

Abstract
The efficient and mild copper-catalyzed synthesis of unsymmetrical diorganyl chalcogenides under ligand- and solvent-free conditions is described. The cross-coupling reaction was performed using aryl boric acids and 0.5 equiv. of diorganyl dichalcogenides (Te/Se/S) in the presence of 3 mol % of CuI and 3 equiv. of DMSO, under microwave irradiation. This new protocol allowed the preparation of several unsymmetrical diorganyl chalcogenides in good to excellent yields.
Keywords: selenide, telluride, boronic acid, solvent-free, cross-coupling, CuI, selenium, tellurium
1. Introduction
Transition metal-catalyzed coupling reactions are among the most commonly applied protocols for the preparation of various target molecules under mild conditions [1,2]. In this regard, the construction of new carbon-carbon and carbon-heteroatom bonds have been described, notably through catalytic processes employing a metal as the catalyst [3,4,5,6,7].
In addition, catalytic transformations have been widely applied in organochalcogen chemistry for the preparation of diorganyl-tellurides, -selenides and -sulfides, which are interesting target molecules and valuable synthetic intermediates for several transformations in modern organic synthesis [8,9,10]. Besides the synthetic applications, these types of molecules have shown relevant biological properties, such as antitumor, antioxidant, antiviral, antimicrobial and neuroprotective effects [11,12,13,14,15,16,17,18,19]. Organochalcogen compounds also have noteworthy applications in materials science [20,21,22].
Due to their considerable importance, the development of new and greener methods for the preparation of organochalcogenides (S, Se and Te) has been widely investigated by several research groups, and a number of methodologies have been reported [23,24,25,26,27,28,29,30]. The most common procedures described for the preparation of alkyl/aryl chalcogenides involve: (a) the reaction between alkyl/aryl dichalcogenide precursors and organometallic reagents [31,32,33,34]; (b) the transition metal-catalyzed reaction of halides with chalcogenols or diorganyl dichalcogenides in the presence of a base or reducing agents [35,36,37]; and (c) synthesis through the C–H activation of arenes [38,39,40,41].
On the other hand, organoboronic acids have been used as suitable reagents for various cross-coupling reactions since they are stable, readily available and compatible with different functional groups. They can also be employed in the boron-to-metal exchange reaction using a catalytic amount of metal [42,43,44]. Thus, organoboronic acids have become interesting and appropriate alternative compounds for the preparation of symmetrical and unsymmetrical diaryl organochalcogenides via cross-coupling transformations.
In this regard, Wang and Tanigushi, independently reported the preparation of diorganyl chalcogenides from organoboronic acids and diorganyl dichalcogenides using copper as a catalyst, in the presence of a ligand and solvent [45,46]. Since their reports, modern studies have been described to improve this kind of transformation. In particular, metals have been efficiently applied as catalysts for the cross-coupling of diorganyl chalcogenides with boronic acids, for instance, employing indium [47], iron [48], copper [49,50,51,52,53,54,55] and silver [56].
However, most of these procedures have their particular drawbacks, such as the use of ligands, reducing agents, air sensitivity, long reaction time, and/or the use of toxic solvents. In addition, some of these methods are not applicable for the synthesis of unsymmetrical organotellurides, which are also very important from the synthetic point of view.
Alternatively, we have recently reported suitable methods for the synthesis of unsymmetrical chalcogenides, using the iodine/DMSO system under microwave irradiation to generate, in situ, electrophilic species of chalcogenyl iodide (RYI) [57]. Similarly, Park and coworkers reported an ultrasound-assisted synthesis of diaryl ditellurides [58]. Nonetheless, the development of new methodologies employing boronic acids and dichalcogenides as starting materials, associated with metal catalysis instead of use RYI species, is still highly desirable. Furthermore, the use of diorganyl dichalcogenides avoids the use of toxic RXH compounds (X = Te, Se, S).
Thus, in connection with our continuing interest in designing and developing eco-friendly processes and cross-coupling reactions, [59,60,61,62,63,64], herein we describe a straightforward method for the synthesis of unsymmetrical diorganyl-chalcogenides (S, Se and Te) through the reaction of boronic acids and half equiv. of diorganyl dichalcogenides, employing CuI as a catalyst, under solvent- and ligand-free conditions (Scheme 1).
Scheme 1.

Solvent- and ligand-free synthesis of unsymmetrical diorganyl chalcogenides catalyzed by CuI.
2. Results and Discussion
In order to optimize the reaction conditions, diphenyl ditelluride (1a) and 4-methoxyphenyl boronic acid (2a) were used as model substrates (Table 1). Firstly, we evaluated the catalyst loading in the reaction system (entries 1–4). When the reaction was carried out with 1.0 mol % of catalyst the desired product was obtained with only 43% yield (entry 1). However, on increasing the catalyst to 2.0 mol %, the yield of the reaction increased to 71% (entry 2). Notably, when the amount of CuI was increased to 3.0 mol % the corresponding product 3a was achieved with 90% yield (entry 3). No change in the yield was observed when the catalyst loading was increased to 4.0 mol % (entry 4).
Table 1.
Optimization of reaction conditions a.
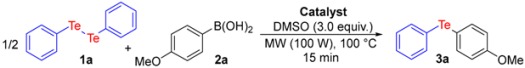
| Entry | Catalyst | Amount | Yield (%) b,c |
|---|---|---|---|
| 1 | CuI | 1.0 mol % | 43 |
| 2 | CuI | 2.0 mol % | 71 |
| 3 | CuI | 3.0 mol % | 90 |
| 4 | CuI | 4.0 mol % | 90 |
| 5 | CuBr | 3.0 mol % | 56 |
| 6 | CuCl | 3.0 mol % | 67 |
| 7 | CuCl2 | 3.0 mol % | 72 |
| 8 | Cu(OAc)2 | 3.0 mol % | 46 |
| 9 | CuO | 3.0 mol % | 61 |
| 10 | nano-CuO | 3.0 mol % | 65 |
| 11 | I2 | 3.0 mol % | 69 |
| 12 | - | - | Traces |
a Reaction conditions: 1a (0.25 mmol), 2a (2.0 equiv.), DMSO (3.0 equiv.) under MW irradiation; b Isolated yield; c This reaction works similarly in 0.125 mmol scale with less reproducibility.
Next, we investigated the influence of the copper source on the reaction system (entries 5–10). In general, copper halides showed better catalytic activity. For instance, when we employed CuCl2 and CuO under the same reaction conditions, the desired organotelluride was synthesized in yields of 72 and 61%, respectively (entries 7 vs. 9). Furthermore, CuO nanoparticles were less effective than copper iodide, affording the desired product in lower yield (entry 10). Considering our previous work [57], when 3% molecular iodine was used as catalyst, 3a was obtained in 69% yield (entry 11). The reaction in the absence of catalyst provided 3a in only trace amounts, which emphasizes the notable activity of CuI in this kind of transformation (entry 12).
With the best catalyst in hand, we investigated the influence of other reaction parameters (temperature, time and power) as well as the effect of the additive/oxidant (Table 2).
Table 2.
Optimization of reaction conditions a.
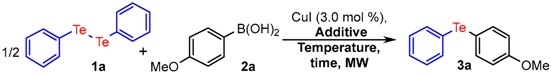
| Entry | Additive (equiv.) | MW (W) | T (°C) | Time (min) | Yield (%) b |
|---|---|---|---|---|---|
| 1 | DMSO (3.0) | 100 | 100 | 15 | 90 |
| 2 | CH3CN (3.0) | 100 | 100 | 15 | 35 |
| 3 | EtOH (3.0) | 100 | 100 | 15 | 25 |
| 4 | Dioxane (3.0) | 100 | 100 | 15 | 40 |
| 5 | H2O (3.0) | 100 | 100 | 15 | - |
| 6 | TBHP (3.0) | 100 | 100 | 15 | - |
| 7 | - | 100 | 100 | 15 | 10 |
| 8 c | DMSO (3.0) | 100 | 100 | 15 | 29 |
| 9 | DMSO (2.0) | 100 | 100 | 15 | 78 |
| 10 | DMSO (4.0) | 100 | 100 | 15 | 90 |
| 11 | DMSO (3.0) | 100 | 100 | 10 | 73 |
| 12 | DMSO (3.0) | 100 | 100 | 20 | 88 |
| 13 | DMSO (3.0) | 100 | 80 | 15 | 68 |
| 14 | DMSO (3.0) | 100 | 120 | 15 | 77 |
| 15 | DMSO (3.0) | 120 | 100 | 15 | 86 |
| 16 | DMSO (3.0) | 80 | 100 | 15 | 70 |
| 17 d | DMSO (3.0) | - | 100 | 24 h | 57 |
a Reaction conditions: 1a (0.25 mmol), 2a (2.0 equiv.), CuI (3.0 mol %), DMSO (3.0 equiv.) under MW irradiation; b Isolated yield; c Reaction under argon atmosphere; d Conventional heating.
In this regard, we initially evaluated the influence of a series of additives (CH3CN, 1,4-dioxane, EtOH, H2O and TBHP). Of these, DMSO was established as the best option, providing the desired product in very high yield in comparison with other additives (entry 1 vs. 2–6). Furthermore, a considerable decrease in the yield was observed when the reaction was carried out either in the absence of an additive or under inert atmosphere, highlighting the need for an oxidant medium for this cross-coupling transformation (entries 7 and 8). In relation to the loading of DMSO, it was observed that 3.0 equiv. was the best alternative, since no improvement in the yield was observed when the amount of this additive was modified (entries 9 and 10).
The influence of the reaction time and temperature was also evaluated (entries 11–14). The screening of these parameters revealed that 15 min and 100 °C were the most appropriate choice for this protocol, as seen in entry 1.
Finally, the effect of the radiation power of the microwave was investigated and increasing the power to 120 W did not change the reaction yield significantly (entry 15). However, when the level of power was decreased to 80 W the desired product 3a was delivered in lower yield (entry 16). In addition, we carried out the reaction under conventional heating and, even after a long reaction time, a decrease in the yield of the product 3a was observed (entry 17). This result indicates that microwave irradiation offers advantages over conventional heating, for this transformation.
Having determined the best reaction conditions, we explored the scope of our protocol by employing various diorganyl ditellurides 1 and arylboronic acids 2 (Scheme 2). Firstly, we evaluated the effect of different groups attached to the aryl ring of boronic acid (Scheme 2).
Scheme 2.

Synthesis of unsymmetrical organotellurides a,b. a Reaction conditions: 1 (0.25 mmol), 2 (0.5 mmol) in the presence of CuI (3.0 mol %) and DMSO (3.0 equiv.) applied for 15 min at 100 °C with 100 W of MW-irradiation; b Isolated yields.
The electronic characteristics of the substituent of boronic acid attached to the aromatic ring (electron donor or acceptor) and its steric hindrance did not appear to affect the performance of the transformation, and the corresponding products 3a–e were obtained in very good yields. Also, 2-naphthylboronic acid reacted very well with diphenyl ditelluride (1a) under the optimized conditions, affording the corresponding product 3f in very good yield.
We further evaluated the generality of the reaction regarding the ditellurides 1, employing electron-withdrawing or electron-donating groups. In general, the reaction proceeded very well in both cases, furnishing the corresponding unsymmetrical tellurides 3g–j in 83–87% yields. For instance, the reaction of para-chlorobenzene ditelluride with meta-substituted aryl boronic acid delivered the respective compound 3j in 83% yield. Moreover, an aliphatic ditelluride, in this case dibutyl ditelluride, was also a suitable substrate for this cross-coupling process, furnishing the product 3k in 87% yield.
The successful preparation of unsymmetrical diorganyl tellurides 3 by a copper-catalyzed transformation prompted us to expand this approach to the synthesis of unsymmetrical diorganyl selenides (Scheme 3). To evaluate the electronic effects, we first verified the influence of the substituent attached at the para position of the aromatic ring of the boronic acid. The treatment of diphenyl diselenide with different para-substituted aryl boronic acids provided the respective products 6a and 6b in 87% and 85% yields, respectively.
Scheme 3.

Synthesis of unsymetrical organoselenides and sulfides a,b. a Reaction conditions: 4 or 5 (0.25 mmol), 2 (0.5 mmol) in the presence of CuI (3.0 mol %) and DMSO (3.0 equiv.) for 15 min at 100 °C and 100 W of MW-irradiations; b Isolated yields.
Similarly, the reaction was not sensitive to steric effects, since the treatment of diphenyl diselenide with aryl boronic acid containing a methyl group at the ortho position of the aromatic ring provided the respective product 6c in 89% yield. Subsequently, a series of different diselenides, including aliphatic and aromatics diselenides, were converted into the corresponding products in very good yields. For example, when we reacted p-chlorobenzene diselenide with 4-methoxyphenylboronic acid the product 6e was obtained in 89% yield.
We also evaluated the applicability of our protocol to the preparation of unsymmetrical sulfides, under the same reaction conditions. Remarkably, diphenyl disulfide and m-chlorophenyl disulfide reacted smoothly with p-methoxyphenyl boronic acid, leading to the respective products 7a and 7b in 71% and 68% yields, respectively. This slight decrease in the yield values could be associated with the lower reactivity of diaryl disulfides when compared to ditellurides or diselenides analogues, which are much more easily cleaved than disulfides [65].
In addition, in order to investigate the synthetic utility of this methodology, we evaluated whether the reaction could be performed with boronic acid at the gram scale under the optimized conditions (Scheme 4). To our delight, the corresponding product 3a was obtained in 85% yield, which is very significant from a synthesis point of view, since this methodology can thus be used to prepare unsymmetrical diorganyl chalcogenides on a larger scale.
Scheme 4.

Scale-Up of the reaction.
In order to gain further insight into the reaction mechanism, control experiments were performed (Scheme 5). Firstly, to evaluate the possibility of a radical path, we performed the standard reaction in the presence of 3.0 equiv. of 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) as a radical inhibitor (Scheme 5a). The presence of TEMPO did not affect the reaction yield, which suggests that radical intermediates are not involved in this protocol. Next, we verified the influence of oxidant species on this transformation. When the reaction was carried out under argon atmosphere, an abrupt decrease was noted and the telluride 3a was delivered in only 29% yield (Scheme 5b). In contrast, when diphenyl ditelluride was treated with phenyl boronic under O2 atmosphere, the corresponding product was obtained in 93% yield (Scheme 5c). These results suggest that oxygen is mainly responsible for the oxidation steps and DMSO is most likely an important additive in terms of improving the reaction yield.
Scheme 5.

Control experiments for reaction mechanism. (a) Reaction in the presence of radical inhibitor; (b) Reaction under inert atmosphere; (c) Reaction under oxygen atmosphere.
On the basis of these results and in accordance with previous reports [66,67], a plausible reaction pathway is illustrated in Scheme 6. In this pathway the catalyst cycle begins with the generation of species A through the reaction between CuI and boronic acid [66]. Subsequently, this specie reacts with diorganyl dichalcogenide to afford the desired product and the intermediate B. The specie B is then oxidized to give the Cu(II) intermediate C [66,67]. In the next step, another equiv. of boronic acid would reacts with this intermediate to furnish the intermediate D. Lastly, a second equiv. of the desired product is obtained through an elimination reaction step and, consequently, the catalyst is regenerated.
Scheme 6.

A plausible reaction pathway.
3. Materials and Methods
3.1. General Information
1H- and 13C-NMR spectra were obtained at 200/50 MHz on an AC-200 NMR spectrometer (Bruker, Rheinstetten, Germany) or at 400/100 MHz on an AS-400 NMR spectrometer (Varian, Palo Alto, CA, USA). Spectra were recorded in CDCl3 solutions. Chemical shifts are reported in ppm, referenced to the solvent peak of CDCl3 or tetramethylsilane (TMS) as the external reference. Data are reported as follows: Chemical shift (δ), multiplicity, coupling constant (J) in hertz and integrated intensity. Abbreviations to denote the multiplicity of a particular signal are: s (singlet), d (doublet), t (triplet), q (quartet), quint (quintet), sext (sextet) and m (multiplet). The NMR spectra are found in the Supplementary Materials. The melting points were determined using a model MQRPF-301 digital apparatus (Microquimica, Palhoça, Brazil) equipped with a heating plate. Column chromatography was performed using silica gel (230–400 mesh). Thin layer chromatography (TLC) was performed using silica gel GF254 plates with 0.25 mm thickness (Merck, Darmstadt, Germany). For visualization, TLC plates were either placed under ultraviolet light or stained with iodine vapor and acidic vanillin. Most reactions were monitored by TLC for the disappearance of the starting material.
Unless otherwise stated, all reactions were carried out in an open atmosphere. All reagents and solvents were obtained from commercial sources and used without further purification. All reactions were performed in 10 mL sealed glass tubes in a commercially available monomode microwave reactor (CEM, Matthews, NC, USA) with IR monitoring and a non-invasive pressure transducer. Reagents and solvents were handled using standard syringe techniques. The yields are based on isolated compounds after purification.
3.2. General Procedure for the Synthesis of Unsymmetrical Organochalcogenides under MW Irradiation
A mixture of aryl boronic acid (0.5 mmol), dichalcogenide (0.25 mmol), CuI (3 mol %, 1.5 mg), and DMSO (3 equiv., 59 mg) was placed in a glass tube. The tube was sealed with a pressure lock and the mixture was heated in air at 100 °C for 15 min with the aid of an initial MW irradiation of 100 W in a CEM Discover MW reactor. When the reaction was finished, the crude mixture was cooled to room temperature, diluted with ethyl acetate (10 mL), and washed with water (3 × 5 mL). The organic phase was separated, dried over MgSO4, and concentrated under vacuum. The residue was purified by flash chromatography on silica gel using hexane or a mixture of ethyl hexane/acetate (99:1) as the eluent.
Analytical Data of Products 3a–k, 6a–h and 7a,b
(4-Methoxyphenyl)(phenyl)tellane (3a). Yield: 0.140 g (90%); white solid; m.p.: 59–61 °C (lit.: [48] 60–62 °C); 1H-NMR (200 MHz, CDCl3, ppm) δ = 7.73 (d, J = 8.8 Hz, 2H, H-Ar), 7.62–7.42 (m, 2H, H-Ar), 7.30–7.07 (m, 3H, H-Ar), 6.80 (d, J = 8.8 Hz, 2H, H-Ar), 3.80 (s, 3H, -OCH3); 13C-NMR (50 MHz, CDCl3, ppm) δ = 160.1, 141.3, 136.5, 129.4, 127.4, 116.0, 115.6, 103.3, 55.3.
(4-Chlorophenyl)(phenyl)tellane (3b). Yield: 0.133 g (84%); yellow oil; [68] 1H-NMR (200 MHz, CDCl3, ppm) δ = 7.63–7.57 (m, 2H, H-Ar), 7.52–7.46 (m, 2H, H-Ar), 7.21–7.05 (m, 5H, H-Ar); 13C-NMR (50 MHz, CDCl3, ppm) δ = 139.3, 138.2, 134.4, 129.8, 129.7, 128.2, 114.5, 112.5.
3-(Phenyltellanyl)aniline (3c). Yield: 0.128 g (86%); yellow oil; [57] 1H-NMR (200 MHz, CDCl3, ppm) δ = 7.57 (d, J = 8.4 Hz, 2H, H-Ar), 7.56–6.99 (m, 6H, H-Ar), 6.60–6.55 (m, 1H, H-Ar), 3.62 (s, 2H, -NH2); 13C-NMR (50 MHz, CDCl3, ppm) δ = 147.3, 138.0, 130.3, 129.6, 128.2, 127.8, 124.4, 115.4, 114.9, 114.8.
(3-Nitrophenyl)(phenyl)tellane (3d). Yield: 0.137 g (84%); yellow oil; [55] 1H-NMR (400 MHz, CDCl3, ppm) δ = 8.42 (s, 1H, H-Ar), 8.06 (d, J = 8.2 Hz, 1H, H-Ar), 7.86–7.80 (m, 3H, H-Ar), 7.41–7.25 (m, 4H, H-Ar); 13C-NMR (101 MHz, CDCl3, ppm) δ = 148.5, 142.4, 139.6, 131.1, 130.1, 130.0, 129.1, 122.5, 117.0, 113.2.
(2-Methoxyphenyl)(phenyl)tellane (3e). Yield: 0.140 g (90%); white solid; m.p.: 53–55 °C (lit.: [48] 53–54 °C); 1H-NMR (200 MHz, CDCl3, ppm) δ = 7.89 (dd, J = 8.1, 1.4 Hz, 2H, H-Ar), 7.44–7.25 (m, 3H, H-Ar), 7.17 (ddd, J = 8.2, 7.3, 1.6 Hz, 1H, H-Ar), 6.94 (dd, J = 7.6, 1.6 Hz, 1H, H-Ar), 6.80–6.69 (m, 2H, H-Ar), 3.86 (s, 3H, -OCH3); 13C-NMR (50 MHz, CDCl3, ppm) δ = 158.2, 141.3, 133.7, 129.7, 128.7, 128.2, 122.5, 112.2, 109.8, 107.8, 56.0.
Naphthalen-2-yl(phenyl)tellane (3f). Yield: 0.146 g (88%); yellow oil; [55] 1H-NMR (200 MHz, CDCl3, ppm) δ = 8.19–8.14 (m, 1H, H-Ar), 7.97 (dd, J = 7.1, 1.1 Hz, 1H, H-Ar), 7.82–7.74 (m, 2H, H-Ar), 7.62–7.57 (m, 2H, H-Ar), 7.51–7.44 (m, 2H, H-Ar), 7.28–7.08 (m, 4H, H-Ar); 13C-NMR (101 MHz, CDCl3, ppm) δ = 138.9, 137.5, 135.9, 133.8, 131.8, 129.6, 128.9, 127.8, 127.1, 126.6, 126.4, 117.9, 114.9.
1-(4-((4-Methoxyphenyl)tellanyl)phenyl)ethan-1-one (3g). Yield: 0.152 g (86%); yellow oil; [57] 1H-NMR (200 MHz, CDCl3, ppm) δ = 7.78 (d, J = 8.8 Hz, 2H, H-Ar), 7.68 (d, J = 8.5 Hz, 2H, H-Ar), 7.48 (d, J = 8.5 Hz, 2H, H-Ar), 6.83 (d, J = 8.8 Hz, 2H, H-Ar), 3.82 (s, 3H, -OCH3), 2.52 (s, 3H, -COCH3); 13C-NMR (50 MHz, CDCl3, ppm) δ = 197.5, 160.4, 142.2, 135.5, 134.5, 128.6, 125.5, 115.8, 102.1, 55.2, 26.4.
(3-Nitrophenyl)(p-tolyl)tellane (3h). Yield: 0.142 g (83%); yellow oil; [57] 1H-NMR (200 MHz, CDCl3, ppm) δ = 8.41–8.29 (m, 1H, H-Ar), 8.06–8.01 (m, 1H, H-Ar), 7.82–7.72 (m, 3H, H-Ar), 7.30 (td, J = 8.3, 2.2 Hz, 1H, H-Ar), 7.12 (d, J = 7.1 Hz, 2H, H-Ar), 2.38 (s, 3H, -CH3); 13C-NMR (50 MHz, CDCl3, ppm) δ = 148.5, 141.8, 140.1, 139.5, 131.0, 130.4, 129.8, 122.2, 117.6, 109.1, 21.4.
1-(4-((4-Chlorophenyl)tellanyl)phenyl)ethanone (3i). Yield: 0.156 g (87%); yellow solid; m.p.: 67–68 °C (lit.: [57] 67–70 °C); 1H-NMR (200 MHz, CDCl3, ppm) δ = 7.76–7.72 (m, 4H, H-Ar), 7.61 (d, J = 7.9 Hz, 2H, H-Ar), 7.24 (d, J = 8.4 Hz, 2H, H-Ar), 2.56 (s, 3H, -COCH3); 13C-NMR (50 MHz, CDCl3, ppm) δ = 197.6, 140.9, 139.5, 136.3, 136.3, 136.2, 135.5, 130.3, 129.0, 123.5, 111.1, 26.6.
3-((4-Chlorophenyl)tellanyl)aniline (3j). Yield: 0.137 g (83%); brown solid; m.p.: 60–61 °C (lit.: [57] 58–60 °C); 1H-NMR (200 MHz, CDCl3, ppm) δ = 7.58 (d, J = 8.2 Hz, 2H, H-Ar), 7.18–6.95 (m, 5H, H-Ar), 6.59 (dt, J = 7.5, 1.9 Hz, 1H, H-Ar), 3.62 (s, 2H, -NH2); 13C-NMR (50 MHz, CDCl3, ppm) δ = 147.4, 139.2, 134. 2, 130.4, 129.7, 128.2, 124.4, 115.0, 112.6.
1-(4-(Butyltellanyl)phenyl)ethan-1-one (3k). Yield: 0.127 g (87%); yellow oil; [69] 1H-NMR (200 MHz, CDCl3, ppm) δ = 7.67 (d, J = 8.8 Hz, 2H, H-Ar), 6.76 (d, J = 8.8 Hz, 2H, H-Ar), 3.79 (s, 3H, -OCH3), 2.86–2.78 (m, 2H, -CH2-), 1.73 (p, J = 7.9, 7.4 Hz, 2H, -CH2-), 1.46–1.287 (m, 2H, -CH2-), 0.92–0.851 (m, 3H, -CH3); 13C-NMR (101 MHz, CDCl3, ppm) δ = 159.7, 141.0, 115.2, 100.7, 55.2, 34.0, 25.1, 13.5, 8.9.
(4-Methoxyphenyl)(phenyl)selane (6a). Yield: 0.114 g (87%); yellow oil; [54] 1H-NMR (200 MHz, CDCl3, ppm) δ = 7.51 (d, J = 8.9 Hz, 2H, H-Ar), 7.35–7.18 (m, 5H, H-Ar), 6.86 (d, J = 8.8 Hz, 2H, H-Ar), 3.81 (s, 3H, -OCH3); 13C-NMR (50 MHz, CDCl3, ppm) δ = 159.7, 136.5, 133.1, 130.9, 129.1, 126.4, 119.9, 115.1, 55.3.
(4-Chlorophenyl)(phenyl)selane (6b). Yield: 0.114 g (85%); transparent oil; [68] 1H-NMR (200 MHz, CDCl3, ppm) δ = 7.47–7.44 (m, 2H, H-Ar), 7.35 (d, J = 8.3 Hz, 2H, H-Ar), 7.27–7.18 (m, 5H, H-Ar); 13C-NMR (50 MHz, CDCl3, ppm) δ = 134.2, 133.6, 133.3, 130.8, 129.7, 129.6, 127.7.
Phenyl(o-tolyl)selane (6c). Yield: 0.110 g (89%); transparent oil; [70] 1H-NMR (200 MHz, CDCl3, ppm) δ = 7.42–7.36 (m, 2H, H-Ar), 7.34–7.14 (m, 6H, H-Ar), 7.09–7.01 (m, 1H, H-Ar), 2.40 (s, 3H, -CH3); 13C-NMR (50 MHz, CDCl3, ppm) δ = 139.9, 133.8, 132.8, 131.8, 130.9, 130.3, 129.4, 127.8, 127.2, 126.8, 22.4.
Naphthalen-2-yl(phenyl)selane (6d). Yield: 0.125 g (88%); yellow solid; m.p.: 68–70°C (lit.: [57] 69–71 °C); 1H-NMR (200 MHz, CDCl3, ppm) δ = 8.35–8.30 (m, 1H, H-Ar), 7.84–7.74 (m, 3H, H-Ar), 7.51–7.15 (m, 8H, H-Ar); 13C-NMR (50 MHz, CDCl3, ppm) δ = 134.2, 133.9, 131.8, 129.4, 129.3, 128.7, 127.8, 127.1, 126.9, 126.5, 126.1.
(4-Chlorophenyl)(4-methoxyphenyl)selane (6e). Yield: 0.132 g (89%); yellow solid; m.p.: 56–57 °C (lit.: [48] 58–59 °C); 1H-NMR (200 MHz, CDCl3, ppm) δ = 7.41 (d, J = 8.8 Hz, 2H, H-Ar), 7.18–7.06 (m, 4H, H-Ar), 6.77 (d, J = 8.8 Hz, 2H, H-Ar), 3.72 (s, 3H, -OCH3); 13C-NMR (50 MHz, CDCl3, ppm) δ = 160.0, 136.6, 132.5, 132.1, 131.6, 129.2, 119.5, 115.2, 55.3.
(4-Chlorophenyl)(o-tolyl)selane (6f). Yield: 0.117 g (83%); transparent oil; [57] 1H-NMR (200 MHz, CDCl3, ppm) δ = 7.36–7.03 (m, 8H, H-Ar), 2.37 (s, 3H, -CH3); 13C-NMR (50 MHz, CDCl3, ppm) δ = 140.2, 134.1, 133.8, 133.4, 131.3, 130.5, 129.6, 129.4, 128.3, 127.0, 22.5.
(4-Chlorophenyl)(o-tolyl)selane (6g). Yield: 0.130 g (89%); transparent oil; [57] 1H-NMR (200 MHz, CDCl3, ppm) δ = 7.55 (d, J = 8.8 Hz, 2H, H-Ar), 7.16–7.05 (m, 1H, H-Ar), 6.88 (d, J = 8.8 Hz, 2H, H-Ar), 6.82–6.72 (m, 3H, H-Ar), 3.88 (s, 3H, -OCH3), 3.81 (s, 3H, -OCH3); 13C-NMR (50 MHz, CDCl3, ppm) δ = 160.3, 156.0, 138.5, 129.1, 127.0, 123.6, 121.7, 117.4, 115.4, 110.2, 55.9, 55.4.
Butyl(4-methoxyphenyl)selane (4-chlorophenyl)(o-tolyl)selane (6h). Yield: 0.094 g (77%); yellow oil; [48] 1H-NMR (200 MHz, CDCl3, ppm) δ = 7.46 (d, J = 8.9 Hz, 2H), 6.81 (d, J = 8.8 Hz, 2H), 3.79 (s, 3H, -OCH3), 2.82 (t, J = 7.6 Hz, 2H, -CH2-), 1.71–1.576 (m, 2H, -CH2-), 1.49–1.34 (m, 2H, -CH2-), 0.89 (t, J = 7.2 Hz, 3H, -CH3); 13C-NMR (50 MHz, CDCl3, ppm) δ = 159.2, 135.6, 120.4, 114.8, 55.4, 32.5, 29.0, 23.0, 13.7.
(4-Methoxyphenyl)(phenyl)sulfane (7a). Yield: 0.077 g (71%); transparent oil; [68] 1H-NMR (200 MHz, CDCl3, ppm) δ = 7.42 (d, J = 8.9 Hz, 2H, H-Ar), 7.28–7.15 (m, 5H, H-Ar), 6.90 (d, J = 8.9 Hz, 2H, H-Ar), 3.82 (s, 3H, -OCH3); 13C-NMR (50 MHz, CDCl3, ppm) δ = 160.0, 138.7, 135.5, 129.0, 128.3, 125.9, 124.5, 115.1, 55.5.
(3-Chlorophenyl)(4-methoxyphenyl)sulfane (7b). Yield: 0.085 g (68%); white solid; m.p.: 59–61 °C (lit.: [57] 59–61 °C); 1H-NMR (400 MHz, CDCl3, ppm) δ = 7.42 (d, J = 8.9 Hz, 2H, H-Ar), 7.13–7.03 (m, 3H, H-Ar), 6.98 (dt, J = 7.7, 1.3 Hz, 1H, H-Ar), 6.91 (d, J = 8.9 Hz, 2H, H-Ar), 3.81 (s, 3H, -OCH3); 13C-NMR (50 MHz, CDCl3, ppm) δ = 160.4, 141.4, 136.2, 134.9, 129.9, 127.2, 125.7, 125.6, 122.8, 115.4, 55.5.
4. Conclusions
In summary, we have developed a straightforward method for the preparation of unsymmetrical diorganyl chalcogenides (S, Se and Te), under solvent- and ligand-free conditions. Remarkably, copper iodide was found to be a highly efficient catalyst for this cross-coupling reaction, affording the desired products in very high yields in the absence of solvent under microwave irradiation. Of particular importance, this new metal-catalyzed process showed good potential applicability within a very broad scope, without the need for the use of ligands or solvents. It appears that the chemistry described herein characterizes a useful alternative approach to the synthesis of organochalcogen compounds through a copper-catalyzed processes.
Acknowledgments
We gratefully acknowledge the CNPq, CAPES, INCT-Catálise, FAPESP-CERSusChem (grant 2014/50249-8), GSK-CERSusChem (grant 2014/50249-8), HEC (SRGP-161), FAPESC-Pronex, and FAPERGS-Pronex, for financial support. CAPES is also acknowledged for the fellowship for J.R. under CAPES-PNPD scheme. The funding bodies had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Supplementary Materials
Copies of the 1H- and 13C-NMR spectra are available online.
Author Contributions
A.L.B., S.S., J.R., G.V.B. conceived and designed the experiments; S.S., J.R., G.V.B. and T.E.A.F. performed the experiments; S.S., J.R., G.V.B., M.G., T.E.A.F. and F.Z.G. performed the spectral analysis, characterizations and contributed reagents/materials; A.L.B., S.S. and J.R. wrote the paper. All authors have approved the final version of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of the compounds 3a–k, 6a–h and 7a–b are available from the authors.
References
- 1.Liu C., Zhang H., Shi W., Lei A. Bond formations between two nucleophiles: Transition metal catalyzed oxidative cross-coupling reactions. Chem. Rev. 2011;111:1780–1824. doi: 10.1021/cr100379j. [DOI] [PubMed] [Google Scholar]
- 2.Yang L., Huang H. Transition-metal-catalyzed direct addition of unactivated C–H bonds to polar unsaturated bonds. Chem. Rev. 2015;115:3468–3517. doi: 10.1021/cr500610p. [DOI] [PubMed] [Google Scholar]
- 3.Li Y., Nie C., Wang H., Li X., Verpoort F., Duan C. A highly efficient method for the copper-catalyzed selective synthesis of diaryl chalcogenides from easily available chalcogen sources. Eur. J. Org. Chem. 2011;2011:7331–7338. doi: 10.1002/ejoc.201101121. [DOI] [Google Scholar]
- 4.Sun J., Deng L. Cobalt complex-catalyzed hydrosilylation of alkenes and alkynes. ACS Catal. 2017;7:631–651. doi: 10.1021/acscatal.5b02308. [DOI] [Google Scholar]
- 5.Chen Z., Huang J., Wang Z. Transition-metal-catalyzed hydrosulfoximination and oxidation reaction for the synthesis of sulfoximine derivatives. J. Org. Chem. 2016;81:9308–9314. doi: 10.1021/acs.joc.6b01891. [DOI] [PubMed] [Google Scholar]
- 6.Liu C., Szostak M. Twisted amides: From obscurity to broadly useful transtition-metal-catalyzed reaction by N-C amide bond Activation. Chem. Eur. J. 2017;23:7157–7173. doi: 10.1002/chem.201605012. [DOI] [PubMed] [Google Scholar]
- 7.Meng G., Shi S., Szostak M. Cross-coupling of amides by N–C bond activation. Synlett. 2016;27:2530–2540. [Google Scholar]
- 8.Godoi M., Paixão M.W., Braga A.L. Chiral organoselenium-transition-metal catalysts in asymmetric transformations. Dalton Trans. 2011;40:11347–11355. doi: 10.1039/c1dt11022e. [DOI] [PubMed] [Google Scholar]
- 9.Cresswell A.J., Eey S.T.-C., Denmark S.E. Catalytic, stereospecific syn-dichlorination of alkenes. Nat. Chem. 2015;7:146–152. doi: 10.1038/nchem.2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prochnow T., Back D.F., Geni Z. Iron(III) chloride and diorganyl diselenide-promoted nucleophilic closures of 1-benzyl-2-alkynylbenzenes in the preparation of 9-(organoselanyl)-5H-benzo[7]annulenes. Adv. Synth. Catal. 2016;358:1119–1129. doi: 10.1002/adsc.201501055. [DOI] [Google Scholar]
- 11.Rafique J., Canto R.F.S., Saba S., Barbosa F.A.R., Braga A.L. Recent advances in the synthesis of biologically relevant selenium-containing 5-membered heterocycles. Curr. Org. Chem. 2016;20:166–188. doi: 10.2174/1385272819666150810222057. [DOI] [Google Scholar]
- 12.Manna D., Roy G., Mugesh G. Antithyroid drugs and their analogues: Synthesis, structure, and mechanism of action. Acc. Chem. Res. 2013;46:2706–2715. doi: 10.1021/ar4001229. [DOI] [PubMed] [Google Scholar]
- 13.Barbosa F.A.R., Canto R.F.S., Saba S., Rafique J., Braga A.L. Synthesis and evaluation of dihydropyrimidinone-derived selenoesters as multi-targeted directed compounds against Alzheimer’s disease. Bioorg. Med. Chem. 2016;24:5762–5770. doi: 10.1016/j.bmc.2016.09.031. [DOI] [PubMed] [Google Scholar]
- 14.Kumar S., Yan J., Poon J.-F., Singh V.P., Lu X., Ott M.K., Engman L., Kumar S. Multifunctional antioxidants: Regenerable radical-trapping and hydroperoxide-decomposing ebselenols. Angew. Chem. Int. Ed. 2016;55:3729–3733. doi: 10.1002/anie.201510947. [DOI] [PubMed] [Google Scholar]
- 15.Rafique J., Saba S., Canto R.F.S., Frizon T.E.A., Hassan W., Waczuk E.P., Jan M., Back D.F., Rocha J.B.T.D., Braga A.L. Synthesis and biological evaluation of 2-picolylamide-based diselenides with non-bonded interactions. Molecules. 2015;20:10095–10109. doi: 10.3390/molecules200610095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Freudendahl D.M., Santoro S., Shahzad S.A., Santi C., Wirth T. Green chemistry with selenium reagents: Development of efficient catalytic reactions. Angew. Chem. Int. Ed. 2009;48:8409–8411. doi: 10.1002/anie.200903893. [DOI] [PubMed] [Google Scholar]
- 17.Braga A.L., Rafique J. Synthesis of biologically relevant small molecules containing selenium. Part A. Antioxidant compounds. In: Rappoport Z., editor. The Chemistry of Organic Selenium and Tellurium Compounds. Volume 4. Wiley & Sons, Ltd.; West Sussex, UK: 2014. pp. 989–1052. Chapter 13. [Google Scholar]
- 18.Braga A.L., Rafique J. Synthesis of biologically relevant small molecules containing selenium. Part B. Anti-infective and anticancer compounds. In: Rappoport Z., editor. The Chemistry of Organic Selenium and Tellurium Compounds. Volume 4. Wiley & Sons, Ltd.; West Sussex, UK: 2014. pp. 1053–1118. Chapter 14. [Google Scholar]
- 19.Braga A.L., Rafique J. Synthesis of biologically relevant small molecules containing selenium. Part C. Miscellaneous biological activities. In: Rappoport Z., editor. The Chemistry of Organic Selenium and Tellurium Compounds. Volume 4. Wiley & Sons, Ltd.; West Sussex, UK: 2014. pp. 1119–1174. Chapter 15. [Google Scholar]
- 20.Frizon T.E., Rafique J., Saba S., Bechtold I.H., Gallardo H., Braga A.L. Synthesis of functionalized organoselenium materials: Selenides and diselenides containing cholesterol. Eur. J. Org. Chem. 2015;2015:3470–3476. doi: 10.1002/ejoc.201500124. [DOI] [Google Scholar]
- 21.Brutchey R.L. Diorganyl dichalcogenides as useful synthons for colloidal semiconductor nanocrystals. Acc. Chem. Res. 2015;48:2918–2926. doi: 10.1021/acs.accounts.5b00362. [DOI] [PubMed] [Google Scholar]
- 22.Ho P.C., Rafique J., Lee J., Lee L.M., Jenkins H.A., Britten J.F., Braga A.L., Vargas-Baca I. Synthesis and structural characterisation of the aggregates of benzo-1,2-chalcogenazole 2-oxides. Dalton Trans. 2017;46:6570–6579. doi: 10.1039/C7DT00612H. [DOI] [PubMed] [Google Scholar]
- 23.Gu J., Zhao Z.-Q., Ding Y., Chen H.-L., Zhang Y.-W., Yan C.-H. Liquid-phase syntheses and material properties of two-dimensional nanocrystals of rare earth-selenium compound containing planar se layers: RESe2 nanosheets and RE4O4Se3 nanoplates. J. Am. Chem. Soc. 2013;135:8363–8371. doi: 10.1021/ja4028583. [DOI] [PubMed] [Google Scholar]
- 24.Godoi B., Schumacher R.F., Zeni G. Synthesis of heterocycles via electrophilic cyclization of alkynes containing heteroatom. Chem. Rev. 2011;111:2937–2980. doi: 10.1021/cr100214d. [DOI] [PubMed] [Google Scholar]
- 25.Bhabak K.P., Mughesh G. Functional mimics of glutathione peroxidase: Bioinspired synthetic antioxidants. Acc. Chem. Res. 2010;43:1408–1419. doi: 10.1021/ar100059g. [DOI] [PubMed] [Google Scholar]
- 26.Zeni G., Braga A.L., Stefani H.A. Palladium-catalyzed coupling of sp2-hybridized tellurides. Acc. Chem. Res. 2003;36:731–738. doi: 10.1021/ar0202621. [DOI] [PubMed] [Google Scholar]
- 27.Wirth T. Organoselenium Chemistry: Synthesis and Reactions. Wiley-VCH; Weinheim, Germany: 2011. [Google Scholar]
- 28.Rappoport Z., Liebman J.F., Marek I., Patai S. The Chemistry of Organic Selenium and Tellurium Compounds. Volume 4 Wiley & Sons, Ltd.; West Sussex, UK: 2014. [Google Scholar]
- 29.Rappoport Z., Liebman J.F., Marek I., Patai S. The Chemistry of Organic Selenium and Tellurium Compounds. Volume 3 Wiley & Sons, Ltd.; West Sussex, UK: 2012. [Google Scholar]
- 30.Zeni G., Lüdtke D.S., Panatieri R.B., Braga A.L. Vinylic tellurides: From preparation to their applicability in organic synthesis. Chem. Rev. 2006;106:1032–1076. doi: 10.1021/cr0505730. [DOI] [PubMed] [Google Scholar]
- 31.Mugesh G., Singh H.B. Heteroatom-directed aromatic lithiation: A versatile route to the synthesis of organochalcogen (Se, Te) compounds. Acc. Chem. Res. 2002;35:226–236. doi: 10.1021/ar010091k. [DOI] [PubMed] [Google Scholar]
- 32.Kumar S., Singh H.B., Wolmershäuser G. Protection against peroxynitrite-mediated nitration reaction by intramolecularly coordinated diorganoselenides. Organometallics. 2006;25:382–393. doi: 10.1021/om050353c. [DOI] [Google Scholar]
- 33.Malysher D.A., Scott N.M., Marion N., Stevens E.D., Ananikov V.P., Beletskaya I.P., Nolan S.P. Homogeneous nickel catalysts for the selective transfer of a single arylthio group in the catalytic hydrothiolation of alkynes. Organometallics. 2006;25:4462–4470. doi: 10.1021/om060302v. [DOI] [Google Scholar]
- 34.Cohen R.J., Fox D.L., Salvatore R.N. A novel and highly efficient synthetic route to unsymmetrical organoselenides using cesium bases. J. Org. Chem. 2004;69:4265–4268. doi: 10.1021/jo0401265. [DOI] [PubMed] [Google Scholar]
- 35.Bates C.G., Gujadhur R.K., Venkataraman D. A general method for the formation of aryl-sulfur bonds using copper (I) catalysts. Org. Lett. 2002;4:2803–2806. doi: 10.1021/ol0264105. [DOI] [PubMed] [Google Scholar]
- 36.Sperotto E., van Klink G.P.M., de Vries J.G., van Koten G. Ligand-free copper-catalyzed c-s coupling of aryl iodides and thiols. J. Org. Chem. 2008;73:5625–5628. doi: 10.1021/jo800491k. [DOI] [PubMed] [Google Scholar]
- 37.Tiecco M., Testaferri L., Bagnoli L., Mariani F., Temperini A., Tomassini C., Santi C. Electrophilic 2-thienylselenenylation of thiophene. Preparation of oligo(seleno-2,5-thienylenes) Tetrahedron. 2000;56:3255–3260. doi: 10.1016/S0040-4020(00)00245-3. [DOI] [Google Scholar]
- 38.Jin W., Zheng P., Wong W.-T., Law G.-L. Efficient palladium-catalyzed direct C−H phenylselenylation of (hetero)arenes in water. Asian J. Org. Chem. 2015;4:875–878. doi: 10.1002/ajoc.201500192. [DOI] [Google Scholar]
- 39.Cheng J.-H., Yi C.-L., Liu T.-J., Lee C.-F. Highly regioselective synthesis of aryl chalcogenides through C–H functionalization of arenes. Chem. Commun. 2012;48:8440–8442. doi: 10.1039/c2cc33950a. [DOI] [PubMed] [Google Scholar]
- 40.Chu L., Yue X., Qing F.-L. Cu (II)-mediated methylthiolation of aryl C−H bonds with DMSO. Org. Lett. 2010;12:1644–1647. doi: 10.1021/ol100449c. [DOI] [PubMed] [Google Scholar]
- 41.Zhao X., Dimitrijević E., Dong V.M. Palladium-catalyzed C−H Bond functionalization with arylsulfonyl chlorides. J. Am. Chem. Soc. 2009;131:3466–3467. doi: 10.1021/ja900200g. [DOI] [PubMed] [Google Scholar]
- 42.Candeias N.R., Montlbano F., Cal P.M.S.D., Gois P.M.P. Boronic acids and esters in the petasis-borono Mannich multicomponent reaction. Chem. Rev. 2010;110:6169–6193. doi: 10.1021/cr100108k. [DOI] [PubMed] [Google Scholar]
- 43.Zhu C., Falck J.R. Transition-Metal-Free ipso-Functionalization of Arylboronic Acids and Derivatives. Adv. Synth. Catal. 2014;356:2395–2410. doi: 10.1002/adsc.201400305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hall D.G. Boronic Acids: Preparation and Applications in Organic Synthesis Medicine and Materials. 2nd ed. Volume 2 Wiley-VCH; Weinheim, Germany: 2011. [Google Scholar]
- 45.Taniguchi N. Aryl- or alkylation of diaryl disulfides using organoboronic acids and a copper catalyst. Synlett. 2006;14:1351–1354. doi: 10.1055/s-2006-939707. [DOI] [Google Scholar]
- 46.Wang L., Wang M., Huang F. A simple copper salt-catalyzed synthesis of unsymmetrical diaryl selenides and tellurides from arylboronic acids with diphenyl diselenide and ditelluride. Synlett. 2005;13:2007–2010. doi: 10.1055/s-2005-871936. [DOI] [Google Scholar]
- 47.Ren K., Wang M., Wang L. Lewis acid InBr3-catalyzed arylation of diorgano diselenides and ditellurides with arylboronic acids. Org. Biomol. Chem. 2009;7:4858–4861. doi: 10.1039/b914533h. [DOI] [PubMed] [Google Scholar]
- 48.Wang M., Ren K., Wang L. Iron-catalyzed ligand-free carbon-selenium (or tellurium) coupling of arylboronic acids with diselenides and ditellurides. Adv. Synth. Catal. 2009;351:1586–1594. doi: 10.1002/adsc.200900095. [DOI] [Google Scholar]
- 49.Yu J.-T., Guo H., Yi Y., Fei H., Jiang Y. The Chan-Lam reaction of chalcogen elements leading to aryl chalcogenides. Adv. Synth. Catal. 2014;356:749–752. doi: 10.1002/adsc.201300853. [DOI] [Google Scholar]
- 50.Zheng B., Gong Y., Xu H.-H. Copper-catalyzed C–Se coupling of diphenyl diselenide with arylboronic acids at room temperature. Tetrahedron. 2013;69:5342–5347. doi: 10.1016/j.tet.2013.04.124. [DOI] [Google Scholar]
- 51.Reddy K.H., Satish G., Rames K., Karnakar K., Nageswar Y.V.D. Magnetically separable CuFe2O4 nanoparticle catalyzed C–Se cross coupling in reusable PEG medium. Chem. Lett. 2012;41:585–587. doi: 10.1246/cl.2012.585. [DOI] [Google Scholar]
- 52.Mohan B., Yoon C., Jang S. Copper nanoparticles catalyzed Se(Te)-Se(Te) bond activation: A straightforward route towards unsymmetrical organochalcogenides from boronic acids. Chemcatchem. 2015;7:405–412. doi: 10.1002/cctc.201402867. [DOI] [Google Scholar]
- 53.Guo Y., Quan Z.-J., Da Y.-X., Zhang Z., Wang X.-C. (2-Chlorobenzoyloxy)copper(I) catalyzed C-S cross-coupling of di(hetero)aryl disulfides with aryl boronic acis under base-free conditions. RSC Adv. 2015;4:45479–45483. doi: 10.1039/C5RA05594F. [DOI] [Google Scholar]
- 54.Alves D., Santos C.G., Paixão M.W., Soares L.C., de Souza D., Rodrigues O.E.D., Braga A.L. CuO nanoparticles: An efficient and recyclable catalyst for cross-coupling reactions of organic diselenides with aryl boronic acids. Tetrahedron Lett. 2009;50:6635–6638. doi: 10.1016/j.tetlet.2009.09.052. [DOI] [Google Scholar]
- 55.Kumar A., Kumar S. A convenient and efficient copper-catalyzed synthesis of unsymmetrical and symmetrical diaryl chalcogenides from arylboronic acids in ethanol. Tetrahedron. 2014;70:1763–1772. doi: 10.1016/j.tet.2014.01.030. [DOI] [Google Scholar]
- 56.Goldani B., Ricordi V.G., Seus N., Lenardao E.J., Schumacher R.F., Alves D. Silver-catalyzed synthesis of diaryl selenides by reaction of diaryl diselenides with aryl boronic acids. J. Org. Chem. 2016;81:11472–11476. doi: 10.1021/acs.joc.6b02108. [DOI] [PubMed] [Google Scholar]
- 57.Saba S., Rafique J., Braga A.L. Synthesis of unsymmetrical diorganyl chalcogenides under greener conditions: Use of an iodine/DMSO system, solvent- and metal-free approach. Adv. Synth. Catal. 2015;357:1446–1452. doi: 10.1002/adsc.201500024. [DOI] [Google Scholar]
- 58.Mohan B., Hwang S., Jang S., Park K.H. Ultrasound-assisted transition-metal-free synthesis of diaryl tellurides from aryl boronic acids: A possible free-radical mechanism. Syntlett. 2014;25:2078–2083. doi: 10.1002/chin.201510252. [DOI] [Google Scholar]
- 59.Rafique J., Saba S., Rosário A.R., Braga A.L. Regioselective, solvent- and metal-free chalcogenation of imidazo[1,2-a]pyridines by employing I2/DMSO as the catalytic oxidation system. Chem. Eur. J. 2016;22:11854–11862. doi: 10.1002/chem.201600800. [DOI] [PubMed] [Google Scholar]
- 60.Saba S., Rafique J., Braga A.L. DMSO/iodine-catalyzed oxidative C–Se/C–S bond formation: A regioselective synthesis of unsymmetrical chalcogenides with nitrogen- or oxygen-containing arenes. Catal. Sci. Technol. 2016;6:3087–3098. doi: 10.1039/C5CY01503K. [DOI] [Google Scholar]
- 61.Rafique J., Saba S., Rosário A.R., Zeni G., Braga A.L. K2CO3-mediated, direct C–H bond selenation and thiolation of 1,3,4-oxadiazoles in the absence of metal catalyst: An eco-friendly approach. RSC Adv. 2014;4:51648–51652. doi: 10.1039/C4RA10490K. [DOI] [Google Scholar]
- 62.Silva L.T., Azeredo J.B., Saba S., Rafique J., Bortoluzzi A.J., Braga A.L. Solvent- and metal-free chalcogenation of bicyclic arenes using I2/DMSO as non-metallic catalytic system. Eur. J. Org. Chem. 2017 doi: 10.1002/ejoc.201700744. [DOI] [Google Scholar]
- 63.Rocha M.S.T., Rafique J., Saba S., Azeredo J.B., Back D., Godoi M., Braga A.L. Regioselective hydrothiolation of terminal acetylene catalyzed by magnetite (Fe3O4) nanoparticles. Synth. Commun. 2017;47:291–298. doi: 10.1080/00397911.2016.1262421. [DOI] [Google Scholar]
- 64.Rafique J., Saba S., Schneider A.R., Franco M.S., Silva S.M., Braga A.L. Metal- and solvent-free approach to access 3-Se/S-chromones from the cyclization of enaminones in the presence of dichalcogenides catalyzed by KIO3. ACS Omega. 2017;2:2280–2290. doi: 10.1021/acsomega.7b00445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ananikov V.P., Gayduk K.A., Belestskaya I.P., Khrustalve V.N., Antipin M.Y. Remarkable ligand effect in Ni- and Pd-catalyzed bisthiolation and bisselenation of terminal alkynes: Solving the problem of stereoselective dialkyldichalcogenide addition to the C≡C bond. Chem. Eur. J. 2008;14:2420–2434. doi: 10.1002/chem.200701278. [DOI] [PubMed] [Google Scholar]
- 66.Taniguchi N. Diarylation of chalcogen elements using arylboronic acids via copper- or palladium-catalyzed oxidative coupling. Tetrahedron. 2016;72:5818–5823. doi: 10.1016/j.tet.2016.08.012. [DOI] [Google Scholar]
- 67.Zhao H., Jiang Y., Chen Q., Cai M. A highly efficient and reusable MCM-41-immobilized bipyridine copper (I) catalyst for the C–Se coupling of organoboronic acids with diaryl diselenides. New J. Chem. 2015;39:2106–2115. doi: 10.1039/C4NJ01687D. [DOI] [Google Scholar]
- 68.Taniguchi N. Convenient synthesis of unsymmetrical organochalcogenides using organoboronic acids with dichalcogenides via cleavage of the S−S, Se−Se, or Te−Te bond by a copper catalyst. J. Org. Chem. 2007;72:1241–1245. doi: 10.1021/jo062131+. [DOI] [PubMed] [Google Scholar]
- 69.Cella R., Cunha R.L.O.R., Reis A.E.S., Pimenta D.C., Klitzke C.F., Stefani H.A. Suzuki-Miyaura cross-coupling reactions of aryl tellurides with potassium aryltrifluoroborate salts. J. Org. Chem. 2006;71:244–250. doi: 10.1021/jo052061r. [DOI] [PubMed] [Google Scholar]
- 70.Ricordi V.G., Freitas C.S., Perin G., Lenardão E.J., Jacob R.G., Savegnago L., Alves D. Glycerol as a recyclable solvent for copper-catalyzed cross-coupling reactions of diaryl diselenides with aryl boronic acids. Green Chem. 2012;14:1030–1034. doi: 10.1039/c2gc16427b. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.