

Abstract
While cytokines such as TNF have long been recognized as essential to normal cerebral physiology, the implications of their chronic excessive production within the brain are now also increasingly appreciated. Syndromes as diverse as malaria and lead poisoning, as well as non‐infectious neurodegenerative diseases, illustrate this. These cytokines also orchestrate changes in tau, α‐synuclein, amyloid‐β levels and degree of insulin resistance in most neurodegenerative states. New data on the effects of salbutamol, an indirect anti‐TNF agent, on α‐synuclein and Parkinson's disease, APOE4 and tau add considerably to the rationale of the anti‐TNF approach to understanding, and treating, these diseases. Therapeutic advances being tested, and arguably useful for a number of the neurodegenerative diseases, include a reduction of excess cerebral TNF, whether directly, with a specific anti‐TNF biological agent such as etanercept via Batson's plexus, or indirectly via surgically implanting stem cells. Inhaled salbutamol also warrants investigating further across the neurodegenerative disease spectrum. It is now timely to integrate this range of new information across the neurodegenerative disease spectrum, rather than keep seeing it through the lens of individual disease states.
Abbreviations
- AD
Alzheimer's disease
- APOE
apolipoprotein E
- Aβ
amyloid‐β
- DAMPs
damage‐associated molecular patterns
- FTD
frontotemporal dementia
- HMGB1
high‐mobility box 1 protein
- IR
insulin resistance
- IRS‐1
insulin receptor substrate 1
- PAMPs
pathogen‐associated molecular patterns
- PD
Parkinson's disease
- Pin‐1
peptidyl prolyl1 isomerase
- PRRs
pattern recognition receptors
- P‐tau
phosphorylated tau
- T2DM
type‐2 diabetes mellitus
- TBI
traumatic brain injury
- TLR
toll‐like receptor
Introduction
Neuroinflammation is gaining traction in explaining most neurodegenerative diseases. The proteins http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4865 and tau, respectively, visible in brain sections as plaques and neurofibrillary tangles, have long been the hallmark cerebral histological findings for a diagnosis of Alzheimer's disease (AD). In the absence of serious competitors, the amyloid theory rapidly took the lead as the most promoted actual cause of this condition, with early spectacular transgenic mouse experiments (Janus et al., 2000) leading directly to large human trials, ensuring that for many years the aim of almost all effort towards developing a treatment for AD involved Aβ removal. Despite all this expenditure, the past decades of focusing on the amyloid theory are now widely accepted to have been unrewarding. As reviewed 8 years ago (Clark et al., 2010), this theory unaccountably continued to dominate long after publication that the ability of Aβ to inhibit long‐term potentiation (LTP; Wang et al., 2005; Rowan et al., 2007) in mouse hippocampal slices depends on downstream activity of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5074, a key pro‐inflammatory cytokine by then well‐known in the disease pathogenesis literature (Clark et al., 2004). These key observations (Wang et al., 2005; Rowan et al., 2007), as well as evidence that Aβ is an agonist of http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1752 and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1754 (Reed‐Geaghan et al., 2009), were largely ignored, even though the inflammatory theory of AD had been well‐aired by this time (Lee et al., 2007). More recently, 3,6′‐dithiothalidomide, a new TNF‐α synthesis inhibitor, was reported to attenuate the effect of i.c.v. injection of Aβ1‐42 on hippocampal neurogenesis and memory deficits (Russo et al., 2012). In addition, microglial activation, the primary cerebral source of pro‐inflammatory cytokines, has proved to coincide with neuronal loss (Wright et al., 2013), and synaptic deficits have been prevented by inhibiting TNF production (Cavanagh et al., 2016), in each case this happened before amyloidosis was evident.
Furthermore, research on the genotypic variants of the apolipoprotein E (APOE) gene (Lynch et al., 2001; Vitek et al., 2009) and on the effects of stimulating a receptor expressed on myeloid cells 2 (see references Ulland et al., 2017 and Ulrich et al., 2017 for recent reviews) has drawn attention towards the primacy of the inflammatory theory of neurodegenerative diseases. Hence, it is now seen as a research focus that promises to develop well, with practical consequences. An early report of side effects observed during the first experimental therapeutic TNF infusions in tumour patients 40 years ago, when the future of this cytokine was thought to lie in this field (Sherman et al., 1988; Spriggs et al., 1988), is worth recalling. Although these authors did not specifically study brain function, they reported unintended side effects that included lethargy, loss of recent memory and aphasia, as often seen in stroke, traumatic brain injury (TBI), Parkinson's disease (PD) and AD, soon after they commenced treatment with TNF.
More recent milestones have been passed, with three large retrospective studies being consistent with the centrality of TNF in the development of AD and PD. In brief, the first two were a lower AD incidence in a longitudinal study of the chronic s.c. use of anti‐TNF biologicals to treat rheumatoid arthritis (Chou et al., 2016), and the reduced development of PD in a population chronically treating asthma with inhaled salbutamol, its well‐established indirect anti‐TNF properties seemingly unappreciated by the authors (Mittal et al., 2017). More recently, systemic anti‐TNF therapy of inflammatory bowel disease has also been associated with a highly significant 78% reduction in the onset of PD (Peter et al., 2018). All three of these reports are extremely significant events for researchers concerned with finding practical treatments for AD and PD, since they demonstrate, for the first time, that chronically reducing just one parameter, cerebral TNF levels, can alter the frequency of onset of these two important neurodegenerative states. Clearly, these outcomes render agents with TNF neutralizing properties, as well as the innate or conferred capacity to enter the brain, prime molecules of interest in developing therapy for these two presently intractable neurodegenerative conditions. These studies of course inevitably carry the caveat that these patients are not normal populations but already bear the burden of a chronic cytokine excess. Nevertheless, the general experience regarding any two chronic inflammatory states is that they predispose, not protect, against each other. In passing, we also note that these systemic injections tend to be assumed to act via removing systemic TNF. It is conceivable, from experience with perispinal administration, that the smaller amounts that manage to enter the CSF are more active. We feel it is now timely to integrate this new information into a review of the larger picture, rather than keep seeing it through the lens of individual neurodegenerative diseases in isolation.
For clarity, we note that the term TNF‐α, still commonly seen in new literature, became an obsolete synonym for TNF some years ago. It is a relic from the short period, a decade after TNF was first described, when lymphotoxin (LT) had been renamed TNF‐β. Accordingly, TNF became TNF‐α. Subsequently, LT was divided into subgroups separate from TNF. Thus, the terms TNF‐β and TNF‐α had no further purpose and were officially discarded at an annual international TNF conference. Accordingly, the TNF field has long reverted to the original term, and we use it in this text.
Toll‐like receptor dependency of chronic inflammatory diseases
The implications of the failure to help patients by removing cerebral Aβ have finally begun to drive home the message that, until the pathogenesis of AD is clearly understood, and research interest in lowering amyloid consequently declines, researchers will remain ill‐equipped to advise on successfully treating this disease (Karran and De Strooper, 2016). We have recently (Clark and Vissel, 2015) reviewed the literature demonstrating that increased Aβ, soluble or not, does not cause direct damage but is merely one of the pro‐inflammatory cytokine‐inducing damage‐associated molecular patterns (DAMPs) that are agonists of toll‐like receptors (TLRs) on or in most cells, including throughout the brain. In particular, Aβ fits the pattern of being one of the secondary, or cytokine‐inducible, DAMPs that induce the cytokines that induce them, thus generating increased severity as well as chronicity. Other TLRs and functionally similar http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=302&familyType=CATALYTICRECEPTOR (PRRs) recognize pathogen‐associated molecular patterns (PAMPs) present on the surface of, for example, the microbes now widely agreed to be sometimes associated with AD (Itzhaki et al., 2016). This is entirely consistent with the relationship between total infectious burden and PD (Bu et al., 2015) (Figure 1).
Figure 1.

An illustration of how a range of chronic external stimuli that present DAMPs or PAMPs to various TLRs on different cerebral cellular components and thus induce TNF and similar cytokines. These in turn generate P‐tau, α‐synuclein (α‐Syn) and Aβ, proteins known to have the capacity to be secondary DAMPs (i.e. induced by TNF, and also act as a TLR agonist, thereby inducing more TNF). Clinical manifestations and therefore diagnoses vary, but the broad edifice, we argue, rests on a foundation of chronic pro‐inflammatory cytokines generated in this manner.
Although excess TNF can be a prime mover in inflammatory responses, at physiological levels, this cytokine is vital to the signalling involved in normal brain function, being required for essentials such as normal transmission via modulating excitatory neurotransmission (Pickering et al., 2005), homeostatic synaptic scaling (Stellwagen and Malenka, 2006) and LTP (Cumiskey et al., 2007). Remarkably, as well as controlling pathogens via innate immunity, these cytokines, when excessively increased, also cause disease (Clark, 1987; Clark and Rockett, 1994; Clark et al., 2004). Compared to the rest of the body, the brain develops negligible tolerance to continuing production of TNF (Steinshamn and Waage, 2000; Qin et al., 2007). Logically, therefore, the affected brain is particularly likely to develop chronic disease if these cytokines are generated in the brain in chronic excess in either infectious or non‐infectious conditions. Such illnesses are now referred to as chronic, or non‐resolving (Nathan and Ding, 2010), inflammatory diseases. Exploration of the interplay of cellular mediators, particularly cytokines allied to TNF, that binds the worlds of brain physiology, innate immunity and neurodegenerative disease into a single conceptual entity is expanding spectacularly at present (Ahmed et al., 2018; Rankin and Artis, 2018; Wendeln et al., 2018).
To paraphrase Fernandez et al. (2008) from a decade ago, what matters is not whether a particular danger signal or PRR is acting. Instead, it is whether the total sum of their activity and persistence, and thus the chronic level of pro‐inflammatory cytokines, including the http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4974 and TNF they induce, are sufficient and chronic enough for homeostasis to be lost, at which point they initiate and drive neurodegenerative disease within neural circuits. The literature on IL‐1β in neurodegenerative disease is much less developed than that of TNF, but the advent of the first specific anti‐human IL‐1β biological, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6773 (Rondeau et al., 2015), may well change this. As in a recent example (Calderon‐Garciduenas and de la Monte, 2017) that discusses a mix that includes inflammation, obesity and air pollution, the likelihood of AD onset depends on the critical mass of relevant signalling inputs being present. Importantly, these same PAMP and DAMP‐induced cytokines are required for many normal physiological functions that fail because homeostasis is lost (Figure 1). This is in part why removing soluble Aβ or its plaque, until recently the goal of many large clinical trials (Salloway et al., 2014), cannot, of itself, be expected to retard sporadic human AD disease progression. As we have summarized (Clark and Vissel, 2015), cytokines generated by other secondary DAMPs, S100 proteins (Foell et al., 2007) and high‐mobility group box 1 protein (HMGB1) (van Zoelen et al., 2009), are still present to continue to induce cytokines such as TNF. Not unexpectedly, removing Aβ was successful in mouse transgenic models designed to generate artificially high levels of Aβ (Janus et al., 2000). Since these mice can be expected to have merely physiological levels of these other secondary DAMPs known to be increased in human AD, the artificially induced Aβ is what creates the critical mass in these animals.
Therapeutic ambition is best focused on very early indicators of disease
Clearly, any indicator of neurodegenerative disease likely to be a successful therapeutic target will be among those reliably present preclinically, so the condition can be nipped in the bud. Here, we address the interactions of four such indicators: pro‐inflammatory cytokines, alterations within http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1800 components, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5012 resistance (IR) itself and the cis isomer of phosphorylated tau (P‐tau). All appear before frank illness and are, therefore, much more likely to be underlying pathogenic mechanisms than subsequent symptoms or signs. Regarding TNF and IL‐1β, plasma levels of clusterin (apolipoprotein J), are intimately associated with the onset, progression and severity of AD (Thambisetty et al., 2010). These authors employed a novel and impressive proteomic neuroimaging paradigm that allowed them to show that clusterin increases 10 years earlier than fibrillar Aβ deposits occur. Unfortunately, the authors refer only to the amyloid chaperone function of clusterin and seem to have been unaware of its role as an acute phase protein (Hardardottir et al., 1994) and, therefore, a marker of inflammation. A subsequent meta‐study reports that CLU, the clusterin gene, is the second highest of a list of the 15 top‐rated genes linked to AD on the Alzgene web‐based collection (Olgiati et al., 2011). This forms an impressive link between inflammatory cytokines and the primary instigation of AD. Likewise, a dysfunctional change in insulin receptor substrate 1 (IRS‐1) that leads to insulin resistance, and therefore glucose hypometabolism, has been shown to be present for up to 10 years before AD onset (Kapogiannis et al., 2017).
Insulin resistance
Insulin resistance in systemic and neurodegenerative diseases
Long‐known as an important regulator of blood glucose, insulin is also, on much evidence, a signalling molecule crucial for normal brain function. Malfunction of this aspect of brain physiology in neurodegenerative diseases, typically arising through cerebral IR caused by insulin receptor dysregulation, has been widely researched and reviewed (de la Monte and Wands, 2008; Talbot and Wang, 2014). As reviewed previously (Clark et al., 2012), excess levels of TNF mediate the development of cerebral IR and consequent glucose hypometabolism, overactive http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=509 and tau hyperphosphorylation. In essence, this constellation of changes comprises the main components of the inflammatory theory of neurodegenerative disease. For example, a downward spiral of degeneration, including acquired epilepsy (Samokhina et al., 2017), is regarded as being strongly contributed to by this glucose hypometabolism (Zilberter and Zilberter, 2017). In addition, and as we discuss below, much recent knowledge of tau biology allows this protein, as is Aβ, to be appreciated as part of the inflammatory theory of this disease.
Increased TNF causes insulin resistance
The basic science spanning immunity and metabolism largely accepts that chronic pro‐inflammatory cytokines cause the cerebral IR that is instrumental to co‐existing cognitive and metabolic impairments in various diseases (see Chawla et al., 2011 for an extensive review). This consensus includes a chronic excess of anti‐inflammatory cytokines promoting insulin sensitivity. Indeed, the prototype TNF inducer, bacterial wall http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5019, reproduces the IR seen in human sepsis within 7 h of its administration (Carlson, 2004). Feinstein et al. (1993) appear to have been the first to argue that TNF exerts a major part of its anti‐insulin effect by interrupting insulin‐stimulated tyrosine phosphorylation. This concept has been repeatedly investigated, initially in adipocytes, with TNF strongly inhibiting insulin‐stimulated glucose uptake (Hotamisligil et al., 1994). Soon afterwards, now over 20 years ago, this cytokine proved to cause IR through inhibiting signalling by IRS‐1 (Hotamisligil et al., 1996). In addition, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5004, the first commercial, specific anti‐TNF biological agent, was shown to improve insulin signal transduction in muscle, liver and hypothalamus in obese diabetic mice. In doing so, it restored the activity of insulin‐induced insulin receptor IRS1, receptor substrate‐2 tyrosine phosphorylation and http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=285 and forkhead box protein O1 (FOXO1) serine phosphorylation (Araujo et al., 2007). Intracerebral infliximab has also been reported to improved insulin signal transduction through IRS‐1 (Arruda et al., 2011). IRS‐1 has since been shown to be abnormally phosphorylated, accounting for IR, in post‐mortem brains from patients with tauopathies, including AD (Yarchoan et al., 2014).
A landmark 2011 study compared frontal cortex activity of the insulin–P13K–Akt signalling pathway in type 2 diabetes mellitus (T2DM) and AD in brains during autopsy (Liu et al., 2011). Not only were the same in‐principle changes found in T2DM and AD brains but insulin signalling apparently contributed to the observed hyperphosphorylation of tau. This approach to understanding IR in neurodegenerative diseases has been continued and refined, particularly in the hands of Bomfim et al. (2012), Talbot et al. (2012) and Talbot and Wang (2014). Similar recent studies into the cognitive impairment in a post‐sepsis mouse model (Neves et al., 2018) showed the same detailed IR pathophysiology, including changes surrounding IRS‐1 and Akt, as well as a positive response to a glucagon‐like protein‐1 mimetic that reverses IR, as reported in AD in this study. In particular, infliximab completely blocks the Aβ‐induced inhibition of IRS‐1, the insulin receptor component discussed below in a tau context (Bomfim et al., 2012). As noted earlier, it is now undisputed that TNF is an essential step in this brain pathophysiology, setting the scene for a series of novel observations in tau biology, as described below. Sometimes, this is couched in terms of Aβ directly provoking increased tau pathology (Karran and De Strooper, 2016), which unfortunately obscures the essential intermediary roles of TNF and IR.
Reduced neurogenesis, via insulin resistance, in neurodegenerative diseases
It has been clear, since a seminal publication in 2003 (Ekdahl et al., 2003), that inflammation is detrimental to neurogenesis in the adult human brain. As we have reviewed previously (Clark et al., 2012), the clock genes that set cells' rates of division are inhibited by increased levels of TNF or IL‐1β (Cavadini et al., 2007). Among these are the period genes, Per1, Per2 and Per3 and the central, interconnecting, response element clock gene, rev‐erbα. Many such genes have been demonstrated to be regulated by insulin (Tahara et al., 2011), including in the progenitors of adult neurogenesis in the hippocampus (Moriya et al., 2007; Borgs et al., 2009; Kimiwada et al., 2009). Therefore, the reduced neurogenesis seen in the subventricular zone is plausibly caused by IR in T2DM (Bachor et al., 2017), PD and AD, which share cerebral IR (Talbot et al., 2012; Aviles‐Olmos et al., 2013), and thus its consequences, one of which is inhibited neurogenesis (Haughey et al., 2002; Marxreiter et al., 2013).
TNF causing tau phosphorylation by initially inducing insulin resistance
A logical corollary of the above is that induction of IR by TNF rationalizes the link between this cytokine and tau phosphorylation, with IR as an intermediate step. In vitro generation or addition of TNF has been reported to promote tau phosphorylation, and TNF neutralization to reduce it (Gorlovoy et al., 2009). Likewise, i.c.v. administration for 3 days of infliximab to APP/PS1 transgenic mice reduced tau phosphorylation (Shi et al., 2011). It also reduced amyloid plaque, presumably because of the capacity of excess TNF to enhance promoter activity of the Aβ (APP) gene (Ge and Lahiri, 2002). The intermediary role of GSK‐3 in these steps is discussed in our 2012 review (Clark et al., 2012).
APOE
Effects of APOE genotype on peripheral compared to cerebral inflammation
While discussing hyperphosphorylated tau (or P‐tau, often written simply as tau when the context is clear), it is appropriate to introduce the influence of APOE proteins on our understanding of neurodegenerative diseases. Briefly, the relevant APOE gene is polymorphic, with three major alleles, APOE‐ε2, APOE‐ε3 and APOE‐ε4, carriers of the latter possessing the best‐known genetic susceptibility to many neurodegenerative diseases (Strittmatter et al., 1993). As reviewed previously (Tai et al., 2015), APOE4 is the greatest genetic risk factor for AD, increasing the risk by up to 12‐fold compared to APOE3, with APOE4‐specific neuroinflammation being an important component of this risk. Reports on the influence of APOE genotypes in neuroinflammation are more fragmentary, since in vivo models are a challenge. In brief, LPS induced pro‐inflammatory cytokines from microglia in vitro in a APOE4 > APOE3 > APOE2 pattern (Maezawa et al., 2006b), whereas the APOE order was reversed to APOE2 > APOE3 > APOE4 when astrocytes were examined (Maezawa et al., 2006a). Adding to the complexity one might encounter in a functioning brain, these authors also found that dendrites of hippocampal neurons damaged by i.c.v. LPS regenerated successfully in an APOE2, but not in an APOE4, environment. Tai et al. (2015) have systematically summarized the field, including their recent studies in which they discuss a more complex set of interactions than previously described in this setting. This involved amplification of detrimental TLR receptor pathways, suppression of the beneficial anti‐inflammatory cytokine http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4996 and loss of positive functions of APOE2 protein when APOE4 is generated instead. They propose that these studies point towards an in vivo pro‐inflammatory effect of APOE4 in the neurodegenerative diseases, chiefly by influencing receptor signalling pathways.
Pro‐inflammatory interaction between APOE4 and tau
Interest in the interactions between APOE and tau has a long history (Strittmatter et al., 1994). Recent impressive data on this topic in a brain injury context (Cao et al., 2017) are entirely compatible with the reasoning in the above paragraphs. So too is research into a mouse tauopathy model, derived from transgenic mice overexpressing the frontotemporal dementia (FTD) tau mutation P301S tau on a background of the various APOE genotypes (Shi et al., 2017). These authors reported that co‐culturing P301S tau‐expressing neurons with APOE4‐expressing mixed glial cells resulted in a significantly higher level of TNF secretion and markedly reduced neuronal viability compared with these same neurons co‐cultured with APOE2 or APOE3 glial cells. Moreover, mice generating APOE4 protein exhibited greatly enhanced brain atrophy and neurodegenerative change. This effect was independent of Aβ, a link previously championed for many years. Thus, APOE4, in all likelihood, has a strong pro‐inflammatory effect in the brain as well as peripherally.
New insights in tau biology – cis/trans isomerism, DAMP activity and insulin resistance control
Discussion of the cis/trans forms of tau requires a synoptic preamble about Pin‐1, a peptidyl‐prolyl cis/trans isomerase. As reviewed previously (Lim and Lu, 2005), Pin‐1 binds to and isomerizes specific pSer/Thr‐Pro motifs and, in a novel regulatory mechanism, catalytically induces conformational changes following phosphorylation. Pin‐1 is reported to minimize the accumulation of the pathogenic cis pT231‐tau conformation by converting it to the non‐pathogenic trans form (Nakamura et al., 2012). Through hippocampal Pin‐1 being oxidized and largely inactivated in the AD brain (Sultana et al., 2006), the pathogenic (see next paragraph) cis P‐tau form increases. In a similar vein, two single nucleotide polymorphisms of Pin‐1, which were in linkage disequilibrium and combined to form haplotypes, increased the risk of developing AD (Segat et al., 2007). For these reasons, impaired Pin‐1 has been argued to be one of the driving forces for the initiation and progression of AD.
This development in tau biology explains much and has now given tau a considerably firmer foothold within the literature on neurodegenerative disease pathogenesis. Two additional advances have further strengthened the case for its relevance. First, it is now evident that the harmfulness of P‐tau resides in the cis, as distinct from the trans, conformation of the molecule (Nakamura et al., 2013b). The anti‐cis form antibody that this group developed has been used to argue the case for this P‐tau variant being an early driver of neurodegeneration in human AD (Nakamura et al., 2013a) and in human TBI as well as in a mouse model of TBI (Kondo et al., 2015). Additionally, this antibody has proved to be therapeutically useful in this TBI model (Albayram et al., 2016). Second, an unrelated set of recent observations leads to the conclusion that at least one form of P‐tau, seen in FTD patients, P301L, can act as a DAMP in a mouse tauopathy model, enhancing the signalling message for the production of TNF and IL‐1β (Cook et al., 2015). Similarly, while investigating the hippocampal infiltration of CD38 T cells – also seen in FTD – in another tauopathy model, at the part of the mouse lifespan when deficits and tau pathology are maximal, others (Laurent et al., 2017) reported an up‐regulation of TLR2, CD68 and TNF, all of which are associated with activation of innate immunity, in the hippocampus.
New TNF‐based insights
Insights from the recognition of the A1 astrocyte subset
Microglia and astrocytes both become reactive during neuroinflammation, and knowledge about the interactions of these cells, as well as the cytokines and other molecules they release when in this state, is still limited. Five years ago, a novel line of reasoning in astrocyte biology began with the discovery that LPS, as distinct from ischaemia, induces an increase in the astrocyte subtype, later termed A1. These cells, in which some 50% of gene expression is different from in the A2 subtype, have been demonstrated to be detrimental, rather than protective, in the mouse brain (Zamanian et al., 2012). Arguably, they are part of a spectrum of astrocyte subtypes (Liddelow and Barres, 2017). Generation of these detrimental A1 astrocytes (Liddelow et al., 2017; Liddelow and Barres, 2017) requires the joint action of TNF, IL‐1α and complement component 1q, and they release an as yet unidentified toxin(s) that causes in vitro apoptosis of neurons and oligodendrocytes, as well as of axotomized neurons in vivo.
Hence, introducing an anti‐TNF specific biological into the brain that is generating many of these noxious astrocytes would be a constructive start in reducing the formation of this harmful subtype. Ideally, it could even be administered in conjunction with a neutralizer of the as yet unidentified toxin released by A1 astrocytes, which are present in damaged regions in AD, PD, amyotrophic lateral sclerosis and multiple sclerosis brains (Liddelow et al., 2017). Indeed, these authors have drawn from these lines of evidence to propose that any treatment that would prevent the formation of A1 astrocytes or reduce their effects can reasonably be predicted to increase new synapse formation (through reducing the release of harmful complement components), reduce neurodegeneration and stimulate spontaneous remyelination. Thus, from this initial post‐LPS observation (Zamanian et al., 2012), a narrative is beginning to emerge that provides an additional rationale for reducing the excess cerebral TNF present in neurodegenerative diseases. It is yet to be determined whether this novel astrocyte subtype operates in parallel with the apparent capacity of TNF of microglial origin to cause apoptosis of neurons directly (Burguillos et al., 2011; Kaur et al., 2014) or is an essential downstream component for this process.
Insights from infectious disease
Clearly, discovering causes of disease is retarded by artificial barriers within medical research. Clinical versus non‐clinical, systemic versus cerebral and non‐infectious versus infectious are examples. When clinical features and markers overlap, ignoring these barriers is surely the only way to stand far enough away from the minutiae to see a big enough picture to gain a therapeutically useful perspective. Fortunately, our initial collaboration, while working on haemoprotozoa, with the group who first identified TNF in their tumour research (Carswell et al., 1975) gave us a bigger picture from the start. As reviewed previously (Clark, 2007a; Clark, 2007b), they helped us to pioneer expanding TNF towards the pathogenesis malaria and sepsis disease (Clark, 1978; Clark et al., 1981). In this earlier work, we also assayed for, and theorized about, what came to be called IL‐1β. As the concept of pro‐inflammatory and anti‐inflammatory cytokines evolved, TNF proved to be near the top of a cascade of the former, with anti‐TNF, in the hands of arthritis researchers, inhibiting IL‐1 production as well as neutralizing TNF (Brennan et al., 1989). In addition, recombinant TNF, but not IL‐1β, became readily available as a research tool. We focused on TNF thenceforth, although for many years, only in a cerebral context when a systemic infectious disease influenced brain function, as in malaria (Clark et al., 1989). We were quite oblivious to the work of others who were, at this time, far‐sightedly arguing the case that IL‐1 was responsible for the astrogliosis central to AD and Down syndrome (Griffin et al., 1989).
Since TNF is extremely pleiotropic, it has proven to be a very useful avenue to understanding physiology and the pathophysiology of disease. For example, research into the roles of this cytokine has allowed us to understand why systemic infectious diseases caused by protozoa, bacteria and viruses can include strong CNS components. In tropical Africa, falciparum malaria provides an excellent example of a systemic sepsis‐like inflammatory disease that may include an acute encephalopathy, in this case termed cerebral malaria. Once this acute disease has subsided and the pathogen cleared, a chronic state termed the post‐cerebral malaria syndrome may develop (Idro et al., 2005). This condition can exhibit long‐term cognitive impairments, including deficits in memory, attention, visuospatial skills, language and executive function (Carter et al., 2005; Boivin et al., 2007; John et al., 2008a; Kihara et al., 2009) and aggressive behaviour (Idro et al., 2010). As we have reviewed previously (Clark et al., 2010), the literature links TNF with aggression. Similar cerebral consequences, not yet as closely studied as the above, can follow severe sepsis (Iwashyna et al., 2010; Lazosky et al., 2010; Jacob et al., 2011). The pathogenesis of the post‐cerebral malaria syndrome is considered to be consistent with the work of D'Mello et al. (2009), who demonstrated that peripheral TNF can pass through the blood–brain barrier and stimulate microglia to proliferate by generating chemokines that mediate the recruitment of TNF‐generating monocytes into the brain.
Post‐cerebral malaria syndrome correlates with CSF levels of TNF (John et al., 2008b) and has been likened to acquired cerebral palsy (Holding and Snow, 2001; Carter et al., 2005; Idro et al., 2010). Notably, through its neutralization with http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6789, TNF has been incriminated in a rat model of cerebral palsy (Aden et al., 2010). Moreover, glutamate excitotoxicity, a TNF‐induced phenomenon we have explained (Clark and Vissel, 2016) through the combined effects of this cytokine enhancing http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2891 (Takeuchi et al., 1993) and inhibiting glutamate re‐uptake proteins (Fine et al., 1994; Carmen et al., 2009), has been documented in a mouse model of cerebral malaria (Miranda et al., 2010). This model has recently been reported to be highly sensitive to treatment with the glutamine analogue 6‐diazo‐5‐oxo‐L‐norleucine, a glutaminase inhibitor (Gordon et al., 2015). This agent is, therefore, plausibly applicable to non‐infectious neurodegenerative states in which cerebral TNF levels are raised. Likewise, cerebral http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1697 depletion induced by excessive cerebral TNF in malaria is proposed to cause the coma in cerebral malaria and may be implicated in the poor sleep patterns, cognition and motor control in certain neurodegenerative states (Clark and Vissel, 2014). It has recently been demonstrated that intranasal orexin reverses both the coma of experimental post‐cardiac arrest and the cardiac arrest‐induced increases in mRNA for inflammatory cytokines (Modi et al., 2017).
Insights from lead toxicity
The harmful public health implications of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2525 exposure are well recognized and, as recently discussed (Taylor et al., 2011) have been campaigned against, with incomplete success, for over a century (Turner, 1909). From the beginning, this knowledge included an awareness of an associated encephalopathy, and parallels with AD started to be published over 40 years ago (Niklowitz and Mandybur, 1975). More contemporary reviews describe these manifestations in some detail, including negative effects of Pb2+ on intelligence, learning, memory, executive function, processing speed, language, visuospatial skills and affect (Mason et al., 2014), glutamate release, LTP and synaptic plasticity, increased amyloid precursor protein and increased Aβ (Basha et al., 2005; White et al., 2007) in some detail. Others describe α‐synuclein accumulation (Zhang et al., 2012) and increased tau phosphorylation (Li et al., 2010; Zhang et al., 2012; Gassowska et al., 2016). Publications on these topics were contemporary with others linking Pb2+ exposure with increased TNF production, initially using peripheral cells (Guo et al., 1996; Cheng et al., 2006) but later microglia (Li et al., 2009; Kumawat et al., 2014; Li et al., 2014).
The sequence of events from Pb2+ exposure to increased cerebral TNF production, and thence the generation of the surprisingly wide array of altered cerebral function outlined above, currently appears to be best explained through the DNA hypomethylation literature. As we have previously summarized (Clark and Vissel, 2013), the background for this understanding was set in place by virally transferring an over‐ or under‐expressing gene for Dnmt3a2, a DNA methyltransferase, directly into the hippocampus. This was shown to cause local changes in global DNA methylation that greatly improved or reduced, respectively, the cognitive abilities of old or young mice (Oliveira et al., 2012). These same enzyme changes occur in the hippocampus of young animals exposed to lead (Dosunmu et al., 2012; Schneider et al., 2013), rationalizing the epigenetic changes seen in the ageing primate brain, with implications for AD development (Bihaqi et al., 2011). Hypomethylated DNA, whether in lead‐poisoned brains, bacterial DNA or mitochondrial DNA, is a DAMP, an agonist for http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1759, generating a TNF‐initiated pro‐inflammatory cytokine cascade. Although LPS operates through TLR4, one would therefore expect, as has been reported (Cheng et al., 2004), signalling for cerebral TNF regulation to proceed along the same kinase pathway when either LPS or Pb2+ is the initiator.
Insights from recent α‐synuclein literature
α‐Synuclein, a product of one of the three known snc genes, is widely known in basic biology. It was first identified in the torpedo fish (Maroteaux et al., 1988) and has more recently studied roles in frog embryology (Wang et al., 2011). α‐Synuclein is also present in its soluble form in normal human macrophages, T cells, beta cells, NK cells and monocytes (Shin et al., 2000), and has a significant role in innate immunity (Beatman et al., 2015; Stolzenberg et al., 2017). Historically, neurodegenerative diseases have been categorized by which of several proteins are produced in unusually high amounts, those relevant to this section of this text being termed the synucleopathies. While the archetypal example is PD, this protein has been correlated closely with AD outcomes (Larson et al., 2012; Korff et al., 2013) and been described in both TBI (Mondello et al., 2013; Acosta et al., 2015) and Pb2+ toxicity (Zhang et al., 2012). As well as in the brain, α‐synuclein also has a predilection for forming in the gut wall (Natale et al., 2011; Cersosimo et al., 2013). Additionally, it is strongly induced by H5N1 influenza virus (Jang et al., 2009), which is a strong PAMP (Geeraedts et al., 2008), and therefore TNF generator.
Several strands of research have begun to develop in the relationship between α‐synuclein and TNF over the last 5 to 10 years. They co‐exist with a general increased interest in inflammation within the PD research world (Ferger et al., 2004; Bartels and Leenders, 2007; Cebrian et al., 2014). First, for motives unassociated with TNF, the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=29 agonist, albutamol (a synonym for http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=558) was studied in two small open trials in PD patients, some years apart (Alexander et al., 1994; Uc et al., 2003). Next, with dependence on original concepts from 1995 (Ignatowski and Spengler, 1995), http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=559, another β2‐adrenoceptor agonist, was reported to reduce levels of cytokines such as TNF and also to reduce dopaminergic neurotoxicity in vitro and in vivo (Qian et al., 2011). A short‐acting equivalent, salbutamol, was subsequently shown to share this anti‐TNF activity (Romberger et al., 2016; Keranen et al., 2017). This agent, brain‐penetrant when inhaled, is a widely used asthma medication. Importantly, in a recent 11‐year study in over 4 million Norwegians, it was associated with significantly reduced α‐synuclein levels and associated risk of developing PD (Mittal et al., 2017). As a control, the use of the β2‐adrenoceptor antagonist, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=564, was monitored in this population for some years, as well as used in vivo and in vitro experiments, and was found to have the opposite effect on PD incidence. Although not mentioned by these authors, this outcome is entirely consistent with the capacity of salbutamol to reduce TNF, as noted above. This provides evidence for chronically enhanced levels of this cytokine, conceivably aided by the cascade of chemokines and cytokines it induces, to increase α‐synuclein levels. This outcome also sits strikingly alongside α‐synuclein possessing agonist activity for TLR2 (Kim et al., 2013), that is, being a TNF‐inducing DAMP. Accordingly, a considerable literature shows that this protein, particularly in its aggregated state, induces TNF (Alvarez‐Erviti et al., 2011; Beraud et al., 2013; Hoffmann et al., 2016). Thus, as for S100 proteins, HMGB1 and Aβ in an AD context (Clark and Vissel, 2015), it seems clear that α‐synuclein can function as a secondary, or positive feedback DAMP (de Haan et al., 2013) (Figure 1). As such, it further enhances levels of the cytokines that induced it in the first place, thus increasing its presence, as well the concentration of these harmful cytokines. Among other consequences, excitotoxicity, acknowledged to be present in PD (Ambrosi et al., 2014), can ensue (Clark and Vissel, 2016). Equally importantly, these developments will open minds to the likelihood of salbutamol, given its anti‐TNF properties, plus the increasing awareness of excess TNF in the pathogenesis of such conditions as post‐stroke syndromes, TBI and AD, as being a plausible treatment for these other clinical variants within the neurodegenerative disease stable.
Insights from the basic similarities of certain neurodegenerative states
Superficially, post‐stroke syndromes and TBI develop very differently than do PD and AD. Clinically, their origins seem a world apart: not a gradual onset over years, as with AD or PD, but typically absent one day and dramatically present the next. But this does not necessarily mean a fundamentally different mechanism is operating. Abruptness in onset is what one would expect from an ischaemic event(s) in a highly oxygen‐dependent site, or from instantaneous external trauma. Also, while AD and PD continue to worsen as the months pass, post‐stroke syndromes and TBI generally do not. Yet histology and laboratory assays identify all these conditions as a close‐knit group. Like PD and AD, post‐stroke syndromes and TBI both exhibit enhanced TNF production (Taupin et al., 1993; Cui et al., 2012), increases in tau (Ihle‐Hansen et al., 2017; Kulbe and Hall, 2017), IR (Brooks et al., 1984; Mowery et al., 2009), α‐synuclein (Impellizzeri et al., 2016; Kim et al., 2016) and Aβ (Lee et al., 2005; Acosta et al., 2017). One study (Simpkins et al., 2009) collected much relevant data, reporting, for example, that some 70% of brains of 55 patients who died within 24 h of head injury had Aβ precursor protein accumulations, with the earliest detectable at 2 h of survival (McKenzie et al., 1996).
Also, these conditions predispose to one another, for example, stroke to AD (Henon et al., 2001) and TBI to PD in older individuals (Gardner et al., 2015), with a more recent study (Camacho‐Soto et al., 2017) finding the link to be stronger during the prodromal period. Similarly, TBI predisposes to AD (Fleminger et al., 2003). As with AD and PD, post‐stroke syndromes and TBI also manifest a predisposition to be more severe in APOE4 individuals (Eramudugolla et al., 2014; Das et al., 2016). Another set of observations transgressing these traditional clinical divides concerns the ability of SB623 stem cells, which manifest an anti‐TNF activity (see below) being efficacious for the PD model generated by injecting methylphenyltetrahydropyridine (MPTP) (Tate et al., 2017) and in post‐stroke patients (Steinberg et al., 2016b).
It is also illuminating to recall here, as we have discussed earlier in this text, that AD (Chou et al., 2016) and PD (Mittal et al., 2017; Peter et al., 2018) have been inadvertently reduced in incidence when patients were treated with anti‐TNF agents, on a grand scale, for another purpose. It therefore seems most plausible that post‐stroke syndromes and TBI, AD and PD eventually result in similar clinical outcomes through the effects of increased levels of TLR agonists, chronically excessive TNF generation, and thus cellular and cytokine activity that is clinically recognized as post‐TNF P‐tau, IR, α‐synuclein and Aβ activity. Logically, therefore, what sets post‐stroke syndromes and TBI apart, in a pathophysiological sense, from AD and PD is simply where, and at what rate, this excess TNF is initially produced.
Therapeutic implications of these insights
The financial interest of the pharmaceutical industry in anti‐TNF biologicals is enormous. The gross income from Humira (http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4860), Enbrel (etanercept) and Remicade (infliximab) seen on the graph at https://www.forbes.com/sites/matthewherper/2015/07/29/the-top-drug-launches-of-all-time/#46db4cdf6512 succinctly demonstrates that this class of agents is the most commercially successful therapeutic agents of recent decades. It is difficult to doubt, from this observation as well as the number of biosimilars becoming available, that they can be remarkably efficacious. For two decades, this class of drug has been approved, and commonly used, to treat a number of non‐cerebral disease states, which is what this graph depicts. Considering the number of people afflicted with neurodegenerative diseases, these totals have the potential to much more than double.
Evidence of the usefulness of infliximab in a small open trial on rheumatoid arthritis (Elliott et al., 1993) was successfully expanded to formal, identical protocol testing within a year (Elliott et al., 1994). Testing and adoption across clinical specialty boundaries followed in due course, fairly soon encompassing psoriasis and Crohn's disease. Once TNF also began to appear in the literature on the pathogenesis of neurodegenerative disease, it seemed perfectly reasonable, to those with a broad interest in roles of TNF in disease pathogenesis, that a similar story would soon unfold in these non‐infectious chronic cerebral disease states. It was therefore scientifically unaccountable to see the use of another anti‐TNF biological, etanercept, for a neurodegenerative disease, AD, attracting negligible neutral or positive comment, but much strong scepticism, on Alzforum, and unprecedented industrial‐scale criticism on the web. Remarkably, this negativity persisted throughout a 2006 AD pilot study (Tobinick et al., 2006), 2008 case reports (Tobinick and Gross, 2008) and subsequent invited reviews on its logic (Tobinick, 2009; Tobinick, 2010; Tobinick, 2018). Nowadays, however, the general perception has mellowed, the UK Alzheimer's Society seeing an anti‐TNF approach as their main research interest (https://www.alzheimers.org.uk/research/our-research/research-projects/understanding-whether-drugs-rheumatoid-arthritis-can-reduce-risk-alzheimers-disease).
In these earlier studies, initially in AD but subsequently mainly in post‐stroke syndromes, etanercept was administered by a novel route (Tobinick and Vega, 2006; Tobinick, 2016) into Batson's plexus (the cerebrospinal venous system) plus a short period head‐down tilt (Trendelenburg position) to overcome its large molecular size and take advantage of the valveless nature of this vascular system (the perispinal route). A proposal was made from Southampton to replicate this perispinal approach in a formal trial, but in the event, both etanercept and placebo were administered s.c. (Butchart et al., 2015). From the limited information available in this text, published in a journal owned by the American Academy of Neurology (AAN), this Southampton trial can appear to have had a completely negative outcome. Unfortunately, this is often misleadingly quoted as though it had replicated, and negated, the original technique of 9 years earlier (Tobinick et al., 2006; Tobinick and Vega, 2006), in which treatments were administered via Batson's plexus. However, other positive and significant outcomes of the Southampton trial are presented and discussed in the first author's PhD thesis (Butchart, 2017), which covers outcomes of this trial more extensively, is consistent with enough s.c. administered etanercept having entered the brain to have had some significant clinical effects. These extended data are consistent with a report, drawn from a particularly large database over a long period, of s.c. chronic use of anti‐TNF biologicals to treat rheumatoid arthritis significantly reducing AD incidence (Chou et al., 2016).
In the current absence of funding for a controlled replication of the use of the perispinal route, studies of treating post‐stroke syndromes and TBI with etanercept administered by this route continue to appear as case reports (Tobinick, 2011; Tobinick et al., 2012; Tobinick et al., 2014), with the 2012 publication involving over 600 cases. Unaccountably, this approach, and therefore controlled trials, has been actively discouraged by the AAN through issuing a Practice Advisory notice that, unusually, attracted a rebuttal editorial (Clark, 2017). As noted, such clinical advice ultimately needs to be based on observation by AAN members, which remains absent after years of invitations. Also, this Advisory, in the face of the literature and related FDA approvals, profoundly over‐states the toxicity of etanercept.
The prospect of routine perispinal anti‐TNF therapy for post‐stroke syndromes is greatly strengthened by another treatment, from the literature effectively anti‐TNF, that has had a strikingly similar outcome to etanercept in an open trial (Steinberg et al., 2016b). The rationale for this approach began when human marrow stromal cells provided useful therapy in a rat model of stroke in 2002 (Li et al., 2002). Some years later (Liu et al., 2009), rat mesenchymal stem cells were shown, when introduced into a cerebral ventricle in a rat stroke model, to significantly improve function while enhancing production of the anti‐inflammatory cytokine http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4975 and decreasing TNF levels. These studies, and others, laid the groundwork for the above open trial, with a positive outcome, of surgically transplanting SB623 stem cells into brains of patients suffering from post‐stroke syndromes (Steinberg et al., 2016b). Evidently this generates essentially the same outcome, and from the literature for the same reasons, as does perispinal etanercept (Tobinick, 2011; Tobinick et al., 2012). This is consistent with data from experimental PD, in which both SB623 cells (Tate et al., 2017) and an anti‐TNF agent (Ferger et al., 2004) have attenuated the toxicity of MPTP. As we have noted in our published correspondence regarding this text (Clark, 2016), with acceptance of the possibility from the authors (Steinberg et al., 2016a), the capacity of these stem cells to improve post‐stroke disabilities is most likely through creating a strong anti‐inflammatory milieu, generated in this case by http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4924 (Aizman et al., 2015). Among other activities, this cytokine counters that of TNF (Yu et al., 2016). Hence, injecting these stem cells and appropriately administering etanercept evidently have the same fundamental mechanism (Figure 2), which will be easy enough to confirm experimentally in due course.
Figure 2.
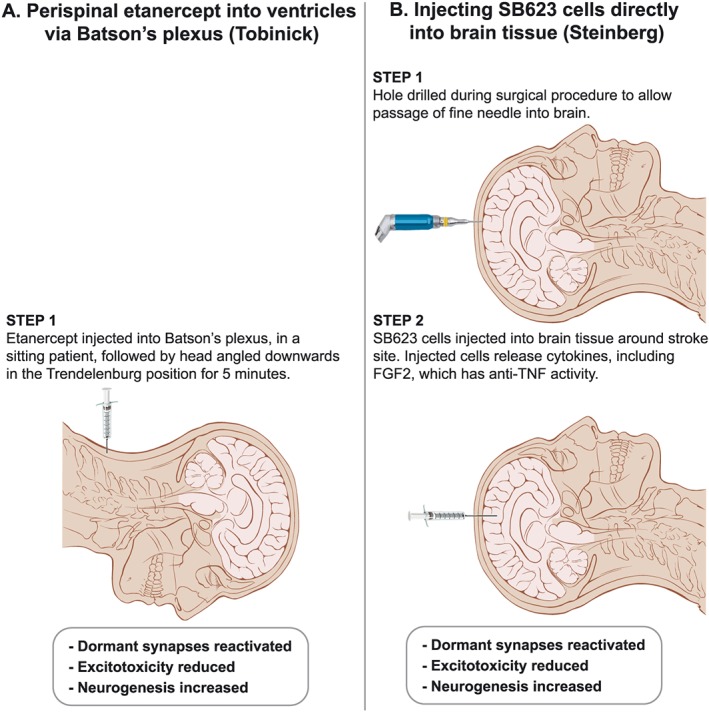
A visual comparison of complexity of methods designed to generate similar clinical improvements in post‐stroke syndromes by reducing cerebral TNF. To date, both have been done in open trials only. Illustration adapted from ‘Head anatomy anterior lateral views’; Patrick J. Lynch; illustrator; C. Carl Jaffe; MD; cardiologist Yale University Center for Advanced Instructional Media; Creative Commons Attribution 2.5 Licence 2006.
Speaking after acceptance of a 2017 Smithsonian Ingenuity Award (https://www.smithsonianmag.com/science-nature/neurosurgeon-remarkable-plan-stroke-victims-stem-cells-180967211), this researcher quite reasonably – considering the now abundant evidence from perispinal etanercept from 2011 as well as his intra‐cerebral injection of SB623 cells – argued for reactivating circuits historically considered dead rather than dormant being the future for achieving sustained improvement in a number of neurological diseases. It is relevant to note here that the logic of the perispinal route has recently been greatly enhanced by new studies on the anatomy of the cranial extensions of the spinal venous plexuses at the cranio‐cervical junction (Tubbs et al., 2018). The readiness of 51 neurosurgical centres across the US to be involved in a formal expansion of this SB623 approach (Steinberg et al., 2016b) (https://clinicaltrials.gov/show/NCT02448641) is most encouraging, since it implies that the stroke research community is finally comfortable with discarding the traditional tenet of neurology that if post‐stroke functional loss is still present some months after the event, it inevitably signifies irreversibility through cerebro‐cellular death. In passing, we recall this acceptance could have begun years earlier, when the striking similar open trial consequences of administering etanercept perispinally to post‐stroke patients were first available in widely accessible literature (Tobinick, 2011; Tobinick et al., 2012).
Conclusions
In conclusion, the logic of treating neurodegenerative diseases through reducing chronically increased cerebral levels of TNF is becoming a competitive field and rapidly gaining credibility. As discussed in this text, the links between TNF and APOE4 predisposition, P‐tau, α‐synuclein, Aβ and IR in these diseases have now generated a compelling literature on harmful sequences of events that can disappear or be reduced to the degree to which homeostasis can return once excess TNF is therapeutically removed. This degree is governed by the proportion of reversible dormancy compared to permanent damage this excess TNF has caused in that patient's neural circuits. In effect, we have argued here that the ubiquity of excess cerebral TNF in post‐stroke syndromes, TBI, PD and AD appears to have gathered all these clinical descriptors under the one pharmacological roof.
Unfortunately, treatments that pass the test of being logical, can, through poor communication delaying consensus on the details of a condition's pathophysiology, be long‐delayed. This does not need to be so: malarial researchers do not, and may never, agree on the pathogenesis of the encephalopathy termed cerebral malaria (Clark and Rockett, 1994), but this did not prevent quinine providing a reasonably successful therapy through killing the PAMP‐generating parasite. Indeed, through the centuries before malaria was realized to be an infectious disease, quinine was equally successful while thought to work through reducing fever. Similarly, gaps still exist in the PD literature, such as precisely how α‐synuclein aggregates, and why its capacity to harm eventually focuses on dopaminogenic neurons (Mor et al., 2017).
Hence, however scientifically important it is to fully understand details of pathophysiology, these may plausibly be resolved in parallel with finding a useful treatment through applying what is already known. Most advances arise from logical small open trials on re‐positioning medication, such as etanercept, that is already approved for a number of related conditions and very widely used. Indeed, this is already what is happening with etanercept, SB623 cells and salbutamol, as discussed in this review. In the same vein, much is not yet known about the detailed pathogenesis of rheumatoid arthritis, Crohn's disease and psoriasis, yet their anti‐TNF treatment is, overall, a success. This is despite these diseases being much more dissimilar clinically than post‐stroke syndromes, TBI, PD and AD. What is now decades of experience in treating these three unalike peripheral diseases thus provides valuable experience for the neurodegenerative disease world. It therefore now seems valid to suggest that enough is now known about the pathophysiology of mainstream versions of these neurodegenerative conditions to foster comparisons in outcomes and costs between the virtues of treating patients by injecting specific anti‐TNF biologicals perispinally (Tobinick et al., 2006; Tobinick et al., 2012), achieving the same end by injecting into the brain cells that generate FGF2 (Steinberg et al., 2016b) and administering salbutamol (Mittal et al., 2017). Variants of each condition may respond only slightly, but this is no less true of, for example, rheumatoid arthritis. As antagonists of other pro‐inflammatory cytokines show more promise, they may well join the list of rational target molecules.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a,b,c)
Author contributions
I.A.C. proposed the scope of the review. Both authors were involved in planning and editing the manuscript, blending their complementary expertises. Both authors read and approved the final manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
The authors are most grateful to Sharyn Wragg for the artwork in Figure 2. No funding was received for this work.
Clark, I. A. , and Vissel, B. (2018) Therapeutic implications of how TNF links apolipoprotein E, phosphorylated tau, α‐synuclein, amyloid‐β and insulin resistance in neurodegenerative diseases. British Journal of Pharmacology, 175: 3859–3875. 10.1111/bph.14471.
References
- Acosta SA, Tajiri N, Sanberg PR, Kaneko Y, Borlongan CV (2017). Increased amyloid precursor protein and tau expression manifests as key secondary cell death in chronic traumatic brain injury. J Cell Physiol 232: 665–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acosta SA, Tajiri N, de la Pena I, Bastawrous M, Sanberg PR, Kaneko Y et al (2015). Alpha‐synuclein as a pathological link between chronic traumatic brain injury and Parkinson's disease. J Cell Physiol 230: 1024–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aden U, Favrais G, Plaisant F, Winerdal M, Felderhoff‐Mueser U, Lampa J et al (2010). Systemic inflammation sensitizes the neonatal brain to excitotoxicity through a pro‐/anti‐inflammatory imbalance: key role of TNF pathway and protection by etanercept. Brain Behav Immun 24: 747–758. [DOI] [PubMed] [Google Scholar]
- Ahmed RM, Ke YD, Vucic S, Ittner LM, Seeley W, Hodges JR et al (2018). Physiological changes in neurodegeneration – mechanistic insights and clinical utility. Nat Rev Neurol 14: 259–271. [DOI] [PubMed] [Google Scholar]
- Aizman I, Vinodkumar D, McGrogan M, Bates D (2015). Cell injury‐induced release of fibroblast growth factor 2: relevance to intracerebral mesenchymal stromal cell transplantations. Stem Cells Dev 24: 1623–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albayram O, Herbert MK, Kondo A, Tsai CY, Baxley S, Lian X et al (2016). Function and regulation of tau conformations in the development and treatment of traumatic brain injury and neurodegeneration. Cell Biosci 6: 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017a). The Concise Guide To PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide To PHARMACOLOGY 2017/18: Catalytic receptors. Br J Pharmacol 174: S225–S271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017c). The Concise Guide To PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander GM, Schwartzman RJ, Nukes TA, Grothusen JR, Hooker MD (1994). Beta 2‐adrenergic agonist as adjunct therapy to levodopa in Parkinson's disease. Neurology 44: 1511–1513. [DOI] [PubMed] [Google Scholar]
- Alvarez‐Erviti L, Couch Y, Richardson J, Cooper JM, Wood MJ (2011). Alpha‐synuclein release by neurons activates the inflammatory response in a microglial cell line. Neurosci Res 69: 337–342. [DOI] [PubMed] [Google Scholar]
- Ambrosi G, Cerri S, Blandini F (2014). A further update on the role of excitotoxicity in the pathogenesis of Parkinson's disease. J Neural Transm (Vienna) 121: 849–859. [DOI] [PubMed] [Google Scholar]
- Araujo EP, De Souza CT, Ueno M, Cintra DE, Bertolo MB, Carvalheira JB et al (2007). Infliximab restores glucose homeostasis in an animal model of diet‐induced obesity and diabetes. Endocrinology 148: 5991–5997. [DOI] [PubMed] [Google Scholar]
- Arruda AP, Milanski M, Coope A, Torsoni AS, Ropelle E, Carvalho DP et al (2011). Low‐grade hypothalamic inflammation leads to defective thermogenesis, insulin resistance, and impaired insulin secretion. Endocrinology 152: 1314–1326. [DOI] [PubMed] [Google Scholar]
- Aviles‐Olmos I, Limousin P, Lees A, Foltynie T (2013). Parkinson's disease, insulin resistance and novel agents of neuroprotection. Brain 136: 374–384. [DOI] [PubMed] [Google Scholar]
- Bachor TP, Karbanova J, Buttner E, Bermudez V, Marquioni‐Ramella M, Carmeliet P et al (2017). Early ciliary and prominin‐1 dysfunctions precede neurogenesis impairment in a mouse model of type 2 diabetes. Neurobiol Dis 108: 13–28. [DOI] [PubMed] [Google Scholar]
- Bartels AL, Leenders KL (2007). Neuroinflammation in the pathophysiology of Parkinson's disease: evidence from animal models to human in vivo studies with [11C]‐PK11195 PET. Mov Disord 22: 1852–1856. [DOI] [PubMed] [Google Scholar]
- Basha MR, Wei W, Bakheet SA, Benitez N, Siddiqi HK, Ge YW et al (2005). The fetal basis of amyloidogenesis: exposure to lead and latent overexpression of amyloid precursor protein and beta‐amyloid in the aging brain. J Neurosci 25: 823–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatman EL, Massey A, Shives KD, Burrack KS, Chamanian M, Morrison TE et al (2015). Alpha‐synuclein expression restricts RNA viral infections in the brain. J Virol 90: 2767–2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beraud D, Hathaway HA, Trecki J, Chasovskikh S, Johnson DA, Johnson JA et al (2013). Microglial activation and antioxidant responses induced by the Parkinson's disease protein alpha‐synuclein. J Neuroimmune Pharmacol 8: 94–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bihaqi SW, Huang H, Wu J, Zawia NH (2011). Infant exposure to lead (Pb) and epigenetic modifications in the aging primate brain: implications for Alzheimer's disease. J Alzheimers Dis 27: 819–833. [DOI] [PubMed] [Google Scholar]
- Boivin MJ, Bangirana P, Byarugaba J, Opoka RO, Idro R, Jurek AM et al (2007). Cognitive impairment after cerebral malaria in children: a prospective study. Pediatr 119: E360–E366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomfim TR, Forny Germano L, Sathler LB, Brito Moreira J, Houzel JC, Decker H et al (2012). An anti‐diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer's disease‐associated Aβ oligomers. J Clin Invest 122: 1339–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgs L, Beukelaers P, Vandenbosch R, Nguyen L, Moonen G, Maquet P et al (2009). Period 2 regulates neural stem/progenitor cell proliferation in the adult hippocampus. BMC Neurosci 10: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan FM, Chantry D, Jackson A, Maini R, Feldmann M (1989). Inhibitory effect of TNF alpha antibodies on synovial cell interleukin‐1 production in rheumatoid arthritis. Lancet 2: 244–247. [DOI] [PubMed] [Google Scholar]
- Brooks DC, Bessey PQ, Black PR, Aoki TT, Wilmore DW (1984). Post‐traumatic insulin resistance in uninjured forearm tissue. J Surg Res 37: 100–107. [DOI] [PubMed] [Google Scholar]
- Bu XL, Wang X, Xiang Y, Shen LL, Wang QH, Liu YH et al (2015). The association between infectious burden and Parkinson's disease: a case‐control study. Parkinsonism Relat Disord 21: 877–881. [DOI] [PubMed] [Google Scholar]
- Burguillos MA, Deierborg T, Kavanagh E, Persson A, Hajji N, Garcia‐Quintanilla A et al (2011). Caspase signalling controls microglia activation and neurotoxicity. Nature 472: 319–324. [DOI] [PubMed] [Google Scholar]
- Butchart J, Brook L, Hopkins V, Teeling J, Puntener U, Culliford D et al (2015). Etanercept in Alzheimer disease: a randomized, placebo‐controlled, double‐blind, phase 2 trial. Neurology 84: 2161–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butchart JW. (2017). Systemic inflammation and sickness behaviour in Alzheimer's disease. PhD Thesis University of Southhampton.
- Calderon‐Garciduenas L, de la Monte SM (2017). Apolipoprotein E4, gender, body mass index, inflammation, insulin resistance, and air pollution interactions: recipe for Alzheimer's disease development in Mexico City young females. J Alzheimers Dis 58: 613–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho‐Soto A, Warden MN, Searles Nielsen S, Salter A, Brody DL, Prather H et al (2017). Traumatic brain injury in the prodromal period of Parkinson's disease: a large epidemiological study using medicare data. Ann Neurol 82: 744–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, Gaamouch FE, Meabon JS, Meeker KD, Zhu L, Zhong MB et al (2017). ApoE4‐associated phospholipid dysregulation contributes to development of Tau hyper‐phosphorylation after traumatic brain injury. Sci Rep 7: 11372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson GL (2004). Hunterian lecture: insulin resistance in human sepsis: implications for the nutritional and metabolic care of the critically ill surgical patient. Ann R Coll Surg Engl 86: 75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmen J, Rothstein JD, Kerr DA (2009). Tumor necrosis factor‐alpha modulates glutamate transport in the CNS and is a critical determinant of outcome from viral encephalomyelitis. Brain Res 31: 143–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B (1975). An endotoxin‐induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci U S A 72: 3666–3670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter JA, Mung'ala‐Odera V, Neville BG, Murira G, Mturi N, Musumba C et al (2005). Persistent neurocognitive impairments associated with severe falciparum malaria in Kenyan children. J Neurol Neurosurg Psychiatry 76: 476–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavadini G, Petrzilka S, Kohler P, Jud C, Tobler I, Birchler T et al (2007). TNF alpha suppresses the expression of clock genes by interfering with E‐box‐mediated transcription. Proc Natl Acad Sci U S A 104: 12843–12848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanagh C, Tse YC, Nguyen HB, Krantic S, Breitner JC, Quirion R et al (2016). Inhibiting tumor necrosis factor‐α before amyloidosis prevents synaptic deficits in an Alzheimer's disease model. Neurobiol Aging 47: 41–49. [DOI] [PubMed] [Google Scholar]
- Cebrian C, Zucca FA, Mauri P, Steinbeck JA, Studer L, Scherzer CR et al (2014). MHC‐I expression renders catecholaminergic neurons susceptible to T‐cell‐mediated degeneration. Nat Commun 5: 3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cersosimo MG, Raina GB, Pecci C, Pellene A, Calandra CR, Gutierrez C et al (2013). Gastrointestinal manifestations in Parkinson's disease: prevalence and occurrence before motor symptoms. J Neurol 260: 1332–1338. [DOI] [PubMed] [Google Scholar]
- Chawla A, Nguyen KD, Goh YP (2011). Macrophage‐mediated inflammation in metabolic disease. Nat Rev Immunol 11: 738–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng YJ, Yang BC, Liu MY (2006). Lead increases lipopolysaccharide‐induced liver‐injury through tumor necrosis factor‐α overexpression by monocytes/macrophages: role of protein kinase C and P42/44 mitogen‐activated protein kinase. Environ Health Perspect 114: 507–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng YJ, Liu MY, Wu TP, Yang BC (2004). Regulation of tumor necrosis factor‐alpha in glioma cells by lead and lipopolysaccharide: involvement of common signaling pathway. Toxicol Lett 152: 127–137. [DOI] [PubMed] [Google Scholar]
- Chou RC, Kane M, Ghimire S, Gautam S, Gui J (2016). Treatment for rheumatoid arthritis and risk of Alzheimer's disease: A nested case‐control analysis. CNS Drugs 30: 1111–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark I, Atwood C, Bowen R, Paz‐Filho G, Vissel B (2012). Tumor necrosis factor‐induced cerebral insulin resistance in Alzheimer's disease links numerous treatment rationales. Pharmacol Rev 64: 1004–1026. [DOI] [PubMed] [Google Scholar]
- Clark IA (1978). Does endotoxin cause both the disease and parasite death in acute malaria and babesiosis? Lancet 312: 75–77. [DOI] [PubMed] [Google Scholar]
- Clark IA (1987). Cell‐mediated immunity in protection and pathology of malaria. Parasitol Today 3: 300–305. [DOI] [PubMed] [Google Scholar]
- Clark IA (2007a). The advent of the cytokine storm. Immunol Cell Biol 85: 271–273. [DOI] [PubMed] [Google Scholar]
- Clark IA (2007b). How TNF was recognized to be a key mechanism of disease. Cytokine Growth FR 18: 335–343. [DOI] [PubMed] [Google Scholar]
- Clark IA (2016). Letter by Clark regarding article, “Clinical Outcomes of Transplanted Modified Bone Marrow‐Derived Mesenchymal Stem Cells in Stroke: A Phase 1/2a Study”. Stroke 47: e268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark IA (2017). Editorial: an unsound AAN practice advisory on poststroke etanercept. Expert Rev Neurother 17: 215–217. [DOI] [PubMed] [Google Scholar]
- Clark IA, Rockett KA (1994). The cytokine theory of human cerebral malaria. Parasitol Today 10: 410–412. [DOI] [PubMed] [Google Scholar]
- Clark IA, Vissel B (2013). Treatment implications of the altered cytokine‐insulin axis in neurodegenerative disease. Biochem Pharmacol 86: 862–871. [DOI] [PubMed] [Google Scholar]
- Clark IA, Vissel B (2014). Inflammation‐sleep interface in brain disease: TNF, insulin, orexin. J Neuroinflammation 11: 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark IA, Vissel B (2015). Amyloid β: one of three danger‐associated molecules that are secondary inducers of the proinflammatory cytokines that mediate Alzheimer's disease. Br J Pharmacol 172: 3714–3727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark IA, Vissel B (2016). Excess cerebral TNF causing glutamate excitotoxicity rationalizes treatment of neurodegenerative diseases and neurogenic pain by anti‐TNF agents. J Neuroinflammation 13: 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark IA, Chaudhri G, Cowden WB (1989). Roles of tumour necrosis factor in the illness and pathology of malaria. Trans R Soc Trop Med Hyg 83: 436–440. [DOI] [PubMed] [Google Scholar]
- Clark IA, Alleva LM, Vissel B (2010). The roles of TNF in brain dysfunction and disease. Pharmacol Ther 128: 519–548. [DOI] [PubMed] [Google Scholar]
- Clark IA, Virelizier J‐L, Carswell EA, Wood PR (1981). Possible importance of macrophage‐derived mediators in acute malaria. Infect Immun 32: 1058–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark IA, Alleva LE, Mills AC, Cowden WB (2004). Pathogenesis of malaria and clinically similar conditions. Clin Microbiol Rev 17: 509–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook C, Kang SS, Carlomagno Y, Lin WL, Yue M, Kurti A et al (2015). Tau deposition drives neuropathological, inflammatory and behavioral abnormalities independently of neuronal loss in a novel mouse model. Hum Mol Genet 24: 6198–6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui G, Wang H, Li R, Zhang L, Li Z, Wang Y et al (2012). Polymorphism of tumor necrosis factor alpha (TNF‐alpha) gene promoter, circulating TNF‐alpha level, and cardiovascular risk factor for ischemic stroke. J Neuroinflammation 9: 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cumiskey D, Butler MP, Moynagh PN, O'Connor JJ (2007). Evidence for a role for the group I metabotropic glutamate receptor in the inhibitory effect of tumor necrosis factor‐α on long‐term potentiation. Brain Res 1136: 13–19. [DOI] [PubMed] [Google Scholar]
- D'Mello C, Le T, Swain MG (2009). Cerebral microglia recruit monocytes into the brain in response to tumor necrosis factoralpha signaling during peripheral organ inflammation. J Neurosci 29: 2089–2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Kaul S, Jyothy A, Munshi A (2016). Association of APOE (E2, E3 and E4) gene variants and lipid levels in ischemic stroke, its subtypes and hemorrhagic stroke in a South Indian population. Neurosci Lett 628: 136–141. [DOI] [PubMed] [Google Scholar]
- de Haan JJ, Smeets MB, Pasterkamp G, Arslan F (2013). Danger signals in the initiation of the inflammatory response after myocardial infarction. Mediators Inflamm 2013: 206039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Monte SM, Wands JR (2008). Alzheimer's disease is type 3 diabetes‐evidence reviewed. J Diabetes Sci Technol 2: 1101–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dosunmu R, Alashwal H, Zawia NH (2012). Genome‐wide expression and methylation profiling in the aged rodent brain due to early‐life Pb exposure and its relevance to aging. Mech Ageing Dev 133: 435–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekdahl CT, Claasen JH, Bonde S, Kokaia Z, Lindvall O (2003). Inflammation is detrimental for neurogenesis in adult brain. Proc Natl Acad Sci U S A 100: 13632–13637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott MJ, Maini RN, Feldmann M, Long Fox A, Charles P, Katsikis P et al (1993). Treatment of rheumatoid arthritis with chimeric monoclonal antibodies to tumor necrosis factor alpha. Arthr Rheum 36: 1681–1690. [DOI] [PubMed] [Google Scholar]
- Elliott MJ, Maini RN, Feldmann M, Kalden JR, Antoni C, Smolen JS et al (1994). Randomised double‐blind comparison of chimeric monoclonal antibody to tumour necrosis factor alpha (cA2) versus placebo in rheumatoid arthritis. Lancet 344: 1105–1110. [DOI] [PubMed] [Google Scholar]
- Eramudugolla R, Bielak AA, Bunce D, Easteal S, Cherbuin N, Anstey KJ (2014). Long‐term cognitive correlates of traumatic brain injury across adulthood and interactions with APOE genotype, sex, and age cohorts. J Int Neuropsychol Soc 20: 444–454. [DOI] [PubMed] [Google Scholar]
- Feinstein R, Kanety H, Papa MZ, Lunenfeld B, Karasik A (1993). Tumor necrosis factor‐alpha suppresses insulin‐induced tyrosine phosphorylation of insulin receptor and its substrates. J Biol Chem 268: 26055–26058. [PubMed] [Google Scholar]
- Ferger B, Leng A, Mura A, Hengerer B, Feldon J (2004). Genetic ablation of tumor necrosis factor‐alpha (TNF‐alpha) and pharmacological inhibition of TNF‐synthesis attenuates MPTP toxicity in mouse striatum. J Neurochem 89: 822–833. [DOI] [PubMed] [Google Scholar]
- Fernandez JA, Rojo L, Kuljis RO, Maccioni RB (2008). The damage signals hypothesis of Alzheimer's disease pathogenesis. J Alzheimers Dis 14: 329–333. [DOI] [PubMed] [Google Scholar]
- Fine JS, Macosko HD, Grace MJ, Narula SK (1994). Interleukin‐10 enhances gamma delta T cell development in the murine fetal thymus. Cell Immunol 155: 111–122. [DOI] [PubMed] [Google Scholar]
- Fleminger S, Oliver DL, Lovestone S, Rabe‐Hesketh S, Giora A (2003). Head injury as a risk factor for Alzheimer's disease: the evidence 10 years on; a partial replication. J Neurol Neurosurg Psychiatry 74: 857–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foell D, Wittkowski H, Vogl T, Roth J (2007). S100 proteins expressed in phagocytes: a novel group of damage‐associated molecular pattern molecules. J Leukoc Biol 81: 28–37. [DOI] [PubMed] [Google Scholar]
- Gardner RC, Burke JF, Nettiksimmons J, Goldman S, Tanner CM, Yaffe K (2015). Traumatic brain injury in later life increases risk for Parkinson disease. Ann Neurol 77: 987–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gassowska M, Baranowska‐Bosiacka I, Moczydlowska J, Tarnowski M, Pilutin A, Gutowska I et al (2016). Perinatal exposure to lead (Pb) promotes Tau phosphorylation in the rat brain in a GSK‐3beta and CDK5 dependent manner: Relevance to neurological disorders. Toxicology 347‐349: 17–28. [DOI] [PubMed] [Google Scholar]
- Ge YW, Lahiri DK (2002). Regulation of promoter activity of the APP gene by cytokines and growth factors: implications in Alzheimer's disease. Ann N Y Acad Sci 973: 463–467. [DOI] [PubMed] [Google Scholar]
- Geeraedts F, Goutagny N, Hornung V, Severa M, de Haan A, Pool J et al (2008). Superior immunogenicity of inactivated whole virus H5N1 influenza vaccine is primarily controlled by Toll‐like receptor signalling. PLoS Pathog 4: e1000138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon EB, Hart GT, Tran TM, Waisberg M, Akkaya M, Kim AS et al (2015). Targeting glutamine metabolism rescues mice from late‐stage cerebral malaria. Proc Natl Acad Sci U S A 112: 13075–13080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorlovoy P, Larionov S, Pham TT, Neumann H (2009). Accumulation of tau induced in neurites by microglial proinflammatory mediators. FASEB J 23: 2502–2513. [DOI] [PubMed] [Google Scholar]
- Griffin WS, Stanley LC, Ling C, White L, MacLeod V, Perrot LJ et al (1989). Brain interleukin 1 and S‐100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc Natl Acad Sci U S A 86: 7611–7615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo TL, Mudzinski SP, Lawrence DA (1996). The heavy metal lead modulates the expression of both TNF‐alpha and TNF‐alpha receptors in lipopolysaccharide‐activated human peripheral blood mononuclear cells. J Leukoc Biol 59: 932–939. [DOI] [PubMed] [Google Scholar]
- Hardardottir I, Kunitake ST, Moser AH, Doerrler WT, Rapp JH, Grunfeld C et al (1994). Endotoxin and cytokines increase hepatic messenger RNA levels and serum concentrations of apolipoprotein J (clusterin) in Syrian hamsters. J Clin Invest 94: 1304–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haughey NJ, Nath A, Chan SL, Borchard AC, Rao MS, Mattson MP (2002). Disruption of neurogenesis by amyloid beta‐peptide, and perturbed neural progenitor cell homeostasis, in models of Alzheimer's disease. J Neurochem 83: 1509–1524. [DOI] [PubMed] [Google Scholar]
- Henon H, Durieu I, Guerouaou D, Lebert F, Pasquier F, Leys D (2001). Poststroke dementia: incidence and relationship to prestroke cognitive decline. Neurology 57: 1216–1222. [DOI] [PubMed] [Google Scholar]
- Hoffmann A, Ettle B, Bruno A, Kulinich A, Hoffmann AC, von Wittgenstein J et al (2016). Alpha‐synuclein activates BV2 microglia dependent on its aggregation state. Biochem Biophys Res Commun 479: 881–886. [DOI] [PubMed] [Google Scholar]
- Holding PA, Snow RW (2001). Impact of Plasmodium falciparum malaria on performance and learning: review of the evidence. Am J Trop Med Hyg 64: 68–75. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS, Murray DL, Choy LN, Spiegelman BM (1994). Tumor necrosis factor alpha inhibits signaling from the insulin receptor. Proc Natl Acad Sci U S A 91: 4854–4858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF, Spiegelman BM (1996). IRS‐1‐mediated inhibition of insulin receptor tyrosine kinase activity in TNF‐alpha‐ and obesity‐induced insulin resistance. Science 271: 665–668. [DOI] [PubMed] [Google Scholar]
- Idro R, Jenkins NE, Newton CR (2005). Pathogenesis, clinical features, and neurological outcome of cerebral malaria. Lancet Neurol 4: 827–840. [DOI] [PubMed] [Google Scholar]
- Idro R, Kakooza‐Mwesige A, Balyejjussa S, Mirembe G, Mugasha C, Tugumisirize J et al (2010). Severe neurological sequelae and behaviour problems after cerebral malaria in Ugandan children. BMC Res Notes 3: 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignatowski TA, Spengler RN (1995). Regulation of macrophage‐derived tumor necrosis factor production by modification of adrenergic receptor sensitivity. J Neuroimmunol 61: 61–70. [DOI] [PubMed] [Google Scholar]
- Ihle‐Hansen H, Hagberg G, Fure B, Thommessen B, Fagerland MW, Oksengard AR et al (2017). Association between total‐Tau and brain atrophy one year after first‐ever stroke. BMC Neurol 17: 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Impellizzeri D, Campolo M, Bruschetta G, Crupi R, Cordaro M, Paterniti I et al (2016). Traumatic brain injury leads to development of Parkinson's disease related pathology in mice. Front Neurosci 10: 458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itzhaki RF, Lathe R, Balin BJ, Ball MJ, Bearer EL, Braak H et al (2016). Microbes and Alzheimer's disease. J Alzheimers Dis 51: 979–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwashyna TJ, Ely EW, Smith DM, Langa KM (2010). Long‐term cognitive impairment and functional disability among survivors of severe sepsis. JAMA 304: 1787–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob A, Brorson JR, Alexander JJ (2011). Septic encephalopathy: inflammation in man and mouse. Neurochem Int 58: 472–476. [DOI] [PubMed] [Google Scholar]
- Jang H, Boltz D, Sturm‐Ramirez K, Shepherd KR, Jiao Y, Webster R et al (2009). Highly pathogenic H5N1 influenza virus can enter the central nervous system and induce neuroinflammation and neurodegeneration. Proc Natl Acad Sci U S A 106: 14063–14068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janus C, Pearson J, McLaurin J, Mathews PM, Jiang Y, Schmidt SD et al (2000). A beta peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer's disease. Nature 408: 979–982. [DOI] [PubMed] [Google Scholar]
- John CC, Bangirana P, Byarugaba J, Opoka RO, Idro R, Jurek AM et al (2008a). Cerebral malaria in children is associated with long‐term cognitive impairment. Pediatr. 122: 92–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John CC, Panoskaltsis Mortari A, Opoka RO, Park GS, Orchard PJ, Jurek AM et al (2008b). Cerebrospinal fluid cytokine levels and cognitive impairment in cerebral malaria. Am J Trop Med Hyg 78: 198–205. [PMC free article] [PubMed] [Google Scholar]
- Kapogiannis D, Boxer A, Schwartz JB, Abner EL, Biragyn A, Masharani U et al (2017). Dysfunctionally phosphorylated type 1 insulin receptor substrate in neural‐derived blood exosomes of preclinical Alzheimer's disease. FASEB J 29: 589–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karran E, De Strooper B (2016). The amyloid cascade hypothesis: are we poised for success or failure? J Neurochem 139 (Suppl. 2): 237–252. [DOI] [PubMed] [Google Scholar]
- Kaur C, Sivakumar V, Zou Z, Ling EA (2014). Microglia‐derived proinflammatory cytokines tumor necrosis factor‐alpha and interleukin‐1beta induce Purkinje neuronal apoptosis via their receptors in hypoxic neonatal rat brain. Brain Struct Funct 219: 151–170. [DOI] [PubMed] [Google Scholar]
- Keranen T, Hommo T, Moilanen E, Korhonen R (2017). β2‐Receptor agonists salbutamol and terbutaline attenuated cytokine production by suppressing ERK pathway through cAMP in macrophages. Cytokine 94: 1–7. [DOI] [PubMed] [Google Scholar]
- Kihara M, Carter JA, Holding PA, Vargha Khadem F, Scott RC, Idro R et al (2009). Impaired everyday memory associated with encephalopathy of severe malaria: the role of seizures and hippocampal damage. Malar J 8: 273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C, Ho DH, Suk JE, You S, Michael S, Kang J et al (2013). Neuron‐released oligomeric alpha‐synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun 4: 1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T, Mehta SL, Kaimal B, Lyons K, Dempsey RJ, Vemuganti R (2016). Poststroke induction of alpha‐synuclein mediates ischemic brain damage. J Neurosci 36: 7055–7065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimiwada T, Sakurai M, Ohashi H, Aoki S, Tominaga T, Wada K (2009). Clock genes regulate neurogenic transcription factors, including NeuroD1, and the neuronal differentiation of adult neural stem/progenitor cells. Neurochem Int 54: 277–285. [DOI] [PubMed] [Google Scholar]
- Kondo A, Shahpasand K, Mannix R, Qiu J, Moncaster J, Chen CH et al (2015). Antibody against early driver of neurodegeneration cis P‐tau blocks brain injury and tauopathy. Nature 523: 431–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korff A, Liu C, Ginghina C, Shi M, Zhang J (2013). α‐Synuclein in cerebrospinal fluid of Alzheimer's disease and mild cognitive impairment. J Alzheimers Dis 36: 679–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulbe JR, Hall ED (2017). Chronic traumatic encephalopathy‐integration of canonical traumatic brain injury secondary injury mechanisms with tau pathology. Prog Neurobiol 158: 15–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumawat KL, Kaushik DK, Goswami P, Basu A (2014). Acute exposure to lead acetate activates microglia and induces subsequent bystander neuronal death via caspase‐3 activation. Neurotoxicology 41: 143–153. [DOI] [PubMed] [Google Scholar]
- Larson ME, Sherman MA, Greimel S, Kuskowski M, Schneider JA, Bennett DA et al (2012). Soluble alpha‐synuclein is a novel modulator of Alzheimer's disease pathophysiology. J Neurosci 32: 10253–10266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent C, Dorothee G, Hunot S, Martin E, Monnet Y, Duchamp M et al (2017). Hippocampal T cell infiltration promotes neuroinflammation and cognitive decline in a mouse model of tauopathy. Brain 140: 184–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazosky A, Young GB, Zirul S, Phillips R (2010). Quality of life after septic illness. J Crit Care 25: 406–412. [DOI] [PubMed] [Google Scholar]
- Lee HG, Zhu X, Castellani RJ, Nunomura A, Perry G, Smith MA (2007). Amyloid‐beta in Alzheimer disease: the null versus the alternate hypotheses. J Pharmacol Exp Ther 321: 823–829. [DOI] [PubMed] [Google Scholar]
- Lee PH, Bang OY, Hwang EM, Lee JS, Joo US, Mook‐Jung I et al (2005). Circulating beta amyloid protein is elevated in patients with acute ischemic stroke. J Neural Transm (Vienna) 112: 1371–1379. [DOI] [PubMed] [Google Scholar]
- Li N, Liu F, Song L, Zhang P, Qiao M, Zhao Q et al (2014). The effects of early life Pb exposure on the expression of IL1‐β, TNF‐α and Aβ in cerebral cortex of mouse pups. J Trace Elem Med Biol 28: 100–104. [DOI] [PubMed] [Google Scholar]
- Li N, Yu ZL, Wang L, Zheng YT, Jia JX, Wang Q et al (2009). Early‐life lead exposure affects the activity of TNF‐alpha and expression of SNARE complex in hippocampus of mouse pups. Biol Trace Elem Res 132: 227–238. [DOI] [PubMed] [Google Scholar]
- Li N, Yu ZL, Wang L, Zheng YT, Jia JX, Wang Q et al (2010). Increased tau phosphorylation and beta amyloid in the hippocampus of mouse pups by early life lead exposure. Acta Biol Hung 61: 123–134. [DOI] [PubMed] [Google Scholar]
- Li Y, Chen J, Chen XG, Wang L, Gautam SC, Xu YX et al (2002). Human marrow stromal cell therapy for stroke in rat: neurotrophins and functional recovery. Neurology 59: 514–523. [DOI] [PubMed] [Google Scholar]
- Liddelow SA, Barres BA (2017). Reactive astrocytes: production, function, and therapeutic potential. Immunity 46: 957–967. [DOI] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L et al (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541: 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J, Lu KP (2005). Pinning down phosphorylated tau and tauopathies. Biochim Biophys Acta 1739: 311–322. [DOI] [PubMed] [Google Scholar]
- Liu N, Chen R, Du H, Wang J, Zhang Y, Wen J (2009). Expression of IL‐10 and TNF‐alpha in rats with cerebral infarction after transplantation with mesenchymal stem cells. Cell Mol Immunol 6: 207–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Liu F, Grundke Iqbal I, Iqbal K, Gong CX (2011). Deficient brain insulin signalling pathway in Alzheimer's disease and diabetes. J Pathol 225: 54–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch JR, Morgan D, Mance J, Matthew WD, Laskowitz DT (2001). Apolipoprotein E modulates glial activation and the endogenous central nervous system inflammatory response. J Neuroimmunol 114: 107–113. [DOI] [PubMed] [Google Scholar]
- Maezawa I, Maeda N, Montine TJ, Montine KS (2006a). Apolipoprotein E‐specific innate immune response in astrocytes from targeted replacement mice. J Neuroinflammation 3: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maezawa I, Nivison M, Montine KS, Maeda N, Montine TJ (2006b). Neurotoxicity from innate immune response is greatest with targeted replacement of E4 allele of apolipoprotein E gene and is mediated by microglial p38MAPK. FASEB J 20: 797–799. [DOI] [PubMed] [Google Scholar]
- Maroteaux L, Campanelli JT, Scheller RH (1988). Synuclein: a neuron‐specific protein localized to the nucleus and presynaptic nerve terminal. J Neurosci 8: 2804–2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marxreiter F, Regensburger M, Winkler J (2013). Adult neurogenesis in Parkinson's disease. Cell Mol Life Sci 70: 459–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason LH, Harp JP, Han DY (2014). Pb neurotoxicity: neuropsychological effects of lead toxicity. Biomed Res Int 2014: 840547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie KJ, McLellan DR, Gentleman SM, Maxwell WL, Gennarelli TA, Graham DI (1996). Is beta‐APP a marker of axonal damage in short‐surviving head injury? Acta Neuropathol 92: 608–613. [DOI] [PubMed] [Google Scholar]
- Miranda AS, Vieira LB, Lacerda‐Queiroz N, Souza AH, Rodrigues DH, Vilela MC et al (2010). Increased levels of glutamate in the central nervous system are associated with behavioral symptoms in experimental malaria. Braz J Med Biol Res 43: 1173–1177. [DOI] [PubMed] [Google Scholar]
- Mittal S, Bjornevik K, Im DS, Flierl A, Dong X, Locascio JJ et al (2017). β2‐Adrenoreceptor is a regulator of the α‐synuclein gene driving risk of Parkinson's disease. Science 357: 891–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modi HR, Wang Q, Gd S, Sherman D, Greenwald E, Savonenko AV et al (2017). Intranasal post‐cardiac arrest treatment with orexin‐A facilitates arousal from coma and ameliorates neuroinflammation. PLoS One 12: e0182707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondello S, Buki A, Italiano D, Jeromin A (2013). α‐Synuclein in CSF of patients with severe traumatic brain injury. Neurology 80: 1662–1668. [DOI] [PubMed] [Google Scholar]
- Mor DE, Tsika E, Mazzulli JR, Gould NS, Kim H, Daniels MJ et al (2017). Dopamine induces soluble α‐synuclein oligomers and nigrostriatal degeneration. Nat Neurosci 20: 1560–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriya T, Hiraishi K, Horie N, Mitome M, Shinohara K (2007). Correlative association between circadian expression of mousePer2 gene and the proliferation of the neural stem cells. Neurosci 146: 494–498. [DOI] [PubMed] [Google Scholar]
- Mowery NT, Gunter OL, Guillamondegui O, Dossett LA, Dortch MJ, Morris JA Jr et al (2009). Stress insulin resistance is a marker for mortality in traumatic brain injury. J Trauma 66: 145–151. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Zhou XZ, Lu KP (2013a). Distinct functions of cis and trans phosphorylated tau in Alzheimer's disease and their therapeutic implications. Curr Mol Med 13: 1098–1109. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Zhen Zhou X, Ping Lu K (2013b). Cis phosphorylated tau as the earliest detectable pathogenic conformation in Alzheimer disease, offering novel diagnostic and therapeutic strategies. Prion 7: 117–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, Greenwood A, Binder L, Bigio EH, Denial S, Nicholson L et al (2012). Proline isomer‐specific antibodies reveal the early pathogenic tau conformation in Alzheimer's disease. Cell 149: 232–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natale G, Pasquali L, Paparelli A, Fornai F (2011). Parallel manifestations of neuropathologies in the enteric and central nervous systems. Neurogastroenterol Motil 23: 1056–1065. [DOI] [PubMed] [Google Scholar]
- Nathan C, Ding A (2010). Nonresolving inflammation. Cell 140: 871–882. [DOI] [PubMed] [Google Scholar]
- Neves FS, Marques PT, Barros‐Aragao F, Nunes JB, Venancio AM, Cozachenco D et al (2018). Brain‐defective insulin signaling is associated to late cognitive impairment in post‐septic mice. Mol Neurobiol 55: 435–444. [DOI] [PubMed] [Google Scholar]
- Niklowitz WJ, Mandybur TI (1975). Neurofibrillary changes following childhood lead encephalopathy. J Neuropathol Exp Neurol 34: 445–455. [DOI] [PubMed] [Google Scholar]
- Olgiati P, Politis AM, Papadimitriou GN, De Ronchi D, Serretti A (2011). Genetics of late‐onset Alzheimer's disease: update from the alzgene database and analysis of shared pathways. Int J Alzheimer's Dis 2011: 832379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira AM, Hemstedt TJ, Bading H (2012). Rescue of aging‐associated decline in Dnmt3a2 expression restores cognitive abilities. Nat Neurosci 15: 1111–1113. [DOI] [PubMed] [Google Scholar]
- Peter I, Dubinsky M, Bressman S, Park A, Lu C, Chen N et al (2018). Anti‐tumor necrosis factor therapy and incidence of Parkinson disease among patients with inflammatory bowel disease. JAMA Neurol 75: 939–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickering M, Cumiskey D, O'Connor JJ (2005). Actions of TNF‐alpha on glutamatergic synaptic transmission in the central nervous system. Exp Physiol 90: 663–670. [DOI] [PubMed] [Google Scholar]
- Qian L, Wu HM, Chen SH, Zhang D, Ali SF, Peterson L et al (2011). β2‐adrenergic receptor activation prevents rodent dopaminergic neurotoxicity by inhibiting microglia via a novel signaling pathway. J Immunol 186: 4443–4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin LY, Wu XF, Block ML, Liu YX, Bresse GR, Hong JS et al (2007). Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 55: 453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rankin LC, Artis D (2018). Beyond host defense: emerging functions of the immune system in regulating complex tissue physiology. Cell 173: 554–567. [DOI] [PubMed] [Google Scholar]
- Reed‐Geaghan EG, Savage JC, Hise AG, Landreth GE (2009). CD14 and toll‐like receptors 2 and 4 are required for fibrillar A{beta}‐stimulated microglial activation. J Neurosci 29: 11982–11992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romberger DJ, Heires AJ, Nordgren TM, Poole JA, Toews ML, West WW et al (2016). β2‐Adrenergic agonists attenuate organic dust‐induced lung inflammation. Am J Physiol Lung Cell Mol Physiol 311: L101–L110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rondeau JM, Ramage P, Zurini M, Gram H (2015). The molecular mode of action and species specificity of canakinumab, a human monoclonal antibody neutralizing IL‐1β. MAbs 7: 1151–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowan MJ, Klyubin I, Wang Q, Hu NW, Anwyl R (2007). Synaptic memory mechanisms: Alzheimer's disease amyloid beta‐peptide‐induced dysfunction. Biochem Soc Trans 35: 1219–1223. [DOI] [PubMed] [Google Scholar]
- Russo I, Caracciolo L, Tweedie D, Choi SH, Greig NH, Barlati S et al (2012). 3,6′‐Dithiothalidomide, a new TNF‐α synthesis inhibitor, attenuates the effect of Aβ1‐42 intracerebroventricular injection on hippocampal neurogenesis and memory deficit. J Neurochem 122: 1181–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salloway S, Sperling R, Fox NC, Blennow K, Klunk W, Raskind M et al (2014). Two phase 3 trials of bapineuzumab in mild‐to‐moderate Alzheimer's disease. N Engl J Med 370: 322–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samokhina E, Popova I, Malkov A, Ivanov AI, Papadia D, Osypov A et al (2017). Chronic inhibition of brain glycolysis initiates epileptogenesis. J Neurosci Res 95: 2195–2206. [DOI] [PubMed] [Google Scholar]
- Schneider JS, Kidd SK, Anderson DW (2013). Influence of developmental lead exposure on expression of DNA methyltransferases and methyl cytosine‐binding proteins in hippocampus. Toxicol Lett 217: 75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segat L, Pontillo A, Annoni G, Trabattoni D, Vergani C, Clerici M et al (2007). PIN1 promoter polymorphisms are associated with Alzheimer's disease. Neurobiol Aging 28: 69–74. [DOI] [PubMed] [Google Scholar]
- Sherman ML, Spriggs DR, Arthur KA, Imamura K, Frei EF, Kufe DW (1988). Recombinant human tumor necrosis factor administered as a five‐day continuous infusion in cancer patients: phase 1 toxicity and effects on lipid metabolism. J Clin Oncol 6: 344–350. [DOI] [PubMed] [Google Scholar]
- Shi JQ, Shen W, Chen J, Wang BR, Zhong LL, Zhu YW et al (2011). Anti‐TNF‐α reduces amyloid plaques and tau phosphorylation and induces CD11c‐positive dendritic‐like cell in the APP/PS1 transgenic mouse brains. Brain Res 1368: 239–247. [DOI] [PubMed] [Google Scholar]
- Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao L, Luo W et al (2017). ApoE4 markedly exacerbates tau‐mediated neurodegeneration in a mouse model of tauopathy. Nature 549: 523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin EC, Cho SE, Lee DK, Hur MW, Paik SR, Park JH et al (2000). Expression patterns of alpha‐synuclein in human hematopoietic cells and in Drosophila at different developmental stages. Mol Cells 10: 65–70. [DOI] [PubMed] [Google Scholar]
- Simpkins JW, Gatson JW, Wigginton JG (2009). Commentary on “a roadmap for the prevention of dementia II. Leon Thal Symposium 2008.” Rationale and recommendations for first evaluating anti‐Alzheimer's disease medications in acute brain injury patients. Alzheimers Dement 5: 143–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spriggs DR, Sherman ML, Michie H, Arthur KA, Imamura K, Wilmore D et al (1988). Recombinant human tumor necrosis factor administered as a 24‐hour intravenous infusion. A phase 1 and pharmacologic study. J. Natl Cancer Inst 80: 1039–1044. [DOI] [PubMed] [Google Scholar]
- Steinberg GK, Kondziolka D, Bates D (2016a). Response by Steinberg et al to letter regarding article, “Clinical Outcomes of Transplanted Modified Bone Marrow‐Derived Mesenchymal Stem Cells in Stroke: A Phase 1/2A Study”. Stroke 47: e269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg GK, Kondziolka D, Wechsler LR, Lunsford LD, Coburn ML, Billigen JB et al (2016b). Clinical outcomes of transplanted modified bone marrow‐derived mesenchymal stem cells in stroke: a phase 1/2a study. Stroke 47: 1817–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinshamn S, Waage A (2000). Lack of endotoxin tolerance with respect to TNF alpha production in the subarachnoid space. APMIS 108: 107–112. [DOI] [PubMed] [Google Scholar]
- Stellwagen D, Malenka RC (2006). Synaptic scaling mediated by glial TNF‐α. Nature 440: 1054–1059. [DOI] [PubMed] [Google Scholar]
- Stolzenberg E, Berry D, Yang LEY, Kroemer A, Kaufman S et al (2017). A role for neuronal alpha‐synuclein in gastrointestinal immunity. J Innate Immun 9: 456–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strittmatter WJ, Saunders AM, Schmechel D, Pericak‐Vance M, Enghild J, Salvesen GS et al (1993). Apolipoprotein E: high‐avidity binding to beta‐amyloid and increased frequency of type 4 allele in late‐onset familial Alzheimer disease. Proc Natl Acad Sci U S A 90: 1977–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strittmatter WJ, Saunders AM, Goedert M, Weisgraber KH, Dong LM, Jakes R et al (1994). Isoform‐specific interactions of apolipoprotein E with microtubule‐associated protein tau: implications for Alzheimer disease. Proc Natl Acad Sci U S A 91: 11183–11186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultana R, Boyd‐Kimball D, Poon HF, Cai J, Pierce WM, Klein JB et al (2006). Oxidative modification and down‐regulation of Pin1 in Alzheimer's disease hippocampus: a redox proteomics analysis. Neurobiol Aging 27: 918–925. [DOI] [PubMed] [Google Scholar]
- Tahara Y, Otsuka M, Fuse Y, Hirao A, Shibata S (2011). Refeeding after fasting elicits insulin‐dependent regulation of Per2 and Rev‐erbα with shifts in the liver clock. J Biol Rhythms 26: 230–240. [DOI] [PubMed] [Google Scholar]
- Tai LM, Ghura S, Koster KP, Liakaite V, Maienschein‐Cline M, Kanabar P et al (2015). APOE‐modulated Aβ‐induced neuroinflammation in Alzheimer's disease: current landscape, novel data, and future perspective. J Neurochem 133: 465–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi K, Ohuchi T, Kato S, Okabe S (1993). Cytoprotective action of L‐arginine against HCl‐Induced gastric injury in rats – involvement of nitric oxide. Jpn J Pharmacol 61: 13–21. [DOI] [PubMed] [Google Scholar]
- Talbot K, Wang HY (2014). The nature, significance, and glucagon‐like peptide‐1 analog treatment of brain insulin resistance in Alzheimer's disease. Alzheimers Dement 10: S12–S25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot K, Wang HY, Kazi H, Han LY, Bakshi KP, Stucky A et al (2012). Demonstrated brain insulin resistance in Alzheimer's disease patients is associated with IGF‐1 resistance, IRS‐1 dysregulation, and cognitive decline. J Clin Invest 122: 1316–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tate CC, Chou VP, Campos C, Moalem AS, Di Monte DA, McGrogan M et al (2017). Mesenchymal stromal SB623 cell implantation mitigates nigrostriatal dopaminergic damage in a mouse model of Parkinson's disease. J Tissue Eng Regen Med 11: 1835–1843. [DOI] [PubMed] [Google Scholar]
- Taupin V, Toulmond S, Serrano A, Benavides J, Zavala F (1993). Increase in IL‐6, IL‐1 and TNF levels in rat brain following traumatic lesion. Influence of pre‐ and post‐traumatic treatment with Ro5 4864, a peripheral‐type (p site) benzodiazepine ligand. J Neuroimmunol 42: 177–185. [DOI] [PubMed] [Google Scholar]
- Taylor MP, Schniering CA, Lanphear BP, Jones AL (2011). Lessons learned on lead poisoning in children: one‐hundred years on from Turner's declaration. J Paediatr Child Health 47: 849–856. [DOI] [PubMed] [Google Scholar]
- Thambisetty M, Simmons A, Velayudhan L, Hye A, Campbell J, Zhang Y et al (2010). Association of plasma clusterin concentration with severity, pathology, and progression in Alzheimer's disease. Arch Gen Psychiatry 67: 739–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobinick E (2009). Tumour necrosis factor modulation for treatment of Alzheimer's disease: rationale and current evidence. CNS Drugs 23: 713–725. [DOI] [PubMed] [Google Scholar]
- Tobinick E (2010). Perispinal etanercept: a new therapeutic paradigm in neurology. Expert Rev Neurother 10: 985–1002. [DOI] [PubMed] [Google Scholar]
- Tobinick E (2011). Rapid improvement of chronic stroke deficits after perispinal etanercept: three consecutive cases. CNS Drugs 25: 145–155. [DOI] [PubMed] [Google Scholar]
- Tobinick E (2018). Perispinal etanercept advances as a neurotherapeutic. Expert Rev Neurother 18: 453–455. [DOI] [PubMed] [Google Scholar]
- Tobinick E, Vega CP (2006). The cerebrospinal venous system: anatomy, physiology, and clinical implications. MedGenMed 8: 53. [PubMed] [Google Scholar]
- Tobinick E, Kim NM, Reyzin G, Rodriguez‐Romanacce H, DePuy V (2012). Selective TNF inhibition for chronic stroke and traumatic brain injury: an observational study involving 629 consecutive patients treated with perispinal etanercept. CNS Drugs 26: 1051–1070. [DOI] [PubMed] [Google Scholar]
- Tobinick E, Rodriguez‐Romanacce H, Levine A, Ignatowski TA, Spengler RN (2014). Immediate neurological recovery following perispinal etanercept years after brain injury. Clin Drug Investig 34: 361–366. [DOI] [PubMed] [Google Scholar]
- Tobinick EL (2016). Perispinal delivery of CNS drugs. CNS Drugs 30: 469–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobinick EL, Gross H (2008). Rapid cognitive improvement in Alzheimer's disease following perispinal etanercept administration. J Neuroinflammation 5: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobinick EL, Gross H, Weinberger A, Cohen H (2006). TNF‐alpha modulation for treatment of Alzheimer's disease: a 6‐month pilot study. MedGenMed Neurol Neurosurg 8: 25. [PMC free article] [PubMed] [Google Scholar]
- Tubbs RS, Demerdash A, Loukas M, Cure J, Oskouian RJ, Ansari S et al (2018). Intracranial connections of the vertebral venous plexus: anatomical study with application to neurosurgical and endovascular procedures at the craniocervical junction. Oper Neurosurg (Hagerstown, Md) 14: 51–57. [DOI] [PubMed] [Google Scholar]
- Turner AJ (1909). On lead poisoning in children. Br Med J 1: 895–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uc EY, Lambert CP, Harik SI, Rodnitzky RL, Evans WJ (2003). Albuterol improves response to levodopa and increases skeletal muscle mass in patients with fluctuating Parkinson disease. Clin Neuropharmacol 26: 207–212. [DOI] [PubMed] [Google Scholar]
- Ulland TK, Song WM, Huang SC, Ulrich JD, Sergushichev A, Beatty WL et al (2017). TREM2 maintains microglial metabolic fitness in Alzheimer's disease. Cell 170: 649–663.e613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich JD, Ulland TK, Colonna M, Holtzman DM (2017). Elucidating the role of TREM2 in Alzheimer's disease. Neuron 94: 237–248. [DOI] [PubMed] [Google Scholar]
- van Zoelen MA, Yang H, Florquin S, Meijers JC, Akira S, Arnold B et al (2009). Role of toll‐like receptors 2 and 4, and the receptor for advanced glycation end products in high‐mobility group box 1‐induced inflammation in vivo. Shock 31: 280–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitek MP, Brown CM, Colton CA (2009). APOE genotype‐specific differences in the innate immune response. Neurobiol Aging 30: 1350–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Liu Y, Chan WY, Chan SO, Grunz H, Zhao H (2011). Characterization of three synuclein genes in Xenopus laevis. Dev Dyn 240: 2028–2033. [DOI] [PubMed] [Google Scholar]
- Wang QW, Wu JQ, Rowan MJ, Anwyl R (2005). Beta‐amyloid inhibition of long‐term potentiation is mediated via tumor necrosis factor. Eur J Neurosci 22: 2827–2832. [DOI] [PubMed] [Google Scholar]
- Wendeln AC, Degenhardt K, Kaurani L, Gertig M, Ulas T, Jain G et al (2018). Innate immune memory in the brain shapes neurological disease hallmarks. Nature 556: 332–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White LD, Cory‐Slechta DA, Gilbert ME, Tiffany‐Castiglioni E, Zawia NH, Virgolini M et al (2007). New and evolving concepts in the neurotoxicology of lead. Toxicol Appl Pharmacol 225: 1–27. [DOI] [PubMed] [Google Scholar]
- Wright AL, Zinn R, Hohensinn B, Konen LM, Beynon SB, Tan RP et al (2013). Neuroinflammation and neuronal loss precede Aβ plaque deposition in the hAPP‐J20 mouse model of Alzheimer's disease. PLoS One 8: e59586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarchoan M, Toledo JB, Lee EB, Arvanitakis Z, Kazi H, Han LY et al (2014). Abnormal serine phosphorylation of insulin receptor substrate 1 is associated with tau pathology in Alzheimer's disease and tauopathies. Acta Neuropathol 128: 679–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, He J, Li S, Song L, Guo X, Yao W et al (2016). Fibroblast growth factor 21 (FGF21) inhibits macrophage‐mediated inflammation by activating Nrf2 and suppressing the NF‐κB signaling pathway. Int Immunopharmacol 38: 144–152. [DOI] [PubMed] [Google Scholar]
- Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG et al (2012). Genomic analysis of reactive astrogliosis. J Neurosci 32: 6391–6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Cai T, Zhao F, Yao T, Chen Y, Liu X et al (2012). The role of alpha‐synuclein and tau hyperphosphorylation‐mediated autophagy and apoptosis in lead‐induced learning and memory injury. Int J Biol Sci 8: 935–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilberter Y, Zilberter M (2017). The vicious circle of hypometabolism in neurodegenerative diseases: ways and mechanisms of metabolic correction. J Neurosci Res 95: 2217–2235. [DOI] [PubMed] [Google Scholar]