

Abstract
Gaussia princeps luciferase (GLuc) generates an intense burst of blue light when exposed to coelenterazine in the absence of ATP. Here we show that this 5‐disulfide containing enzyme can be used as a facile and convenient substrate for studies of oxidative protein folding. Reduced GLuc (rGLuc), with 10 free cysteine residues, is completely inactive as a luciferase but >60% bioluminescence activity, compared to controls, can be recovered using a range of oxidizing regimens in the absence of the exogenous shuffling activity of protein disulfide isomerase (PDI). The sulfhydryl oxidase QSOX1 can be assayed using rGLuc in a simple bioluminescence plate reader format. Similarly, low concentrations of rGLuc can be oxidized by millimolar levels of dehydroascorbate, hydrogen peroxide or much lower concentrations of sodium tetrathionate. The oxidative refolding of rGLuc in the presence of a range of glutathione redox buffers is only marginally accelerated by micromolar levels of PDI. This modest rate enhancement probably results from a relatively simple disulfide connectivity in native GLuc; reflecting two homologous domains each carrying two disulfide bonds with a single interdomain disulfide. When GLuc is reoxidized under denaturing conditions the resulting scrambled protein (sGLuc) can be used in a sensitive bioluminescence assay for reduced PDI in the absence of added exogenous thiols. Finally, the general facility by which rGLuc can recover bioluminescent activity in vitro provides a sensitive method for the assessment of inhibitors of oxidative protein folding.
Keywords: bioluminescence assay, disulfide, Gaussia luciferase, oxidative protein folding, protein disulfide isomerase, quiescin sulfhydryl oxidase, redox buffer, thiol‐disulfide exchange, thiol oxidation
Introduction
Approximately one quarter of mammalian proteins contain disulfide bonds that often markedly stabilize proteins destined for secretion. Oxidative protein folding occurs in two conceptual steps that are typically interdigitated during the emergence of the native fold (Fig. 1). In the oxidative step, pairs of proximal cysteines are oxidized with the removal of two electrons for each disulfide introduced. This reaction is typically error‐prone and mispaired disulfides are corrected by a series of thiol‐disulfide exchange reactions catalyzed by the protein disulfide isomerases.
Figure 1.

Oxidative protein folding. Emergence of the native fold requires oxidation of cysteine residues and isomerization of mispaired disulfides. For simplicity, these steps are shown as sequential processes, but they are likely to be interdigitated during the oxidative folding of realistically complex protein clients.
Oxidative protein folding was first studied systematically in the laboratories of Anfinsen1 and Straub2 using pancreatic ribonuclease A (RNase). This 4‐disulfide hydrolase can be reduced under forcing conditions to yield a tractable reduced protein that is still widely employed in investigations of oxidative folding. Egg white lysozyme was also used in early experiments3 but the marked tendency of the reduced protein to aggregate4, 5 complicates its use as a general substrate for oxidative folding. Bovine pancreatic inhibitor (BPTI) with three disulfides would be expected to provide a simpler folding pathway than the 4‐disulfide containing hydrolases (15 vs. 105 disulfide isomers for the fully oxidized protein); as such the oxidative folding of BPTI has received particular attention.1, 6, 7, 8, 9 A number of other disulfide‐containing peptides and small proteins9, 10 have served as substrates for oxidative folding studies including hirudin11 and other protease inhibitors,9 alpha‐lactalbumin,12 proinsulin,13 and cyclic cystine knot proteins.14
We earlier found that egg white riboflavin binding protein (RfBP) was a useful in vitro substrate for oxidative protein folding studies.15 The corresponding apoprotein contains nine disulfides and rapidly binds riboflavin with a K d of 1 nM.16 Binding is associated with both a significant change in the absorbance envelope of the isoalloxazine ring and with an almost complete loss of flavin fluorescence.15, 16 This allows the oxidative folding of reduced RfBP to be followed continuously by the decline in flavin fluorescence from that of free riboflavin.15 While the opportunity to monitor refolding continuously is an advantage, the sensitivity of the RfBP assay is limited because it relies on a disappearance of signal. As a complementary approach, we sought a robust method to screen the oxidative folding abilities of redox environments wherein a signal appears from a minimal basal level. Here we describe a simple system based on the appearance of bioluminescence when the 10 cysteine residues of reduced Gaussia princeps luciferase (rGLuc) are paired to generate active protein. GLuc is a very thermodynamically stable, small 5‐disulfide protein that generates an intense, ATP‐independent, burst of luminescence in the presence of coelenterazine and molecular oxygen:17, 18, 19
coelenterazine + O2 ‐> coelenteramide + CO2 + hυ (475 nm)
Herein, we characterize fully reduced and scrambled GLuc as new substrates for the in vitro interrogation of both oxidation and isomerization phases of oxidative protein folding.
Results and Discussion
Preparation of oxidized and reduced GLuc
Both domains of GLuc contain five cysteine residues as depicted in Figure 2, 17 yielding a protein with five disulfides including at least one inter‐domain crosslink.18, 20 GLuc has been expressed in Escherichia coli using a variety of constructs.17, 21, 22, 23 Here, we utilize a GLuc (Fig. 2) incorporating a C‐terminal HIS tag for affinity purification. GLuc was expressed at 20°C and purified as a soluble protein without the involvement of inclusion bodies as described in Materials and Methods. Purity was assessed via SDS‐PAGE (Fig. S1) and the pooled fractions were examined by mass spectrometry yielding a mass of 21,833 as expected for the presence of 5‐disulfides (see Methods and Fig. S2). Routinely GLuc assays were conducted by integrating the luminescence for 10 s after the addition of coelenterazine using the built‐in injector in the plate reader (Fig. S3; see Methods). In this burst mode light output was linear over a wide range of GLuc concentration (Fig. S4) as reported previously.21
Figure 2.

Sequence of the GLuc construct used in this work. The 10 cysteine residues are highlighted in red. The bolded N‐terminal MD dipeptide replaces the signal peptide in the GLuc sequence (GenBank AAG54095.1). The bolded C‐terminal region adds a (GSGGG)2 linker, a sortase recognition sequence and a hexaHIS tag. The sequence used in this work has an I164T mutation which does not impact bioluminescence activity (see Materials and Methods). The N and C terminal GLuc domains are shaded blue and pink, respectively.
GLuc was exhaustively reduced with dithiothreitol (DTT) and quantitatively freed from excess reductant by size exclusion or via repeated centrifuge ultrafiltration steps (see Methods). The resulting protein showed the expected gain of 10 mass units corresponding to the reduction of five disulfides. Further a sample of the reduced protein was stoichiometrically alkylated with N‐ethylmaleimide (NEM) with a gain of 1250 (10 × 125) amu (Fig. S2). Reduced protein reproducibly gave residual luciferase activities of < 0.03% (see later).
Reoxidation of reduced GLuc by glutathione redox buffers
Figure 3 shows that negligible activity is recovered on incubation of rGLuc for 2 h in aerobic potassium phosphate buffer, pH 8.0, containing 1 mM EDTA showing that the basal level of autoxidation over this time interval does not complicate data analysis of subsequent experiments. Activity was then assessed in mixtures of oxidized and reduced glutathione (GSSG and GSH, respectively) with an aggregate concentration of 5 mM (see Methods). Such glutathione redox buffers were first introduced by Saxena and Wetlaufer24 in the oxidative refolding of lysozyme. The GSSG component provides the oxidizing equivalents for disulfide bond insertion, and GSH contributes to the shuffling of mispaired disulfides via the generation and resolution of mixed disulfide species. The marked decrease in bioluminescence activity under more oxidizing glutathione redox buffers in Figure 3 suggests that the rate of GLuc thiol oxidation outstrips the disulfide shuffling needed for regain of bioluminescence (see later). Protein disulfide isomerases are expected to catalyze both the oxidative and isomerization phases of oxidative folding in the presence of redox buffers. However, in vitro they are often modest accelerants of oxidative folding; Gilbert and coworkers found that 1.4 µM PDI was only able to accelerate oxidative folding of fully reduced RNase by about 4‐fold.25 The oxidative folding of reduced hirudin (6 cysteines) is enhanced more strongly (by 2880‐fold) using 50 µM PDI 9, 26 but again these accelerations are paltry compared to the typical 1010‐fold found in many enzymes. With rGLuc, human PDI (PDIA1: the most widely studied human protein disulfide isomerase) was found to be a very poor catalyst of oxidative folding only accelerating the regain of activity by <2‐fold using 0.1 µM PDI (Fig. S5).
Figure 3.
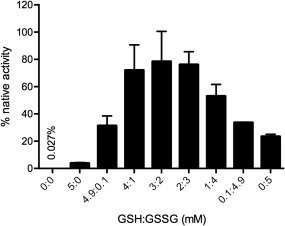
Incubation of rGLuc in aerobic buffer in the absence or presence of a glutathione redox buffer. rGLuc was incubated for 2 h in phosphate buffer, pH 8.0, containing 1 mM EDTA (see Methods) with the indicated concentration of glutathione (in mM). Recovery of activity compared to the same amount of native GLuc was evaluated as in Methods.
We next assessed the ability of PDI to shuffle mispaired disulfides in completely oxidized GLuc. Scrambled GLuc (sGLuc) was conveniently prepared by rapid oxidation of reduced unfolded GLuc in GuHCl using sodium tetrathionate (see Methods) rather than the traditional procedure of slow air oxidation of reduced client proteins. The resulting material showed 0.08 ± 0.01% of native GLuc activity. Figure 4 shows only a 2‐ to 3‐fold enhancement using 1 µM PDI over the use of redox buffer alone consistent with the very modest effects seen earlier. However in the absence of redox buffer we were able to devise a sensitive bioluminescence assay using reduced PDI alone. Under these conditions there is a linear increase in bioluminescence from 30 to 200 nM PDI (Fig. 5). For perspective reduced PDI, was previously shown to be capable of catalyzing the unscrambling of sRNase in the absence of redox buffer but at rates that were significantly slower than those with the additional presence of redox buffer.27, 28 With sGLuc 100 nM rPDI leads to a 1600‐fold enhancement in the bioluminescent activity over a 2 h incubation. This unscrambling assay sGLuc should provide a convenient bioluminescent screening assay for the isomerase activity of rPDI to complement the more widely employed reductase‐based assays.29, 30, 31, 32, 33
Figure 4.
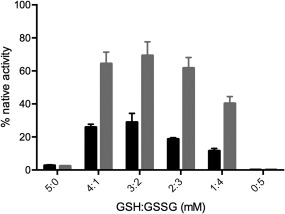
Refolding of sGLuc in glutathione redox buffer. The activity of GLuc was measured 2 h after incubation with the indicated glutathione buffers in the absence or presence of 1 µM PDI (black and grey columns, respectively).
Figure 5.
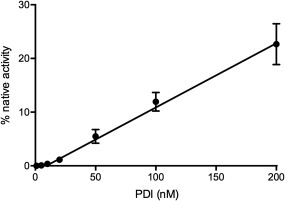
Assaying reduced PDI using sGluc. The scrambled luciferase (100 nM in 50 mM phosphate buffer, pH 8.0) was incubated for 2 h with the indicated concentrations of PDI indicated prior to assay.
Oxidation of rGLuc by QSOX1
Quiescin sulfhydryl oxidases (QSOX) can catalyze rapid and indiscriminate oxidation of cysteine side chains in flexible regions of proteins and peptides to generate the corresponding disulfides with the concomitant reduction of oxygen to hydrogen peroxide (typical k cat and K m values per protein thiol oxidized are 30/s and 200 µM).34, 35, 36, 37 Importantly, QSOX enzymes show no evidence of PDI‐like isomerase activity; either correct cysteine pairings arise de novo, or result from self‐shuffling within partially oxidized rGLuc. Figure 6(A) shows the regain of bioluminescence on incubation of 1 µM rGLuc thiols (100 nM protein) with 10 nM human QSOX1. Reactivation continues over 3 h and is preceded by a distinct lag phase. Nevertheless, the net regain of activity over 2 h is linearly dependent on QSOX1 concentration up to 20 nM [Fig. 6(B), inset]. This provides the option of a bioluminescent assay for this interesting family of sulfhydryl oxidases.38, 39 At higher QSOX1 concentrations, however, the total activity regained markedly declines reaching only ∼25% at 500 nM of the oxidase [Fig. 6(B)]. This biphasic response with oxidant concentration is observed consistently with rGLuc (see later) and likely reflects the balance between the rate for the net insertion of disulfide bridges and the ability of thiol‐disulfide self‐shuffling to secure the correct pairings. At higher oxidant concentrations incorrect pairings persist because thiols required for thiol‐disulfide exchange are depleted too rapidly.
Figure 6.
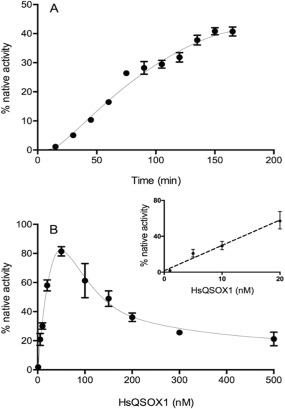
Oxidative refolding of rGLuc using human QSOX1. Panel A: rGLuc (100 nM in phosphate buffer, pH 8.0, 25°C) was incubated with 10 nM QSOX1 with activity regained over 3 h. Panel B plots activity recovered in a 2 h incubation using 10–500 nM QSOX1.
Oxidative refolding of rGLuc using additional small molecule oxidants
If self‐shuffling is an effective route to the recovery of biological activity of rGLuc then a range of chemical oxidants should also be effective. We chose three reagents that, unlike glutathione, would not be expected to directly participate in thiol/disulfide exchange reactions.40, 41 Dehydroascorbate (DHA) has been found to be a useful in vitro oxidant for unfolded reduced proteins42, 43, 44 starting with the seminal studies of Anfinsen and Straub and their coworkers. DHA is an effective oxidant for rGLuc with a maximal recovery of ∼60% activity at 1 mM DHA over 2 h [Fig. 7(A)]. Under these conditions, maximal regain of activity was obtained using 1 mM DHA. Higher concentrations up to 10 mM lead to sharply lowered activity recoveries presumably because thiol oxidation again outstrips the self‐shuffling needed for regain of bioluminescence.
Figure 7.
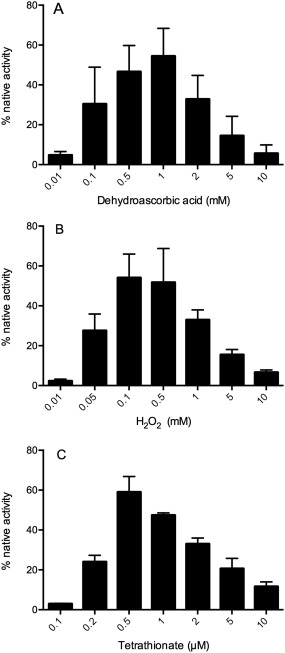
Treatment of rGLuc with three oxidizing agents. rGLuc (100 nM in phosphate buffer, pH 8.0) was incubated for 2 h with dehydroascorbate, hydrogen peroxide and tetrathionate at the concentrations indicated in panels A–C, respectively.
Hydrogen peroxide has been suggested as an efficient direct oxidant of protein dithiols via the generation of sulfenic acid intermediates.45 Figure 7(B) again shows a biphasic response with about 60% bioluminescence activity recovered using 100 µM H2O2 followed by a decline at higher oxidant concentrations. Panel C utilizes a more potent, but less widely employed, thiol oxidant. Sodium tetrathionate, Na2S4O6, 46 shows a standard redox potential of approximately +198 mV47, 48 and should drive the following reaction to completion:
Indeed, stoichiometric levels of tetrathionate (0.5 µM for 1 µM protein thiols) yielded the maximal recovery of bioluminescence under these conditions [Fig. 7(C)].
Screening for inhibitors of the oxidative protein folding of rGLuc
The regain of bioluminescent activity following exposure of rGLuc to a range of oxidants allows a simple assessment of the ability of thiol‐reactive reagents to disrupt oxidative folding pathways. We illustrate this first with the arsenical, p‐succinylamidophenylarsine oxide (PSAO).49 PSAO captures vicinal thiols and has been shown to inhibit oxidative protein folding of reduced RNase, lysozyme, and reduced riboflavin binding protein.50 Figure 8 shows that PSAO at 1 µM (equimolar with rGLuc protein thiols) effects an approximately 60‐fold decrease in bioluminescence recovery with both tetrathionate and QSOX1 oxidation. In contrast, concentrations of PSAO up to 10 µM are ineffective when oxidative protein folding is catalyzed by a glutathione buffer (Fig. 8). Here, the arsenical is sequestered effectively in vitro by mM levels of reduced glutathione leaving oxidative protein folding unimpaired as observed for other proteins.33, 50, 51
Figure 8.
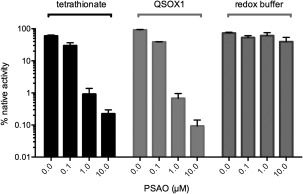
Inhibition of oxidative protein folding by PSAO. rGLuc (100 nM) was incubated for 2 h with the indicated concentrations of PSAO in the presence of either 1 µM tetrathionate, 50 nM QSOX1 or a redox buffer of 3 mM GSH, and 2 mM GSSG.
Christen and colleagues have reported that Cd2+, Hg2+, and Pb2+ are inhibitors of the folding of several cysteine‐containing cytosolic proteins via the formation of multidentate complexes.52, 53 We were interested in assessing whether Zn2+, another thiophilic metal ion, would inhibit the oxidative refolding of rGLuc. Indeed, at stoichiometric levels (100 nM of both Zn2+ and rGLuc protein) >90% inhibition of folding is observed over 2h (Fig. 9). Higher zinc concentrations provide even more dramatic inhibition once again highlighting the need to carefully regulate the cellular concentrations of metal ions that could otherwise disrupt folding pathways of unstructured nascent chains.52, 53
Figure 9.
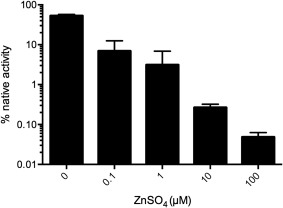
Inhibition of oxidative folding of rGLuc by zinc ions. rGLuc (100 nM in 50 mM Tris buffer, pH 8.0, containing 50 mM NaCl) was incubated for 2 h in the presence of 1 µM sodium tetrathionate and the indicated concentrations of zinc sulfate.
Conclusions
This work presents the first in vitro bioluminescence‐based assays for oxidative protein folding. In terms of the net disulfide generation phase of this key post‐translational modification, QSOX1 and a variety of non‐enzymatic oxidants produce a substantial recovery of GLuc activity. The regain of luminescence from a very low residual background represents a sensitive assay that can be conducted over a single time interval. The method could, for example, be explored to screen small molecule inhibitors of oxidative folding. In terms of the isomerization aspect of oxidative protein folding, scrambled RNase has been the substrate of choice for assessing the isomerase activity of rPDI. However, the relative insensitivity of the older assays for RNase have limited its widespread utilization. sGLuc now provides a sensitive alternative protein substrate for the unscrambling activity of rPDI. This new method may provide a convenient complement to the widely employed assays based on the ability of the isomerase to promote reduction of disulfide‐containing terminal oxidants.29, 30, 31, 32, 33
While reduced PDI strongly catalyzes the unscrambling of sGLuc in the absence of exogenous thiols, the additional presence of a glutathione redox buffer largely obscures this effect. As noted earlier, high background isomerization rates have also been observed with glutathione redox buffers in early work with sRNase; there is only a several‐fold increase rate in unscrambling of sRNase when glutathione redox buffers are supplemented with low micromolar levels of PDI.28 Similarly, this very modest catalytic rate enhancement is found when the oxidative refolding of reduced RNase is examined in the presence of a glutathione redox buffer which serves as the ultimate oxidant in the presence or absence of PDI.25 Again, Gaussia luciferase provides a close parallel in that oxidative refolding is only marginally accelerated by 1 µM PDI (Fig. 4).
Finally, why is the bioluminescent activity of GLuc readily recovered when rGLuc is incubated with nanomolar levels of QSOX, or higher concentrations of a range of chemical oxidants, without the obligatory requirement of an added isomerase? These results initially appeared surprising because there are 975 ways to arrange the 5‐disulfide crosslinks in GLuc. However, since GLuc represents the duplication of two very similar domains, each carrying four conserved cysteine residues (Fig. S6),17 the independent oxidative folding of each domain could drastically simplify the number of disulfide isomers formed before the appearance of native GLuc (there are only three ways to pair four cysteine residues in disulfide linkages). In this instance, self‐shuffling may provide an efficient route to the native fold of GLuc provided that oxidation is not overly rapid.
Materials and Methods
Materials
Coelenterazine and Gaussia luciferase were purchased from NanoLight Technology. All bacterial culture media components were from Fisher Scientific. Dithiothreitol, glutathione disulfide and glutathione were purchased from Sigma. Zinc sulfate was from Mallickrodt. ProBond nickel‐chelating Ni‐IDA resin was from Gold Biotechnology. PSAO was synthesized as described by Cline et al.49
General methods
Unless otherwise stated, phosphate buffer contained 50 mM potassium phosphate supplemented with 50 mM NaCl and 1 mM EDTA adjusted to pH 8.0. UV‐Vis spectra were recorded using HP8452 or HP8453 instruments. Protein concentrations were determined at 280 nm using extinction coefficients calculated using ProtParam for native and reduced GLuc of 7615 and 6990 M−1 cm−1, respectively.54
Expression and purification of Gaussia luciferase protein
The GLuc construct is shown in Figure 2; following the two GLuc domains is a C terminal region with two G4S linkers, a spacer region, and a hexa HIS‐tag. The construct used in this work has an I164T mutation in the flexible region following the second domain (Fig. 2). This protein shows comparable activity (1.05 ± 0.03 fold) to the wild‐type sequence (see later; data not shown). Both constructs show ∼1.2 ± 0.1 fold higher activity compared to a commercial sample of GLuc from NanoLight Technology.
Single colonies of BL21(DE3) cells were used to inoculate tubes containing 10 mL media supplemented with 30 µg/mL kanamycin. After overnight incubation at 37°C the contents were transferred to flasks containing 1 L of media and the cells were grown at this temperature until the suspension reached an optical density of 0.8 at 600 nm. The flasks were then transferred to a shaker at 20°C and expression of GLuc was induced by 1 mM IPTG. After 24 h, cells were harvested by centrifugation at 5,000g for 10 min at 4°C. The cell pellets were resuspended by rocking in nine volumes of lysis buffer (50 mM potassium phosphate, 50 mM NaCl, with 10 mM imidazole, 1% NP‐40, 1 mM phenylmethyl sulfonyl fluoride, and 0.1 mg/mL lysozyme adjusted to pH 8.0). Resuspended cells were subject to two passages through a French press chamber (10,000 psi), and the broken cells treated for five cycles of 15 s sonication separated by 10 s cooling on ice. The suspension was centrifuged at 4°C for 30 min at 17,000g and the supernatant was combined with 2 mL of pre‐equilibrated Ni‐IDA resin and rocked overnight in the cold room. The slurry was loaded into a column and rinsed with three volumes of wash solution (containing 50 mM KPi, 50 mM NaCl, with 20 mM imidazole; final pH 8.0). The column was developed with four 5‐mL volumes supplemented with increasing imidazole concentrations (50, 100, 200, and 300 mM). Fractions were evaluated by SDS‐PAGE and pooled as appropriate before overnight dialysis against 1 L of 50 mM KPi, pH 8.0, containing 50 mM NaCl. The GLuc was reabsorbed from the dialysate using a second 2 mL volume of Ni‐IDA resin, eluted as before, and the purified GLuc was dialyzed with the same buffer supplemented with 1 mM EDTA. The luciferase (typical yield of 40 mg/L of culture; Fig. S1) was then concentrated to ∼1 mM protein using a 10,000 MWCO Centricon centrifugal ultrafiltration device (Millipore). GLuc was quantitated by absorbance at 280 nm and stored at 4°C.
Gaussia luciferase bioluminescence assay
A coelenterazine stock solution (50 µL of 0.3 mM in ethanol) was used to prepare a 3 µM working solution in phosphate buffer that was subsequently stored for 30 min in the dark prior to use. Luciferase assays were conducted using a BMG POLARstar OMEGA plate reader with 96‐well white flat‐bottomed polystyrene plates from Corning. Each well contained 200 µL of 50 mM phosphate buffer with 50 mM sodium chloride and 1 mM EDTA, pH 8.0, together with additional reagents as needed. Each well was assayed by injecting 50 µL of the working coelenterazine solution using the built‐in injector followed by a 1 s shake cycle to mix reagents and 10 s of integration of the bioluminescence signal. Representative standard curves for the activity of recombinant GLuc prepared in this work are shown in Figure S4.
Expression and purification of human PDI and QSOX1
Human PDIA1 was expressed, purified and handled as described earlier15 with minor modifications.33 The previous protocol37 for the expression of HsQSOX in E. coli was modified as summarized below. The QSOX1 gene in a pET28a plasmid was introduced into Rosetta‐gami2 (DE3) cells and 5 mL of starter cultures in LB media supplemented with antibiotics were grown overnight at 37°C. These cultures were used to inoculate four 0.5 L batches of the same media (omitting the 5 µM riboflavin supplement used previously37). Cultures were grown at 37°C to an OD600 of ∼2.0, cooled over 45 min to 15°C and induced overnight with 1 mM IPTG. Cells were harvested by centrifugation at 5000g for 10 min at 4°C and resuspended in 30 mL of 50 mM phosphate buffer, pH 7.5, containing 1 mM phenylmethyl sulfonyl fluoride, 1 µM leupeptin, 100 µM FAD, and 300 mM NaCl. The suspension was homogenized with two passes through a French Pressure cell (at 10,000 psi) followed by five cycles of 15 s of sonication with 10 s intervals of cooling. The extract was then clarified by centrifugation (17,000g, 30 min, 4°C) and the supernatant rocked overnight at 4°C with 2 mL of Ni‐IDA resin. The resin was washed with phosphate buffer containing 300 mM NaCl pH 7.5, and then eluted with four column volumes of 50, 100, 300, and 500 mM imidazole in phosphate buffer containing 500 mM NaCl, adjusted to pH 7.5. Subsequent purification was as described previously37 except that the second Ni‐affinity and cation exchange column steps were omitted. QSOX1‐containing fractions were identified by SDS‐PAGE, pooled, and brought to 75% ammonium sulfate. The yellow precipitate was redissolved in a minimal volume of phosphate buffer containing 1 mM EDTA and 20% v/v glycerol and dialyzed overnight against the same buffer. QSOX1 was stored in this buffer at 4 or −80°C.
Mass spectrometry analysis of GLuc
GLuc (1 mM in 50 mM phosphate buffer containing 6 M GuHCl) was incubated for 2 h in the presence or absence of 10 mM DTT. The solution was then diluted 50‐fold into phosphate buffer and 1 µL applied to a Waters MassPrep online desalting column prior to injection into a Waters XEVO G2‐S QToF instrument. To assess the extent of alkylation of GLuc by NEM, the reduced and oxidized proteins were diluted 50‐fold into phosphate buffer containing 4 mM NEM, incubated for 10 min at room temperature and then analyzed as above.
Preparation of rGLuc and oxidative protein folding of the reduced luciferase
The reduced protein was prepared as needed daily by incubating 0.1 mM GLuc with 10 mM DTT for 2 h in 50 mM phosphate buffer, pH 8.0, containing 6 M GuHCl. Aliquots (0.1 mL) were applied to a NAP‐5 size‐exclusion column (GE Healthcare) equilibrated with 50 mM phosphate buffer, pH 8.0 containing 1 mM EDTA and 0.5 mL fractions were collected. Small volumes from each tube were monitored for UV absorbance and thiol‐titer using 5,5‐dithio‐bis‐(2‐nitrobenzoic acid) to ensure baseline removal of excess from the reduced protein. Alternatively, reductant could be removed using a 3000 MWCO Centricon ultrafiltration device following washing 0.5 mL reaction mixture with seven successive aliquots of 0.5 mL of 1 mM HCl containing 3 M GuHCl.
Oxidative folding experiments using 100 nM rGLuc in 200 µL of 50 mM phosphate containing 50 mM NaCl and 1 mM EDTA, adjusted to pH 8.0, were routinely conducted over a 2 h time interval in the presence of the oxidants/redox buffers listed in this study. Where appropriate, GSH solutions were carefully adjusted to pH 8 with KOH before use. When inhibitors of oxidative protein folding were included, they were mixed with rGLuc before the addition of oxidants. Controls containing 100 nM GLuc were used to set the gain on the plate reader, and to normalize for instability of the coelenterazine in the phosphate buffer working solution. Time course studies dispensed 200 µL aliquots from a larger volume containing 100 nM luciferase and paired each time interval with a contemporaneous control using GLuc to set the 100% response for that sample.
Preparation of scrambled GLuc (sGLuc)
Reduced GLuc (0.1 mM, 100 µL) was incubated with 900 µL of 6.7 M GuHCl in phosphate buffer, pH 8.0, containing 10 mM sodium tetrathionate and 1 mM EDTA. After 1 h, sGLuc was separated from excess reagents using a PD10 size exclusion column equilibrated with 50 mM phosphate buffer, pH 8.0, containing 1 mM EDTA. The residual activity of sGLuc was 0.0075 ± .0022%.
Supplementary Material
Figures S1–S6 show: the purity and characterization of GLuc by SDS‐PAGE and mass spectrometry; a representative burst assay for bioluminescence; the linearity of luminescence with GLuc concentration; the refolding of rGLuc in GSH/GSSG redox buffer; and the amino acid sequence aligning the domains of GLuc.
Conflict of Interest Statement
The authors have no competing financial interests.
Supporting information
Supporting Information
Figure S1. SDS‐PAGE gel of fractions eluting from Ni‐affinity purification of the GLuc construct used in this work (see Methods). The left lane represents BioRad standards. Lanes 1–3 are consecutive fractions eluting from the second ProBond nickel‐chelating Ni‐IDA resin (see Methods).
Figure S2. Mass Spectra of the treated Gaussia luciferase construct used in this work. Panels A and B represent oxidized and reduced protein respectively. The expected mass of the native 5‐disulfide protein is 21,833 from the sequence shown in Figure 2. Panels C and D are the mass spectra observed after treatment of oxidized and reduced GLuc with NEM. The mass resolution is 0.5 Daltons (see Materials and Methods).
Figure S3. Sample output from the GLuc assay started by the addition of 0.6 µM coelenterazine as described in Methods section. Coelenterazine was added from 0 to 1 s followed by 1 s shaking. Light emission is integrated from 2‐12 s.
Figure S4. Calibration curves for GLuc assays under the conditions described in Methods. Each point in both data sets is the average of 3 determinations. Panels A and B cover 0‐1 and 0‐100 nM Gluc respectively. Under these instrumental conditions (see Materials and Methods) the relative luminescence units per mg of GLuc is 1.8 x 1010/s.
Figure S5. The effect of PDI on the recovery of GLuc activity in glutathione redox buffer. Panel A shows reactivation of rGLuc over 1 h in the presence of 3 mM GSH and 2 mM GSSG in the absence or presence of 100 nM PDI (circles and squares, respectively). Panel B shows the luminescence activity reached after 10 min incubation under the same conditions but with PDI levels increased to 10 µM.
Figure S6. Alignment of amino acid sequence of first and second domains of Gaussia luciferase. The domain boundaries from Inouye and Sahara [17] were aligned using Multalin [53]. Four pairs of conserved cysteine residues are identified with arrows. One cysteine in each of the first and second domains is not conserved (smaller yellow boxes).
Acknowledgments
We thank Ms. Celia Foster for gifts of PSAO and human QSOX1. This work was funded in part by National Institutes of Health (GM26643 to C.T.); National Science Foundation (CBET‐1351302 CAREER award to J.A.P.) and BEST IGERT program (NSF award DGE‐1144901 to J.R.L.). Mass spectrometry experiments were supported by the Delaware COBRE programs with grants from the National Institute of General Medical Sciences [5P30 GM110758 and P20 GM104316].
Broader statement: Here we study protein folding coupled to the formation of disulfide crosslinks using a luciferase from the marine shrimp Gaussia princeps. Scission of its five disulfide bonds by chemical reduction abolishes its ability to generate light and permits the conditions that facilitate oxidative protein folding to be studied by following the regain of bioluminescence activity. Gaussia luciferase represents a convenient and sensitive new tool to assist in the identification of inhibitors of oxidative protein folding.
References
- 1. Goldberger RF, Epstein CJ, Anfinsen CB (1963) Acceleration of reactivation of reduced bovine pancreatic ribonuclease by a microsomal system from rat liver. J Biol Chem 238:628–635. [PubMed] [Google Scholar]
- 2. Venetianer P, Straub FB (1963) The enzymic reactivation of reduced ribonuclease. Biochim Biophys Acta 67:166–168. [DOI] [PubMed] [Google Scholar]
- 3. Anderson WL, Wetlaufer DB (1976) The folding pathway of reduced lysozyme. J Biol Chem 251:3147–3153. [PubMed] [Google Scholar]
- 4. Goldberg ME, Rudolph R, Jaenicke R (1991) A kinetic study of the competition between renaturation and aggregation during the refolding of denatured‐reduced egg white lysozyme. Biochemistry 30:2790–2797. [DOI] [PubMed] [Google Scholar]
- 5. van den Berg B, Chung EW, Robinson CV, Mateo PL, Dobson CM (1999) The oxidative refolding of hen lysozyme and its catalysis by protein disulfide isomerase. EMBO J 18:4794–4803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Creighton TE, Goldenberg DP (1984) Kinetic role of a meta‐stable native‐like two‐disulphide species in the folding transition of bovine pancreatic trypsin inhibitor. J Mol Biol 179:497–526. [DOI] [PubMed] [Google Scholar]
- 7. Weissman JS, Kim PS (1991) Reexamination of the folding of BPTI: predominance of native intermediates. Science 253:1386–1393. [DOI] [PubMed] [Google Scholar]
- 8. Weissman JS, Kim PS (1992) Kinetic role of nonnative species in the folding of bovine pancreatic trypsin inhibitor. Proc Natl Acad Sci USA 89:9900–9904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chang JY (2011) Diverse pathways of oxidative folding of disulfide proteins: underlying causes and folding models. Biochemistry 50:3414–3431. [DOI] [PubMed] [Google Scholar]
- 10. Arolas JL, Aviles FX, Chang JY, Ventura S (2006) Folding of small disulfide‐rich proteins: clarifying the puzzle. Trends Biochem Sci 31:292–301. [DOI] [PubMed] [Google Scholar]
- 11. Chatrenet B, Chang JY (1992) The folding of hirudin adopts a mechanism of trial and error. J Biol Chem 267:3038–3043. [PubMed] [Google Scholar]
- 12. Ewbank JJ, Creighton TE (1993) Structural characterization of the disulfide folding intermediates of bovine alpha‐lactalbumin. Biochemistry 32:3694–3707. [DOI] [PubMed] [Google Scholar]
- 13. Hua QX, Mayer JP, Jia W, Zhang J, Weiss MA (2006) The folding nucleus of the insulin superfamily: a flexible peptide model foreshadows the native state. J Biol Chem 281:28131–28142. [DOI] [PubMed] [Google Scholar]
- 14. Cemazar M, Gruber CW, Craik DJ (2008) Oxidative folding of cyclic cystine knot proteins. Antioxid Redox Signal 10:103–111. [DOI] [PubMed] [Google Scholar]
- 15. Rancy PC, Thorpe C (2008) Oxidative protein folding in vitro: a study of the cooperation between quiescin‐sulfhydryl oxidase and protein disulfide isomerase. Biochemistry 47:12047–12056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Becvar J, Palmer G (1982) The binding of flavin derivatives to the riboflavin‐binding protein of egg white. A kinetic and thermodynamic study. J Biol Chem 257:5607–5617. [PubMed] [Google Scholar]
- 17. Inouye S, Sahara Y (2008) Identification of two catalytic domains in a luciferase secreted by the copepod Gaussia princeps . Biochem Biophys Res Commun 365:96–101. [DOI] [PubMed] [Google Scholar]
- 18. Hunt EA, Moutsiopoulou A, Ioannou S, Ahern K, Woodward K, Dikici E, Daunert S, Deo SK (2016) Truncated variants of Gaussia luciferase with tyrosine linker for site‐specific bioconjugate applications. Sci Rep 6:26814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shimomura O. The photoproteins In: Daunert S, Deo SK, Ed. (2006) Photoproteins in bioanalysis. Weinheim: Wiley, pp 1–20. [Google Scholar]
- 20. Wu N, Rathnayaka T, Kuroda Y (2015) Bacterial expression and re‐engineering of Gaussia princeps luciferase and its use as a reporter protein. Biochim Biophys Acta 1854:1392–1399. [DOI] [PubMed] [Google Scholar]
- 21. Verhaegen M, Christopoulos TK (2002) Recombinant Gaussia luciferase. Overexpression, purification, and analytical application of a bioluminescent reporter for DNA hybridization. Anal Chem 74:4378–4385. [DOI] [PubMed] [Google Scholar]
- 22. Rathnayaka T, Tawa M, Nakamura T, Sohya S, Kuwajima K, Yohda M, Kuroda Y (2011) Solubilization and folding of a fully active recombinant Gaussia luciferase with native disulfide bonds by using a SEP‐Tag. Biochim Biophys Acta 1814:1775–1778. [DOI] [PubMed] [Google Scholar]
- 23. Tannous BA (2009) Gaussia luciferase reporter assay for monitoring biological processes in culture and in vivo. Nat Protoc 4:582–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Saxena VP, Wetlaufer DB (1970) Formation of three‐dimensional structure in proteins. I. Rapid nonenzymic reactivation of reduced lysozyme. Biochemistry 9:5015–5023. [DOI] [PubMed] [Google Scholar]
- 25. Lyles MM, Gilbert HF (1991) Catalysis of the oxidative folding of ribonuclease A by protein disulfide isomerase: dependence of the rate on the composition of the redox buffer. Biochemistry 30:613–619. [DOI] [PubMed] [Google Scholar]
- 26. Chang JY (1994) Controlling the speed of hirudin folding. Biochem J 300:643–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fuchs S, De Lorenzo F, Anfinsen CB (1967) Studies of mechanism of enzymic catalysis of disulfide interchange in proteins. J Biol Chem 242:398–402. [PubMed] [Google Scholar]
- 28. Schwaller M, Wilkinson B, Gilbert HF (2003) Reduction‐reoxidation cycles contribute to catalysis of disulfide isomerization by protein‐disulfide isomerase. J Biol Chem 278:7154–7159. [DOI] [PubMed] [Google Scholar]
- 29. Raturi A, Mutus B (2007) Characterization of redox state and reductase activity of protein disulfide isomerase under different redox environments using a sensitive fluorescent assay. Free Radic Biol Med 43:62–70. [DOI] [PubMed] [Google Scholar]
- 30. Hatahet F, Ruddock LW (2009) Protein disulfide isomerase: a critical evaluation of its function in disulfide bond formation. Antioxid Redox Signal 11:2807–2850. [DOI] [PubMed] [Google Scholar]
- 31. Watanabe MM, Laurindo FR, Fernandes DC (2014) Methods of measuring protein disulfide isomerase activity: a critical overview. Front Chem 2:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xu S, Sankar S, Neamati N (2014) Protein disulfide isomerase: a promising target for cancer therapy. Drug Discov Today 19:222–240. [DOI] [PubMed] [Google Scholar]
- 33. Foster CK, Thorpe C (2017) Challenges in the evaluation of thiol‐reactive inhibitors of human protein disulfide isomerase. Free Radic Biol Med 108:741–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hoober KL, Sheasley SS, Gilbert HF, Thorpe C (1999) Sulfhydryl oxidase from egg white: a facile catalyst for disulfide bond formation in proteins and peptides. J Biol Chem 274:22147–22150. [DOI] [PubMed] [Google Scholar]
- 35. Codding JA, Israel BA, Thorpe C (2012) Protein substrate discrimination in the Quiescin Sulfhydryl Oxidase (QSOX) family. Biochemistry 51:4226–4235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hoober KL, Joneja B, White HB III, Thorpe C (1996) A sulfhydryl oxidase from chicken egg white. J Biol Chem 271:30510–30516. [DOI] [PubMed] [Google Scholar]
- 37. Heckler EJ, Alon A, Fass D, Thorpe C (2008) Human quiescin‐sulfhydryl oxidase, QSOX1: probing internal redox steps by mutagenesis. Biochemistry 47:4955–4963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Heckler EJ, Rancy PC, Kodali VK, Thorpe C (2008) Generating disulfides with the Quiescin‐sulfhydryl oxidases. Biochim Biophys Acta 1783:567–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kodali VK, Thorpe C (2010) Oxidative protein folding and the Quiescin‐sulfhydryl oxidase family of flavoproteins. Antioxid Redox Signal 13:1217–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Winther JR, Thorpe C (2014) Quantification of thiols and disulfides. Biochim Biophys Acta 1840:838–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fass D, Thorpe C (2018) Chemistry and enzymology of disulfide cross‐linking in proteins. Chem Rev 118:1169–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Saaranen MJ, Karala AR, Lappi AK, Ruddock LW (2010) The role of dehydroascorbate in disulfide bond formation. Antiox Redox Signal 12:15–25. [DOI] [PubMed] [Google Scholar]
- 43. Givol D, Anfinsen CB, Goldberger RF (1964) Oxidation + disulfide interchange in reactivation of reduced ribonuclease. J Biol Chem 239:3114–3116. [PubMed] [Google Scholar]
- 44. Venetianer P, Straub FB (1964) Mechanism of action of ribonuclease‐reactivating enzyme. Biochim Biophys Acta 89:189–190. [DOI] [PubMed] [Google Scholar]
- 45. Karala AR, Lappi AK, Saaranen M, Ruddock LW (2009) Efficient peroxide mediated oxidative refolding of a protein at physiological pH and implications for oxidative folding in the endoplasmic reticulum. Antioxid Redox Signal 11:963–970. [DOI] [PubMed] [Google Scholar]
- 46. Pihl A, Lange R (1962) Interaction of oxidized glutathione, cystamine monosulfoxide, and tetrathionate with –SH groups of rabbit muscle D‐glyceraldehyde 3‐phosphate dehydrogenase. J Biol Chem 237:1356–1362. [PubMed] [Google Scholar]
- 47. Kurth JM, Dahl C, Butt JN (2015) Catalytic protein film electrochemistry provides a direct measure of the tetrathionate/thiosulfate reduction potential. J Am Chem Soc 137:13232–13235. [DOI] [PubMed] [Google Scholar]
- 48. Jocelyn PC (1972) Biochemistry of the SH group. London: Academic Press. [Google Scholar]
- 49. Cline DJ, Thorpe C, Schneider JP (2003) Effects of As(III) binding on alpha‐helical structure. J Am Chem Soc 125:2923–2929. [DOI] [PubMed] [Google Scholar]
- 50. Ramadan D, Rancy PC, Nagarkar RP, Schneider JP, Thorpe C (2009) Arsenic(III) species inhibit oxidative protein folding in vitro. Biochemistry 48:424–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sapra A, Ramadan D, Thorpe C (2015) Multivalency in the inhibition of oxidative protein folding by arsenic(III) species. Biochemistry 54:612–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sharma SK, Goloubinoff P, Christen P (2008) Heavy metal ions are potent inhibitors of protein folding. Biochem Biophys Res Commun 372:341–345. [DOI] [PubMed] [Google Scholar]
- 53. Tamas MJ, Sharma SK, Ibstedt S, Jacobson T, Christen P (2014) Heavy metals and metalloids as a cause for protein misfolding and aggregation. Biomolecules 4:252–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A (2003) ExPASy: the proteomics server for in‐depth protein knowledge and analysis. Nucleic Acids Res 31:3784–3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Figure S1. SDS‐PAGE gel of fractions eluting from Ni‐affinity purification of the GLuc construct used in this work (see Methods). The left lane represents BioRad standards. Lanes 1–3 are consecutive fractions eluting from the second ProBond nickel‐chelating Ni‐IDA resin (see Methods).
Figure S2. Mass Spectra of the treated Gaussia luciferase construct used in this work. Panels A and B represent oxidized and reduced protein respectively. The expected mass of the native 5‐disulfide protein is 21,833 from the sequence shown in Figure 2. Panels C and D are the mass spectra observed after treatment of oxidized and reduced GLuc with NEM. The mass resolution is 0.5 Daltons (see Materials and Methods).
Figure S3. Sample output from the GLuc assay started by the addition of 0.6 µM coelenterazine as described in Methods section. Coelenterazine was added from 0 to 1 s followed by 1 s shaking. Light emission is integrated from 2‐12 s.
Figure S4. Calibration curves for GLuc assays under the conditions described in Methods. Each point in both data sets is the average of 3 determinations. Panels A and B cover 0‐1 and 0‐100 nM Gluc respectively. Under these instrumental conditions (see Materials and Methods) the relative luminescence units per mg of GLuc is 1.8 x 1010/s.
Figure S5. The effect of PDI on the recovery of GLuc activity in glutathione redox buffer. Panel A shows reactivation of rGLuc over 1 h in the presence of 3 mM GSH and 2 mM GSSG in the absence or presence of 100 nM PDI (circles and squares, respectively). Panel B shows the luminescence activity reached after 10 min incubation under the same conditions but with PDI levels increased to 10 µM.
Figure S6. Alignment of amino acid sequence of first and second domains of Gaussia luciferase. The domain boundaries from Inouye and Sahara [17] were aligned using Multalin [53]. Four pairs of conserved cysteine residues are identified with arrows. One cysteine in each of the first and second domains is not conserved (smaller yellow boxes).