

Abstract
Research for three decades and major recent advances have provided crucial insights into how neurotransmitters are released by Ca2+‐triggered synaptic vesicle exocytosis, leading to reconstitution of basic steps that underlie Ca2+‐dependent membrane fusion and yielding a model that assigns defined functions for central components of the release machinery. The soluble N‐ethyl maleimide sensitive factor attachment protein receptors (SNAREs) syntaxin‐1, SNAP‐25, and synaptobrevin‐2 form a tight SNARE complex that brings the vesicle and plasma membranes together and is key for membrane fusion. N‐ethyl maleimide sensitive factor (NSF) and soluble NSF attachment proteins (SNAPs) disassemble the SNARE complex to recycle the SNAREs for another round of fusion. Munc18‐1 and Munc13‐1 orchestrate SNARE complex formation in an NSF‐SNAP‐resistant manner by a mechanism whereby Munc18‐1 binds to synaptobrevin and to a self‐inhibited “closed” conformation of syntaxin‐1, thus forming a template to assemble the SNARE complex, and Munc13‐1 facilitates assembly by bridging the vesicle and plasma membranes and catalyzing opening of syntaxin‐1. Synaptotagmin‐1 functions as the major Ca2+ sensor that triggers release by binding to membrane phospholipids and to the SNAREs, in a tight interplay with complexins that accelerates membrane fusion. Many of these proteins act as both inhibitors and activators of exocytosis, which is critical for the exquisite regulation of neurotransmitter release. It is still unclear how the actions of these various proteins and multiple other components that control release are integrated and, in particular, how they induce membrane fusion, but it can be expected that these fundamental questions can be answered in the near future, building on the extensive knowledge already available.
Keywords: neurotransmitter release, synaptic vesicle fusion, SNAREs, Munc18, Munc13, synaptotagmin, complexin, NSF, SNAPs
Introduction
The amazing variety of functions performed by the brain is made possible by the ability of neurons to communicate with each other, which occurs primarily via chemical synaptic transmission. This process involves neurotransmitters that are packaged in vesicles at presynaptic nerve terminals and are released by Ca2+‐evoked synaptic vesicle exocytosis. The vesicles are tethered to specialized sites on the presynaptic plasma membrane called active zones, undergo one or more priming reactions that leaves them ready for release, and fuse with the plasma membrane when an action potential triggers Ca2+ influx into the terminal.1 Release exhibits a fast, synchronous component that appears in less than 0.5 ms after Ca2+ influx and a slower, asynchronous component.1 Each of the steps that lead to release can be regulated during a variety of short‐ and long‐term presynaptic plasticity processes that alter the probability of vesicle release, thus modulating the properties of neural networks and underlying multiple forms of information processing in the brain.2 Hence, neurotransmitter release represents an exquisitely regulated form of membrane fusion. Correspondingly, comprehensive studies of the release machinery for almost three decades have shown that release is controlled by core components that have homologues in most types of intracellular membrane traffic and underlie a general mechanism of membrane fusion, as well as by specialized factors that help to confer the tight regulation of synaptic vesicle fusion.3 Clearly, determining the mechanism of membrane fusion is critical to understand how neurotransmitters are released but, from the point of view of neuroscience, it is equally or perhaps even more important to elucidate how release is regulated and how the ancient membrane fusion machinery evolved during billions of years to provide such a powerful signaling mechanism that enables in part the many wonders of the human brain.
Many mechanistic insights into neurotransmitter release have been obtained progressively over the years, but multiple fundamental questions remained just a few years ago.4 Recent advances have been bringing into focus the blurry image that was emerging from the available information and, although membrane fusion itself remains enigmatic, it seems plausible that a reasonably detailed understanding of the mechanism of release may be reachable in the near future. In this review I outline important concepts that emerged from earlier studies and emphasize recent key advances, trying to make a clear distinction between the notions that I consider well established and those that remain debatable. I focus primarily on N‐ethyl maleimide sensitive factor (NSF), soluble NSF attachment proteins (SNAPs), SNAP receptors (SNAREs), Munc18‐1 and Munc13s as core components of the presynaptic fusion machinery, and on synaptotagmin‐1 (Syt1) and complexins as key regulators, but I will also mention briefly some other important regulators of release such as Rab3s, RIMs and CAPS. Note however that some of the core components themselves are also intimately involved in the regulation of release, and that most of these proteins perform both active and inhibitory roles in release, as expected from the very nature of this biological process. Another noteworthy feature of many of these proteins is the presence in their sequence of C2 domains, which often function as Ca2+‐dependent phospholipid binding modules but can also perform other functions5 and can be considered critical regulators of neurotransmitter release. I will describe some results from studies of other types of intracellular membrane fusion that have brought seminal insights but, unfortunately, it is impossible to cover all the vast amount of literature available in this field, and I apologize to the authors of many studies that I will not describe. Excellent recent reviews have covered some aspects not discussed here (e.g., 3, 4, 6, 7, 8, 9).
The SNARE Complex
SNARE proteins are characterized by sequences of about 65 residues called SNARE motifs that have propensity to form coiled cols. The neuronal SNAREs involved in neurotransmitter release are syntaxin‐1, SNAP‐25 (no relation to SNAPs) and synaptobrevin‐2 (also called VAMP2; below referred to as synaptobrevin). Syntaxin‐1 and synaptobrevin each contain one SNARE motif preceding a transmembrane (TM) region, and are anchored to the plasma and vesicle membranes, respectively [Fig. 1(A,B)]. SNAP‐25 includes two SNARE motifs and is attached to the plasma membrane through palmitoylation. The three SNAREs form a tight SNARE complex10 that consists of a four‐helix bundle11, 12 and brings the membranes into close proximity13 [Fig. 1(C,D)], which is critical for membrane fusion (see below). Synaptobrevin is often referred to as a v‐SNARE because of its vesicle localization, while syntaxin‐1 and SNAP‐25 are called t‐SNAREs because they reside on the target membrane.14 The observation of a conserved polar layer in the middle of the SNARE complex that is formed by three glutamines (from syntaxin‐1 and SNAP‐25) and one arginine (from synaptobrevin) led to a more general classification that designates syntaxin‐1 and SNAP‐25 as Q‐SNAREs, and synatobrevin as an R‐SNARE.15
Figure 1.
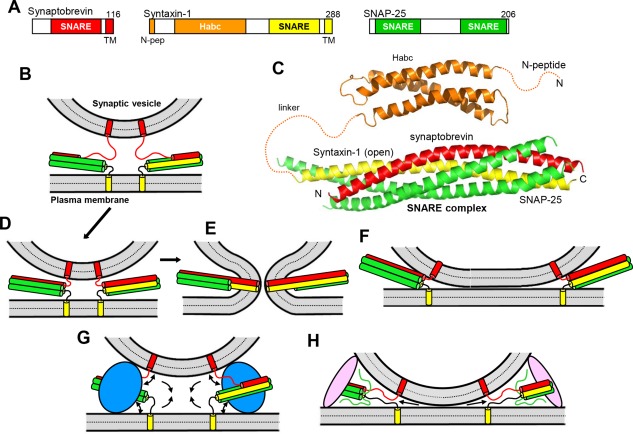
Models of SNARE‐dependent membrane fusion. (A) Domain structures of synaptobrevin, syntaxin‐1 and SNAP‐25. SNARE indicates SNARE motif and N‐pep indicates the N‐peptide of syntaxin‐1. Numbers on the right above the diagrams indicate the length of the protein. The same color coding for the SNAREs is used in all figures. (B) Diagram illustrating the topology of the neuronal SNAREs, with synaptobrevin anchored on a synaptic vesicle and syntaxin‐1 anchored on the plasma membrane, and showing how the SNAREs can form partially assembled trans‐SNARE complexes between the two membranes. In this model, the N‐terminal half of the four‐helix bundle is assembled and the C‐terminal half of the synaptobrevin SNARE motif is unstructured; the syntaxin‐1 and SNAP‐25 SNARE motifs are often assumed to be fully helical, as shown in the diagram, but their C‐termini may actually be unstructured.164 (C) Ribbon diagrams representing the three‐dimensional structures of the syntaxin‐1 H abc domain19 (PDB accession code 1BR0) and the neuronal SNARE complex12 (PDB accession code 1SFC). N and C indicate the N‐ and C‐terminus, respectively. Dashed curves represent flexible regions that were not present in the elucidated structures. (D–E) Together with panel (B), these diagrams illustrate the widespread model whereby the SNARE complex is initially formed at the N‐terminus (B), it zippers toward the C‐terminus, bringing the two membranes in close proximity (D), and causes membrane fusion as continuous helices are formed by the SNARE motifs and TM regions of syntaxin‐1 and synaptobrevin, as well as the short linkers between them.12, 27, 38 (E). For simplicity, only the SNARE motifs, TM regions and these short linkers are shown. (F) Diagram showing how SNARE complex assembly can lead to extended membrane‐membrane interfaces without fusion.32 (G) Diagram illustrating how a bulky protein(s) (blue) bound to the SNARE four‐helix bundle could play a key role in membrane fusion by pushing the membranes away from each other at the same time that assembly of the SNARE complex pulls the membranes together, which could cause a torque (see arrows) that helps to bend the membranes and initiate fusion.45, 46 (H) Diagram showing how membrane bridging by an elongated tethering factor (pink) could bring the vesicle and plasma membranes into proximity. In this arrangement, the SNARE complex would assemble in the periphery of the membrane‐membrane interface, perhaps bound to the bridging protein and/or another factor, and C‐terminal zippering of the SNARE complex could pull the membranes radially (see arrows), thus perturbing the packing of the lipids and catalyzing membrane fusion.48
SNARE complexes that are formed between two membranes before fusion are referred to as trans‐SNARE complexes, whereas those residing on the same membrane after fusion are called cis SNARE complexes. Synaptic vesicle priming to a readily‐releasable state is widely believed to involve formation of trans SNARE complexes that are partially assembled at the N‐terminus (e.g., 16). Trans SNARE complexes are also assumed to be partially assembled in vesicles that are morphologically docked using the definition of docking that is now commonly used, that is, contact or very close proximity (≤5 nm) between a vesicle and the plasma membrane at the active zone (e.g., 17, 18). This definition, which I adopt below, is more stringent than those used earlier and implies that docking and priming may be equivalent (but see below). I will use the term tethering to indicate vesicle proximity to the plasma membrane that involves longer distances and occurs before SNARE complex assembly.
In addition to a SNARE motif and a TM region, syntaxin‐1 contains an N‐terminal region that includes a three‐helix bundle called H abc domain19 and a short N‐terminal sequence referred to as N‐peptide20 [Fig. 1(A,C)]. The H abc domain binds intramolecularly to the SNARE motif, forming a self‐inhibited structure known as closed conformation that prevents SNARE complex formation21 and binds to Munc18‐1, in cooperation with the N‐peptide (see below). SNARE complex assembly is also influenced by the tendency of SNARE motifs to promiscuously form four‐helix bundles that constitute kinetic traps (reviewed in22). For instance, syntaxin‐1 and SNAP‐25 form heterodimers called t‐SNARE complexes that are heterogenous and exhibit predominantly a 2:1 stoichiometry because the missing synaptobrevin SNARE motif is replaced by a second copy of syntaxin‐1.23
Mechanics of SNARE‐Dependent Membrane Fusion
The notion that the high energy released by SNARE complex assembly provides the energy for membrane fusion13 is now widely accepted, and models whereby SNARE complexes induce fusion without the assistance of any other factors [e.g., Fig. 1(B,D,E)] abound in the literature. However, it is still unclear whether the SNAREs alone can induce physiological membrane fusion, and alternatively views are still considered (e.g., 24). Important insights into this question have been obtained with diverse types of reconstitution studies where fluorescent probes were used to monitor lipid and/or content mixing between liposomes containing R‐SNAREs and liposomes or supported bilayers containing Q‐SNAREs (reviewed in8), but two issues confound the available literature. First, many papers use the term “fusion” when only lipid mixing was observed, but it is well established that content mixing also needs to be observed to demonstrate true membrane fusion, as close to quantitative lipid mixing between liposomes can occur with very little content mixing (e.g., 25). This finding and the asymmetry observed in lipid mixing experiments where the fluorescence probes where placed on the vacuolar R‐ or Q‐SNARE liposomes26 show that lipid mixing does not even prove the existence of hemifusion (where only the proximal leaflets of the two bilayers have fused) and could occur by other mechanisms (e.g., lipid transfer catalyzed by perturbation of closely apposed membranes). The second confounding issue is that the observation of membrane fusion in reconstitutions assays is often assumed to be of physiological significance. However, much like a protein‐protein interaction observed in vitro can be completely irrelevant in vivo, membrane fusion in reconstitution assays could occur by a mechanism that is distinct from that occurring in vivo. Hence, to demonstrate that fusion observed in vitro is physiologically relevant, it is crucial to establish clear correlations of the results of the fusion assays with physiological data, as well as to develop a detailed mechanistic understanding that gives confidence that the results obtained in the fusion assays recapitulate at least to some degree the molecular events that occur in vivo.
Pioneering reconstitution experiments showing lipid and content mixing between synaptobrevin‐liposomes and syntaxin‐1‐SNAP‐25‐liposomes suggested that the neuronal SNAREs constitute a minimal membrane fusion machinery,27, 28 and a single neuronal SNARE complex was reported to be sufficient for fusion.29 However, there is also evidence for stable formation of trans‐SNARE complexes between two membranes without fusion.30, 31 Moreover, cryo‐electron microscopy (cryo‐EM) analyses of liposome fusion reactions mediated by the neuronal SNAREs revealed the formation of wide, closely apposed membrane‐membrane interfaces [Fig. 1(F)] that presumably proceed to fusion through hemifusion intermediates.32 This mechanism is markedly different from the stalk mechanism that is generally believed to mediate physiological membrane fusion and involves initial merger of the proximal leaflets to yield a stalk intermediate, followed by fusion pore formation when the distal leaflets merge.33 Although it is plausible that physiological membrane fusion does not really proceed through the stalk intermediate, it is also possible that the SNAREs alone cannot induce fusion through a biologically relevant pathway, or perhaps can do so but not very efficiently.
At the core of this unresolved question are the mechanical properties of the SNARE complex. Physiological studies have suggested that at least two or three SNARE complexes are required for Ca2+‐evoked exocytosis,34, 35 and estimates of the energy of SNARE complex assembly suggest that one or a few SNARE complexes are sufficient to provide the energy required to fuse membranes.36 However, it is unclear how this energy is applied to the membranes to fuse them. Multiple lines of evidence indicate that the SNARE complex assembles in stages, starting at the N‐terminus and “zippering” toward the C‐terminus16, 36, 37 [Fig. 1(B,D)]. A widespread model assumes that, for both synaptobrevin and syntaxin‐1, the SNARE motif, the TM region and the short linker between them can form a continuous helix, and mechanical force to induce fusion is exerted by zippering of the entire synaptobrevin and syntaxin‐1 helices, beyond the four‐helix bundle and reaching the end of their TM regions12, 27, 38 [Fig. 1(E)]. Such continuous helices were observed in a crystal structure of the neuronal SNARE complex in detergent.39 However, there is also evidence for flexibility in the linkers40 and the introduction of a flexible sequence within the synaptobrevin linker impairs Ca2+‐evoked but not spontaneous neurotransmitter release.41 Moreover, SNAREs bearing lipid anchors instead of TM regions can support yeast vacuolar fusion42 and neurotransmitter release.43 Thus, it appears that helix continuity including the TM regions is not a critical aspect of SNARE function. Note also that most of the energy of SNARE complex assembly is consumed by formation of the four‐helix bundle,36 which is expected to bring the two membranes within a few nanometers of each other but not necessarily into direct contact44 [Fig. 1(D)]. This observation indicates that SNARE complexes can form without inducing fusion, as observed in some studies.30, 31 Conversely, it is also worth noting that interactions between the linkers36 and between their multiple basic residues and negatively charged phospholipids can bring the membranes closer together than depicted in Figure 1(D), but this proximity may not necessarily induce fusion and might lead to the wide membrane‐membrane interfaces observed by cryo‐EM32 [Fig. 1(F)].
To ascertain whether or not the neuronal SNAREs alone can induce physiological membrane fusion, it is crucial to understand how the SNAREs work together with other central components of the neurotransmitter release machinery. Many authors assume that other components act merely as “accessory” or “regulatory” proteins that control SNARE complex assembly, but, even if they are involved in SNARE complex assembly, some proteins may actually have direct roles in fusion; if that is the case, they should be considered as intrinsic components of the macromolecular assembly that induces fusion. For instance, the energy of SNARE complex assembly may be applied more efficiently to the membranes if the SNAREs are bound to a bulky protein(s) (e.g., Munc18‐1) because the bulky protein would keep the membranes apart while the SNAREs pull them together, which would generate a torque that would help to bend the membranes and induce fusion45, 46 [Fig. 1(G)]. Recent studies of yeast vacuolar fusion and molecular dynamics simulations have provided support for this model.47 Another model proposed that an elongated protein that can bridge two membranes (e.g., Munc13‐1) could by itself bring the synaptic vesicle and plasma membranes into close apposition and assembly of SNARE complexes bound to this and potentially other proteins would exert force not to bring the membranes together but rather to pull the lipids outward, thus perturbing the packing of the lipids in the bilayers and catalyzing fusion48 [Fig. 1(H)]. Syt1 has also been proposed to cooperate with the SNAREs in causing membrane fusion by a variety of mechanisms, and strongly stimulates SNARE‐dependent fusion in reconstitutions assays (see below). Moreover, it has been postulated that formation of trans SNARE complexes does not fuse the membranes but rather sets up the stage for other proteins such as Syt1 or Sec17 to insert hydrophobic loops in the membranes to catalyze fusion24 (see below). Further research will be required to test the validity of these various models.
SNARE Complex Disassembly by NSF and SNAPs
The fully zippered cis SNARE complexes residing on the plasma membrane after synaptic vesicle fusion are highly stable, and their disassembly to recycle the SNAREs for another round of fusion requires the energy of ATP hydrolysis by NSF with the help of SNAPs (Sec18 and Sec17, respectively, in yeast).10, 49, 50 NSF contains an N‐terminal domain and two nucleotide‐binding domains from the AAA family (called D1 and D2). The N‐terminal domain contains two subdomains, one with a double‐ψ β‐barrel fold and another that forms an α/β roll51 [Fig. 2(A)], and is responsible for binding to SNAPs, which in turn bind to the SNARE complex and thus serve as adaptors.52 The D1 and D2 domains assemble into hexamers [illustrated by the crystal structure of D2,53 Fig. 2(B)]. ATP hydrolysis by the D1 domain is key for SNARE complex disassembly, whereas D2 has little ATPase activity.54 SNAPs adopt an elongated structure formed mostly by α‐helical hairpins and a globular helical domain at the C‐terminus [Fig. 2(C)], with an overall twist that allows binding to the also elongated surface of the SNARE complex.55
Figure 2.
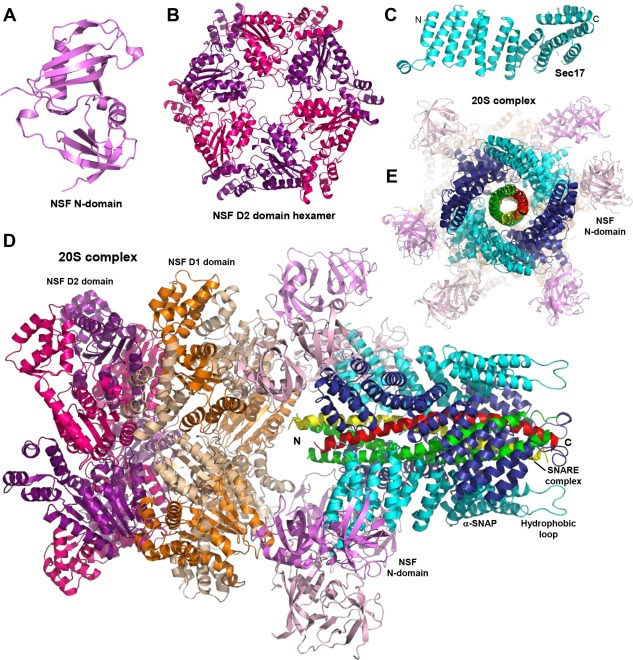
Structures of NSF, SNAP/Sec17 and the 20S complex. (A–E) Ribbon diagrams showing the three‐dimensional structures of the N‐terminal domain of NSF51 (A), the NSF D2 domain hexamer53 (B), Sec1755 (C), and the 20S complex56 (D,E). The PDB accession codes are 1QDN, 1NSF, 1QQE, and 3J96, respectively. In (C) and (D), N and C indicate the N‐ and C‐termini of Sec17 (C) and the SNARE four‐helix bundle (D), respectively. Panels (D,E) show two different views of the 20S complex rotated approximately 90°. In (E), the C‐terminus of the SNARE four‐helix bundle is pointing to the front. For alternate subunits of NSF, the N‐terminal domain is shown in violet or light pink, the D1 domain in orange or wheat, and the D2 domain in magenta or purple. Alternate αSNAP subunits are shown in cyan or deep blue. The N‐terminal hydrophobic loop of one of the αSNAP subunits is indicated in (D).
A seminal study by cryo‐EM recently reported the structure of the NSF hexamer in ADP‐ and ATP‐bound states, as well as four structures of the 20S complex formed by NSF, αSNAP and the neuronal SNARE complex, representing four states of the complex bound to a non‐hydrolyzable ATP analogue that co‐existed in the same sample.56 One of the four structures is shown in Figure 2(D,E) and illustrates how four αSNAP molecules bind to the SNARE four‐helix bundle while making contact at the same time with N‐terminal domains of NSF molecules. Among several important insights, these structures show how the hexameric symmetry of NSF transitions to the pseudo four‐fold symmetry of the SNARE complex and reveal that the twist of the αSNAP molecules is opposite to that of the SNARE complex surface. This finding suggests that the SNARE complex is disassembled by an unwinding mechanism when ATP hydrolysis causes structural changes in NSF that are transferred via its N‐terminal domains to the αSNAP‐SNARE assembly. The largely electrostatic nature of the interactions between αSNAP and the SNARE complex explains why NSF and SNAPs can disassemble distinct SNARE complexes.56 It is also important to note that αSNAP contains an N‐terminal hydrophobic loop that mediates a dramatic increase in the rate of disassembly of membrane‐anchored cis SNARE complexes by NSF‐αSNAP, compared to soluble SNARE complexes.57 The structures of the 20S complex nicely show how this hydrophobic loop is close to the SNARE C‐termini [Fig. 2(D)] and therefore should facilitate assembly of the 20S complex on the membrane.
In addition to disassembling cis SNARE complexes, NSF and SNAPs have additional roles that have been particularly well studied in yeast vacuolar fusion. Sec18 and Sec17 disassemble trans SNARE complexes,58 thus preventing fusion mediated by the SNAREs alone, but also play positive synergistic roles with the HOPS tethering factor.59 This synergy may arise at least in part because Sec18‐Sec17 disassemble t‐SNAREs complexes,60 as t‐SNARE complexes with wrong stoichiometry can hinder trans SNARE complex formation (see above). Reconstitution assays using a mutant SNARE with a deletion at the C‐terminus, which allows formation of trans SNARE complexes between liposomes without fusion, showed that Sec17 by itself can trigger fusion of this arrested state, and that this activity requires the hydrophobic N‐terminal loop of Sec17.61, 62 Moreover, ATP‐bound NSF can further stimulate fusion (without ATP hydrolysis).63 These results have led to the proposal that Sec17 actively participates in inducing fusion through membrane insertion of the N‐terminal hydrophobic loop,62 or stimulates fusion because it releases an inhibitory action of the HOPS complex after the SNARE complex is assembled.64 Note also that the stimulatory activity of Sec17 occurs at the late stages of the reconstitution assays, but at the early stages Sec17 inhibits fusion.64
It is unclear to what extent these properties are conserved in the neurotransmitter release machinery. Thus, NSF‐αSNAP disassemble t‐SNARE complexes,65 which was proposed to underlie the inhibition of SNARE‐dependent fusion by NSF‐αSNAP,66, 67 while some evidence suggested that NSF‐αSNAP do not disassemble trans SNARE complexes66 (but see68). No stimulatory activity in membrane fusion has been reported for αSNAP, but it has been shown that αSNAP by itself impairs SNARE‐dependent fusion, which was attributed to interference in SNARE zippering.69 Conversely, experiments with optical tweezers suggested that αSNAP enhances zippering.70 Clearly, much needs to be learned still about the various functions of NSF and αSNAP.
Orchestration of SNARE Complex Assembly by Munc18‐1 and Munc13s
Munc18‐1 is a member of the Sec1/Munc18 (SM) family of proteins that are critical for most types of intracellular membrane traffic,46 and contains three domains that form an arched‐shaped structure71 [Fig. 3(A)]. Munc13s are large (ca. 200 kDa) multidomain proteins from pre‐synaptic active zones with a variable N‐terminal region and a conserved C‐terminal region. Munc13‐1, the major neuronal isoform in mammals, contains a C2 domain and a calmodulin‐binding (CaMb) sequence in the N‐terminal region, whereas its C‐terminal region contains a C1 domain, two C2 domains and a MUN domain, a highly elongated module that is homologous to tethering factors involved in diverse forms of membrane traffic48, 72, 73, 74 (Fig. 4). Ca2+‐triggered neurotransmitter release, spontaneous release and sucrose‐induced release, which measures the readily‐releasable pool of vesicles, are totally abrogated in the absence of Munc18‐1 or of Munc13‐1 and −2 in mice75, 76 (and also in invertebrates lacking their homologue, unc1377, 78). These findings not only revealed the critical nature of Munc18‐1 and Munc13s for synaptic vesicle priming (and potentially also in downstream events), but also suggested that their functions are related. The basis for this critical nature remained for a long time as one of the most enigmatic questions in this field,22 but major advances in recent years have yielded defined models for the mechanism of actions of Munc18‐1 and Munc13s.
Figure 3.
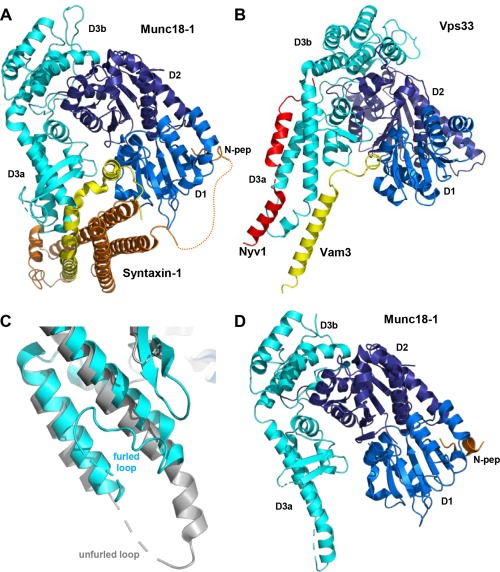
Structures of SM protein‐SNARE complexes. (A) Ribbon diagram showing the three‐dimensional structure of Munc18‐1 bound to closed syntaxin‐171, 79 (PDB accession code 3C98). The domains of Munc18‐1 are colored in blue (D1), deep blue (D2) and cyan (D3a and D3b). Syntaxin‐1 is colored in orange (N‐terminal region) and yellow (SNARE motif). The dashed curve represents the flexible sequence linking the N‐peptide (N‐pep) to the H abc domain of syntaxin‐1. (B) Superposition of the crystal structures of Vps33 (blue, deep blue and cyan) bound to the SNARE motifs of the synaptobrevin homologue Nyv1 (red) and the syntaxin‐1 homolog Vam396 (yellow) (PDB accession codes 5BUZ and 5BV0, respectively). Vps33 is shown for only the Vps33/Nyv1 complex. Note that the Vam3 SNARE motif is in a similar location as the syntaxin‐1 SNARE motif in the Munc18‐1/syntaxin‐1 complex (A) and that simultaneous binding of the Vam3 and Nyv1 SNARE motifs to the Vps33 sites observed in the two structures would place the SNARE motifs in register to form the SNARE complex. (C) Close up of a superposition showing the helix‐loop‐helix of domain 3a of Munc18‐1 in the crystal structure of the Munc18‐1‐closed syntaxin‐1 complex (cyan) and the crystal structure of Munc18‐1 bound to the syntaxin‐4 N‐peptide98 (gray) (PDB accession code 3PUJ). Note that the loop is furled in the former and unfurled in the latter. (D) Ribbon diagram of the full Munc18‐1‐syntaxin‐4 N‐peptide structure.
Figure 4.
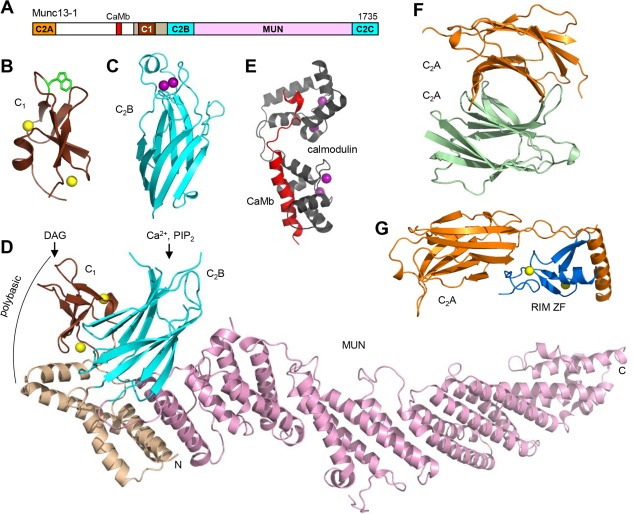
Structure of Munc13‐1. (A) Domain diagram of Munc13‐1. The calmodulin‐binding sequence is labeled CaMb. The number on the right above the diagram indicates the length of the protein. (B–G) Ribbon diagrams of the three‐dimensional structures of the C1 domain110 (B), the Ca2+‐bound C2B domain111 (C) and the C1C2BMUN fragment48 (D) of Munc13‐1, as well as calmodulin (purple) bound to the Munc13‐1 CaMb sequence (red)114 (E), the Munc13‐1 C2A domain homodimer116 (F) and the heterodimer of the Munc13‐1 C2A domain (orange) with the RIM2α ZF domain (blue)116 (G). The PDB accession codes are 1Y8F, 3KWU, 5UE8, 2KDU, 2CJT and 2CJS, respectively. Ca2+ ions are shown as purple spheres and zinc ions are shown as yellow spheres. In (D), the locations of the DAG/phorbol ester‐binding site in the C1 domain, the Ca2+/PIP2‐binding site in the C2B domain, and a polybasic region formed by the C1 domain, the C2B domain and the linker sequence between them are indicated.
Early research (reviewed in more detail in4, 22) showed that Munc18‐1 binds very tightly through the cavity of its arch‐shape to the closed conformation of syntaxin‐121, 71 and binding involves also the syntaxin‐1 N‐peptide79 [Fig. 3(A)]. Munc18‐1 binding stabilizes the closed conformation80 and hinders SNARE complex assembly,79, 81 but also has a positive role by stabilizing both proteins in vivo.75, 82 As a consequence, a so‐called LE mutation that destabilizes the closed conformation of syntaxin‐1 impairs priming but increases the vesicular release probability.82 Biochemical data83 and the observation that syntaxin‐1 bearing the LE mutation partially rescues neurotransmitter release in unc13 nulls in Caenorhabditis elegans 84 suggested that Munc13s/unc13 mediate syntaxin‐1 opening. Indeed, the MUN domain was later shown to be critical for Munc13‐1 function,72 and biophysical studies showed that the MUN domain facilitates the transition from the Munc18‐1‐closed syntaxin‐1 complex to the SNARE complex.81 Munc18‐1 also binds to the assembled SNARE complex though interactions with the syntaxin‐1 N‐peptide, the H abc domain and the SNARE four‐helix bundle.20, 85, 86, 87, 88 The interaction with the four‐helix bundle may be alternative to binding to the H abc domain89 and was proposed to underlie a direct role of Munc18‐1 in fusion45 [Fig. 1(G)]. The functional importance of the syntaxin‐1 N‐peptide‐Munc18‐1 interaction has been supported by some studies,20, 86, 90, 91 but not by others.92, 93 It seems likely that the N‐peptide provides an anchor point to retain Munc18‐1 attached to syntaxin‐1 when conformational changes lead to syntaxin‐1 opening [Fig. 5(A)]. Note that some of these properties likely arose to meet specific regulatory requirements of neurotransmitter release, as Vam3, the syntaxin involved in yeast vacuolar fusion, does not adopt a closed conformation and does not contain an N‐peptide.94
Figure 5.
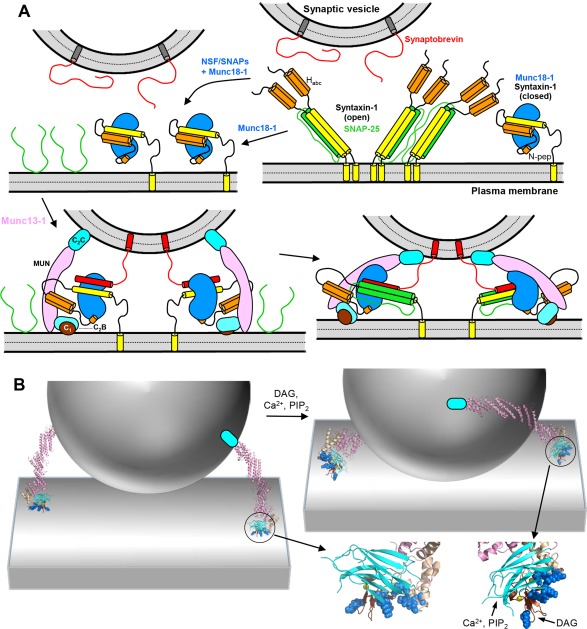
Orchestration of SNARE complex formation by Munc18‐1 and Munc13‐1. (A) Model of the mechanism of SNARE complex assembly.48, 67, 95, 99 The model postulates that the starting point for the productive pathway that leads to neurotransmitter release is the complex of closed syntaxin‐1 (yellow and orange) with Munc18‐1 (blue), which may be formed directly or after NSF‐SNAPs disassemble syntaxin‐1‐SNAP‐25 complexes (upper diagrams). SNARE complex assembly occurs when Munc13‐1 (pink, cyan and brown) bridges the synaptic vesicle and plasma membranes through interactions of the C2C domain with the former and the C1‐C2B region with the latter, and catalyzes opening of syntaxin‐1, which facilitates binding of synaptobrevin (red) to Munc18‐1 (lower left diagram). By placing the N‐terminal halves of the syntaxin‐1 and synaptobrevin SNARE motifs into proximity and in the correct register, Munc18‐1 acts as a template that initiates assembly of the SNARE four‐helix bundle,96, 99 which later requires incorporation of SNAP‐25 (green). The N‐peptide of syntaxin‐1 (N‐pep) is proposed to help keeping syntaxin‐1 bound to Munc18‐1 during the conformational changes that take place. Munc13‐1 is proposed to initially bind to the plasma membrane through multiple basic residues in the C1‐C2B region, which leads to an approximately perpendicular orientation with respect to the membrane (lower left). DAG binding to the C1 domain and Ca2+/PIP2 binding to the C2B domain are proposed to favor a more slanted orientation of Munc13‐1 that helps to bring the two membranes into closer proximity and facilitates SNARE complex assembly (lower right diagram). This orientation is expected to be more favored when DAG and intracellular Ca2+ levels increase during repetitive stimulation, thus increasing the release probability. The model also predicts that Munc18‐1 and Munc13‐1 remain bound to the SNARE complex after assembly, perhaps helping in fusion as proposed in Figure 1(G,H), but this feature is unclear. It is also unclear whether such binding is compatible with interactions of the SNARE complex with Syt1 and Cpx1, which are not shown for simplicity. (B) Three‐dimensional model to better illustrate the notion that Munc13‐1 can bridge the synaptic vesicle and plasma membranes in two different orientations. The model includes only a vesicle, the plasma membrane and a ribbon diagram representing the structure of the Munc13‐1 C1C2BMUN fragment attached to a cyan ellipsis that represents the C2C domain. Below the right diagram are close‐up views of the Munc13‐1 C1‐C2B region. The residues that form the polybasic region are shown as blue spheres. On the right, the DAG‐ and Ca2+/PIP2‐binding sites are indicated.
Altogether, these and other observations led to a model postulating that, in contrast to the widespread textbook model whereby the syntaxin‐1‐SNAP‐25 constitutes the starting point for synaptic vesicle fusion, the real starting point is instead the Munc18‐1‐closed syntaxin‐1 complex, which then requires Munc13‐1 to open syntaxin‐1 and coordinate SNARE complex formation81 (see also Refs. 21, 71, 84 for earlier related models). An attractive feature of this model is that the Munc18‐1‐closed syntaxin‐1 complex provides a much better defined starting point for an exquisitely regulated process such as neurotransmitter release than the syntaxin‐1‐SNAP‐25 complex, which is heterogeneous due to the promiscuity of the SNARE motifs (see above). In support of this model, competition assays showed that Munc18‐1 can quantitatively displace SNAP‐25 bound to syntaxin‐1, and reconstitution assays using syntaxin‐1‐Munc18‐1 liposomes revealed efficient fusion with synaptobrevin‐liposomes that required SNAP‐25 and a Munc13‐1 fragment spanning the C1, C2B and MUN domains (C1C2BMUN), and that was enhanced by Syt1.67 However, these results still did not clarify why Munc18‐1 and Munc13s are so crucial for neurotransmitter release in vivo, as extensive reconstitution experiments that used syntaxin‐1‐SNAP‐25 liposomes had shown that SNARE complex assembly and fusion with synaptobrevin‐liposomes could also occur in the absence of Munc18‐1 and Munc13s (see above). The key to solve this conundrum was the inclusion of NSF and αSNAP, which abolish fusion between syntaxin‐1‐SNAP‐25 liposomes and synaptobrevin liposomes,66 even in the presence of the strong stimulation provided by Syt1,67 because NSF‐αSNAP disassemble syntaxin‐1‐SNAP‐25 complexes and perhaps trans SNARE complexes (see above). Under these conditions, Munc18‐1 and the Munc13‐1 C1C2BMUN are essential for liposome fusion because they orchestrate SNARE complex assembly in an NSF‐αSNAP resistant manner.67 This notion provides a clear explanation for the essential nature of Munc18‐1 and Munc13s in vivo and is reminiscent of earlier results obtained in seminal reconstitution studies of yeast vacuolar fusion showing that SNARE complex assembly and fusion in the presence of Sec17‐Sec18 require the HOPS tethering complex, which includes the SM protein Vps33 and five additional components.58, 59 Since the machineries that control yeast vacuolar fusion and synaptic vesicle exocytosis are very distantly related in evolution, this convergence of results suggests that the critical function that makes SM proteins and tethering factors so fundamental for intracellular membrane fusion in general is the organization of SNARE complex assembly in an environment that strongly favors SNARE complex disassembly.
Recent results from both systems have also provided key clues on how SNARE complex assembly is orchestrated. On one hand, a fragment spanning the conserved C‐terminal region of Munc13‐1 (C1C2BMUNC2C, which includes the C2C domain) was shown to be much more efficient that the C1C2BMUN fragment in mediating liposome fusion because the C1‐C2B region can bind to one membrane while the C2C binds to another membrane, which correlated with the need to include both the C1‐C2B region and the C2C domain (in addition to the MUN domain) to efficiently rescue neurotransmitter release in Munc13‐1/2 double KO mice.95 These findings indicate that, in addition to helping to open syntaxin‐1, a crucial function of Munc13‐1 is to provide a bridge between the synaptic vesicle and plasma membranes, thus facilitating SNARE complex assembly (Fig. 5). This notion is supported by the finding that Munc13s are critical for synaptic vesicle docking.18 On the other hand, two crystal structures of Vps33 bound to the syntaxin‐1 homologue Vam3 or to the synaptobrevin homologue Nyv1 showed how simultaneous binding of Vam3 and Nyv1 to Vps33 would place their SNARE motifs in close proximity and in register to form the SNARE complex [Fig. 3(B)], strongly suggesting that SM proteins form a template to facilitate SNARE complex assembly.96 In support of this proposal, the location of the Vam3 SNARE motif on Vps33 is similar to that observed for the syntaxin‐1 SNARE motif in the structure of the Munc18‐1‐closed syntaxin‐1 complex [Fig. 3(A)], and Munc18‐1 was also found to bind to synaptobrevin.89, 97 This interaction is weak because the helical hairpin that forms the synaptobrevin‐binding site in domain 3a of Munc18‐1 adopts a furled conformation where the binding site is occluded by the loop connecting the two helices [Fig. 3(C)], but a crystal structure of Munc18‐1 bound to the N‐peptide of another syntaxin isoform, syntaxin‐4, showed that this loop can also adopt an unfurled conformation98 [Fig. 3(C,D)] that resembles the conformation of the loop in Nyv1‐bound Vps33 [Fig. 3(B)]. Moreover, a mutation in the helical hairpin of Munc18‐1 that disrupts synaptobrevin binding impairs the activity of Munc18‐1 in stimulating fusion in reconstitution assays and a mutation that perturbs the furled loop conformation increases synaptobrevin binding as well as Munc18‐1 activity.97, 99
In summary, the available data lead to a model whereby Munc18‐1 templates SNARE complex assembly by first binding to closed syntaxin‐1 and later binding to synaptobrevin when Munc13‐1 opens syntaxin‐1 while at the same time bridges the vesicle and plasma membranes [Fig. 5(A)]. The observation that Munc18‐1 and Munc13‐1 prevent antiparallel binding modes between the SNAREs100 show that, in addition to providing resistance to NSF‐αSNAP, this mechanism improves the fidelity of SNARE complex assembly. There are however important issues to be resolved, particularly regarding the incorporation of SNAP‐25 into the SNARE complex. Some data suggested that a tripartite Munc18‐1‐closed syntaxin‐1‐SNAP‐25 complex can be formed,101, 102 but NMR experiments indicate a clear competition as long as syntaxin‐1 is closed,67 as expected from the structures of the SNARE complex and the closed syntaxin‐1‐Munc18‐1 complex. The mechanism of syntaxin‐1 opening by the Munc13‐1 MUN domain is also unclear, as interactions of the MUN domain with the linker between the syntaxin‐1 H abc domain and its SNARE motif,103 or with the SNARE motif itself,81 have been implicated in this activity. The latter interaction and the finding that the Munc13‐1 MUN domain helps to prevent the formation of antiparallel SNARE complexes100 suggest that the MUN domain also helps to template SNARE complex assembly. Note in this context that the orientations of the synaptobrevin and syntaxin‐1 SNARE motifs bound to Munc18‐1 according to the available structural data (Fig. 3) imply that these interactions with Munc18‐1 must be released in order to allow SNAP‐25 binding. Perhaps the most critical unanswered question is whether Munc18‐1 and/or Munc13‐1 remain bound to the SNARE complex after assembly. There is indeed evidence for binding of the SNARE four‐helix bundle to Munc18‐185, 86, 88 (see above) and to the Munc13‐1 MUN domain.104, 105 Such binding might prevent disassembly of trans SNARE complexes by NSF‐αSNAP and thus stabilize the primed state of readily releasable vesicles, as suggested by a recent study,106 and could also mediate a direct involvement of Munc18‐1 and/or Munc13‐1 in membrane fusion as proposed in the models of Figure 1(G,H), but these models need further testing.
Munc13‐1 as a Master Regulator of Neurotransmitter Release
The increasing progress in our understanding of the mechanisms underlying the steps that lead to neurotransmitter release facilitates the elucidation of the molecular basis for diverse short‐ and long‐term presynaptic plasticity processes that underlie varied forms of information processing in the brain. This notion is epitomized by advances made in understanding the functions of Munc13‐1, which acts as a master regulator of release through its multidomain architecture [Fig. 4(A)], in addition to being critical for release itself. It is interesting to note that a gain‐of‐function mutation that is believed to unfurl the loop in domain 3a of unc18 can partially rescue the severe phenotypes observed in unc13 nulls in C. elegans,107 which is reminiscent of the rescue of these nulls by the open syntaxin‐1 mutant.84 These findings show that, without the energy barriers caused by the closed conformation of syntaxin‐1 and the furled conformation of the Munc18‐1/unc18 loop, Munc13s/unc13 would not be essential for neurotransmitter release; in wild type animals, the presence of these energy barriers leads to the essential nature of Munc13s/unc13 and enables the multiple forms of regulation that depend on Munc13s/unc13. The varied regulatory functions of Munc13‐1 are most likely related to modulation of its activity in mediating synaptic vesicle docking‐priming. In the conserved C‐terminal region, the Munc13‐1 C1 domain mediates DAG‐ and phorbol ester‐dependent augmentation of neurotransmitter release.108, 109 A point mutation that unfolds the C1 domain (H567K) practically abolishes this augmentation in mice and leads to death within 2–3 hours after birth despite leaving Ca2+‐evoked release unaltered,108 illustrating the critical importance of the regulation of release for survival. DAG‐phorbol ester binding to the Munc13‐1 C1 domain is hindered by a tryptophane side chain that blocks the phorbol ester‐binding site [Fig. 4(B)], which may allow activation at high local concentrations of DAG caused by repetitive stimulation without responding to smaller perturbations in DAG levels that mediate PKC‐dependent signaling.110 The adjacent C2B domain of Munc13‐1 mediates short‐term plasticity that depends on Ca2+ and phosphatidylinositide phosphates, in part through an unusual α‐helix present in its Ca2+‐ binding region111 [Fig. 4(C)].
The crystal structure of the Munc13‐1 C1C2BMUN fragment recently revealed how the DAG‐binding region of the C1 domain and the Ca2+‐PIP2‐binding region of the C2B domain are close to each other and point in the same orientation48 [Fig. 4(D)], showing that the two domains can cooperate in binding to the plasma membrane in a mode that is enhanced by DAG, Ca2+ and PIP2 [Fig. 5(B), right]. However, both domains form a polybasic region [Fig. 4(D)] that can mediate binding to the plasma membrane in a different orientation in the absence of these agents [Fig. 5(B), left]. Note also that the Munc13‐1 C2C domain can bind to membranes in a Ca2+‐independent manner and that the C1C2BMUNC2C fragment can bridge synaptobrevin‐liposomes with syntaxin‐1‐SNAP‐25 liposomes containing DAG and PIP2 in the absence and presence of Ca2+, but Ca2+ is required for fusion between these liposomes in the presence of C1C2BMUNC2C, Munc18‐1, NSF and αSNAP.95 These findings have led to a model of short‐term presynaptic plasticity whereby the conserved C‐terminal region of Munc13‐1 can bridge the synaptic vesicle and plasma membranes in at least two different orientations [Fig. 5(B)], one present in the absence of Ca2+ that primes the release machinery but inhibits membrane fusion, and another that facilitates fusion and is favored by Ca2+, PIP2 and DAG.48 It is also plausible that the two different orientations differ in their ability to promote SNARE complex assembly and that the orientation activated by Ca2+, PIP2 and DAG leads to formation of a larger number of trans SNARE complexes, which then dictate a larger release probability. Regardless of these possibilities, these ideas correlate with the findings that the H567K mutation that unfolds the Munc13‐1 C1 domain leads to a higher release probability in mice,108 and that deletion of either the C1 domain or the C2B domain of unc13 enhances evoked release in C. elegans.112 The latter study also showed that that Ca2+‐binding to the unc13 C2B domain releases the inhibition caused by this domain. Note also that deletion of the entire C1‐C2B region practically abolishes release,95, 112 supporting the notion that this region is critical to bind to the plasma membrane to enable bridging of the vesicle and plasma membranes by Munc13‐1.
The N‐terminal region of Munc13‐1 is also involved in diverse forms of regulation of release. Deletion of this region (residues 1–520) leads to a 50% decrease in evoked release, while deletion of only the C2A domain (residues 1–150) causes a much more dramatic impairment of release, and deletion of the linker region spanning residues 151–520 has mild effects on release.113 These results indicate that the linker region plays an inhibitory role and that the C2A domain is critical to relieve this inhibition. This linker region contains a CaMb sequence [Fig. 4(A)], which includes two mobile structural modules114 [Fig. 4(E)] and mediates some forms of Ca2+‐dependent short‐term plasticity.115 Because the N‐terminal region emerges next to the MUN domain in the structure of Munc13‐1 C1C2BMUN [Fig. 4(D)], it is tempting to speculate that the linker region inhibits release by blocking interactions of the MUN domain with the SNAREs and that these inhibitory interactions are released by an activity of the C2A domain and/or by calmodulin binding to the CaMB sequence.
The Munc13‐1 C2A domain is Ca2+ independent and acts as a protein‐protein interaction domain that forms a homodimer116 [Fig. 4(F)], which inhibits neurotransmitter release,117 and alternatively forms a heterodimer with αRIMs118, 119 [Fig. 4(G)]. αRIMs are large (ca. 180 kDa) Rab3 effectors that play critical roles in synaptic vesicle priming and in organizing the active zone,120, 121 and are also involved in tethering Ca2+ channels to the active zone122 and in long‐term potentiation at mossy fibers.123 The priming role is primarily performed by a zinc finger (ZF) domain at the N‐terminus of αRIMs,122 which is responsible for binding to the Munc13‐1 C2A domain [Fig. 4(F)] and activates vesicle priming by disrupting the C2A domain homodimer.116, 117 In addition, heterodimerization of αRIMs with the Munc13‐1 C2A domain plays an important role in docking‐priming, as a mutation that disrupts both homo and heterodimerization leads to a 50% decrease in vesicle priming and in the number of docked vesicles, similar to that observed upon deletion of the entire N‐terminal region.113 However, a mutation that disrupts only the heterodimer has a similar effect in docking but a much stronger effect on priming,113 suggesting that homodimerization may allow docking while hindering fusion. Note also that sequences adjacent to the αRIM ZF domain bind to Rab3,124 which is believed to provide a mechanism for initial tethering of synaptic vesicles to the active zone. However, the mild phenotype observed upon deletion of the four Rab3 isoforms,125 compared to the much stronger effects observed in the absence of Rab proteins in other membrane traffic systems,126 suggests that there may be some redundancy in synaptic vesicle tethering, perhaps involving additional Rab isoforms. Regardless of this possibility, the observations that Rab3A and Munc13‐1 are also involved in mossy fiber long‐term potentiation,127, 128 and that Munc13‐1, αRIMs and Rab3A can form a tripartite complex, suggest that this complex provides a connection between synaptic vesicle priming and Rab3/αRIM‐dependent long‐term presynaptic plasticity.119 The molecular mechanisms underlying these different regulatory processes are still unclear. Nevertheless, it seems likely that the Munc13‐1 N‐terminal region increases docking‐priming because it helps to bring Munc13‐1 to the active zone, but this region is not essential because the conserved C‐terminal region can by itself perform its functions in bridging the vesicle and plasma membranes and opening syntaxin‐1.
Dual Roles of Complexins in Neurotransmitter Release
Complexins are small soluble proteins that bind tightly to the SNARE complex.129 Analyses in mice bearing a double knockout of the two major complexin isoforms, complexin‐1 (Cpx1) and −2, or a triple knockout of Cpx1, 2 and 3, as well as experiments where Cpx1 and 2 where knocked down, revealed marked impairments in evoked neurotransmitter release130, 131, 132 (reviewed in 133, 134). Spontaneous release was decreased, unaltered or increased in these different studies, a variability that may arise in part from compensatory increases in Cpx3 levels in the knockdown experiments.135 In the absence of complexins, sucrose‐induced release was impaired but to a lesser extent than evoked release, leading to a decreased release probability,130, 136, 137 with no defect in vesicle docking.18 These findings suggest that mammalian complexins play an important role in the Ca2+‐triggering step of release and, although they are not essential for docking‐priming, they stabilize the primed state and/or enhance the fusogeneicity of the primed state (sometimes called superpriming). The distinct effects on spontaneous release likely arise because of a different balance between this positive role on priming and an inhibitory activity of complexins in the different preparations used. An inhibitory role for complexins was originally indicated by physiological and reconstitution experiments that suggested an interplay with the Ca2+ sensor Syt1 whereby the inhibition caused by complexins is released by Syt1 (see below).138, 139, 140 The dual role of complexins was further illustrated by the increased spontaneous release and the decreased evoked release observed in invertebrate complexin nulls.141, 142, 143 Docking was severely impaired in C. elegans complexin nulls,143 suggesting that there might be some functional differences between mammalian and invertebrate complexins.
Mammalian Cpx1 is largely unstructured in solution144 but binds to the SNARE complex by forming a central α‐helix that inserts into the groove between syntaxin‐1 and synaptobrevin, and is preceded by an accessory α‐helix that does not contact the SNAREs145 [Fig. 6(A,B)]. The central helix is critical for all Cpx1 functions, in part by placing other Cpx1 sequences in the right position to execute their roles, while the accessory helix plays an inhibitory function132, 146, 147 by a mechanism that is still under debate (see below). The N‐terminal region preceding the accessory helix plays a stimulatory role in mice, likely by helping to release the inhibition caused by the accessory helix and interacting with membranes and/or the SNARE complex.132, 136, 146, 148, 149 In both mammalian Cpx1 and invertebrate complexin, the C‐terminal region beyond the central helix contains a membrane‐binding motif that senses membrane curvature and helps to localize them to synaptic vesicles, which is important for inhibition of spontaneous release.150, 151, 152, 153 Indeed, sequence differences in the C‐terminal region underlie the stronger suppression of spontaneous release by invertebrate complexins than mammalian Cpx1,154 and the strong activity of Drosophila complexin in inhibiting spontaneous release depends on a C‐terminal farnesylation motif that mediates membrane localization and is present in mammalian Cpx3 and Cpx4 but not Cpx1 and Cpx2.155 Note however that the C‐terminal region of Cpx1 contributes to both active and inhibitory roles in neurotransmitter release,156 and may simply help to localize Cpx1 near the sites of fusion without having a direct active or inhibitory action.
Figure 6.
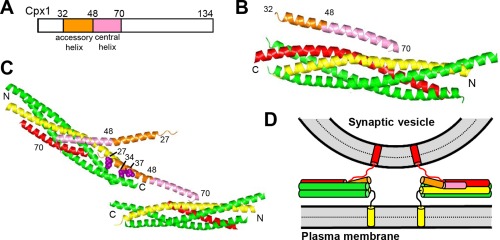
Structure and function of complexins. (A) Domain diagram of Cpx1. Numbers above the diagram indicate the domain boundaries and the length of the protein. (B) Ribbon diagram showing the three‐dimensional structure of the Cpx1(26–83)/SNARE complex145 (PDB accession code 1KIL). (C) Ribbon diagram illustrating the three‐dimensional structure of the complex between Cpx1(26–83) bearing the superclamp mutation and a SNARE complex that was truncated at the synaptobrevin C‐terminus160 (PDB accession code 3RK3). Two copies of Cpx1(26–83) and of the truncated SNARE complex are displayed to show how one Cpx1(26–83) molecule binds to one SNARE complex through the central helix and to another SNARE complex through the mutated accessory helix, resulting in a zigzag array. The three mutated residues (shown as brown spheres) are hydrophobic and bind to the hydrophobic groove left by the synaptobrevin truncation, but these three residues are charged in WT Cpx1. In (B,C), N and C indicate the N‐ and C‐termini of the SNARE complex, and selected residue numbers of Cpx1 are indicated. (D) Model illustrating how, upon binding of Cpx1 to SNARE complexes partially assembled between two membranes, the accessory helix would hinder closer membrane‐membrane proximity due to steric and/or electrostatic hindrance with the vesicle. For simplicity, only the SNARE motifs, TM regions and linkers between them are shown. In panels (B–D), Cpx1(26–83) is color coded as in panel (A), syntaxin‐1 is yellow, synaptobrevin red and SNAP‐25 green.
The mechanisms underlying the dual functions of complexins are still unclear. It seems likely that the active function(s) arises because binding of the Cpx1 central helix stabilizes the SNARE complex in the prime state145 and/or because it helps setting up the Ca2+ sensing action of Syt1138 by forming a tripartite macromolecular assembly with Syt1 and the SNARE complex157 (see below). Single‐vesicle reconstitution assays indicated that Cpx1 plays an active role by increasing the on‐rate of vesicle docking,158 but other reconstitution assays where Cpx1 was pre‐incubated with t‐SNARE vesicles suggested that Cpx1 hinders docking because it hinders SNARE complex formation,149, 159 and both of these conclusions contrast with the lack of docking defects in the absence of complexins in mice.18
The accessory helix is responsible at least in part for the inhibitory role of Cpx1, and a model that was widely accepted proposed that this role arises because this helix inserts into the C‐terminus of partially assembled SNARE complexes, replacing part of the synaptobrevin SNARE motif.146, 147 This proposal was supported by the finding that so‐called superclamp and poor‐clamp mutations in the accessory helix enhanced or decreased, respectively, the inhibitory activity of Cpx1 in cell‐cell fusion assays.147 The crystal structure of a SNARE complex with a truncated synaptobrevin C‐terminus bound to a Cpx1 fragment bearing the superclamp mutation revealed how the accessory helix inserts into the groove left by the synaptobrevin truncation in one SNARE complex while the central helix binds to another SNARE complex, yielding a zigzag array that was proposed to inhibit neurotransmitter release160 [Fig. 6(C)]. Other biophysical experiments, including isothermal titration calorimetry (ITC) data, supported the notion that this interaction occurs in solution for both WT and superclamp Cpx1.160, 161, 162, 163 However, all supporting data could have alternative interpretations, and NMR experiments did not detect any interaction of truncated SNARE complexes with the accessory helix of WT or superclamp Cpx1 despite the very high sensitivity of these experiments to detect protein‐protein interactions.164 Moreover, the observation of heat in the ITC data presented in support of the zigzag model160, 163 requires the highly basic juxtamembrane region of syntaxin‐1, strongly suggesting that this heat arises from an ionic interaction involving this region rather than insertion of the accessory helix into the SNARE complex.165 Note also that the superclamp mutation replaces three charged residues with hydrophobic residues [Fig. 6(C)]; hence, it is highly unlikely that WT Cpx1 can interact with the truncated SNARE complex in the same mode observed for the superclamp mutant in the crystal structure.
The superclamp mutation was reported to increase the ability of Cpx1 to inhibit spontaneous neurotransmitter release in mice,137 but the data presented in this study did not really show a statistically significant effect and in fact are consistent with a subsequent study showing no significant effect of the superclamp mutation in spontaneous or evoked release.164 Rescue experiments with mouse Cpx1 in Drosophila complexin nulls did show a lower rate of spontaneous release for rescue with the superclamp mutant than for rescue with WT Cpx1,166 but another way to look at these data is that both WT and superclamp mutant Cpx1 exhibited a similarly strong ability to suppress spontaneous release, considering the very high rate observed for the complexin nulls. Note also that rescue with a Cpx1 mutant that behaved as a poor‐clamp in cell‐cell fusion assays160 yielded similar spontaneous release to that observed with the WT Cpx1 rescue,166 and another poor‐clamp mutant actually impaired release in mice.164 Additional mutagenesis experiments in the latter study led to a model whereby the inhibitory activity of the accessory helix arises because it is oriented towards the membranes at the site of fusion, hindering fusion because of electrostatic and/or steric hindrance with the membranes164 [Fig. 6(D)]. The observation that the inhibition of spontaneous release by complexin in C. elegans is unaltered when the accessory helix is replaced by a non‐native α‐helix with a completely unrelated, neutral sequence167 strongly supports the conclusion that the inhibitory role of the accessory helix does not involve specific interactions with the SNAREs or any other protein, and is consistent with the steric hindrance model.
Regardless of the underlying mechanism, beautiful experiments using the surface force apparatus162 and single‐molecule FRET studies using nanodisc‐anchored SNAREs168 showed that complexin indeed hinders C‐terminal zippering of trans‐SNARE complexes, arresting them in a half‐zippered state. Interestingly, another single‐molecule FRET study showed that Cpx1 can actually hinder C‐terminal zippering of cis‐SNARE complexes to some degree,169 although this activity was strongly hindered by a no‐clamp mutation that did not have significant effects in the rescue experiments in Drosophila.166 The same single‐molecule study also showed that Cpx1 bound to a SNARE complex via its central helix can bind in trans to the syntaxin‐1‐SNAP‐25 complex, and that this interaction is strengthened by the superclamp mutation and weakened by the no‐clamp mutation,169 in correlation with other biophysical experiments.149 However, the significance of these data is also unclear given the lack of correlation with the effects of these mutations in neurons.137, 164, 166 In summary, there is a large amount of interesting data on complexins, but the mechanism underlying its inhibitory role is still unclear. I favor the steric hindrance model, but further research establishing clear correlations between in vitro and in vivo data will be required to fully understand this mechanism.
Ca2+ Sensing by Synaptotagmin‐1 and Interplay with Complexins
Synaptotagmins constitute a family of membrane trafficking proteins characterized by the presence of tandem C2 domains in their cytoplasmic region.170 Syt1 is essential for synchronous neurotransmitter release but asynchronous release is increased in its absence,171 and knockdown of Syt7 in the Syt1 KO background strongly impairs this enhanced asynchronous release.172 These and other results indicate that Syt1 and Syt7 act as Ca2+ sensors for synchronous and asynchronous release, respectively, and that different Ca2+ sensors may compete for a common target (e.g., the SNARE complex) at release sites (reviewed in1). There is also evidence for competition between different Ca2+ sensors that mediate spontaneous release, including Syt1.172, 173 Such potential competition complicates the interpretation of the functional effects of mutations in a given sensor. Although it was initially thought that synaptotagmins are not involved in priming, more recent data indicated that deletion of Syt1 or of Syt1 together with Syt7 leads to a considerable decrease in the readily‐releasable pool without affecting the rate of priming.174, 175 In hippocampal synapses from Syt1 KO mice, vesicle docking was reported to be unaltered18 or decreased by 35%175 (but see176). These results suggest that, similar to complexins, synaptotagmins are not essential for docking‐priming or for fusion, but they stabilize the primed state of synaptic vesicles; most importantly, synaptotagmins accelerate fusion to increase the release probability within the short time window when the Ca2+ concentration increases at release sites upon arrival of an action potential. Below I focus largely on Syt1, which has been studied most extensively.
Syt1 is a synaptic vesicle protein that includes an intravesicular sequence, a TM region, a linker and tandem C2 domains [Fig. 7(A)]. The Syt1 C2A and C2B domains adopt characteristic β‐sandwich structures that bind three and two Ca2+ ions, respectively, through loops at the top177, 178, 179 [Fig. 7(B)]. Ca2+ binding does not induce substantial conformational changes on the Syt1 C2 domains but drastically changes the electrostatic potential in the Ca2+‐binding region, suggesting that these domains function as electrostatic switches that inhibit fusion before Ca2+ influx and activate fusion upon Ca2+ binding.179, 180, 181 The Ca2+‐binding loops mediate Ca2+‐dependent binding to membranes182, 183 and mutations that increase or decrease the apparent Ca2+ affinity in phospholipid binding lead to parallel changes in the Ca2+‐dependence of neurotransmitter release, which demonstrated the functional importance of this activity and showed that Syt1 is the major Ca2+ sensor that triggers neurotransmitter release.184, 185 Mutations in the C2B domain Ca2+‐binding sites practically abolish release while mutations in the C2A domain Ca2+‐binding sites can also impair release strongly, although not as drastically,186, 187, 188, 189 suggesting that the C2B domain plays a preponderant role. However, these results need to be examined with caution, as mutations in the C2B domain Ca2+‐binding sites cause dominant negative effects186, 190 and the relative functional importance of Ca2+‐binding to the C2 domains is switched for Syt7172, which may arise because the C2A domain instead of the C2B domain dominates Ca2+‐dependent phospholipid‐binding to Syt7191. For Syt1, the C2B domain can also interact with membranes via a polybasic region on the side of the β‐sandwich that binds to PIP2 192, 193 and through two arginines at the bottom of the β‐sandwich (R398‐R399)194 [Fig. 7(B)], and mutations in both regions strongly disrupt release.175, 195, 196, 197
Figure 7.
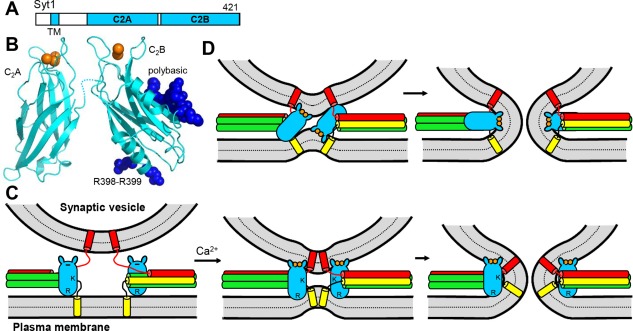
Structure and function of Syt1. (A) Domain diagram of Syt1. The number on the right above the diagram indicates the length of the protein. (B) Ribbon diagrams showing the three‐dimensional structures of the Ca2+‐bound Syt1 C2A and C2B domains178, 179 (PDB accession codes 1BYN and 1K5W, respectively). Ca2+ ions are shown as orange spheres. The side chains of R398‐R399 and the polybasic region of the C2B domain are shown as deep blue spheres. (C,D) Potential models of Syt1 function. In (C), the C2B domain is proposed to cooperate with the SNAREs in bringing the two membranes together in a Ca2+‐dependent manner by binding to the plasma membrane through R398‐R399 and to the vesicle membrane through its Ca2+‐binding loops (small protuberances at the top of the domain).194 R indicates the location of R398‐R399, and K the location of the polybasic region. Note that the Ca2+‐binding region is negatively charged before Ca2+ binding and would thus have electrostatic repulsion with the vesicle membrane (left), but binding of the Ca2+ ions (orange circles) would promote membrane binding. This model also proposes that, at the same time, the highly positive electrostatic potential of the C2B domain would help to bend the membranes (middle) and induce membrane fusion (right).194 Note that the orientation of the C2B domain could be reversed. (D) Model whereby insertion of the Ca2+‐binding loops of the C2B domain into the bilayers induces membrane curvature to catalyze fusion.198 In both (C) and (D), syntaxin‐1 is yellow, synaptobrevin red and SNAP‐25 green, and only the SNARE motifs, TM regions and linkers between them are shown. The C2A domain is not shown for simplicity and because its location with respect to the C2B domain is unclear, but the C2A domain could cooperate with the C2B domain in bringing the membranes together and in inducing membrane curvature. Note that in both models interactions of Syt1 with the SNAREs are not necessary, but Syt1 and the SNARE complex would still cooperate in inducing membrane fusion.
These and other findings led to various models of how Syt1 triggers fast Ca2+‐dependent membrane fusion. For instance, the finding that the Ca2+‐binding loops at the top of the C2B domain can bind to one membrane while R398‐R399 at the bottom bind to another membrane suggested that Syt1 can bring the vesicle and plasma membranes together, much like the SNAREs do, but in a Ca2+ dependent manner194 [Fig. 7(C)]. In addition, the C2B domain could also facilitate fusion by promoting membrane curvature through electrostatic interactions194 [Fig. 7(C)]. Membrane curvature can also be favored by insertion of the Ca2+‐binding loops of the Syt1 C2 domains into the membranes198 [e.g., Fig. 7(D)], which was also proposed to cause tension199 or perform work200 on the membranes, or to perturb the packing of the lipids in the bilayers to induce fusion.24 These various concepts are not necessarily incompatible with each other. Binding of the C2B domain polybasic region to PIP2 was proposed to function upstream of fusion by steering Syt1 to the plasma membrane.192 Reconstitution experiments have shown that Syt1 can stimulate SNARE‐dependent lipid and content mixing, and have yielded some support for these models, particularly the notion that Syt1 brings the two membranes into close proximity (e.g.,30, 195, 201, 202, 203, 204, 205, 206). However, it has been difficult to validate these models, in part because of two key hurdles.
First, many papers reported interactions of the Syt1 C2 domains with syntaxin‐1, SNAP‐25, the t‐SNARE complex and/or the SNARE complex that could coordinate their functions, but it has been difficult to distinguish whether these interactions are functionally relevant or may result from the promiscuity of these proteins (reviewed in 4, 200). For instance, sequences of the Syt1 C2B domain that bind to lipids such as the polybasic region and R398‐R399 have also been implicated in SNARE binding (e.g., 207, 208, 209, 210, 211) and it is still unclear which are the true targets of these sequences in vivo. Note also that the strong Ca2+‐dependent stimulation of lipid mixing between synaptobrevin‐liposomes and syntaxin‐1‐SNAP‐25 liposomes caused by a soluble fragment containing both Syt1 C2 domains (C2AB) in reconstitution experiments202 might arise simply from its ability to bring two membranes into close proximity.195, 205, 212 Incorporation of full‐length Syt1 into synaptobrevin‐liposomes leads to a Ca2+‐independent enhancement of lipid mixing201 that does involve interactions of Syt1 with the t‐SNAREs and bridges the v‐ and t‐vesicles,209 but such bridging is likely caused in vivo by binding of Munc13‐1 to the two membranes when syntaxin‐1 is still bound to Munc18‐1 rather than SNAP‐25 (see above). Among the described Syt1‐SNARE interactions, those involving the SNARE complex are the most likely to be functionally important based on the mechanism of SNARE complex formation proposed in Figure 5 and the natural expectation that Syt1 and the SNAREs cooperate in the final step that leads to membrane fusion. It has been suggested that Syt1‐SNARE complex interactions are abolished at physiological ionic strength,213 but such interactions should be favored at the high local concentrations of Syt1 and the SNAREs present in the primed state even if they are weak. Moreover, the recent structural studies discussed below have provided compelling evidence for the relevance of Syt1‐SNARE complex binding.
A second major hurdle to elucidate how Syt1 functions has been to understand its interplay with complexins. The finding that the Syt1 C2AB fragment competes with a Cpx1 fragment spanning its accessory and central helices [Cpx1(26–83) for binding to membrane‐anchored SNARE complexes138, 214 provided a natural basis for functional, cell‐cell fusion and reconstitution experiments suggesting that Syt1 releases the inhibition caused by complexins,138, 139, 140 but it was puzzling that Syt1 C2AB and Cpx1(26–83) can bind simultaneously to the SNARE complex in solution.215 Subsequent reconstitution assays have further supported the notion that the functions of Syt1 and complexins are coupled, for instance showing that the Ca2+‐independent stimulation of lipid mixing caused by Syt1 was inhibited by CpxII.216 Moreover, single vesicle‐single vesicle fusion assays have indicated that Syt1 and Cpx1 cooperate in accelerating Ca2+‐dependent fusion, thus synchronizing fusion with Ca2+ arrival.217 Another study using similar experiments showed that the low fusion efficiency commonly observed in these assays at 1–5 μM Cpx1 can be dramatically enhanced by using lower Cpx1 concentrations (100–200 nM), suggesting that excess Cpx1 can unduly inhibit fusion.218 These assays also showed that Cpx1 can stimulate fusion in a Ca2+‐dependent manner even in the absence of Syt1,218 which raises the possibility that Cpx1 may act as a Ca2+ sensor, although no Ca2+‐binding sites have been identified in Cpx1 so far. Clearly, understanding how Syt1 binds to the SNARE complex and how such binding affects Cpx1‐SNARE complex interactions is critical to rationalize these interesting results.
Structures of Syt1‐SNARE Complex Assemblies
Key insights have been brought by three structural studies of Syt1‐SNARE complexes recently reported. First, an analysis by NMR spectroscopy in the presence of Ca2+ revealed the predominant binding mode between Syt1 and the SNARE complex in solution, which is highly dynamic and involves the polybasic region of the C2B domain and a polyacidic patch formed by SNAP‐25 and syntaxin‐1 in the SNARE complex196 [Fig. 8(A)]. This dynamic structure is consistent with extensive evidence implicating the C2B domain polybasic region in SNARE complex binding207, 208, 209, 210 and explains the puzzling biochemical interplay with Cpx1(26–83). Thus, the C2AB and Cpx1 binding sites on the SNARE complex are distinct, consistent with simultaneous binding in solution, but, in a putative C2AB‐SNARE‐Cpx1 complex anchored on a membrane, insertion of the C2B domain Ca2+‐binding loops into the membrane would cause steric clashes of the Cpx1 accessory helix with the membrane, consistent with the competition observed on membranes [Fig. 8(B)]. Note that Syt1 C2AB could not displace full‐length Cpx1 from membrane‐anchored SNARE complexes,215 suggesting that Syt1 binding to the membrane and the SNARE complex in this mode may lead to “melting” of the Cpx1 accessory helix without full displacement of Cpx1 due to membrane interactions involving its N‐ and/or C‐termini.196 Importantly, the orientation of the C2B domain in the structure would allow interaction of its Ca2+‐binding loops with the synaptic vesicle membrane while R398‐R399 bind to the plasma membrane, supporting the notion that Syt1 could cooperate with the SNAREs in bringing the two membranes together to accelerate membrane fusion [Fig. 8(F)]. The functional relevance of the structure was supported by the finding that different mutations in the polybasic region of the C2B domain caused strong or mild impairments in neurotransmitter release that correlated with their effects in disrupting C2AB‐SNARE complex binding but not with their more indiscriminate effects on PIP2 binding.196
Figure 8.
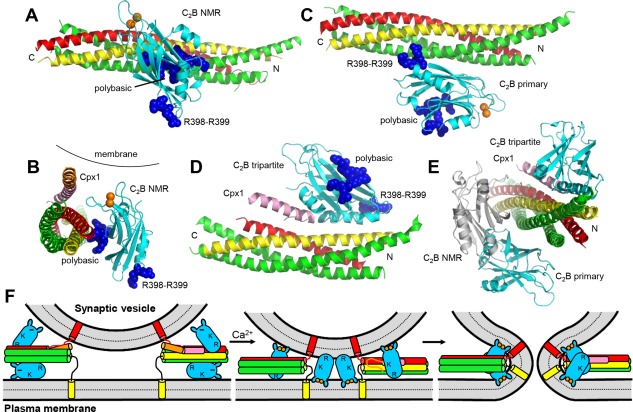
Structures of Syt1‐SNARE complexes. (A,C,D) Ribbon diagrams illustrating the dynamic structure of the Syt1 C2B domain‐SNARE complex assembly determined in solution by NMR spectroscopy196 (A), the crystal structure of the Syt1 C2AB‐SNARE complex assembly197 (C) and the crystal structure of the Syt1 C2AB‐Cpx1‐SNARE complex assembly157 (D) (PDB accession codes 2N1T, 5KJ7, and 5W5C, respectively). For all structures, only the Syt1 C2B domain is shown (cyan), with R398‐R399 and the polybasic region represented as blue spheres and bound Ca2+ ions as orange spheres. Syntaxin‐1 is yellow, synaptobrevin red, SNAP‐25 green, and Cpx1 pink. Note that the structure shown in (A) represents just one snapshot of the many closely‐related binding modes that form this dynamic ensemble. N and C indicate the N‐ and C‐termini of the SNARE complex. (B) Ribbon diagram showing a superposition of the NMR structure shown in (A) with the Cpx1(26–83)‐SNARE complex shown in Figure 6(B). Both structures have been rotated to better show how, if Cpx1 and the Syt1 C2B domain are bound simultaneously to the SNARE complex in these modes, Ca2+‐dependent binding of the C2B domain to a membrane would induce strong steric clashes of the Cpx1 accessory helix with the membrane.196 (E) Superposition of the structures shown in (A,C,D) illustrating how the binding modes of (A) and (C) overlap partially but they are both compatible with the binding mode of (D). (F) Diagrams illustrating a model that attempts to integrate the three structures of Syt1‐SNARE complex assemblies shown in (A,C,D). As in Figure 7(C,D), the Syt1 C2B domain is represented by cyan ellipses with protuberances that represent the Ca2+‐binding loops, Ca2+ ions are represented by orange circles and the C2A domain is not shown for simplicity. R indicates the location of R398‐R399, and K the location of the polybasic region. Cpx1 is shown in orange (accessory helix) and pink (central helix). The model postulates that, before Ca2+ influx, two Syt1 molecules bind through their C2B domain to each partially assembled SNARE complex according to the crystal structures shown in (C,D). In this arrangement, the C2B domain at the bottom binds also to PIP2 on the plasma membrane while the C2B domain at the top is expected to have electrostatic repulsion with the vesicle membrane, and steric hindrance between the vesicle and the Cpx1 accessory helix also contributes to hinder membrane fusion (left). Ca2+ binding to the Syt1 C2B domain is proposed to cause rearrangements that allow, for some of the Syt1 molecules, simultaneous binding of the C2B domain polybasic region to the SNARE complex as in the NMR structure shown in (A) while the Ca2+‐binding loops bind to the vesicle and R398‐R399 bind to the plasma membrane; the C2B domain of other Syt1 molecules may just bridge the two membranes without contacting the SNAREs, perhaps in an opposite orientation (middle). Concomitantly, the SNARE complex zippers and the Cpx1 accessory helix melts because of steric hindrance. The position of Syt1 with respect to the membranes is proposed to further rearrange to facilitate membrane bending and fusion (right).
Subsequently, two similar crystal structures of Syt1 C2AB bound to the SNARE complex revealed three binding modes197 that were drastically different from each other and from that defined by NMR spectroscopy, consistent FRET and NMR data showing that there are multiple types of Syt1‐SNARE complex interactions.196, 219 One of the binding modes observed in the crystal structures, referred to as the primary interface, was validated by mutagenesis and electrophysiological data, while the other two binding modes most likely arise from crystal contacts (see also220, 221). The primary interface involves the two SNARE motifs of SNAP‐25, partially overlapping with the C2B domain‐binding site defined by NMR spectroscopy, but the interacting region of the C2B domain is located on the convex side of the β‐sandwich, opposite to the concave side containing the polybasic region [Fig. 8(C,E)]. As a result, this region is available for binding to PIP2 on the plasma membrane. Two additional crystal structures of the SNARE complex bound to a Cpx1 fragment and the Syt1 C2B domain or C2AB fragment later revealed yet another C2B domain‐SNARE complex interface, referred to as tripartite interface, that does not overlap with the primary interface or the binding mode defined by NMR157 [Fig. 8(D,E)]. The Cpx1 binding mode is analogous to that observed in the binary Cpx1‐SNARE complex assembly145 and the Cpx1 central helix is “continued” by an α‐helix of the Syt1 C2B domain that is unique to C2B domains in tandem C2 domain proteins222 and binds to the same groove of the SNARE complex as Cpx1 [Fig. 8(D)]. Because there are also C2B domain‐Cpx1 contacts, Cpx1 binding to the SNARE complex enhances its affinity for the C2B domain at this binding site.157 This finding provides an explanation for the active role of Cpx1 in neurotransmitter release, as it helps to position Syt1 next to the SNARE complex, and also suggests that Syt1 and Cpx1 cooperate in forming a “locked” state that inhibits release before Ca2+ influx but is ready for fast release upon Ca2+ binding to Syt1. Mutagenesis and electrophysiological experiments supported this notion and the functional relevance of the tripartite interface.157 It is also worth noting that, although the Syt1‐SNARE complex‐complexin stoichiometry is 1:1:1 in the crystals, the SNARE complex has other interfaces with the C2B domain of symmetry mates, including the primary interface observed in the first crystal structure of the C2AB‐SNARE complex. This finding illustrates that two Syt1 molecules can bind simultaneously to the SNARE complex through these two binding modes.
Figure 8(F) presents a model that incorporates the three structures of Syt1‐SNARE complexes and attempts to integrate much (but not all) of the existing data. The two binding modes observed in the crystal structures appear to be Ca2+‐independent and likely occur in the primed state that exists before Ca2+ influx, helping to bring Syt1 next to the SNARE complex to generate the release‐ready state. The finding that disruption of either binding mode strongly impairs release suggests that it is functionally important to have more than one Syt1 molecule next to the SNAREs to cooperate in triggering fusion, consistent with the known high cooperativity of release with Ca2+. Fusion may be inhibited in the primed state by the steric and/or electrostatic repulsion of the Cpx1 accessory helix and the Ca2+‐binding region of one of the C2B domain molecules with the synaptic vesicle membrane [Fig. 8(F)]. The two Syt1‐SNARE binding modes are unlikely to remain unaltered upon Ca2+‐binding to Syt1, as they do not explain how Syt1 releases the inhibition caused by Cpx1 and helps to induce membrane fusion. The model of Figure 8(F) proposes that Ca2+ binding to Syt1 induces rearrangements such that both Syt1 C2B domains bridge the vesicle and plasma membranes through their Ca2+‐binding loops at the top and R398‐R399 at the bottom, with one of the C2B domains binding to the SNARE complex through the polybasic region as in the mode defined by NMR spectroscopy. Note that, although the polybasic region is not involved in Ca2+ binding, the affinity of this largely electrostatic binding mode is increased by Ca2+ binding to the top loops,210 likely because of the resulting increase in the positive electrostatic potential. In this model, the Cpx1 accessory helix becomes unstructured due to steric clashes as binding of the C2B domains to the two membranes and C‐terminal zippering of the SNARE complex bring the membranes together.196 This model also accounts for the strong impairment of release caused by mutations in R398‐R399 together or individually,157, 195 and for functional data suggesting that these residues are important for triggering fusion.223 Note in this context that both arginines participate in the primary interface defined by X‐ray crystallography, but R398 does not appear to make strong contacts and mutation of R398‐R399 did not significantly affect the co‐immunoprecipitation of Syt1 with the SNAREs,197 suggesting that R398‐R399 interact with another key target [e.g., the membranes; Fig. 8(F)].
From the available structural information, the position of the Syt1 C2A domain is unclear, but there is no doubt that Ca2+ binding to this domain is also functionally important.184, 188, 189 The C2A domain may cooperate with the C2B domain in binding to the same membrane, help in bridging the two membranes,194, 204, 212, 224 and/or contribute to induce membrane curvature [as proposed in the model of Fig. 8(F)]. The model also proposes that, after bridging the two membranes, the insertion of the Ca2+‐binding loops into the membranes induces membrane curvature198 and/or perturbs the lipid packing.24 Note that the time scale of neurotransmitter release (<0.5 ms1) is fast for a biological process but provides ample time for the molecular rearrangements involved in this cascade of Syt1 actions.
Clearly, this model needs further testing, and multiple questions about Syt1 function remain. First, it is highly unclear how Syt1 helps to induce membrane fusion. Second, the multiplicity of Syt1‐SNARE complex binding modes [Fig. 8(F)] seems counterintuitive and it is plausible that some of the data of the structural studies might need re‐interpretation. Third, Syt1 was shown to form ring‐like oligomers that were proposed to inhibit neurotransmitter release before Ca2+ influx and to be disassembled upon Ca2+‐dependent membrane binding to the C2 domains,225, 226 but it is unclear whether such oligomers are compatible with the crystal structures of Syt1‐SNARE complex assemblies. It is worth noting that the observed rings led to a hypothesis whereby a buttressed ring of Syt1s is surrounded by a ring of Munc13s227 and, interestingly, supramolecular assemblies containing multiple Munc13‐1 molecules have been observed by super‐resolution imaging of synapses.228 Fourth, and more generally, it is unclear how this model can be integrated with the notion that Munc18‐1 and/or Munc13‐1 may also bind to the SNARE complex as intrinsic components of the complex that triggers membrane fusion (Fig. 5).
Perspectives
The research summarized above has yielded a wealth of information on the mechanism of neurotransmitter release, establishing that: (i) SNARE complexes are critical to bring membranes together and catalyze membrane fusion; (ii) NSF and αSNAP disassemble SNARE complexes and play additional roles; (iii) Munc18‐1 and Munc13‐1 play key roles in orchestrating SNARE complex assembly in an NSF‐αSNAP‐resistant manner, the former by binding to synaptobrevin and closed syntaxin‐1, thus forming a template to assemble the SNARE complex, and the latter by bridging the vesicle and plasma membranes and helping to open syntaxin‐1; and (iv) Syt1 functions as a Ca2+‐sensor by binding to the membranes and the SNAREs, in a tight interplay with complexin that accelerates membrane fusion. However, fundamental questions still remain, particularly with regard to the mechanism of membrane fusion. It seems clear that the SNAREs, Syt1 and complexin are part of the macromolecular assembly that triggers fusion, but how do they actually execute fusion? Are Munc18‐1 and/or Munc13‐1 part of this macromolecular complex, thus being also involved in fusion? Are αSNAP and perhaps NSF also involved in fusion, as suggested by studies of the roles of Sec17 and Sec18 in yeast vacuolar fusion? Which are the initial interactions that tether synaptic vesicles to the plasma membrane at the active zone and what is the cascade of events that bring the membranes progressively closer through the action of proteins that can bridge membranes such as the SNAREs, Munc13‐1 and Syt1?
Structural studies have been fundamental to develop many of the key concepts underlying our current understanding of neurotransmitter release, but most of these studies were performed without membranes. To address the questions listed above, it will be crucial to obtain structural information on complexes assembled between two membranes. It can be expected that major advances in this area may be obtained with cryo‐electron microscopy and tomography, techniques that have already provided important information in this field194, 212, 229 and have experienced a dramatic technical revolution in recent years.230, 231 Note that these techniques also allow observation of membrane fusion intermediates,32, 217, 232 and tomography can be used to observe presynaptic complexes in actual neuronal synapses.233
Reconstitutions experiments will continue to play a major role in complementing the structural studies and providing a means to probe the functions of the components of the release machinery in vitro, but it is important to understand the limitations of the approaches used so far in order to make further progress. Thus, bulk reconstitution assays have provided crucial information and allowed reconstitution of Ca2+‐dependent membrane fusion with the three SNAREs, Munc18‐1, Munc13‐1, Syt1, NSF and αSNAP,67 but these assays have low time resolution and do not distinguish between docking and fusion. Moreover, much of the Ca2+ dependence in these assays arises from Ca2+ binding to the Munc13 C2B domain,95 which is known to be important for release during repetitive stimulation but not for release evoked by the first action potential.111 This apparent discrepancy may arise because of partial functional redundancy with another protein such as CAPS, which also contains a Ca2+‐dependent C2 domain and a MUN domain, and likely has similar but not completely overlapping functions with Munc13s.22, 234, 235 Moreover, it is likely that the major function of Syt1 is to accelerate fusion to time scales well beyond those probed in bulk fusion assays. Single vesicle‐single vesicle fusion assays have also made important contributions to this field, can distinguish between docking and fusion, and have better time resolution than bulk assays,8 although the time resolution is still limited. Studies that monitor fusion of single vesicles with supported planar bilayers afford faster speed, having been able to reveal fusion within 20 ms,236 and are also very useful to study the neurotransmitter release machinery.237 However, even higher time resolution will be required to monitor fusion at the speeds occurring in vivo. Novel methods, including for instance amperometric recordings on artificial cells,238 may be necessary for this purpose. Of course, electrophysiological studies will continue to be essential to test the models emerging from structural and reconstitution studies, as well as to study the functions of the release machinery.
A detailed understanding of the molecular mechanism underlying a wide variety of presynaptic plasticity processes that make our brain so powerful has become increasingly feasible with the progress made in elucidating the basic mechanisms of docking, priming and fusion, particularly with regarding to Munc13 function. However, the presynaptic active zone is a complex supramolecular assembly that, in addition to Munc13s, include multiple large proteins such as RIMs, RIM‐binding proteins, bassoon, piccolo and liprins among others.239 The functional importance of these proteins was emphasized by the dramatic disruption of active zone formation and of neurotransmitter release caused by deletion of RIMs and RIM‐binding proteins.240 Hence, structural and reconstitution studies need to gradually incorporate these proteins, together with Rab3s and CAPS. Moreover, post‐translation modifications such as phosphorylation can also play key roles in regulating release (e.g., 241), and long‐term changes in release efficiency likely involve also transcriptional regulation. We have come a long way, but we still have a long way to go.
Acknowledgments
I thank Karolina Stepien, Eric Prinslow, Thomas Südhof and Axel Brunger for critical reading of the manuscript, and all the members of my laboratory, as well as Thomas Südhof, Christian Rosenmund, Diana Tomchick, Shuzo Sugita, William Wickner, Reinhard Jahn, Axel Brunger, Yeon‐Kyun Shin, James Rothman, Frederic Pincet and Cameron Gundersen for fruitful discussions.
References
- 1. Sudhof TC (2013) Neurotransmitter release: the last millisecond in the life of a synaptic vesicle. Neuron 80:675–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Regehr WG (2012) Short‐term presynaptic plasticity. Cold Spring Harb Perspect Biol 4:a005702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sudhof TC, Rothman JE (2009) Membrane fusion: Grappling with SNARE and SM proteins. Science 323:474–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rizo J, Xu J (2015) The synaptic vesicle release machinery. Annu Rev Biophys 44:339–367. [DOI] [PubMed] [Google Scholar]
- 5. Rizo J, Sudhof TC (1998) C2‐domains, structure and function of a universal Ca2+‐binding domain. J Biol Chem 273:15879–15882. [DOI] [PubMed] [Google Scholar]
- 6. Jahn R, Fasshauer D (2012) Molecular machines governing exocytosis of synaptic vesicles. Nature 490:201–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sudhof TC (2014) The molecular machinery of neurotransmitter release (Nobel lecture). Angew Chem Int Ed Engl 53:12696–12717. [DOI] [PubMed] [Google Scholar]
- 8. Brunger AT, Cipriano DJ, Diao J (2015) Towards reconstitution of membrane fusion mediated by SNAREs and other synaptic proteins. Crit Rev Biochem Mol Biol 50:231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pinheiro PS, Houy S, Sorensen JB (2016) C2‐domain containing calcium sensors in neuroendocrine secretion. J Neurochem 139:943–958. [DOI] [PubMed] [Google Scholar]
- 10. Sollner T, Bennett MK, Whiteheart SW, Scheller RH, Rothman JE (1993) A protein assembly‐disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell 75:409–418. [DOI] [PubMed] [Google Scholar]
- 11. Poirier MA, Xiao W, Macosko JC, Chan C, Shin YK, Bennett MK (1998) The synaptic SNARE complex is a parallel four‐stranded helical bundle. Nat Struct Biol 5:765–769. [DOI] [PubMed] [Google Scholar]
- 12. Sutton RB, Fasshauer D, Jahn R, Brunger AT (1998) Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 A resolution. Nature 395:347–353. [DOI] [PubMed] [Google Scholar]
- 13. Hanson PI, Roth R, Morisaki H, Jahn R, Heuser JE (1997) Structure and conformational changes in NSF and its membrane receptor complexes visualized by quick‐freeze/deep‐etch electron microscopy. Cell 90:523–535. [DOI] [PubMed] [Google Scholar]
- 14. Sollner T, Whiteheart SW, Brunner M, Erdjument‐Bromage H, Geromanos S, Tempst P, Rothman JE (1993) SNAP receptors implicated in vesicle targeting and fusion. Nature 362:318–324. [DOI] [PubMed] [Google Scholar]
- 15. Fasshauer D, Sutton RB, Brunger AT, Jahn R (1998) Conserved structural features of the synaptic fusion complex: SNARE proteins reclassified as Q‐ and R‐SNAREs. Proc Natl Acad Sci USA 95:15781–15786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sorensen JB, Wiederhold K, Muller EM, Milosevic I, Nagy G, de Groot BL, Grubmuller H, Fasshauer D (2006) Sequential N‐ to C‐terminal SNARE complex assembly drives priming and fusion of secretory vesicles. EMBO J 25:955–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hammarlund M, Palfreyman MT, Watanabe S, Olsen S, Jorgensen EM (2007) Open syntaxin docks synaptic vesicles. PLoS Biol 5:e198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Imig C, Min S‐W, Krinner S, Arancillo M, Rosenmund C, Südhof TC, Rhee JSeop, Brose N, Cooper BH (2014) The morphological and molecular nature of synaptic vesicle priming at presynaptic active zones. Neuron 84:416–431. [DOI] [PubMed] [Google Scholar]
- 19. Fernandez I, Ubach J, Dulubova I, Zhang X, Sudhof TC, Rizo J (1998) Three‐dimensional structure of an evolutionarily conserved N‐terminal domain of syntaxin 1A. Cell 94:841–849. [DOI] [PubMed] [Google Scholar]
- 20. Khvotchev M, Dulubova I, Sun J, Dai H, Rizo J, Sudhof TC (2007) Dual modes of Munc18‐1/SNARE interactions are coupled by functionally critical binding to syntaxin‐1 N terminus. J Neurosci 27:12147–12155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dulubova I, Sugita S, Hill S, Hosaka M, Fernandez I, Sudhof TC, Rizo J (1999) A conformational switch in syntaxin during exocytosis: Role of munc18. EMBO J 18:4372–4382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rizo J, Sudhof TC (2012) The membrane fusion enigma: SNAREs, Sec1/Munc18 proteins, and their accomplices‐guilty as charged?. Annu Rev Cell Dev Biol 28:279–308. [DOI] [PubMed] [Google Scholar]
- 23. Xiao W, Poirier MA, Bennett MK, Shin YK (2001) The neuronal t‐SNARE complex is a parallel four‐helix bundle. Nat Struct Biol 8:308–311. [DOI] [PubMed] [Google Scholar]
- 24. Wickner W, Rizo J (2017) A cascade of multiple proteins and lipids catalyzes membrane fusion. Mol Biol Cell 28:707–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chan YH, van LB, Boxer SG (2009) Effects of linker sequences on vesicle fusion mediated by lipid‐anchored DNA oligonucleotides. Proc Natl Acad Sci USA 106:979–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zick M, Wickner WT (2014) A distinct tethering step is vital for vacuole membrane fusion. eLife 3:e03251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Weber T, Zemelman BV, McNew JA, Westermann B, Gmachl M, Parlati F, Sollner TH, Rothman JE (1998) SNAREpins: Minimal machinery for membrane fusion. Cell 92:759–772. [DOI] [PubMed] [Google Scholar]
- 28. Nickel W, Weber T, McNew JA, Parlati F, Sollner TH, Rothman JE (1999) Content mixing and membrane integrity during membrane fusion driven by pairing of isolated v‐SNAREs and t‐SNAREs. Proc Natl Acad Sci USA 96:12571–12576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. van den Bogaart G, Holt MG, Bunt G, Riedel D, Wouters FS, Jahn R (2010) One SNARE complex is sufficient for membrane fusion. Nat Struct Mol Biol 17:358–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kyoung M, Srivastava A, Zhang Y, Diao J, Vrljic M, Grob P, Nogales E, Chu S, Brunger AT (2011) In vitro system capable of differentiating fast Ca2+‐triggered content mixing from lipid exchange for mechanistic studies of neurotransmitter release. Proc Natl Acad Sci USA 108:E304–E313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shin J, Lou X, Kweon DH, Shin YK (2014) Multiple conformations of a single SNAREpin between two nanodisc membranes reveal diverse pre‐fusion states. Biochem J 459:95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hernandez JM, Stein A, Behrmann E, Riedel D, Cypionka A, Farsi Z, Walla PJ, Raunser S, Jahn R (2012) Membrane fusion intermediates via directional and full assembly of the SNARE complex. Science 336:1581–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chernomordik LV, Kozlov MM (2008) Mechanics of membrane fusion. Nat Struct Mol Biol 15:675–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mohrmann R, de WH, Verhage M, Neher E, Sorensen JB (2010) Fast vesicle fusion in living cells requires at least three SNARE complexes. Science 330:502–505. [DOI] [PubMed] [Google Scholar]
- 35. Sinha R, Ahmed S, Jahn R, Klingauf J (2011) Two synaptobrevin molecules are sufficient for vesicle fusion in central nervous system synapses. Proc Natl Acad Sci USA 108:14318–14323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gao Y, Zorman S, Gundersen G, Xi Z, Ma L, Sirinakis G, Rothman JE, Zhang Y (2012) Single reconstituted neuronal SNARE complexes zipper in three distinct stages. Science 337:1340–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Min D, Kim K, Hyeon C, Cho YH, Shin YK, Yoon TY (2013) Mechanical unzipping and rezipping of a single SNARE complex reveals hysteresis as a force‐generating mechanism. Nat Commun 4:1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hanson PI, Heuser JE, Jahn R (1997) Neurotransmitter release—Four years of SNARE complexes. Curr Opin Neurobiol 7:310–315. [DOI] [PubMed] [Google Scholar]
- 39. Stein A, Weber G, Wahl MC, Jahn R (2009) Helical extension of the neuronal SNARE complex into the membrane. Nature 460:525–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kim CS, Kweon DH, Shin YK (2002) Membrane topologies of neuronal SNARE folding intermediates. Biochemistry 41:10928–10933. [DOI] [PubMed] [Google Scholar]
- 41. Deak F, Shin OH, Kavalali ET, Sudhof TC (2006) Structural determinants of synaptobrevin 2 function in synaptic vesicle fusion. J Neurosci 26:6668–6676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Xu H, Zick M, Wickner WT, Jun Y (2011) A lipid‐anchored SNARE supports membrane fusion. Proc Natl Acad Sci USA 108:17325–17330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhou P, Bacaj T, Yang X, Pang ZP, Sudhof TC (2013) Lipid‐anchored SNAREs lacking transmembrane regions fully support membrane fusion during neurotransmitter release. Neuron 80:470–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rizo J (2012) Cell biology. Staging membrane fusion. Science 337:1300–1301. [DOI] [PubMed] [Google Scholar]
- 45. Rizo J, Chen X, Arac D (2006) Unraveling the mechanisms of synaptotagmin and SNARE function in neurotransmitter release. Trends Cell Biol 16:339–350. [DOI] [PubMed] [Google Scholar]
- 46. Carr CM, Rizo J (2010) At the junction of SNARE and SM protein function. Curr Opin Cell Biol 22:488–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. D'Agostino M, Risselada HJ, Lurick A, Ungermann C, Mayer A (2017) A tethering complex drives the terminal stage of SNARE‐dependent membrane fusion. Nature 551:634–638. [DOI] [PubMed] [Google Scholar]
- 48. Xu J, Camacho M, Xu Y, Esser V, Liu X, Trimbuch T, Pan Y‐Z, Ma C, Tomchick DR, Rosenmund C, Rizo J (2017) Mechanistic insights into neurotransmitter release and presynaptic plasticity from the crystal structure of Munc13‐1 C1C2BMUN. Elife 6:e22567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mayer A, Wickner W, Haas A (1996) Sec18p (NSF)‐driven release of Sec17p (alpha‐SNAP) can precede docking and fusion of yeast vacuoles. Cell 85:83–94. [DOI] [PubMed] [Google Scholar]
- 50. Banerjee A, Barry VA, DasGupta BR, Martin TF (1996) N‐ethylmaleimide‐sensitive factor acts at a prefusion ATP‐dependent step in Ca2+‐activated exocytosis. J Biol Chem 271:20223–20226. [DOI] [PubMed] [Google Scholar]
- 51. May AP, Misura KM, Whiteheart SW, Weis WI (1999) Crystal structure of the amino‐terminal domain of N‐ethylmaleimide‐sensitive fusion protein. Nat Cell Biol 1:175–182. [DOI] [PubMed] [Google Scholar]
- 52. Nagiec EE, Bernstein A, Whiteheart SW (1995) Each domain of the N‐ethylmaleimide‐sensitive fusion protein contributes to its transport activity. J Biol Chem 270:29182–29188. [DOI] [PubMed] [Google Scholar]
- 53. Yu RC, Hanson PI, Jahn R, Brunger AT (1998) Structure of the ATP‐dependent oligomerization domain of N‐ethylmaleimide sensitive factor complexed with ATP. Nat Struct Biol 5:803–811. [DOI] [PubMed] [Google Scholar]
- 54. Whiteheart SW, Rossnagel K, Buhrow SA, Brunner M, Jaenicke R, Rothman JE (1994) N‐ethylmaleimide‐sensitive fusion protein: a trimeric ATPase whose hydrolysis of ATP is required for membrane fusion. J Cell Biol 126:945–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rice LM, Brunger AT (1999) Crystal structure of the vesicular transport protein Sec17: Implications for SNAP function in SNARE complex disassembly. Mol Cell 4:85–95. [DOI] [PubMed] [Google Scholar]
- 56. Zhao M, Wu S, Zhou Q, Vivona S, Cipriano DJ, Cheng Y, Brunger AT (2015) Mechanistic insights into the recycling machine of the SNARE complex. Nature 518:61–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Winter U, Chen X, Fasshauer D (2009) A conserved membrane attachment site in alpha‐SNAP facilitates N‐ethylmaleimide‐sensitive factor (NSF)‐driven SNARE complex disassembly. J Biol Chem 284:31817–31826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Xu H, Jun Y, Thompson J, Yates J, Wickner W (2010) HOPS prevents the disassembly of trans‐SNARE complexes by Sec17p/Sec18p during membrane fusion. EMBO J 29:1948–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mima J, Hickey CM, Xu H, Jun Y, Wickner W (2008) Reconstituted membrane fusion requires regulatory lipids, SNAREs and synergistic SNARE chaperones. EMBO J 27:2031–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ungermann C, Wickner W (1998) Vam7p, a vacuolar SNAP‐25 homolog, is required for SNARE complex integrity and vacuole docking and fusion. EMBO J 17:3269–3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Schwartz ML, Merz AJ (2009) Capture and release of partially zipped trans‐SNARE complexes on intact organelles. J Cell Biol 185:535–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zick M, Orr A, Schwartz ML, Merz AJ, Wickner WT (2015) Sec17 can trigger fusion of trans‐SNARE paired membranes without Sec18. Proc Natl Acad Sci USA 112:E2290–E2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Song H, Orr A, Duan M, Merz AJ, Wickner W (2017) Sec17/Sec18 act twice, enhancing membrane fusion and then disassembling cis‐SNARE complexes. eLife 6:e26646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Schwartz ML, Nickerson DP, Lobingier BT, Plemel RL, Duan M, Angers CG, Zick M, Merz AJ (2017) Sec17 (alpha‐SNAP) and an SM‐tethering complex regulate the outcome of SNARE zippering in vitro and in vivo. eLife 6:e27396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hayashi T, Yamasaki S, Nauenburg S, Binz T, Niemann H (1995) Disassembly of the reconstituted synaptic vesicle membrane fusion complex in vitro. EMBO J 14:2317–2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Weber T, Parlati F, McNew JA, Johnston RJ, Westermann B, Sollner TH, Rothman JE (2000) SNAREpins are functionally resistant to disruption by NSF and alphaSNAP. J Cell Biol 149:1063–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ma C, Su L, Seven AB, Xu Y, Rizo J (2013) Reconstitution of the vital functions of Munc18 and Munc13 in neurotransmitter release. Science 339:421–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yavuz H, Kattan I, Hernandez JM, Hofnagel O, Witkowska A, Raunser S, Walla PJ, Jahn R (2018) Arrest of trans‐SNARE zippering uncovers loosely and tightly docked intermediates in membrane fusion. J Biol Chem, http://www.jbc.org/cgi/doi/10.1074/jbc.RA118.003313 (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Park Y, Vennekate W, Yavuz H, Preobraschenski J, Hernandez JM, Riedel D, Walla PJ, Jahn R (2014) SNAP interferes with the zippering of the SNARE protein membrane fusion machinery. J Biol Chem 289:16326–16335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ma L, Kang Y, Jiao J, Rebane AA, Cha HK, Xi Z, Qu H, Zhang Y (2016) SNAP enhances SNARE zippering by stabilizing the SNARE four‐helix bundle. Cell Rep 15:531–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Misura KM, Scheller RH, Weis WI (2000) Three‐dimensional structure of the neuronal‐Sec1‐syntaxin 1a complex. Nature 404:355–362. [DOI] [PubMed] [Google Scholar]
- 72. Basu J, Shen N, Dulubova I, Lu J, Guan R, Guryev O, Grishin NV, Rosenmund C, Rizo J (2005) A minimal domain responsible for Munc13 activity. Nat Struct Mol Biol 12:1017–1018. [DOI] [PubMed] [Google Scholar]
- 73. Pei J, Ma C, Rizo J, Grishin NV (2009) Remote homology between Munc13 MUN domain and vesicle tethering complexes. J Mol Biol 391:509–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Yang X, Wang S, Sheng Y, Zhang M, Zou W, Wu L, Kang L, Rizo J, Zhang R, Xu T, Ma C (2015) Syntaxin opening by the MUN domain underlies the function of Munc13 in synaptic‐vesicle priming. Nat Struct Mol Biol 22:547–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Verhage M, Maia AS, Plomp JJ, Brussaard AB, Heeroma JH, Vermeer H, Toonen RF, Hammer RE, van den BTK, Missler M, Geuze JH, Sudhof TC (2000) Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science 287:864–869. [DOI] [PubMed] [Google Scholar]
- 76. Varoqueaux F, Sigler A, Rhee JS, Brose N, Enk C, Reim K, Rosenmund C (2002) Total arrest of spontaneous and evoked synaptic transmission but normal synaptogenesis in the absence of Munc13‐mediated vesicle priming. Proc Natl Acad Sci USA 99:9037–9042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Richmond JE, Davis WS, Jorgensen EM (1999) UNC‐13 is required for synaptic vesicle fusion in Caenorhabditis elegans . Nat Neurosci 2:959–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Aravamudan B, Fergestad T, Davis WS, Rodesch CK, Broadie K (1999) Drosophila UNC‐13 is essential for synaptic transmission. Nat Neurosci 2:965–971. [DOI] [PubMed] [Google Scholar]
- 79. Burkhardt P, Hattendorf DA, Weis WI, Fasshauer D (2008) Munc18a controls SNARE assembly through its interaction with the syntaxin N‐peptide. EMBO J 27:923–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Chen X, Lu J, Dulubova I, Rizo J (2008) NMR analysis of the closed conformation of syntaxin‐1. J Biomol NMR 41:43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ma C, Li W, Xu Y, Rizo J (2011) Munc13 mediates the transition from the closed syntaxin‐Munc18 complex to the SNARE complex. Nat Struct Mol Biol 18:542–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Gerber SH, Rah J‐C, Min S‐W, Liu X, de Wit H, Dulubova I, Meyer AC, Rizo J, Arancillo M, Hammer RE, Verhage M, Rosenmund C, Südhof TC (2008) Conformational switch of syntaxin‐1 controls synaptic vesicle fusion. Science 321:1507–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Betz A, Okamoto M, Benseler F, Brose N (1997) Direct interaction of the rat unc‐13 homologue Munc13‐1 with the N terminus of syntaxin. J Biol Chem 272:2520–2526. [DOI] [PubMed] [Google Scholar]
- 84. Richmond JE, Weimer RM, Jorgensen EM (2001) An open form of syntaxin bypasses the requirement for UNC‐13 in vesicle priming. Nature 412:338–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Dulubova I, Khvotchev M, Liu S, Huryeva I, Sudhof TC, Rizo J (2007) Munc18‐1 binds directly to the neuronal SNARE complex. Proc Natl Acad Sci USA 104:2697–2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Shen J, Tareste DC, Paumet F, Rothman JE, Melia TJ (2007) Selective activation of cognate SNAREpins by Sec1/Munc18 proteins. Cell 128:183–195. [DOI] [PubMed] [Google Scholar]
- 87. Deak F, Xu Y, Chang WP, Dulubova I, Khvotchev M, Liu X, Sudhof TC, Rizo J (2009) Munc18‐1 binding to the neuronal SNARE complex controls synaptic vesicle priming. J Cell Biol 184:751–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Ma L, Rebane AA, Yang G, Xi Z, Kang Y, Gao Y, Zhang Y (2015) Munc18‐1‐regulated stage‐wise SNARE assembly underlying synaptic exocytosis. eLife 4:e09580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Xu Y, Su L, Rizo J (2010) Binding of Munc18‐1 to synaptobrevin and to the SNARE four‐helix bundle. Biochemistry 49:1568–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Rathore SS, Bend EG, Yu H, Hammarlund M, Jorgensen EM, Shen J (2010) Syntaxin N‐terminal peptide motif is an initiation factor for the assembly of the SNARE‐Sec1/Munc18 membrane fusion complex. Proc Natl Acad Sci USA 107:22399–22406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Zhou P, Pang ZP, Yang X, Zhang Y, Rosenmund C, Bacaj T, Sudhof TC (2013) Syntaxin‐1 N‐peptide and Habc‐domain perform distinct essential functions in synaptic vesicle fusion. EMBO J 32:159–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Han L, Jiang T, Han GA, Malintan NT, Xie L, Wang L, Tse FW, Gaisano HY, Collins BM, Meunier FA, Sugita S (2009) Rescue of Munc18‐1 and −2 double knockdown reveals the essential functions of interaction between Munc18 and closed syntaxin in PC12 cells. Mol Biol Cell 20:4962–4975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Meijer M, Burkhardt P, de WH, Toonen RF, Fasshauer D, Verhage M (2012) Munc18‐1 mutations that strongly impair SNARE‐complex binding support normal synaptic transmission. EMBO J 31:2156–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Dulubova I, Yamaguchi T, Wang Y, Sudhof TC, Rizo J (2001) Vam3p structure reveals conserved and divergent properties of syntaxins. Nat Struct Biol 8:258–264. [DOI] [PubMed] [Google Scholar]
- 95. Liu X, Seven AB, Camacho M, Esser V, Xu J, Trimbuch T, Quade B, Su L, Ma C, Rosenmund C, Rizo J (2016) Functional synergy between the Munc13 C‐terminal C1 and C2 domains. eLife 5:e13696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Baker RW, Jeffrey PD, Zick M, Phillips BP, Wickner WT, Hughson FM (2015) A direct role for the Sec1/Munc18‐family protein Vps33 as a template for SNARE assembly. Science 349:1111–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Parisotto D, Pfau M, Scheutzow A, Wild K, Mayer MP, Malsam J, Sinning I, Sollner TH (2014) An extended helical conformation in domain 3a of Munc18‐1 provides a template for SNARE (soluble N‐ethylmaleimide‐sensitive factor attachment protein receptor) complex assembly. J Biol Chem 289:9639–9650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Hu SH, Christie MP, Saez NJ, Latham CF, Jarrott R, Lua LHL, Collins BM, Martin JL (2011) Possible roles for Munc18‐1 domain 3a and Syntaxin1 N‐peptide and C‐terminal anchor in SNARE complex formation. Proc Natl Acad Sci USA 108:1040–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Sitarska E, Xu J, Park S, Liu X, Quade B, Stepien K, Sugita K, Brautigam CA, Sugita S, Rizo J (2017) Autoinhibition of Munc18‐1 modulates synaptobrevin binding and helps to enable Munc13‐dependent regulation of membrane fusion. eLife 6:24278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Lai Y, Choi UB, Leitz J, Rhee HJ, Lee C, Altas B, Zhao M, Pfuetzner RA, Wang AL, Brose N, Rhee JSeop, Brunger AT (2017) Molecular mechanisms of synaptic vesicle priming by Munc13 and Munc18. Neuron 95:591–607.e510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Dawidowski D, Cafiso DS (2016) Munc18‐1 and the syntaxin‐1 N terminus regulate open‐closed states in a t‐SNARE complex. Structure 24:392–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Jakhanwal S, Lee CT, Urlaub H, Jahn R (2017) An activated Q‐SNARE/SM protein complex as a possible intermediate in SNARE assembly. EMBO J 36:1788–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Wang S, Choi UB, Gong J, Yang X, Li Y, Wang AL, Yang X, Brunger AT, Ma C (2017) Conformational change of syntaxin linker region induced by Munc13s initiates SNARE complex formation in synaptic exocytosis. EMBO J 36:816–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Guan R, Dai H, Rizo J (2008) Binding of the Munc13‐1 MUN domain to membrane‐anchored SNARE complexes. Biochemistry 47:1474–1481. [DOI] [PubMed] [Google Scholar]
- 105. Weninger K, Bowen ME, Choi UB, Chu S, Brunger AT (2008) Accessory proteins stabilize the acceptor complex for synaptobrevin, the 1:1 syntaxin/SNAP‐25 complex. Structure 16:308–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. He E, Wierda K, van Westen R, Broeke JH, Toonen RF, Cornelisse N, Verhage M (2017) Munc13‐1 and Munc18‐1 together prevent NSF‐dependent de‐priming of synaptic vesicles. Nat Commun 8:15915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Park S, Bin NR, Yu B, Wong R, Sitarska E, Sugita K, Ma K, Xu J, Tien C‐W, Algouneh A, Turlova E, Wang S, Siriya P, Shahid W, Kalia L, Feng Z‐P, Monnier PP, Sun H‐S, Zhen M, Gao S, Rizo J, Sugita S (2017) UNC‐18 and tomosyn antagonistically control synaptic vesicle priming downstream of UNC‐13 in Caenorhabditis elegans . J Neurosci 37:8797–8815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Rhee JS, Betz A, Pyott S, Reim K, Varoqueaux F, Augustin I, Hesse D, Sudhof TC, Takahashi M, Rosenmund C, Brose N (2002) Beta phorbol ester‐ and diacylglycerol‐induced augmentation of transmitter release is mediated by Munc13s and not by PKCs. Cell 108:121–133. [DOI] [PubMed] [Google Scholar]
- 109. Basu J, Betz A, Brose N, Rosenmund C (2007) Munc13‐1 C1 domain activation lowers the energy barrier for synaptic vesicle fusion. J Neurosci 27:1200–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Shen N, Guryev O, Rizo J (2005) Intramolecular occlusion of the diacylglycerol‐binding site in the C1 domain of munc13‐1. Biochemistry 44:1089–1096. [DOI] [PubMed] [Google Scholar]
- 111. Shin O‐H, Lu J, Rhee J‐S, Tomchick DR, Pang ZP, Wojcik SM, Camacho‐Perez M, Brose N, Machius M, Rizo J, Rosenmund C, Südhof TC (2010) Munc13 C2B domain is an activity‐dependent Ca2+ regulator of synaptic exocytosis. Nat Struct Mol Biol 17:280–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Michelassi F, Liu H, Hu Z, Dittman JS (2017) A C1‐C2 module in Munc13 inhibits calcium‐dependent neurotransmitter release. Neuron 95:577–590.e575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Camacho M, Basu J, Trimbuch T, Chang S, Pulido‐Lozano C, Chang S‐S, Duluvova I, Abo‐Rady M, Rizo J, Rosenmund C (2017) Heterodimerization of Munc13 C2A domain with RIM regulates synaptic vesicle docking and priming. Nat Commun 8:15293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Rodriguez‐Castaneda F, Maestre‐Martinez M, Coudevylle N, Dimova K, Junge H, Lipstein N, Lee D, Becker S, Brose N, Jahn O, Carlomagno T, Griesinger C (2010) Modular architecture of Munc13/calmodulin complexes: dual regulation by Ca2+ and possible function in short‐term synaptic plasticity. EMBO J 29:680–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Junge HJ, Rhee J‐S, Jahn O, Varoqueaux F, Spiess J, Waxham MN, Rosenmund C, Brose N (2004) Calmodulin and Munc13 form a Ca2+ sensor/effector complex that controls short‐term synaptic plasticity. Cell 118:389–401. [DOI] [PubMed] [Google Scholar]
- 116. Lu J, Machius M, Dulubova I, Dai H, Sudhof TC, Tomchick DR, Rizo J (2006) Structural basis for a Munc13‐1 homodimer to Munc13‐1/RIM heterodimer switch. PLoS Biol 4:e192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Deng L, Kaeser PS, Xu W, Sudhof TC (2011) RIM proteins activate vesicle priming by reversing autoinhibitory homodimerization of Munc13. Neuron 69:317–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Betz A, Thakur P, Junge HJ, Ashery U, Rhee J‐S, Scheuss V, Rosenmund C, Rettig J, Brose N (2001) Functional interaction of the active zone proteins Munc13‐1 and RIM1 in synaptic vesicle priming. Neuron 30:183–196. [DOI] [PubMed] [Google Scholar]
- 119. Dulubova I, Lou X, Lu J, Huryeva I, Alam A, Schneggenburger R, Sudhof TC, Rizo J (2005) A Munc13/RIM/Rab3 tripartite complex: from priming to plasticity?. EMBO J 24:2839–2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Schoch S, Castillo PE, Jo T, Mukherjee K, Geppert M, Wang Y, Schmitz F, Malenka RC, Sudhof TC (2002) RIM1alpha forms a protein scaffold for regulating neurotransmitter release at the active zone. Nature 415:321–326. [DOI] [PubMed] [Google Scholar]
- 121. Koushika SP, Richmond JE, Hadwiger G, Weimer RM, Jorgensen EM, Nonet ML (2001) A post‐docking role for active zone protein Rim. Nat Neurosci 4:997–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Kaeser PS, Deng L, Wang Y, Dulubova I, Liu X, Rizo J, Südhof TC (2011) RIM proteins tether Ca2+ channels to presynaptic active zones via a direct PDZ‐domain interaction. Cell 144:282–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Castillo PE, Schoch S, Schmitz F, Sudhof TC, Malenka RC (2002) RIM1alpha is required for presynaptic long‐term potentiation. Nature 415:327–330. [DOI] [PubMed] [Google Scholar]
- 124. Wang Y, Okamoto M, Schmitz F, Hofmann K, Sudhof TC (1997) Rim is a putative Rab3 effector in regulating synaptic‐vesicle fusion. Nature 388:593–598. [DOI] [PubMed] [Google Scholar]
- 125. Schluter OM, Basu J, Sudhof TC, Rosenmund C (2006) Rab3 superprimes synaptic vesicles for release: implications for short‐term synaptic plasticity. J Neurosci 26:1239–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Grosshans BL, Ortiz D, Novick P (2006) Rabs and their effectors: achieving specificity in membrane traffic. Proc Natl Acad Sci USA 103:11821–11827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Castillo PE, Janz R, Sudhof TC, Tzounopoulos T, Malenka RC, Nicoll RA (1997) Rab3A is essential for mossy fibre long‐term potentiation in the hippocampus. Nature 388:590–593. [DOI] [PubMed] [Google Scholar]
- 128. Yang Y, Calakos N (2011) Munc13‐1 is required for presynaptic long‐term potentiation. J Neurosci 31:12053–12057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. McMahon HT, Missler M, Li C, Sudhof TC (1995) Complexins: Cytosolic proteins that regulate SNAP receptor function. Cell 83:111–119. [DOI] [PubMed] [Google Scholar]
- 130. Reim K, Mansour M, Varoqueaux F, McMahon HT, Sudhof TC, Brose N, Rosenmund C (2001) Complexins regulate a late step in Ca2+‐dependent neurotransmitter release. Cell 104:71–81. [DOI] [PubMed] [Google Scholar]
- 131. Xue M, Stradomska A, Chen H, Brose N, Zhang W, Rosenmund C, Reim K (2008) Complexins facilitate neurotransmitter release at excitatory and inhibitory synapses in mammalian central nervous system. Proc Natl Acad Sci USA 105:7875–7880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Maximov A, Tang J, Yang X, Pang ZP, Sudhof TC (2009) Complexin controls the force transfer from SNARE complexes to membranes in fusion. Science 323:516–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Mohrmann R, Dhara M, Bruns D (2015) Complexins: small but capable. Cell Mol Life Sci 72:4221–4235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Trimbuch T, Rosenmund C (2016) Should I stop or should I go? The role of complexin in neurotransmitter release. Nat Rev Neurosci 17:118–125. [DOI] [PubMed] [Google Scholar]
- 135. Yang X, Cao P, Sudhof TC (2013) Deconstructing complexin function in activating and clamping Ca2+‐triggered exocytosis by comparing knockout and knockdown phenotypes. Proc Natl Acad Sci USA 110:20777–20782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Xue M, Craig TK, Xu J, Chao HT, Rizo J, Rosenmund C (2010) Binding of the complexin N terminus to the SNARE complex potentiates synaptic‐vesicle fusogenicity. Nat Struct Mol Biol 17:568–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Yang X, Kaeser‐Woo YJ, Pang ZP, Xu W, Südhof TC (2010) Complexin clamps asynchronous release by blocking a secondary Ca(2+) sensor via its accessory alpha helix. Neuron 68:907–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Tang J, Maximov A, Shin OH, Dai H, Rizo J, Sudhof TC (2006) A complexin/synaptotagmin 1 switch controls fast synaptic vesicle exocytosis. Cell 126:1175–1187. [DOI] [PubMed] [Google Scholar]
- 139. Giraudo CG, Eng WS, Melia TJ, Rothman JE (2006) A clamping mechanism involved in SNARE‐dependent exocytosis. Science 313:676–680. [DOI] [PubMed] [Google Scholar]
- 140. Schaub JR, Lu X, Doneske B, Shin YK, McNew JA (2006) Hemifusion arrest by complexin is relieved by Ca2+‐synaptotagmin I. Nat Struct Mol Biol 13:748–750. [DOI] [PubMed] [Google Scholar]
- 141. Huntwork S, Littleton JT (2007) A complexin fusion clamp regulates spontaneous neurotransmitter release and synaptic growth. Nat Neurosci 10:1235–1237. [DOI] [PubMed] [Google Scholar]
- 142. Martin JA, Hu Z, Fenz KM, Fernandez J, Dittman JS (2011) Complexin has opposite effects on two modes of synaptic vesicle fusion. Curr Biol 21:97–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Hobson RJ, Liu Q, Watanabe S, Jorgensen EM (2011) Complexin maintains vesicles in the primed state in Caenorhabditis elegans . Curr Biol 21:106–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Pabst S, Hazzard JW, Antonin W, Sudhof TC, Jahn R, Rizo J, Fasshauer D (2000) Selective interaction of complexin with the neuronal SNARE complex. Determination of the binding regions. J Biol Chem 275:19808–19818. [DOI] [PubMed] [Google Scholar]
- 145. Chen X, Tomchick DR, Kovrigin E, Arac D, Machius M, Sudhof TC, Rizo J (2002) Three‐dimensional structure of the complexin/SNARE complex. Neuron 33:397–409. [DOI] [PubMed] [Google Scholar]
- 146. Xue M, Reim K, Chen X, Chao H‐T, Deng H, Rizo J, Brose N, Rosenmund C (2007) Distinct domains of complexin I differentially regulate neurotransmitter release. Nat Struct Mol Biol 14:949–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Giraudo CG, Garcia‐Diaz A, Eng WS, Chen Y, Hendrickson WA, Melia TJ, Rothman JE (2009) Alternative zippering as an on‐off switch for SNARE‐mediated fusion. Science 323:512–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Lai Y, Choi UB, Zhang Y, Zhao M, Pfuetzner RA, Wang AL, Diao J, Brunger AT (2016) N‐terminal domain of complexin independently activates calcium‐triggered fusion. Proc Natl Acad Sci USA 113:E4698–4E4707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Zdanowicz R, Kreutzberger A, Liang B, Kiessling V, Tamm LK, Cafiso DS (2017) Complexin binding to membranes and acceptor t‐SNAREs explains its clamping effect on fusion. Biophys J 113:1235–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Seiler F, Malsam J, Krause JM, Sollner TH (2009) A role of complexin‐lipid interactions in membrane fusion. FEBS Lett 583:2343–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Wragg RT, Snead D, Dong Y, Ramlall TF, Menon I, Bai J, Eliezer D, Dittman JS (2013) Synaptic vesicles position complexin to block spontaneous fusion. Neuron 77:323–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Snead D, Wragg RT, Dittman JS, Eliezer D (2014) Membrane curvature sensing by the C‐terminal domain of complexin. Nat Commun 5:4955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Gong J, Lai Y, Li X, Wang M, Leitz J, Hu Y, Zhang Y, Choi UB, Cipriano D, Pfuetzner RA, Sudhof TC, Yang X, Brunger AT, Diao J (2016) C‐terminal domain of mammalian complexin‐1 localizes to highly curved membranes. Proc Natl Acad Sci USA 113:E7590–E7599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Wragg RT, Parisotto DA, Li Z, Terakawa MS, Snead D, Basu I, Weinstein H, Eliezer D, Dittman JS (2017) Evolutionary divergence of the C‐terminal domain of complexin accounts for functional disparities between vertebrate and invertebrate complexins. Front Mol Neurosci 10:146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Xue M, Lin YQ, Pan H, Reim K, Deng H, Bellen HJ, Rosenmund C (2009) Tilting the balance between facilitatory and inhibitory functions of mammalian and Drosophila Complexins orchestrates synaptic vesicle exocytosis. Neuron 64:367–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Kaeser‐Woo YJ, Yang X, Südhof TC (2012) C‐terminal complexin sequence is selectively required for clamping and priming but not for Ca2+ triggering of synaptic exocytosis. J Neurosci 32:2877–2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Zhou Q, Zhou P, Wang AL, Wu D, Zhao M, Sudhof TC, Brunger AT (2017) The primed SNARE‐complexin‐synaptotagmin complex for neuronal exocytosis. Nature 548:420–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Diao J, Cipriano DJ, Zhao M, Zhang Y, Shah S, Padolina MS, Pfuetzner RA, Brunger AT (2013) Complexin‐1 enhances the on‐rate of vesicle docking via simultaneous SNARE and membrane interactions. J Am Chem Soc 135:15274–15277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Yoon TY, Lu X, Diao J, Lee SM, Ha T, Shin YK (2008) Complexin and Ca2+ stimulate SNARE‐mediated membrane fusion. Nat Struct Mol Biol 15:707–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Kummel D, Krishnakumar SS, Radoff DT, Li F, Giraudo CG, Pincet F, Rothman JE, Reinisch KM (2011) Complexin cross‐links prefusion SNAREs into a zigzag array. Nat Struct Mol Biol 18:927–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Krishnakumar SS, Radoff DT, Kummel D, Giraudo CG, Li F, Khandan L, Baguley SW, Coleman J, Reinisch KM, Pincet F, Rothman JE (2011) A conformational switch in complexin is required for synaptotagmin to trigger synaptic fusion. Nat Struct Mol Biol 18:934–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Li F, Pincet F, Perez E, Giraudo CG, Tareste D, Rothman JE (2011) Complexin activates and clamps SNAREpins by a common mechanism involving an intermediate energetic state. Nat Struct Mol Biol 18:941–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Krishnakumar SS, Li F, Coleman J, Schauder CM, Kummel D, Pincet F, Rothman JE, Reinisch KM (2015) Re‐visiting the trans insertion model for complexin clamping. eLife 4:e04463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Trimbuch T, Xu J, Flaherty D, Tomchick DR, Rizo J, Rosenmund C (2014) Re‐examining how complexin inhibits neurotransmitter release. eLife 3:e02391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Prinslow EA, Brautigam CA, Rizo J (2017) Reconciling isothermal titration calorimetry analyses of interactions between complexin and truncated SNARE complexes. eLife 6:e30286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Cho RW, Kummel D, Li F, Baguley SW, Coleman J, Rothman JE, Littleton JT (2014) Genetic analysis of the Complexin trans‐clamping model for cross‐linking SNARE complexes in vivo. Proc Natl Acad Sci USA 111:10317–10322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Radoff DT, Dong Y, Snead D, Bai J, Eliezer D, Dittman JS (2014) The accessory helix of complexin functions by stabilizing central helix secondary structure. eLife 3:e04553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Yin L, Kim J, Shin YK (2016) Complexin splits the membrane‐proximal region of a single SNAREpin. Biochem J 473:2219–2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Choi UB, Zhao M, Zhang Y, Lai Y, Brunger AT (2016) Complexin induces a conformational change at the membrane‐proximal C‐terminal end of the SNARE complex. eLife 5:e16886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Sudhof TC (2002) Synaptotagmins: Why so many?. J Biol Chem 277:7629–7632. [DOI] [PubMed] [Google Scholar]
- 171. Geppert M, Goda Y, Hammer RE, Li C, Rosahl TW, Stevens CF, Sudhof TC (1994) Synaptotagmin I: a major Ca2+ sensor for transmitter release at a central synapse. Cell 79:717–727. [DOI] [PubMed] [Google Scholar]
- 172. Bacaj T, Wu D, Yang X, Morishita W, Zhou P, Xu W, Malenka RC, Sudhof TC (2013) Synaptotagmin‐1 and synaptotagmin‐7 trigger synchronous and asynchronous phases of neurotransmitter release. Neuron 80:947–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Xu J, Pang ZP, Shin OH, Sudhof TC (2009) Synaptotagmin‐1 functions as a Ca2+ sensor for spontaneous release. Nat Neurosci 12:759–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Bacaj T, Wu D, Burre J, Malenka RC, Liu X, Sudhof TC (2015) Synaptotagmin‐1 and −7 are redundantly essential for maintaining the capacity of the readily‐releasable pool of synaptic vesicles. PLoS Biol 13:e1002267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Chang S, Trimbuch T, Rosenmund C (2018) Synaptotagmin‐1 drives synchronous Ca(2+)‐triggered fusion by C2B‐domain‐mediated synaptic‐vesicle‐membrane attachment. Nat Neurosci 21:33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. de Wit H, Walter AM, Milosevic I, Gulyás‐Kovács A, Riedel D, Sørensen JB, Verhage M (2009) Synaptotagmin‐1 docks secretory vesicles to syntaxin‐1/SNAP‐25 acceptor complexes. Cell 138:935–946. [DOI] [PubMed] [Google Scholar]
- 177. Sutton RB, Davletov BA, Berghuis AM, Sudhof TC, Sprang SR (1995) Structure of the first C2 domain of synaptotagmin I: A novel Ca2+/phospholipid‐binding fold. Cell 80:929–938. [DOI] [PubMed] [Google Scholar]
- 178. Shao X, Fernandez I, Sudhof TC, Rizo J (1998) Solution structures of the Ca2+‐free and Ca2+‐bound C2A domain of synaptotagmin I: Does Ca2+ induce a conformational change?. Biochemistry 37:16106–16115. [DOI] [PubMed] [Google Scholar]
- 179. Fernandez I, Arac D, Ubach J, Gerber SH, Shin O‐H, Gao Y, Anderson RGW, Sudhof TC, Rizo J (2001) Three‐dimensional structure of the synaptotagmin 1 c(2)b‐domain. Synaptotagmin 1 as a phospholipid binding machine. Neuron 32:1057–1069. [DOI] [PubMed] [Google Scholar]
- 180. Shao X, Li C, Fernandez I, Zhang X, Sudhof TC, Rizo J (1997) Synaptotagmin‐syntaxin interaction: the C2 domain as a Ca2+‐dependent electrostatic switch. Neuron 18:133–142. [DOI] [PubMed] [Google Scholar]
- 181. Ubach J, Zhang X, Shao X, Sudhof TC, Rizo J (1998) Ca2+ binding to synaptotagmin: How many Ca2+ ions bind to the tip of a C2‐domain?. EMBO J 17:3921–3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Chapman ER, Davis AF (1998) Direct interaction of a Ca2+‐binding loop of synaptotagmin with lipid bilayers. J Biol Chem 273:13995–14001. [DOI] [PubMed] [Google Scholar]
- 183. Zhang X, Rizo J, Sudhof TC (1998) Mechanism of phospholipid binding by the C2A‐domain of synaptotagmin I. Biochemistry 37:12395–12403. [DOI] [PubMed] [Google Scholar]
- 184. Fernández‐Chacón R, Königstorfer A, Gerber SH, García J, Matos MF, Stevens CF, Brose N, Rizo J, Rosenmund C, Südhof TC (2001) Synaptotagmin I functions as a calcium regulator of release probability. Nature 410:41–49. [DOI] [PubMed] [Google Scholar]
- 185. Rhee J‐S, Li LY, Shin O‐H, Rah J‐C, Rizo J, Sudhof TC, Rosenmund C (2005) Augmenting neurotransmitter release by enhancing the apparent Ca2+ affinity of synaptotagmin 1. Proc Natl Acad Sci USA 102:18664–18669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Mackler JM, Drummond JA, Loewen CA, Robinson IM, Reist NE (2002) The C(2)B Ca(2+)‐binding motif of synaptotagmin is required for synaptic transmission in vivo. Nature 418:340–344. [DOI] [PubMed] [Google Scholar]
- 187. Nishiki T, Augustine GJ (2004) Dual roles of the C2B domain of synaptotagmin I in synchronizing Ca2+‐dependent neurotransmitter release. J Neurosci 24:8542–8550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Shin OH, Xu J, Rizo J, Sudhof TC (2009) Differential but convergent functions of Ca2+ binding to synaptotagmin‐1 C2 domains mediate neurotransmitter release. Proc Natl Acad Sci USA 106:16469–16474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Striegel AR, Biela LM, Evans CS, Wang Z, Delehoy JB, Sutton RB, Chapman ER, Reist NE (2012) Calcium binding by synaptotagmin's C2A domain is an essential element of the electrostatic switch that triggers synchronous synaptic transmission. J Neurosci 32:1253–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Wu D, Bacaj T, Morishita W, Goswami D, Arendt KL, Xu W, Chen L, Malenka RC, Sudhof TC (2017) Postsynaptic synaptotagmins mediate AMPA receptor exocytosis during LTP. Nature 544:316–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Voleti R, Tomchick DR, Sudhof TC, Rizo J (2017) Exceptionally tight membrane‐binding may explain the key role of the synaptotagmin‐7 C2A domain in asynchronous neurotransmitter release. Proc Natl Acad Sci USA 114:E8518–E8527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Bai J, Tucker WC, Chapman ER (2004) PIP2 increases the speed of response of synaptotagmin and steers its membrane‐penetration activity toward the plasma membrane. Nat Struct Mol Biol 11:36–44. [DOI] [PubMed] [Google Scholar]
- 193. Li L, Shin O‐H, Rhee J‐S, Arac D, Rah J‐C, Rizo J, Sudhof T, Rosenmund C (2006) Phosphatidylinositol phosphates as co‐activators of Ca2+ binding to C2 domains of synaptotagmin 1. J Biol Chem 281:15845–15852. [DOI] [PubMed] [Google Scholar]
- 194. Arac D, Chen X, Khant HA, Ubach J, Ludtke SJ, Kikkawa M, Johnson AE, Chiu W, Sudhof TC, Rizo J (2006) Close membrane‐membrane proximity induced by Ca(2+)‐dependent multivalent binding of synaptotagmin‐1 to phospholipids. Nat Struct Mol Biol 13:209–217. [DOI] [PubMed] [Google Scholar]
- 195. Xue M, Ma C, Craig TK, Rosenmund C, Rizo J (2008) The Janus‐faced nature of the C(2)B domain is fundamental for synaptotagmin‐1 function. Nat Struct Mol Biol 15:1160–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Brewer KD, Bacaj T, Cavalli A, Camilloni C, Swarbrick JD, Liu J, Zhou A, Zhou P, Barlow N, Xu J, Seven AB, Prinslow EA, Voleti R, Häussinger D, Bonvin AMJJ, Tomchick DR, Vendruscolo M, Graham B, Südhof TC, Rizo J (2015) Dynamic binding mode of a Synaptotagmin‐1‐SNARE complex in solution. Nat Struct Mol Biol 22:555–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Zhou Q, Lai Y, Bacaj T, Zhao M, Lyubimov AY, Uervirojnangkoorn M, Zeldin OB, Brewster AS, Sauter NK, Cohen AE, Soltis SM, Alonso‐Mori R, Chollet M, Lemke HT, Pfuetzner RA, Choi UB, Weis WI, Diao J, Südhof TC, Brunger AT (2015) Architecture of the synaptotagmin‐SNARE machinery for neuronal exocytosis. Nature 525:62–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Martens S, Kozlov MM, McMahon HT (2007) How synaptotagmin promotes membrane fusion. Science 316:1205–1208. [DOI] [PubMed] [Google Scholar]
- 199. Sudhof TC (2004) The synaptic vesicle cycle. Annu Rev Neurosci 27:509–547. [DOI] [PubMed] [Google Scholar]
- 200. Chapman ER (2008) How does synaptotagmin trigger neurotransmitter release? Annu Rev Biochem 77:615–641. [DOI] [PubMed] [Google Scholar]
- 201. Mahal LK, Sequeira SM, Gureasko JM, Sollner TH (2002) Calcium‐independent stimulation of membrane fusion and SNAREpin formation by synaptotagmin I. J Cell Biol 158:273–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Tucker WC, Weber T, Chapman ER (2004) Reconstitution of Ca2+‐regulated membrane fusion by synaptotagmin and SNAREs. Science 304:435–438. [DOI] [PubMed] [Google Scholar]
- 203. Lee HK, Yang Y, Su Z, Hyeon C, Lee T‐S, Lee H‐W, Kweon D‐H, Shin Y‐K, Yoon T‐Y (2010) Dynamic Ca2+‐dependent stimulation of vesicle fusion by membrane‐anchored synaptotagmin 1. Science 328:760–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Hui E, Gaffaney JD, Wang Z, Johnson CP, Evans CS, Chapman ER (2011) Mechanism and function of synaptotagmin‐mediated membrane apposition. Nat Struct Mol Biol 18:813–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. van den BG, Thutupalli S, Risselada JH, Meyenberg K, Holt M, Riedel D, Diederichsen U, Herminghaus S, Grubmuller H, Jahn R (2011) Synaptotagmin‐1 may be a distance regulator acting upstream of SNARE nucleation. Nat Struct Mol Biol 18:805–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Park Y, Hernandez JM, van den Bogaart G, Ahmed S, Holt M, Riedel D, Jahn R (2012) Controlling synaptotagmin activity by electrostatic screening. Nat Struct Mol Biol 19:991–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Rickman C, Jimenez JL, Graham ME, Archer DA, Soloviev M, Burgoyne RD, Davletov B (2006) Conserved prefusion protein assembly in regulated exocytosis. Mol Biol Cell 17:283–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Lai AL, Huang H, Herrick DZ, Epp N, Cafiso DS (2011) Synaptotagmin 1 and SNAREs form a complex that is structurally heterogeneous. J Mol Biol 405:696–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Kim J‐Y, Choi B‐K, Choi M‐G, Kim S‐A, Lai Y, Shin Y‐K, Lee NK (2012) Solution single‐vesicle assay reveals PIP2‐mediated sequential actions of synaptotagmin‐1 on SNAREs. EMBO J 31:2144–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Zhou A, Brewer KD, Rizo J (2013) Analysis of SNARE complex/synaptotagmin‐1 interactions by one‐dimensional NMR spectroscopy. Biochemistry 52:3446–3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Gaffaney JD, Dunning FM, Wang Z, Hui E, Chapman ER (2008) Synaptotagmin C2B domain regulates Ca2+‐triggered fusion in vitro: critical residues revealed by scanning alanine mutagenesis. J Biol Chem 283:31763–31775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Seven AB, Brewer KD, Shi L, Jiang QX, Rizo J (2013) Prevalent mechanism of membrane bridging by synaptotagmin‐1. Proc Natl Acad Sci USA 110:E3243–E3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Park Y, Seo JB, Fraind A, Perez‐Lara A, Yavuz H, Han K, Jung S‐R, Kattan I, Walla PJ, Choi M, Cafiso DS, Koh D‐S, Jahn R (2015) Synaptotagmin‐1 binds to PIP(2)‐containing membrane but not to SNAREs at physiological ionic strength. Nat Struct Mol Biol 22:815–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Dai H, Shen N, Arac D, Rizo J (2007) A quaternary SNARE‐synaptotagmin‐Ca(2+)‐phospholipid complex in neurotransmitter release. J Mol Biol 367:848–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Xu J, Brewer KD, Perez‐Castillejos R, Rizo J (2013) Subtle Interplay between Synaptotagmin and Complexin Binding to the SNARE Complex. J Mol Biol 425:3461–3475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Malsam J, Parisotto D, Bharat TA, Scheutzow A, Krause JM, Briggs JAG, Sollner TH (2012) Complexin arrests a pool of docked vesicles for fast Ca2+‐dependent release. EMBO J 31:3270–3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Diao J, Grob P, Cipriano DJ, Kyoung M, Zhang Y, Shah S, Nguyen A, Padolina M, Srivastava A, Vrljic M, Shah A, Nogales E, Chu S, Brunger AT (2012) Synaptic proteins promote calcium‐triggered fast transition from point contact to full fusion. eLife 1:e00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Kim J, Zhu Y, Shin YK (2016) Preincubation of t‐SNAREs with complexin I increases content‐mixing efficiency. Biochemistry 55:3667–3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Choi UB, Strop P, Vrljic M, Chu S, Brunger AT, Weninger KR (2010) Single‐molecule FRET‐derived model of the synaptotagmin 1‐SNARE fusion complex. Nat Struct Mol Biol 17:318–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Schupp M, Malsam J, Ruiter M, Scheutzow A, Wierda KDB, Sollner TH, Sorensen JB (2016) Interactions between SNAP‐25 and synaptotagmin‐1 are involved in vesicle priming, clamping spontaneous and stimulating evoked neurotransmission. J Neurosci 36:11865–11880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Guan Z, Bykhovskaia M, Jorquera RA, Sutton RB, Akbergenova Y, Littleton JT (2017) A synaptotagmin suppressor screen indicates SNARE binding controls the timing and Ca(2+) cooperativity of vesicle fusion. eLife 6:e28409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Ubach J, Garcia J, Nittler MP, Sudhof TC, Rizo J (1999) Structure of the Janus‐faced C2B domain of rabphilin. Nat Cell Biol 1:106–112. [DOI] [PubMed] [Google Scholar]
- 223. Kedar GH, Munch AS, van Weering JRT, Malsam J, Scheutzow A, de Wit H, Houy S, Tawfik B, Söllner TH, Sørensen JB, Verhage M (2015) A post‐docking role of synaptotagmin 1‐C2B domain bottom residues R398/399 in mouse chromaffin cells. J Neurosci 35:14172–14182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Bai H, Xue R, Bao H, Zhang L, Yethiraj A, Cui Q, Chapman ER (2016) Different states of synaptotagmin regulate evoked versus spontaneous release. Nat Commun 7:10971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Wang J, Bello O, Auclair SM, Wang J, Coleman J, Pincet F, Krishnakumar SS, Sindelar CV, Rothman JE (2014) Calcium sensitive ring‐like oligomers formed by synaptotagmin. Proc Natl Acad Sci USA 111:13966–13971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Wang J, Li F, Bello OD, Sindelar CV, Pincet F, Krishnakumar SS, Rothman JE (2017) Circular oligomerization is an intrinsic property of synaptotagmin. eLife 6:e27441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Rothman JE, Krishnakumar SS, Grushin K, Pincet F (2017) Hypothesis‐buttressed rings assemble, clamp, and release SNAREpins for synaptic transmission. FEBS Lett 591:3459–3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Sakamoto H, Ariyoshi T, Kimpara N, Sugao K, Taiko I, Takikawa K, Asanuma D, Namiki S, Hirose K (2018) Synaptic weight set by Munc13‐1 supramolecular assemblies. Nat Neurosci 21:41–49. [DOI] [PubMed] [Google Scholar]
- 229. Gipson P, Fukuda Y, Danev R, Lai Y, Chen D‐H, Baumeister W, Brunger AT (2017) Morphologies of synaptic protein membrane fusion interfaces. Proc Natl Acad Sci USA 114:9110–9115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Bai XC, McMullan G, Scheres SH (2015) How cryo‐EM is revolutionizing structural biology. Trends Biochem Sci 40:49–57. [DOI] [PubMed] [Google Scholar]
- 231. Beck M, Baumeister W (2016) Cryo‐electron tomography: Can it reveal the molecular sociology of cells in atomic detail?. Trends Cell Biol 26:825–837. [DOI] [PubMed] [Google Scholar]
- 232. Xu Y, Seven AB, Su L, Jiang QX, Rizo J (2011) Membrane bridging and hemifusion by denaturated munc18. PLoS ONE 6:e22012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Fernandez‐Busnadiego R, Zuber B, Maurer UE, Cyrklaff M, Baumeister W, Lucic V (2010) Quantitative analysis of the native presynaptic cytomatrix by cryoelectron tomography. J Cell Biol 188:145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Martin TF (2015) PI(4,5)P(2)‐binding effector proteins for vesicle exocytosis. Biochim Biophys Acta 1851:785–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Jockusch WJ, Speidel D, Sigler A, Sorensen JB, Varoqueaux F, Rhee J‐S, Brose N (2007) CAPS‐1 and CAPS‐2 are essential synaptic vesicle priming proteins. Cell 131:796–808. [DOI] [PubMed] [Google Scholar]
- 236. Kim J, Shin YK (2017) Productive and non‐productive pathways for synaptotagmin 1 to support Ca(2+)‐triggered fast exocytosis. Front Mol Neurosci 10:380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Kreutzberger AJB, Kiessling V, Liang B, Seelheim P, Jakhanwal S, Jahn R, Castle JD, Tamm LK (2017) Reconstitution of calcium‐mediated exocytosis of dense‐core vesicles. Sci Adv 3:e1603208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Keighron JD, Wigstrom J, Kurczy ME, Bergman J, Wang Y, Cans AS (2015) Amperometric detection of single vesicle acetylcholine release events from an artificial cell. ACS Chem Neurosci 6:181–188. [DOI] [PubMed] [Google Scholar]
- 239. Sudhof TC (2012) The presynaptic active zone. Neuron 75:11–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Acuna C, Liu X, Sudhof TC (2016) How to make an active zone: Unexpected universal functional redundancy between RIMs and RIM‐BPs. Neuron 91:792–807. [DOI] [PubMed] [Google Scholar]
- 241. Meijer M, Dorr B, Lammertse HCA, Blithikioti C, van Weering JRT, Toonen RFG, Sollner TH, Verhage M (2018) Tyrosine phosphorylation of Munc18‐1 inhibits synaptic transmission by preventing SNARE assembly. EMBO J 37:300–320. [DOI] [PMC free article] [PubMed] [Google Scholar]