

Abstract
Dysregulation of L-type Ca2+ channels (LTCCs) underlies numerous cardiac pathologies. Understanding their modulation with high fidelity relies on investigating LTCCs in their native environment with intact interacting proteins. Such studies benefit from genetic manipulation of endogenous channels in cardiomyocytes, which often proves cumbersome in mammalian models. Drosophila melanogaster, however, offers a potentially efficient alternative as it possesses a relatively simple heart, is genetically pliable, and expresses well-conserved genes. Fluorescence in situ hybridization confirmed an abundance of Ca-α1D and Ca-α1T mRNA in fly myocardium, which encode subunits that specify hetero-oligomeric channels homologous to mammalian LTCCs and T-type Ca2+ channels, respectively. Cardiac-specific knockdown of Ca-α1D via interfering RNA abolished cardiac contraction, suggesting Ca-α1D represents the primary functioning Ca2+ channel in Drosophila hearts. Moreover, we successfully isolated viable single cardiomyocytes and recorded Ca2+ currents via patch clamping, a feat never before accomplished with the fly model. The profile of Ca2+ currents recorded in individual cells when Ca2+ channels were hypomorphic, absent, or under selective LTCC blockage by nifedipine, additionally confirmed the predominance of Ca-α1D current across all activation voltages. T-type current, activated at more negative voltages, was also detected. Lastly, Ca-α1D channels displayed Ca2+-dependent inactivation, a critical negative feedback mechanism of LTCCs, and the current through them was augmented by forskolin, an activator of the protein kinase A pathway. In sum, the Drosophila heart possesses a conserved compendium of Ca2+ channels, suggesting that the fly may serve as a robust and effective platform for studying cardiac channelopathies.
Introduction
Cardiac action potentials, which drive rhythmic contractions of the heart, are the result of well-choreographed opening and closing of multiple ion channels. Among these are the L-type Ca2+ channels (LTCCs). LTCCs not only permit Ca2+ entry to initiate myocyte shortening, but also set the length of the plateau phase of the cardiac action potential and, thereby, determine its duration [1]. Disruption of these critical channels or their precise regulation underlies numerous pathologies. For example, mutations in CaV1.2, a major LTCC in heart muscle, lead to Timothy syndrome, a multi-system disorder featuring autism, polydactyly, and long-QT syndrome [2-5], while mutations in calmodulin, a regulator of CaV1.2 Ca2+-dependent inactivation (CDI), result in a malignant form of long-QT syndrome [6-9]. Mutations in CaV1.3, an LTCC in nodal cells, can lead to bradycardia and congenital deafness [10, 11]. Additionally, changes in channel density and function are associated with atrial fibrillation [12, 13] and heart failure [14, 15].
Mechanistic dissection of alterations in LTCC regulation is typically conducted using recombinant channels expressed in a heterologous system. Unfortunately, these systems often lack key auxiliary elements that are readily available in the context of cardiac myocytes. Studying Ca2+channel regulation, including CDI and protein kinase A (PKA)-mediated current augmentation in native mammalian myocytes is, however, also challenging due to cellular complexity, including redundancy among genes, and the difficulty of endogenously manipulating the channels. Thus, an intermediate system, which is both genetically pliable and reflective of a cardiac muscle cell, is desirable.
Cardiomyocytes from Drosophila melanogaster, the fruit fly, offer an attractive, yet incompletely explored alternative. Drosophila benefit from a completely sequenced genome, conservation of disease orthologs [16-19], and a host of genetic tools [16, 19-21]. For example, the bipartite Gal4-UAS expression system [22] permits straightforward control of gene expression across space (by means of tissue-specific promoters) and time (via drug- or temperature-inducible expression) [23]. This system readily supports investigation of gain- or loss-of-function of genes of interest, owing to large resources and libraries including thousands of independent GAL4 and UAS Drosophila lines that permit selective transgene overexpression or RNAi (interfering RNA)-mediated gene silencing [24, 25]. Importantly, mutant genes can easily be studied in the same genetic environment as their wild-type counterparts, thus minimizing confounding effects, such as those that may result from insertion of transgenes in different locations throughout the genome [26-28]. Many Drosophila proteins are encoded by a single gene that generates multiple distinct isoforms through alternative splicing of the primary transcript, simplifying knockout and/or gene suppression experiments [21]. Severe manipulation of heart components can also be tolerated because oxygen transport occurs through trachea that invaginate from the cuticle into the interior of the fly [29]. The effects of potentially lethal cardiac mutations can therefore be studied in vivo without necessarily initiating death. Furthermore, the recent development of techniques to image [30-35] and record electrical field potentials [34, 36] of both larval [32-35] and adult [30-33, 36] hearts allows functional assessment of different developmental stages of Drosophila myocardium after manipulation of particular genes. Finally, flies are economical and easy to breed, generate numerous offspring, and feature a short life cycle, making large-scale genetic and small-compound screens, in wild-type and disease models, eminently feasible.
Drosophila have an open circulatory system with a dorsal vessel or “cardiac tube” (Figure 1A), which in many ways functionally and developmentally resembles the embryonic vertebrate heart [21, 37, 38]. The tube is composed of a single layer of two opposing rows of cardiac myocytes [35, 39] whose action potential, as found in higher organisms, is myogenic in origin, i.e. action potentials originate from muscle itself as opposed to in response to neuronal impulses (neurogenic origin) [40-42]. The activity of these myocytes is modulated by a neurohormonal system that features the epinephrine-like compound, octopamine [43]. Action potentials of Drosophila heart tubes have been coarsely measured [34, 36, 44, 45], and the voltage waveforms appear comparable in duration and amplitude to those recorded in vertebrate myocytes. As LTCCs play a major role in shaping vertebrate cardiac action potentials, it is plausible that a Drosophila analog is playing a similar role in the heart tube. Like vertebrates, flies express hetero-oligomeric voltage gated Ca2+ channels (CaV) composed of α1, β, α2δ, and, in some tissues, ϒ subunits [17, 46, 47]. Three distinct genes, Ca-α1D (CG4894), cacophony (CG43368), and Ca-α1T (CG15899) encode the α1-subunits A1D, cac, and T-type, which specify three CaV that correspond to CaV1, CaV2, and CaV3, the main Ca2+ channel families in vertebrates (Supplementary Table 1). Drosophila A1D is similar to the dihydropyridine-sensitive (L-type) channels of vertebrates; cac to N-, P/Q-, R-type channels; and T-type to CaV3 channels [17]. RNA microarray data demonstrate enrichment of Ca-α1D in the hearts of adult fruit flies compared to the whole body, hinting at the potential presence of A1D in Drosophila cardiac tubes [16, 48]. Still, the complete molecular biosignature of heart-tube Ca2+ channels is unknown.
Figure 1. Ca-α1D and Ca-α1T transcripts are abundant in Drosophila heart tubes.

A) Confocal micrograph of a semi-intact wild-type w1118 Drosophila heart tube extending along the dorsal side of the abdomen. The anterior conical chamber is outlined and displayed in (Figure 1B). Note the non-cardiac alary muscles (AM) and the retractors of tergite muscles (RT). Scale bar = 100 μm. B) Micrograph of the conical chamber after Ca-α1D (red) and GAPDH (white) mRNA molecules were labeled with FISH probes. A representative small region of a single cardiomyocyte used for mRNA quantitation is outlined in white. PC, pericardial cell. Scale bar = 25 μm. C) Examples of Cav α1-subunit mRNA particle densities in cardiomyocyte areas of interest (e.g. white box in Figure 1B) and the quantitative determination of the number of subunit messages normalized to the number of GAPDH messages within the same regions of interest. There were significantly more Ca-α1D and Ca-α1T transcripts compared to cac transcripts. *** p < 0.001. (n = 11, 10, 11 animals for Ca-α1D, cac, and Ca-α1T).
Here, we have elucidated the identity of the Ca2+ channels in the adult Drosophila heart. Direct visualization and quantitation of mRNA revealed an abundance of Ca-α1D and Ca-α1T and a paucity of cacophony Ca2+ channel messages in the cardiac tube. Suppression of Ca-α1D effectively eliminated contraction and Ca2+ cycling activity, while knocking down other channels minimally disrupted contraction and Ca2+ activity. Although these two lines of evidence suggest the presence of A1D and T-type channels, definitive validation required direct measurement of Ca2+ currents across the sarcolemma of Drosophila cardiomyocytes. To this end, we devised a method for isolating viable single cardiomyocytes from heart tubes. These myocytes enabled whole-cell patch clamp recordings of Ca2+ currents. Utilizing pharmacological agents and existing Drosophila Ca2+ channel mutants, we verified the presence of both A1D and T-type Ca2+ currents in fly cardiomyocytes with A1D being the major contributor of the Ca2+ influx for contraction. Similar to mammalian LTCCs, Drosophila A1D also exhibits CDI, a critical negative feedback process that helps with channel regulation, and Ca2+ current amplification through PKA signaling. Overall, we resolved the ensemble of Ca2+ channels functioning in the adult Drosophila heart and devised a novel technique for isolating single cardiomyocytes, which highlight the fly as a feasible alternative for studying diseases involving misregulation of cardiac LTCCs.
Materials and Methods
Drosophila strains and maintenance
TinCΔ4-Gal4 [49], TinCΔ4-Gal4;UAS-GCaMP3 (a generous gift from Dr. Rolf Bodmer), and Hand4.2-GS-Gal4 [50] Drosophila were employed to drive UAS-transgene expression in a cardiac-restricted fashion. UAS-RNAi stocks, obtained from the Vienna Drosophila Resource Center (VDRC), included UAS-Ca-α1DRNAi: 51491 (RNAi Ca-α1D #1), 52644 (RNAi Ca-α1D #2), UAS-cacophonyRNAi: 104168, and UAS-Ca-α1TRNAi: 108827. Background (control) RNAi strains included w1118 wild-type and KK injection lines. GFP-Zasp52 (BDSC Stock no. 6838, which expresses a fluorescently labeled scaffold protein that binds α-actinin and localizes to muscle Z-discs), hypomorphic cacophony cacS (a generous gift from Dr. Chun-Fang Wu) [51], and T-type channel knock out (a generous gift from Dr. Carsten Duch) [52] lines were used for electrophysiological studies. All flies were maintained at room temperature (25 °C) on a standard cornmeal-yeast-sucrose-agar medium.
Heart tube morphology
Confocal microscopy was performed as detailed by Alayari et al. (2009) [53]. Briefly, w1118 Drosophila hearts were surgically exposed according to Vogler and Ocorr (2009) [32] and arrested using 10mM EGTA in artificial hemolymph. The relaxed, semi-intact hearts were fixed (4% formaldehyde in 1X PBS) and washed three times with 1X PBST (PBS with 0.1% Triton X-100). Fixed hearts were then stained with Alexa594 TRITC-phalloidin (1:1000 in PBST), rinsed three times in 1X PBST, mounted and imaged with a Leica TCS SPE RGBV confocal microscope.
RNA fluorescence in situ hybridization (FISH)
TinCΔ4-Gal4 virgin females were crossed with w1118 males. Five days post eclosion, the heart tubes of the progeny were surgically exposed under artificial hemolymph [32] and the semi-intact preparations fixed as described above. Cardiac in situ hybridization was performed as reported previously [54] using the QuantiGene® ViewRNA Cell Assay kit from Panomics (Affymetrix Inc.), following the manufacturer’s recommendation. Fluorophore-tagged probes were custom designed to target Drosophila mRNA coding for Ca-α1D (VF1-18656, VF4-18190), cacophony (VF4-18657), and Ca-α1T (VF4-18658), and a house keeping gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH, VF6-18191). The sequences chosen for probe design are present in all splice variants of each channel subunit to ensure complete coverage of mRNA molecules encoding each Ca2+ channel. A pre-designed probe targeting human alpha feto-protein (h-AFP, VA1-10125) was used as a negative control (Supplementary Figure 1).
Following RNA hybridization, heart tubes were mounted on coverslips in Prolong Gold Antifade mounting media with DAPI (Thermo Fisher Scientific, Catalog No. P36941) and imaged using a Leica TCS SPE confocal microscope (Leica Microsystems) at 40X magnification. Micrographs of heart tubes were analyzed with ImageJ software (National Institute of Health). To quantitate transcripts from the confocal micrographs, individual channels were separated following binary conversion of the images using the same intensity threshold for each channel across all samples. Regions of interest, which included only the cardiomyocytes, were outlined using the freehand selection tool. An automated ImageJ particle counting plugin was used to determine the number of mRNA particles. Cav α1-subunit particle numbers were normalized to total area and to GAPDH.
Cardiac functional analysis and Ca2+ transient imaging
Homozygous TinCΔ4-Gal4;UAS-GCaMP3 or Hand4.2-GS-Gal4 virgin females were crossed with males carrying a UAS-RNAi cassette that targeted a specific Ca2+ channel subunit. Two weeks post eclosion, the female progenies’ hearts were surgically exposed under artificial hemolymph and the beating, semi-intact heart tubes were filmed using a Hamamatsu Orca Flash 2.8 CMOS camera at ~120 frames per second on a Leica DM5000B TL microscope with a 10× (NA, 0.30) immersion lens. Cardiac performance was assessed from the videos using the Semi-automated Optical Heartbeat Analysis (SOHA) program [30, 55]. M-modes, which provide an edge trace documenting heart wall movement over time, were generated via the program. Cardiac function metrics used in this study are similar to those described in Fink et al (2009) [55]. Heart rate variability, akin to the previously reported arrhythmicity index [55], was calculated as heart rate standard deviation divided by median heart rate.
After filming, heart tubes were incubated with 5 μM CellTracker Orange CMTR dye (Thermo Fisher Scientific, Catalog No. C2927), a live cell-permeant fluorescence dye, at room temperature for 30 minutes and rinsed with hemolymph thrice at room temperature for 10-15 minutes. Fly hearts were then imaged using a dual-camera Andor Revolution X1 spinning disc confocal on an inverted microscope (Olympus, Inc) in both green (GCaMP3; ex/em. 488/525 nm) and red (CellTracker Orange CMTR dye; ex/em 561/617 nm) channels simultaneously.
Ca2+ transient analysis was performed using in-house Matlab (MathWorks, Inc.)-based algorithms. The green signal was normalized to the red signal and the fractional change of this ratio was used to gauge cardiomyocyte Ca2+ handling activity. A single exponential fit was performed on the decay phase of the Ca2+ transient to estimate the decay kinetics.
Isolation of live cardiomyocytes from Drosophila heart tubes
Heart tubes from 30-40 two-week-old adult flies were surgically removed and placed into modified Ca2+-free hemolymph. The solution was supplemented with collagenase type I to achieve a final concentration of 0.2% w/v and incubated at room temperature on a shaker for 20 minutes. Trypsin was added to the suspension to achieve a final concentration of 0.1% w/v and incubated for 10 minutes with gentle shaking at room temperature. The digesting tissue was gently triturated every 3 minutes after addition of trypsin. The reaction was quenched by adding fetal bovine serum at a 2:1 ratio. The solution was centrifuged at 550 × g for 5 minutes. The supernatant was aspirated and the cell pellet resuspended in Ca2+-free hemolymph. Cell suspensions were plated on glass coverslips 1 hour prior to electrophysiological studies to allow the cardiomyocytes to attach to the substrate.
Assessment of Drosophila cardiomyocyte and sarcomeric dimensions
Heart tubes of GFP-Zasp52 Drosophila were surgically exposed and maintained in artificial hemolymph under low Ca2+ as previously described [32]. The semi-intact, relaxed hearts were either 1) fixed in 4% formaldehyde in 1X PBS, washed, and mounted, 2) completely removed from the abdominal segment, fixed, washed, and mounted, or 3) removed and dissociated into individual cardiomyocytes as outlined above, prior to fixation and mounting. These three sample groups were also prepared under conditions that prevented active myosin crossbridge cycling. Thus, preceding and during fixation, the samples were exposed to 100 μM blebbistatin in 0.1% DMSO v/v or 0.1% DMSO v/v in artificial hemolymph. Individual cardiomyocytes were imaged at 63X with a Leica TCS SPE RGBV confocal microscope and cellular dimensions (length, width, area) determined using ImageJ software. Resting sarcomere lengths were measured from micrographs of semi-intact and detached hearts, also imaged at 63X, and isolated cardiomyocytes, under low Ca2+, blebbistatin-treated, and DMSO-incubated conditions.
Electrophysiology
Whole-cell recordings of individual cardiomyocytes were acquired at room temperature using an Axopatch 200B amplifier (Axon Instruments). Internal solutions contained (in mM): CsMeSO3, 114; CsCl, 5; MgCl2, 1; MgATP, 4; HEPES (pH 7.3), 10; and BAPTA, 10; at 295 mOsm adjusted with CsMeSO3. Seals were formed in artificial hemolymph and following patch rupture, the bath solution was switched to Ca2+- or Ba2+-external solution containing (in mM): TEA-MeSO3, 140; HEPES (pH 7.4), 10; and CaCl2 or BaCl2, 5; at 300 mOsm, adjusted with TEA-MeSO3. Traces were lowpass filtered at 2 kHz, and digitally sampled at 10 kHz. A P/8 leak subtraction protocol, where a leak pulse is 1/8 of the test pulse, was used with series resistances of 1-2 MΩ. 10 μM nifedipine (Sigma-Aldrich, N7634) or 10 μM forskolin (Sigma-Aldrich, F6886) was added to the bath solution in certain experiments to assess the pharmacological specificity of the observed Ca2+ currents.
Ca2+ channel sequence alignment
Amino acid sequences of CACNA1C from Oryctolagus (Gene ID: 100101555; mRNA ID: NM_001136522) and A1D from Drosophila (Gene ID: 34950 or CG4894; mRNA ID: NM_165147) were aligned using Clustal Omega [56] to assess sequence homology.
Statistical Methods
Statistical analysis was performed using GraphPad Prism 7. Data are presented as mean ± SEM. One-way ANOVAs followed by Tukey’s multiple comparisons tests were used to determine whether statistically significant differences existed between the means of 3 or more groups. When data were not normally distributed, Kruskal-Wallis one-way ANOVAs followed by Dunn’s post hoc tests were employed. For comparisons between two unmatched groups, unpaired Student’s t-tests were used to determine if the data sets significantly differed from each other. Significance was assessed at p < 0.05.
Results
Drosophila cardiomyocytes express genes that encode specific voltage-gated Ca2+ channels
The fly genome encodes three α1-subunits (Ca-α1D, cacophony, and Ca-α1T) of CaV. These subunits define three distinct Drosophila hetero-oligomeric channels: A1D, cac, and T-type [17, 57-59] that are homologous to the major mammalian classes CaV1, CaV2, and CaV3, respectively. FISH, which allows direct visualization and relative quantitation of individual mRNA molecules, was utilized to decipher the cardiomyocyte CaV biosignature in control TinCΔ4-Gal4 x w1118 Drosophila. Semi-intact fly hearts, which remain suspended within the dissected abdominal segment (Figure 1A), were fixed, permeabilized, and incubated with fluorescent probes that possess base sequence complementarity to specific α1-subunit mRNAs (Figure 1B). On average, when normalized to GAPDH particles, Drosophila cardiomyocytes showed an abundance of Ca-α1D (0.51±0.03) and Ca-α1T (0.45±0.03) mRNA molecules relative to a limited number of cacophony (0.06±0.01) messages (Figure 1C). Thus, the fly heart expresses significantly higher amounts of Ca-α1D and Ca-α1T vs. cacophony CaV α1-subunit mRNA.
RNA interference reveals the predominant types of functioning voltage-gated Ca2+ channels in Drosophila hearts
The prevalence of Ca-α1D and Ca-α1T mRNA implies that A1D and T-type are the major CaV that orchestrate cardiac contraction in Drosophila. To verify a direct role of A1D, T-type, and potentially of cac CaV, in cardiomyocyte Ca2+ signaling, the functional consequences of heart-specific RNAi-mediated silencing of each gene were evaluated. Unlike what is found with the main classes of mammalian CaV, each Drosophila CaV α1-subunit is encoded by a single gene, thus limiting the number of targets to be tested. Previous studies have shown that transmembrane Ca2+ and not Na+ current substantially contributes to Drosophila cardiac action potentials [40, 60]. Therefore, alteration in heart tube contraction after CaV α1-subunit knockdown could hint at the predominant type(s) of Ca2+ channels operating in fly cardiomyocytes. A cardiac-specific driver line, TinCΔ4-Gal4; UAS-GCaMP3, was crossed with multiple UAS-RNAi lines, yielding progenies with selective reduction of one of the three Ca2+ channels (Figure 2A). A highly significant reduction of Ca-α1D, cacophony, or Ca-α1T transcripts was verified using FISH (Supplementary Figure 3). Spontaneous, myogenic contractions and Ca2+ cycling properties in semi-intact heart tubes were assessed in two-week-old adult offspring.
Figure 2. Cardiac contraction and Ca2+ transients are suppressed by Ca-α1D knockdown.

A) The Drosophila UAS-GAL4 bipartite expression system. Cardiac-specific TinCΔ4-Gal4 drives expression of UAS-transgenes including the simultaneous expression of a Ca2+ biosensor (UAS-GCaMP3) and UAS-RNAi. B) Exemplar M-mode tracings of heart tubes following Ca-α1D, cacophony, or Ca-α1T knockdown (Ca-α1D(−), cac(−), and Ca-α1T(−)). The progenies of w1118 or the injection lines (KK) crossed with the driver line served as controls. Ca-α1D knockdown completely suppressed contraction. C) Quantitative measurements of cardiac physiological parameters. Population data confirmed substantially altered contraction following Ca-α1D knockdown. *** p < 0.0001 compared to w1118; ## p < 0.01 compared to KK. There were no significant differences in cardiac variables between the controls. (n=25-31 animals) D) Representative Ca2+ transient recordings from individuals of the same population of heart tubes examined in Figure 2B–C. Ca-α1D silencing effectively abolished Ca2+ transients in the heart tubes. E) Measurements of Ca2+ transients confirmed a significant reduction in Ca2+ transient frequency and magnitude of the peak Ca2+ transient upon Ca-α1D RNAi expression. The time required to reach the peak Ca2+ transient magnitude (time to peak) and the time constant for the Ca2+ transient decay (tau decay) are not shown for Ca-α1D(−)#1 and Ca-α1D(−)#2 because of inaccurate measurements due to minimal Ca2+ activity in these hearts. Knockdown of cacophony or Ca-α1T produced no significant change in Ca2+ transient frequency, Ca2+ transient magnitude, time to peak, or tau decay. *** p < 0.001 compared to w1118. (n=21-30 animals).
TinCΔ4-Gal4; UAS-GCaMP3 > RNAi Ca-α1D #1 Drosophila exhibited minimal heart tube motion compared to the rhythmic contractions in TinCΔ4-Gal4; UAS-GCaMP3 x w1118 controls as demonstrated by M-mode recordings (Figure 2B). To account for potential confounding positional effects that may result from insertion of the RNAi cassette in discrete locations throughout the Drosophila genome, two different RNAi lines with the same genetic background as the control were evaluated, and the effects of gene silencing on several cardiac parameters were quantified. Compared to control, both TinCΔ4-Gal4; UAS-GCaMP3 > RNAi Ca-α1D #1 and > RNAi Ca-α1D #2 showed significantly decreased heart rates (0.35±0.16 and 0.36±0.10 vs. 1.24±0.10 beats/s in control) and increased heart rate variabilities (0.45±0.12 and 0.22±0.06 vs. 0.10±0.02 in control) (Figure 2C). Moreover, the extent of contraction was significantly diminished in the RNAi-expressing heart tubes as demonstrated by ~4 fold reduction in fractional shortening (0.08±0.01 and 0.05±0.01 vs. 0.42±0.01 in control) and ~4.5 fold reduction in shortening velocities (52.9±17.7 and 69.3±18.7 vs. 872.7±21.3 μm/s in control).
We next ascertained if Ca2+ signaling was altered following Ca-α1D knock down. GCaMP3-based green fluorescence, emitted from actively beating heart cells, was recorded simultaneously with orange fluorescence that originated from CellTracker, a dye that was passively loaded into the cardiomyocytes to monitor cell movement. The relative change in ratio between the two signals was used to determine the Ca2+ cycling properties. The ratiometric approach helped correct for heart contraction motion artifacts and for different amounts of GCaMP3 expression or CellTracker loading within and among the samples. Following Ca-α1D knockdown, fluorescent signal analysis suggested a completely abolished Ca2+ transient (Figure 2D). Moreover, population data of cyclical fluorescent fluctuations confirmed significant reductions of Ca2+ transient rates (RNAi Ca-α1D #1, #2 vs. control: 0.04±0.02, 0.03±0.02 vs. 1.32±0.08 Hz) and peak Ca2+ transient magnitudes (0.005±0.003, 0.001±0.001 vs. 0.182±0.017) upon suppression of Ca-α1D expression (Figure 2E) compared to control. This observation was consistent with the nearly complete cessation of contraction upon Ca-α1D knockdown described above.
These experimental results demonstrate the effects of constitutive Ca-α1D suppression, i.e. both during and after heart tube development. To assess functional changes in hearts with Ca-α1D knockdown post cardiogenesis, the same UAS-RNAi lines were crossed with the inducible, cardiac-specific Hand4.2-GS-Gal4 driver line. Expression of Ca-α1D RNAi in the offspring was activated from two days after eclosion by supplementing the food with RU486 and the heart tubes of two-week-old Drosophila were imaged. Reduced fractional shortening and shortening velocity were observed when Ca-α1D was knocked down post-developmentally, although to a lesser extent compared to flies with cardiac-restricted Ca-α1D knockdown throughout development (Supplementary Figure 2). Overall, these results suggest a key role of A1D CaV in cardiac function in flies, both during and post-development.
In addition to A1D, potential contributions from cac and, given its high expression levels, T-type Ca2+ channels in defining Drosophila cardiac contraction and Ca2+-handling properties, were also explored. Individually suppressing expression of these channels in TinCΔ4-Gal4; UAS-GCaMP3 > cacophonyRNAi and > Ca-α1TRNAi did not significantly alter heart rates (cacophonyRNAi, Ca-α1TRNAi vs. control: 2.50±0.17, 1.70±0.14 vs. 1.95±0.19 beats/s), heart rate variabilities (0.16±0.02, 0.15±0.03 vs. 0.12±0.03), or shortening velocities (1056±79.3, 960±58.4 vs. 982.4±67.1 μm/s) (Figures 2B-C) compared to the TinCΔ4-Gal4; UAS-GCaMP3 x KK control line. Although there was a statistically significant reduction of fractional shortening subsequent to cacophony or Ca-α1T knockdown (0.40±0.01 or 0.40±0.01 vs. 0.44±0.01 in control), the extent of reduction was minimal compared to that following Ca-α1D knockdown. Similarly, cacophony or Ca-α1T knockdown did not yield statistically significant changes in Ca2+ transient rates (cacophonyRNAi, Ca-α1TRNAi vs. control: 2.04±0.15, 1.32±0.11 vs. 1.66±0.10 Hz), Ca2+ transient magnitudes (0.16±0.02, 0.15±0.02 vs. 0.17±0.02), time to peak (165.0±10.8, 214.0±8.5 vs. 184.6±8.5 ms), or decay time constants (261.2±16.9, 332.6±18.6 vs. 303.3±16.6 ms) compared to control (Figures 2E). Collectively, these data illustrate that A1D plays a major role in defining the Drosophila cardiac Ca2+ transient and myocardial contraction with potentially minor contributions from cac and T-type Ca2+ channels.
Isolation and morphological characterization of Drosophila cardiomyocytes
So far, we have examined the sources of plasmalemmal Ca2+ flux in Drosophila cardiomyocytes using multiple indirect approaches. However, voltage clamping individual cells and directly measuring Ca2+ current across the membrane could ultimately confirm the major types of active Ca2+ channels functioning in Drosophila myocardium. Although isolated cardiomyocytes have been a mainstay for cellular electrophysiology in mammalian systems for decades, no published reports of analogous protocols for flies exist. Therefore, we devised a method for isolating viable single cells from Drosophila heart tubes, which consist of a single layer of bilateral rows of opposing cardiomyocytes. Following enzymatic dissociation of GFP-Zasp52 hearts, individual cells, which maintain their curved shape (Figure 3A, Supplementary Figure 4), were successfully obtained. Consistent with a gradual tapering of the heart’s diameter along its length, the cardiomyocytes exhibited moderate variability in dimensions, which on average were 51.3±2.7 μm in length, 32.2±1.0 μm in width, and 1561±115 μm2 in maximally projected area. Sarcomere lengths were determined by measuring the distance between peak fluorescent signals emanating from the Z-disc-associated, Zasp52-GFP. Interestingly, the average resting sarcomere length along myofibrils of myocytes isolated and maintained under low Ca2+ (1.38±0.04 μm) was significantly less than that determined for semi-intact (2.50±0.03 μm) or detached whole hearts (2.54±0.02 μm) maintained under similar conditions (Figure 3B). Despite variability in cellular dimensions, the sarcomere lengths were consistent among isolated cardiomyocytes (Supplementary Figure 5). To determine if the shortened sarcomeres resulted from excessive actomyosin associations activated upon myocyte dissociation, we compared the distance between the middle of consecutive Z-discs along myofibrils of semi-intact hearts, detached whole hearts, and single cells maintained in DMSO or in the presence of blebbistatin, a small-molecule inhibitor of several striated muscle myosins. The sarcomere lengths in semi-intact and detached cardiac tubes in the presence of blebbistatin (2.53±0.02 and 2.53±0.02 μm, respectively) were not significantly different from those determined in DMSO (2.52±0.02 and 2.55±0.02 μm, respectively) or under conditions of low Ca2+ (Figure 3B). Sarcomeres of isolated cells incubated in blebbistatin or in DMSO did not differ in length from each other (1.46±0.04 vs. 1.36±0.04 μm, respectively) or from those incubated under low Ca2+, but were significantly shorter than the sarcomeres of semi-intact or detached whole hearts (Figures 3B and 3C). These data suggest the shortened sarcomeres result from passive processes that accompany cellular separation. Despite reduced sarcomere lengths, the individual myocytes nonetheless remained viable, as demonstrated by rhythmic contractions (Supplementary Movie 3) and the presence of Ca2+ transients (Supplementary Figure 4) for up to 2-3 hours in Ca2+-containing artificial hemolymph at 25°C.
Figure 3. Morphological characterization of isolated Drosophila cardiomyocytes.
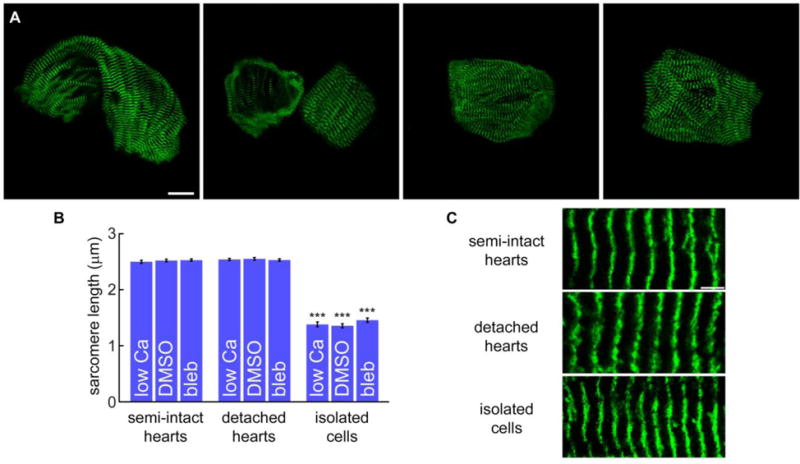
A) GFP-ZASP52 cardiomyocytes after dissociation from detached heart tubes maintained their curved morphology. Note, the left-most cardiomyocyte originated from the conical chamber of the heart. The remaining cells most likely came from the middle one third of the cardiac tube and are representative of those commonly isolated. Scale bar = 20 μm. B) Sarcomere lengths along myofibrils within cardiomyocytes of semi-intact and detached whole hearts were not significantly different under low Ca2+ conditions or following exposure to DMSO or 100 μM blebbistatin. After cardiomyocyte dissociation, the average sarcomere length of isolated cells under all three conditions did not significantly differ; however, the sarcomeres were significantly shorter than those of semi-intact and detached whole hearts. *** p < 0.0001. (n=22-31 cells) C) Enlarged views of myofibrils demonstrating the effect of cardiomyocyte isolation on resting sarcomere length.
Voltage clamp experiments confirm A1D as a predominant mediator of Ca2+ current in Drosophila cardiomyocytes
Isolated Drosophila cardiomyocytes were voltage clamped at −80 mV (resting potential) and the Ca2+ current, induced by various depolarizing voltage steps, was measured. For example, the Ca2+ current evoked by a 0-mV step potential in control (GFP-Zasp52) cells was characterized by a rapid influx of Ca2+ ions, which slowly decayed over time (Figure 4A, black) and resembled the characteristic inactivating mammalian ventricular cardiomyocyte CaV1 Ca2+ current. Peak Ca2+ current density at various test potentials is shown in Figure 4B (black). To explore potential contributions of Ca2+ current from cac and T-type channels, peak Ca2+ current density was measured in cardiomyocytes isolated from hypomorphic cac and null T-type Drosophila lines. No significant differences in the peak current density between cardiomyocytes from the control (Figure 4B, black) and hypomorphic cac hearts (Figure 4B, blue) across all test voltages were observed. For example, the current densities of cac hypomorphic vs. control cardiomyocytes at −30 mV and 0 mV were −9.3±1.7 vs. −11.0±2.6 pA/pF and −13.9±0.9 vs. −12.2±1.4 pA/pF, respectively. These results imply a negligible contribution of cac channels in establishing Drosophila myocardial Ca2+ currents. However, peak Ca2+ current density measured from T-type null cardiomyocytes was significantly reduced relative to control at the lower voltage range (defined here as V < 0 mV with the employed external and internal solutions), e.g. −3.2±0.5 pA/pF at −30 mV, while there was no significant difference in peak current density between T-type null and control cells at the higher voltage range (defined as V > 0 mV), e.g. −13.0±2.3 pA/pF at 0 mV. As both mammalian CaV3 and Drosophila T-type Ca2+ channels are activated mainly at low voltages [61-64], the current density profile from T-type null cells compared to control suggests the presence of functional T-type Ca2+ channels in Drosophila heart tubes.
Figure 4. Voltage clamp recordings confirm A1D channels conduct the predominant transsarcolemmal Ca2+ current in Drosophila cardiomyocytes.

A) Ca2+ current recording of a dissociated fly myocyte. The current in control cardiomyocytes was suppressed by a dihydropyridine, nifedipine, a hallmark of CaV1 channels. The average capacitance of the Drosophila cardiomyocyte membrane was 125±11 pF. B) Mean peak current density in control, hypomorphic cacophony (cacs), and T-type (Ca-α1T) null cardiomyocytes across test potentials. At high voltages, comparable to the plateau phase of mammalian cardiac action potentials, peak currents of cacs hypomorphic and T-type null cardiomyocytes were similar to that observed in control, indicating that A1D is the major high-voltage-activated Ca2+ channel isoform in the Drosophila heart. At low voltages, T-type null cardiomyocytes showed reduced current densities, suggesting a contribution of T-type channels at low activation voltages. * p < 0.5 compared to control. (n=4, 6, and 4 cells for control, cacs, and T-type null). C) Population data of the Ca2+ current response to 10 μM nifedipine as compared to untreated control myocytes. (n=5 cells).
Finally, to unequivocally validate a requirement for A1D CaV in Ca2+ signaling in Drosophila cardiomyocytes, dihydropyridine nifedipine, a specific mammalian CaV1 and Drosophila A1D channel blocker, was added to the bathing solution of control cells. Application of 10 μM nifedipine significantly decreased the magnitude of Ca2+ current (Figure 4A, blue) as confirmed by population data of current reduction after application of nifedipine across multiple test potentials (Figure 4C). In sum, these results corroborate the FISH data (Figure 1), and indicate that both A1D and T-type Ca2+ channels are abundantly present and functionally active in Drosophila cardiomyocytes.
Properties of cardiac A1D channels in Drosophila
Multiple lines of evidence illustrate that A1D is a major contributor to transmembrane Ca2+ currents in fly cardiomyocytes. Because A1D is homologous to mammalian CaV1, it may possess similar properties to its mammalian counterparts, including CDI. CDI is a negative feedback mechanism and crucial feature of CaV1 channels (CaV1.2, CaV1.3, and CaV1.4) that helps regulate the level of intracellular Ca2+. Key components for orchestrating CDI include calmodulin (CaM), a resident Ca2+ sensor molecule that is pre-bound to the C-terminus of the channel [65-67], the N-terminal spatial Ca2+ transforming element (NSCaTE) [68-70] (on the N-terminus of the channel), two EF hands [65, 71, 72], an IQ domain [65, 71, 73], and Ca2+-free CaM (apoCaM) binding sites (on the C-terminus of the channels) [65, 66] (Figure 5A). Since Drosophila A1D possesses domains with homology to all aforementioned CDI components, it also likely exhibits CDI. Ca2+ current recordings of fly cardiomyocytes (Figure 5B, red) showed Ca2+ influx due to channel opening followed by a gradual decrease in the current size or channel inactivation (Figure 5B, red). CDI can be distinguished from voltage-dependent inactivation (VDI), another form of channel feedback regulation, by comparing Ca2+ vs. Ba2+ current (Figure 5B, black) passing through the same channel. Because Ba2+ is unable to efficiently bind to CaM, Ba2+ current inactivation solely represents the degree of VDI. Thus, the true extent of CDI (Figure 5B, shaded pink) can be calculated as f300 = (r300/Ba − r300/Ca)/r300/Ba, where r300/x is the fraction of Ca2+ and Ba2+ currents remaining after 300 ms of channel opening. For example, at a 0-mV test potential the f300 = 0.35±0.14, which confirms that Drosophila A1D demonstrates robust CDI (Figure 5C).
Figure 5. Ca2+-dependent inactivation is a conserved feature of Drosophila A1D.

A) Illustration of the alpha subunit of the vertebrate LTCC with CDI interface regions NSCaTE (yellow), two EF hands (rose, green), and the IQ domain (blue). Sequence comparison of Oryctolagus’s CACNA1C and Drosophila’s A1D with CDI components highlighted in the same color as in the diagram on the left. The binding sites of Ca2+-free CaM (apoCaM) are highlighted in grey with key amino acid residues interacting with N-, C-, and both lobes of apoCaM bolded in blue, red, and black. B) Ca2+ current in fly cardiomyocytes decayed more rapidly than Ba2+ current in the same cell, demonstrating CDI (shaded rose) as Ba2+ cannot effectively bind calmodulin. C) Population data showing the fraction of current that remained after 300 ms of activation (r300). The different degree of decay between Ca2+ and Ba2+ currents represents the extent of CDI (shaded rose). (n=7 cells).
Another key property of mammalian cardiac CaV1.2 is current augmentation in response to PKA-mediated phosphorylation. Adrenergic-like octopamine receptors (OctαRs, and OctβRs) [74], adenyl cyclase (rutabaga) [75], phosphodiesterase (dunce) [75], and both regulatory [76] and catalytic [77] subunits of PKA are also expressed in Drosophila (Figure 6A), suggesting that A1D may also exhibit PKA-mediated current enhancement. Application of 10 μM forskolin, an activator of adenyl cyclase, increased the amplitude of Ca2+ current through the A1D channels (Figure 6B). For example, at 0-mV test potential, the peak Ca2+ current was greatly enhanced by 1.58±0.13 fold after forskolin application (Figure 6C). Similar current amplification by forskolin was observed across multiple voltages, confirming the presence of PKA-mediated current augmentation of A1D channels in Drosophila cardiomyocytes.
Figure 6. Ca2+ current augmentation by protein kinase A is conserved in Drosophila cardiomyocytes.

A) Depiction of the β-adrenergic pathway. Epinephrine binds to a G-protein coupled receptor and activates the enzyme adenyl cyclase receptor, which converts ATP to cyclic AMP (cAMP). This small signaling molecule activates protein kinase A (PKA), which phosphorylates LTCCs, augmenting their current size. Phosphodiesterase (PDE) deactivates cAMP. Forskolin bypasses this signaling cascade by directly activating adenyl cyclase. Drosophila homologs of the β-adrenergic pathway elements are in parentheses. Oct, octopamine. B) Ca2+ current in Drosophila cardiomyocytes increased in amplitude after application of 10 μM forskolin. C) Population data confirm augmentation of Drosophila A1D current by PKA. (n=4 cells).
Discussion
Drosophila represents a potentially ideal platform for studying regulation of, and diseases involving, cardiac Ca2+ channel function and dysfunction. In addition to their genetic pliability, fly cardiomyocytes, as shown here, harbor A1D and T-type Ca2+ channels (orthologs of mammalian CaV1 and CaV3 channels, respectively [17]), which is analogous to the Ca2+ channel ensemble of mammalian cardiomyocytes [78-80]. Moreover, Drosophila A1D also possesses striking conservation of two key properties of CaV1 channels, which are CDI and PKA-dependent current augmentation.
In the current study, we show for the first time, that Drosophila cardiomyocytes express significantly higher amounts of Ca-α1D and Ca-α1T relative to cacophony mRNA. Functional analyses revealed drastically impaired heart tube contraction and decreased Ca2+ transient amplitude when Ca-α1D expression was suppressed by RNAi as opposed to a minimal change in contractile properties resulting from Ca-α1T or cacophony silencing. These results imply that not only is A1D highly enriched in Drosophila cardiomyocytes, but that A1D channels are the major contributor to Ca2+ flux responsible for orchestrating contraction. However, despite an abundance of Ca-α1T mRNA, T-type channels were shown to contribute minimally to heart tube contraction. These results suggest that there is a potential discordance between Ca-α1T mRNA versus protein load, a high transcriptional reserve to ensure an adequate T-type channel stoichiometry, and/or poorly effective RNA interference and gene suppression. Direct assessment of plasmalemmal Ca2+ currents in cardiomyocytes that are genetically devoid of T-type channels could reveal insight into this discrepancy.
Although multiple reports previously described changes in contractions of intact Drosophila hearts [34, 40, 42, 55, 60, 81], at baseline or in response to pharmacological manipulation, approaches to evaluate plasmalemmal currents in adult cardiomyocytes have remained somewhat coarse. Field potentials across the surface of heart tubes were recorded [34, 36] while a rudimentary current clamp recording, performed by inserting electrodes into whole hearts, successfully measured transmembrane voltages [44, 82, 83]. However, the gold standard to directly probe plasmalemmal current is the voltage clamp technique, which yields highly resolved data when performed on single cells as opposed to cellular networks. Voltage clamp recordings have been acquired from neurons of Drosophila embryos [84], pupae [57], and adult [57] brains, motoneurons of adult indirect flight muscle [52] and of larval body-wall muscle [85], larval retinal cells [86], and larval body wall muscles [87-89]. Nonetheless, despite well-established methods for the isolation of cardiomyocytes from various mammalian species and subsequent transsarcolemmal ion current measurements, voltage clamp experiments using single Drosophila cardiomyocytes have not been conducted. Therefore, based on approaches employed for mammalian tissue, we developed a method to enzymatically dissociate viable cardiomyocytes from adult Drosophila heart tubes.
Brief enzymatic digestion of ~35 fly hearts resulted in a population of individual cells that appeared well suited for electrophysiological studies. As reported for vertebrate cardiomyocytes, which show a significant 17% reduction in sarcomere length upon isolation from the intact heart [90], Drosophila sarcomeres likewise displayed reduced lengths following cardiomyocyte dissociation. Comparing the sarcomere lengths from cells of semi-intact relative to those of detached heart tubes revealed that changes in sarcomere length did not transpire in response to heart extraction. Moreover, incubating and digesting the hearts in artificial hemolymph containing either EGTA and cell-permeant EGTA-AM, which chelates and minimizes intracellular Ca2+ (data not shown), or blebbistatin did not prevent sarcomere shortening upon cell-to-cell separation. These findings suggest that the change in sarcomere length is not due to excessive or unregulated Ca2+-mediated tension or active actomyosin-based contraction in general.
The heightened sarcomeric shortening event that occurs upon fly vs. rat cardiomyocyte isolation (i.e. ~40 vs. 17% reduction in sarcomere length) may be related to several factors. Firstly, the extreme geometric constraints that are imposed upon fly cardiomyocytes, due to two opposing cells establishing the tubular nature of the organ, are partially released following proteolytic digestion. Consequently, coinciding with changes in cellular strain/stress upon separation, relatively large changes in Drosophila myofibril and therefore sarcomere deformation are expected to occur. The elastic properties of the fly connecting filaments also likely differ from those of vertebrate cardiac titin isoforms, which are the predominant determinants of cardiomyocyte passive tension [91]. Such differences may uniquely influence cellular and molecular recoil post digestion.
Despite the exaggerated changes in resting sarcomere length that transpire upon dissociation, we show that Drosophila cardiomyocytes are exceptionally amenable to voltage clamp experiments. We confirmed the presence of Ca2+ currents through both A1D and T-type Ca2+ channels, which are remarkably similar to the those observed in mammalian cardiomyocytes [79, 80]. In mammals, T-type channels or CaV3 are activated at lower potentials and are characterized by a small conductance [79, 80]. Cardiac LTCCs or CaV1.2, however, are activated at relatively higher potentials compared to CaV3 with higher pore conductance [79, 80] and, thus, are better-suited to drive the shape and duration of the cardiac action potential [1]. As heart tube contraction is the end-point of a multi-step process, from pacemaker cells firing, action potential transmission to myocytes, and Ca2+-centric conversion of electrical to mechanical activities, alteration in contractile properties could stem from deviations in any of these critical steps. The T-type Ca2+ current was proposed to play a modulatory role in pacemaking, which is predominantly driven by a Na+/Ca2+ exchanger-dependent Ca2+ clock [92, 93]. Therefore, consistent with our findings, suppression of Ca-α1T expression by RNAi minimally affects the contractile properties of the heart tubes. LTCCs, on the other hand, are responsible for the transsarcolemmal Ca2+ flux that triggers Ca2+-induced Ca2+ release from the sarcoplasmic reticulum leading to cardiomyocyte contraction. Hence, it is not surprising that Ca-α1D knock down nearly abolishes heart tube contraction (Figure 2).
Most interestingly, Drosophila A1D also shares key regulatory features with those of mammalian CaV1.2, which include CDI and PKA-mediated current augmentation. After entering through CaV, Ca2+ ions bind to CaM, and Ca2+/CaM induces a conformational change that decreases open probability of individual channels. This leads to a reduction of current amplitude at the whole cell level [69, 94] and, thereby, accounts for CDI. During the fight-or-flight response, epinephrine binds to the β1-adrenergic receptor, a type of G-protein-coupled receptor, and initiates a signaling cascade that results in PKA-mediated CaV1.2 phosphorylation. The phosphorylated channels display higher open probability, which leads to an increased whole cell Ca2+ current size. The larger Ca2+ influx enhances the force of cardiac contraction that is necessary for the flight response. The conservation of these vital features in fly cardiomyocytes hints at the evolutionary importance of LTCCs and, thus, their tightly controlled functions.
Despite the advantages afforded by Drosophila myocytes, there are considerations prior to their widespread use for mechanistic studies of cardiac Ca2+ channels. For example, while we confirmed the presence of CDI and PKA-mediated current enhancement, several additional up- and downstream modifiers of Ca2+ flux, which exist in cardiomyocytes from higher organisms, have yet to be characterized in insects and may be absent. Additionally, the cells that comprise the complex, four-chambered mammalian heart display regional differences in their Ca2+ current and electrophysiological properties78. Myocytes originating from different locations along the Drosophila cardiac tube may also display regional variation in Ca2+ currents; however, resolving such differences would require careful cellular purification and handling procedures. Moreover, limited tissue yields could make the fly model cumbersome for certain applications. Multiple surgeries are required to obtain enough cells for well-powered studies. Nonetheless, our data suggest Drosophila proves to be an effective and novel platform to investigate regulation of and diseases involving cardiac Ca2+ channels. Due to the fly’s genetic versatility, efficient tools, and multiple modalities for functional assessment, screens to dissect physiological or unravel pathological mechanisms are likely eminently feasible using this animal model.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Highlights.
Drosophila cardiomyocytes primarily express mRNA encoding L- and T-type Ca channels
A1D L-type Ca channels are required for contraction of the fly heart
Isolated Drosophila cardiomyocytes are amenable to patch clamp experiments
A1D is the main conduit for sarcolemmal Ca flux in fly cardiomyocytes
Drosophila can serve as an efficient model to study cardiac Ca channel regulation
Acknowledgments
David T. Yue passed away on December 23, 2014. His mentorship, wisdom, and kindness are greatly missed. We thank Hogan Tang and Holly Tang for technical advice and initial training for fruit fly husbandry, Anna Blice-Baum for assistance with fluorescent in situ hybridization assay optimization, and Wanjun Yang for dedicated technical support.
Sources of Funding
This study was supported by AHA 13PRE16500029 and 15PRE25860042 (W.B.L), NHLBI R01HL124091 (A.C.), NIMH R01MH065531 (D.T.Y.), and NHLBI 5R37HL076795 (D.T.Y.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
None.
References
- 1.Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev. 2005;85:1205–53. doi: 10.1152/physrev.00002.2005. [DOI] [PubMed] [Google Scholar]
- 2.Barrett CF, Tsien RW. The Timothy syndrome mutation differentially affects voltage- and calcium-dependent inactivation of CaV1.2 L-type calcium channels. Proc Natl Acad Sci U S A. 2008;105:2157–62. doi: 10.1073/pnas.0710501105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dick IE, Joshi-Mukherjee R, Yang W, Yue DT. Arrhythmogenesis in Timothy Syndrome is associated with defects in Ca(2+)-dependent inactivation. Nat Commun. 2016;7:10370. doi: 10.1038/ncomms10370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Splawski I, Timothy KW, Decher N, Kumar P, Sachse FB, Beggs AH, et al. Severe arrhythmia disorder caused by cardiac L-type calcium channel mutations. Proc Natl Acad Sci U S A. 2005;102:8089–96. doi: 10.1073/pnas.0502506102. discussion 6-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119:19–31. doi: 10.1016/j.cell.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 6.Crotti L, Johnson CN, Graf E, De Ferrari GM, Cuneo BF, Ovadia M, et al. Calmodulin mutations associated with recurrent cardiac arrest in infants. Circulation. 2013;127:1009–17. doi: 10.1161/CIRCULATIONAHA.112.001216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Limpitikul WB, Dick IE, Joshi-Mukherjee R, Overgaard MT, George AL, Jr, Yue DT. Calmodulin mutations associated with long QT syndrome prevent inactivation of cardiac L-type Ca(2+) currents and promote proarrhythmic behavior in ventricular myocytes. J Mol Cell Cardiol. 2014;74:115–24. doi: 10.1016/j.yjmcc.2014.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Limpitikul WB, Dick IE, Tester DJ, Boczek NJ, Limphong P, Yang W, et al. A Precision Medicine Approach to the Rescue of Function on Malignant Calmodulinopathic Long-QT Syndrome. Circ Res. 2017;120:39–48. doi: 10.1161/CIRCRESAHA.116.309283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yin G, Hassan F, Haroun AR, Murphy LL, Crotti L, Schwartz PJ, et al. Arrhythmogenic calmodulin mutations disrupt intracellular cardiomyocyte Ca2+ regulation by distinct mechanisms. J Am Heart Assoc. 2014;3:e000996. doi: 10.1161/JAHA.114.000996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baig SM, Koschak A, Lieb A, Gebhart M, Dafinger C, Nurnberg G, et al. Loss of Ca(v)1.3 (CACNA1D) function in a human channelopathy with bradycardia and congenital deafness. Nat Neurosci. 2011;14:77–84. doi: 10.1038/nn.2694. [DOI] [PubMed] [Google Scholar]
- 11.Platzer J, Engel J, Schrott-Fischer A, Stephan K, Bova S, Chen H, et al. Congenital deafness and sinoatrial node dysfunction in mice lacking class D L-type Ca2+ channels. Cell. 2000;102:89–97. doi: 10.1016/s0092-8674(00)00013-1. [DOI] [PubMed] [Google Scholar]
- 12.Van Wagoner DR, Pond AL, Lamorgese M, Rossie SS, McCarthy PM, Nerbonne JM. Atrial L-type Ca2+ currents and human atrial fibrillation. Circ Res. 1999;85:428–36. doi: 10.1161/01.res.85.5.428. [DOI] [PubMed] [Google Scholar]
- 13.Yue L, Feng J, Gaspo R, Li GR, Wang Z, Nattel S. Ionic remodeling underlying action potential changes in a canine model of atrial fibrillation. Circ Res. 1997;81:512–25. doi: 10.1161/01.res.81.4.512. [DOI] [PubMed] [Google Scholar]
- 14.Balke CW, Shorofsky SR. Alterations in calcium handling in cardiac hypertrophy and heart failure. Cardiovasc Res. 1998;37:290–9. doi: 10.1016/s0008-6363(97)00272-1. [DOI] [PubMed] [Google Scholar]
- 15.Richard S, Leclercq F, Lemaire S, Piot C, Nargeot J. Ca2+ currents in compensated hypertrophy and heart failure. Cardiovasc Res. 1998;37:300–11. doi: 10.1016/s0008-6363(97)00273-3. [DOI] [PubMed] [Google Scholar]
- 16.Chintapalli VR, Wang J, Dow JA. Using FlyAtlas to identify better Drosophila melanogaster models of human disease. Nat Genet. 2007;39:715–20. doi: 10.1038/ng2049. [DOI] [PubMed] [Google Scholar]
- 17.Chorna T, Hasan G. The genetics of calcium signaling in Drosophila melanogaster. Biochim Biophys Acta. 1820:1269–82. doi: 10.1016/j.bbagen.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 18.Reiter LT, Potocki L, Chien S, Gribskov M, Bier E. A systematic analysis of human disease-associated gene sequences in Drosophila melanogaster. Genome Res. 2001;11:1114–25. doi: 10.1101/gr.169101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pandey UB, Nichols CD. Human disease models in Drosophila melanogaster and the role of the fly in therapeutic drug discovery. Pharmacol Rev. 2011;63:411–36. doi: 10.1124/pr.110.003293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pfeiffer BD, Ngo TT, Hibbard KL, Murphy C, Jenett A, Truman JW, et al. Refinement of tools for targeted gene expression in Drosophila. Genetics. 2010;186:735–55. doi: 10.1534/genetics.110.119917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wolf MJ, Rockman HA. Drosophila, genetic screens, and cardiac function. Circ Res. 109:794–806. doi: 10.1161/CIRCRESAHA.111.244897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–15. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- 23.Roman G, Endo K, Zong L, Davis RL. P[Switch], a system for spatial and temporal control of gene expression in Drosophila melanogaster. Proc Natl Acad Sci U S A. 2001;98:12602–7. doi: 10.1073/pnas.221303998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dietzl G, Chen D, Schnorrer F, Su KC, Barinova Y, Fellner M, et al. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature. 2007;448:151–6. doi: 10.1038/nature05954. [DOI] [PubMed] [Google Scholar]
- 25.Neely GG, Kuba K, Cammarato A, Isobe K, Amann S, Zhang L, et al. A global in vivo Drosophila RNAi screen identifies NOT3 as a conserved regulator of heart function. Cell. 2010;141:142–53. doi: 10.1016/j.cell.2010.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rong YS, Golic KG. Gene targeting by homologous recombination in Drosophila. Science. 2000;288:2013–8. doi: 10.1126/science.288.5473.2013. [DOI] [PubMed] [Google Scholar]
- 27.Rong YS, Golic KG. A targeted gene knockout in Drosophila. Genetics. 2001;157:1307–12. doi: 10.1093/genetics/157.3.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Groth AC, Fish M, Nusse R, Calos MP. Construction of transgenic Drosophila by using the site-specific integrase from phage phiC31. Genetics. 2004;166:1775–82. doi: 10.1534/genetics.166.4.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Demerec M. Biology of drosophila. CSHL Press; 1994. [Google Scholar]
- 30.Cammarato A, Ocorr S, Ocorr K. Enhanced assessment of contractile dynamics in Drosophila hearts. Biotechniques. 2015;58:77–80. doi: 10.2144/000114255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wolf MJ, Amrein H, Izatt JA, Choma MA, Reedy MC, Rockman HA. Drosophila as a model for the identification of genes causing adult human heart disease. Proc Natl Acad Sci U S A. 2006;103:1394–9. doi: 10.1073/pnas.0507359103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vogler G, Ocorr K. Visualizing the beating heart in Drosophila. J Vis Exp. 2009 doi: 10.3791/1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Men J, Jerwick J, Wu P, Chen M, Alex A, Ma Y, et al. Drosophila Preparation and Longitudinal Imaging of Heart Function In Vivo Using Optical Coherence Microscopy (OCM) J Vis Exp. 2016 doi: 10.3791/55002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cooper AS, Rymond KE, Ward MA, Bocook EL, Cooper RL. Monitoring heart function in larval Drosophila melanogaster for physiological studies. J Vis Exp. 2009 doi: 10.3791/1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu M, Sato TN. On the mechanics of cardiac function of Drosophila embryo. PLoS One. 2008;3:e4045. doi: 10.1371/journal.pone.0004045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Papaefthmiou C, Theophilidis G. An in vitro method for recording the electrical activity of the isolated heart of the adult Drosophila melanogaster. In Vitro Cell Dev Biol Anim. 2001;37:445–9. doi: 10.1290/1071-2690(2001)037<0445:aivmfr>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 37.Bodmer R. Heart development in Drosophila and its relationship to vertebrates. Trends Cardiovasc Med. 1995;5:21–8. doi: 10.1016/1050-1738(94)00032-Q. [DOI] [PubMed] [Google Scholar]
- 38.Tao Y, Schulz RA. Heart development in Drosophila. Semin Cell Dev Biol. 2007;18:3–15. doi: 10.1016/j.semcdb.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 39.Miller A. The internal anatomy and histology of the image ofDrosophila melanogaster. New York: Harrier; 1950. [Google Scholar]
- 40.Gu GG, Singh S. Pharmacological analysis of heartbeat in Drosophila. J Neurobiol. 1995;28:269–80. doi: 10.1002/neu.480280302. [DOI] [PubMed] [Google Scholar]
- 41.Dowse H, Ringo J, Power J, Johnson E, Kinney K, White L. A congenital heart defect in Drosophila caused by an action-potential mutation. J Neurogenet. 1995;10:153–68. doi: 10.3109/01677069509083461. [DOI] [PubMed] [Google Scholar]
- 42.Johnson E, Ringo J, Dowse H. Native and heterologous neuropeptides are cardioactive in Drosophila melanogaster. J Insect Physiol. 2000;46:1229–36. doi: 10.1016/s0022-1910(00)00043-3. [DOI] [PubMed] [Google Scholar]
- 43.Johnson E, Sherry T, Ringo J, Dowse H. Modulation of the cardiac pacemaker of Drosophila: cellular mechanisms. J Comp Physiol B. 2002;172:227–36. doi: 10.1007/s00360-001-0246-8. [DOI] [PubMed] [Google Scholar]
- 44.Ocorr K, Reeves NL, Wessells RJ, Fink M, Chen HS, Akasaka T, et al. KCNQ potassium channel mutations cause cardiac arrhythmias in Drosophila that mimic the effects of aging. Proc Natl Acad Sci U S A. 2007;104:3943–8. doi: 10.1073/pnas.0609278104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ocorr K, Zambon A, Nudell Y, Pineda S, Diop S, Tang M, et al. Age-dependent electrical and morphological remodeling of the Drosophila heart caused by hERG/seizure mutations. PLoS Genet. 2017;13:e1006786. doi: 10.1371/journal.pgen.1006786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hofmann F, Biel M, Flockerzi V. Molecular basis for Ca2+ channel diversity. Annu Rev Neurosci. 1994;17:399–418. doi: 10.1146/annurev.ne.17.030194.002151. [DOI] [PubMed] [Google Scholar]
- 47.Isom LL, De Jongh KS, Catterall WA. Auxiliary subunits of voltage-gated ion channels. Neuron. 1994;12:1183–94. doi: 10.1016/0896-6273(94)90436-7. [DOI] [PubMed] [Google Scholar]
- 48.Robinson SW, Herzyk P, Dow JA, Leader DP. FlyAtlas: database of gene expression in the tissues of Drosophila melanogaster. Nucleic Acids Res. 2013;41:D744–50. doi: 10.1093/nar/gks1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lo PC, Frasch M. A role for the COUP-TF-related gene seven-up in the diversification of cardioblast identities in the dorsal vessel of Drosophila. Mech Dev. 2001;104:49–60. doi: 10.1016/s0925-4773(01)00361-6. [DOI] [PubMed] [Google Scholar]
- 50.Monnier V, Iche-Torres M, Rera M, Contremoulins V, Guichard C, Lalevee N, et al. dJun and Vri/dNFIL3 are major regulators of cardiac aging in Drosophila. PLoS Genet. 2012;8:e1003081. doi: 10.1371/journal.pgen.1003081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Smith LA, Wang XJ, Peixoto AA, Neumann EK, Hall LM, Hall JC. A Drosophila calcium channel alpha 1 subunit gene maps to a genetic locus associated with behavioral and visual defects. J Neurosci. 1996;16:7868–79. doi: 10.1523/JNEUROSCI.16-24-07868.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ryglewski S, Lance K, Levine RB, Duch C. Ca(v)2 channels mediate low and high voltage-activated calcium currents in Drosophila motoneurons. J Physiol. 2012;590:809–25. doi: 10.1113/jphysiol.2011.222836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Alayari NN, Vogler G, Taghli-Lamallem O, Ocorr K, Bodmer R, Cammarato A. Fluorescent labeling of Drosophila heart structures. J Vis Exp. 2009 doi: 10.3791/1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Viswanathan MC, Blice-Baum AC, Sang TK, Cammarato A. Cardiac-Restricted Expression of VCP/TER94 RNAi or Disease Alleles Perturbs Drosophila Heart Structure and Impairs Function. J Cardiovasc Dev Dis. 2016;3 doi: 10.3390/jcdd3020019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fink M, Callol-Massot C, Chu A, Ruiz-Lozano P, Izpisua Belmonte JC, Giles W, et al. A new method for detection and quantification of heartbeat parameters in Drosophila, zebrafish, and embryonic mouse hearts. Biotechniques. 2009;46:101–13. doi: 10.2144/000113078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McWilliam H, Li W, Uludag M, Squizzato S, Park YM, Buso N, et al. Analysis Tool Web Services from the EMBL-EBI. Nucleic Acids Res. 2013;41:W597–600. doi: 10.1093/nar/gkt376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gu H, Jiang SA, Campusano JM, Iniguez J, Su H, Hoang AA, et al. Cav2-type calcium channels encoded by cac regulate AP-independent neurotransmitter release at cholinergic synapses in adult Drosophila brain. J Neurophysiol. 2009;101:42–53. doi: 10.1152/jn.91103.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hara Y, Koganezawa M, Yamamoto D. The Dmca1D channel mediates Ca(2+) inward currents in Drosophila embryonic muscles. J Neurogenet. 2015;29:117–23. doi: 10.3109/01677063.2015.1054991. [DOI] [PubMed] [Google Scholar]
- 59.Iniguez J, Schutte SS, O’Dowd DK. Cav3-type alpha1T calcium channels mediate transient calcium currents that regulate repetitive firing in Drosophila antennal lobe PNs. J Neurophysiol. 2013;110:1490–6. doi: 10.1152/jn.00368.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Johnson E, Ringo J, Bray N, Dowse H. Genetic and pharmacological identification of ion channels central to the Drosophila cardiac pacemaker. J Neurogenet. 1998;12:1–24. doi: 10.3109/01677069809108552. [DOI] [PubMed] [Google Scholar]
- 61.Hagiwara N, Irisawa H, Kameyama M. Contribution of two types of calcium currents to the pacemaker potentials of rabbit sino-atrial node cells. J Physiol. 1988;395:233–53. doi: 10.1113/jphysiol.1988.sp016916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Perez-Reyes E. Molecular physiology of low-voltage-activated t-type calcium channels. Physiol Rev. 2003;83:117–61. doi: 10.1152/physrev.00018.2002. [DOI] [PubMed] [Google Scholar]
- 63.Perez-Reyes E, Cribbs LL, Daud A, Lacerda AE, Barclay J, Williamson MP, et al. Molecular characterization of a neuronal low-voltage-activated T-type calcium channel. Nature. 1998;391:896–900. doi: 10.1038/36110. [DOI] [PubMed] [Google Scholar]
- 64.Rossier MF. T-Type Calcium Channel: A Privileged Gate for Calcium Entry and Control of Adrenal Steroidogenesis. Front Endocrinol (Lausanne) 2016;7:43. doi: 10.3389/fendo.2016.00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zuhlke RD, Pitt GS, Deisseroth K, Tsien RW, Reuter H. Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature. 1999;399:159–62. doi: 10.1038/20200. [DOI] [PubMed] [Google Scholar]
- 66.Adams PJ, Ben-Johny M, Dick IE, Inoue T, Yue DT. Apocalmodulin itself promotes ion channel opening and Ca(2+) regulation. Cell. 2014;159:608–22. doi: 10.1016/j.cell.2014.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Imredy JP, Yue DT. Mechanism of Ca(2+)-sensitive inactivation of L-type Ca2+ channels. Neuron. 1994;12:1301–18. doi: 10.1016/0896-6273(94)90446-4. [DOI] [PubMed] [Google Scholar]
- 68.Ben Johny M, Yang PS, Bazzazi H, Yue DT. Dynamic switching of calmodulin interactions underlies Ca2+ regulation of CaV1.3 channels. Nat Commun. 2013;4:1717. doi: 10.1038/ncomms2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dick IE, Tadross MR, Liang H, Tay LH, Yang W, Yue DT. A modular switch for spatial Ca2+ selectivity in the calmodulin regulation of CaV channels. Nature. 2008;451:830–4. doi: 10.1038/nature06529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Taiakina V, Boone AN, Fux J, Senatore A, Weber-Adrian D, Guillemette JG, et al. The calmodulin-binding, short linear motif, NSCaTE is conserved in L-type channel ancestors of vertebrate Cav1.2 and Cav1.3 channels. PLoS One. 2013;8:e61765. doi: 10.1371/journal.pone.0061765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Qin N, Olcese R, Bransby M, Lin T, Birnbaumer L. Ca2+-induced inhibition of the cardiac Ca2+ channel depends on calmodulin. Proc Natl Acad Sci U S A. 1999;96:2435–8. doi: 10.1073/pnas.96.5.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhou J, Olcese R, Qin N, Noceti F, Birnbaumer L, Stefani E. Feedback inhibition of Ca2+ channels by Ca2+ depends on a short sequence of the C terminus that does not include the Ca2+ -binding function of a motif with similarity to Ca2+ -binding domains. Proc Natl Acad Sci U S A. 1997;94:2301–5. doi: 10.1073/pnas.94.6.2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bazzazi H, Ben Johny M, Adams PJ, Soong TW, Yue DT. Continuously tunable Ca(2+) regulation of RNA-edited CaV1.3 channels. Cell reports. 2013;5:367–77. doi: 10.1016/j.celrep.2013.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Evans PD, Maqueira B. Insect octopamine receptors: a new classification scheme based on studies of cloned Drosophila G-protein coupled receptors. Invert Neurosci. 2005;5:111–8. doi: 10.1007/s10158-005-0001-z. [DOI] [PubMed] [Google Scholar]
- 75.Pavot P, Carbognin E, Martin JR. PKA and cAMP/CNG Channels Independently Regulate the Cholinergic Ca(2+)-Response of Drosophila Mushroom Body Neurons(1,2,3) eNeuro. 2015:2. doi: 10.1523/ENEURO.0054-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Taylor SS, Buechler JA, Yonemoto W. cAMP-dependent protein kinase: framework for a diverse family of regulatory enzymes. Annu Rev Biochem. 1990;59:971–1005. doi: 10.1146/annurev.bi.59.070190.004543. [DOI] [PubMed] [Google Scholar]
- 77.Li W, Tully T, Kalderon D. Effects of a conditional Drosophila PKA mutant on olfactory learning and memory. Learn Mem. 1996;2:320–33. doi: 10.1101/lm.2.6.320. [DOI] [PubMed] [Google Scholar]
- 78.Bodi I, Mikala G, Koch SE, Akhter SA, Schwartz A. The L-type calcium channel in the heart: the beat goes on. J Clin Invest. 2005;115:3306–17. doi: 10.1172/JCI27167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bean BP. Two kinds of calcium channels in canine atrial cells. Differences in kinetics, selectivity, and pharmacology. J Gen Physiol. 1985;86:1–30. doi: 10.1085/jgp.86.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nilius B, Hess P, Lansman JB, Tsien RW. A novel type of cardiac calcium channel in ventricular cells. Nature. 1985;316:443–6. doi: 10.1038/316443a0. [DOI] [PubMed] [Google Scholar]
- 81.Zornik E, Paisley K, Nichols R. Neural transmitters and a peptide modulate Drosophila heart rate. Peptides. 1999;20:45–51. doi: 10.1016/s0196-9781(98)00151-x. [DOI] [PubMed] [Google Scholar]
- 82.Dulcis D, Levine RB. Glutamatergic innervation of the heart initiates retrograde contractions in adult Drosophila melanogaster. J Neurosci. 2005;25:271–80. doi: 10.1523/JNEUROSCI.2906-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ocorr KA, Crawley T, Gibson G, Bodmer R. Genetic variation for cardiac dysfunction in Drosophila. PLoS One. 2007;2:e601. doi: 10.1371/journal.pone.0000601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Peng IF, Wu CF. Drosophila cacophony channels: a major mediator of neuronal Ca2+ currents and a trigger for K+ channel homeostatic regulation. J Neurosci. 2007;27:1072–81. doi: 10.1523/JNEUROSCI.4746-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Worrell JW, Levine RB. Characterization of voltage-dependent Ca2+ currents in identified Drosophila motoneurons in situ. J Neurophysiol. 2008;100:868–78. doi: 10.1152/jn.90464.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sullivan KM, Scott K, Zuker CS, Rubin GM. The ryanodine receptor is essential for larval development in Drosophila melanogaster. Proc Natl Acad Sci U S A. 2000;97:5942–7. doi: 10.1073/pnas.110145997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bhattacharya A, Gu GG, Singh S. Modulation of dihydropyridine-sensitive calcium channels in Drosophila by a cAMP-mediated pathway. J Neurobiol. 1999;39:491–500. doi: 10.1002/(sici)1097-4695(19990615)39:4<491::aid-neu3>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 88.Gielow ML, Gu GG, Singh S. Resolution and pharmacological analysis of the voltage-dependent calcium channels of Drosophila larval muscles. J Neurosci. 1995;15:6085–93. doi: 10.1523/JNEUROSCI.15-09-06085.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ren D, Xu H, Eberl DF, Chopra M, Hall LM. A mutation affecting dihydropyridine-sensitive current levels and activation kinetics in Drosophila muscle and mammalian heart calcium channels. J Neurosci. 1998;18:2335–41. doi: 10.1523/JNEUROSCI.18-07-02335.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bub G, Camelliti P, Bollensdorff C, Stuckey DJ, Picton G, Burton RA, et al. Measurement and analysis of sarcomere length in rat cardiomyocytes in situ and in vitro. Am J Physiol Heart Circ Physiol. 2010;298:H1616–25. doi: 10.1152/ajpheart.00481.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Irving T, Wu Y, Bekyarova T, Farman GP, Fukuda N, Granzier H. Thick-filament strain and interfilament spacing in passive muscle: effect of titin-based passive tension. Biophys J. 2011;100:1499–508. doi: 10.1016/j.bpj.2011.01.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Maltsev VA, Lakatta EG. Normal heart rhythm is initiated and regulated by an intracellular calcium clock within pacemaker cells. Heart Lung Circ. 2007;16:335–48. doi: 10.1016/j.hlc.2007.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Maltsev VA, Vinogradova TM, Bogdanov KY, Lakatta EG, Stern MD. Diastolic calcium release controls the beating rate of rabbit sinoatrial node cells: numerical modeling of the coupling process. Biophys J. 2004;86:2596–605. doi: 10.1016/S0006-3495(04)74314-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yue DT, Backx PH, Imredy JP. Calcium-sensitive inactivation in the gating of single calcium channels. Science. 1990;250:1735–8. doi: 10.1126/science.2176745. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.