during ischaemia– reperfusion or severe infection). These molecules, called DAMPs, include reactive oxygen species (ROS) from damaged and unrecycled mitochondria, extra-cellular nucleotides such as ATP, oxidized cardiolipin, free nuclear and mitochondrial DNA fragments or histones, high-mobility group protein B1, oxidized LDL, amyloid-β, islet amyloid polypeptide, and particulates such as monosodium urate and cholesterol crystals, in addition to many others114. If notpromptly removed, these molecules accumulate and possibly contribute to inflammageing115,116. Accordingly, inflammageing is proposed to originate from an imbalance between the production and disposal of cellular debris, misfolded proteins, and/or misplaced self-molecules that develops with age8. For example, accumulation of DAMPs is sensed by the NLRP3 inflammasome and causes NLRP3 oligomerization, resulting in caspase 1-dependent secretion of the inflammatory cytokines IL-1β and IL-18. In humans, IL-18 blood levels increase with ageing, and strong evidence from mouse studies indicates that blockade of the NLRP3 inflammasome extends healthspan, attenuating multiple age-related degenerative changes that have been linked to inflammageing, including insulin resistance, thymic involution, T cell senescence, and bone loss as well as physical and cognitive function decline114,117,118. Of note, ROS produced by dysfunctional mitochondria can also trigger an inflammatory response by activating the NF-κB signalling pathway119.
Intrinsic defects in immune cells and chronic infections.
Studies conducted in isolated immune cells, mostly lymphocytes, suggest that an intrinsic defect in immune cells also contributes to inflammageing. For example, gene-expression studies show that CD4+ lymphocytes from older individuals have higher intrinsic activation of the NF-κB pathways than those from younger individuals120. After stimulation with anti-CD3, the production of pro-inflammatory cytokines in vitro is lower in cells from older individuals than in cells from younger individuals120, a phenomenon that might be related to altered metabolic activity121. However, because these studies have been performed only in small populations, their relevance to inflammageing is unknown.
Subclinical and clinically evident chronic infections can chronically stimulate immune function and result in changes in levels of inflammatory markers that are indistinguishable from those of the inflammageing signature. Particularly relevant for inflammageing are human CMV and HIV infections. Human CMV infection is herpesvirus present in a latent state in more than half of the adult population122, where intermittent transcription episodes cause antigen reactivation throughout the life course. This situation might explain why the human CMV-specific memory T cells can comprise up to 50% of the total memory T cell compartment in older individuals123,124 and leads to the hypothesis that persistent human CMV infection has a role in immunosenescence and inflammageing. However, evidence to support this hypothesis remains controversial109. Some studies have found that human CMV infection in older individuals is associated with increased cardiovascular and all-cause mortality, negative immune risk profile, inflammageing, and lower antibody responses to influenza125–129. However, evidence that the association between CMV infection and CVD and mortality is mediated by inflammageing is scant at best109,130. The theoretical CMV-associated impaired capacity to control heterologous infections in old age and the association with high circulating levels of pro-inflammatory cytokines have also been challenged109. Ultimately, whether human CMV infections cause accelerated immune senescence is controversial.
In the era of highly active antiretroviral therapy (HAART), patients with HIV infection have a life expectancy that is, on average, only slightly lower than in the general population. However, this therapy does not protect patients from the persistent immune activation, chronic inflammation, and excess risk of developing CVD and frailty131–135. Chronic inflammation in HIV is mediated by depletion of memory CD41+ T cells, resulting in increased permeability in the gut epithelium and translocation of microbial products into the circulation, which causes inflammation136. Patients with HIV are also affected by antiretroviral-associated lipodystrophy and visceral obesity that can further contribute to inflammation and cause insulin resistance137.
Finally, chronic infections, such as oral infection, asymptomatic chronic infection in the urinary and biliary tracts, and hidden intestinal infections, are associated with the release of PAMPs into the circulation, which elicits a persistent inflammatory state. Treatment of these infections can reduce inflammageing and potentially has many long-term beneficial effects beyond immediate local resolution of symptoms.
As explained above, the possible causes of inflammageing are numerous and very heterogeneous (Fig. 1). These different mechanisms are likely to be additive and interconnected, acting in different combinations and with different relevance in selected individuals. Therefore, effectively reducing inflammation without weakening the surveillance and defensive functions of the immune system requires individualized approaches as well as an accurate diagnosis of the underlying causes of inflammation.
Fig. 1 |. Potential causes of inflammageing.
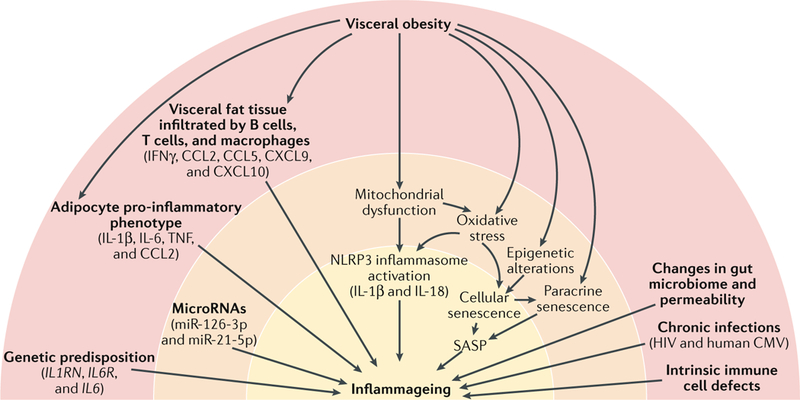
Several genetic variants associated with high levels of inflammatory markers or increased response to inflammatory stimuli have been identified; the most relevant factors are indicated in parentheses. In central obesity, visceral fat tissue is infiltrated by T cells, macrophages, and monocytes. T cells secrete IFNγ, which stimulates the production of several chemokines by adipocytes, including C-C motif chemokine 2 (CCL2), CCL5, C-X-C motif chemokine 9 (CXCL9), and CXCL10, which further amplify tissue T cell infiltration. The number of B cells and macrophages in visceral adipose tissue from obese individuals is also increased and is correlated with BMI55. A specific subset of B cells expressing the tumour necrosis factor (TNF) superfamily ligand superfamily member 9 and producing TNF, IFNγ, and granzyme B accumulates in the abdominal cavity of older individuals56. Cytokines released by B cells contribute to the phenotypic change of adipocytes in the visceral cavity, causing them to release adipokines, other pro-inflammatory factors, and cell debris52. Activated monocytes that give rise to M1 and M2 macrophages produce even more inflammatory compounds57. Damaged mitochondria that cannot be repaired by repeated cycles of fission and fusion and are not recycled owing to defective autophagy release damage-associated molecular patterns (DAMPs) that trigger the NLRP3 inflammasome and lead to caspase 1-dependent production of IL-1β and IL-18. Oxidative stress is one of the possible triggers of cell senescence, which can be induced by several other stressors, including epigenetic alterations. Senescent cells, through the senescence-associated secretory phenotype (SASP), secrete large quantities of cytokines, chemokines, and other molecules, locally triggering more cell senescence (paracrine senescence) and contributing to inflammageing. Studies have emphasized the role of age-related changes in the microbiome and increases in the gut mucosa permeability that lead to bacterial product release into the blood and stimulate an inflammatory response, in part through the NLRP3 inflammasome. In addition, part of inflammageing is probably caused by chronic infections (for example, human immunodeficiency virus (HIV) or human Cytomegalovirus (CMV) infection) and intrinsic defective mechanisms in immune cells that might involve metabolic stress as well as age-related changes in microRNA transcription. Of note, other important triggers of cell senescence, such as genomic instability, the activation of oncogenes, and the inhibition of tumour-suppressor genes, are not shown in the figure but might be part of the same mechanism.
Consequences of inflammageing for CVD
Although strong epidemiological evidence indicates that inflammation is a powerful risk factor for CVD, non-cardiovascular comorbidities and conditions that are often associated with CVD, as well as with frailty, disability, and mortality in older individuals, and the mechanisms that underlie these associations have only just begun to be elucidated. Controversy exists on whether high levels of pro-inflammatory compounds in the circulation and tissues causally contribute to associated pathological conditions or whether inflammation is a reactive marker of underlying pathology. These two mechanisms are not mutually exclusive; for example, early damage that occurs during vascular endothelial cell inflammation participates in the pathogenesis of atherosclerotic plaques, whereas atherosclerosis itself produces antigens that trigger and sustain an inflammatory response, and senescent cells are found often in large quantities in atherosclerotic plaques. Therefore, multiple mechanisms amplify the role of inflammation in atherogenesis.
In this section of the Review, we summarize available evidence to suggest that chronic inflammation is both a risk factor and a pathogenic mechanism in CVD. Moreover, because inflammation also contributes to the pathogenesis of other chronic non-CVDs — such as anaemia, cancer, type 2 diabetes, dementia, osteoporosis, sarcopenia, chronic kidney disease, and depression — CVDs in old age often develop in the context of multimorbidity and frailty3,10,138–143 (Fig. 2). Epidemiological studies have produced insufficient evidence to demonstrate whether inflammation occurs in response to underlying disease pathologies or whether inflammation itself contributes to disease initiation and progression. To address this issue, we combine observational evidence with results from randomized, controlled trials (RCTs) that tested the efficacy of anti-inflammatory drugs in preventing or controlling CVD clinical manifestations (Table 1).
Fig. 2 |. Inflammageing is a risk factor for multiple chronic diseases.
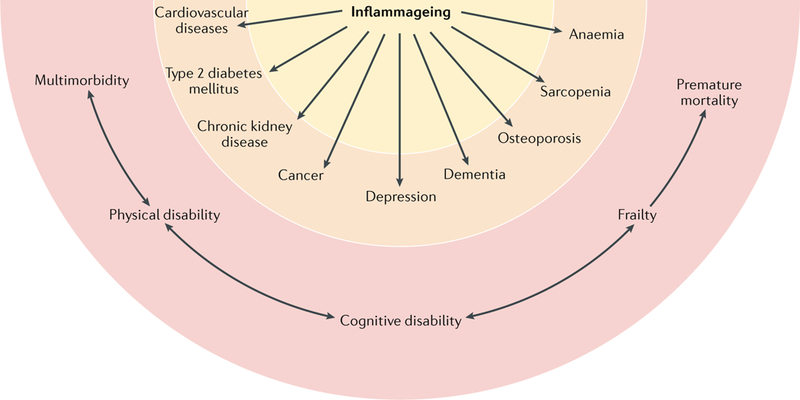
Inflammageing, defined as an age-related increase in the levels of pro-inflammatory markers in blood and tissues, is a strong risk factor for multiple diseases that are highly prevalent and frequent causes of disability in elderly individuals but are pathophysiologically uncorrelated. Mild chronic inflammation is generally considered to be a biomarker of accelerated biological ageing or one of the mechanisms by which the ageing process is associated with increased global susceptibility to all diseases. Cardiovascular diseases, chronic kidney disease, cancer, depression, dementia, osteoporosis, sarcopenia, and anaemia are shown in the figure as examples because extensive evidence indicates that inflammation contributes to the development of these diseases in old age, but the list is far from exhaustive3,138,139,142,143,196. Concordant with this view, elevated blood levels of pro-inflammatory markers (such as IL-6) are a powerful risk factor for multimorbidity (the number of coexisting diseases) and predict future rates of change in multimorbidity. Unsurprisingly, inflammageing is also a strong risk factor for typical geriatric conditions, such as physical and cognitive disability, frailty, and premature death. Although this effect is primarily mediated by multimorbidity, evidence also indicates that inflammation interferes with the maintenance and repair that constantly occur in all tissues, leading to accumulation of damage that contributes to frailty.
Table 1 |.
Clinical trials of anti-inflammatory drugs in chronic inflammatory diseases
| Trial name | Participants | Design | n | Drug | Dosage | Type of treatment |
Outcome | Result | Refs |
|---|---|---|---|---|---|---|---|---|---|
| CANTOS | Patients with previous MI and hsCRP ≥2mg/l |
Randomized | 10,061 | Canakinumab | 150 mg every 3 months |
Secondary prevention |
Cardiovascular events |
Beneficial | 169,293 |
| CIRT | Patients with previous MI and either T2DM or metabolic syndrome |
Randomized | 7,000 | Methotrexate | 15–20 mg weekly |
Secondary prevention |
Cardiovascular events |
Ongoing | 170 |
| LoDoCo | Patients with clinically stable CAD |
Randomized | 532 | Colchicine | 0.5 mg daily | Primary and Secondary prevention |
Cardiovascular events |
Beneficial | 172 |
| LoDoCo2 | Patients with clinically stable CAD |
Randomized | 3,000 | Colchicine | 0.5 mg daily | Primary and Secondary prevention |
Cardiovascular events |
Ongoing | 294 |
| COLCOT | Patients with a Documented acute MI in the past 30 days |
Randomized | 4,500 | Colchicine | 0.5 mg daily | Secondary prevention |
Cardiovascular events |
Ongoing | 173 |
| ENTRACTE | Patients with moderate- to-severe rheumatoid arthritis |
Randomized | 3,080 | Tocilizumab | 8mg/kg every 4 weeks |
Prevention | Cardiovascular events |
Ongoing | 166 |
| PEDRIAN | Patients with T2DM and stage 3–4 CKD |
Randomized | 169 | Pentoxifylline | 1,200 mg daily | Prevention | CKD progression |
Beneficial | 295 |
| NA | Patients aged ≥25 years with T1DM or T2DMa |
Randomized | 416 | Monoclonal anti-TGFβ1 antibody |
2, 10, or 50 mg Monthly (subcutaneous) |
Prevention | CKD progression |
Not beneficial |
296 |
| NA | Patients with a recent TIA or minor ischaemic stroke and no contraindication to aspirin |
Meta- analysis of two randomized trialsb |
5,139+ 2,449 |
Aspirin | 300, 500, or 1,200 mg daily |
Primary prevention |
Colorectal cancer |
Beneficial | 297 |
| NA | Patients with a recent TIA or minor ischaemic stroke and no contraindication to aspirin |
Meta- analysis of four randomized trialsc |
14,033 | Aspirin | 30, 75, 283, 300, 500, or 1,200 mg daily |
Primary prevention |
Colorectal cancer |
Beneficial | 298 |
| CANTOS | Patients with previous MI and hsCRP ≥2mg/l |
Randomized | 10,061 | Canakinumab | 150 or 300 mg every 3 months |
Primary prevention |
Lung cancer | Beneficial | 168 |
| NA | Patients with osteoarthritis |
Meta- Analysis of five randomized trialsd |
1,497 | Ibuprofen, naproxen, or celecoxib |
800 mg three times daily, 500 mg twice daily, or 200 mg daily |
Treatment | Depressive symptoms |
Beneficial | 299 |
| ADAPT | Individuals aged ≥70 years, cognitively healthy , and with a family history of AD-like dementia |
Randomized | 2,528 | Celecoxib or naproxen |
200 mg twice daily or 220 mg twice daily |
Treatment | Depressive symptoms |
Not beneficial |
300 |
| NA | Outpatients with major depression |
Randomized | 60 | Infliximab | 5mg/kg (three infusions) |
Treatment | Depressive symptoms |
Beneficial in patients with high baseline inflammatory blood biomarkers |
301 |
| NA | Patients with moderate-to- severe psoriasis |
Randomized | 96 | Adalimumab | 40 mg every other week |
Treatment | Depressive symptoms |
Beneficial | 302 |
| NA | Patients with moderate- to-severe psoriasis |
Randomized | 618 | Etanercept | 50 mg twice weekly |
Treatment | Depressive symptoms |
Beneficial | 303 |
| NA | Patients with probable AD |
Randomized | 40 | Nimesulide | 100 mg twice daily |
Treatment | AD | Not beneficial |
304 |
| NSAID study | Patients with mild-to-moderate AD |
Randomized | 351 | Rofecoxib or naproxen sodium |
25 mg once daily or 220 mg twice daily |
Treatment | AD | Not beneficial |
305 |
| NA | Patients with mild or moderate AD aged ≥50 years |
Randomized | 692 | Rofecoxib | 25 mg daily | Treatment | AD | Not beneficial |
306 |
| NA | Patients with mild-to-moderate AD |
Randomized | 41 | Diclofenac | 50 mg daily | Treatment | AD | Not beneficial |
307 |
| ADAPT | Individuals aged ≥70 years, cognitively healthy , and with a family history of AD |
Randomized | 2,117 | Celecoxib or naproxen |
200 mg twice daily or 220 mg twice daily |
Primary prevention |
AD | Not beneficial |
308 |
| TOMORROW | Cognitively healthy participants at high risk of developing MCI |
Randomized | 3,500 | Pioglitazone | 0.8 mg daily | Prevention | Onset of MI or MCI owing to AD |
Ongoing | 309,310 |
| Metformin for Preventing Frailty in High-risk Older Adults |
Older adults with impaired glucose tolerance |
Randomized | 120 | Metformin | 1,000 mg twice daily |
Prevention | Frailty | Ongoing | 256 |
| TAME | Individuals aged 65–79 years |
Randomized | 3,000 | Metformin | 850 mg twice daily |
Prevention | Cardiovascular events, cancer, dementia, and mortality |
Ongoing | 277 |
AD, Alzheimer disease; CAD coronary artery disease; CKD, chronic kidney disease; hsCRP, C-reactive protein measured by high-sensitivity assay; MCI, mild cognitive impairment; MI, myocardial infarction; NA, not applicable; T1DM, type 1 diabetes mellitus; T2DM, type 2 diabetes mellitus; TGFβ1, transforming growth factor-β1; TIA, transient ischaemic attack.
Patients also had a serum creatinine level of 1.3–3.3 mg/dl for women or 1.5–3.5 mg/dl for men (or estimated glomerular filtration rate of 20–60 ml/min/1.73 m2) and a 24-h urine protein: creatinine ratio ≥800 mg/g.
British Doctors Aspirin Trial and UK-TIA Aspirin Trial.
Thrombosis Prevention Trial, British Doctors Aspirin Trial, Swedish Aspirin Low Dose Trial, and UK-TIA Aspirin Trial.
Five phase IV development trials conducted by Pfizer.
Atherosclerosis.
Atherosclerosis originates from damaged endothelium that allows the accumulation of cholesterol-containing LDL particles in the arterial wall that tend to be oxidized, which triggers an inflammatory response that fails to resolve144. Activation of both innate and adaptive immunity actively contributes to the initiation and progression of atherogenesis, from early endothelial dysfunction to the development of acute thrombotic complications triggered by plaque rupture or erosion 9,145–149. Monocytes that migrate into the intima of the arterial wall differentiate into macrophages and then transform into foam cells in the lipid necrotic core of the atheroma147,149. Cholesterol crystals and other DAMPs present in the atherosclerotic lesion activate the inflammasomes within macrophages, leading to the release of IL-1β, IL-18, and other pro-inflammatory cytokines150 that are chemotactic for other inflammatory cells, including T cells and B cells, which are major drivers ofatherosclerosis151. Late atherosclerosis is characterized by massive cell apoptosis and accumulation of cells with senescent features, which support a pro-inflammatory status and lead to the formation of a necrotic core that ultimately causes fragility and rupture of the plaque, formation of a thrombus, and acute vascular occlusion.
Cells in advanced atherosclerotic plaques often show markers of senescence, such as p16INK4A and tumour suppressor ARF (commonly known as p14ARF in humans and p19ARF in mice), and express a SASP that further fuels inflammation while producing metalloproteinases that degrade the extracellular matrix, further destabilizing the atherosclerotic plaque152. In turn, the degradation of the extracellular matrix induces the proliferation and phenotypic shift of vascular smooth muscle cells that migrate from the medial layer and, by synthesizing new extracellular matrix, build a fibrous cap that stabilizes atherosclerotic lesions. However, in an inflammatory environment, vascular smooth muscle cells undergo extensive DNA damage and excessive telomere shortening, develop markers of senescence, and might undergo loss of proliferative capacity or even apoptosis153. In addition, the production of metalloproteinases from senescent cells can further weaken the fibrous cap. Therefore, major mechanisms of plaque stabilization are impaired, and additional antigens might be uncovered that further amplify the inflammatory response154. Preclinical studies have shown that activated subtypes of T and B lymphocytes in plaques contribute to plaque instability, leading to an increase in the risk of cardiovascular disease155. miRNAs have emerged as important regulators of cellular adhesion, proliferation, lipid homeostasis, and inflammatory cytokine synthesis, potentially affecting the balance between atherosclerotic plaque progression and regression, although their mechanism of action and relationship with inflammageing is not fully clarified156.
Although the detailed mechanisms that affect the genesis and progression of atherosclerosis are far from being fully understood, evidence is accumulating that inflammation is a major contributor, acting through multiple mechanisms, including a vicious cycle that accelerates clinical progression. Consistent with this view, longitudinal studies demonstrate that high blood levels of pro-inflammatory markers, including high-sensitivity CRP assay and IL-6, predict the risk of cardiovascular disease in both middle-aged and older adults, independent of other CVD risk factors157–160. Moreover, statin therapy with rosuvastatin reduces the incidence of major cardiovascular events in healthy individuals who are free from hyperlipidaemia but who have elevated high-sensitivity CRP levels147,161. Although studies in endothelial cells suggest that CRP directly contributes to CVD by increasing oxidative stress162, other mechanistic studies and Mendelian randomization analyses in large populations suggest that CRP is a predictive biomarker that is not causally related to atherothrombosis163,164. By contrast, IL-6 and IL-1 contribute to atherosclerosis and should be considered to be therapeutic targets158,163. Mendelian randomization studies have shown that polymorphisms that affect IL-6 signalling are associated with lower life-time risk of cardiovascular disease23,24. In the MEASURE trial165, the IL-6 receptor blocker tocilizumab increased the concentration of HDL particles in patients with rheumatoid arthritis compared with placebo, despite an increase multicentre ENTRACTE trial166 compared the cardiovascular safety profile of tocilizumab to that of the TNF inhibitor etanercept in >3,000 patients with moderate-to-severe rheumatoid arthritis, but the final results are not yet published.
In addition, because IL-1β production is a secondary effect of NLRP3 inflammasome activation, which is induced by cholesterol crystals and other DAMPs, the IL-1β signalling pathway has been suggested to be a promising target for atherothrombosis protection. New compounds that interfere with IL-1 and IL-6 signalling are under investigation163,167. The CANTOS trial168 has revealed that anti-inflammatory therapy with canakinumab, a human monoclonal anti-IL-1β antibody, significantly reduced recurrent cardiovascular events in >10,000 stable patients who had residual inflammation after myocardial infarction, independent of lowered lipid levels169. The ongoing CIRT trial170 is testing the hypothesis that low-dose methotrexate, a drug that suppresses IL-1β production by mononuclear cells in addition to other functions171, reduces major vascular events in patients with previous myocardial infarction and either type 2 diabetes or metabolic syndrome. In a small pilot study (the LoDoCo trial)172, anti-inflammatory treatment with colchicine seemed to be effective for secondary prevention of CVD. Larger RCTs, such as the ongoing LoDoCo2 trial and COLCOT trials173, are needed to confirm these findings.
Type 2 diabetes.
The focus of this Review is on inflammageing conceptualized as a shared risk factor and pathophysiological mechanism between CVD and frailty. However, it is important to note that inflammation is associated with the risk and clinical evolution of non-CVD related disease and accelerated decline of physical function. This concept is clearly exemplified by the close connection between inflammation, CVD, and type 2 diabetes. Type 2 diabetes is a strong risk factor for CVD, and both CVD mortality and the effect of diabetes on the risk of CVD increase sharply with older age and frailty status174–176. Strong evidence indicates that insulin resistance and lipotoxicity cause the production of inflammatory mediators that cause neutrophil infiltration, macrophage proliferation, and smooth muscle and endothelial cell activation, which accelerate atherogenesis177. Excessive oxidative stress causes endothelial dysfunction that enables permeation, trapping, and physicochemical modification of circulating lipoprotein particles in the subendothelial space178. Telomeres are, on average, shorter and the number of cells positive for senescence biomarkers is higher in arteries from patients with diabetes than in individuals without diabetes, and this finding might be one of the mechanisms for the increased inflammation and accelerated atherosclerosis in diabetes179,180. Overproduction of angiotensin II amplifies chronic inflammation and can cause mitochondrial dysfunction181. At the same time, inflammation is a risk factor for the development of diabetes and its complications, and these associations are not accounted for by body composition parameters138.
Treating inflammation in non-CVDs.
Of note, a causal role of inflammation in CVD pathogenesis is also suggested by clinical trials that used an anti-inflammatory intervention as treatment for overt, noncardiovascular inflammatory diseases. For example, treatment with TNF inhibitors in rheumatoid arthritis, an overt inflammatory disease, is associated with a decreased risk of cardiovascular events182–187. Treatment with TNF inhibitors in psoriasis is also associated with decreased incidence of major adverse cardiac events186,188,189. However, anti-inflammatory treatment does not always yield beneficial effects. For example, in patients with congestive heart failure, the levels of pro-inflammatory cytokines, especially TNF, IL-6, and IL-1, are markedly elevated, and the TNF level is a negative prognostic factor190–192. However, clinical trials with the TNF inhibitor etanercept yielded no beneficial reductions in mortality or hospitalization due to congestive heart failure193, whereas high doses of infliximab, a TNF antagonist, did not improve and even worsened moderate-to-severe congestive heart failure194.
Multimorbidity and frailty.
The important role of inflammation in CVD, in particular in atherosclerosis, together with the observation that the pro-inflammatory state typical of ageing is a strong risk factor for many age-related chronic diseases, explains why CVD in older individuals often precedes, follows, or develops in the context of multimorbidity and frailty. Patients with CVD tend to have greater multimorbidity than individuals who are free from CVD195. Diseases most often associated with multimorbidity are diabetes, chronic kidney disease, anaemia, chronic pulmonary disease, depression, and dementia, which all involve inflammageing as an important risk factor3,138,139,142,143,196. The presence of comorbid diseases is well established to affect both the response to treatment and the prognosis for hard clinical outcomes, such as cardiovascular and all-cause mortality, as well as hospitalization and health-care utilization197. However, limited data are available on whether CVD comorbidities affect non-traditional outcomes that are still very important for geriatric patients, such as symptom burden, functional capacity, and self-rated health197. Whether these important, non-traditional out-comes respond to anti-inflammatory treatment is also unknown because this information is not commonly collected in most RCTs.
The resulting syndromes present clinical challenges whose complexity is often ignored in clinical practice guidelines for single diseases, which are based on randomized clinical trials in which patients with multimorbidity are under-represented, including most of the trials cited above that targeted inflammation195. The degree of clinical complexity is even higher when the comorbid medical condition is frailty. Age-associated frailty is a medical syndrome characterized by morphological and physiological changes across multiple systems and organs, resulting in a progressive loss of internal homeostasis, reduced physiological reserves, loss of function, reduced resilience, and increased vulnerability to internal and external stresses198,199. We address the general pathophysiology of frailty later in this Review. Here, we limit the discussion to the effect of frailty that emerges in patients with CVD. The prevalence of frailty in older individuals with CVD rises progressively from subclinical CVD, to heart failure, to overt acute syndromes, to cardiac surgery when, depending on the procedure, the rate of frailty can be >60%200. Strong evidence from multiple, large, observational studies indicates that the presence of CVD is a risk factor for frailty and that patients with frailty are more likely to develop CVD than those who are not frail201–203. This observation is not surprising because inflammation, insulin resistance, and coagulation problems have been identified as cardinal factors in the pathophysiology of frailty204–207. Therefore, CVD and frailty can be viewed as diseases arising from similar causal mechanisms, mutually accelerating their clinical course by vicious cycles that amplify inflammation, insulin resistance, and other still-unknown mechanisms, thereby synergistically contributing to adverse health outcomes. In accordance with this theory, independent of age and other risk factors, frailty in patients with CVD is associated with a twofold increase in the risk of death208.
We have previously proposed that a pro-inflammatory state might be caused by most of the described putative biological mechanisms of ageing, such as telomere shortening, cell senescence, mitochondrial dysfunction, altered nutrient sensing, and epigenetic alterations1. Therefore, the coexistence of CVD, diabetes, and frailty might be just one example of a general phenomenon that involves multiple diseases, as exemplified by the following observations. Low-grade chronic inflammation is a primary contributor to the onset and progression of chronic kidney disease11,142,209,210. Inflammation is a causal factor for cancer initiation, promotion, malignancy, and metastatic dissemination211,212. Depression is characterized by increased levels of pro-inflammatory cytokines and acute phase proteins in both peripheral blood and cerebrospinal fluid213,214. Inflammatory diseases and high levels of pro-inflammatory biomarkers in the blood increase the risk of depression143,215–217. Inflammation has a central role in age-related neuro-degeneration and in neurodegenerative diseases, such as Alzheimer disease218. These studies support the hypothesis that neuroinflammatory changes are important pathological components of Alzheimer disease and other neurodegenerative diseases, highlighting the potential clinical importance of cytokines in neurogenesis. Interestingly, most genetic variants associated with late-onset Alzheimer disease are within immunity-related genes219.
Inflammation and age-related frailty
The collective evidence suggests that chronic inflammation is a risk factor across multiple diseases, some of which are traditionally viewed as pathophysiologically unrelated, such as CVD157,159,160,220, diabetes138,180,221, chronic kidney disease11,142,209, cancer211,212,222, depression13,143,217, and dementia12,139,223. In addition, higher levels of inflammatory markers in blood are associated with a greater loss of muscle mass and strength, accelerated loss of mobility, lower-extremity performance and physical activity, and depression in older individuals224–228, all of which are essential elements for defining frailty on the basis of the criteria most often used in the literature229. From this perspective, inflammageing could act as a focal point for ageing mechanisms that are associated with increased susceptibility to stressors and impaired functional reserves. Consistent with this observation, both higher baseline levels and increasing accumulation rates of IL-6 predict accelerated longitudinal accumulation of multiple chronic diseases in older individuals14. In addition, inflammation contributes to accelerated ageing in individuals with multimorbidity230, and partially mediates the association between multimorbidity and functional limitations and disability231. Not surprisingly, most patients with frailty have chronic inflammation, especially those who are affected by sarcopenia, which is defined as a reduction in muscle strength and mass that is abnormally severe for an individual’s age207,232,233.
A variety of hypotheses have been proposed to explain the link between inflammation and sarcopenia and frailty; interestingly, some of these mechanisms are shared with the pathogenesis of CVD. Inflammation is associated with reduced synthesis and activity of insulin-like growth factor I (IGF1), a growth factor that is essential for muscle regeneration and maintenance of muscle integrity and that is protective against plaque instability in atherosclerosis158,234. In vitro studies have shown that IL-1α, IL-6, and TNF inhibit IGF1-mediated anabolism and that IL-6 reduces the production of IGF1 and IGF-binding protein 3 (reF.235). In observational studies, high levels of IL-6 and low levels of IGF1 synergistically correlate with lower muscle strength and power, effectively predicting progressive disability and death236,237. Inflammation impairs endothelial reactivity and muscle perfusion, interfering with the uptake of long branched-chain amino acids that are essential for muscle energetics and protein anabolism238–240. Dysfunctional mitochondria that are not recycled owing to defective mitophagy produce ROS that stimulate the production of pro-inflammatory cytokines and catabolism via increased NF-κB-dependent protein ubiquitylation and proteasome degradation241. Senescent cells that produce inflammatory mediators might also have a role in the pathogenesis of sarcopenia. One study quantified senescent p16INK4A-expressing cells in thigh intramuscular adipose tissue from older women, revealing that senescent cell burden was associated with grip strength, walking speed, and self-perceived mobility242. Moreover, inflammation impairs satellite cell regenerative function243–245. Of note, the emergence of senescence traits in vascular smooth muscle cells has been implicated in the initiation and progression of CVD, specifically atherosclerosis246, again suggesting that atherosclerosis and the resulting CVD is a syndrome of accelerated ageing.
Both aerobic and resistance exercise — as well as dietary supplementation of amino acids or protein, vitamin D, and polyunsaturated fatty acids — have been associated with protection against age-associated sarcopenia, possibly because of their antiinflammatory and antioxidative properties141. In observational studies, adherence to the Mediterranean diet was the only behavioural factor consistently associated with a lower risk of frailty, which might be a result of the anti-inflammatory properties inherent to the diet247,248. Underlying the relationship between CVD and frailty, the Mediterranean diet is also one of the few behavioural interventions that, in both observational studies and clinical trials, was associated with lower cardiovascular morbidity and mortality249,250. Aspirin, a potent anti-inflammatory molecule, is effective in the treatment of acute myocardial infarction and in secondary prevention of CVD, and some evidence indicates that aspirin might be effective in primary prevention of myocardial infarction, at least in high-risk groups251,252. Interestingly, chronic use of NSAIDs is associated with a lower risk of sarcopenia in community-dwelling individuals aged ≥80 years253.
Metformin, an antidiabetic drug that counteracts inflammation and insulin resistance, has been suggested to prevent frailty and attenuate its progression. In a large, observational study conducted in US veterans with type 2 diabetes and stratified according to baseline risk, treatment with metformin reduced the risk of multiple age-related diseases, including CVDs, cancer, depression, and frailty-related diseases254. An RCT conducted in Indonesia revealed that metformin improves gait speed, but not handgrip strength, in nondiabetic, pre-frail, older individuals255. A trial is currently ongoing at the University of Texas Health Science Center at San Antonio, TX, USA, aimed at examining whether metformin prevents frailty development in older individuals with impaired glucose tolerance256.
The evidence reported above suggests that CVD, multimorbidity, frailty, and perhaps other chronic diseases have inflammageing as a common root cause. Unfortunately, although strong evidence shows that targeting inflammation can reduce the risk of CVD, no definitive evidence exists that reducing inflammation can prevent or modify the progression of multimorbidity with frailty or sarcopenia. In part, this situation is because of the lack of adequately sized RCTs to test this hypothesis and because RCTs that have tested the effectiveness of anti-inflammatory treatments for cardiovascular prevention did not collect data on multimorbidity, frailty, or disability. These issues should be considered a priority in setting the future research agenda. The ENRGISE trial257 is an ongoing, multicentre, double-blind, placebo-controlled, randomized pilot study, enrolling older men and women (aged ≥70 years) who have high levels of IL-6 and impaired physical function, to test whether losartan, omega-3 fish oil, or a combination of the two reduce plasma IL-6 levels compared with placebo. Interestingly, results from the CRATUS trial258,259 demonstrated that administration of allogeneic human mesenchymal stem cells improves measures of lower-extremity performance and reduces inflammatory biomarkers in age-related frailty. By contrast, despite their anti-inflammatory effects, statin use in the Women’s Health Initiative survey had no significant effect on the risk of frailty260.
Inflammageing is a pillar of geroscience
Many interventions that increase longevity in animal models cause a reduction in inflammatory markers. For example, calorie restriction is the most powerful life-extension intervention in most animal models and is associated with reduced inflammatory biomarkers261,262. Mechanisms by which calorie restriction reduces chronic inflammation include diminished ROS production and consequent downregulation of NF-κB-induced transcription of pro-inflammatory genes in multiple tissues263. Of note, dietary restriction significantly reduces the risk of CVD in humans, and in animal models, dietary restriction is associated with numerous beneficial changes in arterial walls264.
Rapamycin, a specific inhibitor of mechanistic target of rapamycin (mTOR) signalling with many effects, including anti-inflammatory activity265, has an important role in longevity regulation266 in both animals and humans267, and improves survival and healthspan in animal models268–272. Aspirin improves lifespan in mice273, whereas metformin, which is known to have direct anti-inflammatory effects beyond its canonical glucose-lowering activity274, improves lifespan and healthspan in animal models275. A meta-analysis revealed that metformin reduces all-cause mortality and diseases associated with ageing independent of diabetic control in humans276. The TAME trial277 is designed to examine the effect of metformin on delaying the onset of age-related conditions and diseases and its potential use in expanding human health span. Finally, the clearance of senescent cells by either genetic engineering or the administration of senolytic drugs has been associated with reduced circulating levels of pro-inflammatory markers, increased lifespan, and delayed frailty-related phenotypes in mice111,112. Clinical trials are needed to test the efficacy of these potential treatments in humans278. These examples suggest that interventions that target some of the fundamental mechanisms of ageing affect the general susceptibility to CVD, as well as other age-related diseases, and prevent frailty and disability in older individuals.
Over the past 3 decades, a wealth of evidence has been gathered that suggests that chronic inflammation is one of these mechanisms. Although this Review on the connection between inflammageing, CVD, and frailty is far from comprehensive, the role of chronic inflammation in health and functional deterioration with ageing is clearly emerging. Of course, conceptual problems remain. Throughout this Review, we have focused on inflammation as posing a threat to human health over the course of ageing. However, inflammation has been evolutionarily selected as a fundamental defensive mechanism that protects organisms from microbial invasion, ensures the integrity of a self-recognized inventory of proteins and other macromolecules, prevents cancer by recognizing and removing cells that present tumour antigens, and has an important role in tissue repair. The benefits of inflammation overcome the risks associated with autoimmune disease and inflammageing. As such, inflammation has a positive influence on health when activated transiently — that is, when inflammation deploys quickly and fully armed in response to an adequate stimulation, successfully eliminates the challenge, and recedes quickly to a baseline resting state. When inflammation becomes chronic, however, problems arise and deleterious effects emerge, as first described by Rudolph Virchow (1821–1902)279. Chronic inflammatory diseases, such as certain chronic infections, cancer, congestive heart failure, chronic obstructive pulmonary disease, and HIV, cause syndromes that share many characteristics with accelerated ageing and frailty, including sarcopenia, weight loss, and loss of energy coupled with fatigability. Therefore, chronically elevated levels of circulating pro-inflammatory markers observed with ageing are unsurprisingly also associated with similar signs and symptoms to those of chronic disease, although the age-related symptoms develop progressively and over a longer time frame.
The mechanisms for these actions are not fully understood, but a comprehensive analysis of the literature reveals that inflammation is often associated with a catabolic state (Fig. 3). As described previously in this Review, inflammation is associated with anabolic resistance in muscle, which is caused partly by inhibition of the perfusion adjustment to anabolic stimuli and partly by inhibition of IGF1 production and signalling236–240. Chronic inflammation causes anaemia via direct inhibition of iron absorption and recycling as well as interference with erythropoietin production and signalling3,280. Evidence indicates that inflammation causes insulin resistance. In particular, TNF receptor superfamily member 1A and Toll-like receptor 4 block insulin signalling through Janus kinase activation, which causes serine phosphorylation of insulin receptor substrate 1 and 2, contributing to insulin resistance281–283. Conversely, evidence also indicates that insulin resistance promotes the accumulation of M1 macrophages and fosters inflammation in adipose tissue through the production of CCL2 (reF.284). Pro-inflammatory cytokines — including IL-1β, IL-6, IL-11, IL-15, IL-17, and TNF — stimulate bone resorption and almost certainly contribute to osteoporosis285. For example, bone resorption is increased in patients with inflammatory diseases, such as rheumatoid arthritis286. Studies in cultured cells show that IL-1β, IL-6, and TNF induce mitochondrial dysfunction with reduced ATP synthesis-driven respiration, reduction of the NAD+:NADH ratio, and reduced mRNA levels of PPARGC1A (encoding peroxisome proliferator-activated receptor-γ co-activator 1α; PGC1α), suggesting that mitochondrial biogenesis is impaired287. Studies conducted both in vitro and in animal models suggest that inflammation in general, and IL-1β and IFNα in particular, inhibit neurogenesis and reduce the magnitude of neurogenesis that is normally induced by exercise288,289. These data delineate an overall mechanism by which inflammageing affects multiple physiological systems and phenotypes. During an infection that unleashes an inflammatory response, the physiological and metabolic state of the organism is focused on defence, and all other anabolic activities are paused, including nondefensive functions of the immune system, such as surveillance of damage and continuous repair in tissue, which mostly rely on growth factors. If this condition is temporary, turnover and repair of macromolecules, organelles, and cells can be delayed, avoiding irreversible damage. However, in older individuals, inflammation remains chronically activated, either because of continued stress from the inflammation source or because of a primary immune dysregulation. In the absence of macromolecular and organellar recycling, the accumulation of damage can reach a critical threshold, thereby causing severe functional consequences that become difficult or impossible to reverse, conferring the clinical syndrome of frailty.
Fig. 3 |. Inflammageing induces a catabolic state.
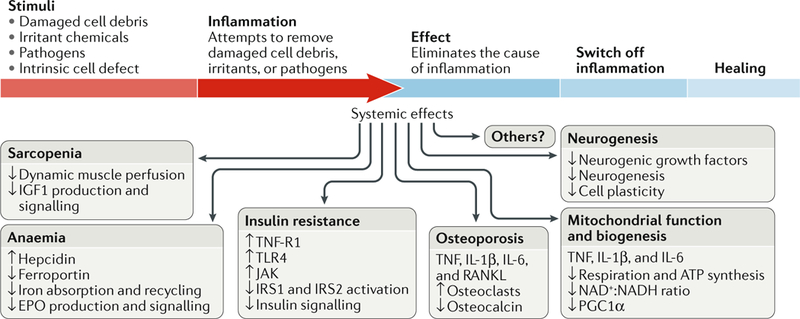
Inflammation causes pathological states linked with frailty, cardiovascular disease, and ageing. Sarcopenia: the induction of anabolic resistance in muscle inhibits the perfusion adjustment to anabolic stimuli as well as insulin-like growth factor (IGF1) production and signalling235–240. Anaemia: chronic elevation of IL-6 levels causes anaemia through the production of hepcidin, reduction of the transmembrane iron transporter ferroportin, and inhibition of iron absorption and recycling as well as interference with erythropoietin (EPO) production and signalling3,280. Insulin resistance: tumour necrosis factor receptor superfamily member 1A (TNF-R1) and Toll-like receptor 4 (TLR4) block insulin signalling through Janus kinase (JAK) activation, which causes serine phosphorylation of insulin receptor substrate 1 (IRS1) and IRS2, contributing to insulin resistance283. Osteoporosis: TNF, IL-1β, IL-6, and TNF ligand superfamily member 11 (RANKL) contribute to osteoporosis by stimulating osteoclast growth and activity and inhibiting the production of osteocalcin290,291. Mitochondria biogenesis: studies in vitro show that TNF, IL-1β, and IL-6 induce mitochondrial dysfunction with reduced ATP synthesis-driven respiration, a reduced NAD+:NADH ratio, and reduced mRNA levels of PPARGC1A (encoding peroxisome proliferator-activated receptor-γ co-activator 1α; PGC1α), suggesting impairment in mitochondrial biogenesis287. Neurogenesis: pro-inflammatory cytokines interfere with the biological activity of neuronal growth factors, such as brain-derived neurotrophic factor, thereby affecting neurogenesis and plasticity292. Accordingly, the addition of IFNα to human hippocampal progenitor cells reduces neurogenesis289. These are just few examples of how chronic inflammation promotes a catabolic state, suggesting a possible unifying hypothesis. During an acute bout of inflammation, induced for example by an infection, the surveillance of damage and continuous repair functions in multiple tissues are chronically inhibited, leading to accumulated damage in organelles and macromolecules. Over time, this damage accumulation across different tissues and organs could become so severe that it cannot be compensated for and causes irreversible frailty.
Conclusions
On the basis of the data and the hypotheses presented in this Review, modulating inflammageing is a promising strategy not only to prevent CVD but also to slow the decline of health that occurs with ageing. Modulating inflammation is likely to be most effective at the early stage of health decline, at a time when the compensatory capacity of the organism is not completely exhausted and might still counteract physiological and functional declines. New pharmacological treatments that selectively affect some of the signalling pathways that regulate inflammation are needed to balance the relationship between risks and benefits. Early treatments will require early diagnosis and availability of a signature biomarker profile that allows for a differential diagnosis between true inflammageing and chronic inflammation sustained by the persistence of an infectious or toxic cause. Ultimately, RCTs are needed to test the hypothesis that modulating inflammation prevents the development of CVD as well as multimorbidity, disability, and frailty.
Key points.
High levels of pro-inflammatory markers in the blood and other tissues are often detected in older individuals and predict the risk of cardiovascular diseases, frailty, multimorbidity, and decline of physical and cognitive function.
In individuals with obesity, visceral fat produces pro-inflammatory and chemotactic compounds and is infiltrated by macrophages, lymphocytes, and senescent cells with a senescence-associated secretory phenotype that contributes to inflammageing.
Mechanisms potentially underlying inflammageing include genomic instability, cell senescence, mitochondria dysfunction, microbiota composition changes, NLRP3 inflammasome activation, primary dysregulation of immune cells, and chronic infections.
Clinical trials suggest that modulating inflammation prevents cardiovascular diseases, but studies to explore the effects on other chronic diseases, frailty, and disability are scarce and controversial.
Inflammageing can complicate the clinical features of cardiovascular disease in older individuals by causing an energetic imbalance towards catabolism and interfering with homeostatic signalling, leading to frailty.
Acknowledgements
The authors received support from the Intramural Research Program of the National Institute on Aging, NIH, Baltimore, MD, USA. The authors thank A. Cornish (National Institute on Aging) for help in editing the manuscript and for the many suggestions that greatly improved the quality of this work, in particular the microbiota section.
Footnotes
Competing interests
The authors declare no competing interests.
Publisher's Disclaimer: Publisher’s note
Publisher's Disclaimer: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Review criteria
The information in this Review is based on a search of the scientific literature published since 2008 using the Medline database and the search terms: “inflammaging”, “inflammation and cardiovascular disease and aging”, “inflammation and frailty”, or “cardiovascular disease and frailty”. The authors reviewed all 3,377 relevant abstracts and selected the manu-scripts for which information is reported in this Review. Of note, some articles >10 years old were also cited because their content was considered critical for the topic addressed.
References
- 1.Bektas A, Schurman SH, Sen R & Ferrucci L Aging, inflammation and the environment. Exp. Gerontol 10.1016/j.exger.2017.12.015 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Franceschi C et al. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. NY Acad. Sci 908, 244–254 (2000). [DOI] [PubMed] [Google Scholar]
- 3.Ferrucci L et al. Proinflammatory state, hepcidin, and anemia in older persons. Blood 115, 3810–3816 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fulop T et al. Immunosenescence and inflamm-aging as two sides of the same coin: friends or foes? Frontiers Immunol 8, 1960 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cohen HJ, Pieper CF, Harris T, Rao KM & Currie MS The association of plasma IL-6 levels with functional disability in community-dwelling elderly. J. Gerontol. A. Biol. Sci. Med. Sci 52, M201–M208 (1997). [DOI] [PubMed] [Google Scholar]
- 6.Newman AB et al. Trajectories of function and biomarkers with age: the CHS All Stars Study. Int. J. Epidemiol 45, 1135–1145 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gerli R et al. Chemokines, sTNF-Rs and sCD30 serum levels in healthy aged people and centenarians. Mech. Ageing Dev 121, 37–46 (2000). [DOI] [PubMed] [Google Scholar]
- 8.Franceschi C, Garagnani P, Vitale G, Capri M & Salvioli S Inflammaging and ‘garb-aging’. Trends Endocrinol. Metab 28, 199–212 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Ruparelia N, Chai JT, Fisher EA & Choudhury RP Inflammatory processes in cardiovascular disease: a route to targeted therapies. Nat. Rev. Cardiol 14, 133–144 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leonardi GC, Accardi G, Monastero R, Nicoletti F & Libra M Ageing: from inflammation to cancer. Immun. Ageing 15, 1 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Salimi S et al. Inflammation and trajectory of renal function in community-dwelling older adults. J. Am. Geriatr. Soc 66, 804–811 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gorelick PB Role of inflammation in cognitive impairment: results of observational epidemiological studies and clinical trials. Ann. NY Acad. Sci 1207, 155–162 (2010). [DOI] [PubMed] [Google Scholar]
- 13.Miller AH & Raison CL The role of inflammation in depression: From evolutionary imperative to modern treatment target. Nat. Rev. Immunol 16, 22–34 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fabbri E et al. Aging and the burden of multimorbidity: associations with inflammatory and anabolic hormonal biomarkers. J. Gerontol. A. Biol. Sci. Med. Sci 70, 63–70 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ferrucci L et al. Serum IL-6 level and the development of disability in older persons. J. Am. Geriatr. Soc 47, 639–646 (1999). [DOI] [PubMed] [Google Scholar]
- 16.Kuo H, Bean JF, Yen C & Leveille SG Linking C-reactive protein to late-life disability in the National Health and Nutrition Examination Survey (NHANES) 1999–2002. J. Gerontol. A. Biol. Sci. Med. Sci 61, 380–387 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schaap LA et al. Higher inflammatory marker levels in older persons: associations with 5-year change in muscle mass and muscle strength. J. Gerontol. A. Biol. Sci. Med. Sci 64, 1183–1189 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Soysal P et al. Inflammation and frailty in the elderly: a systematic review and meta-analysis. Ageing Res. Rev 31, 1–8 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Volpato S et al. Cardiovascular disease, interleukin-6, and risk of mortality in older women: the women’s health and aging study. Circulation 103, 947–953 (2001). [DOI] [PubMed] [Google Scholar]
- 20.Hodes RJ et al. Disease drivers of aging. Ann. NY Acad. Sci 1386, 45–68 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith AJP & Humphries SE Cytokine and cytokine receptor gene polymorphisms and their functionality. Cytokine Growth Factor Rev 20, 43–59 (2009). [DOI] [PubMed] [Google Scholar]
- 22.Rafiq S et al. Common genetic variation in the gene encoding interleukin-1-receptor antagonist (IL-1RA) is associated with altered circulating IL-1RA levels. Genes Immun 8, 344–351 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Sarwar N et al. Interleukin-6 receptor pathways in coronary heart disease: a collaborative meta-analysis of 82 studies. Lancet 379, 1205–1213 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Swerdlow DI et al. The interleukin-6 receptor as a target for prevention of coronary heart disease: a mendelian randomisation analysis. Lancet 379, 1214–1224 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dehghan A et al. Meta-analysis of genome-wide association studies in >80 000 subjects identifies multiple loci for C-reactive protein levels. Circulation 123, 731–738 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reiner AP et al. Polymorphisms of the IL1-receptor antagonist gene (IL1RN) are associated with multiple markers of systemic inflammation. Arterioscler. Thromb. Vasc. Biol 28, 1407–1412 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carrol ED et al. The IL1RN promoter rs4251961 correlates with IL-1 receptor antagonist concentrations in human infection and is differentially regulated by GATA-1. J. Immunol 186, 2329–2335 (2011). [DOI] [PubMed] [Google Scholar]
- 28.Herder C et al. Genetic determinants of circulating interleukin-1 receptor antagonist levels and their association with glycemic traits. Diabetes 63, 4343–4359 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu X et al. IL-1 receptor antagonist gene as a predictive biomarker of progression of knee osteoarthritis in a population cohort. Osteoarthr. Cartil 21, 930–938 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dai L, Liu D, Guo H, Wang Y & Bai Y Association between polymorphism in the promoter region of interleukin 6 (−174 G/C) and risk of Alzheimer’s disease: a meta-analysis. Neurol. J 259, 414–419 (2012). [DOI] [PubMed] [Google Scholar]
- 31.Hou H et al. Association of interleukin-6 gene polymorphism with coronary artery disease: an updated systematic review and cumulative meta-analysis. Inflamm. Res 64, 707–720 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Testa R et al. Interleukin-6–174 G > C polymorphism affects the association between IL-6 plasma levels and insulin resistance in type 2 diabetic patients. Diabetes Res. Clin. Pract 71, 299–305 (2006). [DOI] [PubMed] [Google Scholar]
- 33.Moffett SP et al. Association of the G-174C variant in the interleukin-6 promoter region with bone loss and fracture risk in older women. J. Bone Miner. Res 19, 1612–1618 (2004). [DOI] [PubMed] [Google Scholar]
- 34.Fishman D et al. The effect of novel polymorphisms in the interleukin-6 (IL-6) gene on IL-6 transcription and plasma IL-6 levels, and an association with systemic-onset juvenile chronic arthritis. J. Clin. Invest 102, 1369–1376 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zeng Y et al. Novel loci and pathways significantly associated with longevity. Sci. Rep 6, 21243 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Khandaker GM, Zammit S, Burgess S, Lewis G & Jones PB Association between a functional interleukin 6 receptor genetic variant and risk of depression and psychosis in a population-based birth cohort. Brain. Behav. Immun 69, 264–272 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ferreira RC et al. Functional IL6R 358Ala allele impairs classical IL-6 receptor signaling and influences risk of diverse inflammatory diseases. PLoS Genet 9, e1003444 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lange LA et al. Association of polymorphisms in the CRP gene with circulating C-reactive protein levels and cardiovascular events. JAMA 296, 2703 (2006). [DOI] [PubMed] [Google Scholar]
- 39.Peters MJ et al. The transcriptional landscape of age in human peripheral blood. Nat. Commun 6, 8570 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pilling LC et al. Gene expression markers of age-related inflammation in two human cohorts. Exp. Gerontol 70, 37–45 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fabian MR, Sonenberg N & Filipowicz W Regulation of mRNA translation and stability by microRNAs. Annu. Rev. Biochem 79, 351–379 (2010). [DOI] [PubMed] [Google Scholar]
- 42.Noren Hooten N et al. microRNA expression patterns reveal differential expression of target genes with age. PLoS ONE 5, e10724 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lai C-Y et al. Modulated expression of human peripheral blood microRNAs from infancy to adulthood and its role in aging. Aging Cell 13, 679–689 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Olivieri F et al. Age-related differences in the expression of circulating microRNAs: miR-21 as a new circulating marker of inflammaging. Mech. Ageing Dev 133, 675–685 (2012). [DOI] [PubMed] [Google Scholar]
- 45.Freedman JE et al. Diverse human extracellular RNAs are widely detected in human plasma. Nat. Commun 7, 11106 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Noren Hooten N et al. Age-related changes in microRNA levels in serum. Aging 5, 725–740 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ameling S et al. Associations of circulating plasma microRNAs with age, body mass index and sex in a population-based study. BMC Med. Genomics 8, 61 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang H et al. Investigation of microRNA expression in human serum during the aging process. J. Gerontol. A. Biol. Sci. Med. Sci 70, 102–109 (2015). [DOI] [PubMed] [Google Scholar]
- 49.Dluzen DF, Noren Hooten N & Evans MK Extracellular. RNA in aging. WIREs RNA 10.1002/wrna.1385 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Olivieri F et al. Age- and glycemia-related miR-126–3p levels in plasma and endothelial cells. Aging (Albany. NY) 6, 771–787 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Olivieri F et al. DNA damage response (DDR) and senescence: shuttled inflamma-miRNAs on the stage of inflamm-aging. Oncotarget 6, 35509–35521 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Frasca D, Blomberg BB & Paganelli R Aging, obesity, and inflammatory age-related diseases. Front. Immunol 8, 1–10 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rocha VZ & Libby P Obesity, inflammation, and atherosclerosis. Nat. Rev. Cardiol 6, 399–409 (2009). [DOI] [PubMed] [Google Scholar]
- 54.Vandanmagsar B et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med 17, 179–189 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Frasca D & Blomberg BB Adipose tissue inflammation induces B cell inflammation and decreases B cell function in aging. Front. Immunol 8, 1003 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee-Chang C et al. Accumulation of 4–1BBL+ B cells in the elderly induces the generation of granzyme-B+ CD8+ T cells with potential antitumor activity. Blood 124, 1450–1459 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Weisberg SP et al. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Invest 112, 1796–1808 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Panagiotakos DB, Pitsavos C, Yannakoulia M, Chrysohoou C & Stefanadis C The implication of obesity and central fat on markers of chronic inflammation: The ATTICA study. Atherosclerosis 183, 308–315 (2005). [DOI] [PubMed] [Google Scholar]
- 59.Clément K et al. Weight loss regulates inflammation-related genes in white adipose tissue of obese subjects. FASEB J 18, 1657–1669 (2004). [DOI] [PubMed] [Google Scholar]
- 60.Nicklas BJ, You T & Pahor M Behavioural treatments for chronic systemic inflammation: effects of dietary weight loss and exercise training. Can. Med. Assoc. J 172, 1199–1209 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Illán-Gómez F et al. Obesity and inflammation: change in adiponectin, C-reactive protein, tumour necrosis factor-alpha and interleukin-6 after bariatric surgery. Obes. Surg 22, 950–955 (2012). [DOI] [PubMed] [Google Scholar]
- 62.Meydani SN et al. Long-term moderate calorie restriction inhibits inflammation without impairing cell-mediated immunity: a randomized controlled trial in non-obese humans. Aging 8, 1416–1431 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zomer E et al. Interventions that cause weight loss and the impact on cardiovascular risk factors: a systematic review and meta-analysis. Obes. Rev 17, 1001–1011 (2016). [DOI] [PubMed] [Google Scholar]
- 64.Ma C et al. Effects of weight loss interventions for adults who are obese on mortality, cardiovascular disease, and cancer: systematic review and meta-analysis. BMJ 359, j4849 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Villareal DT et al. Aerobic or resistance exercise, or both, in dieting obese older adults. N. Engl. J. Med 376, 1943–1955 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mariat D et al. The Firmicutes/Bacteroidetes ratio of the human microbiota changes with age. BMC Microbiol 9, 123 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Biagi E et al. Gut microbiota and extreme longevity. Curr. Biol 26, 1480–1485 (2016). [DOI] [PubMed] [Google Scholar]
- 68.Shapiro H, Thaiss CA, Levy M & Elinav E The cross talk between microbiota and the immune system: metabolites take center stage. Curr. Opin. Immunol 30, 54–62 (2014). [DOI] [PubMed] [Google Scholar]
- 69.Rampelli S et al. Functional metagenomic profiling of intestinal microbiome in extreme ageing. Aging 5, 902–912 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Biagi E et al. Through ageing, and beyond: gut microbiota and inflammatory status in seniors and centenarians. PLoS ONE 5, e10667 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zapata HJ & Quagliarello VJ The microbiota and microbiome in aging: potential implications in health and age-related diseases. Am. J. Geriatr. Soc 63, 776–781 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Thevaranjan N et al. Age-associated microbial dysbiosis promotes intestinal permeability, systemic inflammation, and macrophage dysfunction. Cell Host Microbe 21, 455–466.e4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Picca A et al. Gut dysbiosis and muscle aging: searching for novel targets against sarcopenia. Mediators Inflamm 2018, 7026198 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.van Tongeren SP, Slaets JPJ, Harmsen HJM & Welling GW Fecal microbiota composition and frailty. Appl. Environ. Microbiol 71, 6438–6442 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.O’Toole PW & Jeffery IB Gut microbiota and aging. Science 350, 1214–1215 (2015). [DOI] [PubMed] [Google Scholar]
- 76.Mello AM, Paroni G, Daragjati J & Pilotto A Gastrointestinal microbiota and their contribution to healthy aging. Dig. Dis 34, 194–201 (2016). [DOI] [PubMed] [Google Scholar]
- 77.Barrios C et al. Gut-microbiota-metabolite axis in early renal function decline. PLoS ONE 10, e0134311 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Everard A et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc. Natl Acad. Sci. USA 110, 9066–9071 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Turroni F et al. Molecular dialogue between the human gut microbiota and the host: a Lactobacillus and Bifidobacterium perspective. Cell. Mol. Life Sci 71, 183–203 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ott B et al. Effect of caloric restriction on gut permeability, inflammation markers, and fecal microbiota in obese women. Sci. Rep 7, 11955 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Westfall S et al. Microbiome, probiotics and neurodegenerative diseases: deciphering the gut brain axis. Cell. Mol. Life Sci 74, 3769–3787 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kim YA, Keogh JB & Clifton PM Probiotics, prebiotics, synbiotics and insulin sensitivity. Nutr. Res. Rev 31, 35–51 (2018). [DOI] [PubMed] [Google Scholar]
- 83.Liu Y, Gibson GR & Walton GE An in vitro approach to study effects of prebiotics and probiotics on the faecal microbiota and selected immune parameters relevant to the elderly. PLoS ONE 11, e0162604 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nagpal R et al. Gut microbiota in health and disease: an overview focused on metabolic inflammation. Benef. Microbes 7, 181–194 (2016). [DOI] [PubMed] [Google Scholar]
- 85.Turchet P, Laurenzano M, Auboiron S & Antoine JM Effect of fermented milk containing the probiotic Lactobacillus casei DN-114001 on winter infections in free-living elderly subjects: a randomised, controlled pilot study. J. Nutr. Health Aging 7, 75–77 (2003). [PubMed] [Google Scholar]
- 86.López-Otín C, Blasco MA, Partridge L, Serrano M & Kroemer G The hallmarks of aging. Cell 153, 1194–1217 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kennedy BK et al. Geroscience: linking aging to chronic disease. Cell 159, 709–713 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Campisi J & D’Adda Di Fagagna F Cellular senescence: when bad things happen to good cells. Nat. Rev. Mol. Cell. Biol 8, 729–740 (2007). [DOI] [PubMed] [Google Scholar]
- 89.Sharpless NE & Sherr CJ Forging a signature of in vivo senescence. Nat. Rev. Cancer 15, 397–408 (2015). [DOI] [PubMed] [Google Scholar]
- 90.Bernardes de Jesus B & Blasco MA Assessing cell and organ senescence biomarkers. Circ. Res 111, 97–109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jeck WR, Siebold AP & Sharpless NE Review: a meta-analysis of GWAS and age-associated diseases. Aging Cell 11, 727–731 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Johnson SC, Dong X, Vijg J & Suh Y Genetic evidence for common pathways in human age-related diseases. Aging Cell 14, 809–817 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Melzer D et al. A common variant of the p16INK4a genetic region is associated with physical function in older people. Mech. Ageing Dev 128, 370–377 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.van Deursen JM The role of senescent cells in ageing. Nature 509, 439–446 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hernandez-Segura A et al. Unmasking transcriptional heterogeneity in senescent cells. Curr. Biol 27, 2652–2660.e4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Borodkina AV, Deryabin PI, Giukova AA & Nikolsky NN ‘Social life’ of senescent sells: what is SASP and why study it? Acta Naturae 10, 4–14 (2018). [PMC free article] [PubMed] [Google Scholar]
- 97.Herbig U, Ferreira M, Condel L, Carey D & Sedivy JM Cellular senescence in aging primates. Science 311, 1257 (2006). [DOI] [PubMed] [Google Scholar]
- 98.Waaijer MEC et al. The number of p16INK4a positive cells in human skin reflects biological age. Aging Cell 11, 722–725 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Liu Y et al. Expression of p16INK4a in peripheral blood T-cells is a biomarker of human aging. Aging Cell 8, 439–448 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Koppelstaetter C et al. Markers of cellular senescence in zero hour biopsies predict outcome in renal transplantation. Aging Cell 7, 491–497 (2008). [DOI] [PubMed] [Google Scholar]
- 101.Helman A et al. p16Ink4a-induced senescence of pancreatic beta cells enhances insulin secretion. Nat. Med 22, 412–420 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Rossman MJ et al. Endothelial cell senescence with aging in healthy humans: prevention by habitual exercise and relation to vascular endothelial function. Am. J. Physiol. Heart Circ. Physiol 313, H890–H895 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Diekman BO et al. Expression of p16INK4a is a biomarker of chondrocyte aging but does not cause osteoarthritis. Aging Cell 10.1111/acel.12771 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chimenti C et al. Senescence and death of primitive cells and myocytes lead to premature cardiac aging and heart failure. Circ. Res 93, 604–613 (2003). [DOI] [PubMed] [Google Scholar]
- 105.Kajstura J et al. Myocyte turnover in the aging human heart. Circ. Res 107, 1374–1386 (2010). [DOI] [PubMed] [Google Scholar]
- 106.Gregor MF & Hotamisligil GS Inflammatory mechanisms in obesity. Annu. Rev. Immunol 29, 415–445 (2011). [DOI] [PubMed] [Google Scholar]
- 107.Ogrodnik M et al. Cellular senescence drives age-dependent hepatic steatosis. Nat. Commun 8, 15691 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Klenerman P & Oxenius A T cell responses to cytomegalovirus. Nat. Rev. Immunol 16, 367–377 (2016). [DOI] [PubMed] [Google Scholar]
- 109.Sansoni P et al. New advances in CMV and immunosenescence. Exp. Gerontol 55, 54–62 (2014). [DOI] [PubMed] [Google Scholar]
- 110.Fulop T, Larbi A & Pawelec G Human T cell aging and the impact of persistent viral infections. Front. Immunol 4, 271 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Baker DJ et al. Clearance of p16 Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479, 232–236 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Baker DJ et al. Naturally occurring p16 Ink4a-positive cells shorten healthy lifespan. Nature 530, 184–189 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Medina CB & Ravichandran KS Do not let death do us part: ‘find-me’ signals in communication between dying cells and the phagocytes. Cell Death Differ 23, 979–989 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Youm YH et al. Canonical Nlrp3 inflammasome links systemic low-grade inflammation to functional decline in aging. Cell Metab 18, 519–532 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Goldberg EL & Dixit VD Drivers of age-related inflammation and strategies for healthspan extension. Immunol. Rev 265, 63–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Medzhitov R & Janeway CAJ Decoding the patterns of self and nonself by the innate immune system. Science 296, 298–300 (2002). [DOI] [PubMed] [Google Scholar]
- 117.Kepp O, Galluzzi L & Kroemer G Mitochondrial control of the NLRP3 inflammasome. Nat. Immunol 12, 199–200 (2011). [DOI] [PubMed] [Google Scholar]
- 118.Ferrucci L et al. The origins of age-related proinflammatory state. Blood 105, 2294–2299 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sies H, Berndt C & Jones DP Oxidative stress. Annu. Rev. Biochem 86, 715–748 (2017). [DOI] [PubMed] [Google Scholar]
- 120.Bektas A et al. Age-associated alterations in inducible gene transcription in human CD4+ T lymphocytes. Aging 5, 18–36 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bektas A et al. Age-associated changes in basal NF-κB function in human CD4+ T lymphocytes via dysregulation of PI3 kinase. Aging 6, 957–974 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Cannon MJ, Schmid DS & Hyde TB Review of cytomegalovirus seroprevalence and demographic characteristics associated with infection. Rev. Med. Virol 20, 202–213 (2010). [DOI] [PubMed] [Google Scholar]
- 123.Vescovini R et al. Massive load of functional effector CD4+ and CD8+ T cells against cytomegalovirus in very old subjects. J. Immunol 179, 4283–4291 (2007). [DOI] [PubMed] [Google Scholar]
- 124.Simon CO et al. CD8 T cells control cytomegalovirus latency by epitope-specific sensing of transcriptional reactivation. J. Virol 80, 10436–10456 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Roberts ET, Haan MN, Dowd JB & Aiello AE Cytomegalovirus antibody levels, inflammation, and mortality among elderly Latinos over 9 years of follow-up. Am. J. Epidemiol 172, 363–371 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Spyridopoulos I et al. CMV seropositivity and T-cell senescence predict increased cardiovascular mortality in octogenarians: results from the Newcastle 85+ study. Aging Cell 15, 389–392 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Adriaensen W et al. CD4:8 ratio above 5 is associated with all-cause mortality in CMV-seronegative very old women: results from the BELFRAIL study. J. Gerontol. A. Biol. Sci. Med. Sci 72, 1155–1162 (2017). [DOI] [PubMed] [Google Scholar]
- 128.Brodin P et al. Variation in the human immune system is largely driven by non-heritable influences. Cell 160, 37–47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Aiello AE, Chiu Y-L & Frasca D How does cytomegalovirus factor into diseases of aging and vaccine responses, and by what mechanisms? GeroScience 39, 261–271 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Goldeck D et al. No strong correlations between serum cytokine levels, CMV serostatus and hand-grip strength in older subjects in the Berlin BASE-II cohort. Biogerontology 17, 189–198 (2016). [DOI] [PubMed] [Google Scholar]
- 131.Nasi M et al. Ageing and inflammation in patients with HIV infection. Clin. Exp. Immunol 187, 44–52 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Neuhaus J et al. Markers of inflammation, coagulation, and renal function are elevated in adults with HIV infection. J. Infect. Dis 201, 1788–1795 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Losina E et al. Projecting 10-year, 20-year, and lifetime risks of cardiovascular disease in persons living with human immunodeficiency virus in the United States. Clin. Infect. Dis 65, 1266–1271 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Brothers TD et al. Frailty in people aging with human immunodeficiency virus (HIV) infection. J. Infect. Dis 210, 1170–1179 (2014). [DOI] [PubMed] [Google Scholar]
- 135.Stein JH & Hsue PY Inflammation, immune activation, and CVD risk in individuals with HIV infection. JAMA 308, 405–406 (2012). [DOI] [PubMed] [Google Scholar]
- 136.Brenchley JM et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat. Med 12, 1365–1371 (2006). [DOI] [PubMed] [Google Scholar]
- 137.Grunfeld C et al. Association of upper trunk and visceral adipose tissue volume with insulin resistance in control and HIV-infected subjects in the FRAM study. J. Acquir. Immune Def. Syndr 46, 283–290 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Odegaard AO et al. Oxidative stress, inflammation, endothelial dysfunction and incidence of type 2 diabetes. Cardiovasc. Diabetol 15, 51 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lai KSP et al. Peripheral inflammatory markers in Alzheimer’s disease: a systematic review and meta-analysis of 175 studies. J. Neurol. Neurosurg. Psychiatry 88, 876–882 (2017). [DOI] [PubMed] [Google Scholar]
- 140.Iseme RA et al. Is osteoporosis an autoimmune mediated disorder? Bone Rep 7, 121–131 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Dalle S, Rossmeislova L & Koppo K The role of inflammation in age-related sarcopenia. Front. Physiol 8, 1045 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Amdur RL et al. Inflammation and progression of CKD: the CRIC study. Clin. J. Am. Soc. Nephrol 11, 1546–1556 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kohler O, Krogh J, Mors O & Eriksen Benros M Inflammation in depression and the potential for anti-inflammatory treatment. Curr. Neuropharmacol 14, 732–742 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Libby P, Ridker PM & Hansson GK Progress and challenges in translating the biology of atherosclerosis. Nature 473, 317–325 (2011). [DOI] [PubMed] [Google Scholar]
- 145.Andreou DE & Andreadou I Atherosclerosis:an inflammatory disease. Pharmakeftiki 22, 83–96 (2009). [Google Scholar]
- 146.Hansson GK Inflammation, atherosclerosis and coronary artery disease. N. Engl. J. Med 352, 1685–1695 (2005). [DOI] [PubMed] [Google Scholar]
- 147.Libby P, Ridker PM & Hansson GK Inflammation in atherosclerosis. from pathophysiology to practice. Am. J. Coll. Cardiol 54, 2129–2138 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.De Caterina R, D’Ugo E & Libby P Inflammation and thrombosis – Testing the hypothesis with anti-inflammatory drug trials. Thromb. Haemost 116, 1012–1021 (2016). [DOI] [PubMed] [Google Scholar]
- 149.Libby P, Okamoto Y, Rocha VZ & Folco E Inflammation in atherosclerosis. Circ. J 74, 213–220 (2010). [DOI] [PubMed] [Google Scholar]
- 150.Duewell P et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464, 1357–1361 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Warnatsch A, Ioannou M, Wang Q & Papayannopoulos V Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science 349, 316–320 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Wang M, Kim SH, Monticone RE & Lakatta EG Matrix metalloproteinases promote arterial remodeling in aging, hypertension, and atherosclerosis. Hypertension 65, 698–703 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Matthews C et al. Vascular smooth muscle cells undergo telomere-based senescence in human atherosclerosis: effects of telomerase and oxidative stress. Circ. Res 99, 156–164 (2006). [DOI] [PubMed] [Google Scholar]
- 154.Grootaert MOJ et al. Vascular smooth muscle cell death, autophagy and senescence in atherosclerosis. Cardiovasc. Res 114, 622–634 (2018). [DOI] [PubMed] [Google Scholar]
- 155.Ketelhuth DFJ & Hansson GK Adaptive response of T and B cells in atherosclerosis. Circ. Res 118, 668–678 (2016). [DOI] [PubMed] [Google Scholar]
- 156.Feinberg MW & Moore KJ MicroRNA regulation of atherosclerosis. Circ. Res 118, 703–720 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ridker PM, Cushman M, Stampfer MJ, Tracy RP & Hennekens CH Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N. Engl. J. Med 336, 973–979 (1997). [DOI] [PubMed] [Google Scholar]
- 158.Ridker PM, Hennekens CH, Buring JE & Rifai N C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N. Engl. J. Med 342, 836–843 (2000). [DOI] [PubMed] [Google Scholar]
- 159.Cushman M et al. C-reactive protein and the 10-year incidence of coronary heart disease in older men and women: the cardiovascular health study. Circulation 112, 25–31 (2005). [DOI] [PubMed] [Google Scholar]
- 160.Cesari M et al. Inflammatory markers and onset of cardiovascular events: results from the Health ABC study. Circulation 108, 2317–2322 (2003). [DOI] [PubMed] [Google Scholar]
- 161.Levinson SS Rosuvastatin to prevent vascular events in men and women with elevated C.-reactive protein – an analysis. Clin. J. Ligand Assay 31, 25–28 (2008). [Google Scholar]
- 162.Noren Hooten N, Ejiogu N, Zonderman AB & Evans MK Association of oxidative DNA damage and C-reactive protein in women at risk for cardiovascular disease. Arterioscler. Thromb. Vasc. Biol 32, 2776–2784 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Ridker PM From C-reactive protein to interleukin-6 to interleukin-1: moving upstream to identify novel targets for atheroprotection. Circ. Res 118, 145–156 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Elliott P et al. Genetic loci associated with C-reactive protein levels and risk of coronary heart disease. JAMA - J. Am. Med. Assoc 302, 37–48 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.McInnes IB et al. Effect of interleukin-6 receptor blockade on surrogates of vascular risk in rheumatoid arthritis: MEASURE, a randomised, placebo-controlled study. Ann. Rheum. Dis 74, 694–702 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/show/NCT01331837 (2011).
- 167.Libby P Interleukin-1 β as a target for atherosclerosis therapy: biological basis of CANTOS and beyond. J. Am. Coll. Cardiol 70, 2278–2289 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Ridker PM et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med 377, 1119–1131 (2017). [DOI] [PubMed] [Google Scholar]
- 169.Ridker PM et al. Effects of interleukin-1β inhibition with canakinumab on hemoglobin A1c, lipids, C-reactive protein, interleukin-6, and fibrinogen. Circulation 126, 2739–2748 (2012). [DOI] [PubMed] [Google Scholar]
- 170.Everett BM et al. Rationale and design of the cardiovascular inflammation reduction trial: a test of the inflammatory hypothesis of atherothrombosis. Am. Heart J 166, 199–207.e15 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Daniels LB Pretenders and contenders: inflammation, C-reactive protein and interleukin-6. Am. J. Heart Assoc 6, e007490 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Nidorf SM, Mbbs JWE, Hons CAB & Thompson PL Low-dose colchicine for secondary prevention of cardiovascular disease. Am. J. Coll. Cardiol 61, 404–410 (2013). [DOI] [PubMed] [Google Scholar]
- 173.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/show/NCT02551094 (2015).
- 174.Cigolle CT, Blaum CS & Halter JB Diabetes and cardiovascular disease prevention in older adults. Clin. Geriatr. Med 25, 607–641 (2009). [DOI] [PubMed] [Google Scholar]
- 175.Stout MB, Justice JN, Nicklas BJ & Kirkland JL Physiological aging: Links among adipose tissue dysfunction, diabetes, and frailty. Physiology 32, 9–19 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Halter JB et al. Diabetes and cardiovascular disease in older adults: current status and future directions. Diabetes 63, 2578–2589 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Nahrendorf M & Swirski FK Immunology. Neutrophil-macrophage communication in inflammation and atherosclerosis. Science 349, 237–238 (2015). [DOI] [PubMed] [Google Scholar]
- 178.Gimbrone MAJ & Garcia-Cardena G Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ. Res 118, 620–636 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Shakeri H, Lemmens K, Gevaert AB, De Meyer GRY & Segers V Cellular senescence links aging and diabetes in cardiovascular disease. Am. J. Physiol. Heart Circ. Physiol 10.1152/ajpheart.00287.2018 (2018). [DOI] [PubMed] [Google Scholar]
- 180.Aryan Z et al. Baseline high-sensitivity C-reactive protein predicts macrovascular and microvascular complications of type 2 diabetes: a population-pased study. Ann. Nutr. Metab 72, 287–295 (2018). [DOI] [PubMed] [Google Scholar]
- 181.Eguchi S, Kawai T, Scalia R & Rizzo V Understanding Angiotensin II type 1 receptor signaling in vascular pathophysiology. Hypertension 71, 804–810 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Jacobsson LT et al. Treatment with tumor necrosis factor blockers is associated with a lower incidence of first cardiovascular events in patients with rheumatoid arthritis. J. Rheumatol 32, 1213–1218 (2005). [PubMed] [Google Scholar]
- 183.Greenberg JD et al. Tumour necrosis factor antagonist use and associated risk reduction of cardiovascular events among patients with rheumatoid arthritis. Ann. Rheum. Dis 70, 576–582 (2011). [DOI] [PubMed] [Google Scholar]
- 184.Solomon DH et al. Cardiovascular risk in rheumatoid arthritis: comparing TNF-α blockade with nonbiologic DMARDs. Am. J. Med 126, 730.e9–730.e17 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Bili A et al. Tumor necrosis factor α inhibitor use and decreased risk for incident coronary events in rheumatoid arthritis. Arthritis Care Res 66, 355–363 (2014). [DOI] [PubMed] [Google Scholar]
- 186.Roubille C et al. The effects of tumour necrosis factor inhibitors, methotrexate, non-steroidal anti-inflammatory drugs and corticosteroids on cardiovascular events in rheumatoid arthritis, psoriasis and psoriatic arthritis: a systematic review and meta-analysis. Ann. Rheum. Dis 74, 480–489 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Low ASL et al. Relationship between exposure to tumour necrosis factor inhibitor therapy and incidence and severity of myocardial infarction in patients with rheumatoid arthritis. Ann. Rheum. Dis 76, 654–660 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Shaaban D & Al-Mutairi N The effect of tumor necrosis factor inhibitor therapy on the incidence of myocardial infarction in patients with psoriasis: a retrospective study. J. Dermatolog. Treat 29, 3–7 (2018). [DOI] [PubMed] [Google Scholar]
- 189.Yang Z, Lin N, Li L & Li Y The effect of TNF inhibitors on cardiovascular events in psoriasis and psoriatic arthritis: an updated meta-analysis. Clin. Rev. Allergy Immunol 51, 240–247 (2016). [DOI] [PubMed] [Google Scholar]
- 190.Anker SD Inflammatory mediators in chronic heart failure: an overview. Heart 90, 464–470 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Levine B, Kalman J, Mayer L, Fillit HM & Packer M Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N. Engl. J. Med 323, 236–241 (1990). [DOI] [PubMed] [Google Scholar]
- 192.Torre-Amione G et al. Proinflammatory cytokine levels in patients with depressed left ventricular ejection fraction: A report from the studies of left ventricular dysfunction (SOLVD). J. Am. Coll. Cardiol 27, 1201–1206 (1996). [DOI] [PubMed] [Google Scholar]
- 193.Mann DL et al. Targeted anticytokine therapy in patients with chronic heart failure: results of the randomized etanercept worldwide evaluation (RENEWAL). Circulation 109, 1594–1602 (2004). [DOI] [PubMed] [Google Scholar]
- 194.Chung ES Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-α, in patients with moderate-to-severe heart failure: results of the anti-TNF therapy against congestive heart failure. Circulation 107, 3133–3140 (2003). [DOI] [PubMed] [Google Scholar]
- 195.Forman DE et al. Multimorbidity in older adults with cardiovascular disease. J. Am. Coll. Cardiol 71, 2149–2161 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Brandenberger C & Muhlfeld C Mechanisms of lung aging. Cell Tissue Res 367, 469–480 (2017). [DOI] [PubMed] [Google Scholar]
- 197.Tisminetzky M, Goldberg R & Gurwitz JH Magnitude and impact of multimorbidity on clinical outcomes in older adults with cardiovascular aisease: a literature review. Clin. Geriatr. Med 32, 227–246 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Fried LP et al. Frailty in older adults: evidence for a phenotype. J. Gerontol. A. Biol. Sci. Med. Sci 56, M146–M156 (2001). [DOI] [PubMed] [Google Scholar]
- 199.Bergman H et al. Frailty: An emerging research and clinical paradigm — issues and controversies. J. Gerontol. A. Biol. Sci. Med. Sci 62, 731–737 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Afilalo J et al. Frailty assessment in the cardiovascular care of older adults. J. Am. Coll. Cardiol 63, 747–762 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Woods NF et al. Frailty: emergence and consequences in women aged 65 and older in the Women’s Health Initiative Observational Study. J. Am. Geriatr. Soc 53, 1321–1330 (2005). [DOI] [PubMed] [Google Scholar]
- 202.Corti MC, Salive ME & Guralnik JM Serum albumin and physical function as predictors of coronary heart disease mortality and incidence in older persons. Clin. J. Epidemiol 49, 519–526 (1996). [DOI] [PubMed] [Google Scholar]
- 203.McDermott MM et al. Decline in functional performance predicts later increased mobility loss and mortality in peripheral arterial disease. J. Am. Coll. Cardiol 57, 962–970 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Walker KA et al. Midlife systemic inflammation is associated with frailty in later life: the ARIC Study. J. Gerontol. A Biol. Sci. Med. Sci 10.1093/gerona/gly045 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Abbatecola AM & Paolisso G Is there a relationship between insulin resistance and frailty syndrome? Curr. Pharm. Des 14, 405–410 (2008). [DOI] [PubMed] [Google Scholar]
- 206.Barzilay JI et al. Insulin resistance and inflammation as precursors of frailty: the Cardiovascular Health Study. Arch. Intern. Med 167, 635–641 (2007). [DOI] [PubMed] [Google Scholar]
- 207.Walston J et al. Frailty and activation of the inflammation and coagulation systems with and without clinical comorbidities: results from the Cardiovascular Health Study. Arch. Intern. Med 162, 2333–2341 (2002). [DOI] [PubMed] [Google Scholar]
- 208.Afilalo J, Karunananthan S, Eisenberg MJ, Alexander KP & Bergman H Role of frailty in patients with cardiovascular disease. Am. J. Cardiol 103, 1616–1621 (2009). [DOI] [PubMed] [Google Scholar]
- 209.Gupta J et al. Association between albuminuria, kidney function, and inflammatory biomarker profile in CKD in CRIC. Clin. J. Am. Soc. Nephrol 7, 1938–1946 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Mc Causland FR et al. C-reactive protein and risk of ESRD: results from the trial to reduce cardiovascular events with aranesp therapy (TREAT). Am. J. Kidney Dis 68, 873–881 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Grivennikov SI, Greten FR & Karin M Immunity. inflammation, and cancer. Cell 140, 883–899 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Todoric J, Antonucci L & Karin M Targeting inflammation in cancer prevention and therapy. Cancer Prev. Res 9, 895–905 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Howren MB, Lamkin DM & Suls J Associations of depression with C-reactive protein, IL-1, and IL-6: a meta-analysis. Psychosom. Med 71, 171–186 (2009). [DOI] [PubMed] [Google Scholar]
- 214.Miller AH, Maletic V & Raison CL Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol. Psychiatry 65, 732–741 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Matthews KA et al. Are there bi-directional associations between depressive symptoms and C-reactive protein in mid-life women? Brain. Behav. Immun 24, 96–101 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Zalli A, Jovanova O, Hoogendijk WJG, Tiemeier H & Carvalho LA Low-grade inflammation predicts persistence of depressive symptoms. Psychopharmacology 233, 1669–1678 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Lamers F, Milaneschi Y, de Jonge P, Giltay EJ & Penninx BWJH Metabolic and inflammatory markers: associations with individual depressive symptoms. Psychol. Med 48, 1102–1110 (2018). [DOI] [PubMed] [Google Scholar]
- 218.Heneka MT et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol 14, 388–405 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Eikelenboom P et al. Innate immunity and the etiology of late-onset Alzheimer’s disease. Neurodegener. Dis 10, 271–273 (2012). [DOI] [PubMed] [Google Scholar]
- 220.Hansen PR Chronic inflammatory diseases and atherosclerotic cardiovascular disease: Innocent bystanders or partners in crime? Curr. Pharm. Des 24, 281–290 (2018). [DOI] [PubMed] [Google Scholar]
- 221.Goldfine AB & Shoelson SE Therapeutic approaches targeting inflammation for diabetes and associated cardiovascular risk. J. Clin. Invest 127, 83–93 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Zitvogel L, Pietrocola F & Kroemer G Nutrition, inflammation and cancer. Nat. Immunol 18, 843–850 (2017). [DOI] [PubMed] [Google Scholar]
- 223.Morgan AR et al. The correlation between inflammatory biomarkers and polygenic risk score in Alzheimer’s Disease. J. Alzheimers. Dis 56, 25–36 (2017). [DOI] [PubMed] [Google Scholar]
- 224.Schlegel TF, Hawkins RJ, Lewis CW, Motta T & Turner AS The effects of augmentation with swine small intestine submucosa on tendon healing under tension: histologic and mechanical evaluations in sheep. Am. J. Sports Med 34, 275–280 (2006). [DOI] [PubMed] [Google Scholar]
- 225.Ferrucci L et al. Change in muscle strength explains accelerated decline of physical function in older women with high interleukin-6 serum levels. J. Am. Geriatr. Soc 50, 1947–1954 (2002). [DOI] [PubMed] [Google Scholar]
- 226.Visser M et al. Relationship of interleukin-6 and tumor necrosis factor-α with muscle nass and muscle strength in elderly men and women: the Health ABC Study. J. Gerontol. A Biol. Sci. Med. Sci 57, M326–M332 (2002). [DOI] [PubMed] [Google Scholar]
- 227.Cesari M et al. Inflammatory markers and physical performance in older persons: the InCHIANTI Study. J. Gerontol. A Biol. Sci. Med. Sci 59, M242–M248 (2004). [DOI] [PubMed] [Google Scholar]
- 228.Santos-Eggimann B, Cuénoud P, Spagnoli J & Junod J Prevalence of frailty in middle-aged and older community-dwelling Europeans living in 10 countries. J. Gerontol. A Biol. Sci. Med. Sci 64, 675–681 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Walston J et al. Research agenda for frailty in older adults: toward a better understanding of physiology and etiology: summary from the American Geriatrics Society/National Institute on Aging research conference on frailty in older adults. J. Am. Geriatr. Soc 54, 991–1001 (2006). [DOI] [PubMed] [Google Scholar]
- 230.Stepanova M, Rodriguez E, Birerdinc A & Baranova A Age-independent rise of inflammatory scores may contribute to accelerated aging in multi-morbidity. Oncotarget 6, 1414–1421 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Friedman EM, Montez JK, Sheehan CMD, Guenewald TL & Seeman TE Childhood adversities and adult cardiometabolic health: does the quantity, timing, and type of adversity matter? J. Aging Health 27, 1311–1338 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Hubbard RE, O’Mahony MS, Savva GM, Calver BL & Woodhouse KW Inflammation and frailty measures in older people. J. Cell Mol. Med 13, 3103–3109 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Newman AB et al. Weight change in old age and its association with mortality. J. Am. Geriatr. Soc 49, 1309–1318 (2001). [DOI] [PubMed] [Google Scholar]
- 234.Higashi Y et al. Insulin-like growth factor-1 receptor deficiency in macrophages accelerates atherosclerosis and induces an unstable plaque phenotype in apolipoprotein E-deficient mice. Circulation 133, 2263–2278 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Lazarus DD, Moldawer LL & Lowry SF Insulin-like growth factor-1 activity is inhibited by interleukin-1α, tumor necrosis factor-α, and interleukin-6. Lymphokine Cytokine Res 12, 219–223 (1993). [PubMed] [Google Scholar]
- 236.Barbieri M et al. Chronic inflammation and the effect of IGF-I on muscle strength and power in older persons. Am. J. Physiol. Metab 284, E481–E487 (2003). [DOI] [PubMed] [Google Scholar]
- 237.Cappola AR et al. Insulin-like growth factor I and interleukin-6 contribute synergistically to disability and mortality in older women. J. Clin. Endocrinol. Metab 88, 2019–2025 (2003). [DOI] [PubMed] [Google Scholar]
- 238.Timmerman KL et al. Pharmacological vasodilation improves insulin-stimulated muscle protein anabolism but not glucose utilization in older adults. Diabetes 59, 2764–2771 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Moaddel R et al. Plasma biomarkers of poor muscle quality in older men and women from the Baltimore Longitudinal Study of Aging. J. Gerontol. A Biol. Sci. Med. Sci 71, 1266–1272 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Fichtlscherer S et al. Elevated C-reactive protein levels and impaired endothelial vasoreactivity in patients with coronary artery disease. Circulation 102, 1000–1006 (2000). [DOI] [PubMed] [Google Scholar]
- 241.Bar-Shai M, Carmeli E & Reznick AZ The role of NF-κB in protein breakdown in immobilization, aging, and exercise: From basic processes to promotion of health. Ann. NY Acad. Sci 1057, 431–447 (2005). [DOI] [PubMed] [Google Scholar]
- 242.Justice JN et al. Cellular senescence biomarker p16INK4a+ cell burden in thigh adipose is associated with poor physical function in older women. J. Gerontol. A Biol. Sci. Med. Sci 73, 939–945 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Roth SM, Metter EJ, Ling S & Ferrucci L Inflammatory factors in age-related muscle wasting. Curr. Opin. Rheumatol 18, 625–630 (2006). [DOI] [PubMed] [Google Scholar]
- 244.Jo E, Lee S-R, Park B-S & Kim J-S Potential mechanisms underlying the role of chronic inflammation in age-related muscle wasting. Aging Clin. Exp. Res 24, 412–422 (2012). [DOI] [PubMed] [Google Scholar]
- 245.Walston JD Connecting age-related biological decline to frailty and late-life vulnerability. Nestle Nutr. Inst. Workshop Ser 83, 1–10 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Wang J et al. Vascular smooth muscle cell senescence promotes atherosclerosis and features of plaque vulnerability. Circulation 132, 1909–1919 (2015). [DOI] [PubMed] [Google Scholar]
- 247.Ntanasi E et al. Adherence to mediterranean diet and frailty. J. Am. Med. Dir. Assoc 19, 315–322.e2 (2017). [DOI] [PubMed] [Google Scholar]
- 248.Talegawkar SA et al. A higher adherence to a mediterranean-style diet is inversely associated with the development of frailty in community-dwelling elderly men and women. J. Nutr 142, 2161–2166 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Rosato V et al. Mediterranean diet and cardiovascular disease: a systematic review and meta-analysis of observational studies. Eur. J. Nutr 10.1007/s00394-017-1582-0 (2017). [DOI] [PubMed] [Google Scholar]
- 250.Dinu M, Pagliai G, Casini A & Sofi F Mediterranean diet and multiple health outcomes: an umbrella review of meta-analyses of observational studies and randomized trials. Nutr. Metab. Cardiovasc. Dis 27, e21 (2017). [DOI] [PubMed] [Google Scholar]
- 251.Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 324, 71–86 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.De Caterina R [Aspirin for primary cardiovascular disease prevention - an update]. G. Ital. Cardiol 18, 1–6 (2017). [DOI] [PubMed] [Google Scholar]
- 253.Landi F et al. Nonsteroidal anti-inflammatory drug (NSAID) use and sarcopenia in older people: results from the ilsirente study. J. Am. Med. Dir. Assoc 14, 626.e9–626.e13 (2013). [DOI] [PubMed] [Google Scholar]
- 254.Wang C-P, Lorenzo C, Habib SL, Jo B & Espinoza SE Differential effects of metformin on age related comorbidities in older men with type 2 diabetes. J. Diabetes Complications 31, 679–686 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Laksmi PW, Setiati S, Tamin TZ & Soewondo P Effect of metformin on handgrip strength, gait speed, myostatin serum level, and health-related quality of life: a double blind randomized controlled trial among non-diabetic pre-frail elderly patients. Acta Med. Indones 49, 118–127 (2017). [PubMed] [Google Scholar]
- 256.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02570672 (2017).
- 257.Manini TM et al. ENabling reduction of low-grade inflammation in SEniors pilot study: concept, rationale, and design. J. Am. Geriatr. Soc 65, 1961–1968 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Golpanian S et al. Allogeneic human mesenchymal stem cell infusions for aging frailty. J. Gerontol. A. Biol. Sci. Med. Sci 72, 1505–1512 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Tompkins BA et al. Allogeneic mesenchymal stem cells ameliorate aging frailty: a phase II randomized, double-blind, placebo-controlled clinical trial. J. Gerontol. A. Biol. Sci. Med. Sci 72, 1513–1522 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.LaCroix AZ et al. Statin use and incident frailty in women aged 65 years or older: prospective findings from the Women’s Health Initiative Observational Study. J. Gerontol. A. Biol. Sci. Med. Sci 63, 369–375 (2008). [DOI] [PubMed] [Google Scholar]
- 261.Barzilai N, Huffman DM, Muzumdar RH & Bartke A The critical role of metabolic pathways in aging. Diabetes 61, 1315–1322 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Fontana L Neuroendocrine factors in the regulation of inflammation: excessive adiposity and calorie restriction. Exp. Gerontol 44, 41–45 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Kim HJ et al. Modulation of redox-sensitive transcription factors by calorie restriction during aging. Mech. Ageing Dev 123, 1589–1595 (2002). [DOI] [PubMed] [Google Scholar]
- 264.LaRocca TJ, Martens CR & Seals DR Nutrition and other lifestyle influences on arterial aging. Ageing Res. Rev 39, 106–119 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Johnson SC, Rabinovitch PS & Kaeberlein M MTOR is a key modulator of ageing and age-related disease. Nature 493, 338–345 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Li J, Kim SG & Blenis J Rapamycin: one drug, many effects. Cell Metab 19, 373–379 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Harries LW et al. Advancing age is associated with gene expression changes resembling mTOR inhibition: evidence from two human populations. Mech. Ageing Dev 133, 556–562 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Blagosklonny MV From rapalogs to anti-aging formula. Oncotarget 8, 35492–35507 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Halloran J et al. Chronic inhibition of mammalian target of rapamycin by rapamycin modulates cognitive and non-cognitive components of behavior throughout lifespan in mice. Neuroscience 223, 102–113 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Zhang Y et al. Rapamycin extends life and health in C57BL/6 mice. J. Gerontol. A. Biol. Sci. Med. Sci 69A, 119–130 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Harrison DE et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460, 392–395 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Liao CY et al. Rapamycin reverses metabolic deficits in lamin A/C-deficient mice. Cell Rep 17, 2542–2552 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Strong R et al. Nordihydroguaiaretic acid and aspirin increase lifespan of genetically heterogeneous male mice. Aging Cell 7, 641–650 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Saisho Y Metformin and inflammation: Its potential beyond glucose-lowering effect. Endocr. Metab. Immune Disord. Drug Targets 15, 196–205 (2015). [DOI] [PubMed] [Google Scholar]
- 275.Martin-Montalvo A et al. Metformin improves healthspan and lifespan in mice. Nat. Commun 4, 2192 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Campbell JM, Bellman SM, Stephenson MD & Lisy K Metformin reduces all-cause mortality and diseases of ageing independent of its effect on diabetes control: a systematic review and meta-analysis. Ageing Res. Rev 40, 31–44 (2017). [DOI] [PubMed] [Google Scholar]
- 277.Barzilai N, Crandall JP, Kritchevsky SB & Espeland MA Metformin as a tool to target aging. Cell Metab 23, 1060–1065 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Robbins PD & Niedernhofer LJ Advances in therapeutic approaches to extend healthspan: a perspective from the 2nd Scripps symposium on the biology of aging. Aging Cell 16, 610–614 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Walter E & Scott M The life & work of Rudolf Virchow 1821–1902: ‘Cell theory, thrombosis and the sausage duel’. J. Intensive Care Soc 18, 234–235 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Ferrucci L et al. Proinflammatory state and circulating erythropoietin in persons with and without anemia. Am. J. Med 118, 1288.e11–1288.e19 (2005). [DOI] [PubMed] [Google Scholar]
- 281.de Luca C & Olefsky JM Inflammation and insulin resistance. FEBS Lett 582, 97–105 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Abbatecola AM et al. Diverse effect of inflammatory markers on insulin resistance and insulin-resistance syndrome in the elderly. J. Am. Geriatr. Soc 52, 399–404 (2004). [DOI] [PubMed] [Google Scholar]
- 283.Hotamisligil GS The role of TNFα and TNF receptors in obesity and insulin resistance. J. Intern. Med 245, 621–625 (1999). [DOI] [PubMed] [Google Scholar]
- 284.Shimobayashi M et al. Insulin resistance causes inflammation in adipose tissue. J. Clin. Invest 128, 1538–1550 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Abdelmagid SM, Barbe MF & Safadi FF Role of inflammation in the aging bones. Life Sci 123, 25–34 (2015). [DOI] [PubMed] [Google Scholar]
- 286.Goldring SR Pathogenesis of bone erosions in rheumatoid arthritis. Curr. Opin. Rheumatol 14, 406–410 (2002). [DOI] [PubMed] [Google Scholar]
- 287.Hahn WS et al. Proinflammatory cytokines differentially regulate adipocyte mitochondrial metabolism, oxidative stress, and dynamics. Am. J. Physiol. Endocrinol. Metab 306, E1033–E1045 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Lezi E, Burns JM & Swerdlow RH Effect of high-intensity exercise on aged mouse brain mitochondria, neurogenesis, and inflammation. Neurobiol. Aging 35, 2574–2583 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Borsini A et al. Interferon-α reduces human hippocampal neurogenesis and increases apoptosis via activation of distinct STAT1-dependent mechanisms. Int. J. Neuropsychopharmacol 21, 187–200 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Li YP & Stashenko P Proinflammatory cytokines tumor necrosis factor-α and IL-6, but not IL-1, down-regulate the osteocalcin gene promoter. J. Immunol 148, 788–794 (1992). [PubMed] [Google Scholar]
- 291.Ginaldi L, Di Benedetto MC & De Martinis M Osteoporosis, inflammation and ageing. Immun. Ageing 2, 14 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Audet M-C & Anisman H Interplay between pro-inflammatory cytokines and growth factors in depressive illnesses. Front. Cell. Neurosci 7, 68 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Ridker PM et al. Relationship of C-reactive protein reduction to cardiovascular event reduction following treatment with canakinumab: a secondary analysis from the CANTOS randomised controlled trial. Lancet 391, 319–328 (2017). [DOI] [PubMed] [Google Scholar]
- 294.EU Clinical Trial Register. ClinicalTrialsRegister.eu https://www.clinicaltrialsregister.eu/ctr-search/search?query=LoDoCo2 (2016).
- 295.Navarro-Gonzalez JF et al. Effect of pentoxifylline on renal function and urinary albumin excretion in patients with diabetic kidney disease: the PREDIAN trial. J. Am. Soc. Nephrol 26, 220–229 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Voelker J et al. Anti–TGF-β 1 antibody therapy in patients with diabetic nephropathy. J. Am. Soc. Nephrol 28, 953–962 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Flossmann E & Rothwell PM Effect of aspirin on long-term risk of colorectal cancer: consistent evidence from randomised and observational studies. Lancet 369, 1603–1613 (2007). [DOI] [PubMed] [Google Scholar]
- 298.Rothwell PM et al. Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-year follow-up of five randomised trials. Lancet 376, 1741–1750 (2010). [DOI] [PubMed] [Google Scholar]
- 299.Iyengar RL et al. NSAIDs are associated with lower depression scores in patients with osteoarthritis. Am. J. Med 126, 1017.e11–1017.e18 (2013). [DOI] [PubMed] [Google Scholar]
- 300.Fields C, Drye L, Vaidya V & Lyketsos C Celecoxib or naproxen treatment does not benefit depressive symptoms in persons age 70 and older: findings from a randomized controlled trial. Am. J. Geriatr. Psychiatry 20, 505–513 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Raison CL et al. A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression: the role of baseline inflammatory biomarkers. Arch. Gen. Psychiatry 70, 31–41 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Menter A et al. The effect of adalimumab on reducing depression symptoms in patients with moderate to severe psoriasis: a randomized clinical trial. J. Am. Acad. Dermatol 62, 812–818 (2010). [DOI] [PubMed] [Google Scholar]
- 303.Tyring S et al. Etanercept and clinical outcomes, fatigue, and depression in psoriasis: double-blind placebo-controlled randomised phase III trial. Lancet 367, 29–35 (2006). [DOI] [PubMed] [Google Scholar]
- 304.Aisen PS, Schmeidler J & Pasinetti GM Randomized pilot study of nimesulide treatment in alzheimer’s disease. Neurology 58, 1050–1054 (2002). [DOI] [PubMed] [Google Scholar]
- 305.Aisen PS et al. Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression. JAMA 289, 2819 (2003). [DOI] [PubMed] [Google Scholar]
- 306.Reines SA et al. Rofecoxib: No effect on Alzheimer’s disease in a 1-year, randomized, blinded, controlled study. Neurology 62, 66–71 (2004). [DOI] [PubMed] [Google Scholar]
- 307.Scharf S, Mander A, Ugoni A, Vajda F & Christophidis N A double-blind, placebo-controlled trial of diclofenac/misoprostol in Alzheimer’s disease. Neurology 53, 197–197 (1999). [DOI] [PubMed] [Google Scholar]
- 308.Martin BK et al. Cognitive function over time in the Alzheimer’s disease anti-inflammatory prevention trial (ADAPT): results of a randomized, controlled trial of naproxen and celecoxib. Arch. Neurol 65, 896–905 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/show/NCT02284906 (2014).
- 310.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/show/NCT01931566 (2013).