

Abstract
Protein conformations play crucial roles in most, if not all, biological processes. Here we show that the current carried through a nanopore by ions allows monitoring conformational changes of single and native substrate-binding domains (SBD) of an ATP-Binding Cassette importer in real-time. Comparison with single-molecule Förster Resonance Energy Transfer and ensemble measurements revealed that proteins trapped inside the nanopore have bulk-like properties. Two ligand-free and two ligand-bound conformations of SBD proteins were inferred and their kinetic constants were determined. Remarkably, internalized proteins aligned with the applied voltage bias, and their orientation could be controlled by the addition of a single charge to the protein surface. Nanopores can thus be used to immobilize proteins on a surface with a specific orientation, and will be employed as nanoreactors for single-molecule studies of native proteins. Moreover, nanopores with internal protein adaptors might find further practical applications in multianalyte sensing devices.
Introduction
Protein conformational dynamics play key roles in molecular recognition1−3 and regulation of protein activity.4 The lack of understanding of macromolecule dynamics is a fundamental problem in today’s biochemistry. Single-molecule techniques allow probing the conformational heterogeneity and structural dynamics of proteins in real-time. However, such approaches typically require labeling with fluorophores and/or tethering proteins to a surface, which in turn may perturb the very same dynamic behavior that is intended to be studied.5
Ionic currents through individual nanopores can be employed to sample the activity of unlabeled molecules at the single-molecule level. Initial work focused on observing the traversing of polymers across single nanopores,6,7 or the binding8 or reacting of small molecules with a nanopore in real-time.9 More recently, nanopore currents have been employed to sample enzymatic reactions.10 Notable examples are the visualization of the intermediate mechanical steps during an helicase catalyzed unwinding of DNA,11 or the sampling of the kinetic intermediates of the GroEL-assisted protein refolding reaction with an engineered co-chaperon GroES-nanopore.12
A more generic approach to nanopore enzymology, however, is sampling proteins inside a nanopore. Work with solid-state nanopores showed that many features of proteins that are rapidly diffusing across a nanopore, including their approximate shape, dipole moment and conformation can be identified.13,14 We reported that single proteins can be incorporated and sampled inside Cytolysin A (ClyA) biological nanopores from Salmonella typhi (Figure 1) for tens of seconds,15−19 which allowed sampling the ubiquitination of proteins in solution19 or the binding of ligands to the proteins inside the nanopore.15,20
Figure 1.

Trapping proteins inside the ClyA nanopore. (Left) Surface representation of Type I ClyA-AS (blue) in the bilayer (gray) with SBD1 (PDB-ID = 4KPT) in open conformation lodged inside the nanopore. (Right) SBD2 in open (PDB-ID = 4KR5) and closed (PDB-ID = 4KQP) conformation. SBDs are colored according to the residue type: basic residues are colored blue, acidic residues red, polar residues green, and nonpolar residues white. Created with VMD24 while ClyA was created by homology modeling using the E. coli ClyA crystal structure.16,25
Here, we use substrate-binding domains (SBD1 and SBD2) of the ABC importer Gln PQ from Lactococcus lactis (Figure 1) as model systems to probe the conformational dynamics of proteins within a nanopore. SBD1 and SBD2 differ in amino acid specificity and dynamics of closing and opening upon ligand binding as described previously.21 Substrate-binding proteins undergo large conformational changes upon binding to their ligands. SBD1 and SBD2 have been previously characterized by several analytical techniques,22,23 including single-molecule Förster resonance energy transfer (smFRET),21 making them ideal to characterize this new experimental setup. We found that nanopore currents can efficiently report the conformational changes of SBD1 and SBD2. Remarkably, the kinetic constants for ligand binding to the proteins inside the nanopore are almost identical to those measured in solution. Thus, nanopores can be used as nanoreactors to report the conformational dynamics of proteins lodged inside the nanopore in real-time. Our technique does not require bulky tags and proteins are sampled with high bandwidth for tens of seconds, which is challenging in single-molecule fluorescence experiments.
Results and Discussion
Conformational Dynamics of SBD1 Inside ClyA Nanopores
SBD1 (74 nM) added to the cis side of single type I ClyA-AS nanopores (hereafter ClyA, Figure 1) entered the nanopore under negative applied potentials. The longest average residence time of SBD1 was under −60 mV (4.2 ± 1.8 s, n = 291 events, Figure 2A). Individual blockades showed a main current level LO with a residual current percent (Ires% = IB /IO × 100, with IB the blocked pore and IO the open pore current level) equal to 67.6 ± 0.1% (N = 5 independent nanopore experiments). The blockades switched with a frequency of 0.7 ± 0.2 s–1 to a second level (LC, Ires% = 66.8 ± 0.4%, Figure 2B). Then LC switched back to LO with an average rate of 15.1 ± 4.1 s–1 (n = 441). The addition of asparagine (0.4 μM, Figure 2B) to the cis side of the nanopore increased the frequency of LC blockades; and at 50 μM asparagine LC was the most prominent current blockade (Figure 2B). By contrast, SBD1(E184W), a variant that does not show any asparagine transport in L. lactis cells (Figure S1 of the Supporting Information, SI), showed only one single level (Ires% = 66.8 ± 0.1%, N = 3) that did not respond to asparagine (up to 1 mM, Figure 2C). Together, these results suggest that LO corresponds to the open conformation of SBD1 and LC to the closed asparagine-bound state of the protein.21 The current blockades allowed calculating the equilibrium dissociation constant (Kdapp) by plotting the relative dwell times of the closed population [LC/(LC + LO), Figure 2D]. The Kdapp calculated by this method (0.47 ± 0.03 μM) is similar to the values obtained by isothermal titration calorimetry (ITC, 0.2 μM) and smFRET (0.35 μM).21 The ligand-induced closing rate constant (kclosing = 1.4 ± 0.8 × 107 s–1 M–1) could be determined from the LO to LC transition and was similar to the values measured by smFRET (2.2 × 107 s–1 M–1, Figure 2E).21 The LC to LO transition showed a slight variation with the ligand concentration (Figure 2E). At high asparagine concentration the transition rate (4.8 ± 0.5 s–1, 2.8 μM asparagine) was similar to the rate constants measured by smFRET (4.2 s–1).21
Figure 2.

Conformational dynamics of SBD1 inside ClyA-AS. (A) Typical ionic current blockades provoked by the capture of SBD1 (74 nM, cis) by the ClyA-AS nanopore at −60 mV. The open pore (IO) and SBD-blocked (L) current levels are indicated, with LO and LC corresponding to the open and closed state of SBD1. (B) Details of SBD1 current blockade before and after addition of 0.40 μM and 50 μM asparagine (added cis). (C) Typical current blockades of the inactive SBD1(E184W) variant before and after addition of 50 μM asparagine showing no ligand-induced current blockades. (D) The Kdapp value of SBD1 for asparagine was obtained from the areas of the histograms of the open (LO) and ligand-bound (LC) populations as [LC/(LC + LO)]. The Kdapp is the concentration of substrate at 50% signal saturation. (E) Dependency of the opening and closing rates of SBD1 on asparagine concentration. The data in E were fitted to eq S5C (opening rates) and S6C (closing rates) as described in the SI. Current traces were collected in 150 mM NaCl, 15 mM Tris-HCl, pH 7.5 at 24 °C by applying a Bessel low-pass filter with a 2 kHz cutoff and sampled at 10 kHz. A postacquisition Gaussian filter of 100 Hz was applied. Experiments were performed at −60 mV.
Conformational Dynamics of SBD2 Inside ClyA Nanopores
The average residence time of SBD2 inside ClyA (3.9 ± 0.7 s, n = 225) was similar to that of SBD1, albeit at −100 mV. Contrary to SBD1, however, individual blockades showed three levels: LOA (Ires% = 61.9 ± 0.3%, rate = 3.6 ± 0.4 s–1, n = 2900, N = 3), LOB (Ires% = 59.3 ± 0.2%, rate = 6.1 ± 1.3 s–1, n = 2900, N = 3, Figure 3A), and LC (Ires% = 60.7 ± 0.2%, kopening = 55.6 ± 3.1 s–1, n = 856, N = 3). The addition of the substrate glutamine increased the frequency of LC current levels, and reduced the dwell time of the LOA and LOB levels (Figure 3A). When the inactive D417F variant of SBD221 was used instead of SBD2, LC events were not observed at any glutamine concentration tested (up to 200 μM, Figure 3B). Therefore, LC reflects the glutamine-bound closed conformation of SBD2 from which the equilibrium dissociation constant (Kdapp = 0.83 ± 0.10 μM, Figure 3C) could be measured. The Kdapp for internalized SBD2 proteins corresponds well to the values measured in solution by smFRET and ITC experiments (Kdapp = 1.1 ± 0.1 μM and Kdapp = 0.9 ± 0.2 μM, respectively).21 As observed for SBD1, the resulting opening rate showed a slight decrease with the ligand concentration (Figure 3D). At high ligand concentration the opening rate (kopening = 31.8 ± 3.7 s–1, 2.3 μM glutamine) was about 2-fold faster than the value measured by smFRET (kopening = 17.2 s–1). The rate constants measured from LOA and LOB were almost identical (4.3 ± 0.1 × 107 s–1 M–1 and 4.2 ± 0.1 × 107 s–1 M–1, respectively), and corresponded well to the closing rate constant measured with smFRET (3.8 × 107 s–1 M–1), suggesting that they are related to the closing of SBD2.21
Figure 3.

Orientation and dynamics of SBD2 measured by nanopore experiments. (A) Typical current blockade provoked by the capture of SBD2 (72 nM, cis) by the ClyA-AS nanopore. Left, apo-SBD2, middle current blockades after the addition of 0.40 μM of glutamine (cis); and right, blockades after adding 50 μM glutamine (cis). (B) Typical current blockades provoked by the capture of inactive SBD2(D417F) (70 nM) before and after the addition of 200 μM glutamine to the cis side. Red asterisks represent the restoration of the open pore current after SBD2(D417F) exited from the pore. (C) Kdapp value of SBD2 for glutamine obtained by fitting to a binding isotherm, using the relative closed population [LC/(LOA + LOB + LC)] at the indicated substrate concentrations. (D) Opening and closing rates of SBD2 versus the glutamine concentration. The data was fitted by eq S5C (opening rates) and S6C (closing rates) as described in the SI. Experiments were performed at −100 mV in 150 mM NaCl, 15 mM Tris-HCl, pH 7.5 at 24 °C by applying a Bessel low-pass filter with a 2 kHz cutoff and sampled at 10 kHz. A postacquisition Gaussian filter of 100 Hz was then applied.
SBD2 Has a Fixed Orientation Inside ClyA That Can Be Controlled
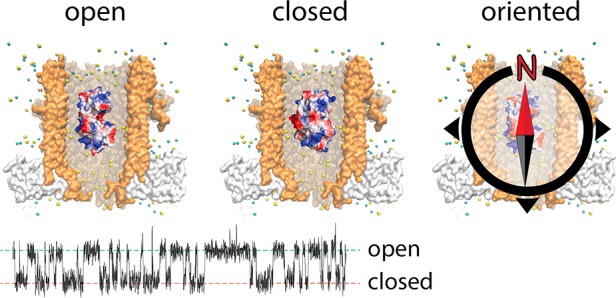
LOA and LOB could reflect the protein lodging inside ClyA at a deep and a shallow residence site within the nanopore, as it was previously shown for Dendra2 fluorescent protein,15 AlkB26 and thrombin.15,17 However, for these proteins the relative time spent between the two sites was strongly voltage-dependent, and the difference in residual current between the two levels was large (between 13.6% and 56.5%). By contrast, the relative distribution of LOA and LOB showed no voltage dependence (Figure S2) and relatively small ΔIres values (2.6 ± 0.4%, −100 mV). Alternatively, LOA and LOB might reflect two different SBD2 open conformations. However, previous smFRET experiments21 did not show any indication for the presence of two open conformations. In addition, glutamine binding rate constants measured from LOA and LOB were identical (Figure 3C), which suggests they both reflect the same open conformation. Further, the inactive SBD2(D417F), which rests solely in the open conformation21 still showed LOA and LOB blockades. Hence, it is unlikely that LOA and LOB correspond to two different open conformations of SBD2. We propose that LOA and LOB reflect two orientations of SBD2 inside the nanopore. SBD2 has two globular domains or lobes (lobe A, residues 253–343 and 441–481, lobe B, residues 348–437) connected by a flexible linker (residues 344–347 and 438–440, Figure 4A). The two lobes of SBD2 do not show a large imbalance in the spatial distribution of the surface charges as shown by the protein dipole moment that is aligned to the latitudinal axis of the protein (Figures 4B and S3). Thus, within the electric field inside ClyA the probability of SBD2 to be oriented with either lobes toward the cis or trans opening is likely similar (Figure 4A). In contrast, SBD1 has its dipole moment aligned to the longitudinal axis of the protein (Figure S3), suggesting that inside the nanopore SBD1 may be oriented. This is consistent with the observation that SBD1 shows one main current level (Figure 2A, B). Note that as measured from the ratio of the area of the histogram of LOA and LOB in a SBD2 blockade, orientation A is about 1.7-fold more represented than orientation B (Figure 4B, top), despite the dipole of SBD2 would predict a 50% distribution between the two orientations. This effect is possibly related to the asymmetry of the ClyA nanocavity, which is formed by chiral amino acids that might favor one of the two orientations of SBD2.
Figure 4.

Tuning the orientation of SBD2 in the ClyA nanopore. (A) SBD2 inside the ClyA nanopore showing two possible orientations. The red arrows indicate the electric field lines upon negative applied voltage. (B) Typical ionic current blockades provoked by the capture of SBD2 and its variants (∼70 nM, cis) by Type I ClyA-AS nanopore at −100 mV. The conformation of SBD2 is shown on the left of the current trace with the arrow indicating the dipole moment of the protein. The latter was calculated using the dipole watcher plugin of VMD.24 The LOA, LOB, and LC current levels are indicated. The histograms show the distribution of LOA and LOB. The additional current spikes observed for SBD2 T256K; S358E and T256K+S358E variants did not depend on the concentration of ligand, suggesting they do not represent an additional conformation of SBD2. The asterisk represents the restoration of the open pore current after SBD2 exited from the pore. (C) Table showing the amino acids in lobe A and lobe B that were considered for substitutions and selected after supercharging and MD simulations. The residues are arranged from the least to the most conserved as indicated by the gray arrows. The values indicate the distances in angstrom between the two respective residues.
Next, we imposed asymmetry between the lobes of SBD2 via point mutations that grafted a charged residue at the apical positions in each lobe of the protein surface. We reasoned that tuning the dipole moment of the protein would allow the electric field inside the nanopore to align the protein with one preferred orientation. Residues in both lobes were selected in silico by supercharging of SBD2, followed by molecular dynamics simulations in order to minimize the impact on protein structure (Figure 4C and SI). Two nonconserved residues that were furthest apart from the center of mass of SBD2 were selected: T256 on lobe A and S358 on lobe B (Figure 4C). In remarkable agreement to our view, when a negative charged residue was introduced to lobe A (T256E) or a positive charged residue was introduced in lobe B (S358 K), or both (T256E+S358 K), blockades provoked by SBD2 variants were mainly LOA (Figure 4B, Tables S1 and S2). Accordingly, inverse arrangement of the additional charges resulted in mainly LOB blockades (Figure 4B, Tables S1 and S2). All tested variants could bind glutamine, although the SBD2 variants that favored LOB orientation, showed a slightly reduced affinity (Figures S4 and S5, additional discussion in SI). Therefore, LOA and LOB current levels describe two orientations of SBD2 relative to ClyA, with LOA corresponding to lobe A facing the cis side of ClyA (orientation A) and LOB corresponding to the opposite orientation (orientation B, Figure 4A, B).
SBD1 and SBD2 Adopt Multiple Open and Closed Conformations
Two textbook mechanisms are often evoked to explain molecular recognition in proteins: Koshland’s “induced fit” hypothesis27 and the “conformational selection” model.28,29 The “induced fit” model treats the unliganded protein as if it exists in a low-energy, single, and stable conformation that switches to the liganded-conformation upon binding the substrate. However, NMR and crystallographic studies revealed that proteins are inherently dynamic and may sample a vast ensemble of conformations even in the absence of ligands,30 which may play important roles in molecular recognition.28 The “conformational selection” model accommodates this thermally accessed conformational heterogeneity by arguing that weakly populated, higher energy conformations recognize and bind ligands with high(er) affinity with subsequent population shift toward these conformers.31 However, experimental evidence to prove models of substrate recognition is difficult, mainly because the ability of measuring the affinity of ligands to multiple protein structures can only be obtained using single-molecule methods.
Several crystal structures of substrate-binding proteins revealed that they may exist in at least four states or conformations: a ligand-free (O) or ligand-bound (OL) open state, and a ligand-free (C) or ligand-bound (CL) closed state.32−36 The O, CL, and the C states of SBD1 and SBD2 were directly observed by both single-molecule FRET21 and nanopore analysis (Figures 2 and 3). At saturating substrate concentrations (50 μM) an open state of SBD1 and SBD2 could still be observed (Figures 2B, 3A, and S6), suggesting that the OL state also exists for SBD1 and SBD2. Open-liganded states were not observed by smFRET at 50 μM ligand concentration, possibly because the sampling rate was close to the resolution of the event.
Notably, the nanopore analysis also revealed that that the opening rates of SBD1 and SBD2 changed with the concentration of the substrate (Figures 2E and 3D). A decrease in kopening with increasing the substrate concentration was also observed in smFRET experiments; however it was ignored and the small range of substrate concentrations sampled (from 0 to 0.4 or 1.2 μM of ligands) masked the effect.21 ITC experiments revealed that SBD1 and SBD2 have one binding site21 ruling out the existence of an allosteric effect. It is also unlikely that the change in binding affinity of SBD proteins is the result of the electrophoretic field or due to an artifact induced by the filtering of the data. This is because the studied substrates carry no net charge and alternative data analysis that did not require filtering yielded similar results (Figure S7). The observed ligand concentration dependence of kopening is compatible with a kinetic model where the experimentally observed CL and C conformations have a different thermodynamic stability, and the presence of the ligand stabilizes the closed conformation (Scheme 1 and SI). Further, for SBD1 we observed a nonlinear increase of the closing rates with the concentration of ligands (Figure 2E), suggesting there is an upper limit for the closing rate of the protein. This is compatible with a kinetic scheme that includes the binding of the ligand to an open state (OL) followed by a relatively slow conformational change to the closed state (CL). The latter becomes the rate-limiting step at high ligand concentrations.
Scheme 1. Kinetic Schemes for the Binding of Ligands to SBD1 (A) and SBD2 (B).

C represents a closed state, O an open state, and L is the ligand. The kinetics rates are obtained as shown in the SI. 95% confidence intervals are shown in Tables S3 and S4 in the SI.
The simplest model that could accommodate all observations and kinetic data for SBD1 and SBD2 includes two open and two closed states (Scheme 1 and SI). Fitting the kinetic data for SBD1 and SBD2 to kinetic equations derived from such model (SI) revealed that both SBD proteins open about two-folds slower and close about 100- to 1000-folds faster in the presence of the ligand (Scheme 1). In turn this suggests a mechanism in which the ligands induce a conformational change that results in the closing of the protein around the ligand.
ClyA Nanopores As Nanoreactors for Single Molecule Enzymology
The results presented in this work show that nanopore currents can be used to sample the conformational dynamics of proteins. This approach samples native proteins, does not require expensive equipment or lengthy sample preparations, and allows long recordings at high sampling frequency. Molecular dynamics simulations suggested that the stability and activity of a folded macromolecule in a confined space are different from bulk because of entropic effects.37−41 Therefore, the kinetics and thermodynamics of proteins trapped inside the nanopore might be different from bulk, although experimental evidence for these predictions is scarce. Surprisingly, we found that inside the nanopore the conformational dynamics of SBD1 and SBD2 were similar to that in solution, suggesting that the effect of confinement for these proteins is negligible. In any case, even if other proteins inside the nanopore will show a different dynamics behavior than in bulk, it should be noticed that the cell is highly crowded, and the interaction of proteins with nearby macromolecules can impact protein stability and influence the recognition of ligands.42,43 Thus, the interaction of proteins with the nanopore inner surface may actually reproduce more faithfully the crowded environment that proteins face inside cells than the highly diluted conditions that are sampled with other single-molecule techniques.
Inside the nanopore proteins experience a relatively strong electric field (∼106 V/m). The effect of electric fields on protein stability, activity and orientation is not well understood, with some reports suggesting that proteins might even unfold when subjected to relatively moderate EFs (5 × 102 V/m),44 or inside nanopores.45 However, inside cells high transmembrane fields (2–3 × 107 V/m) preserve the nonequilibrium chemical steady-state or allow the cell-to-cell signaling in complex organisms. Our results suggest that electric fields in the order of 106 V/m do not have a strong effect on the activity and dynamics of proteins with weak dipoles such as SBD1 and SBD2. Nonetheless, we also showed that inside nanopores proteins can be oriented, and switching a single charge can control such orientation. This is intriguing, because recently it has been proposed that a relatively strong (5 × 105–3 × 106 V/m) electric field exists inside the cytoplasm of cells.46,47 Thus, in-cell proteins might be oriented or sorted by intracellular electric fields, with post-translational modification such as phosphorylation controlling such processes.
Conclusions
In summary, we show that nanopore currents can be used to monitor the conformational dynamics of proteins. This approach is simple, low-cost, requires only microgram amounts of proteins, and allows high bandwidth sampling with no intrinsic limitations on the observation time. Importantly, it is also the first method that allows the analysis of native proteins at the single-molecule level. We found that the dynamics, kinetics and equilibrium constants of substrate binding to SBD1 and SBD2 inside the nanopore were almost identical to those in solution, suggesting that the electric field and nanoconfinement of water inside the nanopore most likely have no effect on the activity or stability of proteins. The latter finding is surprising, considering that the transport of water in nanoconfined geometries is expected to differ from bulk phase.48 In any case, proteins inside the nanopore are likely to mimic more faithfully the tightly packed environment inside cells and might be used to sample other biological confined reactions such as the refolding of proteins inside the GroEL nanochamber.
This innovative technology is likely to have applications in basic and applied science. The single-molecule data showed that the proteins adopt multiple open and closed conformations. The kinetic analysis is compatible with a mechanism in which the binding of ligands to the open protein conformation triggers a conformational change that induces the closing of the protein around the substrate. Further, we showed unambiguously that the proteins inside the nanopore align with the electric field, and that a single change in the protein surface charge can switch the orientation. Thus, the orientation and subsequent activity of proteins inside cellular electric fields can be controlled post-translationally by a single modification. Finally, we show here that nanopores are capable of transducing protein conformational changes and ligand binding into electrical signals. Thus, nanopores with a panel of protein adaptors might be integrated in low-cost and portable devices for real-time detection of biologically relevant analytes from biological samples.
Acknowledgments
V.V.M. thanks the Research Foundation Flanders (FWO) for the doctoral fellowship. The work of G.M. was founded by an ERC starting grant (SMEN, # 260884). The work of B.P. was funded by a NWO TOPGO (#L.10.060) and ERC Advanced grant (ABCVolume, #670578). We thank Joris M.H. Goudsmits for performing the autocorrelation analysis.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b10106.
Additional results and discussion, materials and methods, figures, tables of ligands SBD1 and SBD2, and additional references (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Boehr D. D.; Nussinov R.; Wright P. E. Nat. Chem. Biol. 2009, 5 (11), 789. 10.1038/nchembio.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovych N.; Sun S.; Ebright R. H.; Kalodimos C. G. Nat. Struct. Mol. Biol. 2006, 13 (9), 831. 10.1038/nsmb1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll M. J.; Mauldin R. V.; Gromova A. V.; Singleton S. F.; Collins E. J.; Lee A. L. Nat. Chem. Biol. 2012, 8 (3), 246. 10.1038/nchembio.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzeng S.-R.; Kalodimos C. G. Nature 2009, 462 (7271), 368. 10.1038/nature08560. [DOI] [PubMed] [Google Scholar]
- Toseland C. P. J. Chem. Biol. 2013, 6 (3), 85. 10.1007/s12154-013-0094-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezrukov S. M.; Vodyanoy I.; Parsegian V. A. Nature 1994, 370 (6487), 279. 10.1038/370279a0. [DOI] [PubMed] [Google Scholar]
- Kasianowicz J. J.; Brandin E.; Branton D.; Deamer D. W. Proc. Natl. Acad. Sci. U. S. A. 1996, 93 (24), 13770. 10.1073/pnas.93.24.13770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayley H.; Cremer P. S. Nature 2001, 413 (6852), 226. 10.1038/35093038. [DOI] [PubMed] [Google Scholar]
- Shin S. H.; Luchian T.; Cheley S.; Braha O.; Bayley H. Angew. Chem., Int. Ed. 2002, 41 (19), 3707.. [DOI] [PubMed] [Google Scholar]
- Willems K.; Van Meervelt V.; Wloka C.; Maglia G. Philos. Trans. R. Soc., B 2017, 372, 20160230. 10.1098/rstb.2016.0230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig J. M.; Laszlo A. H.; Brinkerhoff H.; Derrington I. M.; Noakes M. T.; Nova I. C.; Tickman B. I.; Doering K.; de Leeuw N. F.; Gundlach J. H. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 11932. 10.1073/pnas.1711282114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho C.-W.; Van Meervelt V.; Tsai K.-C.; De Temmerman P.-J.; Mast J.; Maglia G. Sci. Adv. 2015, 1 (11), e1500905. 10.1126/sciadv.1500905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusko E. C.; Bruhn B. R.; Eggenberger O. M.; Houghtaling J.; Rollings R. C.; Walsh N. C.; Nandivada S.; Pindrus M.; Hall A. R.; Sept D.; Li J.; Kalonia D. S.; Mayer M. Nat. Nanotechnol. 2016, 12 (4), 360. 10.1038/nnano.2016.267. [DOI] [PubMed] [Google Scholar]
- Waduge P.; Hu R.; Bandarkar P.; Yamazaki H.; Cressiot B.; Zhao Q.; Whitford P. C.; Wanunu M.. ACS Nano 2017, 11, 5706. 10.1021/acsnano.7b01212 [DOI] [PubMed] [Google Scholar]
- Soskine M.; Biesemans A.; Moeyaert B.; Cheley S.; Bayley H.; Maglia G. Nano Lett. 2012, 12 (9), 4895. 10.1021/nl3024438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soskine M.; Biesemans A.; De Maeyer M.; Maglia G. J. Am. Chem. Soc. 2013, 135 (36), 13456. 10.1021/ja4053398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Meervelt V.; Soskine M.; Maglia G. ACS Nano 2014, 8 (12), 12826. 10.1021/nn506077e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biesemans A.; Soskine M.; Maglia G. Nano Lett. 2015, 15 (9), 6076. 10.1021/acs.nanolett.5b02309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wloka C.; Van Meervelt V.; Van Gelder D.; Danda N.; Jager N.; Williams C. P.; Maglia G. ACS Nano 2017, 11 (5), 4387. 10.1021/acsnano.6b07760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soskine M.; Biesemans A.; Maglia G. J. Am. Chem. Soc. 2015, 137 (17), 5793. 10.1021/jacs.5b01520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouridis G.; Schuurman-Wolters G. K.; Ploetz E.; Husada F.; Vietrov R.; de Boer M.; Cordes T.; Poolman B. Nat. Struct. Mol. Biol. 2014, 22 (1), 57. 10.1038/nsmb.2929. [DOI] [PubMed] [Google Scholar]
- Schuurman-Wolters G. K.; Poolman B. J. Biol. Chem. 2005, 280 (25), 23785. 10.1074/jbc.M500522200. [DOI] [PubMed] [Google Scholar]
- Fulyani F.; Schuurman-Wolters G. K.; Žagar A. V.; Guskov A.; Slotboom D. J.; Poolman B. Structure 2013, 21 (10), 1879. 10.1016/j.str.2013.07.020. [DOI] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. J. Mol. Graphics 1996, 14 (1), 33. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Mueller M.; Grauschopf U.; Maier T.; Glockshuber R.; Ban N. Nature 2009, 459 (7247), 726. 10.1038/nature08026. [DOI] [PubMed] [Google Scholar]
- Soskine M.; Biesemans A.; Maglia G. J. Am. Chem. Soc. 2015, 137 (17), 5793. 10.1021/jacs.5b01520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshland D. E. Proc. Natl. Acad. Sci. U. S. A. 1958, 44, 98. 10.1073/pnas.44.2.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frauenfelder H.; Sligar S. G.; Wolynes P. G. Science (Washington, DC, U. S.) 1991, 254 (5038), 1598. 10.1126/science.1749933. [DOI] [PubMed] [Google Scholar]
- Ma B.; Kumar S.; Tsai C. J.; Nussinov R. Protein Eng., Des. Sel. 1999, 12 (9), 713. 10.1093/protein/12.9.713. [DOI] [PubMed] [Google Scholar]
- Boehr D. D.; Nussinov R.; Wright P. E. Nat. Chem. Biol. 2009, 5 (11), 789. 10.1038/nchembio.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai C. J.; Ma B.; Nussinov R. Proc. Natl. Acad. Sci. U. S. A. 1999, 96 (18), 9970. 10.1073/pnas.96.18.9970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shouldice S. R.; Skene R. J.; Dougan D. R.; Snell G.; McRee D. E.; Schryvers A. B.; Tari L. W. J. Bacteriol. 2004, 186 (12), 3903. 10.1128/JB.186.12.3903-3910.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan X.; Quiocho F. A. Biochemistry 2002, 41 (3), 706. 10.1021/bi015784n. [DOI] [PubMed] [Google Scholar]
- Berntsson R. P.-A.; Smits S. H. J.; Schmitt L.; Slotboom D.-J.; Poolman B. FEBS Lett. 2010, 584 (12), 2606. 10.1016/j.febslet.2010.04.043. [DOI] [PubMed] [Google Scholar]
- Flocco M. M.; Mowbray S. L. J. Biol. Chem. 1994, 269 (12), 8931. [PubMed] [Google Scholar]
- Oswald C.; Smits S. H. J.; Höing M.; Sohn-Bösser L.; Dupont L.; Le Rudulier D.; Schmitt L.; Bremer E. J. Biol. Chem. 2008, 283 (47), 32848. 10.1074/jbc.M806021200. [DOI] [PubMed] [Google Scholar]
- Zhou H. X. J. Mol. Recognit. 2004, 17 (5), 368. 10.1002/jmr.711. [DOI] [PubMed] [Google Scholar]
- Klimov D. K.; Newfield D.; Thirumalai D. Proc. Natl. Acad. Sci. U. S. A. 2002, 99 (12), 8019. 10.1073/pnas.072220699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung M. S.; Thirumalai D. J. Mol. Biol. 2006, 357 (2), 632. 10.1016/j.jmb.2005.12.048. [DOI] [PubMed] [Google Scholar]
- Marino K. A.; Bolhuis P. G. J. Phys. Chem. B 2012, 116, 11872. 10.1021/jp306727r. [DOI] [PubMed] [Google Scholar]
- Vaitheeswaran S.; Thirumalai D. Proc. Natl. Acad. Sci. U. S. A. 2008, 105 (46), 17636. 10.1073/pnas.0803990105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Berg J.; Boersma A. J.; Poolman B. Nat. Rev. Microbiol. 2017, 15 (5), 309. 10.1038/nrmicro.2017.17. [DOI] [PubMed] [Google Scholar]
- Boersma A. J.; Zuhorn I. S.; Poolman B. Nat. Methods 2015, 12 (3), 227. 10.1038/nmeth.3257. [DOI] [PubMed] [Google Scholar]
- Bekard I.; Dunstan D. E. Soft Matter 2014, 10 (3), 431. 10.1039/C3SM52653D. [DOI] [PubMed] [Google Scholar]
- Talaga D. S.; Li J. J. Am. Chem. Soc. 2009, 131 (26), 9287. 10.1021/ja901088b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyner K. M.; Kopelman R.; Philbert M. Biophys. J. 2007, 93 (4), 1163. 10.1529/biophysj.106.092452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreev V. P.PLoS One 2013, 8 ( (4), ), e61884. 10.1371/journal.pone.0061884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majolino D.; Corsaro C.; Crupi V.; Venuti V.; Wanderlingh U. J. Phys. Chem. B 2008, 112 (13), 3927. 10.1021/jp711433d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.