

Regimen adherence remains a major hurdle to the success of daily oral drug regimens for the treatment and prevention of human immunodeficiency virus (HIV) infection. Long-acting drug formulations requiring less-frequent dosing offer an opportunity to improve adherence and allow for more forgiving options with regard to missed doses.
KEYWORDS: HIV prevention, HIV treatment, extended duration, human immunodeficiency virus, long-acting implant
ABSTRACT
Regimen adherence remains a major hurdle to the success of daily oral drug regimens for the treatment and prevention of human immunodeficiency virus (HIV) infection. Long-acting drug formulations requiring less-frequent dosing offer an opportunity to improve adherence and allow for more forgiving options with regard to missed doses. The administration of long-acting formulations in a clinical setting enables health care providers to directly track adherence. MK-8591 (4′-ethynyl-2-fluoro-2′-deoxyadenosine [EFdA]) is an investigational nucleoside reverse transcriptase translocation inhibitor (NRTTI) drug candidate under investigation as part of a regimen for HIV treatment, with potential utility as a single agent for preexposure prophylaxis (PrEP). The active triphosphate of MK-8591 (MK-8591-TP) exhibits protracted intracellular persistence and, together with the potency of MK-8591, supports its consideration for extended-duration dosing. Toward this end, drug-eluting implant devices were designed to provide prolonged MK-8591 release in vitro and in vivo. Implants, administered subcutaneously, were studied in rodents and nonhuman primates to establish MK-8591 pharmacokinetics and intracellular levels of MK-8591-TP. These data were evaluated against pharmacokinetic and pharmacodynamic models, as well as data generated in phase 1a (Ph1a) and Ph1b clinical studies with once-weekly oral administration of MK-8591. After a single administration in animals, MK-8591 implants achieved clinically relevant drug exposures and sustained drug release, with plasma levels maintained for greater than 6 months that correspond to efficacious MK-8591-TP levels, resulting in a 1.6-log reduction in viral load. Additional studies of MK-8591 implants for HIV treatment and prevention are warranted.
INTRODUCTION
The advent of highly active antiretroviral therapy (HAART) in the 1990s transformed the clinical care of human immunodeficiency virus type 1 (HIV-1) infection (1, 2). HAART regimens have proven to be highly effective treatments, significantly decreasing HIV load in HIV-infected patients and thereby slowing evolution of the illness and reducing HIV-related morbidity and mortality. Despite this progress, HIV and AIDS continue to pose a significant and global health care challenge. In 2016, approximately 36.7 million people were living with HIV globally, with approximately 30% unaware of their infection status, and approximately 20% of people on treatment were not virally suppressed, in part due to low regimen adherence (3). Even with a continual reduction in the number of AIDS-related deaths since 2005, the numbers remain alarmingly high, with an estimated 1 million AIDS-related deaths and 1.8 million new HIV infections in 2016, which demands continued efforts and new approaches toward controlling the epidemic (4).
Preexposure prophylaxis (PrEP), an approach leveraging the preemptive use of antiretroviral drugs to prevent HIV infection, has emerged as a promising method to combat new infections. Currently, the only drug approved for HIV PrEP is a once-daily oral tablet of the drug combination tenofovir disoproxil fumarate and emtricitabine (Truvada; Gilead Sciences). When taken consistently, oral PrEP has been shown to reduce the risk of HIV infection in people who are at high risk by 90% or more (5–7). Nonetheless, adherence rates to PrEP regimens continue to be far from optimal. Social, lifestyle, and psychological factors have all been shown to negatively impact adherence, particularly with current PrEP regimens consisting of daily oral pills (8–10), or more recently, intravaginal rings intended for monthly administration (11, 12).
New HIV treatment and prevention interventions that aim to improve adherence by reducing the complexity of regimens, the frequency of the dosages, and/or the side effects of the medications are thus required. Long-acting injectable (LAI) drug formulations that permit less-frequent dosing, on the order of a month or longer, are an increasingly attractive option to address adherence challenges (13–16). Owing to this potential, the investigational HIV integrase strand transfer inhibitor (InSTI) cabotegravir (GSK1265744; GlaxoSmithKline) has recently advanced into phase 3 clinical studies as a long-acting injectable formulation for PrEP, and the combination of cabotegravir and the nonnucleoside reverse transcriptase inhibitor (NNRTI) rilpivirine (RPV; TMC278LA; Janssen Pharmaceuticals) has recently advanced into phase 3 clinical studies as long-acting injectable formulations for HIV treatment maintenance (17–21). The intrinsic physicochemical properties of cabotegravir and rilpivirine enable their presentation as insoluble solid crystalline drug nanoparticles, and when administered in patients as an aqueous suspension, they release the drugs from the intramuscular injection site depots over a protracted period of time. Injections are typically given every 4 or 8 weeks (17–20).
However, the majority of the approved and investigational antiretroviral agents are not well suited for formulation as a long-acting injectable. In large part, this is due to suboptimal physicochemical properties limiting their formulation as conventional drug suspensions, as well as insufficient antiviral potency resulting in high monthly dosing requirements. Even for cabotegravir and rilpivirine, large injection volumes and multiple injections are required to achieve pharmacokinetic profiles supportive of monthly dosing (17–20). It is thus clear that novel formulation approaches capable of achieving extended-duration pharmacokinetics for molecules of diverse physicochemical properties, at practical injection volumes and with a limited number of injections, are highly desirable (15, 22–24).
In this work, we describe the preclinical development of implant devices as an alternative to the reported injectable drug suspensions for long-acting delivery of antiretroviral drugs. Our implants consist of drug dispersed within bioerodible and nonerodible polymers to generate monolithic matrices of dimensions suitable for subcutaneous administration. The drug loading, polymer chemical properties, and polymer degradation kinetics are tuned to achieve a wide range of drug release rates, offering the potential to extend the duration of drug dosing beyond currently achievable monthly paradigms. The designed implants are compatible with molecules having a broad spectrum of physicochemical properties, including those with high aqueous solubility and amorphous phases which are unsuitable to formulation as solid drug suspensions. Implants are deployed to deliver MK-8591, a nucleoside reverse transcriptase translocation inhibitor (NRTTI) with subnanomolar antiviral activity and long half-life, which presents an opportunity for extended-duration dosing for HIV treatment and prevention (25, 26).
In pilot studies, we establish in vivo feasibility of MK-8591 extended-duration dosing with drug-eluting polymer implants and demonstrate the controlled release of MK-8591 in animal models resulting in potentially efficacious MK-8591-TP levels over durations equal to or greater than 6 months. To our knowledge, this is the first study to report on the use of monolithic implants for long-acting delivery of an antiretroviral agent and the first to demonstrate in vivo drug release for greater than 6 months following a single administration. While our development work continues to advance, it is believed that the implant devices described in this work represent a promising approach for extended-duration dosing of antiretroviral drugs useful for the treatment and prevention of HIV infection. These devices have the potential to address critical adherence gaps and improve real-world effectiveness.
RESULTS
Characterization of drug distribution and implant porosity.
Implants were characterized by microcomputed tomography (microCT) and scanning electron microscopy (SEM) to assess implant quality postextrusion (representative images shown in Fig. 1). microCT imaging provides a nondestructive characterization technique, with resolution in the micrometer range, yielding an image of the internal structure of the implant, and is capable of characterizing the entire three-dimensional (3D) structure (videos not shown), whereas SEM provides additional surface characterization of a deliberately created cross-section of the implant. All implants studied in this series were shown to have a low porosity (or high density), with the crystalline MK-8591 drug distributed homogeneously throughout the implant. It is hypothesized that a low-porosity matrix will result in a decreased drug release rate, compared to that of a porous matrix. This could occur by limiting the water influx and therefore the amount of solubilized drug available for diffusive release, while also minimizing the amount of water-filled channels available for drug release, resulting in an increased diffusion path and/or necessitating that the drug diffuse through the polymer.
FIG 1.

(a and b) Low-resolution (a) and high-resolution (b) representative microCT reconstructions showing a single cross-section through the diameter of a 40 wt% MK-8591 in EVA implant (white particles represent drug and gray areas represent polymer). (c and d) Low-resolution (c) and high-resolution (d) representative SEM images showing a single cross-section through the diameter of a 40 wt% MK-8591 in PCL implant (darker phase with rock-like-shaped, anisotropic particles represents drug, and lighter-gray areas represent polymer). (e and f) SEM images of the surface of 40 wt% MK-8591 in EVA (e) and placebo EVA implants (f).
Additional characterization on the implants was obtained using Raman imaging. Raman enables the visualization of drug distribution within the implant. The method provides complementary chemical information confirming the phase separation of drug and polymer seen in the microCT and SEM images. Figure 2 shows the Raman imaging from the 40 wt% MK-8591 in poly(ethylene vinyl acetate) (EVA) implant, with clear distribution of the drug (Fig. 2a) and polymer (Fig. 2b) through the implant cross-section, where high concentrations are observed in red for each component. It is clear from the Raman, SEM, and microCT imaging that the drug-containing implants prepared by hot melt extrusion (HME) contain crystalline drug distributed consistently throughout the implant with no large agglomerates of either crystalline drug or polymer.
FIG 2.

Raman images of a 40 wt% MK-8591 in EVA implant showing both drug (a) and polymer (b) distribution from a 150 by 150 μm, 14-μm spot, 12 by 12 scan (high concentrations depicted in red, low concentrations depicted in blue).
In vitro drug release from implants.
The in vitro release of MK-8591 from implants made with bioerodible poly(lactic acid) (PLA) and poly(caprolactone) (PCL) and nonerodible EVA at a range of drug loads was examined (Fig. 3). The cumulative in vitro release profiles presented in Fig. 3a to c exhibit first-order release kinetics. The first order rate constant from the in vitro cumulative drug release profiles correlated linearly with the drug loading (Fig. 3d, Spearman correlation, r = 0.97, P < 0.0001 for EVA; and PLA, r = 0.88, P = 0.1 for PCL). The slight differences in the slopes of the linear correlations of the rate constant to drug loading can likely be attributed to differences in drug particle size within the various polymer matrices. The implant compositions tested exhibited in vitro release profiles with extended and tunable drug release kinetics, as the selection of polymer and particularly the drug loading are shown to modulate the drug release kinetics and the duration of release.
FIG 3.
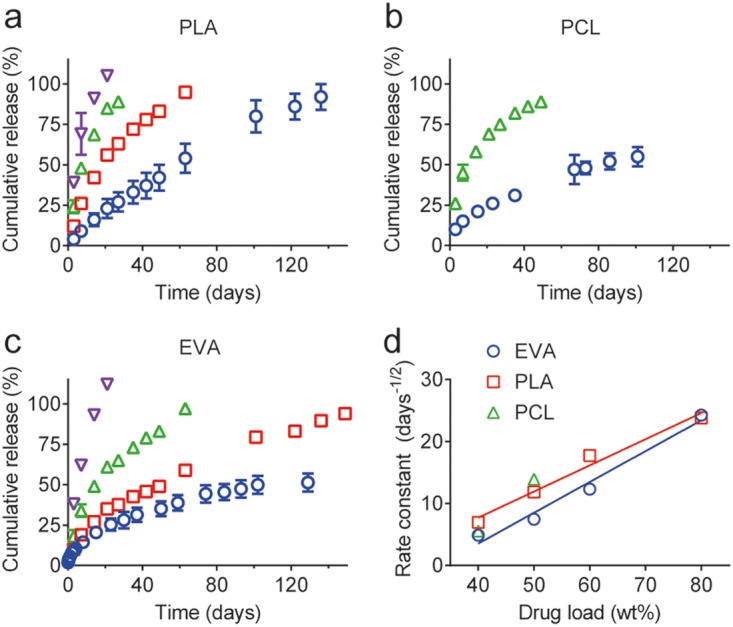
In vitro cumulative drug release profiles from a series of bioerodible, PLA (a) and PCL (b), and nonerodible EVA (c) implants with a range of MK-8591 loadings. (a to c) Blue circles, 40 wt% MK-8591 plus 60 wt% polymer; red squares, 50 wt% MK-8591 plus 50 wt% polymer; green triangles, 60 wt% MK-8591 plus 40 wt% polymer; purple inverted triangles, 80 wt% MK-8591 plus 20 wt% polymer. (d) The linear correlation of the first-order rate constant from the cumulative in vitro release profiles and drug loading.
microCT images of implants as a function of drug release from in vitro experiments at both low and high resolution show the evolution of drug release (Fig. 4a). At the initial time point, before any drug release ensued, drug particles can be seen dispersed throughout the polymer matrix. The drug particles appear lighter in than the continuous gray polymer phase, as confirmed by imaging experiments varying the ratio of drug to polymer, which indicate that the prevalence of lighter domains increases correspondingly with increased drug loading. Rare instances of drug agglomerates were observed. At a cumulative drug release rate of 18%, as measured by in vitro release, the implant cross-section consists of a core of drug and polymer that appears identical to the no-drug release case, with an annulus of a drug-depleted zone. Full drug release is characterized by voids present throughout the continuous polymer phase, as seen at the bottom of Fig. 4a, and in Fig. 4b using helium ion beam microscopy. The drug release mechanism is depicted in Fig. 4c.
FIG 4.

Characterization of 50 wt% MK-8591 in EVA implants undergoing drug release in vitro. (a) microCT images as a function of cumulative drug release from in vitro experiments (scale bar = 1 mm for zoomed-out images on the left; scale bar = 100 μm for higher-resolution images on right). (b) Helium ion microscopy image showing an implant after full drug release. (c) Schematic of drug release via radial diffusion through porous medium.
MK-8591 pharmacokinetics in rodents.
Implants of PLA, PCL, and EVA at multiple MK-8591 drug loads were implanted subcutaneously in rats. The plasma concentration of MK-8591 was measured with time to assess the in vivo release profile. For all implants evaluated, a small and transient burst was observed, producing a rat plasma concentration in the range of 1,000 to 10,000 nM MK-8591. This was likely due to MK-8591 rapidly dissolving from the surface of the implants once exposed to the aqueous biological environment (Fig. 5). The maximum concentration of drug (Cmax) and pseudo-steady-state plasma concentration have a consistent rank order with the drug load of the implant, with higher drug loads showing a higher initial burst and an increased drug release rate. After this small initial release of drug (<5% of the total drug mass), the majority of implant compositions reached their pseudo-steady-state plasma concentration within a few days. Implants tested in vivo showed no test article-related toxicity (either systemically or locally) and were generally well tolerated throughout the duration of the study. Since the potential systemic effects of MK-8591 have been evaluated previously in the context of the oral formulation (27, 28), histopathologic evaluation was limited to implant sites and local draining lymph nodes. All animals survived to the scheduled termination. The MK-8591 implants were easily removed and in one piece. There were no test article-related clinical observations or changes in body weight observed throughout the study. The release rate and the fraction released from the 60 wt% MK-8591 in PCL implant are shown in Fig. 6 as a representative data set for all the above-mentioned formulations. The cumulative drug release rates were approximately 37% at 6 months, 42% at 12 months, and 45% at 17.6 months. The MK-8591 release rate was maintained above 10 μg/day for the study duration.
FIG 5.

In vivo MK-8591 plasma concentration-versus-time profiles in male Wistar Han rats from a series of bioerodible, PLA (a) and PCL (b), and nonerodible EVA (c) implants. Data represent mean ± standard deviation (SD) of n = 4 subjects for all studies. Blue circles, 40 wt% MK-8591 plus 60 wt% polymer; red squares, 50 wt% MK-8591 plus 50 wt% polymer; green triangles, 60 wt% MK-8591 plus 40 wt% polymer; purple inverted triangles, 80 wt% MK-8591 plus 20 wt% polymer.
FIG 6.
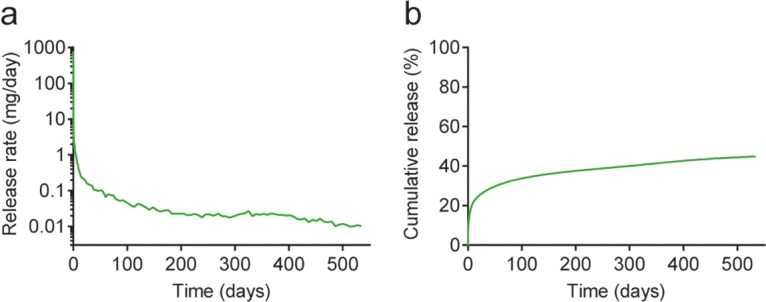
In vivo MK-8591 release rate (a) and cumulative release (b) from a 60 wt% MK-8591 in PCL formulation in rat.
In an attempt to understand the mass balance of drug released in in vivo studies and the accuracy of the deconvolution analysis, a rat study was conducted using 50 wt% MK-8591 in EVA implants, the same implant composition of MK-8591 used in the nonhuman primate study described below. At approximately 1, 2, 3, 4, and 6 months, four animals were euthanized, and the implants were removed and characterized. Drug pharmacokinetic data were collected throughout the study and compared to the residual drug content obtained from the excised implants, as determined by chemical analysis (Table 1). The study demonstrated a mass balance that is consistent with expectations, at approximately 70 to 80%.
TABLE 1.
Mass balance obtained from measuring the plasma concentrations from 50 wt% MK-8591 in EVA implant in rat as well as the amount of drug remaining in the implants at defined timesa
| Time (days) | % in implants | % released in vivo | Mass balance (%) |
|---|---|---|---|
| 0 | 100.0 | 0.0 | 100.0 |
| 28 | 53.1 | 29.0 | 82.1 |
| 56 | 38.4 | 39.7 | 78.0 |
| 84 | 22.8 | 47.5 | 70.3 |
| 112 | 10.9 | <LOQ | <LOQ |
| 182 | 0.4 | <LOQ | <LOQ |
LOQ, limit of quantification.
Evaluation of MK-8591 implants in nonhuman primates.
Implants of 40 wt% and 50 wt% MK-8591 in PLA, 50 wt% MK-8591 in PCL, and 50 wt% MK-8591 in EVA were administered subcutaneously in rhesus macaques. The concentrations of MK-8591 in plasma and MK-8591-TP in peripheral blood mononuclear cells (PBMCs) were measured as a function of time (Fig. 7a and b). Deconvolution of pharmacokinetic data from this in vivo study resulted in release rates from each formulation that were consistent with those obtained from the rat study (Fig. 7c), with similarly low intersubject variability for MK-8591 in the plasma (Fig. 7a). The MK-8591 implants were easily removed at the end of study and remained intact (as one piece). A convolution-based model was developed to predict human pharmacokinetics of an MK-8591 implant based on the data from the nonhuman primate implant study, assuming the drug release rate does not change between species. The predicted MK-8591 plasma pharmacokinetics (PK) in humans from the 50 wt% MK-8591 in the PCL implant were compared to the clinical PK of once-weekly oral doses of MK-8591 at either 2 mg or 10 mg, assuming no accumulation following repeated weekly dosing. The projected MK-8591 plasma concentration exceeds the trough concentration projected for 2-mg once-weekly oral dosing and is similar to the trough concentration projected for 10-mg once-weekly oral dosing (Fig. 7d, projection is a nonparametric superposition of data from a single oral dose of 2 mg or 10 mg).
FIG 7.

Concentration-versus-time profiles of MK-8591 in plasma (a) and MK-8591-TP in PBMC (b) for 40 wt% or 50 wt% MK-8591 in PLA, 50 wt% MK-8591 in PCL, and 50 wt% MK-8591 in EVA (mean ± SD). (c) MK-8591 release rate from a series of both bioerodible and nonerodible MK-8591-containing implants in nonhuman primates. (d) The projected human MK-8591 plasma profile compared to once-weekly (QW) 2 and 10 mg MK-8591 oral dosing.
DISCUSSION
Several key studies demonstrate the potential utility of MK-8591 as a low-dose extended-duration formulation for HIV treatment and prevention (25). The active triphosphate of MK-8591 (MK-8591-TP) exhibits protracted intracellular persistence in human PBMCs, protecting cells from infection in the presence of continued viral exposure in vitro. The virologic activity of MK-8591 in a simian immunodeficiency virus (SIV)-infected macaque was evaluated at once-weekly oral doses ranging from 1.3 to 18.2 mg/kg of body weight. The maximal virologic decline, an approximate 1.5 log decrease in viral load, was correlated with intracellular MK-8591-TP concentrations of ≥0.53 pmol/106 PBMCs (27). In early clinical studies in adults with HIV-1 infection, a single oral dose of MK-8591 at 0.5 mg, 1 mg, 2 mg, 10 mg, or 30 mg resulted in a >1.2-log viral load decline by day seven, associated with MK-8591-TP levels of ≥0.12 pmol/106 PBMCs, suggesting the potential for once-weekly antiviral efficacy at a low dose (27, 28). In a related study, weekly oral dosing of MK-8591 completely protected rhesus macaques from repeated rectal viral challenges. The intracellular MK-8591-TP concentration at the time of simian-human immunodeficiency virus (SHIV) challenge, 6 days after MK-8591 dosing, was approximately 0.8 pmol/106 PBMCs (29). Together, these data support the evaluation of low-dose MK-8591 for HIV-1 treatment and prevention in individuals at a high risk of acquiring infection, which is a key requirement for MK-8591 to be delivered as an implant.
Unlike rilpivirine and cabotegravir, MK-8591 formulated as an aqueous crystalline suspension for injection will not achieve sustained drug release due to its high intrinsic aqueous solubility. As such, alternative formulation approaches are required to achieve long-acting drug release of antiretroviral molecules from different mechanistic classes that cover a wide range of physicochemical properties. Drug-eluting polymer-based implants offer one such approach. Although no clinical proof of concept has been achieved for a long-acting formulation of a hydrophilic antiretroviral drug, there are two examples of preclinical proof-of-concept tenofovir alafenamide-eluting reservoir implants. Gunawardana et al. report a nonerodible subcutaneous reservoir-type implant containing the nucleoside reverse transcriptase inhibitor (NRTI) tenofovir alafenamide, demonstrating sustained drug release in beagle dogs over 40 days (30). The implant consisted of a cylindrical silicone tube with predetermined pores coated with poly(vinyl alcohol) and filled with drug powder. The number and cross-sectional diameter of the pores, coupled with the physicochemical properties of the outer polymer membrane and drug, determined the release kinetics of the drug from the implant through the poly(vinyl alcohol) “windows.” Schlesinger et al. described the in vitro assessment of a degradable implant composed of a PCL capsule filled with tenofovir alafenamide with and without low-molecular-weight polyethylene glycol (31). These initial studies are promising, with minimal burst observed and a linear release of drug for up to 90 days (30, 31), but additional work must target longer durations that achieve a relevant release rate for the specific drug with a limited implant size.
The MK-8591 implant design used herein is a polymeric monolithic matrix. Drug release from the implant is driven by drug dissolution and diffusion from the polymer matrix in the case of a nonerodible polymer and both diffusion- and degradation-facilitated release when the implant matrix is composed of bioerodible polymers. The cumulative in vitro release profiles (Fig. 3a to c) exhibit first-order release kinetics suggesting a primarily diffusion-based release and minimal to no degradation-based release on the time scales studied (32, 33). The MK-8591 implants were prepared by hot melt extrusion (HME), a continuous and solvent-free processing method. Several examples of pharmaceutical commercial combination products prepared using HME include Implanon, Nexplanon, and NuvaRing. For the implants prepared of MK-8591 described herein, the drug and polymer powders were first blended. The blend was fed into a twin screw extruder and melt compounded. The barrel temperature was above the glass transition or melting temperature of the polymer but significantly below the melting temperature of the drug, such that the final product consisted of a distribution of solid crystalline drug embedded in a polymer matrix.
In this study, MK-8591 showed negligible miscibility with any of the polymer materials used for the polymer matrix, suggesting that the polymer phase is inaccessible for drug transport. For a cylindrical drug-eluting matrix implant, the drug at the surface of the implant diffuses from the implant first, leading to a concentration gradient across the implant cross-section (Fig. 4c). As the surface drug dissolves, it creates new pores that enable fluid ingress. This leads to the dissolution of additional drug particles and the creation of a porous structure with interconnected channels. The drug loading and drug particle size distribution control the morphology of the porous network. The thickness of the drug-depleted zone increases with time, resulting in an increased diffusional distance. This leads to the continual reduction in the drug release rate over time typical of a matrix design and observed in the release rate profile in both the in vitro and in vivo studies described herein. Furthermore, the mechanism of release for these matrix implants requires the initial drug load to be above the percolation threshold in order for full drug release to be realized.
The drug release performances of implants consisting of erodible (PLA and PCL), and nonerodible (EVA) polymers were evaluated in rats. A range of drug loadings were selected based on differentiated in vitro release profiles. Consistent with the in vitro drug release testing, the MK-8591 plasma profiles are consistent with first-order drug release kinetics (Fig. 5). For each implant composition evaluated in this study, release occurs through drug dissolution and then diffusion through the polymer matrix. The bioerodible implants can also have a secondary release mechanism due to polymer degradation, and both of these mechanisms (diffusion facilitated and degradation facilitated) can occur simultaneously and cooperatively. Based on the literature for the polymers selected in this study, the PLA backbone should exhibit a faster degradation onset and higher rate than those of PCL. For the PLA implants tested, we hypothesized that the cooperative effect of both diffusion- and degradation-facilitated release of MK-8591 began around 200 days, where an increase in the plasma concentration of MK-8591 was observed for the 40 wt% MK-8591 in the PLA implant. Beyond day 246, the plasma concentration of MK-8591 was below the limit of quantification for the analytical method. The other bioerodible polymer, PCL, appears to be releasing drug in a strictly diffusion-mediated release mechanism up to day 536, the longest time point tested in our study. It is likely that the onset of significant degradation exceeds this time frame. The nonerodible implant consisting of EVA shows a pseudo-steady-state concentration of MK-8591 that was maintained for extended durations (months).
By varying the drug loading in the polymers, the release rate and duration of MK-8591 can be effectively modulated to the target efficacious dose and duration. A number of implant compositions achieved >180-day extended release after a single implantation (Fig. 5), with 40 wt% MK-8591 in PLA, 40 wt% and 60 wt% MK-8591 in PCL, and 50 wt% MK-8591 in EVA. In fact, pseudo-steady-state concentrations of MK-8591 in plasma were maintained for more than 12 months for the 60 wt% MK-8591 in PCL implants (Fig. 6). Based on these data, it is clear that the MK-8591 in PCL and EVA implants should maintain its pseudo-steady-state concentrations for a duration of at least 12 months, given that it has only released approximately half of its drug load after 1 year in vivo, and that up to the 12-month time point, there is no evidence of PCL degradation. Implants prepared from the PLA matrix should maintain their steady-state concentrations for at least 6 months but would not be recommended to target durations longer than that, as PLA degradation was evident before the 12-month time point (Fig. 5a). Potentially longer duration could be achieved with PLA-based implants with a higher molecular weight or with more crystalline PLA (32, 33).
Implants consisting of 40 or 50 wt% MK-8591 in PLA, PCL, and EVA were evaluated in nonhuman primates (Fig. 7), an important animal model for HIV treatment and prevention (34, 35). The nonhuman primate study enabled the quantification of MK-8591-TP in PBMCs (Fig. 7b) and demonstrated sustained MK-8591-TP concentrations far in excess of the in vitro 50% effective concentration (EC50) (36). The PCL and EVA implants achieved intracellular MK-8591-TP concentration at the same order of magnitude that conferred complete protection in the rectal SHIV challenge study, 0.8 pmol/106 PBMCs (29). From the nonhuman primate study, we predicted the MK-8591 plasma PK in humans from the 50 wt% MK-8591 in PCL implants and compared to clinical PK of once-weekly oral doses of MK-8591. The predicted plasma MK-8591 concentration for the MK-8591 implant was similar to the trough concentration projected for a 10-mg MK-8591 once-weekly oral dosing (Fig. 7d). A single 10-mg oral dose was associated with 1.6-log viral load decline in humans (28). Projected MK-8591-TP levels in humans were extrapolated from the MK-8591-TP/MK-8591 ratio in nonhuman primates, and accounting for the 10× species difference in uptake and phosphorylation between humans and macaques (25), are further supportive of the extended-duration dosing in humans.
Conclusion.
In this work, bioerodible and nonerodible drug-eluting implants were evaluated for extended-duration dosing of MK-8591, an investigational NRTTI under development for the treatment of HIV. Implants are designed to achieve a broad range of drug release characteristics and durations, achieved through optimization of drug loading and polymer composition of the implants. After a single subcutaneous administration in rodents and nonhuman primates, optimized MK-8591 implants achieved sustained drug release at clinically relevant drug concentrations for greater than 6 months. Additional studies of MK-8591 implants for HIV treatment and prevention are warranted.
MATERIALS AND METHODS
Materials.
MK-8591 was obtained from Process Chemistry (Merck & Co., Inc., Rahway, NJ, USA). All biodegradable polymers tested were supplied from Evonik Industries AG (Essen, Germany). These included acid-terminated poly(lactic acid) (PLA; polymer name, 100 DL 8A), and ester-terminated poly(caprolactone) (PCL; polymer name, 100 CL 7.5E). Poly(ethylene vinyl acetate) (EVA; polymer name, Ateva 1070, 9% vinyl acetate) was obtained from Celanese (Dallas, TX). Tetrahydrofuran (THF) and methanol (MeOH) were obtained from Sigma-Aldrich, and phosphate-buffered saline was obtained from GE Healthcare Lifesciences (HyClone, catalog no. SH30256.02).
Preparation of implants by hot melt extrusion.
Implant devices were prepared using an extrusion process. PLA and EVA were supplied as pellets and were cryomilled prior to use. The MK-8591 and polymer powders were mixed using a Turbula T2F mixer. Drug and polymer blends were prepared at 40, 50, 60, and 80 wt% drug loads. The drug and polymer blends were hot melt extruded using a custom 7.5-mm corotating twin-screw extruder with a 16:1 length-to-diameter (L/D) ratio, through a 2-mm-diameter die, and pulled to a diameter of approximately 1.9 to 2.3 mm. The screws contained predominately conveying elements with a single 90° mixing section containing 3 paddles. The zone where the drug-polymer blends were introduced was water-cooled and maintained at room temperature. The temperatures for the remaining zones varied based on the polymer, at 65 to 75°C for PCL, 100°C for PLA, and 120°C for EVA.
In vitro drug release studies.
The in vitro release rate of MK-8591 was determined by incubating implant segments, approximately 1 cm in length, in phosphate-buffered saline (PBS) at 37°C and 50 rpm shaking in an Innova 42 incubator. The volume of PBS was sufficient to maintain sink conditions for MK-8591. Sink conditions were defined as the drug concentration at or below 1/3 of the maximum solubility, i.e., an MK-5891 concentration of ≤0.45 mg/ml in PBS at 37°C. Aliquots (0.5 ml) were removed at selected time points, replaced with PBS, and centrifuged at 20,800 × g for 8 min. The supernatant was removed, diluted 4-fold, and vortexed. Samples were assayed by high-performance liquid chromatography (HPLC). See the supplemental material for method details.
Analysis of MK-8591 content in implants.
MK-8591 content was determined by liquid-phase extraction and HPLC analysis. For implants containing polymers soluble in THF (i.e., PLA and PCL), approximately 50 mg of extrudate was dissolved in 8 ml of THF by stirring. The polymer was precipitated with 50:50 MeOH-H2O (1:5) allowed to stir for 24 h, followed by centrifugation at 20,800 × g for 8 min. For implants composed of EVA polymer, which is not soluble in THF, approximately 125 mg extrudate was stirred in 50 ml of 40:40:20 MeOH-H2O-THF in a 125-ml Erlenmeyer flask. Using a handheld Polytron PT 2100 grinder, the sample was mixed for 2 h and transferred to a 100-ml volumetric flask, diluted to volume with 50:50 MeOH-H2O, allowed to stir for 24 h, and followed by centrifugation at 20,800 × g for 8 min. For both extraction methods, the supernatant was removed and analyzed by HPLC (see the supplemental material).
Microcomputed tomography imaging.
Scans of the internal structure of the implants were performed on an XRadia XRM 500 microcomputed tomography machine. Samples were mounted on an adjustable collet to prevent movement during scanning. The X-ray source settings were 80 kV and 7 W. The scans were performed with ×4 and ×20 optical objective lens magnifications, resulting in resolutions of 3.38 and 0.69 μm/pixel, respectively. Images were acquired over 360° of rotation with a rotational step of 0.225 degrees per image, totaling 1,600 images. The total acquisition time per data set was approximately 1 h. Images were reconstructed with the XRadia reconstruction software, and visualization was performed with ORS Dragonfly Pro and ImageJ.
Raman imaging.
Raman mapping was used to investigate drug distribution in the 40 wt% MK-8591 in EVA implant. The Raman mapping study was performed using a RXN Raman station (Kaiser Optical Systems) with a 785-nm laser source. In the Raman experiment, the laser spot size was tuned to be approximately 14 μm under the ×100 objective by reducing laser power to 50 mW. The mapping area has dimensions of 150 μm by 150 μm and was divided into 144 (12 by 12) mapping spots. The mapping step size (center to center) was 12.5 μm. At each mapping spot, Raman spectrum data were collected with a 10-s exposure time and 3 accumulations. A two-dimensional (2D) color-contrast image was obtained from spectrum data collected on all mapping spots in MATLAB to demonstrate the surface distribution of target component based on relative peak intensity from each mapping spot.
SEM imaging.
Scanning electron microscopy (SEM) images were obtained on a freeze-fractured surface to study the internal microstructure using a Quanta FEG250 instrument (FEI Co.). Implants were soaked in liquid nitrogen for 1 min to freeze, followed by a quick snap. The implant formulations evaluated were 40 wt% MK-8591 in EVA, 40 wt% MK-8591 in PCL, and a placebo implant consisting of 100% EVA. The drug-loaded implants showed sufficient resolution at 20 kV, whereas the placebo implant had to be examined at 10 kV to reduce the surface overcharging effect.
Helium ion microscopy imaging.
The helium ion microscopy images were acquired with a Zeiss Orion helium ion microscope. The ion source consisted of a sharpened needle held at high positive voltage and low temperature in the presence of helium gas. The ion beam was transmitted through a two-lens electrostatic ion optical column onto the sample surface. The beam energy was 25 to 35 keV, and the beam current was 0.1 to 10 pA. The image presented here was collected with secondary electrons at 20-nm spatial resolution.
In vivo studies of MK-8591 implants.
All animal studies were conducted using protocols in accordance with the Institutional Animal Care and Use Committee (IACUC) at Merck Sharpe and Dohme (MSD), which adhere to the regulations outlined in the USDA Animal Welfare Act. For each implantation in the rat study, a Wistar Han rat was anesthetized using isoflurane to effect prior to subcutaneous dose administration. Using a 12-gauge trocar needle, the implant (various lengths based on the body weight of the individual animal to achieve the appropriate dose) was placed in the scapular region. Dose selection was as follows: 100 mg/kg for 40 wt% MK-8591 implants, 250 mg/kg for 50 wt% MK-8591 implants, 300 mg/kg for 60 wt% MK-8591 implants, and 400 mg/kg for 80 wt% MK-8591 implants. Animals were monitored until they recovered. At indicated time points, blood samples were obtained from alert animals and processed to plasma for determination of MK-8591 concentrations. Four rats (2 males and 2 females) were used for each implant composition. For each implantation in the nonhuman primate study, a rhesus macaque was sedated with ketamine-HCl (100 mg/ml) prior to subcutaneous implant administration. Using an injector device, the implant, 38 to 39 mm in length, was placed subcutaneously in the interscapular region. Animals were monitored until they recovered. Two animals (both male) were used for 50 wt% MK-8591 in PCL and 40 wt% MK-8591 in PLA formulations, and three animals (all male) were used for 50 wt% MK-8591 in PLA and EVA formulations. At the indicated time points, samples of blood were obtained and processed to plasma or peripheral blood mononuclear cells (PBMCs) for the determination of MK-8591 and MK-8591-TP concentrations, respectively (see the supplemental material).
Deconvolution analysis and human pharmacokinetics projection.
All deconvolution analyses were performed by employing the deconvolution module in the Phoenix WinNonlin 6.3 software (Pharsight, Certara Company). A unit impulse response (UIR) function was first established using intravenous bolus pharmacokinetic (PK) data from rats and macaques (data not shown). Next, mean implant PK profiles were deconvolved using these UIR parameters to yield absorption-time profiles, including both input rate and cumulative percent release.
Human pharmacokinetics projection was conducted as follows: (i) the in vivo release of implants was calculated by deconvolution analysis, as described above; (ii) UIR parameters in humans were obtained from fitting of human PK data following the oral administration of MK-8591 dry filled capsules; (iii) prediction of pharmacokinetic profile in human subjects following the administration of implants was done by convolution-based analysis by assuming the same in vivo release rates in humans and rhesus macaques for the same size implants.
Supplementary Material
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.01058-18.
REFERENCES
- 1.Deeks SG, Smith M, Holodniy M, Kahn JO. 1997. HIV-1 protease inhibitors–a review for clinicians. JAMA 277:145–153. doi: 10.1001/jama.1997.03540260059037. [DOI] [PubMed] [Google Scholar]
- 2.Hammer SM, Saag MS, Schechter M, Montaner JSG, Schooley RT, Jacobsen DM, Thompson MA, Carpenter CCJ, Fischl MA, Gazzard BG, Gatell JM, Hirsch MS, Katzenstein DA, Richman DD, Vella S, Yeni PG, Volberding PA, International AIDS Society-USA panel. 2006. Treatment for adult HIV infection–2006 recommendations of the International AIDS Society-USA panel. JAMA 296:827–843. doi: 10.1001/jama.296.7.827. [DOI] [PubMed] [Google Scholar]
- 3.UNAIDS. 2017. Ending AIDS: progress towards the 90-90–90 targets. Joint United Nations Programme on HIV/AIDS (UNAIDS), Geneva, Switzerland: http://www.unaids.org/sites/default/files/media_asset/Global_AIDS_update_2017_en.pdf. [Google Scholar]
- 4.UNAIDS. 2017. Fact sheet–latest statistics on the status of the AIDS epidemic. Joint United Nations Programme on HIV/AIDS (UNAIDS), Geneva, Switzerland: http://www.unaids.org/en/resources/fact-sheet. [Google Scholar]
- 5.Anderson PL, Glidden DV, Liu A, Buchbinder S, Lama JR, Guanira JV, McMahan V, Bushman LR, Casapia M, Montoya-Herrera O, Veloso VG, Mayer KH, Chariyalertsak S, Schechter M, Bekker LG, Kallas EG, Grant RM, i PrEx Study Team. 2012. Emtricitabine-tenofovir concentrations and pre-exposure prophylaxis efficacy in men who have sex with men. Sci Transl Med 4:151ra125. doi: 10.1126/scitranslmed.3004006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baeten JM, Donnell D, Mugo NR, Ndase P, Thomas KK, Campbell JD, Wangisi J, Tappero JW, Bukusi EA, Cohen CR, Katabira E, Ronald A, Tumwesigye E, Were E, Fife KH, Kiarie J, Farquhar C, John-Stewart G, Kidoguchi L, Coombs RW, Hendrix C, Marzinke MA, Frenkel L, Haberer JE, Bangsberg D, Celum C, Partners PrEP Study Team. 2014. Single-agent tenofovir versus combination emtricitabine plus tenofovir for pre-exposure prophylaxis for HIV-1 acquisition: an update of data from a randomised, double-blind, phase 3 trial. Lancet Infect Dis 14:1055–1064. doi: 10.1016/S1473-3099(14)70937-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grant RM, Lama JR, Anderson PL, McMahan V, Liu AY, Vargas L, Goicochea P, Casapia M, Guanira-Carranza JV, Ramirez-Cardich ME, Montoya-Herrera O, Fernandez T, Veloso VG, Buchbinder SP, Chariyalertsak S, Schechter M, Bekker LG, Mayer KH, Kallas EG, Amico KR, Mulligan K, Bushman LR, Hance RJ, Ganoza C, Defechereux P, Postle B, Wang FR, McConnell JJ, Zheng JH, Lee J, Rooney JF, Jaffe HS, Martinez AI, Burns DN, Glidden DV, iPrEx Study Team. 2010. Preexposure chemoprophylaxis for HIV prevention in men who have sex with men. N Engl J Med 363:2587–2599. doi: 10.1056/NEJMoa1011205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haberer JE, Baeten JM, Campbell J, Wangisi J, Katabira E, Ronald A, Tumwesigye E, Psaros C, Safren SA, Ware NC, Thomas KK, Donnell D, Krows M, Kidoguchi L, Celum C, Bangsberg DR. 2013. Adherence to antiretroviral prophylaxis for HIV prevention: a substudy cohort within a clinical trial of serodiscordant couples in East Africa. PLoS Med 10:e1001511. doi: 10.1371/journal.pmed.1001511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Corneli AL, Deese J, Wang M, Taylor D, Ahmed K, Agot K, Lombaard J, Manongi R, Kapiga S, Kashuba A, Van Damme L. 2014. FEM-PrEP: adherence patterns and factors associated with adherence to a daily oral study product for pre-exposure prophylaxis. J Acquir Immune Defic Syndr 66:324–331. doi: 10.1097/QAI.0000000000000158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van der Straten A, Montgomery ET, Musara P, Etima J, Naidoo S, Laborde N, Hartmann M, Levy L, Bennie T, Cheng H, Piper J, Grossman CI, Marrazzo J, Mensch B, Microbicide Trials-003D Study Team. 2015. Disclosure of pharmacokinetic drug results to understand nonadherence. AIDS 29:2161–2171. doi: 10.1097/QAD.0000000000000801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nel A, van Niekerk N, Kapiga S, Bekker LG, Gama C, Gill K, Kamali A, Kotze P, Louw C, Mabude Z, Miti N, Kusemererwa S, Tempelman H, Carstens H, Devlin B, Isaacs M, Malherbe M, Mans W, Nuttall J, Russell M, Ntshele S, Smit M, Solai L, Spence P, Steytler J, Windle K, Borremans M, Resseler S, Van Roey J, Parys W, Vangeneugden T, Van Baelen B, Rosenberg Z, for the Ring Study Team. 2016. Safety and efficacy of a dapivirine vaginal ring for HIV prevention in women. N Engl J Med 375:2133–2143. doi: 10.1056/NEJMoa1602046. [DOI] [PubMed] [Google Scholar]
- 12.Baeten JM, Palanee-Phillips T, Brown ER, Schwartz K, Soto-Torres LE, Govender V, Mgodi NM, Matovu Kiweewa F, Nair G, Mhlanga F, Siva S, Bekker LG, Jeenarain N, Gaffoor Z, Martinson F, Makanani B, Pather A, Naidoo L, Husnik M, Richardson BA, Parikh UM, Mellors JW, Marzinke MA, Hendrix CW, van der Straten A, Ramjee G, Chirenje ZM, Nakabiito C, Taha TE, Jones J, Mayo A, Scheckter R, Berthiaume J, Livant E, Jacobson C, Ndase P, White R, Patterson K, Germuga D, Galaska B, Bunge K, Singh D, Szydlo DW, Montgomery ET, Mensch BS, Torjesen K, Grossman CI, Chakhtoura N, Nel A, Rosenberg Z, McGowan I, Hillier S, MTN-020-ASPIRE Study Team. 2016. Use of a vaginal ring containing dapivirine for HIV-1 prevention in women. N Engl J Med 375:2121–2132. doi: 10.1056/NEJMoa1506110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Williams J, Sayles HR, Meza JL, Sayre P, Sandkovsky U, Gendelman HE, Flexner C, Swindells S. 2013. Long-acting parenteral nanoformulated antiretroviral therapy: interest and attitudes of HIV-infected patients. Nanomedicine (Lond) 8:1807–1813. doi: 10.2217/nnm.12.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Greene GJ, Swann G, Fought AJ, Carballo-Dieguez A, Hope TJ, Kiser PF, Mustanski B, D'Aquila RT. 2017. Preferences for long-acting pre-exposure prophylaxis (PrEP), daily oral PrEP, or condoms for HIV prevention among US men who have sex with men. AIDS Behav 21:1336–1349. doi: 10.1007/s10461-016-1565-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Owen A, Rannard S. 2016. Strengths, weaknesses, opportunities and challenges for long acting injectable therapies: Insights for applications in HIV therapy. Adv Drug Deliv Rev 103:144–156. doi: 10.1016/j.addr.2016.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luecke EH, Cheng H, Woeber K, Nakyanzi T, Mudekunye-Mahaka IC, van der Straten A, MTN-003D Study Team. 2016. Stated product formulation preferences for HIV pre-exposure prophylaxis among women in the VOICE-D (MTN-003D) study. J Int AIDS Soc 19:20875. doi: 10.7448/IAS.19.1.20875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Spreen WR, Margolis DA, Pottage JC Jr. 2013. Long-acting injectable antiretrovirals for HIV treatment and prevention. Curr Opin HIV AIDS 8:565–571. doi: 10.1097/COH.0000000000000002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rajoli RKR, Back DJ, Rannard S, Meyers CLF, Flexner C, Owen A, Siccardi M. 2015. Physiologically based pharmacokinetic modelling to inform development of intramuscular long-acting nanoformulations for HIV. Clin Pharmacokinetics 54:639–650. doi: 10.1007/s40262-014-0227-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baert L, van 't Klooster G, Dries W, Francois M, Wouters A, Basstanie E, Iterbeke K, Stappers F, Stevens P, Schueller L, Van Remoortere P, Kraus G, Wigerinck P, Rosier J. 2009. Development of a long-acting injectable formulation with nanoparticles of rilpivirine (TMC278) for HIV treatment. Eur J Pharm Biopharm 72:502–508. doi: 10.1016/j.ejpb.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 20.Van 't Klooster G, Hoeben E, Borghys H, Looszova A, Bouche M-P, van Velsen F, Baert L. 2010. Pharmacokinetics and disposition of rilpivirine (TMC278) nanosuspension as a long-acting injectable antiretroviral formulation. Antimicrob Agents Chemother 54:2042–2050. doi: 10.1128/AAC.01529-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Margolis DA, Gonzalez-Garcia J, Stellbrink HJ, Eron JJ, Yazdanpanah Y, Podzamczer D, Lutz T, Angel JB, Richmond GJ, Clotet B, Gutierrez F, Sloan L, Clair MS, Murray M, Ford SL, Mrus J, Patel P, Crauwels H, Griffith SK, Sutton KC, Dorey D, Smith KY, Williams PE, Spreen WR. 2017. Long-acting intramuscular cabotegravir and rilpivirine in adults with HIV-1 infection (LATTE-2): 96-week results of a randomised, open-label, phase 2b, non-inferiority trial. Lancet 390:1499–1510. doi: 10.1016/S0140-6736(17)31917-7. [DOI] [PubMed] [Google Scholar]
- 22.Karmon SL, Markowitz M. 2013. Next-generation integrase inhibitors where to after raltegravir? Drugs 73:213–228. doi: 10.1007/s40265-013-0015-5. [DOI] [PubMed] [Google Scholar]
- 23.Lykins WR, Luecke E, Johengen D, van der Straten A, Desai TA. 2017. Long acting systemic HIV pre-exposure prophylaxis: an examination of the field. Drug Deliv Transl Res 7:805–816. doi: 10.1007/s13346-017-0391-6. [DOI] [PubMed] [Google Scholar]
- 24.McGowan I. 2015. Injectable and implantable antiretroviral strategies for HIV prevention. Future Virol 10:1163–1176. doi: 10.2217/fvl.15.83. [DOI] [Google Scholar]
- 25.Markowitz M, Sarafianos SG. 2018. 4′-Ethynyl-2-fluoro-2′-deoxyadenosine, MK-8591: a novel HIV-1 reverse transcriptase translocation inhibitor. Curr Opin HIV AIDS 13:294–299. doi: 10.1097/COH.0000000000000467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stoddart CA, Galkina SA, Joshi P, Kosikova G, Moreno ME, Rivera JM, Sloan B, Reeve AB, Sarafianos SG, Murphey-Corb M, Parniak MA. 2015. Oral administration of the nucleoside EFdA (4′-ethynyl-2-fluoro-2′-deoxyadenosine) provides rapid suppression of HIV viremia in humanized mice and favorable pharmacokinetic properties in mice and the rhesus macaque. Antimicrob Agents Chemother 59:4190–4198. doi: 10.1128/AAC.05036-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grobler J, Friedman E, Barrett SE, Wood SL, Ankrom W, Fillgrove KL, Lai M, Gindy M, Iwamoto M, Hazuda DJ. 2016. Long-acting oral and parenteral dosing of MK-8591 for HIV treatment or prophylaxis. Conference on retroviruses and opportunistic infections, 22 to 25 February 2016, Boston, MA. [Google Scholar]
- 28.Matthews RP, Schurmann D, Rudd DJ, Levine V, Fox-Bosetti S, Zhang S, Robberechts M, Lepeleire ID, Huser A, Hazuda DJ, Iwamoto M, Grobler JA. 2017. Single doses as low as 0.5 mg of the novel NRTTI MK-8591 suppress HIV for at least seven days. 9th International AIDS Society Conference on HIV Science, 23 to 26 July 2017, Paris, France. [Google Scholar]
- 29.Markowitz M, Gettie A, St Bernard L, Hazuda DJ, Fillgrove KL, Sun L, Grobler JA. 2017. Weekly oral MK-8591 Prpects male rhesus macaques against repeated low dose intrarectal challenge with SHIV109CP3. 9th International AIDS Society Conference on HIV Science, Paris, France, 23 to 26 July 2017. [Google Scholar]
- 30.Gunawardana M, Remedios-Chan M, Miller CS, Fanter R, Yang F, Marzinke MA, Hendrix CW, Beliveau M, Moss JA, Smith TJ, Baum MM. 2015. Pharmacokinetics of long-acting tenofovir alafenamide (GS-7340) subdermal implant for HIV prophylaxis. Antimicrob Agents Chemother 59:3913–3919. doi: 10.1128/AAC.00656-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schlesinger E, Johengen D, Luecke E, Rothrock G, McGowan I, van der Straten A, Desai T. 2016. A tunable, biodegradable, thin-film polymer device as a long-acting implant delivering tenofovir alafenamide fumarate for HIV pre-exposure prophylaxis. Pharm Res 33:1649–1656. doi: 10.1007/s11095-016-1904-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen C-C, Chueh J-Y, Tseng H, Huang H-M, Lee S-Y. 2003. Preparation and characterization of biodegradable PLA polymeric blends. Biomaterials 24:1167–1173. doi: 10.1016/S0142-9612(02)00466-0. [DOI] [PubMed] [Google Scholar]
- 33.Makadia HK, Siegel SJ. 2011. Poly lactic-co-glycolic acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers 3:1377. doi: 10.3390/polym3031377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Evans DT, Silvestri G. 2013. Nonhuman primate models in AIDS research. Curr Opin HIV AIDS 8:255–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Henning TR, McNicholl JM, Vishwanathan SA, Kersh EN. 2015. Macaque models of enhanced susceptibility to HIV. Virol J 12:90. doi: 10.1186/s12985-015-0320-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Michailidis E, Marchand B, Kodama EN, Singh K, Matsuoka M, Kirby KA, Ryan EM, Sawani AM, Nagy E, Ashida N, Mitsuya H, Parniak MA, Sarafianos SG. 2009. Mechanism of inhibition of HIV-1 reverse transcriptase by 4′-ethynyl-2-fluoro-2′-deoxyadenosine triphosphate, a translocation-defective reverse transcriptase inhibitor. J Biol Chem 284:35681–35691. doi: 10.1074/jbc.M109.036616. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.