

Abstract
Functioning DNA repair capabilities are vital for organisms to ensure that the biological information is preserved and correctly propagated. Disruptions in DNA repair pathways can result in the accumulation of DNA mutations, which may lead to onset of complex disease such as cancer. The discovery and characterization of cancer-related biomarkers may allow early diagnosis and targeted treatment, which could significantly contribute to the survival rates of cancer patients. To this end, we have applied a hypothesis driven bioinformatics approach to identify biomarkers related to 25 different DNA repair enzymes, in combination with structural analysis of six selected missense mutations of newly discovered SNPs that are associated with cancer phenotypes. Our search on 8 distinct cancer databases uncovered 43 missense SNPs that statistically significantly associated at least one phenotype. Moreover, nine of these missense SNPs are statistically significantly associated with two or more cancers. In addition, we have performed classical molecular dynamics to characterize the impact of rs10018786 on POLN, which results in the M310L Pol ν variant, and rs3218784 on POLI, which results in the I236M Polι. Our results suggest that both of these cancer-associated variants result in noticeable structural and dynamical changes compared with their respective wild-type proteins.
Introduction
Cancer is a major healthcare challenge as the complexity and diversity of the disease inhibits easy solutions in developing effective treatments. Currently, cancer is the 2nd leading cause of death in the USA1. In 2016, the number of diagnosed cancer cases in the USA was estimated at 1,685,210, while the number of deaths from cancer was reported to be 595,6902. Cancers with high rates of incidence include prostate for men (21% of new cases), and breast for women (29%), while lung and bronchus cancers result in the largest number of cancer caused deaths for both men (27% of all cancer deaths) and women (26%)2. While the treatment for some types of cancers has a relatively high success rate, e.g. prostate cancer has 98.6% survival rate, others remain a challenge, e.g. pancreatic cancer has 8.2% survival rate3.
The search for genes and mutations that are involved in carcinogenesis is an ongoing endeavor. As cancer has its roots in genetic information, finding the genetic variants that are responsible for carcinogenesis would be beneficial for cancer prevention, diagnostics, and treatment. Cancer causing genetic mutations can be of both germline and somatic origin4. Of particular interest are the genes involved in cell proliferation, differentiation, death, and DNA repair as their disruption is associated with carcinogenesis4. Cancer population genetic information is accumulating thanks to a number of initiatives, such as the Cancer Genome Atlas (TCGA) Research Network5, and the NCBI database of genotypes and phenotypes (dbGAP)6.
Taking into consideration personal genetic data in identifying patients’ cancer treatment can significantly contribute to finding the best interventions at the right time and thus increase survival rates. Personalized medicine, the term that has been used in the literature, can allow to foresee the possibility of development of a particular disease and to implement preventive measures, as well as to find the most effective medications tailored individually to a patient7. In addition to individual genomic profile, transcriptomics, proteomics, and metabolomics data may also be used for diagnostic decisions7.
Nevertheless, challenges still remain, although biomarkers’ development have been facilitated by the progress in genotyping and data processing technologies. The search for biomarkers is complicated by the fact that out of many possible mutations in cancer linked genes only a few are actually cancer driving mutations, while most are passengers8. Also, genes confer their role as constituents of various pathways and thus when analyzed individually may not represent the full picture of biological processes. Therefore, methods such as gene sets analysis have been developed to account for biological complexity and interplay of various genes and pathways9. Furthermore, genetics composition of neoplasms are heterogeneous, complicating pinpointing a causative mutation. Methods have been developed to detect low frequency somatic mutations10. More recently, methods that analyze a small to moderate number of SNPs on a several phenotypes have been developed to investigate the possibility of these SNPs for a common underlying genetic predisposition5, 11.
DNA repair genes serve a genome maintenance function and are important for preventing carcinogenesis12, 13. Mutations that disrupt the function of DNA repair genes can result in increased rates of somatic mutations in other genes and thus significantly increase susceptibility to cancer. Without maintenance and repair, cells retain the damaged DNA, continue their growth cycle, divide, and thus propagate and accumulate the damage, eventually turning into cancerous cells. Some of us developed a new method called hypothesis driven-SNP search (HyDn-SNP-S), and used it to uncover SNPs on DNA polymerases 14, 15. Our method uncovered over 75 cancer—associated SNPs on various DNA polymerases. In addition, we characterized the structural/functional impact on the structure of a specific mutation on DNA Polymerase λ resulting from a breast cancer-associated SNP15.
In this work we extend our investigation to uncover and characterize SNPs related to other cancer phenotypes on DNA repair genes (including DNA polymerases) based on cancer case-control studies available in the dbGAP database (project access request #12236)14. Following the HyDn-SNP-S method15, we apply a hypothesis driven approach and uncover SNPs in the genes of interest, i.e. DNA repair genes. By narrowing the gene pool tested, the error of finding false positive associations and the multiple testing penalty are reduced when compared to GWAS. Therefore, a targeted approach provides useful SNP leads for further investigation. Subsequently, the uncovered SNPs are classified as intronic or exonic. SNPs resulting in missense mutations in the protein product are further investigated by all-atom classical molecular dynamcis (MD) methods if high-resolution crystal structures of that particular protein are available. This subsequent characterization allows the determination of whether and/or how the mutation arising from the cancer-related SNP affects the protein structure and/or function.
Methods
SNP Search and Statistical Analysis
Data for analysis was obtained from the NCBI dbGAP database (project access #12236)14. Disease phenotypes analyzed include breast cancer (study reference id: phs00014716, 17; cases: 1145; controls: 1142), prostate cancer (study reference id: phs00020718; cases:1172; controls:1157), lung cancer (study reference id: phs00033619; cases: 5739; controls: 5848), pancreatic cancer (study reference id: phs00020620–22; cases: 5533; controls: 3904), chronic lymphocytic leukemia (CLL) (study reference id: phs00080223 and phs00081823; cases: 2178; controls: 2685), diffuse large B-cell lymphoma (DLBCL) (study reference id: phs00088924 and phs00081823; cases: 2661; controls: 2685), follicular lymphoma (FL) (study reference id: phs00089025 and phs00081823; cases: 2142; controls: 2685), and marginal zone lymphoma (MZL) (study reference id: phs00089126 and phs00081823; cases: 825; controls: 2685).
SNPs in the DNA repair genes were selected utilizing previous compilations27–29, in particular the following gene families were included in the present analysis: Alkb human homologs (ALKBH1, ALKBH2, ALKBH3, ALKBH4, ALKBH5, ALKBH6, ALKBH7, ALBH8, FTO), mismatch repair (EXO1, MLH1, MLH3, MSH2, MSH6, PMS1, PMS2, MSH4, MSH5), DNA polymerases (DNTT, MAD2L2, POLA1, POLA2, POLB, POLD1, POLD2, POLD3, POLD4, POLE, POLE2, POLE3, POLE4, POLG, POLG2, POLH, POLK, POLL, POLM, POLN, POLQ, REV1, REV3L), ten-eleven translocation enzymes (TET1, TET2, TET3, APO1, APO2, APO3A, APO3B, APO3C, APO3D, APO3F, APO3G, APO3H, APO4), base excision repair (UNG, SMUG1, MBD4, TDG, OGG1, MUTYH, MPG, NEIL1, NEIL2, NEIL3, APEX1, APEX2, LIG3, XRCC1, PNKP, APLF), and poly ADP-ribose polymerases (PARP1, PARP2, PARP3). Statistical analysis to detect associations between the SNPs of interest and cancer phenotypes was performed with the logistic regression model. Four inheritance models were considered: multiplicative, additive, dominant, and recessive30. Haplotype analysis for a selected group of SNPs was performed with the haplo.stats package31.
Structural and Dynamics Analysis of Missense Mutations
Two enzymes with SNPs resulting in missense mutations were selected to characterize the impact of the cancer variant on the structure/function using classical molecular dynamics (MD) simulations. We have previously reported the characterization of the ALKBH7 variant resulting from rs654032. Here we present the characterization of the cancer variants for two DNA polymerases: Pol ν (SNPs rs9328764, rs10011549, and rs10018786) and Pol ι (SNP rs3218784). The corresponding mutations for each variant were introduced with the tleap tool of AMBER1633.
Molecular dynamics simulations were performed in triplicate for the wild type and all mutant structures. All systems were prepared as apo- or holo- binary structures, that is, the holo-structures include the DNA substrate without the incoming nucleotide triphosphate. Crystal structures for Pol ν (PDB ID: 4XVK34) and for Pol ι (PDB ID: 3GV835) were obtained from Protein Data Bank36. Coordinates of missing loops in the crystal structures were modeled by MODELLER37. For Pol ν, a magnesium ion was added to the active site of the crystal structure; the structures with DNA (holo-) and without DNA (apo-) were made by either leaving or deleting DNA atoms from crystal structures.
Hydrogen atoms were added to the structures using Molprobity38, 39. The pmemd.cuda40 program of AMBER1633 with protein.ff14SB and DNA.OL15 force fields41, 42 was used for molecular dynamics simulations. All systems were subjected to MD for 200 ns for each replicate trajectory, with a 1 fs time step. Water molecules were modeled with the TIP3P force field using periodic boundary conditions. All systems were created with a minimum distance from the surface of the protein to the edge of the box of 15 Å43. The smooth particle mesh Ewald (sPME) method with an 8 Å real space cutoff was used for modeling the electrostatic interactions44. The water density was brought to 1 g/ml by a restrained MD in the NPT ensemble. Subsequently, calculations were performed in the NVT ensemble at 300K temperature with a Langevin thermostat. The cpptraj tool of AMBER1633 was used for trajectory and hydrogen bonds analyses. Chimera software45 was used for visualization and structure analysis and editing.
Results and Discussion
Statistical Analysis
Five hundred and sixty two SNPs on DNA repair genes are found to have a statistically significant (p<0.05) association with cancer phenotypes for at least one of the inheritance models (see Tables S.1 to S.8). Thirty one of these SNPs are associated with the breast cancer phenotype, 26 with prostate, 134 with lung, 64 with pancreatic, 116 with CLL, 82 with DLBL, 69 with FL, and 40 with MZL. Out of these 564 SNPs only 43, i.e. around 8%, correspond to missense mutations (see Table 1).
TABLE 1.
Missense mutation SNPs linked to cancer phenotypes (p-values of at least one inheritance model < 0.05). Data for polymerase genes for breast and prostate cancers are from Swett et al15.
| breast | prostate | lung | pancreatic | CLL | DLBCL | FL | MZL | |
|---|---|---|---|---|---|---|---|---|
| ABH7 | rs7540 | |||||||
| MSH3 | rs184967 rs26279 |
|||||||
| POLE | rs4883543 rs4883544 |
|||||||
| POLA2 | rs487989 | |||||||
| POLD1 | rs1726801 | |||||||
| POLG | rs2307441 | rs3087374 | ||||||
| POLG2 | rs1427463 | |||||||
| POLI | rs8305 | |||||||
| POLL | rs3730463 | rs3730476 | rs3730463 rs3730476 |
rs3730476 | ||||
| POLN | rs9328764 rs10011549 rs1001878 |
rs11937432 rs2353552 |
||||||
| POLQ | rs3218651 | rs3218651 | rs3218651 | rs3218642 | ||||
| REV3L | rs462779 | |||||||
| TET1 | rs12221107 | rs12221107 rs16925541 |
rs3998860 | rs3998860 | ||||
| APO2 | rs2076472 | rs2076472 | ||||||
| APO3G | rs8177832 | |||||||
| APO3H | rs139298 | rs139297 | rs139297 | |||||
| APO4 | rs1174657 rs1174658 |
rs16861394 | ||||||
| MBD4 | rs140693 | rs2307293 | ||||||
| MUTYH | rs3219484 | rs3219484 | rs3219484 | |||||
| NEIL2 | rs8191613 rs8191664 |
|||||||
| NEIL3 | rs13112390 rs13112358 |
rs13112358 | rs1876268 | rs1876268 | rs13112358 rs13112390 rs1876268 |
|||
| APEX1 | rs1130409 | |||||||
| APEX2 | rs2301416 | |||||||
| PARP1 | rs1136410 | rs1136410 | ||||||
| PARP2 | rs3093921 | rs3093921 |
The POLN gene was found to have three missense mutations linked to breast cancer (Table 1). Haplotype analysis of POLN and POLI genes based on the patients’ data used in this study has been previously published and did not detect haplotypes of genetic variants from these genes15. To check if the combinations of the missense mutations in the POLN gene constitute haplotypes linked to breast cancer all double, triple and quadruple combinations of the POLN genes that were individually found to be linked to breast cancer were analyzed using the haplo.stats package, (Table 2). Each of the haplotypes contained only one missense causing variant.
TABLE 2.
Haplotypes of POLN gene associated with the breast cancer phenotype. SNP reference IDs marked with * are missense mutations.
| SNPs: rs9328764* rs2353552 | SNPs rs10011549* rs11937432 | ||||||||
| Haplotype Effect Model: additive | Haplotype Effect Model: additive | ||||||||
| global-stat = 6.83406, df = 2, p-val = 0.03281 | global-stat = 6.3784, df = 2, p-val = 0.0412 | ||||||||
| Haplotype-specific Scores | Haplotype-specific Scores | ||||||||
| loc-1 | loc-2 | Hap-Freq | Hap-Score | p-val | loc-1 | loc-2 | Hap-Freq | Hap-Score | p-val |
| A | C | 0.12 | −2.36 | 0.02 | A | T | 0.07 | −2.37 | 0.02 |
| G | A | 0.12 | −0.83 | 0.41 | G | T | 0.05 | −0.73 | 0.46 |
| G | C | 0.76 | 2.38 | 0.02 | A | C | 0.88 | 2.3 | 0.02 |
| SNPs: rs9328764* rs11937432 | SNPs rs10011549* rs11937432 | ||||||||
| Haplotype Effect Model: additive | Haplotype Effect Model: recessive | ||||||||
| global-stat = 7.92266, df = 3, p-val = 0.04764 | global-stat = 7.99672, df = 3, p-val = 0.04608 | ||||||||
| Haplotype-specific Scores | Haplotype-specific Scores | ||||||||
| loc-1 | loc-2 | Hap-Freq | Hap-Score | p-val | loc-1 | loc-2 | Hap-Freq | Hap-Score | p-val |
| A | A | 0.06 | −2.45 | 0.01 | G | T | 0.05 | −1.66 | 0.1 |
| G | A | 0.05 | −0.83 | 0.41 | A | T | 0.07 | −1.48 | 0.14 |
| A | G | 0.88 | 2.39 | 0.02 | A | C | 0.88 | 2.15 | 0.03 |
| SNPs rs9328764* rs11937432 | SNPs rs10018786* rs2353552 | ||||||||
| Haplotype Effect Model: recessive | Haplotype Effect Model: additive | ||||||||
| global-stat = 8.03298, df = 3, p-val = 0.04534 | global-stat = 6.21471, df = 2, p-val = 0.04472 | ||||||||
| Haplotype-specific Scores | Haplotype-specific Scores | ||||||||
| loc-1 | loc-2 | Hap-Freq | Hap-Score | p-val | loc-1 | loc-2 | Hap-Freq | Hap-Score | p-val |
| G | A | 0.05 | −1.74 | 0.08 | G | C | 0.12 | −2.23 | 0.03 |
| A | A | 0.06 | −1.1 | 0.27 | T | A | 0.12 | −0.83 | 0.41 |
| A | G | 0.88 | 2.27 | 0.02 | T | C | 0.76 | 2.29 | 0.02 |
A significant finding from our statistical analysis is the relatively large number of missense SNPs on DNA repair enzymes that are found to be associated with more than one cancer phenotype. A total of 109 SNPs were found to be statistically significantly associated with multiple cancer types (TABLE S.9), with 43 of those corresponding to exonic-nonsynonymous mutations. Out of these, SNPs on POLL, POLQ, TET1, APO2, APO3H, MUTYH, NEIL3, PARP1 and PARP2 all have at least one exonic-nonsynonymous SNP that is statistically significantly associated with two or more cancer phenotypes. That is, over 30% of the genes that have a cancer-associated missense mutation, share this mutation with more than one cancer phenotype. Moreover, our results indicate that there are four different enzymes, DNA polymerase θ, APOBEC3H, MUTY and NEIL1, with the same SNP statistically significantly associated with three different cancer phenotypes (see Table 1). It should be noted that our search procedure produces results that are consistent with recently-developed approaches such as phenome-wide association studies, which have been developed to evaluate the impact of one or a small number of genetic variants with phenotypic data46.
Some of these SNPs have been previously reported to be statistically significantly associated with one of a subset of the cancer phenotypes (breast, lung, prostate or melanoma) 15 including two SNPs on the POLL gene, which codes for DNA polymerase λ (Pol λ). One of these SNPs rs3730463, translated to T221P on Pol λ, was previously linked to breast cancer. Our new analysis reveals that this same SNP is also linked with DLBCL (TABLE 1). A haplotype of this SNP coupled with rs3730477, translated to R438W, was previously identified to be associated with breast cancer15. Moreover, SNP rs3730477 has been experimentally shown to be present in significantly higher ratios in germline DNA of breast cancer patients and has been confirmed to be a risk factor for estrogen-driven breast cancer 47.
Structural and Dynamics Analysis of Missense Mutations on Polymerase ν Gene
The SNPs found to be linked to cancer phenotypes in the POLN, and POLI genes were selected for further investigation of the impact of the mutation on the protein structure by computational simulations based on deposited crystal structures available in the protein data bank36. This analysis has been applied previously to cancer variants of Pol λ and ALKBH7.15,32 Our previously reported computational simulations on the ALKBH7 variant resulting from the prostate cancer associated SNP predicted that the structural changes would preclude co-substrate binding, which was confirmed experimentally in the same work as discussed in Ref32.
Three SNPs that result in missense mutation on the POLN gene, which codes for DNA Polymerase ν (Pol ν), were found to be statistically linked to breast cancer15 (Table 1). These missense SNPs, rs10018786, rs10011549, and rs9328764, are located within the available crystal structures of Pol v. These three SNPs result in Pol ν variants M310L, G336S, and R425C respectively. (Figure 1).
Figure 1.
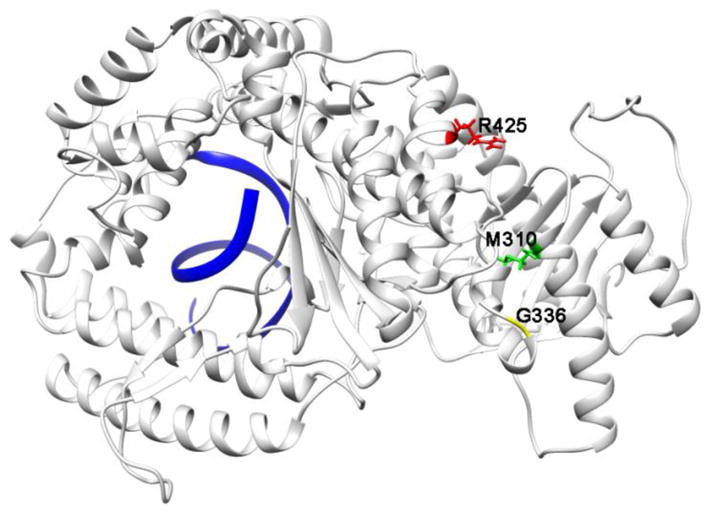
Missense mutations resulting from rs9328764, rs10011549, and rs10018786 mapped onto the structure of Pol ν with DNA bound (pdbid 4XVK). DNA is shown in blue.
The molecular dynamics simulations of the holo and apo structures for each of the three mutants revealed changes in the fluctuation of specific regions with respect to the wild type (WT) Pol ν (Figure 2). In particular, a significant increase in the fluctuation of residues 493-509 is observed in the holo structures of R425C and M310L mutants (Figure 2a). This is an area adjacent to the DNA (Figure 2c). In the same region of the apo structures the wild type and R425C mutant show higher fluctuations with larger change in the magnitude of the fluctuations, as well as the number of the residues involved (Figure 2, b and d). There is also a difference in fluctuation for both the holo and apo structures in the loop comprising residues 256-270. Residues in this region in the holo structure of wild type Pol ν and apo structures of R425C and M310L mutants have higher RMSF. Higher fluctuation was also revealed in the 593-603 region of the holo structures of the wild type and R425C mutant.
Figure 2.

Root mean square fluctuation (RMSF) per residue for the a) holo and b) apo systems of WT Pol ν and three cancer variants. c) holo and d) apo structures with regions exhibiting altered fluctuations highlighted in red.
Interestingly, the regions with altered fluctuations compared with WT Pol ν are located on opposite ends of the protein relative to the sites of the cancer-related mutations, and include loop structures. The area comprising residues 482-515 is located in proximity to the DNA, with T514 forming hydrogen bonds to the DNA backbone in the holo structure. The M310L and G336C mutants having decreased fluctuations in this region in the apo structures, suggesting that these variants may have hindered or incorrect DNA binding. The altered fluctuations in the loops could be indicative of tertiary structure changes caused by the mutations. The change in fluctuations is not limited to the protein structure, indeed, an increase in the mobility of the DNA is observed for the M310L variant compared to the WT structure (Figure S2).
Although all three variants show changes with respect to WT, the M310L mutant shows larger changes in RMSF and RMSD for certain regions compared with the other variants. In particular, residue-wise correlation analysis suggests a change in the motion of several residues in the M310L variant compared with the WT in both the holo and apo structures, consistent with the RMSF results. As evident from the brighter red hue in the difference correlation matrix for the holo structure (Figure 3a), the area near residue 493, which also exhibits higher fluctuation in the M310L and R425 variants (Figure 2a), has a change in the dynamics. In addition, a region comprising residues 678-753 (Figure 3) shows a change in correlation relative to the residues 468-518 with respect to WT (Figure 3b). The two regions with altered correlations correspond to areas in the fingers and thumb domains, which contact the DNA in the holo structure (Figure 4).
Figure 3.

Difference correlation plots, comparing correlation between residues in M310L mutant and the wild type structures for a) holo and b) apo Pol ν.
Figure 4.

Regions with changes in correlation for M310L Pol ν. Residues 468-518 are shown in orange, 678-753 in green, and DNA is shown in blue.
Structural and Dynamics Analysis of Missense Mutations on Polymerase ι Gene
The POLI gene codes for DNA polymerase ι (Pol ι), and was found to have a SNP linked to melanoma, rs3218784, as previously reported in Ref. 15. This genetic variant results in a I236M missense mutation (Figure 5).
Figure 5.
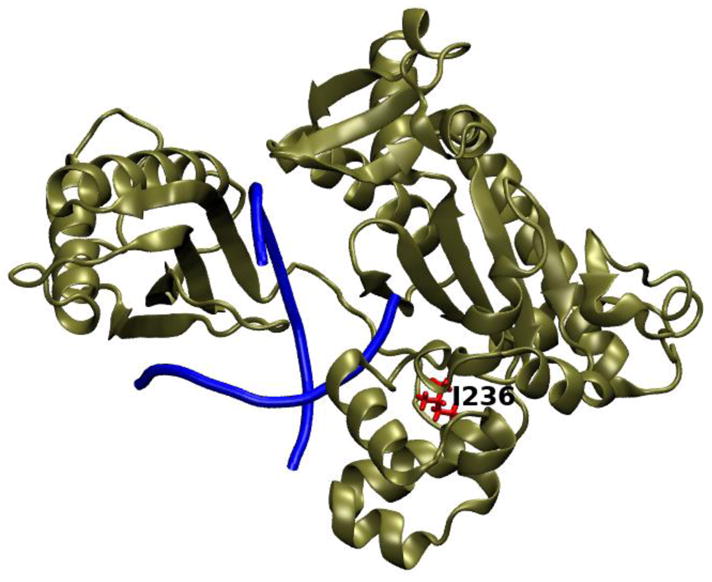
Pol ι crystal structure 3GV8 with the mutation site, I236, highlighted in red.
Our MD simulations revealed changes in the dynamics of Pol ι I236M. As can be seen from Figure 6, several residues exhibit higher fluctuations in the cancer-related variant including regions comprising residues 142-152, 225-234, and 240-277 (highlighted in green in Figure 7). Similarly, the melanoma mutation results in decreased fluctuations in two regions, namely residues 350-357, and 369-377 (highlighted in purple in Figure 7). The residues with increased fluctuations in the I236M variant are located in the thumb domain and are close to the mutation site. RMSF analysis for the DNA substrate for the wild type and Pol ι I236M binary complexes indicates that the DNA has significantly smaller fluctuations in the cancer variant structure (Figure 8). Thus, the I236M Pol ι variant arising from the melanoma-associated mutation affects the dynamics of the protein in two regions, which in turn results in a decrease in the fluctuation of the DNA in the binary structure. This change in fluctuations could result in a negative effect on the protein’s function.
Figure 6.

RMSF plots for wild type and I236M mutant Pol ι.
Figure 7.
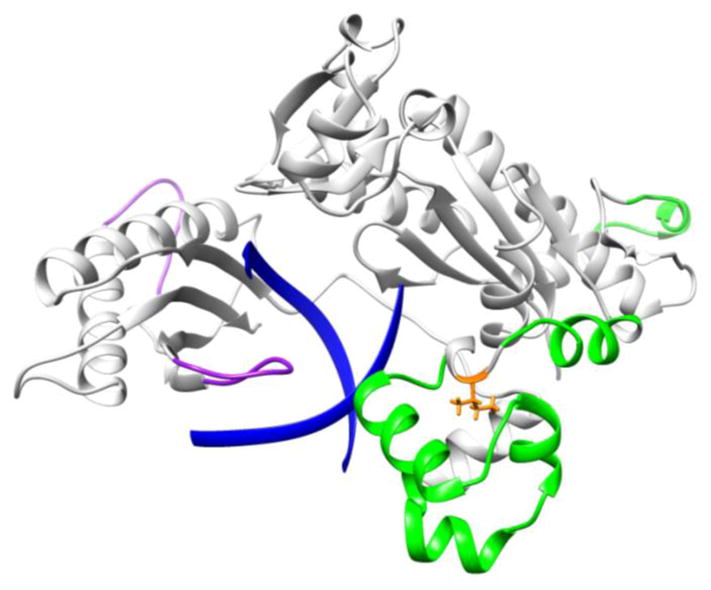
RMSF difference between WT and I236M Pol ι. Regions that show an increase(decrease) in RMSF in I236M Pol ι with respect to WT are shown in green(purple). The DNA substrate is shown in blue ribbons and the mutation site in orange sticks.
Figure 8.

RMSF for DNA for the wild type and mutant Pol ι complexes.
Taken together, our computational simulations for these two enzymes with four cancer variants suggest that these mutations affect the dynamics of the protein, which in turn could affect the function of these important enzymes. It would be interesting to experimentally confirm whether these predicted effects indeed result in a change in the function of the cancer variants.
Conclusions
We have used our previously developed HyDn-SNP-S method to search for cancer mutations on 25 different DNA repair genes related to 8 different cancer phenotypes. Our analysis uncovered 562 total SNPs on DNA repair genes that are statistically significantly associated with at least one of the tested cancer phenotypes, 43 of which result in missense mutations on the final protein product. Moreover, our analysis uncovered that out of the 562 total SNPs, 109 are associated with more than one cancer phenotype, and nine out of the 25 tested DNA repair genes involve a missense SNP that is associated with two or more cancer types, and four of these genes (Polθ, APOBEC3H, MUTY and NEIL1) have the same SNP associated with three cancer phenotypes. The subsequent all-atom MD characterization of four cancer variants on Pol ν and Pol ι provide insights into the possible effects of these cancer mutations on the structure and function of the respective enzymes. In particular, we investigated three breast cancer-related missense mutations on Pol ν: M310L, R425C, and G336S. In all cases, the mutations are observed to affect the fluctuations and correlation of various regions of the thumb and finger domains of Pol ν. One more melanoma-related missense mutation on Pol ι, resulting in the I236M variant, also exhibits altered dynamics. In particular, a decrease in fluctuations in the fingers domain, which in turn also results in a decrease in the observed motion of the DNA substrate in the binary structure. Overall, our results underscore the importance of DNA repair genes and their relation to cancer as observed by the large number of single mutations related to multiple cancer phenotypes, as well as the possible effects of the resulting mutations on the protein structure and function.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (Grants GM108583 and GM118501) and NVIDIA Foundations’ “Compute the Cure” to GAC. The authors thank dbGaP for access to the cancer databases, access request #12236. Assistance with phenotype harmonization, SNP selection, data cleaning, meta-analyses, data management and dissemination, and general study coordination, was provided by the GENEVA Coordinating Center (U01HG004789-01). Funding support for the GENEVA Prostate Cancer study was provided through the National Cancer Institute (R37CA54281, R01CA6364, P01CA33619, U01CA136792, and U01CA98758) and the National Human Genome Research Institute (U01HG004726). Assistance with phenotype harmonization, SNP selection, data cleaning, meta-analyses, data management and dissemination, and general study coordination, was provided by the GENEVA Coordinating Center (U01HG004789-01). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. This project was funded in whole or in part with federal funds from the National Cancer Institute (NCI), US National Institutes of Health (NIH) under contract number HHSN261200800001E. Additional support was received from NIH/NCI K07 CA140790, the American Society of Clinical Oncology Conquer Cancer Foundation, the Howard Hughes Medical Institute, the Lustgarten Foundation, the Robert T. and Judith B. Hale Fund for Pancreatic Cancer Research and Promises for Purple. A full list of acknowledgments for each participating study is provided in the Supplementary Note of the manuscript with PubMed ID: 25086665. The datasets have been accessed through the NIH database for Genotypes and Phenotypes (dbGaP) under accession #phs000206. The Genome-Wide Association Study (GWAS) of Non-Hodgkin Lymphoma (NHL) project was supported by the intramural program of the Division of Cancer Epidemiology and Genetics (DCEG), National Cancer Institute (NCI), National Institutes of Health (NIH). The datasets have been accessed through the NIH database for Genotypes and Phenotypes (dbGaP) under accession #phs000801. A full list of acknowledgements can be found in the supplementary note (Berndt SI et al., Nature Genet., 2013, PMID: 23770605).
References
- 1.Weir HK, Anderson RN, Coleman King SM, Soman A, Thompson TD, Hong Y, Moller B, Leadbetter S. Heart Disease and Cancer Deaths—Trends and Projections in the United States, 1969–2020. Preventing Chronic Disease. 2016;13:E157. doi: 10.5888/pcd13.160211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA: A Cancer Journal for Clinicians. 2016;66(1):7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 3.Howlader N, Noone AM, Krapcho M, Miller D, Bishop K, Kosary CL, Yu M, Ruhl J, Tatalovich Z, Mariotto A, Lewis DR, Chen HS, Feuer EJ, Cronin KA. SEER Cancer Statistics Review, 1975–2014. National Cancer Institute; Bethesda, MD: 2017. [Google Scholar]
- 4.Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, Rahman N, Stratton MR. A census of human cancer genes. Nature Reviews Cancer. 2004;4:177. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weinstein JN, Collisson EA, Mills GB, Shaw KM, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, Stuart JM Cancer Genome Atlas Research N. The Cancer Genome Atlas Pan-Cancer Analysis Project. Nature genetics. 2013;45(10):1113–1120. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wheeler DL, Barrett T, Benson DA, Bryant SH, Canese K, Chetvernin V, Church DM, DiCuccio M, Edgar R, Federhen S, Feolo M, Geer LY, Helmberg W, Kapustin Y, Khovayko O, Landsman D, Lipman DJ, Madden TL, Maglott DR, Miller V, Ostell J, Pruitt KD, Schuler GD, Shumway M, Sequeira E, Sherry ST, Sirotkin K, Souvorov A, Starchenko G, Tatusov RL, Tatusova TA, Wagner L, Yaschenko E. Database resources of the National Center for Biotechnology Information. Nucleic Acids Research. 2008;36(suppl_1):D13–D21. doi: 10.1093/nar/gkm1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chan IS, Ginsburg GS. Personalized Medicine: Progress and Promise. Annual Review of Genomics and Human Genetics. 2011;12(1):217–244. doi: 10.1146/annurev-genom-082410-101446. [DOI] [PubMed] [Google Scholar]
- 8.Liu Y, Tian F, Hu Z, DeLisi C. Evaluation and integration of cancer gene classifiers: identification and ranking of plausible drivers. Scientific Reports. 2015;5:10204. doi: 10.1038/srep10204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khatri P, Sirota M, Butte AJ. Ten Years of Pathway Analysis: Current Approaches and Outstanding Challenges. PLOS Computational Biology. 2012;8(2):e1002375. doi: 10.1371/journal.pcbi.1002375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nature Biotechnology. 2013;31:213. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zheng W, Rao S. Knowledge-based analysis of genetic associations of rheumatoid arthritis to inform studies searching for pleiotropic genes: a literature review and network analysis. Arthritis Research & Therapy. 2015;17(1):202. doi: 10.1186/s13075-015-0715-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hoeijmakers JHJ. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 13.Berwick M, Vineis P. Markers of DNA Repair and Susceptibility to Cancer in Humans: an Epidemiologic Review. JNCI: Journal of the National Cancer Institute. 2000;92(11):874–897. doi: 10.1093/jnci/92.11.874. [DOI] [PubMed] [Google Scholar]
- 14.Mailman MD, Feolo M, Jin Y, Kimura M, Tryka K, Bagoutdinov R, Hao L, Kiang A, Paschall J, Phan L, Popova N, Pretel S, Ziyabari L, Shao Y, Wang ZY, Sirotkin K, Ward M, Kholodov M, Zbicz K, Beck J, Kimelman M, Shevelev S, Preuss D, Yaschenko E, Graeff A, Ostell J, Sherry ST. The NCBI dbGaP database of genotypes and phenotypes. Nature genetics. 2007;39(10):1181–1186. doi: 10.1038/ng1007-1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Swett RJ, Elias A, Miller JA, Dyson GE, Andrés Cisneros G. Hypothesis driven single nucleotide polymorphism search (HyDn-SNP-S) DNA Repair. 2013;12(9):733–740. doi: 10.1016/j.dnarep.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hunter DJ, Kraft P, Jacobs KB, Cox DG, Yeager M, Hankinson SE, Wacholder S, Wang Z, Welch R, Hutchinson A, Wang J, Yu K, Chatterjee N, Orr N, Willett WC, Colditz GA, Ziegler RG, Berg CD, Buys SS, McCarty CA, Feigelson HS, Calle EE, Thun MJ, Hayes RB, Tucker M, Gerhard DS, Fraumeni JF, Jr, Hoover RN, Thomas G, Chanock SJ. A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nature Genetics. 2007;39:870. doi: 10.1038/ng2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haiman CA, Chen GK, Vachon CM, Canzian F, Dunning A, Millikan RC, Wang X, Ademuyiwa F, Ahmed S, Ambrosone CB, Baglietto L, Balleine R, Bandera EV, Beckmann MW, Berg CD, Bernstein L, Blomqvist C, Blot WJ, Brauch H, Buring JE, Carey LA, Carpenter JE, Chang-Claude J, Chanock SJ, Chasman DI, Clarke CL, Cox A, Cross SS, Deming SL, Diasio RB, Dimopoulos AM, Driver WR, Dünnebier T, Durcan L, Eccles D, Edlund CK, Ekici AB, Fasching PA, Feigelson HS, Flesch-Janys D, Fostira F, Försti A, Fountzilas G, Gerty SM, Giles GG, Godwin AK, Goodfellow P, Graham N, Greco D, Hamann U, Hankinson SE, Hartmann A, Hein R, Heinz J, Holbrook A, Hoover RN, Hu JJ, Hunter DJ, Ingles SA, Irwanto A, Ivanovich J, John EM, Johnson N, Jukkola-Vuorinen A, Kaaks R, Ko Y-D, Kolonel LN, Konstantopoulou I, Kosma V-M, Kulkarni S, Lambrechts D, Lee AM, Le Marchand L, Lesnick T, Liu J, Lindstrom S, Mannermaa A, Margolin S, Martin NG, Miron P, Montgomery GW, Nevanlinna H, Nickels S, Nyante S, Olswold C, Palmer J, Pathak H, Pectasides D, Perou CM, Peto J, Pharoah PDP, Pooler LC, Press MF, Pylkäs K, Rebbeck TR, Rodriguez-Gil JL, Rosenberg L, Ross E, Rüdiger T, Silva IdS, Sawyer E, Schmidt MK, Schulz-Wendtland R, Schumacher F, Severi G, Sheng X, Signorello LB, Sinn H-P, Stevens KN, Southey MC, Tapper WJ, Tomlinson I, Hogervorst FBL, Wauters E, Weaver J, Wildiers H, Winqvist R, Berg DVD, Wan P, Xia LY, Yannoukakos D, Zheng W, Ziegler RG, Siddiq A, Slager SL, Stram DO, Easton D, Kraft P, Henderson BE, Couch FJ The Gene Environment Interaction, Breast Cancer in Germany C. A common variant at the TERT-CLPTM1L locus is associated with estrogen receptor–negative breast cancer. Nature Genetics. 2011;43:1210. doi: 10.1038/ng.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yeager M, Orr N, Hayes RB, Jacobs KB, Kraft P, Wacholder S, Minichiello MJ, Fearnhead P, Yu K, Chatterjee N, Wang Z, Welch R, Staats BJ, Calle EE, Feigelson HS, Thun MJ, Rodriguez C, Albanes D, Virtamo J, Weinstein S, Schumacher FR, Giovannucci E, Willett WC, Cancel-Tassin G, Cussenot O, Valeri A, Andriole GL, Gelmann EP, Tucker M, Gerhard DS, Fraumeni JF, Jr, Hoover R, Hunter DJ, Chanock SJ, Thomas G. Genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Nature Genetics. 2007;39:645. doi: 10.1038/ng2022. [DOI] [PubMed] [Google Scholar]
- 19.Landi MT, Chatterjee N, Yu K, Goldin LR, Goldstein AM, Rotunno M, Mirabello L, Jacobs K, Wheeler W, Yeager M, Bergen AW, Li Q, Consonni D, Pesatori AC, Wacholder S, Thun M, Diver R, Oken M, Virtamo J, Albanes D, Wang Z, Burdette L, Doheny KF, Pugh EW, Laurie C, Brennan P, Hung R, Gaborieau V, McKay JD, Lathrop M, McLaughlin J, Wang Y, Tsao M-S, Spitz MR, Wang Y, Krokan H, Vatten L, Skorpen F, Arnesen E, Benhamou S, Bouchard C, Metsapalu A, Vooder T, Nelis M, Välk K, Field JK, Chen C, Goodman G, Sulem P, Thorleifsson G, Rafnar T, Eisen T, Sauter W, Rosenberger A, Bickeböller H, Risch A, Chang-Claude J, Wichmann HE, Stefansson K, Houlston R, Amos CI, Fraumeni JF, Savage SA, Bertazzi PA, Tucker MA, Chanock S, Caporaso NE. A Genome-wide Association Study of Lung Cancer Identifies a Region of Chromosome 5p15 Associated with Risk for Adenocarcinoma. The American Journal of Human Genetics. 2009;85(5):679–691. doi: 10.1016/j.ajhg.2009.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wolpin BM, Rizzato C, Kraft P, Kooperberg C, Petersen GM, Wang Z, Arslan AA, Beane-Freeman L, Bracci PM, Buring J, Canzian F, Duell EJ, Gallinger S, Giles GG, Goodman GE, Goodman PJ, Jacobs EJ, Kamineni A, Klein AP, Kolonel LN, Kulke MH, Li D, Malats N, Olson SH, Risch HA, Sesso HD, Visvanathan K, White E, Zheng W, Abnet CC, Albanes D, Andreotti G, Austin MA, Barfield R, Basso D, Berndt SI, Boutron-Ruault M-C, Brotzman M, Büchler MW, Bueno-de-Mesquita HB, Bugert P, Burdette L, Campa D, Caporaso NE, Capurso G, Chung C, Cotterchio M, Costello E, Elena J, Funel N, Gaziano JM, Giese NA, Giovannucci EL, Goggins M, Gorman MJ, Gross M, Haiman CA, Hassan M, Helzlsouer KJ, Henderson BE, Holly EA, Hu N, Hunter DJ, Innocenti F, Jenab M, Kaaks R, Key TJ, Khaw K-T, Klein EA, Kogevinas M, Krogh V, Kupcinskas J, Kurtz RC, LaCroix A, Landi MT, Landi S, Le Marchand L, Mambrini A, Mannisto S, Milne RL, Nakamura Y, Oberg AL, Owzar K, Patel AV, Peeters PHM, Peters U, Pezzilli R, Piepoli A, Porta M, Real FX, Riboli E, Rothman N, Scarpa A, Shu X-O, Silverman DT, Soucek P, Sund M, Talar-Wojnarowska R, Taylor PR, Theodoropoulos GE, Thornquist M, Tj⊘nneland A, Tobias GS, Trichopoulos D, Vodicka P, Wactawski-Wende J, Wentzensen N, Wu C, Yu H, Yu K, Zeleniuch-Jacquotte A, Hoover R, Hartge P, Fuchs C, Chanock SJ, Stolzenberg-Solomon RS, Amundadottir LT. Genome-wide association study identifies multiple susceptibility loci for pancreatic cancer. Nature genetics. 2014;46(9):994–1000. doi: 10.1038/ng.3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Petersen GM, Amundadottir L, Fuchs CS, Kraft P, Stolzenberg-Solomon RZ, Jacobs KB, Arslan AA, Bueno-de-Mesquita HB, Gallinger S, Gross M, Helzlsouer K, Holly EA, Jacobs EJ, Klein AP, LaCroix A, Li D, Mandelson MT, Olson SH, Risch HA, Zheng W, Albanes D, Bamlet WR, Berg CD, Boutron-Ruault M-C, Buring JE, Bracci PM, Canzian F, Clipp S, Cotterchio M, de Andrade M, Duell EJ, Gaziano JM, Giovannucci EL, Goggins M, Hallmans G, Hankinson SE, Hassan M, Howard B, Hunter DJ, Hutchinson A, Jenab M, Kaaks R, Kooperberg C, Krogh V, Kurtz RC, Lynch SM, McWilliams RR, Mendelsohn JB, Michaud DS, Parikh H, Patel AV, Peeters PHM, Rajkovic A, Riboli E, Rodriguez L, Seminara D, Shu X-O, Thomas G, Tj⊘nneland A, Tobias GS, Trichopoulos D, Van Den Eeden SK, Virtamo J, Wactawski-Wende J, Wang Z, Wolpin BM, Yu H, Yu K, Zeleniuch-Jacquotte A, Fraumeni JF, Hoover RN, Hartge P, Chanock SJ. A genome-wide association study identifies pancreatic cancer susceptibility loci on chromosomes 13q22.1, 1q32.1 and 5p15.33. Nature genetics. 2010;42(3):224–228. doi: 10.1038/ng.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Amundadottir L, Kraft P, Stolzenberg-Solomon RZ, Fuchs CS, Petersen GM, Arslan AA, Bueno-de-Mesquita HB, Gross M, Helzlsouer K, Jacobs EJ, LaCroix A, Zheng W, Albanes D, Bamlet W, Berg CD, Berrino F, Bingham S, Buring JE, Bracci PM, Canzian F, Clavel-Chapelon F, Clipp S, Cotterchio M, de Andrade M, Duell EJ, Fox JW, Gallinger S, Gaziano JM, Giovannucci EL, Goggins M, González CA, Hallmans G, Hankinson SE, Hassan M, Holly EA, Hunter DJ, Hutchinson A, Jackson R, Jacobs KB, Jenab M, Kaaks R, Klein AP, Kooperberg C, Kurtz RC, Li D, Lynch SM, Mandelson M, McWilliams RR, Mendelsohn JB, Michaud DS, Olson SH, Overvad K, Patel AV, Peeters PHM, Rajkovic A, Riboli E, Risch HA, Shu X-O, Thomas G, Tobias GS, Trichopoulos D, Van Den Eeden SK, Virtamo J, Wactawski-Wende J, Wolpin BM, Yu H, Yu K, Zeleniuch-Jacquotte A, Chanock SJ, Hartge P, Hoover RN. Genome-wide association study identifies variants in the ABO locus associated with susceptibility to pancreatic cancer. Nature genetics. 2009;41(9):986–990. doi: 10.1038/ng.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berndt SI, Skibola CF, Joseph V, Camp NJ, Nieters A, Wang Z, Cozen W, Monnereau A, Wang SS, Kelly RS, Lan Q, Teras LR, Chatterjee N, Chung CC, Yeager M, Brooks-Wilson AR, Hartge P, Purdue MP, Birmann BM, Armstrong BK, Cocco P, Zhang Y, Severi G, Zeleniuch-Jacquotte A, Lawrence C, Burdette L, Yuenger J, Hutchinson A, Jacobs KB, Call TG, Shanafelt TD, Novak AJ, Kay NE, Liebow M, Wang AH, Smedby KE, Adami H-O, Melbye M, Glimelius B, Chang ET, Glenn M, Curtin K, Cannon-Albright LA, Jones B, Diver WR, Link BK, Weiner GJ, Conde L, Bracci PM, Riby J, Holly EA, Smith MT, Jackson RD, Tinker LF, Benavente Y, Becker N, Boffetta P, Brennan P, Foretova L, Maynadie M, McKay J, Staines A, Rabe KG, Achenbach SJ, Vachon CM, Goldin LR, Strom SS, Lanasa MC, Spector LG, Leis JF, Cunningham JM, Weinberg JB, Morrison VA, Caporaso NE, Norman AD, Linet MS, De Roos AJ, Morton LM, Severson RK, Riboli E, Vineis P, Kaaks R, Trichopoulos D, Masala G, Weiderpass E, Chirlaque M-D, Vermeulen RCH, Travis RC, Giles GG, Albanes D, Virtamo J, Weinstein S, Clavel J, Zheng T, Holford TR, Offit K, Zelenetz A, Klein RJ, Spinelli JJ, Bertrand KA, Laden F, Giovannucci E, Kraft P, Kricker A, Turner J, Vajdic CM, Ennas MG, Ferri GM, Miligi L, Liang L, Sampson J, Crouch S, Park J-h, North KE, Cox A, Snowden JA, Wright J, Carracedo A, Lopez-Otin C, Bea S, Salaverria I, Martin D, Campo E, Fraumeni JF, de Sanjose S, Hjalgrim H, Cerhan JR, Chanock SJ, Rothman N, Slager SL. Genome-wide Association Study Identifies Multiple Risk Loci for Chronic Lymphocytic Leukemia. Nature genetics. 2013;45(8):868–876. doi: 10.1038/ng.2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cerhan JR, Berndt SI, Vijai J, Ghesquières H, McKay J, Wang SS, Wang Z, Yeager M, Conde L, de Bakker PIW, Nieters A, Cox D, Burdett L, Monnereau A, Flowers CR, De Roos AJ, Brooks-Wilson AR, Lan Q, Severi G, Melbye M, Gu J, Jackson RD, Kane E, Teras LR, Purdue MP, Vajdic CM, Spinelli JJ, Giles GG, Albanes D, Kelly RS, Zucca M, Bertrand KA, Zeleniuch-Jacquotte A, Lawrence C, Hutchinson A, Zhi D, Habermann TM, Link BK, Novak AJ, Dogan A, Asmann YW, Liebow M, Thompson CA, Ansell SM, Witzig TE, Weiner GJ, Veron AS, Zelenika D, Tilly H, Haioun C, Molina TJ, Hjalgrim H, Glimelius B, Adami H-O, Bracci PM, Riby J, Smith MT, Holly EA, Cozen W, Hartge P, Morton LM, Severson RK, Tinker LF, North KE, Becker N, Benavente Y, Boffetta P, Brennan P, Foretova L, Maynadie M, Staines A, Lightfoot T, Crouch S, Smith A, Roman E, Diver WR, Offit K, Zelenetz A, Klein RJ, Villano DJ, Zheng T, Zhang Y, Holford TR, Kricker A, Turner J, Southey MC, Clavel J, Virtamo J, Weinstein S, Riboli E, Vineis P, Kaaks R, Trichopoulos D, Vermeulen RCH, Boeing H, Tjonneland A, Angelucci E, Di Lollo S, Rais M, Birmann BM, Laden F, Giovannucci E, Kraft P, Huang J, Ma B, Ye Y, Chiu BCH, Sampson J, Liang L, Park J-H, Chung CC, Weisenburger DD, Chatterjee N, Fraumeni JF, Slager SL, Wu X, de Sanjose S, Smedby KE, Salles G, Skibola CF, Rothman N, Chanock SJ. Genome-wide association study identifies multiple susceptibility loci for diffuse large B-cell lymphoma. Nature genetics. 2014;46(11):1233–1238. doi: 10.1038/ng.3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Skibola Christine F, Berndt Sonja I, Vijai J, Conde L, Wang Z, Yeager M, de Bakker Paul I, Birmann Brenda M, Vajdic Claire M, Foo J-N, Bracci Paige M, Vermeulen Roel C, Slager Susan L, de Sanjose S, Wang Sophia S, Linet Martha S, Salles G, Lan Q, Severi G, Hjalgrim H, Lightfoot T, Melbye M, Gu J, Ghesquières H, Link Brian K, Morton Lindsay M, Holly Elizabeth A, Smith A, Tinker Lesley F, Teras Lauren R, Kricker A, Becker N, Purdue Mark P, Spinelli John J, Zhang Y, Giles Graham G, Vineis P, Monnereau A, Bertrand Kimberly A, Albanes D, Zeleniuch-Jacquotte A, Gabbas A, Chung Charles C, Burdett L, Hutchinson A, Lawrence C, Montalvan R, Liang L, Huang J, Ma B, Liu J, Adami H-O, Glimelius B, Ye Y, Nowakowski Grzegorz S, Dogan A, Thompson Carrie A, Habermann Thomas M, Novak Anne J, Liebow M, Witzig Thomas E, Weiner George J, Schenk M, Hartge P, De Roos Anneclaire J, Cozen W, Zhi D, Akers Nicholas K, Riby J, Smith Martyn T, Lacher M, Villano Danylo J, Maria A, Roman E, Kane E, Jackson Rebecca D, North Kari E, Diver WR, Turner J, Armstrong Bruce K, Benavente Y, Boffetta P, Brennan P, Foretova L, Maynadie M, Staines A, McKay J, Brooks-Wilson Angela R, Zheng T, Holford Theodore R, Chamosa S, Kaaks R, Kelly Rachel S, Ohlsson B, Travis Ruth C, Weiderpass E, Clavel J, Giovannucci E, Kraft P, Virtamo J, Mazza P, Cocco P, Ennas Maria G, Chiu Brian C, Fraumeni Joseph F, Nieters A, Offit K, Wu X, Cerhan James R, Smedby Karin E, Chanock Stephen J, Rothman N. Genome-wide Association Study Identifies Five Susceptibility Loci for Follicular Lymphoma outside the HLA Region. American Journal of Human Genetics. 2014;95(4):462–471. doi: 10.1016/j.ajhg.2014.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vijai J, Wang Z, Berndt SI, Skibola CF, Slager SL, de Sanjose S, Melbye M, Glimelius B, Bracci PM, Conde L, Birmann BM, Wang SS, Brooks-Wilson AR, Lan Q, de Bakker PIW, Vermeulen RCH, Portlock C, Ansell SM, Link BK, Riby J, North KE, Gu J, Hjalgrim H, Cozen W, Becker N, Teras LR, Spinelli JJ, Turner J, Zhang Y, Purdue MP, Giles GG, Kelly RS, Zeleniuch-Jacquotte A, Ennas MG, Monnereau A, Bertrand KA, Albanes D, Lightfoot T, Yeager M, Chung CC, Burdett L, Hutchinson A, Lawrence C, Montalvan R, Liang L, Huang J, Ma B, Villano DJ, Maria A, Corines M, Thomas T, Novak AJ, Dogan A, Liebow M, Thompson CA, Witzig TE, Habermann TM, Weiner GJ, Smith MT, Holly EA, Jackson RD, Tinker LF, Ye Y, Adami H-O, Smedby KE, De Roos AJ, Hartge P, Morton LM, Severson RK, Benavente Y, Boffetta P, Brennan P, Foretova L, Maynadie M, McKay J, Staines A, Diver WR, Vajdic CM, Armstrong BK, Kricker A, Zheng T, Holford TR, Severi G, Vineis P, Ferri GM, Ricco R, Miligi L, Clavel J, Giovannucci E, Kraft P, Virtamo J, Smith A, Kane E, Roman E, Chiu BCH, Fraumeni JF, Wu X, Cerhan JR, Offit K, Chanock SJ, Rothman N, Nieters A. A genome-wide association study of marginal zone lymphoma shows association to the HLA region. Nature Communications. 2015;6:5751. doi: 10.1038/ncomms6751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liesegang TJ. In: Human DNA repair genes. Wood RD, editor. University of Pittsburgh Cancer Institute; S867 Scaife Hall, 3550 Terrace Street, Pittsburgh, PA 15261: ** [Google Scholar]; Mitchell M, Sgouros J, Lindahl T. Science. 2001;291:1284–1289. doi: 10.1126/science.1056154. [DOI] [PubMed] [Google Scholar]; American Journal of Ophthalmology. 2001;132(2):298. [Google Scholar]
- 28.Wood RD, Mitchell M, Lindahl T. Human DNA repair genes, 2005. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis. 2005;577(1):275–283. doi: 10.1016/j.mrfmmm.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 29.Human DNA repair genes. MD Anderson; 2014. [Google Scholar]
- 30.Clarke GM, Anderson CA, Pettersson FH, Cardon LR, Morris AP, Zondervan KT. Basic statistical analysis in genetic case-control studies. Nature protocols. 2011;6(2):121–133. doi: 10.1038/nprot.2010.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sinnwell JP, Schaid DJ. R package. v1.7.7. haplo.stats: statistical analysis of haplotypes with traits and covariates when linkage phase is ambiguous. [Google Scholar]
- 32.Walker AR, Silvestrov P, Müller TA, Podolsky RH, Dyson G, Hausinger RP, Cisneros GA. ALKBH7 Variant Related to Prostate Cancer Exhibits Altered Substrate Binding. PLOS Computational Biology. 2017;13(2):e1005345. doi: 10.1371/journal.pcbi.1005345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Case DA, Cerutti DS, Cheatham I, TE, Darden TA, Duke RE, Giese TJ, Gohlke H, Goetz AW, Greene D, Homeyer N, Izadi S, Kovalenko A, Lee TS, LeGrand S, Li P, Lin C, Liu J, Luchko T, Luo R, Mermelstein D, Merz KM, Monard G, Nguyen H, Omelyan I, Onufriev A, Pan F, Qi R, Roe DR, Roitberg A, Sagui C, Simmerling CL, Botello-Smith WM, Swails J, Walker RC, Wang J, Wolf RM, Wu X, Xiao L, York DM, Kollman PA. AMBER 2017. University of California; San Francisco: 2017. [Google Scholar]
- 34.Lee Y-S, Gao Y, Yang W. How a homolog of high-fidelity replicases conducts mutagenic DNA synthesis. Nature Structural & Molecular Biology. 2015;22:298. doi: 10.1038/nsmb.2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kirouac KN, Ling H. Structural basis of error-prone replication and stalling at a thymine base by human DNA polymerase ι. The EMBO Journal. 2009;28(11):1644. doi: 10.1038/emboj.2009.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Research. 2000;28(1):235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fiser A, Do RKG, šAli A. Modeling of loops in protein structures. Protein Science. 2000;9(9):1753–1773. doi: 10.1110/ps.9.9.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen VB, Arendall WB, III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallographica Section D. 2010;66(1):12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall IIIWB, Snoeyink J, Richardson JS, Richardson DC. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Research. 2007;35(suppl_2):W375–W383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Salomon-Ferrer R, Götz AW, Poole D, Le Grand S, Walker RC. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. Journal of Chemical Theory and Computation. 2013;9(9):3878–3888. doi: 10.1021/ct400314y. [DOI] [PubMed] [Google Scholar]
- 41.Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. Journal of Chemical Theory and Computation. 2015;11(8):3696–3713. doi: 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zgarbová M, Šponer J, Otyepka M, Cheatham TE, Galindo-Murillo R, Jurečka P. Refinement of the Sugar–Phosphate Backbone Torsion Beta for AMBER Force Fields Improves the Description of Z- and B-DNA. Journal of Chemical Theory and Computation. 2015;11(12):5723–5736. doi: 10.1021/acs.jctc.5b00716. [DOI] [PubMed] [Google Scholar]
- 43.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. The Journal of chemical physics. 1983;79(2):926–935. [Google Scholar]
- 44.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A smooth particle mesh Ewald method. The Journal of chemical physics. 1995;103(19):8577–8593. [Google Scholar]
- 45.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–12. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 46.Verma A, Lucas A, Verma SS, Zhang Y, Josyula N, Khan A, Hartzel DN, Lavage DR, Leader J, Ritchie MD, Pendergrass SA. PheWAS and Beyond: The Landscape of Associations with Medical Diagnoses and Clinical Measures across 38,662 Individuals from Geisinger. The American Journal of Human Genetics. 2018;102(4):592–608. doi: 10.1016/j.ajhg.2018.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nemec AA, Bush KB, Towle-Weicksel JB, Taylor BF, Schulz V, Weidhaas JB, Tuck DP, Sweasy JB. Estrogen Drives Cellular Transformation and Mutagenesis in Cells Expressing the Breast Cancer–Associated R438W DNA Polymerase Lambda Protein. Molecular Cancer Research. 2016;14(11):1068. doi: 10.1158/1541-7786.MCR-16-0209. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.