

Abstract
Expression of the transmembrane pseudokinase ROR1 is required for survival of t(1;19)-pre-B-cell acute lymphoblastic leukemia (t(1;19) pre-B-ALL), chronic lymphocytic leukemia, and many solid tumors. However, targeting ROR1 with small-molecules has been challenging due to the absence of ROR1 kinase activity. To identify genes that regulate ROR1 expression and may, therefore, serve as surrogate drug targets, we employed an siRNA screening approach and determined that the epigenetic regulator and E3 ubiquitin ligase, UHRF1, is required for t(1;19) pre-B-ALL cell viability in a ROR1-dependent manner. Upon UHRF1 silencing, ROR1 protein is reduced without altering ROR1 mRNA, and ectopically expressed UHRF1 is sufficient to increase ROR1 levels. Additionally, proteasome inhibition rescues loss of ROR1 protein after UHRF1 silencing, suggesting a role for the proteasome in the UHRF1-ROR1 axis. Finally, we show that ROR1-positive cells are twice as sensitive to the UHRF1-targeting drug, naphthazarin, and undergo increased apoptosis compared to ROR1-negative cells. Naphthazarin elicits reduced expression of UHRF1 and ROR1, and combination of naphthazarin with inhibitors of pre-B cell receptor signaling results in further reduction of cell survival compared with either inhibitor alone. Therefore, our work reveals a mechanism by which UHRF1 stabilizes ROR1, suggesting a potential targeting strategy to inhibit ROR1 in t(1;19) pre-B-ALL and other malignancies.
Keywords: leukemia, ROR1, UHRF1, ALL
Introduction
Approximately 1-5% of patients with acute lymphoblastic leukemia carry a translocation between the long arm of chromosome 1 and the short arm of chromosome 19, leading to the arrest of normal B cell differentiation and development of pre-B cell acute lymphoblastic leukemia (t(1;19) pre-B-ALL) (1–4). The fusion protein resulting from this translocation, TCF3-PBX1, promotes aberrant expression and re-localization of WNT16B and β-catenin, respectively (5–7). Consequently, t(1;19) pre-B-ALL cases uniformly exhibit differentiation arrest at an intermediate stage of B lineage maturation where blasts express the pre-B cell receptor (pre-BCR) (8, 9). Recent studies have shown that signaling from the pre-BCR through tyrosine kinases such as BTK and SRC-family kinases (SFKs) is important to drive proliferation and survival of t(1;19) pre-B-ALL cells (10, 11). We and others have shown that the pseudokinase ROR1 is also critical for leukemic cell survival through cross-talk with the pre-BCR (10, 12, 13). Furthermore, t(1;19) pre-B-ALLs are sensitive to dasatinib, an inhibitor of pre-BCR effector kinases BTK and SFKs, while ROR1 is upregulated as a compensatory rescue response. Consequently, t(1;19) pre-B-ALL cell lines are maximally sensitive to silencing of ROR1 in combination with kinase inhibitors that block pre-BCR signaling, such as dasatinib (10).
ROR1 is highly expressed on the surface of both t(1;19) pre-B-ALL and chronic lymphocytic leukemia (CLL) cells in addition to multiple types of solid tumors including prostate, breast, and pancreatic cancer (14–19). ROR1 is thought to be rarely expressed in post-natal tissues, although a recent study revealed ROR1 may be present in several normal tissues (14, 20). Multiple groups are currently developing and testing methods of targeting ROR1-expressing tumor cells, some of which have seen initial success in clinical trials (21–24). However, the targeting strategies to date have largely revolved around immunologic agents, since ROR1-specific small-molecule inhibitors have remained elusive due to the absence of ROR1 kinase activity (10). Thus, we sought to develop an improved understanding of the regulation of ROR1 expression in an effort to discover surrogate small-molecule targeting strategies. Previously, groups have shown that STAT3 directly binds the ROR1 locus to promote its expression in CLL (25) and NKX2-1 has been reported to induce ROR1 expression in lung adenocarcinoma (26). In addition to transcriptional activation, ROR1 is thought to be post-translationally modified through glycosylation and ubiquitination (27), but the mediators of these modifications have yet to be elucidated.
The RING E3 ligase UHRF1 (also known as ICBP90) ubiquitinates several substrates, including p53 and histone H3, to mediate protein function and chromatin structure, respectively (28, 29). UHRF1 also has ubiquitin ligase-independent roles interacting with DNA and histones through its Tudor-like, PHD, and SRA/YDG domains (29–37). Both UHRF1 functions can be inhibited through direct binding to or downregulation of UHRF1 expression by a number of small-molecule compounds, including NSC232003 and naphthazarin (38, 39). Despite evidence that UHRF1 promotes solid tumor formation and progression and is associated with low-risk AML (31, 40–45), UHRF1 has not been thoroughly investigated in ALL.
Therefore, we sought to find new mechanisms that regulate ROR1 and, more importantly, may have therapeutic potential that can be targeted by small-molecule inhibitors. We utilized an siRNA approach and identified UHRF1 as a regulator of levels of ROR1 protein in t(1;19) pre-B-ALL. Targeting the UHRF1-ROR1 axis in combination with readily available pre-BCR targeting strategies, such as dasatinib, may prove to be a useful alternative regimen for ROR1-expressing cancers.
Results
UHRF1 is required for t(1;19) pre-B-ALL in a ROR1-dependent manner
To identify genes required for t(1;19) pre-B-ALL viability that also regulate ROR1 expression we performed an siRNA screen targeting a broad range of transcription factors and epigenetic regulators using the t(1;19)-positive pre-B ALL cell line, RCH-ACV. Gene targets were prioritized according to effects on overall cell viability after siRNA knockdown. Upon silencing, siRNA targets that reduced viability by at least one standard deviation were further investigated. UHRF1 and RUNX1 were among the gene targets that, when silenced, significantly reduced RCH-ACV cell viability (Figure 1A, Supplementary Table 1). RUNX1 has previously been shown to be a key regulator of pre-BCR expression (46), consistent with the importance of the pre-BCR in t(1;19) pre-B-ALL cells. In contrast, UHRF1 has not been previously implicated in ALL pathogenesis.
Figure 1. UHRF1 is a potential regulator of ROR1 in t(1;19) pre B-ALL.
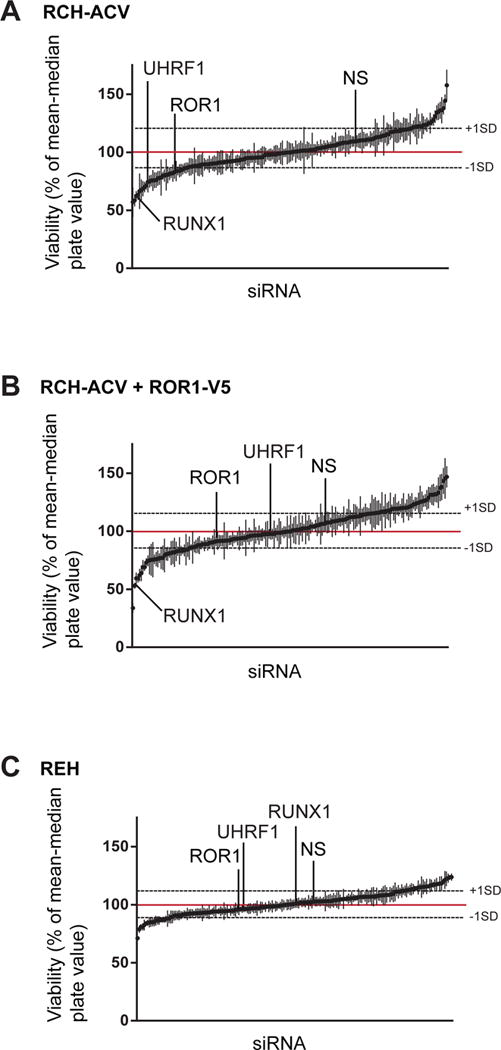
(A) Parental RCH-ACV cells (N=4), (B) RCH-ACV stably expressing ROR1-V5 (N=3), or (C) REH (N=3) cells were electroporated with siRNAs targeting transcription factors or chromatin modifiers/epigenetic regulators. Cell viability was measured by MTS assay. siRNA gene targets were ranked based on their effects on viability upon silencing. Targets that reduced viability by at least one standard deviation among all biological replicates were further investigated. Error bars = S.D. NS = non-specific siRNA.
To determine whether these siRNA targets were important for t(1;19) pre-B-ALL cell survival in a ROR1-dependent or ROR1-independent manner, we repeated the screen with RCH-ACV cells that stably overexpress ROR1 with a V5 tag (RCH+ROR1-V5). These cells retained sensitivity to RUNX1 silencing, once again consistent with the role of RUNX1 in regulating the pre-BCR, which is a pathway orthogonal to ROR1 in t(1;19) cells (10). However, these cells did not exhibit sensitivity to UHRF1 silencing, suggesting that ectopically expressed ROR1 mitigates UHRF1-sensitivity in t(1;19) cells and, therefore, UHRF1 is important for maintaining t(1;19) cell viability in a ROR1-dependent manner (Figure 1B). As a final control, we applied the same siRNA screen to REH cells, an ALL cell line that lacks the 1;19 translocation and do not express ROR1. REH cells generated a very different list of putative targets and, importantly, these cells were not sensitive to silencing of UHRF1 (Figure 1C). These data suggest UHRF1 specifically mediates t(1;19) cell viability through a mechanism associated with ROR1 expression.
UHRF1 is required for the maintenance of ROR1 protein, not mRNA, levels
To determine whether UHRF1 plays a direct role in regulating ROR1 expression, we measured cell viability and levels of ROR1 mRNA and protein after UHRF1 silencing. After confirming that UHRF1 is required for RCH-ACV cell viability (Supplementary Figure 1A), we observed that levels of ROR1 protein, but not mRNA, were significantly reduced after UHRF1 silencing using pooled siRNA or two independent UHRF1 siRNA duplexes (Figure 2A-B, Supplementary Figure 1B). Furthermore, we observed similar patterns with a second t(1;19) pre-B-ALL cell line, Kasumi-2 (Supplementary Figure 1C-D). Together, these results indicate that UHRF1 regulates ROR1 protein levels through a post-transcriptional mechanism. This was unexpected, since the RCH+ROR1-V5 cells were not sensitive to UHRF1 silencing, which initially suggested a transcriptional mechanism. To reconcile these results, we hypothesized that RCH+ROR1-V5 cells are rescued from UHRF1 sensitivity because ROR1 protein levels are present at highly elevated levels in these cells compared with parental RCH-ACV cells. In this case, UHRF1 silencing would also reduce the levels of ectopic RCH+ROR1-V5 in these cells but would still be maintained above a threshold required to maintain cell viability. Consistent with this, we observed that upon UHRF1 silencing, ectopically expressed ROR1 was reduced in RCH+ROR1-V5 cells, but only to levels comparable to those levels seen at baseline in RCH-ACV parental cells (Figure 2C).
Figure 2. UHRF1 mediates ROR1 protein but not mRNA expression in RCH-ACV cells.
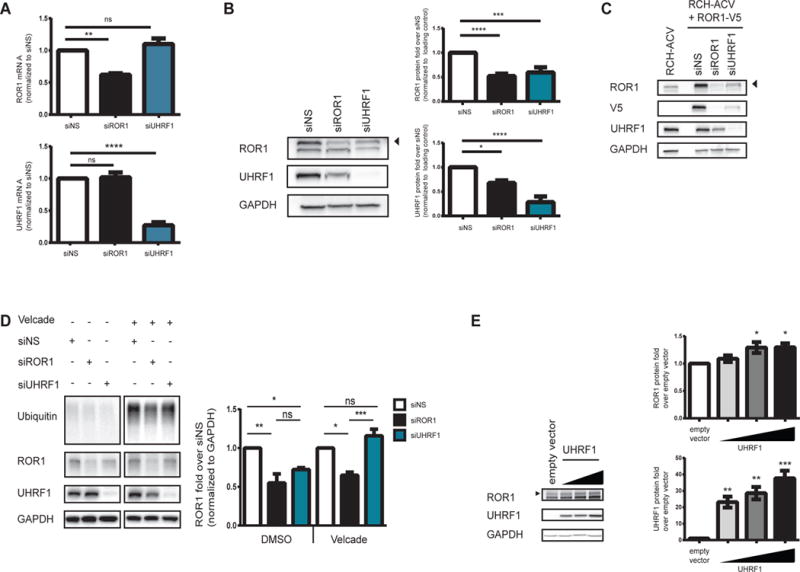
Cells were electroporated with siRNA and (A) 72 hours after electroporation, total RNA and whole protein lysates were harvested and tested for mRNA expression by qRT-PCR (N=3 **p=0.005, ****p<0.0001) and protein levels by (B) immunoblot (N=6 ****p<0.0001, ***p<0.0005, *p<0.05). mRNA and protein levels were normalized to a non-specific siRNA control (siNS). (C) RCH+ROR1-V5 cells were treated with siRNA and whole cell lysates were subjected to immunoblot (N=4). (D) RCH-ACV cells were treated with siRNA for 48 hours prior to treatment with DMSO or Velcade for 16 hours. Whole cell lysates were subjected to immunoblot. ROR1 protein was quantified and normalized to siNS samples treated with DMSO (*p<0.05, **p<0.005) or Velcade (*p<0.05, ***p<0.0005). UHRF1 protein was quantified and nomalized to siNS samples treated with DMSO (*p<0.05). Note that these data are from a contiguous blot (N=3). (E) HEK293T17 cells were transfected with either empty vector or a plasmid encoding UHRF1, and whole cell lysates were harvested 48 hours post-transfection. Levels of ROR1 (p<0.05) and UHRF1 (**p<0.005, ***p<0.0005) were detected by immunoblot. Error bars represent S.E.M. “ns” = not significant.
Next, we asked whether the reduced levels of ROR1 protein that we observe after UHRF1 silencing could be attributed to one of the defined functions of UHRF1. Since UHRF1 can function as an E3 ubiquitin ligase to modulate protein turnover, we suspected that regulation of ROR1 by UHRF1 is associated with the proteasome. We, therefore, measured ROR1 protein levels after UHRF1 silencing in the absence or presence of the proteasome inhibitor Velcade. Treatment with Velcade rescued ROR1 protein levels after UHRF1 silencing compared to vehicle-treated cells where UHRF1 silencing resulted in reduced levels of ROR1 protein (Figure 2D). Collectively, these data suggest that loss of ROR1 protein following UHRF1 silencing is mediated by the proteasome.
To complement the gene silencing experiments, we transiently expressed wild-type UHRF1 in HEK293T17 cells, which express very low levels of ROR1 and UHRF1. As shown in Figure 2E, ectopically expressed UHRF1 correlates with an increase in ROR1 protein levels in a dose-dependent manner compared to the empty vector control. Therefore, these data suggest that overexpression of UHRF1 is sufficient to induce ROR1 protein levels, further support of UHRF1-mediated regulation of ROR1.
UHRF1 indirectly regulates ROR1
Since silencing of UHRF1 resulted in reduced levels of ROR1 protein, we next asked whether UHRF1 and ROR1 are associated in a common complex that would allow UHRF1 to interact with or modify ROR1 directly. Co-immunoprecipitation experiments with either ROR1 or UHRF1 show no detectable endogenous interaction (Figure 3A), which is consistent with previously published mass spectrometry studies (10). These observations were confirmed by co-immunoprecipitation from HEK293T cells with ectopically expressed wild-type ROR1 (Figure 3B). Furthermore, these proteins do not co-localize in the same subcellular compartments in RCH-ACV or RCH+ROR1-V5 cells as UHRF1 is abundant in the nucleus while ROR1 is enriched in the cytoplasmic and membrane-bound fraction (Figure 3C). Therefore, we conclude that UHRF1 regulates ROR1 indirectly.
Figure 3. UHRF1 indirectly regulates ROR1.
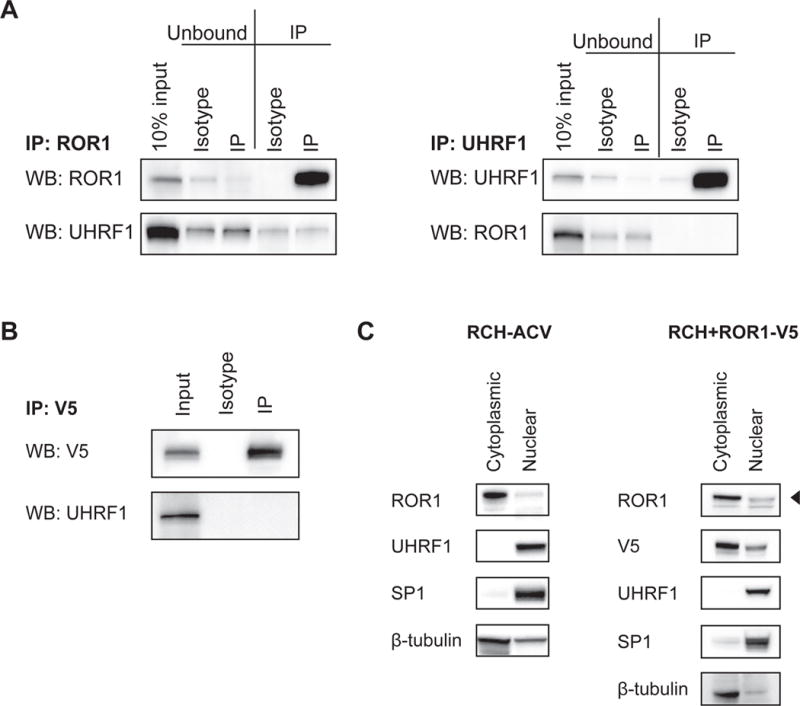
(A) RCH-ACV cell lysate was enriched for either ROR1 (left, IP: ROR1) or UHRF1 (right, IP: UHRF1) and tested for co-immunoprecipitation by immunoblot. (B) HEK293T17 cells were transfected with plasmids encoding V5-tagged ROR1 and UHRF1. Whole cell lysate was enriched using a V5 antibody and tested for co-immunoprecipitation by immunoblot. (C) RCH-ACV (left) and RCH+ROR1-V5 (right) cells were fractionated and localization was detected by immunoblot. “Cytoplasmic” fraction includes membrane-bound proteins. Note that β-tubulin signal in the nuclear fraction is likely contamination between fractions.
UHRF1 silencing sensitizes t(1;19) pre-B-ALL to dasatinib
Since targeting UHRF1 with siRNA led to reduced levels of ROR1 protein, we next investigated whether silencing of UHRF1 would cooperatively reduce t(1;19) cell viability when combined with inhibitors of pre-BCR signaling, as we had previously seen with direct ROR1 silencing (10). To address this, we silenced UHRF1 expression before treating RCH-ACV cells with dasatinib, which inhibits SFKs and BTK that signal as part of the pre-BCR signaling complex. As expected, cells treated with non-specific siRNA responded to dasatinib in a dose-dependent manner. We also observed an enhanced loss of cell viability when ROR1 expression was silenced in combination to dasatinib, which is consistent with previous studies (10). Similarly, silencing UHRF1 significantly reduced cell viability in the presence of dasatinib more than that observed with dasatinib in the presence of non-specific siRNA (Figure 4A). These data demonstrate the potential of inhibiting UHRF1 in combination with dasatinib is comparable to targeting ROR1 directly in combination with the same kinase inhibitor to maximize loss of cell viability.
Figure 4. Targeting the UHRF1-ROR1 axis with naphthazarin and dasatinib inhibits t(1;19) pre B-ALL.
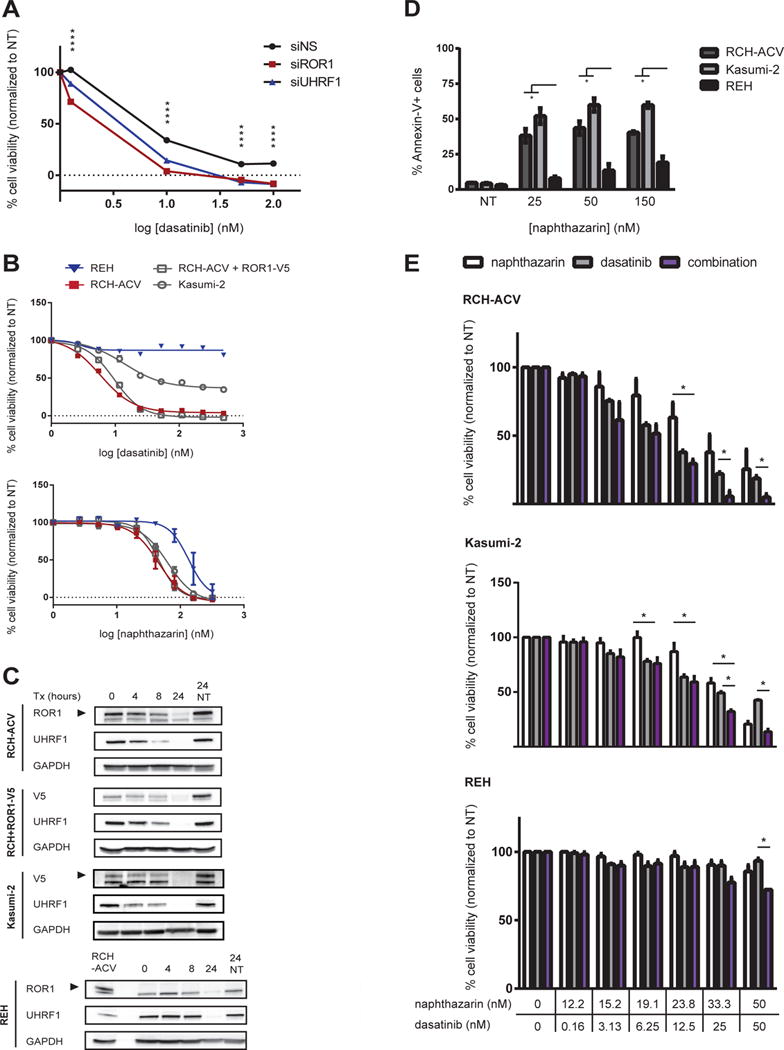
(A) RCH-ACV cells were treated with siRNA for 24 hours followed by exposure to dasatinib for 72 hours. Cell viability was measured by MTS assay and normalized to cells treated with siRNA only (****p<0.0001). (B) Cell lines were treated with the indicated drugs (0-0.5 μM) for 72 hours and cell viability was measured by MTS assay. (C) Cells were treated with naphthazarin for the indicated times and whole cell lysates were tested by immunoblot. RCH-ACV cell lysate was used as a positive control for REH immunoblot. (D) Cells were treated with naphthazarin for 8 hours and Annexin-V was measured by GuavaNexin Assay (*p<0.05). (E) Cells were treated with naphthazarin, dasatinib, or a combination of both for 72 hours (*p<0.05). Cell viability was measured and normalized to non-treated cells. Data shown is an average of three independent experiments. NT = non-treated Error bars = S.E.M.
Naphthazarin inhibits t(1;19) pre-B-ALL viability by downregulating the UHRF1-ROR1 axis
To further investigate the potential of targeting UHRF1, we screened t(1;19) positive and negative cell lines against several putative UHRF1 small-molecule compounds (38) and dasatinib. As expected, dasatinib was specifically effective against RCH-ACV cells. Of all other tested compounds, t(1;19) pre-B-ALL cells were twice as sensitive to the UHRF1 inhibitor naphthazarin (5,8-dihydroxy-1,4-naphthoquinone) compared to REH cells, which lack the 1;19 translocation and do not express ROR1 (Figure 4B, Supplementary Figure 2A). In contrast, other putative UHRF1 inhibitors, including anisomycin, were observed to be toxic since their effects were similar in ROR1-expressing and ROR1-non-expressing cells. Since these other agents also did not result in discernible impact on UHRF1 expression levels, we could not rule out UHRF1-independent effects and did not pursue these other agents further (Supplementary Figure 2A-B).
Naphthazarin has been reported to induce cell cycle arrest and apoptosis in lung and gastric cancers and reduce UHRF1 expression, sensitizing breast cancer to irradiation (47–49). To test the specificity of naphthazarin cytotoxicity on the UHRF1-ROR1 axis, cells were treated for up to 24 hours and levels of protein were measured. Within 24 hours of naphthazarin exposure, the t(1;19)-positive pre-B-ALL RCH-ACV and Kasumi-2 cell lines showed reduced levels of UHRF1 and ROR1 protein (Figure 4C). To further confirm that the effects of naphthazarin mimicked the phenotype we previously observed with UHRF1 silencing, we also treated RCH+ROR1-V5 cells with naphthazarin and observed a similar reduction in ectopically expressed ROR1. In contrast, this result was not observed in t(1;19)-negative REH cells, which correlates with REH insensitivity to naphthazarin. Moreover, naphthazarin induced higher amounts of apoptosis in t(1;19)-positive, but not negative, cell lines as demonstrated by Annexin-V staining (Figure 4D). In parallel to induced apoptosis, we also noticed that these cells underwent cell cycle arrest (Supplementary Figure 3). To determine whether naphthazarin can cooperate with inhibition of the pre-BCR pathway via dasatinib, we measured effects of cell viability in the presence of a naphthazarin-dasatinib combination compared to either agent alone. t(1;19)-positive RCH-ACV and Kasumi-2 cells were sensitive to naphthazarin and dasatinib as single agents, but this effect is enhanced when these small-molecule inhibitors are combined (Figure 4E). This data were in contrast to REH cells, which exhibited insensitivity to single agent or combination treatment. Together, these data suggest that naphthazarin inhibits UHRF1-dependent protein levels of ROR1 and can be used in combination with dasatinib to maximize loss of t(1;19) pre-B-ALL cell viability.
Discussion
ROR1 is a pseudokinase that is required for the viability of t(1;19) pre-B-ALL and other hematologic and solid tumor malignancies. Despite ongoing efforts to utilize ROR1 for targeted therapies, there are no FDA-approved ROR1 small-molecule inhibitors. To identify regulators of ROR1 expression that could be exploited to target ROR1, we screened 190 genes including transcription and chromatin modifying factors. Several candidates were generated including UHRF1. Sensitivity to UHRF1 siRNA was abrogated in RCH+ROR1-V5 cells, suggesting that UHRF1-mediated cell viability is associated with ROR1 expression and function.
Follow-up siRNA experiments demonstrate that UHRF1 is critical for maintenance of levels of ROR1 protein, but not mRNA, indicating that UHRF1 regulates ROR1 through a post-transcriptional regulatory mechanism. This mechanism may involve UHRF1-mediated maintenance of ROR1 through ubiquitination. Indeed, others have demonstrated ROR1 ubiquitination in CHO cells (27), and we have also confirmed this in HEK293 cells (Supplemental Figure 4). Although we show that UHRF1 is sufficient to increase levels of ROR1, we have presented evidence suggesting that UHRF1 indirectly regulates ROR1 and experiments to identify intermediaries of this mechanism will be essential for a complete understanding of the pathway. To preliminarily address this question, we performed mass spectrometry and identified several potential mediators of the UHRF1-ROR1 mechanism including nucleophosmin 1, which is implicated in AML and is an interesting candidate, given its ability to shuttle between the nucleus and cytoplasm (50) (Table 1). These putative UHRF1-ROR1 intermediaries merit further investigation.
Table 1. Candidate UHRF1-ROR1 intermediaries.
Whole cell lysates from t(1;19) pre-B-ALL cell lines (Kasumi-2 and RCH-ACV) were incubated with a ROR1-specific, UHRF1-specific antibody or an isotype matched control. Protein names, UniProt accession number, and corrected MS/MS spectral counts are shown for each identified protein.
| Protein | Description | Accession # | Isotype | ROR1 Antibody | ||
|---|---|---|---|---|---|---|
| Kasumi-2 | RCH-ACV | Kasumi-2 | RCH-ACV | |||
| ALDOA | Fructose-bisphosphate aldolase A | P04075 | 0 | 0 | 3 | 3 |
| DSG1 | Desmoglein-1 | Q02413 | 0 | 0 | 6 | 1 |
| ENPL | Endoplasmin | P14625 | 0 | 0 | 1.471 | 7.78 |
| IMA1 | Importin subunit alpha-1 | P52292 | 0 | 0 | 1 | 3 |
| MCM5 | DNA replication licensing factor MCM5 | P33992 | 0 | 0 | 3 | 5 |
| NPM | Nucleophosmin | P06748 | 0 | 0 | 7 | 7 |
| SRSF3 | Serine/arginine-rich splicing factor 3 | P84103 | 0 | 0 | 3 | 4 |
| XRCC6 | X-ray repair cross-complementing protein 6 | P12956 | 0 | 0 | 5 | 6 |
| Isotype | UHRF1 Antibody | |||||
|---|---|---|---|---|---|---|
| Kasumi-2 | RCH-ACV | Kasumi-2 | RCH-ACV | |||
| ALDOA | Fructose-bisphosphate aldolase A | P04075 | 0 | 0 | 4 | 1 |
| CE170 | Centrosomal protein of 170 kDa | Q5SW79 | 0 | 0 | 10 | 3 |
| DESP | Desmoplakin | P15924 | 0 | 0 | 6 | 4 |
| DSG1 | Desmoglein-1 | Q02413 | 0 | 0 | 5 | 2 |
| FILA2 | Filaggrin-2 | Q5D862 | 0 | 0 | 4 | 1 |
| IGHA1 | Ig alpha-1 chain C region | P01876 | 0 | 0 | 4 | 1 |
| SFPQ | Splicing factor, proline- and glutamine-rich | P23246 | 0 | 0 | 3.027 | 2.533 |
While performing these siRNA studies, we also observed a significant siROR1-mediated loss of UHRF1 (Figure 2B). One potential explanation for these data is that ROR1 stability or function may be critical for UHRF1 expression in a positive feedback loop. This would not be surprising, given the roles of ROR1-associated AKT signaling and UHRF1 in cell proliferation (10, 13, 40, 51). However, this hypothesis will need to be further evaluated to fully elucidate the ROR1-mediated effects on UHRF1.
In addition to characterizing the UHRF1-ROR1 mechanism, we were interested in identifying agents that could target this pathway, since previous studies established the importance of ROR1 in t(1;19) pre-B-ALL and its ability to cross-talk with the pre-BCR (10). First, we have shown that silencing UHRF1 sensitizes t(1;19) pre-B-ALL to dasatinib to levels similar to that of ROR1 silencing, indicative of a potential therapeutic strategy through targeting of UHRF1. Next, we investigated candidate inhibitors of UHRF1 activity and demonstrated that naphthazarin leads to reduced levels of UHRF1 and ROR1 protein without significantly affecting levels of the ROR1 transcript, phenocopying the effect of UHRF1 silencing on ROR1. Furthermore, naphthazarin induces significant apoptosis in t(1;19)-positive pre-B-ALL and can work in parallel with dasatinib to further reduce cell survival, suggesting that UHRF1-ROR1-pre-BCR combination therapy may be effective in treating t(1;19) pre-B-ALL (Figure 5). Together, these preclinical results provide opportunities to further investigate the mechanism by which the combination of dasatinib and naphthazarin suppress t(1;19) pre-B-ALL viability in future in vitro and in vivo studies.
Figure 5. Model of UHRF1-ROR1 regulatory mechanism.
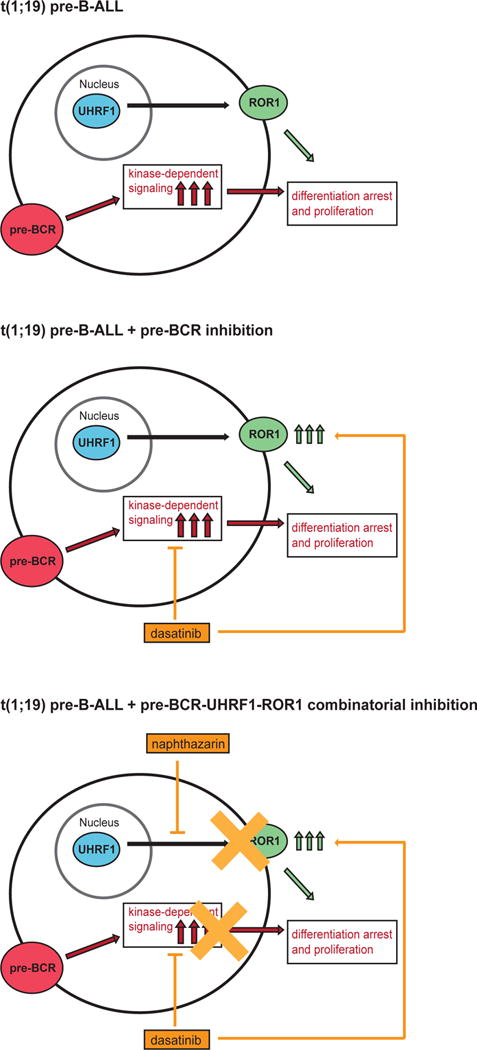
The 1;19 fusion product leads to cell arrest and constitutively active pre-BCR signaling (top). This signaling can be suppressed by small-molecule inhibitors, such as dasatinib, but kinase inhibition is rescued by increased ROR1 expression (middle). Targeting the UHRF1-ROR1 mechanism with compounds such as naphthazarin can suppress baseline ROR1 levels. In combination with pre-BCR inhibition (via dasatinib), naphthazarin helps to prevent upregulation of ROR1 to maximize t(1;19) pre B-ALL apoptotic cell death (bottom).
Currently, few studies suggest a role for UHRF1 in leukemia (45, 52), and this is the first report for an oncogenic role of UHRF1 in t(1;19) pre-B-ALL. However, UHRF1 is an established oncogene in solid tumors, many of which overlap with those that overexpress ROR1 (18, 53). Indeed, it will be interesting to see if the UHRF1-ROR1 mechanism is conserved in other types of cancers.
Although UHRF1 was prioritized after the results of the screen with RCH+ROR1-V5 cells, other candidates are being investigated for their roles in maintaining t(1;19) pre-B-cell ALL through ROR1-independent mechanisms. In addition to RUNX1, RhoH and KAT7 were required for t(1;19) pre-B-cell viability. RhoH is a hematopoietic-specific member of the Rho GTPase family and has roles in promoting CLL through cell intrinsic mechanisms and interactions with the microenvironment (54, 55). KAT7, also known as HBO1 and MYST2, is a lysine acetyltransferase that is critical for H3K14 and histone H4 acetylation during normal embryonic development and plays a role in DNA replication and cell cycle progression (56, 57). However, the roles of RhoH and KAT7 in t(1;19) pre-B-ALL have yet to be elucidated.
Collectively, this work suggests a new regulatory mechanism in which UHRF1 is necessary for maintaining the levels of ROR1 protein. When this mechanism is inhibited by siRNA or naphthazarin, the UHRF1-dependent loss of ROR1 leads to reduced t(1;19) pre-B-ALL viability. UHRF1 has been well-established as an oncogene that impairs the expression of various tumor suppressor genes (38), yet here we show that UHRF1 regulates ROR1, a tumor-promoter. Thus, our findings propose that targeting ROR1 through UHRF1 inhibition, potentially in combination with dasatinib, is a promising novel therapeutic strategy for t(1;19) pre B-cell-ALL and other ROR1-expressing cancers.
Materials and Methods
Cell culture
RCH-ACV and Kasumi-2 cells were purchased from DSMZ (Germany). RCH-ACV+ROR1V5, SupB15, and REH cells were described previously (10). U937 cells were purchased from ATCC. All cell lines were cultured in RPMI1640 supplemented with 10% FBS, 2% L-Glutamine, 1% penicillin/streptinomycin, and 0.1% Fungizone (R10). All cells were grown at 37°C with 5% CO2.
Reagents
Commercially available antibodies used in this study are: ROR1, UHRF1 (#4102, #12387, Cell Signaling, Danvers MA), ROR1 (#AF2000, R&D Systems, Minneapolis MN), UHRF1 (#612264, BD Biosciences, San Jose CA), goat IgG serum (#005-000-003, Jackson ImmunoResearch, West Grove PA), mouse IgG serum (#sc-2025, Santa Cruz Biotechnology, Santa Cruz CA), actin, GAPDH, SP1 (#AM4300, #07-645, Millipore, Burlington MA), V5 (#46-0705, Invitrogen, Carlsbad CA), and ubiquitin (#A-104, Boston Biochem, Cambridge MA). The ROR1 antibody has been validated by the manufacturer and we have also tested this antibody in multiple cell lines to correlate immunoblot data with cell lines known to express or not express ROR1 at the transcript level (Supplementary Figure 5). Commercially available inhibitors used in this study are: Velcade (Bortezomib), dasatinib (SelleckChem, Houston TX), anisomycin (Cayman Chemical, Ann Arbor MI), naphthazarin, thymoquinone (Sigma, St. Louis MO), and flavopiridol (LC Laboratories, Woburn MA).
Plasmids
UHRF1 wild-type was cloned into the pCS2-MT 6XMYC vector using In-Fusion cloning (Clonetech) and ClaI restriction sites. The construct was confirmed by sequencing before use.
siRNA
siGENOME SMARTpools (and the equivalent individual siRNA duplexes) were purchased from Dharmacon (Lafayette CO) with the following sequences:
Non-specific pool #1: UAGCGACUAAACACAUCAA, UAAGGCUAUGAAGAGAUAC, AUGUAUUGGCCUGUAUUAG, AUGAACGUGAAUUGCUCAA
ROR1: GCGUGGCAACAAACGGCAA, CGGAUUAGAAACCUCGACA, UGACUUGUGUCGCGAUGAA, GAGAAUGUCCUGUGUCAAA
UHRF1: (siU#1) GCCAUACCCUCUUGCACUA, (siU#2) GGAACAGUCUUGUGAUCAG, UGGAGGAGGACCUCAUUUA, GAACGGCGUGGUCCAGAUG
Gene silencing studies were performed as previously described (10, 58, 59). In brief, 800,000 cells were electroporated with 1-2 μg SMARTpool siRNA duplexes (Dharmacon) using the BioRad Gene Pulser XCell (305 V, 2 ms, 2 pulses). Cells were briefly centrifuged then resuspended in the appropriate growth medium. Cells were harvested after 72 hours and cell viability was measured by MTS assay 96 hours post-electroporation.
siRNA screen
RCH-ACV (305 V, 2 ms, 2 pulses), RCH-ACV+ROR1-V5 (250 V, 3 ms, 2 pulses), and REH (300 V, 2 ms, 2 pulses) cells were electroporated with 1-2 μg siRNA in siPORT buffer (120 mM Trehalose, 20 mM HEPES pH8, 1 mM Myo-Inositol, 1 mM KCl, 1mM MgCl2, 1 mM K2HPO4, 0.4 mM KH2PO4, 2.14 mM KOH, 1 mM Glutathione). Electroporation was performed using a 96-well BioRad Gene Pulser MXCell (BioRad, Hercules CA). Cells were re-plated in R10 media and incubated at 37°C for 96 hours. Total cell viability was measured using the Cell Titer96® AQueous One Solution Cell Proliferation Assay (MTS) according to the manufacturer’s instructions (Promega, Madison WI).
RNA extraction and qRT-PCR
Total cell RNA was extracted using RNeasy according to the manufacturer’s instructions (QIAGEN), eluted in 30 μL of water and quantified using the TAKE3 plate reader (BioTek Synergy2, Winooski VT). cDNA was synthesized according to the manufacturer’s instructions using equal amounts of RNA (Invitrogen Superstrand III/Superscript). qRT-PCR was performed using primer sets with Taqman FAM probes (ThermoFisher Scientific, Waltham MA). The Opticon2 thermocycler (MJ Research Inc., Waltham MA) and QuantStudio7 (Life Technologies, Carlsbad CA) were used to run the following protocol: 50°C 2 min, 95°C 10 min, 40 cycles of 95°C 15 sec, 60°C 1 min. Technical replicate C(t) values were averaged and normalized to GUSB before further analysis.
Protein extraction and immunoblot
1X cell lysis buffer was made using 10X cell lysis buffer with Triton X-100 (Cell Signaling), 1% phosphatase inhibitor cocktail 2 (Sigma), 1% PMSF (Sigma), and a complete mini protease inhibitor tablet (Roche, Basel Switzerland). Whole cell protein extract was quantified by BCA assay (ThermoScientific) and subjected to SDS-PAGE. Membranes were incubated with antibodies at 4°C overnight. Images were obtained by chemiluminescence according to the manufacturer’s instructions (BioRad ECL Clarity Substrate, ChemiDocMP) and densitometry was performed using ImageJ (NIH, Bathesda MD). Bands were normalized to the loading control (Actin or GAPDH) before further analysis.
Immunoprecipitation
Whole cell protein lysate was extracted in an NP-40-based buffer (1% NP-40, 137 mM NaCl, 20 mM Tris-HCl, 20 mM HEPES pH 8, 2 mM EDTA pH 8, 10% Glycerol). 0.5-1 mg of total whole protein lysate was incubated with 4 μg antibody overnight at 4°C with rotation. Protein G beads (Millipore) were blocked with 0.05% BSA/1XPBS for 1 hour at 4°C with rotation. Beads were added and incubated for 1 hour at 4°C with rotation. The unbound fraction was saved before beads were washed three times in NP-40 buffer without NaCl. Beads were boiled at 95°C in loading dye before performing SDS-PAGE.
Cellular Fractionation
5e6-10e6 cells were pelleted at 500xg for 5 min. Nuclear and cytoplasmic fractions were isolated using the NE-PER kit according to the manufacturer’s instructions (ThermoScientific).
HEK293 Transfections
6-well plates were seeded with 3e5 cells overnight. 1-2 μg DNA was transfected using Fugene6 (Promega) according to the manufacturer’s protocol. Cells were harvested for protein lysate extraction 48-72 hours after transfection.
Inhibitor assays
5e5 –1e6 cells were treated with dasatinib, naphthazarin, thymoquinone, anisomycin, or a combination for 72 hours. Cell viability was measured by MTS assay and IC50 values were calculated using Prism software package (GraphPad, La Jolla CA).
Apoptosis Assay
1.25e6 cells were treated with 25-150 nM naphthazarin for 8 hours. GuavaNexin reagent (Millipore) was added and apoptosis was measured according to the manufacturer’s instructions. Annexin-V positive staining was normalized to unstained cells.
Cell Cycle Analysis
7.5e5-1e6 RCH-ACV cells were treated with inhibitor for to 8 hours and subsequently fixed with pre-chilled ethanol for 2 hours at 4°C. Cells were washed with 1XPBS and stained with propidium iodide (Sigma-Aldrich, 0.5mg/mL) and RNAse A (Sigma-Aldrich, 0.5ug/ml) for 30 minutes in the dark at 37°C. Cell cycle distribution was analyzed by flow cytometry and FlowJo.
Mass Spectrometry
1 mg of RCH-ACV or Kasumi-2 whole cell extracts were incubated with 4 μg ROR1, UHRF1, or a matched isotype control antibody overnight at 4°C. Samples were eluted in 1% SDS and submitted for mass spectrometry. Mass spectrometric analysis was performed by the OHSU Proteomics Shared Resource with partial support from the NIH instrument grant S10OD012246 (Orbitrap Fusion) and core grants P30EY010572 and P30CA069533. Only proteins identified with the ROR1-specific or UHRF1-specific antibody and not the isotype control were considered as candidate interacting proteins.
Statistical Analysis
All statistical analyses on MTS cell viability, qRT-PCR, immunoblot, and apoptosis assays were performed using Prism software package (GraphPad). Data shown in figure 2, 4A, and S1A were analyzed by ANOVA and adjusted p-values were reported to reflect multiple comparisons used in statistical analyses. ANOVA analysis includes statistical comparisons of variance between groups and were all found to be similar (residual ss values less than 1). T-tests were used to analyze data shown in figure 4D-E and S1C. T-tests assume equal variance between groups. Sample sizes listed in figure legends correspond to independent, biological replicates, and at least three independent experiments were performed for data to be analyzed for statistical significance. Error bars are noted in figure legends to represent as either S.D. = standard deviation or S.E.M. = standard error mean.
Supplementary Material
Acknowledgments
The pCS2-6XMYC vector was a generous gift from Dr. Monika Davare. The authors would like to thank David K. Edwards V, Samantha L. Savage, Anna M. Reister Schultz, Dr. Haijiao Zhang, and Janet Pittsenbarger for their technical expertise and Dr. Kevin M. Watanabe-Smith for editing the manuscript.
M.C. was supported in part by the Oregon Clinical and Translational Research Institute (OCTRI) (TL1TR000129) from the National Center for Advancing Translational Sciences (NCATS) at the National Institutes of Health (NIH). B.H.C. is supported by the Hyundai Hope on Wheels. J.W.T. is supported by The Leukemia & Lymphoma Society, the V Foundation for Cancer Research, Gabrielle’s Angel Foundation for Cancer Research, and the National Cancer Institute (5R00CA151457-04; 1R01CA183947-01).
Footnotes
Conflict of Interest
Dr. Tyner’s work has received research support from Agios Pharmaceuticals, Array Biopharma, Aptose Biosciences, AstraZeneca, Constellation Pharmaceuticals, Genentech, Gilead, Incyte Corporation, Janssen Pharmaceutica, Seattle Genetics, Syros, Takeda Pharmaceutical Company and is a co-founder of Leap Oncology. All other authors declare no conflicts of interest.
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc).
References
- 1.Foa R, Vitale A, Mancini M, Cuneo A, Mecucci C, Elia L, et al. E2A-PBX1 fusion in adult acute lymphoblastic leukaemia: biological and clinical features. British journal of haematology. 2003;120(3):484–7. doi: 10.1046/j.1365-2141.2003.04113.x. [DOI] [PubMed] [Google Scholar]
- 2.Loh ML, Mullighan CG. Advances in the genetics of high-risk childhood B-progenitor acute lymphoblastic leukemia and juvenile myelomonocytic leukemia: implications for therapy. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18(10):2754–67. doi: 10.1158/1078-0432.CCR-11-1936. [DOI] [PubMed] [Google Scholar]
- 3.Tasian SK, Hunger SP. Genomic characterization of paediatric acute lymphoblastic leukaemia: an opportunity for precision medicine therapeutics. British journal of haematology. 2017;176(6):867–82. doi: 10.1111/bjh.14474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moorman AV, Chilton L, Wilkinson J, Ensor HM, Bown N, Proctor SJ. A population-based cytogenetic study of adults with acute lymphoblastic leukemia. Blood. 2010;115(2):206–14. doi: 10.1182/blood-2009-07-232124. [DOI] [PubMed] [Google Scholar]
- 5.McWhirter JR, Neuteboom ST, Wancewicz EV, Monia BP, Downing JR, Murre C. Oncogenic homeodomain transcription factor E2A-Pbx1 activates a novel WNT gene in pre-B acute lymphoblastoid leukemia. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(20):11464–9. doi: 10.1073/pnas.96.20.11464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mazieres J, You L, He B, Xu Z, Lee AY, Mikami I, et al. Inhibition of Wnt16 in human acute lymphoblastoid leukemia cells containing the t(1;19) translocation induces apoptosis. Oncogene. 2005;24(34):5396–400. doi: 10.1038/sj.onc.1208568. [DOI] [PubMed] [Google Scholar]
- 7.Nygren MK, Dosen-Dahl G, Stubberud H, Walchli S, Munthe E, Rian E. beta-catenin is involved in N-cadherin-dependent adhesion, but not in canonical Wnt signaling in E2A-PBX1-positive B acute lymphoblastic leukemia cells. Experimental hematology. 2009;37(2):225–33. doi: 10.1016/j.exphem.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 8.Hunger SP. Chromosomal translocations involving the E2A gene in acute lymphoblastic leukemia: clinical features and molecular pathogenesis. Blood. 1996;87(4):1211–24. [PubMed] [Google Scholar]
- 9.Chen D, Zheng J, Gerasimcik N, Lagerstedt K, Sjogren H, Abrahamsson J, et al. The Expression Pattern of the Pre-B Cell Receptor Components Correlates with Cellular Stage and Clinical Outcome in Acute Lymphoblastic Leukemia. PloS one. 2016;11(9):e0162638. doi: 10.1371/journal.pone.0162638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bicocca VT, Chang BH, Masouleh BK, Muschen M, Loriaux MM, Druker BJ, et al. Crosstalk between ROR1 and the Pre-B cell receptor promotes survival of t(1;19) acute lymphoblastic leukemia. Cancer cell. 2012;22(5):656–67. doi: 10.1016/j.ccr.2012.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feldhahn N, Klein F, Mooster JL, Hadweh P, Sprangers M, Wartenberg M, et al. Mimicry of a constitutively active pre-B cell receptor in acute lymphoblastic leukemia cells. The Journal of experimental medicine. 2005;201(11):1837–52. doi: 10.1084/jem.20042101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fukuda T, Chen L, Endo T, Tang L, Lu D, Castro JE, et al. Antisera induced by infusions of autologous Ad-CD154-leukemia B cells identify ROR1 as an oncofetal antigen and receptor for Wnt5a. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(8):3047–52. doi: 10.1073/pnas.0712148105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Widhopf GF, 2nd, Cui B, Ghia EM, Chen L, Messer K, Shen Z, et al. ROR1 can interact with TCL1 and enhance leukemogenesis in Emu-TCL1 transgenic mice. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(2):793–8. doi: 10.1073/pnas.1308374111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baskar S, Kwong KY, Hofer T, Levy JM, Kennedy MG, Lee E, et al. Unique cell surface expression of receptor tyrosine kinase ROR1 in human B-cell chronic lymphocytic leukemia. Clinical cancer research : an official journal of the American Association for Cancer Research. 2008;14(2):396–404. doi: 10.1158/1078-0432.CCR-07-1823. [DOI] [PubMed] [Google Scholar]
- 15.Daneshmanesh AH, Mikaelsson E, Jeddi-Tehrani M, Bayat AA, Ghods R, Ostadkarampour M, et al. Ror1, a cell surface receptor tyrosine kinase is expressed in chronic lymphocytic leukemia and may serve as a putative target for therapy. International journal of cancer Journal international du cancer. 2008;123(5):1190–5. doi: 10.1002/ijc.23587. [DOI] [PubMed] [Google Scholar]
- 16.Shabani M, Asgarian-Omran H, Vossough P, Sharifian RA, Faranoush M, Ghragozlou S, et al. Expression profile of orphan receptor tyrosine kinase (ROR1) and Wilms’ tumor gene 1 (WT1) in different subsets of B-cell acute lymphoblastic leukemia. Leukemia & lymphoma. 2008;49(7):1360–7. doi: 10.1080/10428190802124000. [DOI] [PubMed] [Google Scholar]
- 17.Broome HE, Rassenti LZ, Wang HY, Meyer LM, Kipps TJ. ROR1 is expressed on hematogones (non-neoplastic human B-lymphocyte precursors) and a minority of precursor-B acute lymphoblastic leukemia. Leukemia research. 2011;35(10):1390–4. doi: 10.1016/j.leukres.2011.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang S, Chen L, Wang-Rodriguez J, Zhang L, Cui B, Frankel W, et al. The onco-embryonic antigen ROR1 is expressed by a variety of human cancers. The American journal of pathology. 2012;181(6):1903–10. doi: 10.1016/j.ajpath.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang S, Chen L, Cui B, Chuang HY, Yu J, Wang-Rodriguez J, et al. ROR1 is expressed in human breast cancer and associated with enhanced tumor-cell growth. PLoS One. 2012;7(3):e31127. doi: 10.1371/journal.pone.0031127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Balakrishnan A, Goodpaster T, Randolph-Habecker J, Hoffstrom BG, Jalikis FG, Koch LK, et al. Analysis of ROR1 Protein Expression in Human Cancer and Normal Tissues. Clinical cancer research : an official journal of the American Association for Cancer Research. 2017;23(12):3061–71. doi: 10.1158/1078-0432.CCR-16-2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hudecek M, Schmitt TM, Baskar S, Lupo-Stanghellini MT, Nishida T, Yamamoto TN, et al. The B-cell tumor-associated antigen ROR1 can be targeted with T cells modified to express a ROR1-specific chimeric antigen receptor. Blood. 2010;116(22):4532–41. doi: 10.1182/blood-2010-05-283309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baskar S, Wiestner A, Wilson WH, Pastan I, Rader C. Targeting malignant B cells with an immunotoxin against ROR1. mAbs. 2012;4(3):349–61. doi: 10.4161/mabs.19870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mani R, Mao Y, Frissora FW, Chiang CL, Wang J, Zhao Y, et al. Tumor antigen ROR1 targeted drug delivery mediated selective leukemic but not normal B-cell cytotoxicity in chronic lymphocytic leukemia. Leukemia. 2015;29(2):346–55. doi: 10.1038/leu.2014.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choi MY, Widhopf GF, 2nd, Wu CC, Cui B, Lao F, Sadarangani A, et al. Pre-clinical Specificity and Safety of UC-961, a First-In-Class Monoclonal Antibody Targeting ROR1. Clinical lymphoma, myeloma & leukemia. 2015;15(Suppl):S167–9. doi: 10.1016/j.clml.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li P, Harris D, Liu Z, Liu J, Keating M, Estrov Z. Stat3 activates the receptor tyrosine kinase like orphan receptor-1 gene in chronic lymphocytic leukemia cells. PLoS One. 2010;5(7):e11859. doi: 10.1371/journal.pone.0011859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamaguchi T, Yanagisawa K, Sugiyama R, Hosono Y, Shimada Y, Arima C, et al. NKX2-1/TITF1/TTF-1-Induced ROR1 is required to sustain EGFR survival signaling in lung adenocarcinoma. Cancer Cell. 2012;21(3):348–61. doi: 10.1016/j.ccr.2012.02.008. [DOI] [PubMed] [Google Scholar]
- 27.Kaucka M, Krejci P, Plevova K, Pavlova S, Prochazkova J, Janovska P, et al. Post-translational modifications regulate signalling by Ror1. Acta physiologica. 2011;203(3):351–62. doi: 10.1111/j.1748-1716.2011.02306.x. [DOI] [PubMed] [Google Scholar]
- 28.Ma J, Peng J, Mo R, Ma S, Wang J, Zang L, et al. Ubiquitin E3 ligase UHRF1 regulates p53 ubiquitination and p53-dependent cell apoptosis in clear cell Renal Cell Carcinoma. Biochemical and biophysical research communications. 2015;464(1):147–53. doi: 10.1016/j.bbrc.2015.06.104. [DOI] [PubMed] [Google Scholar]
- 29.Karagianni P, Amazit L, Qin J, Wong J. ICBP90, a novel methyl K9 H3 binding protein linking protein ubiquitination with heterochromatin formation. Molecular and cellular biology. 2008;28(2):705–17. doi: 10.1128/MCB.01598-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim JK, Esteve PO, Jacobsen SE, Pradhan S. UHRF1 binds G9a and participates in p21 transcriptional regulation in mammalian cells. Nucleic acids research. 2009;37(2):493–505. doi: 10.1093/nar/gkn961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang F, Yang YZ, Shi CZ, Zhang P, Moyer MP, Zhang HZ, et al. UHRF1 promotes cell growth and metastasis through repression of p16(ink(4)a) in colorectal cancer. Ann Surg Oncol. 2012;19(8):2753–62. doi: 10.1245/s10434-011-2194-1. [DOI] [PubMed] [Google Scholar]
- 32.Arita K, Ariyoshi M, Tochio H, Nakamura Y, Shirakawa M. Recognition of hemi-methylated DNA by the SRA protein UHRF1 by a base-flipping mechanism. Nature. 2008;455(7214):818–21. doi: 10.1038/nature07249. [DOI] [PubMed] [Google Scholar]
- 33.Avvakumov GV, Walker JR, Xue S, Li Y, Duan S, Bronner C, et al. Structural basis for recognition of hemi-methylated DNA by the SRA domain of human UHRF1. Nature. 2008;455(7214):822–5. doi: 10.1038/nature07273. [DOI] [PubMed] [Google Scholar]
- 34.Sharif J, Muto M, Takebayashi S, Suetake I, Iwamatsu A, Endo TA, et al. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature. 2007;450(7171):908–12. doi: 10.1038/nature06397. [DOI] [PubMed] [Google Scholar]
- 35.Unoki M, Nishidate T, Nakamura Y. ICBP90, an E2F-1 target, recruits HDAC1 and binds to methyl-CpG through its SRA domain. Oncogene. 2004;23(46):7601–10. doi: 10.1038/sj.onc.1208053. [DOI] [PubMed] [Google Scholar]
- 36.Rottach A, Frauer C, Pichler G, Bonapace IM, Spada F, Leonhardt H. The multi-domain protein Np95 connects DNA methylation and histone modification. Nucleic acids research. 2010;38(6):1796–804. doi: 10.1093/nar/gkp1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rajakumara E, Wang Z, Ma H, Hu L, Chen H, Lin Y, et al. PHD finger recognition of unmodified histone H3R2 links UHRF1 to regulation of euchromatic gene expression. Molecular cell. 2011;43(2):275–84. doi: 10.1016/j.molcel.2011.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alhosin M, Omran Z, Zamzami MA, Al-Malki AL, Choudhry H, Mousli M, et al. Signalling pathways in UHRF1-dependent regulation of tumor suppressor genes in cancer. Journal of experimental & clinical cancer research : CR. 2016;35(1):174. doi: 10.1186/s13046-016-0453-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Myrianthopoulos V, Cartron PF, Liutkeviciute Z, Klimasauskas S, Matulis D, Bronner C, et al. Tandem virtual screening targeting the SRA domain of UHRF1 identifies a novel chemical tool modulating DNA methylation. European journal of medicinal chemistry. 2016;114:390–6. doi: 10.1016/j.ejmech.2016.02.043. [DOI] [PubMed] [Google Scholar]
- 40.Mousli M, Hopfner R, Abbady AQ, Monte D, Jeanblanc M, Oudet P, et al. ICBP90 belongs to a new family of proteins with an expression that is deregulated in cancer cells. British journal of cancer. 2003;89(1):120–7. doi: 10.1038/sj.bjc.6601068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Daskalos A, Oleksiewicz U, Filia A, Nikolaidis G, Xinarianos G, Gosney JR, et al. UHRF1-mediated tumor suppressor gene inactivation in nonsmall cell lung cancer. Cancer. 2011;117(5):1027–37. doi: 10.1002/cncr.25531. [DOI] [PubMed] [Google Scholar]
- 42.Mudbhary R, Hoshida Y, Chernyavskaya Y, Jacob V, Villanueva A, Fiel MI, et al. UHRF1 overexpression drives DNA hypomethylation and hepatocellular carcinoma. Cancer cell. 2014;25(2):196–209. doi: 10.1016/j.ccr.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Abu-Alainin W, Gana T, Liloglou T, Olayanju A, Barrera LN, Ferguson R, et al. UHRF1 regulation of the Keap1-Nrf2 pathway in pancreatic cancer contributes to oncogenesis. J Pathol. 2016;238(3):423–33. doi: 10.1002/path.4665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jia Y, Li P, Fang L, Zhu H, Xu L, Cheng H, et al. Negative regulation of DNMT3A de novo DNA methylation by frequently overexpressed UHRF family proteins as a mechanism for widespread DNA hypomethylation in cancer. Cell Discovery. 2016;2:16007. doi: 10.1038/celldisc.2016.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qu X, Davison J, Du L, Storer B, Stirewalt DL, Heimfeld S, et al. Identification of differentially methylated markers among cytogenetic risk groups of acute myeloid leukemia. Epigenetics. 2015;10(6):526–35. doi: 10.1080/15592294.2015.1048060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Niebuhr B, Kriebitzsch N, Fischer M, Behrens K, Gunther T, Alawi M, et al. Runx1 is essential at two stages of early murine B-cell development. Blood. 2013;122(3):413–23. doi: 10.1182/blood-2013-01-480244. [DOI] [PubMed] [Google Scholar]
- 47.Kim MY, Park SJ, Shim JW, Yang K, Kang HS, Heo K. Naphthazarin enhances ionizing radiation-induced cell cycle arrest and apoptosis in human breast cancer cells. International journal of oncology. 2015;46(4):1659–66. doi: 10.3892/ijo.2015.2857. [DOI] [PubMed] [Google Scholar]
- 48.Acharya BR, Bhattacharyya S, Choudhury D, Chakrabarti G. The microtubule depolymerizing agent naphthazarin induces both apoptosis and autophagy in A549 lung cancer cells. Apoptosis : an international journal on programmed cell death. 2011;16(9):924–39. doi: 10.1007/s10495-011-0613-1. [DOI] [PubMed] [Google Scholar]
- 49.Kim JA, Lee EK, Park SJ, Kim ND, Hyun DH, Lee CG, et al. Novel anti-cancer role of naphthazarin in human gastric cancer cells. International journal of oncology. 2012;40(1):157–62. doi: 10.3892/ijo.2011.1195. [DOI] [PubMed] [Google Scholar]
- 50.Rau R, Brown P. Nucleophosmin (NPM1) mutations in adult and childhood acute myeloid leukaemia: towards definition of a new leukaemia entity. Hematological oncology. 2009;27(4):171–81. doi: 10.1002/hon.904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fernandez NB, Lorenzo D, Picco ME, Barbero G, Dergan-Dylon LS, Marks MP, et al. ROR1 contributes to melanoma cell growth and migration by regulating N-cadherin expression via the PI3K/Akt pathway. Mol Carcinog. 2016;55(11):1772–85. doi: 10.1002/mc.22426. [DOI] [PubMed] [Google Scholar]
- 52.Alhosin M, Leon-Gonzalez AJ, Dandache I, Lelay A, Rashid SK, Kevers C, et al. Bilberry extract (Antho 50) selectively induces redox-sensitive caspase 3-related apoptosis in chronic lymphocytic leukemia cells by targeting the Bcl-2/Bad pathway. Scientific reports. 2015;5:8996. doi: 10.1038/srep08996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Borcherding N, Kusner D, Liu GH, Zhang W. ROR1, an embryonic protein with an emerging role in cancer biology. Protein & cell. 2014;5(7):496–502. doi: 10.1007/s13238-014-0059-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sanchez-Aguilera A, Rattmann I, Drew DZ, Muller LU, Summey V, Lucas DM, et al. Involvement of RhoH GTPase in the development of B-cell chronic lymphocytic leukemia. Leukemia. 2010;24(1):97–104. doi: 10.1038/leu.2009.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Troeger A, Johnson AJ, Wood J, Blum WG, Andritsos LA, Byrd JC, et al. RhoH is critical for cell-microenvironment interactions in chronic lymphocytic leukemia in mice and humans. Blood. 2012;119(20):4708–18. doi: 10.1182/blood-2011-12-395939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Doyon Y, Cayrou C, Ullah M, Landry AJ, Cote V, Selleck W, et al. ING tumor suppressor proteins are critical regulators of chromatin acetylation required for genome expression and perpetuation. Molecular cell. 2006;21(1):51–64. doi: 10.1016/j.molcel.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 57.Kueh AJ, Dixon MP, Voss AK, Thomas T. HBO1 is required for H3K14 acetylation and normal transcriptional activity during embryonic development. Molecular and cellular biology. 2011;31(4):845–60. doi: 10.1128/MCB.00159-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tyner JW, Deininger MW, Loriaux MM, Chang BH, Gotlib JR, Willis SG, et al. RNAi screen for rapid therapeutic target identification in leukemia patients. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(21):8695–700. doi: 10.1073/pnas.0903233106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sanda T, Tyner JW, Gutierrez A, Ngo VN, Glover J, Chang BH, et al. TYK2-STAT1-BCL2 pathway dependence in T-cell acute lymphoblastic leukemia. Cancer discovery. 2013;3(5):564–77. doi: 10.1158/2159-8290.CD-12-0504. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.