

The utility of the azole antifungals for the treatment of invasive candidiasis is severely hampered by azole resistance in Candida glabrata. This resistance is mediated almost exclusively by activating mutations in the zinc cluster transcription factor Pdr1, which controls the genes encoding the multidrug resistance transporters Cdr1, Pdh1, and Snq2.
KEYWORDS: Candida glabrata, antifungal resistance
ABSTRACT
The utility of the azole antifungals for the treatment of invasive candidiasis is severely hampered by azole resistance in Candida glabrata. This resistance is mediated almost exclusively by activating mutations in the zinc cluster transcription factor Pdr1, which controls the genes encoding the multidrug resistance transporters Cdr1, Pdh1, and Snq2. However, the specific relative contributions of these transporters to resistance are not known. To address this question, the SAT1 flipper method was used to delete CDR1, PDH1, and SNQ2 in a strain of C. glabrata engineered to carry a clinically relevant activating mutation in PDR1. Susceptibility testing was performed according to the CLSI guidelines, with minor modifications, and confirmed with Etest strips. Of the single-transporter-deletion strains, only the CDR1 deletion resulted in a decreased azole MIC. The deletion of PDH1 in combination with CDR1 resulted in a moderate decrease in MIC compared to that observed with the deletion of CDR1 alone. SNQ2 deletion only decreased the MIC in the triple-deletion strain in the absence of both CDR1 and PDH1. The deletion of all three transporters in combination decreased the MIC to the level observed in the PDR1 deletion strains for some, but not all, azoles tested, which indicates that additional Pdr1 targets likely play a minor role in this process. These results indicate that while Cdr1 is the most important Pdr1-mediated multidrug resistance transporter for azole resistance in this clinical isolate, all three of these transporters contribute to its high-level resistance to the azole antifungals.
INTRODUCTION
The ATP-binding cassette (ABC) transporters are a large family of clinically relevant proteins found across prokaryotic and eukaryotic organisms. ABC transporters act as drug efflux pumps in Candida glabrata, as well as in the related yeasts Saccharomyces cerevisiae and Candida albicans. Additional members of the ABC transporter family play a role in lipid homeostasis, which may also influence antifungal treatment, as the drug targets are influenced by lipid content in the cell (1).
In C. albicans, the contributions of different mechanisms of resistance to the azole class of antifungals have been well characterized (2). There are two ABC transporters known to contribute to the resistance phenotype in C. albicans, Cdr1 and Cdr2. The deletion of each transporter in a clinical isolate with an activating mutation in the gene encoding the transcription factor Tac1, which results in increased expression of CDR1 and CDR2, established Cdr1 as most important in azole resistance (3). In C. glabrata, resistance in clinical isolates is due primarily to activating mutations in the transcription factor Pdr1 that lead to increased expression of the genes encoding one of three ABC transporters, Cdr1, Pdh1, or Snq2 (4–9). CDR1 was originally found to be upregulated in a resistant clinical isolate of C. glabrata compared to the susceptible paired isolate. The deletion of CDR1 in the resistant isolate resulted in increased susceptibility to the level of the susceptible matched isolate. Reintegration of CDR1 into the deletion mutant restored the azole-resistant phenotype (4). PDH1 was also first implicated when its expression was found to be increased in azole-resistant clinical isolates of C. glabrata (6), and further study showed that the deletion of PDH1 results in increased susceptibility to fluconazole (5). The most recent ABC transporter that has been shown to contribute to azole resistance is Snq2. It was found to have increased expression in two azole-resistant clinical isolates that did not overexpress CDR1 or PDH1 (8) and was subsequently shown in one of the same isolates to be required for the azole-resistant phenotype (9).
In order to overcome azole resistance in C. glabrata, it is first important to thoroughly understand the mechanisms by which it exhibits resistance to the azoles. While it is known that in C. glabrata, the vast majority of clinical resistance has been attributed to upregulation of ABC transporters, the relative importance of each of these remains unclear. Based on previous work in C. glabrata and C. albicans, we predicted that Cdr1 plays a significant role in azole resistance compared to Pdh1 and Snq2. In this study, we used a strain engineered to carry an activating mutation in PDR1 from an azole-resistant clinical isolate that results in the upregulation of all three ABC transporters that have been shown to influence azole susceptibility in C. glabrata. By generating deletion strains of each transporter alone and in combination, we were able to characterize their individual contributions to the phenotype.
RESULTS
Deletion of CDR1, but not PDH1 or SNQ2, alone alters fluconazole susceptibility in a resistant C. glabrata strain.
The SAT1 flipper strategy for gene disruption was used to generate deletion strains in C. glabrata. Single-gene-deletion mutants were made for each ABC transporter known to contribute to azole resistance (CDR1, SNQ2, and PDH1). The azole-resistant strain SM1RPDR1(SM3) was used as the parent strain for the mutants. This strain was made in our laboratory previously by replacing the PDR1 allele in fluconazole susceptible-dose-dependent (SDD) clinical isolate SM1 with the PDR1 allele from azole-resistant isolate SM3, which contains the well-characterized L946S activating mutation (10). Isolate SM1 was recovered from a patient with an abdominal abscess prior to antifungal treatment. SM3 was recovered from an abscess from the same patient 46 days later after a course of fluconazole, followed by caspofungin and then amphotericin B (11).
Susceptibility testing was performed by broth dilution for a panel of azoles, as well as Etest for fluconazole. Both assays confirmed susceptibility data published previously for SM1, SM3, and SM1RPDR1(SM3). The fluconazole MIC for SM1 is 8 μg/ml (Table 1 and Fig. 1A), which would be considered susceptible-dose dependent according to the most recent CLSI breakpoints. The fluconazole MIC for both SM3 and SM1RPDR1(SM3) is 256 μg/ml. The deletion of CDR1 in strain SM1RPDR1(SM3) resulted in a decreased fluconazole MIC from 256 μg/ml to 16 μg/ml. However, neither PDH1 nor SNQ2 single-deletion mutants exhibited a change in fluconazole MIC. Fluconazole Etest susceptibility testing confirmed the results from the broth microdilution assay (Fig. 1C). The same trend was seen with additional azoles tested (Table 1). Among single-ABC-transporter-deletion mutants, CDR1 alone was able to alter azole susceptibility.
TABLE 1.
Azole susceptibilities performed by broth microdilution for selected strains
| Strain | MIC50 (μg/ml) by antifungala |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| FLC |
ITC |
KTC |
MIC |
VRC |
||||||
| 24 h | 48 h | 24 h | 48 h | 24 h | 48 h | 24 h | 48 h | 24 h | 48 h | |
| SM1 | 8 | 8 | 0.5 | 0.5 | 0.125 | 0.063 | 0.125 | 0.125 | 0.125 | 0.125 |
| SM3 | 128 | 256 | 32 | 32 | 2 | 4 | 1 | 1 | 4 | 8 |
| SM1RPDR1(SM3) | 128 | 256 | 32 | 32 | 2 | 4 | 1 | 1 | 4 | 8 |
| Δcdr1 mutant | 16 | 16 | 1 | 0.5 | 0.25 | 0.25 | 0.125 | 0.25 | 0.125 | 0.25 |
| Δpdh1 mutant | 128 | 256 | 32 | 32 | 2 | 4 | 1 | 1 | 4 | 8 |
| Δsnq2 mutant | 128 | 256 | 32 | 32 | 2 | 4 | 1 | 1 | 4 | 8 |
| Δcdr1 Δpdh1 mutant | 8 | 8 | 0.125 | 0.125 | ≤0.031 | ≤0.031 | ≤0.016 | 0.063 | 0.031 | 0.063 |
| Δcdr1 Δsnq2 mutant | 16 | 16 | 0.5 | 0.5 | 0.125 | 0.25 | 0.125 | 0.25 | 0.125 | 0.25 |
| Δpdh1 Δsnq2 mutant | 128 | 256 | 32 | 32 | 2 | 4 | 1 | 1 | 4 | 8 |
| Δcdr1 Δpdh1 Δsnq2 mutant | 4 | 2 | 0.125 | 0.125 | ≤0.031 | ≤0.031 | ≤0.016 | ≤0.016 | 0.031 | 0.031 |
Values are representative data from experiments completed in duplicate. FLC, fluconazole; ITC, itraconazole; KTC, ketoconazole; MIC, miconazole; VRC, voriconazole.
FIG 1.

Altered fluconazole sensitivities in strains lacking the genes encoding the ABC transporters CDR1, PDH1, and SNQ2. (A and B) Fluconazole susceptibilities for the parent and single-deletion strains (A) or the parent, double-, and triple-deletion strains (B) for the ABC transporters CDR1, PDH1, and SNQ2, were measured by broth microdilution as described. Absorbance at 600 nm was measured after incubation at 35°C for 48 h. Background due to medium alone was subtracted, and data were plotted as percentage of the no-drug control. Experiments were completed in triplicate. (C) Susceptibility testing was performed using Etest strips, as described. Images were taken at 48 h. Experiments were performed in duplicate, with representative images shown.
PDH1 and SNQ2 contribute to fluconazole resistance to a lesser extent than CDR1.
Previous studies have demonstrated a role for Pdh1 and Snq2 in resistance to azoles. However, we show here that the deletion of PDH1 or SNQ2 in an azole-resistant strain overexpressing all three transporters resulted in no change in fluconazole susceptibility (Table 1 and Fig. 1A). In order to determine if PDH1 and SNQ2 impact fluconazole resistance in the absence of CDR1, double- and triple-deletion strains were made using the SAT1 flipper method described above.
When PDH1 and CDR1 were deleted in combination, the MIC was consistently one doubling dilution lower than the MIC for the strain in which CDR1 alone is deleted (Table 1 and Fig. 1B). When SNQ2 and CDR1 were deleted in combination, the MIC was the same as that observed for the CDR1 single-deletion strain. To investigate the possibility that the presence of PDH1 is masking the effect of SNQ2, we constructed a strain with all three transporters deleted. The MIC for the triple-deletion strain was consistently one doubling dilution less than that of the CDR1 and PDH1 deletion strain. The patterns seen by broth microdilution were confirmed by Etest susceptibility testing (Fig. 1C).
CDR1, PDH1, and SNQ2 mutants exhibit similar susceptibility patterns to other azole antifungals.
Of interest is the potential for these transporters to affect members of the azole class of antifungals differently. Newer triazoles in the class have improved spectra of activity against additional fungal pathogens. The differences in chemical structure could influence the interaction with efflux pumps, resulting in different ABC transporters preferentially effluxing different azoles. However, overall, the patterns for all azoles tested are similar, with few differences (Table 1). The magnitude of changes in MIC are different among the azoles for the different ABC transporters. In general, however, the differences from the parent strain and the triple-ABC-transporter-deletion strain were 6 to 8 drug dilutions for all azoles tested. Among the single-deletion mutants, Cdr1 was the only transporter able to influence the MIC for all azoles tested. Interestingly, the deletion of SNQ2 had no impact on itraconazole or ketoconazole MIC. No notable differences were observed in the susceptibilities to echinocandins and amphotericin B in the mutant strains (data not shown).
CDR1, PDH1, and SNQ2 explain most, but not all, of Pdr1's contribution to azole resistance.
All three ABC transporters in this study are known targets of Pdr1. All three transporters are upregulated in strains that possess an activating mutation in PDR1 (9, 10, 12). Pdr1 has also been shown to bind to the promoter regions of each transporter by chromatin immunoprecipitation sequencing (ChIP-seq) (13). In order to determine if these three transporters account for the entirety of Pdr1's importance to azole resistance, we compared PDR1 deletion strains to the triple-ABC-transporter-deletion strain. Both the susceptible-dose-dependent and resistant clinical isolates lacking PDR1 exhibit increased sensitivity to fluconazole with a treatment of 2 μg/ml, resulting in >95% growth reduction (Fig. 2). The triple-deletion mutant, however, required treatment with 8 μg/ml fluconazole to exhibit a similar reduction in growth. This indicates that there may be additional genes of the Pdr1 regulon that contribute to azole resistance.
FIG 2.
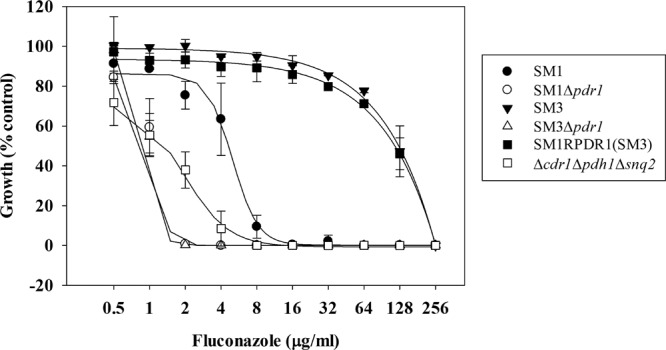
Altered fluconazole sensitivities to strains lacking the genes encoding all three relevant ABC transporters compared to strains lacking the gene encoding the transcription factor Pdr1. Fluconazole susceptibility was measured by broth microdilution, as described. Absorbance at 600 nm was measured after incubation at 35°C for 48 h. Background due to medium alone was subtracted, and data were plotted as percentage of the no-drug control. Experiments were performed in triplicate.
DISCUSSION
Activating mutations in the zinc cluster transcription factor Pdr1 that result in upregulation of ABC transporters are found in the vast majority of azole-resistant clinical isolates of C. glabrata. Similarly to the transcription factors Pdr1 and Pdr3 in the closely related yeast S. cerevisiae (14), Pdr1 in C. glabrata exerts its regulatory effects by binding a pleiotropic drug response element (PDRE) (13, 15). Pdr1 is also autoregulated due to a PDRE in its promoter region (15). Developing a better understanding of this mechanism of azole resistance is important for discovering new treatment strategies. For example, a small molecule that could inhibit efflux by the ABC transporters would allow clinicians to once again successfully treat C. glabrata with azoles.
Many different activating mutations have been found throughout the coding sequence for PDR1 that result in similar decreases in azole susceptibility (10, 12, 15–18). However, there are differences in the gene expression patterns of the Pdr1 regulon for the various mutations. Of particular interest are the patterns of expression of the three ABC transporters known to influence azole susceptibility, which do not appear to correlate with the location of the mutation (12).
ABC transporter expression profiles in clinical isolates reveal a wide distribution of expression patterns. CDR1, PDH1, or both CDR1 and PDH1 were found to be highly expressed in 18 of 20 resistant clinical isolates from one study. The 2 remaining resistant isolates overexpressed SNQ2 alone. The susceptible-dose-dependent isolates overexpressed the ABC transporters to a lesser extent than the resistant isolates (8). Another group showed that 10 of 12 resistant clinical isolates and 6 of 7 laboratory-generated resistant mutants exhibited increased expression of CDR1 or both CDR1 and PDH1 (19). A third study of resistant clinical isolates found CDR1 or PDH1 overexpression in the resistant isolate of 12 of 14 matched clinical isolates (20). A study in which the expression of CDR1, PDH1, and PDR1 was measured in a panel of clinical isolates, with the purpose of developing a quantitative PCR (qPCR)-based assay for the determination of resistance in C. glabrata, found that CDR1 expression alone could be used to predict resistance with 100% sensitivity and 95% specificity. The same was not true for PDH1 expression or PDR1 expression (21).
One might predict the other ABC transporters to overcome the absence of one or two transporters and maintain a high level of resistance. C. albicans, which has only two ABC transporters that have been shown to contribute to the azole-resistant phenotype, does not exhibit compensatory upregulation of the other ABC transporter when one is absent (3). In S. cerevisiae, the deletion of SNQ2 and YOR1 leads to enhanced activity of fluconazole and ketoconazole; however, the SNQ2-YOR1-PDR5 triple-deletion strain demonstrates enhanced sensitivity to the azoles tested. Protein and mRNA levels of expression indicate that there is compensatory upregulation with different combinations of these ABC transporter deletion mutants (22).
The phenomena seen in this study with the deletion of PDH1 and SNQ2 not having a phenotypic effect in the presence of CDR1 or CDR1 and PDH1, respectively, suggests a possible interaction between these ABC transporters. Work in S. cerevisiae would indicate that this is likely. Membrane yeast two-hybrid (MYTH) technology demonstrated a physical interaction between Pdr5 (homolog for C. glabrata Cdr1) and Snq2; however, no changes in protein level, transcript level, or localization of Snq2 in the PDR5 deletion strain were detected (23). Snq2 and Pdr5 in S. cerevisiae have also demonstrated a synergistic effect on estradiol transport. The deletion of SNQ2 alone has no effect on estradiol accumulation, but when deleted in conjunction with PDR5, there is a dramatic increase in intracellular estradiol (24). Pdr5 and Pdr15 (homolog for C. glabrata Pdh1) showed similar additive sensitivity to 2,4-dichlorophenol and various membrane-altering nonionic detergents, such that the double-deletion strains demonstrated marked increased sensitivity compared to the single-deletion strains (25).
All three ABC transporters are found in the plasma membrane. The alteration of their expression alone would affect the composition of the plasma membrane and potentially alter azole uptake in this way. The deletion of PDR5 and PDR15 in S. cerevisiae results in an increase in the amount of phosphatidylethanolamine exposed in the plasma membrane (25). The mechanism by which azoles are taken up into the cell is not well understood at this time, but alteration of the membrane could influence this as-yet-unknown process.
Our experiments show that the three ABC transporters previously implicated in Pdr1-mediated azole resistance in C. glabrata are not able to fully explain the decrease in susceptibility in the absence of Pdr1. In addition to controlling the expression of ABC transporters, Pdr1 also regulates the expression of many other genes both directly and indirectly. We predict that there likely are genes among this group that make small contributions to azole resistance that in combination may explain the phenotypic difference between the PDR1 deletion strain and the triple-ABC-transporter-deletion strain.
Among the list of genes whose promoters were found to be directly bound by Pdr1 are a major facilitator superfamily (MFS) transporter, QDR2, and two additional ABC transporters, YBT1 and YOR1 (13). The deletion of QDR2 in C. glabrata resulted in increased susceptibility to the imidazoles, clotrimazole, ketoconazole, and miconazole but had no effect on fluconazole or itraconazole susceptibility (26). YOR1 and YBT1 are frequently found among genes with altered expression in relation to azole susceptibility, but a direct relationship has not been described. The effects of QDR2, YOR1, and YBT1 on fluconazole susceptibility could potentially be masked by the presence of the more dominant transporters, CDR1, PDH1, and SNQ2, and therefore cannot be ruled out as contributing to azole resistance, albeit likely a minor contribution.
Other genes of interest shown previously to be upregulated in clinical isolates (10, 17) and laboratory-derived strains (27) of C. glabrata with activating mutations in PDR1 are RSB1 and RTA1. Rsb1 is a putative sphingolipid flippase. The homolog for Rta1 in S. cerevisiae is a member of the fungal lipid-translocating exporter family of proteins. The promoter regions of both RSB1 and RTA1 possess a PDRE that is bound directly by Pdr1 (15). Recent work in C. albicans has implicated the putative lipid translocase encoded by RTA3 and the sphingolipid flippase encoded by RTA2 in azole resistance (28, 29). Rsb1 and Rta1 may make similar contributions to azole resistance in C. glabrata.
MATERIALS AND METHODS
Strains and growth media.
All strains used in this study are listed in Table 2. The clinical isolates and the PDR1 replacement strain have been described previously (10, 11). All strains were stored as frozen stocks at −80°C with 40% glycerol. Strains were routinely grown in YPD broth (1% yeast extract, 2% peptone, and 2% dextrose) at 30°C in a shaking incubator, except as indicated for specific experimental conditions.
TABLE 2.
Strains used in this study
| Strain | Parent | Genotype or description | Reference |
|---|---|---|---|
| SM1 | Azole-SDD clinical isolate | 11 | |
| SM3 | Azole-resistant clinical isolate | 11 | |
| SM1RPDR1(SM3) | SM1 Δpdr1 | pdr1Δ::FRT-PDR1SM3 | 10 |
| S1RPS3CDR1M2A and S1RPS3CDR1M2B | SM1RPDR1(SM3) | pdr1Δ::FRT-PDR1SM3/cdr1Δ::FRT | This study |
| S1RPS3PDH1M2A and S1RPS3PDH1M2B | SM1RPDR1(SM3) | pdr1Δ::FRT-PDR1SM3/pdh1Δ::FRT | This study |
| S1RPS3SNQ2M2A and S1RPS3SNQ2M2B | SM1RPDR1(SM3) | pdr1Δ::FRT-PDR1SM3/snq2Δ::FRT | This study |
| S1RPS3CAPDH1M2A | S1RPS3CDR1M2A | pdr1Δ::FRT-PDR1SM3/cdr1Δ::FRT/pdh1Δ::FRT | This study |
| S1RPS3CBPDH1M2A | S1RPS3CDR1M2B | pdr1Δ::FRT-PDR1SM3/cdr1Δ::FRT/pdh1Δ::FRT | This study |
| S1RPS3CASNQ2M2A | S1RPS3CDR1M2A | pdr1Δ::FRT-PDR1SM3/cdr1Δ::FRT/snq2Δ::FRT | This study |
| S1RPS3CBSNQ2M2A | S1RPS3CDR1M2B | pdr1Δ::FRT-PDR1SM3/cdr1Δ::FRT/snq2Δ::FRT | This study |
| S1RPS3SAPDH1M2A | S1RPS3SNQ2M2A | pdr1Δ::FRT-PDR1SM3/pdh1Δ::FRT/snq2Δ::FRT | This study |
| S1RPS3SBPDH1M2A | S1RPS3SNQ2M2B | pdr1Δ::FRT-PDR1SM3/pdh1Δ::FRT/snq2Δ::FRT | This study |
| S1RPS3CAPASNQ2M2A | S1RPS3CAPDH1M2A | pdr1Δ::FRT-PDR1SM3/cdr1Δ::FRT/pdh1Δ::FRT/snq2Δ::FRT | This study |
| S1RPS3CBPASNQ2M2A | S1RPS3CBPDH1M2A | pdr1Δ::FRT-PDR1SM3/cdr1Δ::FRT/pdh1Δ::FRT/snq2Δ::FRT | This study |
Escherichia coli PX5-α chemically competent cells (Protein Express, Cincinnati, OH) were used as the host for plasmid construction and propagation. These strains were grown at 37°C in Luria-Bertani (LB) broth or on LB plates supplemented with 100 μg/ml ampicillin (Sigma, St. Louis, MO) or 50 μg/ml kanamycin (Fisher BioReagents, Fair Lawn, NJ).
Plasmid construction.
For the deletion of CDR1, PDH1, and SNQ2, we modified plasmid pSFS2 (30). Upstream homology regions approximately 800 to 1,000 bp long were amplified using primer pairs CgCDR1A/CgCDR1B, CgPDH1/CgPDH1B, or CgSNQ2A/CgSNQ2B and digested with ApaI/XhoI or KpnI/ApaI as indicated by underlined bases for insertion into their respective plasmids. Downstream homology regions approximately 800 to 1,000 bp long were amplified using primer pairs CgCDR1C/CgCDR1D, CgPDH1C/CgPDH1D, or CgSNQ2C/CgSNQ2D and digested with SacII/SacI or NotI/SacII as indicated by underlined bases for insertion into their respective plasmids. The disruption cassettes consisting of the SAT1 flipper cassette and upstream and downstream flanking sequences of either CDR1, PDH1, or SNQ2 were excised from the final plasmid pCgCDR1, pCgPDH1, or pCgSNQ2 and gel purified. The primers used to construct the cassettes are listed in Table 3.
TABLE 3.
Primers used in this study
| Primer name | Primer sequence (5′–3′)a |
|---|---|
| CgCDR1A | CATAGATCAGGGCCCATTACATTAGCACAG |
| CgCDR1B | CTCAGTGTTGCTCGAGATAGGGTTGATAC |
| CgCDR1C | GTTCTGTTAGTTCCGCGGACTCTCGTAGAT |
| CgCDR1D | GTGAATACAAACAAGAGCTCCACAATAATA |
| CgPDH1A | AAACAGTCTATGGTACCACAAGTTTGCACA |
| CgPDH1B | CCGTATACGTTTCGGGCCCTTGTCATCAA |
| CgPDH1C | ACAGAAGATGCGGCCGCTATGGTATATTTATT |
| CgPDH1D | ATTCCTTAATAACCGCGGAAGTTGACTTTA |
| CgSNQ2A | TTGAGTATCTTAGGGCCCTTGTTTTCAGTT |
| CgSNQ2B | GATAGAATACTCGAGTTGTCGCTGTGCGC |
| CgSNQ2C | GCTATTTATTACCGCGGCCATGTCAGAG |
| CgSNQ2D | AGACAGATATTGAGCTCCACTACTGCTGAG |
Underlined bases indicate the introduction of restriction enzyme cloning sites to allow directional cloning into the SAT1-flipper cassette.
Strain construction.
C. glabrata cells were transformed by the lithium acetate method using approximately 1 μg of DNA. The ApaI/SacI fragments from pCgCDR1 and pCgSNQ2 and the KpnI/SacII fragment from pCgPDH1 were excised and gel purified prior to transformation. The transformed cells were allowed to recover for 6 h in YPD at 30°C before being plated on YPD agar plates containing 200 μg/ml nourseothricin (Jena Biochemical, Germany) and incubated at 30°C. Positive transformants were selected within 24 h, and successful insertion of the disruption cassette at the target gene locus was confirmed by Southern hybridization using gene-specific probes. Subsequently, induction of the flipper recombinase gene in the disruption cassette was performed by overnight growth of the positive transformant clones in YPD at 30°C with shaking (under no selective pressure). Selection for excision of the SAT1 flipper cassette was then performed by plating on YPD agar plates and incubating for up to 24 h at 30°C. Clones were selected and confirmed by Southern hybridization using gene-specific probes.
Isolation of genomic DNA and Southern hybridization.
Genomic DNA from C. glabrata was isolated as described previously (31). For confirmation by Southern hybridization, approximately 10 μg of genomic DNA was digested with the appropriate restriction enzymes, separated on a 1% agarose gel containing ethidium bromide, transferred by vacuum blotting onto a nylon membrane, and fixed by UV-cross-linking. Hybridization was performed with the Amersham AlkPhos direct labeling and detection system (GE Healthcare, Pittsburgh, PA), as per the manufacturer's instructions.
Susceptibility testing.
Susceptibility testing was performed by broth microdilution assay and by Epsilometer test strips (Etest). Broth microdilution was performed according to the CLSI guidelines outlined in document M27-A3, with a few modifications (32). A fluconazole (MP Biomedicals, Solon, OH) stock solution was prepared by reconstitution in water to 5 mg/ml. Itraconazole (0.5 mg/ml), ketoconazole (10 mg/ml), miconazole (10 mg/ml), and voriconazole (10 mg/ml) (Sigma, St. Louis, MO) stock solutions were prepared by reconstitution in dimethyl sulfoxide (DMSO; Sigma) at the indicated concentrations. Colonies grown overnight on Sabouraud dextrose plates were diluted to 2.5 × 103 cells/ml in RPMI 1640 (Sigma) with 2% glucose and morpholinepropanesulfonic acid (MOPS) (pH 7.0). Plates were incubated at 35°C for 48 h. Absorbance at 600 nm was read with a BioTek Synergy 2 microplate reader (BioTek, Winooski, VT); background due to medium was subtracted from all readings. The MIC was defined as the lowest concentration inhibiting growth by at least 50% relative to the drug-free control after incubation with drug for 24 or 48 h. Fluconazole broth microdilution susceptibility testing was performed in triplicate, and additional azole broth microdilution susceptibility testing was performed in duplicate. Fluconazole Etest (bioMérieux) susceptibility assay was performed as per the manufacturer's instructions, with some modifications, in triplicate. Colonies were selected from cultures grown overnight on Sabouraud dextrose plates and diluted in water to a 0.5 McFarland standard. Sterile cotton swabs were used to streak plates prior to Etest strip placement. Plates were incubated at 35°C and read at 24 and 48 h.
ACKNOWLEDGMENTS
This work was supported by NIH NIAID grants R01AI058145 (to P.D.R.) and R01AI131620 (to P.D.R.).
REFERENCES
- 1.Prasad R, Khandelwal NK, Banerjee A. 2016. Yeast ABC transporters in lipid trafficking. Fungal Genet Biol 93:25–34. doi: 10.1016/j.fgb.2016.05.008. [DOI] [PubMed] [Google Scholar]
- 2.MacCallum DM, Coste A, Ischer F, Jacobsen MD, Odds FC, Sanglard D. 2010. Genetic dissection of azole resistance mechanisms in Candida albicans and their validation in a mouse model of disseminated infection. Antimicrob Agents Chemother 54:1476–1483. doi: 10.1128/AAC.01645-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tsao S, Rahkhoodaee F, Raymond M. 2009. Relative contributions of the Candida albicans ABC transporters Cdr1p and Cdr2p to clinical azole resistance. Antimicrob Agents Chemother 53:1344–1352. doi: 10.1128/AAC.00926-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sanglard D, Ischer F, Calabrese D, Majcherczyk PA, Bille J. 1999. The ATP binding cassette transporter gene CgCDR1 from Candida glabrata is involved in the resistance of clinical isolates to azole antifungal agents. Antimicrob Agents Chemother 43:2753–2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sanglard D, Ischer F, Bille J. 2001. Role of ATP-binding-cassette transporter genes in high-frequency acquisition of resistance to azole antifungals in Candida glabrata. Antimicrob Agents Chemother 45:1174–1183. doi: 10.1128/AAC.45.4.1174-1183.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miyazaki H, Miyazaki Y, Geber A, Parkinson T, Hitchcock C, Falconer DJ, Ward DJ, Marsden K, Bennett JE. 1998. Fluconazole resistance associated with drug efflux and increased transcription of a drug transporter gene, PDH1, in Candida glabrata. Antimicrob Agents Chemother 42:1695–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Izumikawa K, Kakeya H, Tsai HF, Grimberg B, Bennett JE. 2003. Function of Candida glabrata ABC transporter gene, PDH1. Yeast 20:249–261. doi: 10.1002/yea.962. [DOI] [PubMed] [Google Scholar]
- 8.Sanguinetti M, Posteraro B, Fiori B, Ranno S, Torelli R, Fadda G. 2005. Mechanisms of azole resistance in clinical isolates of Candida glabrata collected during a hospital survey of antifungal resistance. Antimicrob Agents Chemother 49:668–679. doi: 10.1128/AAC.49.2.668-679.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Torelli R, Posteraro B, Ferrari S, La Sorda M, Fadda G, Sanglard D, Sanguinetti M. 2008. The ATP-binding cassette transporter-encoding gene CgSNQ2 is contributing to the CgPDR1-dependent azole resistance of Candida glabrata. Mol Microbiol 68:186–201. doi: 10.1111/j.1365-2958.2008.06143.x. [DOI] [PubMed] [Google Scholar]
- 10.Caudle KE, Barker KS, Wiederhold NP, Xu L, Homayouni R, Rogers PD. 2011. Genomewide expression profile analysis of the Candida glabrata Pdr1 regulon. Eukaryot Cell 10:373–383. doi: 10.1128/EC.00073-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Magill SS, Shields C, Sears CL, Choti M, Merz WG. 2006. Triazole cross-resistance among Candida spp.: case report, occurrence among bloodstream isolates, and implications for antifungal therapy. J Clin Microbiol 44:529–535. doi: 10.1128/JCM.44.2.529-535.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferrari S, Ischer F, Calabrese D, Posteraro B, Sanguinetti M, Fadda G, Rohde B, Bauser C, Bader O, Sanglard D. 2009. Gain of function mutations in CgPDR1 of Candida glabrata not only mediate antifungal resistance but also enhance virulence. PLoS Pathog 5:e1000268. doi: 10.1371/journal.ppat.1000268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paul S, Bair TB, Moye-Rowley WS. 2014. Identification of genomic binding sites for Candida glabrata Pdr1 transcription factor in wild-type and rho0 cells. Antimicrob Agents Chemother 58:6904–6912. doi: 10.1128/AAC.03921-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Katzmann DJ, Hallstrom TC, Mahe Y, Moye-Rowley WS. 1996. Multiple Pdr1p/Pdr3p binding sites are essential for normal expression of the ATP binding cassette transporter protein-encoding gene PDR5. J Biol Chem 271:23049–23054. doi: 10.1074/jbc.271.38.23049. [DOI] [PubMed] [Google Scholar]
- 15.Paul S, Schmidt JA, Moye-Rowley WS. 2011. Regulation of the CgPdr1 transcription factor from the pathogen Candida glabrata. Eukaryot Cell 10:187–197. doi: 10.1128/EC.00277-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsai HF, Krol AA, Sarti KE, Bennett JE. 2006. Candida glabrata PDR1, a transcriptional regulator of a pleiotropic drug resistance network, mediates azole resistance in clinical isolates and petite mutants. Antimicrob Agents Chemother 50:1384–1392. doi: 10.1128/AAC.50.4.1384-1392.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsai HF, Sammons LR, Zhang X, Suffis SD, Su Q, Myers TG, Marr KA, Bennett JE. 2010. Microarray and molecular analyses of the azole resistance mechanism in Candida glabrata oropharyngeal isolates. Antimicrob Agents Chemother 54:3308–3317. doi: 10.1128/AAC.00535-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gohar AA, Badali H, Shokohi T, Nabili M, Amirrajab N, Moazeni M. 2017. Expression patterns of ABC transporter genes in fluconazole-resistant Candida glabrata. Mycopathologia 182:273–284. doi: 10.1007/s11046-016-0074-8. [DOI] [PubMed] [Google Scholar]
- 19.Vermitsky JP, Edlind TD. 2004. Azole resistance in Candida glabrata: coordinate upregulation of multidrug transporters and evidence for a Pdr1-like transcription factor. Antimicrob Agents Chemother 48:3773–3781. doi: 10.1128/AAC.48.10.3773-3781.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bennett JE, Izumikawa K, Marr KA. 2004. Mechanism of increased fluconazole resistance in Candida glabrata during prophylaxis. Antimicrob Agents Chemother 48:1773–1777. doi: 10.1128/AAC.48.5.1773-1777.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gygax SE, Vermitsky JP, Chadwick SG, Self MJ, Zimmerman JA, Mordechai E, Adelson ME, Trama JP. 2008. Antifungal resistance of Candida glabrata vaginal isolates and development of a quantitative reverse transcription-PCR-based azole susceptibility assay. Antimicrob Agents Chemother 52:3424–3426. doi: 10.1128/AAC.00462-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kolaczkowska A, Kolaczkowski M, Goffeau A, Moye-Rowley WS. 2008. Compensatory activation of the multidrug transporters Pdr5p, Snq2p, and Yor1p by Pdr1p in Saccharomyces cerevisiae. FEBS Lett 582:977–983. doi: 10.1016/j.febslet.2008.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Snider J, Hanif A, Lee ME, Jin K, Yu AR, Graham C, Chuk M, Damjanovic D, Wierzbicka M, Tang P, Balderes D, Wong V, Jessulat M, Darowski KD, San Luis BJ, Shevelev I, Sturley SL, Boone C, Greenblatt JF, Zhang Z, Paumi CM, Babu M, Park HO, Michaelis S, Stagljar I. 2013. Mapping the functional yeast ABC transporter interactome. Nat Chem Biol 9:565–572. doi: 10.1038/nchembio.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mahé Y, Lemoine Y, Kuchler K. 1996. The ATP binding cassette transporters Pdr5 and Snq2 of Saccharomyces cerevisiae can mediate transport of steroids in vivo. J Biol Chem 271:25167–25172. doi: 10.1074/jbc.271.41.25167. [DOI] [PubMed] [Google Scholar]
- 25.Schüller C, Mamnun YM, Wolfger H, Rockwell N, Thorner J, Kuchler K. 2007. Membrane-active compounds activate the transcription factors Pdr1 and Pdr3 connecting pleiotropic drug resistance and membrane lipid homeostasis in Saccharomyces cerevisiae. Mol Biol Cell 18:4932–4944. doi: 10.1091/mbc.e07-06-0610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Costa C, Pires C, Cabrito TR, Renaudin A, Ohno M, Chibana H, Sa-Correia I, Teixeira MC. 2013. Candida glabrata drug:H+ antiporter CgQdr2 confers imidazole drug resistance, being activated by transcription factor CgPdr1. Antimicrob Agents Chemother 57:3159–3167. doi: 10.1128/AAC.00811-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vermitsky JP, Earhart KD, Smith WL, Homayouni R, Edlind TD, Rogers PD. 2006. Pdr1 regulates multidrug resistance in Candida glabrata: gene disruption and genome-wide expression studies. Mol Microbiol 61:704–722. doi: 10.1111/j.1365-2958.2006.05235.x. [DOI] [PubMed] [Google Scholar]
- 28.Whaley SG, Tsao S, Weber S, Zhang Q, Barker KS, Raymond M, Rogers PD. 2016. The RTA3 gene, encoding a putative lipid translocase, influences the susceptibility of Candida albicans to fluconazole. Antimicrob Agents Chemother 60:6060–6066. doi: 10.1128/AAC.00732-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jia XM, Ma ZP, Jia Y, Gao PH, Zhang JD, Wang Y, Xu YG, Wang L, Cao YY, Cao YB, Zhang LX, Jiang YY. 2008. RTA2, a novel gene involved in azole resistance in Candida albicans. Biochem Biophys Res Commun 373:631–636. doi: 10.1016/j.bbrc.2008.06.093. [DOI] [PubMed] [Google Scholar]
- 30.Reuss O, Vik A, Kolter R, Morschhauser J. 2004. The SAT1 flipper, an optimized tool for gene disruption in Candida albicans. Gene 341:119–127. doi: 10.1016/j.gene.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 31.Amberg DC, Burke DJ, Strathern JN. 2006. Isolation of yeast genomic DNA for southern blot analysis. CSH Protoc 2006:pdb.prot4149. doi: 10.1101/pdb.prot4149. [DOI] [PubMed] [Google Scholar]
- 32.CLSI. 2008. Reference method for broth dilution antifungal susceptibility testing of yeasts. Approved standard, 3rd. CLSI document M27-A3. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]