

ABSTRACT
PARK7/DJ-1 is a Parkinson disease- and cancer-associated protein that functions as a multifunctional protein involved in gene transcription regulation and anti-oxidative defense. Although PARK7 lacks the secretory signal sequence, it is secreted and plays important physiological and pathophysiological roles. Whereas secretory proteins that lack the endoplasmic reticulum-targeting signal sequence are secreted from cells by way of what is called the unconventional secretion mechanism, the specific processes responsible for causing PARK7 to be secreted across the plasma membrane have remained unclear. In the present study, we found that PARK7 secretion was increased by treatment with 6-OHDA via the unconventional secretory pathway in human neuroblastoma SH-SY5Y cells and MEF cells. We also found that 6-OHDA-induced PARK7 secretion was suppressed in Atg5-, Atg9-, or Atg16l1-deficient MEF cells or ATG16L1 knockdown SH-SY5Y cells, indicating that the autophagy-based unconventional secretory pathway is involved in PARK7 secretion. We moreover observed that 6-OHDA-derived electrophilic quinone induced oxidative stress as indicated by a decrease in glutathione levels, and that this was suppressed by pretreatment with antioxidant NAC. We further found that NAC treatment suppressed autophagy and PARK7 secretion. We also observed that 6-OHDA-induced autophagy was associated with activation of AMPK and ULK1 via a pathway which was independent of MTOR. Collectively these results suggest that electrophilic 6-OHDA quinone enhances oxidative stress, and that this is followed by AMPK-ULK1 pathway activation and induction of secretory autophagy to produce unconventional secretion of PARK7.
Abbreviations: 6-OHDA: 6-hydroxydopamine; AMPK: AMP-activated protein kinase; ATG: autophagy related; CAV1: caveolin 1; ER: endoplasmic reticulum; FN1: fibronectin 1; GSH: glutathione; IDE: insulin degrading enzyme; IL: interleukin; LDH: lactate dehydrogenase; MAP1LC3B/LC3B: microtubule associated protein 1 light chain 3 beta; MEF: mouse embryonic fibroblast; MTOR: mechanistic target of rapamycin kinase; NAC: N-acetyl-L-cysteine; PARK7/DJ-1: Parkinsonism associated deglycase; PD: Parkinson disease; RPS6KB1/p70S6K: ribosomal protein S6 kinase B1; RPN1: ribophorin I; ROS: reactive oxygen species; ULK1: unc-51 like autophagy activating kinase 1; WT: wild-type
KEYWORDS: AMPK, oxidative stress, PARK7/DJ-1, parkinson disease, secretory autophagy, unconventional secretion
Introduction
Most secretory proteins have a signal sequence at the N-terminus which allows them to be transported from the endoplasmic reticulum (ER) to the Golgi and exported from the cell via the conventional secretion mechanism. There are a smaller number of proteins that lack the ER-targeting signal sequence which are secreted from the cell by what is referred to as the unconventional secretion mechanism [1–3]. Unconventionally secreted proteins include DBI (diazepam binding inhibitor, acyl-CoA binding protein), IL1B (interleukin 1 beta), IL18 (interleukin 18), HMGB1 (high mobility group box 1), and FGF2 (fibroblast growth factor 2) [4–7]. Although it is known that these unconventionally secreted proteins are released from the cell via an ER-/Golgi-independent pathway, the specific mechanism by which the soluble protein is able to translocate across the hydrophobic plasma membrane still remains to be discussed. Multiple pathways have been proposed as possible routes by which such unconventional secretion might be accomplished, these proposed routes including secretory lysosomes, plasma membrane shedding, exosome derived from multivesicular bodies, and secretory autophagy [1–3,8]. Because some of the proteins that are secreted via the unconventional secretory pathway play important extracellular functional roles in developmental processes and inflammation, the unconventional secretion mechanism is being studied extensively.
PARK7/DJ-1 (Parkinsonism associated deglycase) was initially identified as an oncoprotein that cooperates with Ras to promote cell transformation [9]. PARK7 has been implicated as a protein encoded by one of the causative genes (PARK7) in a familial form of Parkinson disease (PD) [10]. It has also been suggested that PARK7 is related to a sporadic form of PD [11,12]. Elevated levels of an oxidized form of PARK7 (oxPARK7) are observed in erythrocytes of PD patients at early stages [11]. oxPARK7 is also observed in the brains of human PD patients and animal PD models [12]. PARK7 is a multifunctional protein involved in several molecular processes such as gene transcription regulation, protein stabilization, and anti-oxidative defense [13,14]. Overexpression of PARK7 can protect dopaminergic neurons against several types of oxidative stress by increasing cellular glutathione (GSH) levels [15]. Knockdown of PARK7 increases susceptibility to oxidative stress [15,16]. Human PARK7 has three cysteine residues at amino acid numbers 46, 53, and 106, and undergoes preferential oxidation at position 106 (Cys106) under oxidative stress [17,18]. Replacement of Cys106 with alanine, serine, or aspartic acid causes PARK7 to lose its anti-oxidative properties.
PARK7 is localized in the cytoplasm and the nucleus; it has also been shown that some PARK7 is localized in the mitochondria [9,10,19,20]. Several lines of evidence suggest that PARK7 is secreted from cells into the circulation despite the fact that PARK7 lacks a secretory signal sequence [21–29]. Increase in PARK7 secretion into serum or plasma has been observed in patients with breast cancer, melanoma, stroke, familial amyloid polyneuropathy, and PD [21–27]. There have been conflicting reports of both increase [28] and decrease [29] in the amount of PARK7 in cerebrospinal fluid in sporadic PD patients as compared with control groups. Secreted PARK7 plays important physiological and pathophysiological roles which include involvement in anti-oxidative effects [30], extracellular signaling between neighboring neuronal cells [31], angiogenic and osteogenic factors [32], and degradation of aggregated protein [26]. Such findings indicate that PARK7 secretion may be implicated in the etiology and/or progression of diseases such as PD and cancer, possibly making it suitable for use as a biomarker. Despite the considerable number of such findings, however, it remains unclear how cytosolic PARK7 protein is able to translocate across the plasma membrane.
In our present study, to investigate the molecular mechanism of PARK7 secretion, we use 6-hydroxydopamine (6-OHDA), which is a catecholaminergic neurotoxin commonly used to generate experimental models for PD in vivo and in vitro [33]. As a result of our study, we find that PARK7 secretion is increased by treatment with 6-OHDA via the unconventional secretory pathway in human neuroblastoma SH-SY5Y cells and mouse embryonic fibroblast (MEF) cells. We demonstrate that 6-OHDA induces oxidative stress as indicated by a decrease in GSH levels, resulting in induction of macroautophagy/autophagy. We further show that 6-OHDA-induced autophagy is associated with activation of AMP-activated protein kinase (AMPK) and its downstream effector ULK1 (unc-51 like autophagy activating kinase 1) and that this occurs via a pathway that is independent of MTOR (mechanistic target of rapamycin kinase). We conclude that AMPK-ULK1-mediated secretory autophagy plays an important role in the unconventional secretion of PARK7.
Results
PARK7 is secreted under non-stress conditions in SH-SY5Y cells
To assess PARK7 secretion from human neuroblastoma SH-SY5Y cells, cells were cultured in serum-free medium for 0–6 h to prevent contamination by serum protein. As controls, FN1 (fibronectin 1) was used as a protein marker secreted via the conventional pathway and RPN1 (ribophorin I) was used as a cell resident protein. Our results showed that PARK7 was secreted in a time-dependent manner similar to the observed secretion of FN1 control, and that RPN1 was present only in the cell lysate fraction (Figure 1(A and (B)). Evaluation was carried out to determine whether LDH (lactate dehydrogenase), an enzyme normally found only in the cytoplasm, was being released from the cell under the conditions tested, as a result of which it was found based on the small amount of LDH released that the PARK7 secretion observed was not due to plasma membrane leakage (Figure 1(B)). To evaluate whether PARK7 secretion was mediated by the conventional ER-/Golgi-dependent secretion mechanism, cells were treated with brefeldin A, an inhibitor of ER-Golgi transport, as a result of which it was found that treatment with brefeldin A inhibited FN1 secretion but not PARK7 secretion (Figure 1(C), suggesting that the conventional secretory pathway was not involved in PARK7 secretion. As previously reported [9,10], most of PARK7 was found to be in the cytosolic protein-enriched fraction obtained by subcellular fractionation (Figure 1(D)), supporting the idea that PARK7 was secreted via an ER-/Golgi-independent secretory pathway. We also used 2D-PAGE to examine the oxidative state of PARK7, as a result of which we found that the ratio of oxPARK7 to total PARK7 in medium was almost the same as that in cells, suggesting that secretion of PARK7 was not induced by its oxidation (Figure 1(E)).
Figure 1.
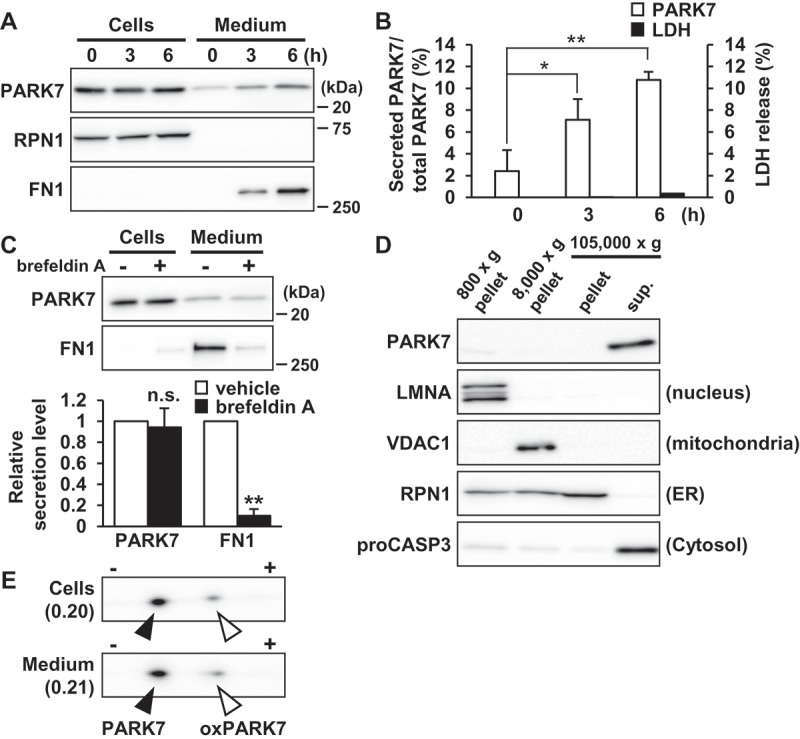
PARK7 was secreted from SH-SY5Y cells. (A and B) SH-SY5Y cells were cultured in serum-free medium for 0–6 h. (A) Whole cell lysates (Cells) and the conditioned medium (Medium) were immunoblotted using antibodies specific for PARK7, RPN1, or FN1. Representative image is shown. (B) PARK7 band intensities were quantified by densitometric scanning and the percentage of secreted PARK7/total PARK7 is shown. LDH release in the conditioned medium was analyzed by LDH assay. n = 3; mean ± S.D.; *, p < 0.05; **, p < 0.01. (C) SH-SY5Y cells were treated with 2 μg/ml brefeldin A in serum-free medium for 3 h. Whole cell lysates and the conditioned medium were immunoblotted with antibodies specific for PARK7 or FN1. PARK7 and FN1 band intensities were quantified by densitometric scanning and relative secretion level to vehicle-treated cells is shown. n = 3; **, p < 0.01; n.s., not significant. (D) SH-SY5Y cells were homogenized by using the Dounce homogenizer and homogenate was sequentially centrifuged as indicated. Equal aliquots from each fraction were immunoblotted using antibodies specific for PARK7, LMNA (lamin A/C), VDAC1, RPN1, or proCASP3 (caspase 3). (E) SH-SY5Y cells were cultured in serum-free medium for 3 h. Whole cell lysates and the conditioned medium were separated by 2D-PAGE and immunoblotted using antibody specific for PARK7. The ratio of oxPARK7 to total PARK7 is shown under each condition.
Treatment with 6-OHDA enhances secretion of PARK7 from SH-SY5Y cells
We then evaluated the effect of 6-OHDA on PARK7 secretion. Because we had noticed that 6-OHDA in medium interfered with protein precipitation during the trichloroacetic acid precipitation procedure, protein secretion was evaluated using the conditioned medium obtained following 6-OHDA treatment as described in Materials and Methods. Results showed that 6-OHDA treatment for 3 h increased PARK7 secretion in a concentration-dependent manner (Figure 2(A)). Significant release of LDH from 100 μM 6-OHDA-treated cells was not observed (Figure 2(B)), suggesting that the increase in PARK7 secretion was not the result of plasma membrane disruption by 6-OHDA. Brefeldin A treatment did not inhibit PARK7 secretion (Figure 2(C)), confirming that 6-OHDA-induced PARK7 secretion was mediated via an ER-/Golgi-independent secretory pathway. Because it had recently been reported that CASP1 (caspase 1) may act as regulator of unconventional protein secretion [34], we decided to assess the respective effects of a pan-caspase inhibitor (zVAD; i.e., Z-VAD[OMe]-FMK) and a specific CASP1 inhibitor (YVAD; i.e., Ac-YVAD-CMK) on PARK7 secretion. Our results indicated that 6-OHDA-induced PARK7 secretion was inhibited by either inhibitor (Figure 2(D)). Taken together, the foregoing series of results suggest that 6-OHDA induces PARK7 secretion via the unconventional secretory pathway.
Figure 2.
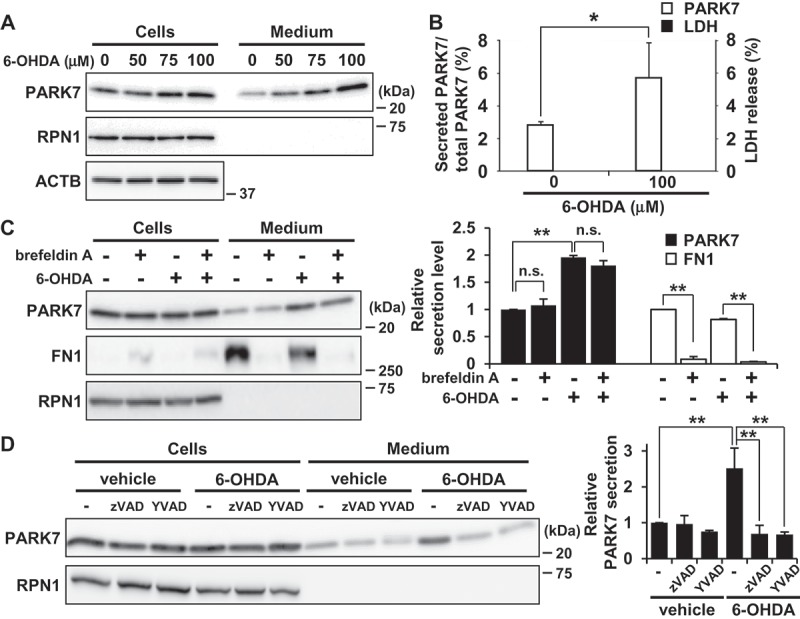
6-OHDA treatment enhanced secretion of PARK7 from SH-SY5Y cells. (A and B) SH-SY5Y cells were treated with 0–100 μM 6-OHDA for 3 h and were then cultured in serum-free medium for 2 h. (A) Whole cell lysates and the conditioned medium were immunoblotted using antibodies specific for PARK7, RPN1, or ACTB. (B) PARK7 band intensities were quantified by densitometric scanning and the percentage of secreted PARK7/total PARK7 is shown. LDH release in the conditioned medium was analyzed by LDH assay. n = 3; mean ± S.D.; *, p < 0.05. (C) SH-SY5Y cells were pre-treated with 2 μg/ml brefeldin A for 3 h and were then treated with 100 μM 6-OHDA for 3 h, followed by culture in serum-free medium for 2 h. Whole cell lysates and the conditioned medium were immunoblotted with antibodies specific for PARK7, FN1, or RPN1. PARK7 and FN1 band intensities were quantified by densitometric scanning and relative secretion level to vehicle-treated cells is shown. n = 3; mean ± S.D.; **, p < 0.01; n.s., not significant. (D) SH-SY5Y cells were pre-treated with or without 20 μM pan-caspase inhibitor (zVAD) or CASP1 inhibitor (YVAD) for 1 h and were then treated with 100 μM 6-OHDA for 3 h, followed by culture in serum-free medium for 2 h. Whole cell lysates and the conditioned medium were immunoblotted using antibodies specific for PARK7 or RPN1. PARK7 band intensities were quantified by densitometric scanning and relative secretion level to vehicle-treated cells is shown. n = 3; mean ± S.D.; **, p < 0.01.
6-OHDA-induced oxidative stress is implicated in PARK7 secretion
Considering the prominent effect of 6-OHDA on PARK7 secretion that we observed as reported above, we then sought to explore the mechanism of 6-OHDA-induced PARK7 secretion. It has been known that 6-OHDA is readily oxidized by molecular oxygen to produce superoxide anion, hydrogen peroxide (H2O2), and 2-hydroxy-5-(2-aminoethyl)-1,4-benzoquinone (6-OHDA quinone), resulting in induction of intracellular oxidative stress [35]. We therefore decided to examine whether 6-OHDA-induced oxidative stress affected PARK7 secretion. We first assessed the effect of 6-OHDA on intracellular antioxidant GSH content, finding as a result that there was a significant decrease in GSH levels in cells treated with 100 μM 6-OHDA for 3 h (Figure 3(A)). We next examined whether the 6-OHDA-induced reduction in GSH level caused PARK7 secretion in the presence or absence of the GSH precursor N-acetyl-L-cysteine (NAC), a well-established cysteine supplier. We found that pretreatment with NAC suppressed the 6-OHDA-induced PARK7 secretion (Figure 3(B)). Since we had previously reported that about 260 μM H2O2 and 600 μM 6-OHDA quinone are formed from 600 µM 6-OHDA in medium [36], we decided to test the effect of H2O2 on PARK7 secretion, finding as a result that treatment with 100 μM H2O2 in the presence or absence of catalase did not increase PARK7 secretion (Figure 3(C)). Based on the foregoing series of results, we hypothesized that electrophilic quinone-induced oxidative stress might be responsible for 6-OHDA-induced PARK7 secretion. To test this possibility, we evaluated the effect of another electrophilic p-quinone, benzoquinone, on PARK7 secretion, finding as a result that, at non-cytotoxic concentrations (i.e., no release of LDH), benzoquinone induced PARK7 secretion in a concentration-dependent manner (Fig. S1A). Furthermore, NAC treatment was found to suppress the increase in 20 μM benzoquinone-induced PARK7 secretion (Fig. S1B). Similar to the effects of 6-OHDA treatment, 20 μM benzoquinone was found to reduce GSH content, and it was moreover found that this reduction could be abrogated by NAC treatment (Fig. S1C). Together, these results suggest that electrophilic quinone-induced oxidative stress underlies 6-OHDA-induced PARK7 secretion. We also examined whether PARK7 secretion was caused by another neurotoxin-induced oxidative stress. We tested the mitochondrial complex I inhibitor rotenone, which can cause reactive oxygen species (ROS) production and a decrease in GSH levels [37]. We found that treatment with rotenone at non-toxic concentrations for 24 h induced PARK7 secretion (Fig. S1D).
Figure 3.
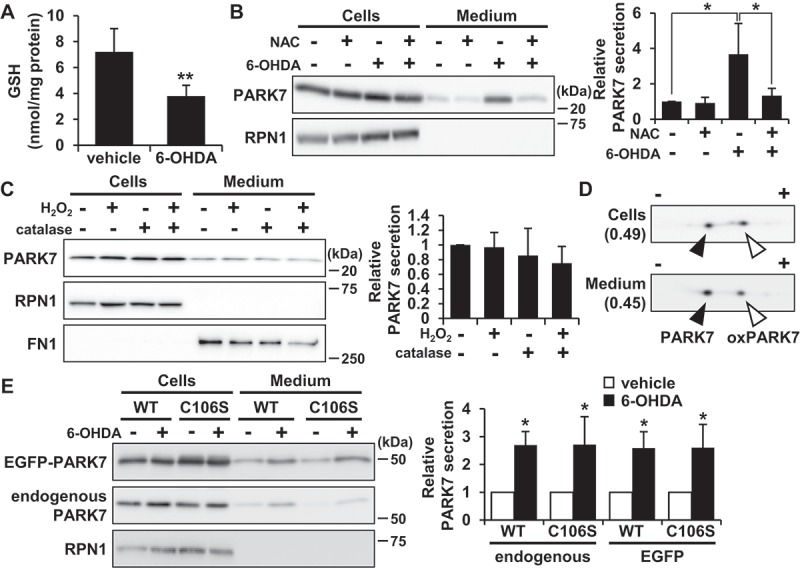
6-OHDA-induced oxidative stress was implicated in PARK7 secretion. (A) SH-SY5Y cells were treated with 100 μM 6-OHDA for 3 h and were subjected to GSH assay. n = 5; mean ± S.D.; **, p < 0.01. (B) SH-SY5Y cells were pre-treated with or without 2 mM NAC for 2 h and were then treated with 100 μM 6-OHDA for 3 h, followed by culture in serum-free medium for 2 h. Whole cell lysates and the conditioned medium were immunoblotted using antibodies specific for PARK7 or RPN1. PARK7 band intensities were quantified by densitometric scanning and relative secretion level to vehicle-treated cells is shown. n = 3; mean ± S.D.; *, p < 0.05. (C) SH-SY5Y cells were treated with 100 μM H2O2 in the presence or absence of 50 U/ml catalase in serum-free medium for 3 h. Whole cell lysates and the conditioned medium were immunoblotted with antibodies specific for PARK7, RPN1, or FN1. PARK7 band intensities were quantified by densitometric scanning and relative secretion level to vehicle-treated cells is shown. n = 3; mean ± S.D. (D) SH-SY5Y cells were treated with 100 μM 6-OHDA for 3 h and were then cultured in serum-free medium for 2 h. Whole cell lysates and the conditioned medium were separated by 2D-PAGE and immunoblotted using antibody specific for PARK7. The ratio of oxPARK7:total PARK7 is shown under each condition. (E) HEK293 cells stably expressing WT or C106S mutant of PARK7 were treated with 100 μM 6-OHDA for 3 h and were then cultured in serum-free medium for 2 h. Whole cell lysates and the conditioned medium were immunoblotted using antibodies specific for PARK7 or RPN1. PARK7 band intensities were quantified by densitometric scanning and relative secretion level to vehicle-treated cells is shown. n = 3; mean ± S.D.; *, p < 0.05.
To evaluate whether the oxidized form of PARK7 was preferentially secreted from cells treated with 6-OHDA, 2D-PAGE analysis was carried out to determine the ratio of reduced:oxidized PARK7. Results showed that the ratio of oxPARK7 to total PARK7 in cells was increased by 6-OHDA treatment (Figs. 1E and 3D). Furthermore, reduced and oxidized forms of PARK7 were both observed in medium, and the ratio of oxPARK7 to total PARK7 in medium was similar to that in cells. To further assess the involvement of Cys106 in PARK7 secretion, HEK293 cells were transfected with either wild-type (WT) PARK7 or PARK7C106S, the latter being a mutant form of PARK7 in which oxidation-sensitive cysteine at amino acid 106 is substituted by serine. Results of transfection indicated that 6-OHDA treatment induced PARK7C106S secretion in a manner similar to that observed with endogenous PARK7 and WT PARK7 (Figure 3(E)). Together, the foregoing series of results suggest that oxidation of PARK7 at Cys106 is not involved in enhancement of PARK7 secretion.
6-OHDA induces autophagy in SH-SY5Y cells and MEF cells
To gain insight into the mechanism by which 6-OHDA treatment increased PARK7 secretion, we examined whether 6-OHDA induced autophagy, since it had recently been shown that the secretion of several proteins lacking a signal sequence is associated with the autophagy-based unconventional secretory pathway [38–40]. To exclude any possible effect of incubation of cells in serum-free medium on monitoring of autophagy induction, autophagic flux was evaluated using the lysate from the cells treated with 6-OHDA for 3 h. By monitoring the conversion of MAP1LC3B/LC3B (microtubule associated protein 1 light chain 3 beta) from a cytosolic form (LC3B-I) to its lipidated phagophore- and autophagosome-associated form (LC3B-II), we found that 6-OHDA treatment induced increase in LC3B-II levels in SH-SY5Y cells (Figure 4(A)).
Figure 4.
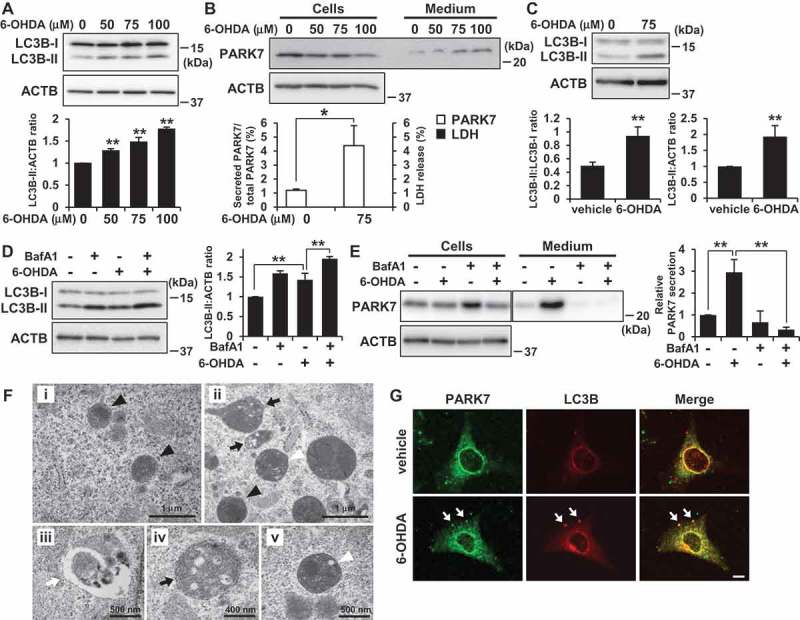
6-OHDA treatment induced autophagy in SH-SY5Y cells and WT MEF cells. (A) SH-SY5Y cells were treated with 0–100 μM 6-OHDA for 3 h. Whole cell lysates were immunoblotted using antibodies specific for LC3B or ACTB. LC3B and ACTB band intensities were quantified by densitometric scanning and LC3B-II:ACTB ratio is shown. n = 3; mean ± S.D.; **, p < 0.01. (B) WT MEF cells were treated with 0–100 μM 6-OHDA for 3 h and were then cultured in serum-free medium for 2 h. Whole cell lysates and the conditioned medium were immunoblotted using antibodies specific for PARK7 or ACTB. PARK7 band intensities were quantified by densitometric scanning and the percentage of secreted PARK7/total PARK7 is shown. LDH release in the conditioned medium was analyzed by LDH assay. n = 3; mean ± S.D.; *, p < 0.05. (C) WT MEF cells were treated with 75 μM 6-OHDA for 3 h. Whole cell lysates were immunoblotted using antibodies specific for LC3B or ACTB. LC3B band intensities were quantified by densitometric scanning and LC3B-II:LC3B-I or LC3B-II:ACTB ratio is shown. n = 3; **, p < 0.01. (D and E) WT MEF cells were pretreated with 100 nM bafilomycin A1 (BafA1) for 1 h and were then treated with 75 μM 6-OHDA for 3 h (D), followed by culture in serum-free medium for 2 h (E). Whole cell lysates and the conditioned medium were immunoblotted using antibodies specific for LC3, PARK7, or ACTB. PARK7, LC3B and ACTB band intensities were quantified by densitometric scanning. LC3B-II:ACTB ratio (D) and relative secretion level to vehicle-treated cells (E) are shown. n = 3; mean ± S.D.; **, p < 0.01. (F) WT MEF cells were treated with vehicle (i) or 75 μM 6-OHDA (ii-v) for 3 h. Cells were subjected to electron microscopy. Representative images are shown. Black arrowheads indicate lysosome. Black arrows indicate autophagosome/amphisome and lysosome. White arrowhead indicates autolysosome. White arrow indicates phagophore. (G) WT MEF cells grown on coverslips were treated with 75 μM 6-OHDA for 3 h. Cells were processed for immunofluorescence staining with antibody against PARK7 or LC3B. Representative confocal images are shown. White arrows indicate co-localization. Scale bar: 10 μm.
In this context, to determine whether autophagy could be induced under the same conditions for which we had observed that 6-OHDA treatment increased PARK7 secretion, we assessed the effects of 6-OHDA on PARK7 secretion and autophagy induction in WT MEF cells, which have been widely used in autophagy research. We found that 6-OHDA treatment increased PARK7 secretion in MEF cells, although the amount of secreted PARK7 from 6-OHDA-treated MEF cells was less than that from 6-OHDA-treated SH-SY5Y cells (Figure 4(B)). Significant LDH release was not observed in cells treated with 100 μM 6-OHDA (data not shown). We further found that significant increase in LC3B-II levels was observed in cells treated with 75 μM 6-OHDA (Figure 4(C)). Since it had previously been indicated that an increase in LC3B-II level may be associated with either enhanced formation of autophagosomes or impairment of autophagosome turnover [41], we decided to evaluate the effect of co-treatment of 6-OHDA with bafilomycin A1, which inhibits acidification inside the lysosome and to block fusion between the autophagosome and the lysosome. Results showed that LC3B-II levels increased in 6-OHDA-treated cells, and that this increase was enhanced by co-treatment of 6-OHDA with bafilomycin A1, indicating that there was increase in autophagic flux during 6-OHDA treatment in MEF cells (Figure 4(D)). We also evaluated the effect of bafilomycin A1 on autophagic degradation and on secretion of PARK7, finding as a result that although LC3B-II levels increased in response to treatment with bafilomycin A1 alone (Figure (4)), this did not result in induction of PARK7 secretion (Figure 4(E)). We further found that this increase in 6-OHDA-induced PARK7 secretion was suppressed by co-treatment with bafilomycin A1, suggesting the possibility that autophagic flux is important for PARK7 secretion and that PARK7 is not a substrate for autophagic degradation. We also used a fluorescent probe for lysosomes, LysoTracker Red, to evaluate the lysosomal membrane integrity. Decreased fluorescence of LysoTracker Red reflects lysosomal membrane permeabilization and/or an increase in lysosomal pH. As control, bafilomycin A1 treatment caused a decrease in LysoTracker Red fluorescence both in SH-SY5Y cells (Fig. S2A) and WT MEF cells (Fig. S2B), as expected. In contrast, the staining pattern of LysoTracker Red puncta in 6-OHDA-treated cells was similar to that in vehicle-treated cells. The lysosomal membrane integrity was further evaluated by subcellular fractionation. Results showed that in vehicle-treated cells, lysosomal protease CTSB (cathepsin B) was found almost exclusively in the membrane organellar protein-rich NP-40 extract but not in the cytosolic protein-rich digitonin extract (Fig. S2C). 6-OHDA treatment did not significantly change the localization of CTSB. Together, these results suggest the conditions in which 6-OHDA induced secretion of PARK7 do not affect lysosomal membrane integrity.
We also performed transmission electron microscopy analysis to elucidate the induction of autophagy in 6-OHDA-treated WT MEF cells. Results indicated that while there were no obvious autophagic structures in the vehicle-treated cells (Figure 4Fi), there were structures (Figure 4Fii) in 6-OHDA-treated cells that seemed to be phagophores (Figure 4Fiii), autophagosomes, amphisomes (Figure 4Fiv), and autolysosomes (Figure 4Fv). We then evaluated intracellular localization of PARK7 by confocal microscopy. Although most of the PARK7 signal was observed in the cytoplasm, 6-OHDA treatment caused the modest but significant localization of PARK7 in LC3B-positive puncta (Figure 4(G)). Together, these results suggest that the same conditions in which 6-OHDA induces secretion of PARK7 also induce autophagy.
PARK7 secretion is suppressed in autophagy-deficient MEF cells and ATG16L1 knockdown SH-SY5Y cells
To further examine the involvement of autophagy in 6-OHDA-induced PARK7 secretion, we investigated the response to 6-OHDA in Atg5-deficient (atg5-/-) MEF cells, as ATG5 is known to play an essential role in autophagosome formation [42]. Results for atg5-/- MEF cells indicated that there was no LC3B-II formation and that 6-OHDA did not induce PARK7 secretion (Figure 5(A)). We further employed Atg9-deficient (atg9-/-) and Atg16l1-deficient (atg16l1-/-) MEF cells, in both of which autophagosome formation was impaired [43,44]. Consistent with the results obtained for atg5-/- MEF cells, 6-OHDA-induced PARK7 secretion and LC3B-II formation were each suppressed in both atg9-/- MEF cells (Figure 5(B)) and atg16l1-/- MEF cells (Figure 5(C)). Furthermore, we performed siRNA knockdown of ATG16L1 in SH-SY5Y cells and confirmed a marked decline in ATG16L1 protein levels (Figure 5(D)). Under these conditions, 6-OHDA-induced increase in LC3B-II levels was suppressed (Figures 5(D)) and 6-OHDA-induced PARK7 secretion was also significantly inhibited (Figure 5(E)) as observed in atg16l1-/- MEF cells. Taken together, these results suggest that autophagic flux is required for unconventional secretion of PARK7.
Figure 5.
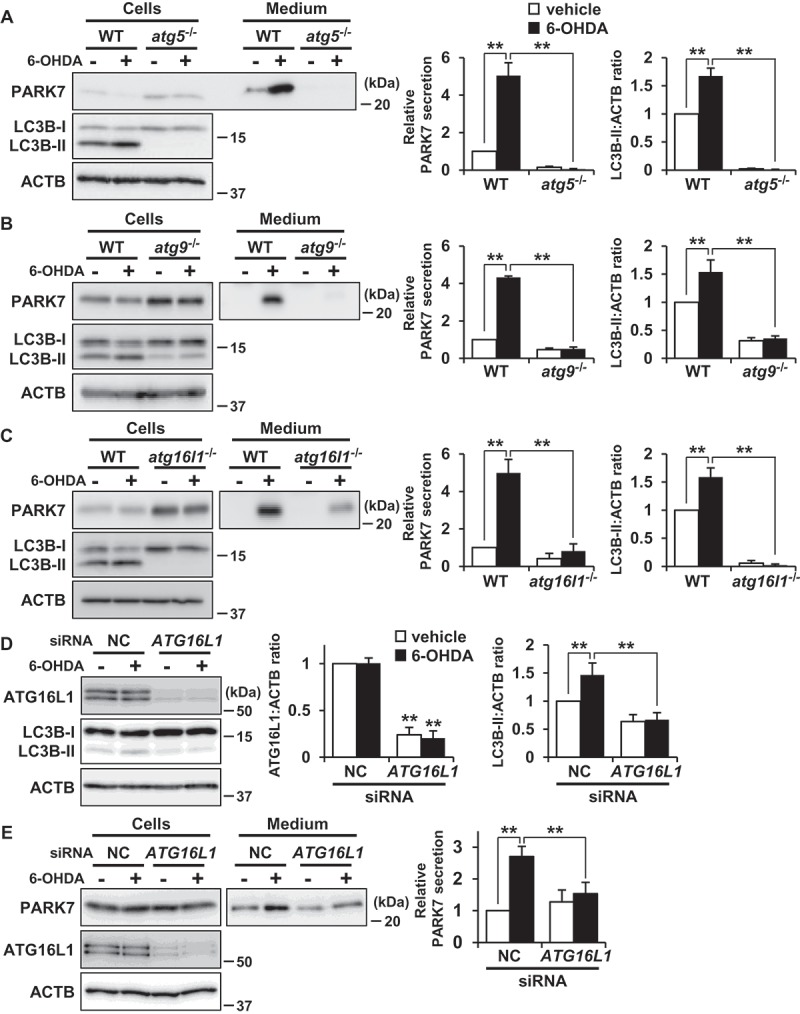
ATG5, ATG9, and ATG16L1 were involved in 6-OHDA-induced PARK7 secretion. (a-c) WT MEF, atg5−/- MEF (A), atg9−/- MEF (B) or atg16l1−/- MEF (C) cells were treated with 75 μM 6-OHDA for 3 h and were then cultured in serum-free medium for 2 h. Whole cell lysates and the conditioned medium were immunoblotted using antibodies specific for PARK7, LC3B, or ACTB. PARK7, LC3B and ACTB band intensities were quantified by densitometric scanning. Relative PARK7 secretion level to vehicle-treated cells and LC3B-II:ACTB ratio are shown. n = 3; mean ± S.D.; **, p < 0.01. (D and E) WT MEF cells were transfected with ATG16L1 or negative control (NC) siRNA oligo for 72 h. The cells were treated with 75 μM 6-OHDA for 3 h (D), followed by culture in serum-free medium for 2 h (E). Whole cell lysates and the conditioned medium were immunoblotted with using antibodies specific for ATG16L1, LC3B, PARK7, or ACTB. ATG16L1, LC3B, PARK7 and ACTB band intensities were quantified by densitometric scanning. ATG16L1:ACTB and LC3B-II:ACTB ratios (d) and relative PARK7 secretion level to vehicle-treated cells (e) are shown. n = 3; mean ± S.D.; **, p < 0.01.
To evaluate whether extracellular PARK7 was associated with small vesicles, we performed ultracentrifugation fractionation of conditioned medium at 100,000 x g to separate vesicle-associated proteins from soluble proteins. CAV1 (caveolin 1), secretion of which has been reported to occur with vesicles [45], was recovered from the pellet fraction, while PARK7 was recovered from the supernatant fraction, in both the medium collected from vehicle and 6-OHDA-treated cells (Fig. S3). These results suggest that extracellular PARK7 exists as soluble protein.
Oxidative stress is implicated in 6-OHDA-induced autophagy and PARK7 secretion
We then sought to test whether 6-OHDA-induced oxidative stress stimulated autophagy and PARK7 secretion. As had been observed in SH-SY5Y cells, we observed that GSH content was significantly reduced by 6-OHDA treatment in WT MEF cells (Figure 6(A)). We also used the fluorescent probe DCFH-DA to monitor intracellular ROS levels, finding as a result that treatment with 6-OHDA caused an increase in ROS levels as compared with vehicle-treated cells, and that this increase could be mitigated by treatment with NAC (Figure 6(B)). We further found that 6-OHDA-induced PARK7 secretion could be suppressed by treatment with NAC (Figure 6(C)) in a similar manner as had been observed in SH-SY5Y cells (Figure 3(B)). Under conditions which were the same as the foregoing, we further noted that 6-OHDA induced an increase in LC3B-II levels, and that this increase could be suppressed by co-treatment with NAC (Figure 6(D)). Together, these results suggest that 6-OHDA-induced oxidative stress is implicated in autophagy-based unconventional secretion of PARK7.
Figure 6.
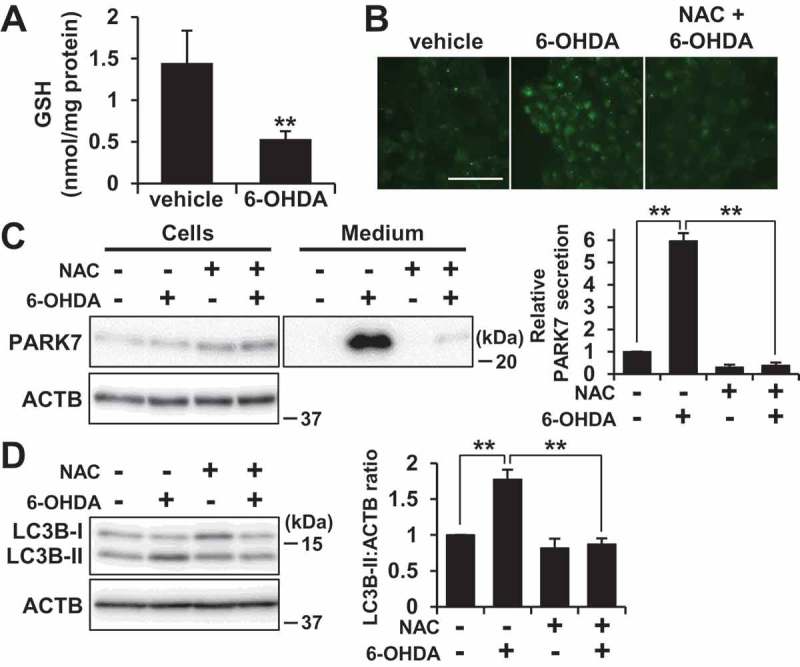
NAC treatment suppressed 6-OHDA-induced autophagy and PARK7 secretion. (A) WT MEF cells were treated with 75 μM 6-OHDA for 3 h and were then subjected to GSH assay. n = 3; mean ± S.D.; **, p < 0.01. (B-D) WT MEF cells were pre-treated with or without 2 mM NAC and were then treated with 75 μM 6-OHDA for 3 h (B and D), followed by culture in serum-free medium for 2 h (C). (B) ROS generation was determined using a fluorescence probe, DCFH-DA. Fluorescence was detected using a fluorescence microscope. Representative fluorescence images are shown. Scale bar: 100 μm. (C and D) Whole cell lysates and the conditioned medium were immunoblotted using antibodies specific for PARK7, LC3B, or ACTB. PARK7, LC3B and ACTB band intensities were quantified by densitometric scanning. Relative PARK7 secretion level to vehicle-treated cells (c) and LC3B-II:ACTB ratio (d) are shown. n = 3; mean ± S.D.; **, p < 0.01.
Activation of AMPK and ULK1 is implicated in 6-OHDA-induced autophagy and PARK7 secretion
Because a cytotoxic concentration of 6-OHDA has been reported to induce AMPK-dependent autophagy in SH-SY5Y cells [46], we examined whether activation of the AMPK pathway could be made to occur under the same conditions as those for which we had observed that 6-OHDA caused secretion of PARK7. We investigated the effect of 6-OHDA on phosphorylation of AMPK at Thr172 (AMPK activation) and on phosphorylation by AMPK of its downstream effector ULK1 at Ser555 (ULK1 activation). Since the inhibitory function of MTOR in autophagy is well-established [41], the phosphorylation of RPS6KB1/p70S6K (ribosomal protein S6 kinase B1) at Thr389 by MTOR was also evaluated to assess MTOR activity. As a control, the MTOR inhibitor rapamycin was employed, this being observed to cause a decrease in RPS6KB1 phosphorylation without causing stimulation of AMPK and ULK1 phosphorylation (Figure 7(A)), as expected. In contrast, 6-OHDA treatment enhanced phosphorylation of AMPK and its substrate ULK1, but did not significantly change the degree of phosphorylation of RPS6KB1. We therefore evaluated the effect of NAC on AMPK phosphorylation, finding as a result that NAC treatment significantly diminished AMPK phosphorylation in a manner roughly paralleling the observed decrease in LC3B-II levels (Figure 7(B)). Because AMPK activation is regulated by the AMP:ATP ratio, we assessed AMP and ATP levels, upon which we found that 6-OHDA treatment increased the AMP:ATP ratio (Figure 7(C)). These results suggest that the same conditions which exist when 6-OHDA induces oxidative stress-mediated PARK7 secretion and autophagy also cause activation of the AMPK-ULK1 pathway without affecting MTOR activity.
Figure 7.
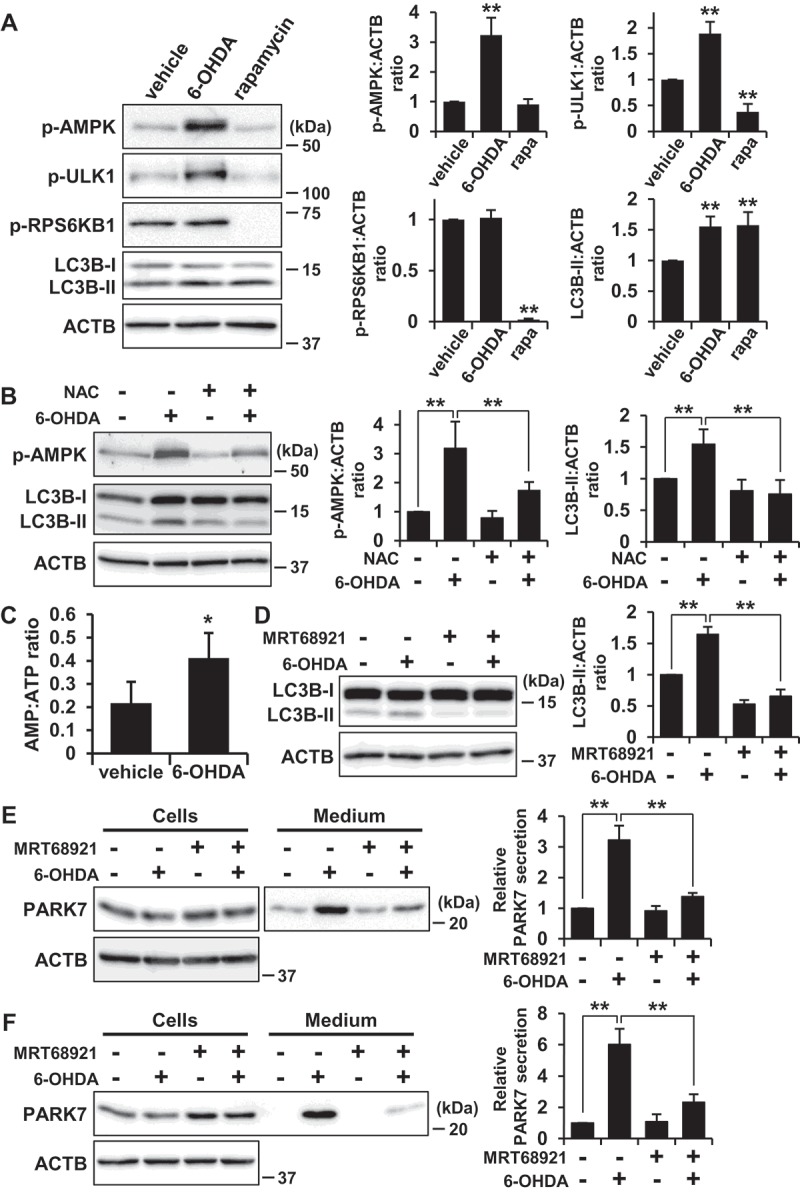
6-OHDA-induced AMPK-ULK1 phosphorylation was associated with PARK7 secretion in WT MEF cells. (A) WT MEF cells were treated with 75 μM 6-OHDA or 2 μM rapamycin for 3 h. Whole cell lysates were immunoblotted using antibodies specific for phospho-AMPK (Thr172), phospho-ULK1 (Ser555), phospho-RPS6KB1 (Thr389), LC3B, or ACTB. Band intensities were quantified by densitometric scanning and p-AMPK:ACTB, p-ULK1:ACTB, p-RPS6KB1:ACTB and LC3B-II:ACTB ratios are shown. n = 3; mean ± S.D.; **, p < 0.01. (B) WT MEF cells were pre-treated with or without 2 mM NAC and were then treated with 75 μM 6-OHDA for 3 h. Whole cell lysates were immunoblotted using antibodies specific for phospho-AMPK, LC3B, or ACTB. Band intensities were quantified by densitometric scanning and p-AMPK:ACTB and LC3B-II:ACTB ratios are shown. n = 3; mean ± S.D.; **, p < 0.01. (c) WT MEF cells were treated with 75 μM 6-OHDA for 3 h and were then subjected to AMP:ATP assay. n = 3; mean ± S.D.; *, p < 0.05. (D and E) SH-SY5Y cells were pre-treated with or without 1 μM MRT68921 for 30 min and were then treated with 75 μM 6-OHDA for 3 h (D), followed by culture in serum-free medium for 2 h (E). Whole cell lysates and the conditioned medium were immunoblotted using antibodies specific for LC3, PARK7, or ACTB. LC3B, PARK7 and ACTB band intensities were quantified by densitometric scanning. LC3B-II:ACTB ratio (d) and relative secretion level to vehicle-treated cells (e) are shown. n = 3; mean ± S.D.; **, p < 0.01. (F) WT MEF cells were pre-treated with or without 2 μM MRT68921 for 30 min and were then treated with 75 μM 6-OHDA for 3 h, followed by culture in serum-free medium for 2 h. Whole cell lysates and the conditioned medium were immunoblotted using antibodies specific for PARK7 or ACTB. PARK7 band intensities were quantified by densitometric scanning and relative secretion level to vehicle-treated cells is shown. n = 3; mean ± S.D.; **, p < 0.01.
To confirm the involvement of the AMPK-ULK1 pathway in 6-OHDA-induced autophagy and PARK7 secretion, SH-SY5Y cells were treated with MRT68921, a specific inhibitor of ULK1 [47], as a result of which it was found that 6-OHDA-induced increase in LC3B-II levels was suppressed by co-treatment with MRT68921 (Figure 7(D)). Furthermore, 6-OHDA-induced PARK7 secretion was suppressed by co-treatment with MRT68921 (Figure 7(E)). Consistent with the results obtained for SH-SY5Y cells, 6-OHDA-induced PARK7 secretion was suppressed in WT MEF cells treated with MRT68921 (Figure 7(F)). Together, these results suggest that AMPK-ULK1 pathway is involved in 6-OHDA-induced autophagy and PARK7 secretion.
Trehalose but not rapamycin promotes PARK7 secretion in WT MEF cells
We then investigated whether another stimulator of autophagy could induce PARK7 secretion. In WT MEF cells, rapamycin treatment increased LC3B-II levels both in the presence and absence of bafilomycin A1 as expected (Figure 8(A)). When the cells were treated with 1–5 μM rapamycin, an increase in PARK7 secretion was observed, but not to the same degree as had been observed with 6-OHDA treatment (Figure 8(B)). We next evaluated the effect of trehalose. Trehalose is a natural disaccharide found in many non-mammalian species that induces MTOR-independent autophagy by inhibiting hexose uptake and subsequently activating the AMPK-ULK1 pathway [48,49]. It should be noted that it has recently been reported that release of SNCA (synuclein alpha) is upregulated by trehalose treatment in rat pheochromocytoma PC12 cells [50]. When WT MEF cells were treated with trehalose, we found that LC3B-II levels were increased in a concentration-dependent manner (Figure 8(C)), confirming that there was activation of autophagic flux under the conditions tested. Furthermore, 200 mM trehalose enhanced PARK7 secretion at the same level with 6-OHDA treatment (Figure 8(D)) without significant LDH release (data not shown). Together, these results suggest the possibility that 6-OHDA-induced PARK7 secretion is mainly mediated by MTOR-independent autophagy.
Figure 8.
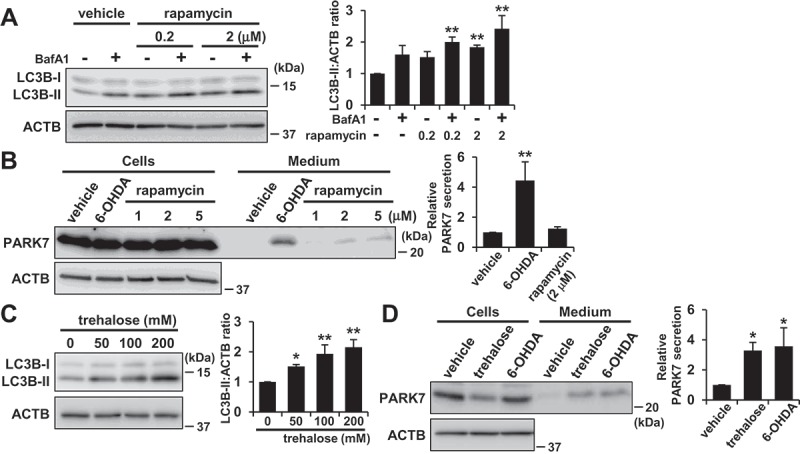
Trehalose but not rapamycin promoted PARK7 secretion in WT MEF cells. (A) WT MEF cells were treated with 100 nM BafA1 for 1 h and were then treated with 0–2 μM rapamycin for 3 h. Whole cell lysates were immunoblotted using antibodies specific for LC3B or ACTB. LC3B and ACTB band intensities were quantified by densitometric scanning and LC3B-II:ACTB ratio is shown. n = 3; mean ± S.D.; **, p < 0.01. (B) WT MEF cells were treated with 1–5 μM rapamycin or 75 μM 6-OHDA for 3 h and were then cultured in serum-free medium for 2 h. Whole cell lysates and the conditioned medium were immunoblotted using antibodies specific for PARK7 or ACTB. PARK7 band intensities were quantified by densitometric scanning and relative secretion level to vehicle-treated cells is shown. n = 3; mean ± S.D.; **, p < 0.01. (C) WT MEF cells were treated with 0–200 mM trehalose for 48 h. Whole cell lysates were immunoblotted using antibodies specific for LC3B or ACTB. LC3B and ACTB band intensities were quantified by densitometric scanning and LC3B-II:ACTB ratio is shown. n = 3; mean ± S.D.; *, p < 0.05; **, p < 0.01. (D) WT MEF cells were treated with 200 mM trehalose for 48 h or 75 μM 6-OHDA for 3 h and were then cultured in serum-free medium for 2 h. Whole cell lysates and the conditioned medium were immunoblotted using antibodies specific for PARK7 or ACTB. PARK7 band intensities were quantified by densitometric scanning and relative secretion level to vehicle-treated cells is shown. n = 3; mean ± S.D.; *, p < 0.05.
Discussion
In our present study, we demonstrate that PARK7 is secreted via the autophagy-mediated unconventional secretion mechanism. Autophagy is generally understood to be the process by which cytosolic proteins and damaged organelles are degraded within lysosomes [41]. During macroautophagy, which is the most prevalent form of autophagy, some cytoplasmic components including organelles are enclosed within a double-membraned structure referred to as an autophagosome. The autophagosome undergoes the fusion with a lysosome (autolysosome) to achieve degradation of its internal contents. The autophagosome can also fuse with an endosome to form an amphisome before fusion with a lysosome. To understand the unconventional secretion mechanism, autophagy may be the mechanism by which cytosolic proteins are secreted into the extracellular space across the hydrophobic environment of the plasma membrane. It is conceivable that some form of autophagic structure (autophagosome, amphisome, or autolysosome) may fuse with the plasma membrane to release the enclosed cytosolic contents into the extracellular space. Indeed, autophagy-mediated unconventional secretion, termed secretory autophagy, has been implicated in the secretion of a subset of unconventionally secreted proteins which include yeast Acb1/DBI, mammalian IL1B, and IDE (insulin degrading enzyme) [8,38–40]. Our study indicates that PARK7 is also secreted via the secretory autophagy. It is however not clear how such a secretory protein might be selected for secretory autophagy and escape lysosomal degradation. One possibility is that the secretion rather than degradation of cytosolic constituents that occurs in secretory autophagy may be induced by a shared but partially divergent autophagic machinery. In our present study, we demonstrated that 6-OHDA-induced PARK7 secretion required the autophagy-related proteins ATG5, ATG9, and ATG16L1, suggesting that ATG proteins which govern biogenesis of autophagic membranes are needed for secretory autophagy-mediated PARK7 secretion. Interestingly, we also showed that 6-OHDA induced MTOR-independent autophagy via the AMPK-ULK1 pathway. We furthermore observed that the MTOR-independent autophagy inducer trehalose caused PARK7 secretion to a degree on the same order as that observed with 6-OHDA treatment, but that the MTOR inhibitor rapamycin only poorly stimulated PARK7 secretion as compared with 6-OHDA treatment. Since MTOR senses cellular nutritional status and is involved in the induction of degradative autophagy that occurs in response to nutrient starvation [41], understanding the role that MTOR plays should assist in elucidating the respective contributions of the MTOR signaling pathway and the MTOR-independent signaling pathway to secretory autophagy.
Several cellular stresses, e.g., inflammation, nutrient stress, and ER stress, have been reported to trigger secretory autophagy [8,38–40]. In the present study, we demonstrated that oxidative stress induced by 6-OHDA also triggered secretory autophagy. Oxidative stress has been implicated in the induction of degradative autophagy [51]. It has been reported that oxidation of Atg4 at cysteine inactivates its delipidating activity on LC3, thereby leading to the induction of autophagosome formation [52]. It has also been proposed that AMPK may be activated in response to increased intracellular ROS levels, particularly through S-glutathionylation [53]. 6-OHDA has been reported to impair the mitochondrial respiratory chain, leading to a decrease in ATP levels [33]. We therefore concluded that 6-OHDA-induced oxidative stress, and the resulting increase in the AMP:ATP ratio and AMPK-ULK1 activation, were implicated in PARK7 secretion at least under the conditions tested. Indeed, we demonstrated in the present study that ULK1 inhibition suppressed 6-OHDA-induced PARK7 secretion. Because the AMPK inhibitor compound C has been reported to induce autophagy in some type of cells [54], it will be necessary to pay attention to evaluate the effect of AMPK inhibition by compound C or AMPK siRNA on PARK7 secretion.
Our study suggests that AMPK-ULK1-mediated but MTOR-independent signaling regulation plays an important role in the distinctive induction of secretory autophagy-mediated PARK7 secretion. In addition, we speculate that a dedicated molecular machinery for formation or intracellular trafficking of autophagic structures may account for the distinctive secretory pathway of PARK7. Further investigations to identify the involvement of other autophagy-related proteins in PARK7 secretion are required for in-depth analysis. It should be noted that Galluzi et al. encourage the use of molecularly-oriented descriptors for non-degradative functions of autophagic machinery [55]. In line with this definition, the mechanisms of PARK7 secretion may need to be referred to as ATG5, ATG9, and ATG16L1-dependent secretion instead of secretory autophagy-mediated secretion. In future studies, it would also be of interest to evaluate whether other known key proteins involved in secretory autophagy, such as RAB8A (RAB8A, member RAS oncogene family) and GORASP2/GRASP55 (golgi reassembly stacking protein 2) [39,40], are implicated in PARK7 secretion, and whether 6-OHDA-induced secretory autophagy is implicated in the secretion of other proteins. It has been recently reported that dedicated SNAREs participate in secretory autophagy-mediated unconventional secretion of IL1B in response to lysosome damage [56]. Interestingly, secretion of IL1B depends on plasma membrane STX3 (syntaxin 3) and STX4 (syntaxin 4) instead of delivery to lysosomes via STX17 (syntaxin 17), which has been identified as a necessary protein for autophagosomal fusion with the endosome/lysosome [57]. To consider how autophagy-mediated PARK7 secretion separates from degradative autophagy, further investigations to elucidate the possible involvement of dedicated SNARE machinery in PARK7 secretion may provide another insight.
With respect to the differences between target proteins for degradative versus secretory autophagy, it is possible that secretory proteins in autophagic vacuoles may possess some protective modification or domain that contributes in some way to prevent lysosomal degradation. It has been demonstrated that the SlyX domain (EKPPHY) of IDE contributes to IDE secretion by preventing its lysosomal degradation [40,58]. Furthermore, KFERQ-like motifs of IL1B are thought to be required for entry of IL1B into the lumen of the phagophore so as to permit secretion as a soluble protein either through a direct fusion of the autophagosome with the plasma membrane or via the multi-vesicular body pathway [59]. It is also known that a KFERQ-like motif is present in substrates of chaperone-mediated autophagy [41]. In the case of PARK7, we found that there are several KFERQ-like motifs in the PARK7 amino acid sequence. Because we found in the present study that PARK7 secretion is mediated by secretory autophagy, studies to elucidate the possible involvement of KFERQ-like motifs in PARK7 secretion may provide further insight.
Electron microscopy analysis showed that 6-OHDA treatment induced formation of structures typically observed where there is involvement of the autophagic pathway, such as autophagosomes, amphisomes, and autolysosomes. Since ATG5, ATG9, and ATG16L1 are known to play essential roles in autophagosome formation [41–44], one possibility is that cytosolic PARK7 may be packaged within LC3-positive autophagic vacuoles for secretion. Indeed, in addition to cytosolic distribution of PARK7 signal, we observed the modest but significant localization of PARK7 in LC3B-positive puncta in the cells treated with 6-OHDA. Since our rough estimate of PARK7 band intensity suggested that less than 10% of PARK7 was secreted from cells in response to 6-OHDA treatment, we posit that this might affect the modest distribution of PARK7 in autophagic vacuoles. Since ultracentrifugation fractionation of conditioned medium showed that extracellular PARK7 was not associated with exosomes or other such small vesicles, we speculate that the PARK7-containing autophagic vacuole, if it exists, is most likely fusing with the plasma membrane to cause release of its soluble contents including PARK7 into the extracellular space, but we cannot exclude the possibility that PARK7 is first secreted via exosomes/ectosomes and then released from vesicle in the extracellular space.
Several lines of evidence suggest that secreted PARK7 may play a functional role in the extracellular space. For example, PARK7 secreted from osteoblasts has been found to promote angiogenesis and osteogenesis [32]. Extracellular deposition of abnormal TTR (transthyretin) protein has been shown to cause familial amyloid polyneuropathy, and secreted PARK7 has been shown to degrade aggregated TTR to protect against the onset of amyloid polyneuropathy [26]. And with respect to PD, PARK7 has been detected in the cerebrospinal fluid of patients suffering from PD [28,29], and it has been found that administration of recombinant PARK7 into the brain of 6-OHDA-induced PD rat model improved PD phenotype [60], suggesting that extracellular PARK7 might exert a protective role against PD. There is a possibility that secreted PARK7 may protect neighboring cells from oxidative stress by means of a process similar to paracrine signaling, which possibility is thought to be supported by reports that PARK7 knockout astrocytes show inferior ability to protect neuronal cells against 6-OHDA-induced cell death both by co-culture and through astrocytes conditioned medium [61]. Since PARK7 has 3 reactive cysteine residues, it is also possible that secreted PARK7 itself receives oxidative modification by scavenging ROS within the extracellular space. Indeed, when the conditioned medium obtained from non-treated cells was incubated in the presence of 6-OHDA, the ratio of oxPARK7 to total PARK7 was increased, suggesting that oxidation of PARK7 occurred in the presence of 6-OHDA within the extracellular space (Fig. S4).
Here we have presented results indicating that PARK7 is secreted via the autophagy-based unconventional secretory pathway. In summary, our data suggest that electrophilic 6-OHDA quinone lowers GSH levels and induces oxidative stress, and that this is followed by AMPK-ULK1 activation to induce secretory autophagy, resulting in unconventional secretion of PARK7 (Figure 9).
Figure 9.
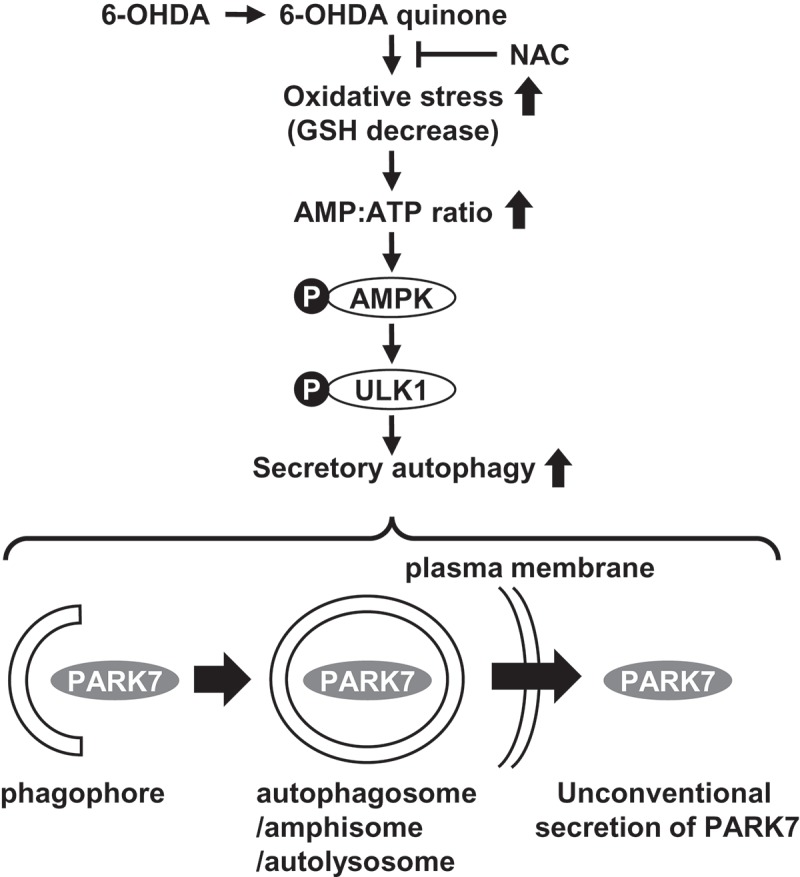
The proposed mechanism of autophagy-based unconventional secretion of PARK7 induced by 6-OHDA. The electrophilic 6-OHDA quinone formed from 6-OHDA lowers GSH levels and induces oxidative stress. Oxidative stress subsequently causes increase in AMP:ATP ratio and activates the AMPK-ULK1 pathway. This induces secretory autophagy, resulting in unconventional secretion of cytosolic PARK7.
Materials and methods
Materials
6-hydroxydopamine hydrochloride (6-OHDA; H4381), catalase (C8531), rotenone (R8875), MRT68921 (SML1644), and trehalose (T9531) were obtained from Sigma-Aldrich. Bafilomycin A1 (023–11641), benzoquinone (171–00242), H2O2 (080–01186), N-acetyl-L-cysteine (NAC; 017–05131), and Z-VAD(OMe)-FMK (zVAD; 269–02071) were from Wako. Brefeldin A (203729) was from Merck. Ac-YVAD-CMK (YVAD; 10014) was from Cayman Chemical Company. Rapamycin (S1039) was obtained from Selleck Chemicals. The following antibodies were from commercial sources: anti-PARK7/DJ-1 (ab4150), anti-CTSB (abcam, ab58802); anti-RPN1 (Santa Cruz Biotechnology, sc-12164); anti-FN1 (610077), anti-CAV1 (BD Biosciences, 610060); anti-VDAC1 (Merck, AP1059); anti-LMNA (2032), anti-CASP3 (9662), anti-ATG16L1 (8089), anti-phospho-AMPK Thr172 (2535), anti-phospho-ULK1 Ser555 (5869), anti-phospho-RPS6KB1/p70S6K Thr389 (Cell Signaling Technology, 9234); and anti-LC3B (L7543), and anti-ACTB (Sigma-Aldrich, A5441). Monoclonal antibody against PARK7 (Clone 3843) was prepared as previously described [62]. All other chemicals, of analytical grade, were obtained from Sigma-Aldrich or Wako.
Cell culture
Human neuroblastoma cells from the SH-SY5Y cell line (CRL-2266) were purchased from ATCC. WT MEF and atg5−/- MEF cells [42] were kindly provided by Dr. Noboru Mizushima (The University of Tokyo). The atg9−/- MEF and atg16l1−/- MEF cells [43,44] were kindly provided by Dr. Shizuo Akira (Osaka University). Cells were maintained in Dulbecco’s Modified Eagle’s Medium/Nutrient Mixture F-12 (DMEM/F12; Thermo Fisher Scientific, 11320–033), to which 10% fetal bovine serum (Hyclone, SH30370.03) and antibiotics (100 U/ml penicillin, 100 μg/ml streptomycin; Thermo Fisher Scientific, 15140122) had been added. Cells were grown at 37°C in an atmosphere of 5% CO2.
Cell treatment
6-OHDA was dissolved in phosphate-buffered saline (PBS; 137 mM NaCl [Wako, 195–01663], 2.7 mM KCl [Wako, 163–03545], 8.1 mM Na2HPO4 [Wako, 193–02845], 7.4 mM KH2PO4 [Wako, 169–04245]). Brefeldin A, zVAD, YVAD, and bafilomycin A1 were dissolved in dimethyl sulfoxide (DMSO; Wako, 043–07216). To examine PARK7 secretion, SH-SY5Y or MEF cells plated on a 6-well dish were treated with 50–100 μM 6-OHDA for 3 h and were then cultured in serum-free medium for 2 h. To evaluate the effects of brefeldin A, zVAD, YVAD, NAC, bafilomycin A1, MRT68921, trehalose, or rapamycin, cells were pretreated with 2 μg/ml brefeldin A for 3 h, or 20 μM zVAD or YVAD for 1 h, or with 2 mM NAC for 2 h, or with 100 nM bafilomycin A1 for 1 h, or 1–2 μM MRT68921 for 30 min, or 200 mM trehalose for 48 h, or 1–5 μM rapamycin for 3 h before further treatments. For benzoquinone treatment, cells were treated with 20 μM benzoquinone in serum-free medium for 3 h. For rotenone treatment, cells were treated with 0.1 or 0.2 μM rotenone for 24 h and were then cultured in serum-free medium for 2 h.
Assessment of protein secretion
Treated cells were harvested and whole cell lysates were prepared in lysis buffer (10 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% NP-40 [Sigma-Aldrich, I3021], 0.1% SDS [Wako, 191–07145], and 5 mM EDTA-2Na [Dojindo, 345–01865]) plus protease inhibitor cocktail (Nacalai Tesque, 25955–11). Protein concentration was determined by BCA assay kit (Thermo Fisher Scientific, 23225). Cultured medium from treated cells was collected and cell debris was removed by centrifugation. To concentrate the protein fraction in medium, 1 ml supernatant was mixed with 10 μg BSA carrier protein (Wako, 011–27055) and 110 μl of 100% w:v trichloroacetic acid and placed on ice for 1 h. Precipitated proteins were pelleted for 5 min at 16,000 x g at 4°C. Pelleted proteins were washed with 500 μl cold acetone and resuspended in 20 μl SDS-PAGE sample buffer (62.5 mM Tris-HCl, pH 6.8, 0.012% bromophenol blue [Wako, 029–02912], 2% SDS, 5% sucrose [Wako, 196–00015]). The protein samples were subjected to SDS-PAGE or 2D-PAGE, and to immunoblotting using appropriate antibodies as indicated in figures. An LDH (lactate dehydrogenase) release assay was performed as described previously [63]. Data are expressed as percentage of total LDH activity, after subtraction of background as determined from the medium alone.
2D-Page
Trichloroacetic acid-precipitated whole cell lysates or medium were mixed with rehydration buffer (9 M urea [Wako, 211–01213], 2% CHAPS [Dojindo, 347–04723], 65 mM dithioerythritol [Sigma-Aldrich, D8255], IPG buffer [pH 4–7; GE Healthcare Bioscience, 17–6000–86)]) and applied to IPG gel strips (pH 4–7, non-linear, 7 cm; GE Healthcare Bioscience, 17–6001–10). Electrophoresis voltage was increased stepwise to reach a final voltage of 5,000 V or 8,000 V at a maximum current of 200 mA for 3–5 h. The second-dimension separation was carried out through SDS-PAGE. Densitometric analysis of reduced PARK7 and oxidized PARK7 spots were quantified by Multi Gauge software (Fujifilm), ratios (oxPARK7:total PARK7) shown being mean values.
Subcellular fractionation
SH-SY5Y cells were homogenized using a Dounce homogenizer with 60 strokes in 200 μl homogenization buffer (250 mM sucrose, 20 mM Tris-HCl, pH 7.4, 1 mM EDTA-2Na) with protease inhibitor cocktail as described previously [64]. The resulting homogenate was centrifuged at 800 x g for 10 min twice at 4°C. The post-nuclear supernatant was pelleted at 8,000 x g for 10 min twice at 4°C. The resulting supernatant was further centrifuged at 100,000 x g for 30 min in a Beckman TLA100.3 rotor at 4°C. Each pellet was resuspended in 200 μl homogenization buffer. Equal aliquots from each fraction were analyzed for immunoblotting.
Crude cytosolic and membrane/organelle fractions were isolated from WT MEF cells by sequential detergent extraction as described previously [65]. Briefly, cells were resuspended by gentle pipetting in 200 μl digitonin buffer (150 mM NaCl, 10 mM Tris-HCl, pH 7.4, 40 μg/ml digitonin [Wako, 043–21371]). After 10 min incubation on ice, the cell suspension was centrifuged at 8,000 x g for 5 min at 4°C. The resulting supernatant was kept as cytosol-enriched digitonin extract. The resulting pellet was resuspended by vortexing in 200 μl NP-40 buffer (150 mM NaCl, 10 mM Tris-HCl, pH 7.4, 1% NP-40). After 30 min incubation on ice, samples were centrifuged at 13,000 x g for 5 min at 4°C. The resulting supernatant was kept as membrane organellar protein-enriched NP-40 extract. Equal aliquots from each fraction were analyzed for immunoblotting.
DNA constructs and transfection
Human WT or C106S PARK7 cDNA was cloned into pEGFP-N1 (Clontech, 6085–1). Human HEK293 cells (ATCC, CRL-1573) were transiently transfected using Lipofectamine 2000 (Thermo Fisher Scientific, 11668). To generate stable transfectant, cells were selected with 0.8 mg/ml G418 (Nacalai Tesque, 16512–94).
Determination of cellular GSH content
Cells were lysed in 0.2 M perchloric acid (Wako, 162–00695). The deproteinized supernatant was adjusted with 1 M CH3COONa (Wako, 192–01075) to approximately pH 3, and this was then subjected to HPLC analysis using a column (Eicom, Eicompak SC-5ODS 3 × 150 mm) equipped with an electrochemical detection system (Eicom, HTEC-500). Flow rate of eluent (99% 0.1 M sodium phosphate buffer, pH 2.5, 1% methanol, 100 mg/l sodium octanesulfonate [Sigma-Aldrich, O0133], and 50 mg/l EDTA-2Na) was 0.5 ml/min. Glutathione (GSH) content was calculated using reduced GSH as standard, this being expressed as nmol per mg of total protein per assay.
Detection of lysosome or intracellular ROS by fluorescence microscopy
For lysosome staining, cells grown on coverslips were treated with 6-OHDA or bafilomycin A1 for 3 h. Cells were then stained with LysoTracker Red DND-99 (Thermo Fisher Scientific, L7528) at a final concentration of 500 nM in serum-free medium for 1 h at 37°C. To detect intracellular ROS generation, cells were pre-incubated with 10 μM of DCFH-DA (Thermo Fisher Scientific, D399) for 1 h before 6-OHDA treatment. Cells were fixed with 4% paraformaldehyde (Wako, 166–2325)/PBS for 20 min and were washed with PBS twice. Cell nuclei were stained with Hoechst 33342 (Dojindo, 346–07951). Fluorescence was detected using a fluorescence microscope (OLYMPUS IX71).
Immunofluorescence staining
Cells grown on glass coverslips were fixed with cold methanol for 10 min at 4°C, and then blocked with 3% BSA in PBS for 1 h. The coverslips were incubated with anti-PARK7 (3843) and anti-LC3B antibodies for 3 h at room temperature. Confocal fluorescence images were obtained using a Zeiss LSM710 confocal microscope with oil objective lens and accompanying software (LSM Software ZEN2009).
Electron microscopy analysis
For electron microscopy, cells were fixed in phosphate-buffered 2% glutaraldehyde (Electron Microscopy Sciences, 16220), and subsequently post-fixed in 2% osmium tetroxide (Nisshin EM, 300–1) for 3 h in an ice bath, following which the cells were dehydrated in ethanol and embedded in epoxy resin. Ultrathin sections were obtained using the ultramicrotome technique. Ultrathin sections stained with uranyl acetate (Cerac) for 10 min and with lead staining solution (Sigma-Aldrich, 18–0875–2) for 5 min were subjected to TEM observation (JEM-1200 EX, JEOL) at Hanaichi Ultrastructure Research Institute (Okazaki, Japan).
Knockdown of ATG16L1 by small interfering RNA
ON-TARGETplus human ATG16L1 siRNA (L-021033) or Non-targeting siRNA (D-001810) were obtained from Dharmacon. SH-SY5Y cells grown in 6-well plates were transfected with siRNA, at a concentration of 30 pmol/well with Lipofectamine RNAiMAX (Thermo Fisher Scientific, 13778) twice according to the manufacturer’s instructions. Seventy-two h after transfection, cells were used for further analyses.
Determination of cellular AMP:ATP ratio
Cells were lysed in 0.25 ml of 0.6 N perchloric acid. Acid-insoluble material was removed by centrifugation at 1,000 x g for 5 min. The supernatant was adjusted with KOH to pH 5.0–7.0 and, after 10 min, was centrifuged at 8,000 x g for 5 min to remove KClO4. HPLC analysis was carried out using an Inertsil ODS-2 column (GL Science). Flow rate of eluent (0.1 M sodium phosphate buffer, pH 6) was 0.5 ml/min. Nucleotides were detected by their absorbances at 260 nm, AMP:ATP ratios being calculated using standards for AMP and ATP.
Statistical analysis
Data are reported as mean ± S.D. of at least 3 independent experiments unless otherwise indicated. Statistical processing was conducted by variance analysis using Tukey test for multiple comparisons and Student t-test for comparison of 2 means. The difference was considered significant when the p value was < 0.05.
Funding Statement
This work was supported by the Ministry of Education, Culture, Sports, Science, and Technology-supported Program for the Strategic Research Foundation at Private Universities in Japan for the years 2012–2016 and 2015–2019, and the Cosmetology Research Foundation.
Acknowledgments
We thank Dr. Noboru Mizushima (The University of Tokyo) and Dr. Shizuo Akira (Osaka University) for providing cell lines. The authors thank Chihiro Satomi, Hitomi Takashima, Daichi Kawashima, Norie Kato for research support and Gerry Peters (JTT K.K., Takatsuki, Japan) for his careful editing of the manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary Material
Supplementary data can be accessd here.
References
- [1].Nickel W, Rabouille C.. Mechanisms of regulated unconventional protein secretion. Nat Rev Mol Cell Biol. 2009;10:148–155. PMID:19122676. [DOI] [PubMed] [Google Scholar]
- [2].Zhang M, Schekman R.. Unconventional secretion, unconventional solutions. Science. 2013;340:559–561. PMID:23641104. [DOI] [PubMed] [Google Scholar]
- [3].Malhotra V. Unconventional protein secretion: an evolving mechanism. EMBO J. 2013;32:1660–1664. PMID:23665917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kinseth MA, Anjard C, Fuller D, et al. The Golgi-associated protein GRASP is required for unconventional protein secretion during development. Cell. 2007;130:524–534. PMID:17655921. [DOI] [PubMed] [Google Scholar]
- [5].Rubartelli A, Cozzolino F, Talio M, et al. A novel secretory pathway for interleukin-1 beta, a protein lacking a signal sequence. EMBO J. 1990;9:1503–1510. PMID:2328723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Gardella S, Andrei C, Ferrera D, et al. The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep. 2002;3:995–1001. PMID:12231511. doi: 10.1093/embo-reports/kvf198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Temmerman K, Ebert AD, Müller HM, et al. A direct role for phosphatidylinositol-4,5-bisphosphate in unconventional secretion of fibroblast growth factor 2. Traffic. 2008;9:1204–1217. PMID:18419755. [DOI] [PubMed] [Google Scholar]
- [8].Ponpuak M, Mandell MA, Kimura T, et al. Secretory autophagy. Curr Opin Cell Biol. 2015;35:106–116. PMID:25988755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Nagakubo D, Taira T, Kitaura H, et al. DJ-1 a novel oncogene which transforms mouse NIH3T3 cells in cooperation with ras. Biochem Biophys Res Commun. 1997;231:509–513. PMID:9070310. [DOI] [PubMed] [Google Scholar]
- [10].Bonifati V, Rizzu P, Van Baren MJ, et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299:256–259. PMID:12446870. [DOI] [PubMed] [Google Scholar]
- [11].Saito Y, Hamakubo T, Yoshida Y, et al. Preparation and application of monoclonal antibodies against oxidized DJ-1. Significant elevation of oxidized DJ-1 in erythrocytes of early-stage Parkinson disease patients. Neurosci Lett. 2009;465:1–5. PMID:19733211. [DOI] [PubMed] [Google Scholar]
- [12].Akazawa YO, Saito Y, Hamakubo T, et al. Elevation of oxidized DJ-1 in the brain and erythrocytes of Parkinson disease model animals. Neurosci Lett. 2010;483:201–205. PMID:20708070. [DOI] [PubMed] [Google Scholar]
- [13].Ariga H, Takahashi-Niki K, Kato I, et al. Neuroprotective function of DJ-1 in Parkinson’s disease. Oxid Med Cell Longevity. 2013;2013:683920 PMID:23766857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Taira T, Saito Y, Niki T, et al. DJ-1 has a role in antioxidative stress to prevent cell death. EMBO Rep. 2004;5:213–218. PMID:14749723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zhou W, Freed CR. DJ-1 up-regulates glutathione synthesis during oxidative stress and inhibits A53T alpha-synuclein toxicity. J Biol Chem. 2005;280:43150–43158. PMID:16227205. [DOI] [PubMed] [Google Scholar]
- [16].Guzman JN, Sanchez-Padilla J, Wokosin D, et al. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature. 2010;468:696–700. PMID:21068725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wilson MA. The role of cysteine oxidation in DJ-1 function and dysfunction. Antioxid Redox Signal. 2011;15:111–122. PMID:20812780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Blackinton J, Lakshminarasimhan M, Thomas KJ, et al. Formation of a stabilized cysteine sulfinic acid is critical for the mitochondrial function of the parkinsonism protein DJ-1. J Biol Chem. 2009;284:6476–6485. PMID:19124468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Miller DW, Ahmad R, Hague S, et al. L166P mutant DJ-1, causative for recessive Parkinson’s disease, is degraded through the ubiquitin-proteasome system. J Biol Chem. 2003;278:36588–36595. PMID:12851414. [DOI] [PubMed] [Google Scholar]
- [20].Canet-Avilés RM, Wilson MA, Miller DW, et al. The Parkinson’s disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proc Natl Acad Sci U S A. 2004;101:9103–9108. PMID:15181200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Le Naour F, Misek DE, Krause MC, et al. Proteomics-based identification of RS/DJ-1 as a novel circulating tumor antigen in breast cancer. Clin Cancer Res. 2001;7:3328–3335. PMID:11705844. [PubMed] [Google Scholar]
- [22].Tsuchiya B, Iwaya K, Kohno N, et al. Clinical significance of DJ-1 as a secretory molecule: retrospective study of DJ-1 expression at mRNA and protein levels in ductal carcinoma of the breast. Histopathology. 2012;61:69–77. PMID:22385318. [DOI] [PubMed] [Google Scholar]
- [23].Oda M, Makita M, Iwaya K, et al. High levels of DJ-1 protein in nipple fluid of patients with breast cancer. Cancer Sci. 2012;103:1172–1176. PMID:22404125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Allard L, Burkhard PR, Lescuyer P, et al. PARK7 and nucleoside diphosphate kinase A as plasma markers for the early diagnosis of stroke. Clin Chem. 2005;51:2043–2051. PMID:16141287. [DOI] [PubMed] [Google Scholar]
- [25].Pardo M, García A, Thomas B, et al. The characterization of the invasion phenotype of uveal melanoma tumour cells shows the presence of MUC18 and HMG-1 metastasis markers and leads to the identification of DJ-1 as a potential serum biomarker. Int J Cancer. 2006;119:1014–1022. PMID:16570276. [DOI] [PubMed] [Google Scholar]
- [26].Koide-Yoshida S, Niki T, Ueda M, et al. degrades transthyretin and an inactive form of DJ-1 is secreted in familial amyloidotic polyneuropathy. Int J Mol Med. 2007;19:885–893. PMID:17487420. [PubMed] [Google Scholar]
- [27].Waragai M, Nakai M, Wei J, et al. Plasma levels of DJ-1 as a possible marker for progression of sporadic Parkinson’s disease. Neurosci Lett. 2007;425:18–22. PMID:17720313. [DOI] [PubMed] [Google Scholar]
- [28].Waragai M, Wei J, Fujita M, et al. Increased level of DJ-1 in the cerebrospinal fluids of sporadic Parkinson’s disease. Biochem Biophys Res Commun. 2006;345:967–972. PMID:16707095. [DOI] [PubMed] [Google Scholar]
- [29].Hong Z, Shi M, Chung KA, et al. DJ-1 and alpha-synuclein in human cerebrospinal fluid as biomarkers of Parkinson’s disease. Brain. 2010;133:713–726. PMID:20157014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Yanagida T, Tsushima J, Kitamura Y, et al. Oxidative stress induction of DJ-1 protein in reactive astrocytes scavenges free radicals and reduces cell injury. Oxid Med Cell Longev. 2009;2:36–42. PMID: 20046643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kaneko Y, Shojo H, Burns J, et al. DJ-1 ameliorates ischemic cell death in vitro possibly via mitochondrial pathway. Neurobiol Dis. 2014;62:56–61. PMID:24060818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kim JM, Shin HI, Cha SS, et al. DJ-1 promotes angiogenesis and osteogenesis by activating FGF receptor-1 signaling. Nat Commun. 2012;3:1296 PMID:23250426. [DOI] [PubMed] [Google Scholar]
- [33].Blum D, Torch S, Lambeng N, et al. Molecular pathways involved in the neurotoxicity of 6-OHDA, dopamine and MPTP: contribution to the apoptotic theory in Parkinson’s disease. Prog Neurobiol. 2001;65:135–172. PMID:11403877. [DOI] [PubMed] [Google Scholar]
- [34].Keller M, Rüegg A, Werner S, et al. Active caspase-1 is a regulator of unconventional protein secretion. Cell. 2008;132:818–831. PMID:18329368. [DOI] [PubMed] [Google Scholar]
- [35].Cohen G, Heikkila RE. The generation of hydrogen peroxide, superoxide radical, and hydroxyl radical by 6-hydroxydopamine, dialuric acid, and related cytotoxic agents. J Biol Chem. 1974;249:2447–2452. PMID:4362682. [PubMed] [Google Scholar]
- [36].Miyama A, Saito Y, Yamanaka K, et al. Oxidation of DJ-1 induced by 6-hydroxydopamine decreasing intracellular glutathione. PLoS One. 2011;6:e27883 PMID:22132160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Watabe M, Nakaki T. Mitochondrial complex I inhibitor rotenone-elicited dopamine redistribution from vesicles to cytosol in human dopaminergic SH-SY5Y cells. J Pharmacol Exp Ther. 2007;323:499–507. PMID:17726156. [DOI] [PubMed] [Google Scholar]
- [38].Duran JM, Anjard C, Stefan C, et al. Unconventional secretion of Acb1 is mediated by autophagosomes. J Cell Biol. 2010;188:527–536. PMID:20156967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Dupont N, Jiang S, Pilli M, et al. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. EMBO J. 2011;30:4701–4711. PMID:22068051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Son SM, Cha MY, Choi H, et al. Insulin-degrading enzyme secretion from astrocytes is mediated by an autophagy-based unconventional secretory pathway in Alzheimer disease. Autophagy. 2016;12:784–800. PMID:26963025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1–222. PMID:26799652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kuma A, Hatano M, Matsui M, et al. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–1036. PMID:15525940. [DOI] [PubMed] [Google Scholar]
- [43].Saitoh T, Fujita N, Hayashi T, et al. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc Natl Acad Sci U S A. 2009;106:20842–20846. PMID:19926846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Saitoh T, Fujita N, Jang MH, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456:264–268. PMID:18849965. [DOI] [PubMed] [Google Scholar]
- [45].Llorente A, De Marco MC, Alonso MA. Caveolin-1 and MAL are located on prostasomes secreted by the prostate cancer PC-3 cell line. J Cell Sci. 2004;117:5343–5351. PMID:15466889. [DOI] [PubMed] [Google Scholar]
- [46].Arsikin K, Kravic-Stevovic T, Jovanovic M, et al. Autophagy-dependent and -independent involvement of AMP-activated protein kinase in 6-hydroxydopamine toxicity to SH-SY5Y neuroblastoma cells. Biochim Biophys Acta. 2012;1822:1826–1836. PMID:22917563. [DOI] [PubMed] [Google Scholar]
- [47].Petherick KJ, Conway OJ, Mpamhanga C, et al. Pharmacological inhibition of ULK1 kinase blocks mammalian target of rapamycin (mTOR)-dependent autophagy. J Biol Chem. 2015;290:11376–11383. PMID: 25833948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Sarkar S, Davies JE, Huang Z, et al. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J Biol Chem. 2007;282:5641–5652. PMID:17182613. [DOI] [PubMed] [Google Scholar]
- [49].DeBosch BJ, Heitmeier MR, Mayer AL, et al. Trehalose inhibits solute carrier 2A (SLC2A) proteins to induce autophagy and prevent hepatic steatosis. Sci Signal. 2016;9:ra21 PMID:26905426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Ejlerskov P, Rasmussen I, Nielsen TT, et al. Tubulin polymerization-promoting protein (TPPP/p25α) promotes unconventional secretion of α-synuclein through exophagy by impairing autophagosome-lysosome fusion. J Biol Chem. 2013;288:17313–17335. PMID:23629650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Filomeni G, De Zio D, Cecconi F. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ. 2015;22:377–388. PMID:25257172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Scherz-Shouval R, Shvets E, Fass E, et al. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26:1749–1760. PMID: 17347651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Zmijewski JW, Banerjee S, Bae H, et al. Exposure to hydrogen peroxide induces oxidation and activation of AMP-activated protein kinase. J Biol Chem. 2010;285:33154–33164. PMID:20729205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Vucicevic L, Misirkic M, Janjetovic K, et al. Compound C induces protective autophagy in cancer cells through AMPK inhibition-independent blockade of Akt/mTOR pathway. Autophagy. 2011;7:40–50. PMID: 20980833 [DOI] [PubMed] [Google Scholar]
- [55].Galluzzi L, Baehrecke EH, Ballabio A, et al. Molecular definitions of autophagy and related processes. EMBO J. 2017;36:1811–1836. PMID: 28596378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Kimura T, Jia J, Kumar S, et al. Dedicated SNAREs and specialized TRIM cargo receptors mediate secretory autophagy. EMBO J. 2017;36:42–60. PMID: 27932448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Itakura E, Kishi-Itakura C, Mizushima N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell. 2012;151:1256–1269. PMID: 23217709. [DOI] [PubMed] [Google Scholar]
- [58].Glebov K, Schütze S, Walter J. Functional relevance of a novel SlyX motif in non-conventional secretion of insulin-degrading enzyme. J Biol Chem. 2011;286:22711–22715. PMID:21576244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Zhang M, Kenny SJ, Ge L, et al. Translocation of interleukin-1β into a vesicle intermediate in autophagy-mediated secretion. Elife. 2015;4:e11205 PMID:26523392. doi: 10.7554/eLife.11205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Inden M, Taira T, Kitamura Y, et al. PARK7 DJ-1 protects against degeneration of nigral dopaminergic neurons in Parkinson’s disease rat model. Neurobiol Dis. 2006;24:144–158. PMID:16860563. [DOI] [PubMed] [Google Scholar]
- [61].Lev N, Barhum Y, Ben-Zur T, et al. Knocking out DJ-1 attenuates astrocytes neuroprotection against 6-hydroxydopamine toxicity. J Mol Neurosci. 2013;50:542–550. PMID:23536331. [DOI] [PubMed] [Google Scholar]
- [62].Saito Y, Akazawa-Ogawa Y, Matsumura A, et al. Oxidation and interaction of DJ-1 with 20S proteasome in the erythrocytes of early stage Parkinson’s disease patients. Sci Rep. 2016;6:30793 PMID:27470541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Yamanaka K, Urano Y, Takabe W, et al. Induction of apoptosis and necroptosis by 24(S)-hydroxycholesterol is dependent on activity of acyl-CoA: cholesterolacyltransferase 1. Cell Death Dis. 2014;5:e990 PMID:24407243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Urano Y, Ochiai S, Noguchi N. Suppression of amyloid-β production by 24S-hydroxycholesterol via inhibition of intracellular amyloid precursor protein trafficking. FASEB J. 2013;27:4305–4315. PMID:23839932. [DOI] [PubMed] [Google Scholar]
- [65].Holden P, Horton WA. Crude subcellular fractionation of cultured mammalian cell lines. BMC Res Notes. 2009;2:243 PMID:20003239. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.