

Abstract
Background
Tetrastigma hemsleyanum is of great medicinal importance and used as a model system to address the evolutionary history of warm-temperate evergreen (WTE) forest biomes in East Asia over Neogene time scales. However, further studies on the neutral and adaptive divergence processes of T. hemsleyanum are currently impeded by a lack of genomic resources. In this study, we de novo assembled and annotated a reference transcriptome for two cpDNA lineages (Central-South-East vs. Southwest) of T. hemsleyanum. We further used comparative genomic and multilocus coalescent approaches to investigate the tempo and mode of lineage diversification in T. hemsleyanum.
Results
A total of 52,838 and 65,197 unigenes with an N50 of 1,667 and 1,841 bp for Central-South-East (CSE) and Southwest (SW) lineages, respectively, were recovered, and 6,692 putative orthologs were identified between the two lineages. Estimation of Ka/Ks ratios for these orthologs revealed that ten genes had Ka/Ks values significantly greater than 0.5 (P < 0.05), whereas 2,099 (Ka/Ks < 0.5, P < 0.05) were inferred to be under purifying selection. Based on three bioinformatic strategies, we identified a total of 1,018 single-copy nuclear genes (SCNGs) from the orthologs. We successfully designed eight nuclear gene primer pairs with high intraspecific variation (e.g. hT = 0.923, πT = 1.68×10-3), when surveyed across a subset of T. hemsleyanum individuals. Concordant with the previous cpDNA data, the haplotype networks constructed for most nuclear gene loci clearly identified the two lineages. A multilocus coalescence analysis suggested that the separation between the two lineages appears to have occurred during the mid-Pliocene. Despite their ancient divergence, both lineages experienced expansion at rather localized scales and have continued to exchange genes at a low rate.
Conclusions
This study demonstrated the utility of transcriptome sequencing as a basis for SCNG development in non-model species and the advantages of integrating multiple nuclear loci for phylogeographic and phylogenetic studies.
Electronic supplementary material
The online version of this article (10.1186/s12870-018-1429-8) contains supplementary material, which is available to authorized users.
Keywords: coalescent-based analyses, demographic history, gene flow, Ka/Ks, single-copy nuclear gene, Tetrastigma hemsleyanum, transcriptome
Background
Tetrastigma hemsleyanum Diels et Gilg (Vitaceae) is a diploid, perennial herb, with red berries dispersed by birds, bats and mammals, and is distinguished as the sole herbaceous climber of a genus that otherwise comprises only woody lianas [1–4]. This species is widely distributed throughout subtropical China, but also occurs rarely farther south on Hainan and Taiwan [5, 6]. The tubers of T. hemsleyanum have long been used in Chinese folk medicines to treat hepatitis, fever, pneumonia, rheumatism, and sore throat [7], along with other uses that exploit its anti-tumor effects [8, 9]. Thus, the species has been the subject of considerable phytochemical and pharmacological studies (e.g. [10]). In addition, since T. hemsleyanum is a typical component of warm-temperate evergreen (WTE) forest habitats in subtropical China, it has emerged as an excellent model species to address the evolutionary history of WTE forest biomes in eastern Asia over Neogene time scales.
Our previous phylogeographic survey using chloroplast (cp) DNA sequences showed that the modern range of T. hemsleyanum comprises two major cpDNA lineages, Southwest (SW) and Central-South-East (CSE) China. Moreover, our recent studies revealed that phenotypic traits, e.g. leaf size, tuber size, and the effect of phytochemical compounds, differ greatly between the two lineages [11]. The two major lineages likely diverged through climate/the uplift of QTP-induced vicariance of an ancestral southern range during the early Pliocene [6]. Nevertheless, given that this climber of the WTE forest is primarily dispersed by frugivorous birds [12, 13] and shows a nearly continuous range at present, we therefore presume that adaptive divergence processes may play an additional role in maintaining the separation of two cpDNA lineages of T. hemsleyanum inhabiting different floristic regions [6]. In fact, it has been recognized that Quaternary climatic changes have affected the demographic and adaptive processes in many species, especially in boreal and temperate regions undergoing glacial cycles [14–16]. However, a paucity of genetic resources such as genomic and transcriptomic sequences has made further studies on the neutral and adaptive divergence processes of T. hemsleyanum and other non-model species a challenging task.
Recent advances in next-generation sequencing (NGS) and bioinformatic tools have generated genome-scale information for both model and non-model species. Access to these massive sequence data provides researchers with exciting opportunities to make large-scale comparisons at genomic, exomic, or transcriptomic levels [17–19] and develop hundreds of informative, taxon-specific loci from nuclear genomes [20]. Massively parallel sequencing of RNA (RNA-Seq), in particular, has emerged as a powerful and cost-efficient tool to obtain the expressed sequences of the genome in non-model species when other genomic resources, such as a sequenced genome, are not yet developed. Through this approach, data are obtained on nucleotide variation as well as transcriptome characteristics and gene expression levels, substantially improving phylogeographic studies of population history, demography, genetic structure, and adaptive evolution [21]. For example, genome-wide scans and comparative transcriptome analysis not only provide the opportunity to estimate transcriptome-wide divergence and identify loci under selection [22, 23], but also enable the mining of polymorphic molecular markers such as single-copy nuclear genes (SCNG) and microsatellites (SSR) for population genetic and phylogeographic studies [24, 25]. With the increasing abundance of sequence data from across the genome, researchers can use multilocus coalescent methods to effectively estimate demographic parameters on a species tree to help disentangle the historical context of divergence and (incipient) speciation.
In this study, using the Illumina HiSeq3000 platform, we obtained RNA sequence data for two individual samples of T. hemsleyanum that correspond to the CSE and SW lineages [6], respectively. Our aims were to: 1) characterize the transcriptomes of the two lineages of T. hemsleyanum; 2) perform pairwise comparisons of the putatively orthologous sequences from these lineages to identify candidate genes under selection that might be involved in local adaptation and maintenance of lineage boundaries; 3) identify large numbers of putative SCNG and expressed sequence tag–simple sequence repeat (EST-SSR) markers and validate the polymorphism of each SCNG locus in a subset of T. hemsleyanum individuals; and 4) analyze SCNG sequence data with coalescent-based methods to estimate the history and timing of lineage divergence and the amount of post-divergence gene flow between the two lineages of T. hemsleyanum.
Results
De novo assembly and functional annotation of unigenes
After filtering and evaluating the raw reads, a total of 47,880,882 and 122,587,340 clean reads were generated by Illumina Sequencing from the cDNA libraries of the CSE and SW lineages, respectively (Table 1). The GC percentage and Q20 percentage (percentage of sequences with sequencing error rate lower than 1%) were 45.13% and 97.98%, respectively, for the CSE lineage and 46.10% and 98.15%, respectively, for the SW lineage (Table 1). Through de novo assembly, we obtained 101,421 contigs with a mean length of 413 bp and an N50 value of 1,001 bp for the CSE lineage, and 138,294 contigs with a mean length of 377 bp and an N50 value of 926 bp for the SW lineage (Table 1). For CSE lineage sequences, the contigs were assembled into 52,838 unigenes with an average length of 1,034 bp and an N50 value of 1,667 bp, while for the SW lineage, 65,197 unigenes were obtained with a mean length of 1,095 bp and an N50 value of 1,841 bp (Table 1). Detailed information on de novo assembly is summarized in Table 1, and the length distributions of the contigs and unigenes of the two lineages are shown in Additional file 1: Figure S1.
Table 1.
Summary statistics for the transcriptomes of CSE and SW lineages of Tetrastigma hemsleyanum.
| CSE lineage | SW lineage | |
|---|---|---|
| Total number of clean reads | 47,880,822 | 122,587,340 |
| Total length of clean reads (bp) | 4,309,273,980 | 12,258,734,000 |
| Total numbers of contigs | 101,421 | 138,294 |
| N50 value of contigs | 1001 | 926 |
| Mean length of contigs (bp) | 413 | 377 |
| Q20 percentage (%) | 97.98% | 98.51% |
| GC percentage (%) | 45.13% | 46.10% |
| Total numbers of unigenes | 52,838 | 65,197 |
| N50 value of unigenes (bp) | 1667 | 1841 |
| Mean length of unigenes (bp) | 1034 | 1095 |
Notes: Q20 percentage denotes the percentage of sequences with sequencing error rate lower than 1%; N50 means that the contig size such that 50% of the entire assembly is contained in contigs equal to or longer than this value, bp = base pair.
The sequence similarity searches found that 48,697 (92.16%) non-redundant unigene sequences from the CSE lineage and 58,622 (89.92%) from the SW lineage had at least one significant match to the priority-ordered protein databases (i.e. Nr, Nt, Swiss-Prot, KEGG, COG) (Table 2). For both lineages, a blastx top-hit species distribution of gene annotations showed the highest similarity to the closely related Vitis vinifera [35,546 of 38,451 hits in the CSE lineage (92.44%) vs. 38,689 of 43,172 hits in the SW lineage (89.62%)], followed by Amygdalus persica (0.98% vs 1.18%).
Table 2.
Annotation results of assembled unigenes from CSE and SW lineages.
| Functional annotations | CDS annotations | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Nr | Nt | Swiss-Prot | KEGG | COG | GO | All | Homolog | estscan | All | |
| CSE lineage | ||||||||||
| Number (N) | 38,451 | 47,387 | 24,383 | 22,887 | 14,520 | 28,064 | 48,697 | 49,915 | 1056 | 50,971 |
| N/All annotated (%) | 78.96 | 97.31 | 50.07 | 47.00 | 29.82 | 57.63 | 100 | |||
| N/All-unigene (%) | 72.77 | 89.68 | 46.15 | 43.32 | 27.48 | 53.11 | 92.16 | 94.47 | 2.00 | 96.47 |
| SW lineage | ||||||||||
| Number (N) | 43,172 | 56,208 | 27,246 | 25,659 | 16,932 | 30,177 | 58,622 | 54,373 | 1449 | 55,822 |
| N/All annotated (%) | 73.64 | 95.88 | 46.48 | 43.77 | 28.88 | 51.48 | 100 | |||
| N/All-unigene (%) | 66.22 | 86.21 | 41.79 | 39.36 | 25.97 | 46.29 | 89.92 | 83.40 | 2.22 | 85.62 |
Based on the Nr annotations, 28,064 (53.11%) unigenes for the CSE lineage and 30,177 (46.29%) unigenes for the SW lineage were assigned to at least one GO term annotation under three GO categories: biological process (CSE: 21,742, 41.15%; SW: 23,371, 35.85%), cellular component (CSE: 22,123, 41.87%; SW: 23,473, 36.00%) and molecular function (CSE: 21,296, 40.30%; SW: 23166, 35.53%) (Additional file 2: Figure S2). The GO category distributions of the unigenes for both lineages were highly similar. For each lineage, the two mostly highly represented level-2 categories of ‘biological process’ were ‘cellular process’ (CSE: 17,548, 62.5%; SW: 18,658, 61.8%) and ‘metabolic process’ (CSE: 17,411, 62.0%; SW: 18,592, 61.61%); of ‘metabolic process’, ‘cell’ (CSE: 20,754, 73.95%; SW: 22,093, 73.21%) and ‘cell part’ (CSE: 20,753, 73.95%; SW: 22,092, 73.21%); and of ‘molecular function’, ‘catalytic activity’ (CSE:13,703, 48.83%; SW: 14,599, 48.38%) and ‘binding’ (CSE: 14,006, 49.91%; SW: 15,749, 52.19%) Additional file 2: (Figure S2).
Prediction of protein-coding sequence (CDS) regions
According to the blast search results against various protein databases, we identified and extracted the CDS regions of 49,915 unigenes in the CSE lineage and 54,373 in the SW lineage, which were then translated into amino sequences with the standard codon table. The respective size distributions of these nucleotide sequences in each lineage (CSE/SW) were as follows: 100–1000 bp (27,219/30,342), 1,000–2,000 bp (7,762/8,660), and > 2000 bp (2,535/3,072) (see Additional file 1: Figure S1). Beyond that, 1,053 unigene sequences in the CSE lineage and 1,499 in the SW lineage with CDS were obtained using the estscan software prediction. The shortest estscan CDS were at least 200 bp, and the most abundant size class was 200–500 bp for both the CSE and SW lineages (831 vs. 1,182, respectively), constituting 78.70% and 81.57% of the total unigenes, respectively (Additional file 1: Figure S1).
Identification of orthologous contigs and comparison of substitution rates between lineages
Based on the predicted protein sequences, we identified 17,738 putative orthologous groups between the two lineages (CSE/SW) using orthomcl. The one-to-one, reciprocal best method of elucidating orthologous proteins generated 9,924 putative orthologous pairs (i.e. two sequences from the different lineages have higher blast scores with each other than with any other sequences in the other genome). After filtering gene pairs annotated with different protein products in the Swiss-Prot database, 6,692 pairs of putative orthologs were finally identified and used in the downstream analyses. Of these, 1,786 pairs had only synonymous or non-synonymous substitutions, and 4,906 pairs had both types of substitutions, for which the Ka/Ks ratio were calculated. The mean values of Ka, Ks, and the Ka/Ks ratio of all orthologous pairs were 0.011 ± 0.034, 0.051 ± 0.259, and 0.326 ± 0.438, respectively. Of the 4,906 pairs of putative orthologs, only 329 had a Ka/Ks ratio >1, and 840 had a Ka/Ks ratio between 0.5 and 1. Of the 1,169 putative orthologous pairs with a Ka/Ks value > 0.5, none had a Ka/Ks ratio significantly greater than 1, and only ten had a ratio significantly greater than 0.5 (see corresponding Ka/Ks ratios and gene functions in Table 3). By contrast, over half of the putative orthologous pairs (3,737, 55.84%) showed a Ka/Ks ratio < 0.5, of which 2,099 pairs had ratios significantly < 0.5 (P < 0.05).
Table 3.
List of candidate orthologs potentially under positive selection in the transcriptomes of CSE and SW lineages
| Gene ID | Ka/Ks value | P-value (Fisher) | Descriptions | |
|---|---|---|---|---|
| CSE lineage | SW lineage | |||
| Unigene21284 | CL1668_Contig1 | 4.13 | 0.002 | Type 2 ribosome-inactivating protein Nigrin l precursor |
| CL1485_Contig2 | CL876_Contig13 | 1.94 | 0.017 | TMV resistance protein N-like |
| Unigene9215 | CL4926_Contig1 | 0.55 | 0.001 | GDSL esterase/lipase EXL3 |
| CL7288_Contig1 | CL7933_Contig2 | 0.5 | 0.006 | IAA-amino acid hydrolase ILR1-like 4 |
| CL2482_Contig1 | CL3284_Contig1 | 0.51 | 0.015 | Probable disease resistance protein RDL6/RF9-like |
| Unigene1115 | Unigene8463 | 0.53 | 0.022 | Proline-rich receptor-like protein kinase PERK9 |
| Unigene22333 | CL1306_Contig1 | 0.52 | 0.029 | Probable glycosyltransferase At3g07620-like |
| CL291_Contig2 | CL4212_Contig1 | 0.59 | 0.036 | Armadillo repeat-containing protein 7 |
| CL6365_Contig1 | Unigene18910 | 0.52 | 0.046 | Glutamyl-tRNA(Gln) amidotransferase subunit A |
| CL1077_Contig1 | Unigene25101 | 0.52 | 0.049 | Probable disease resistance protein At1g15890 |
Taking a more appropriate threshold of 0.5 for the Ka/Ks ratio as an indicator of positive selection [26], ten putative orthologous pairs might have experienced relaxed purifying selection and/or unfixed mutation, whereas 2,099 were inferred to be under purifying selection. We clustered the above 2,109 putative orthologous pairs into three main GO categories: biological process (1,556, 74.13%), cellular component (1,572, 74.94%), and molecular function (1,459, 69.51%). Within the biological process category, the term ‘cellular process’ (1,269, 61.74%) and ‘metabolic process’ (1,249, 59.50%) were the most dominant. Within the cellular component category, ‘cell’ (1,503, 71.61%) and ‘cell part’ (1,502, 71.56%) represented the major subcategories. Within the molecular function category, the main functional subcategories are ‘binding’ (947, 45.12%) and ‘catalytic activity’ (911, 43.40%) (Additional file 2: Figure S2).
Identification and characterization of EST-SSRs
A total of 11,006 and 15,531 EST-SSRs were identified from 9,054 (CSE) and 12,486 (SW) unigenes, accounting for 17.14% and 19.15% of the total unigenes in CSE lineage (52,838) and SW lineage (65,197), respectively. From the SSR-containing unigenes, 1,590 (CSE) and 2,402 (SW) had more than two EST–SSR loci. The most abundant repeat types in the CSE lineage were trinucleotide (3,679, 33.43%), followed by dinucleotide (3,289, 29.88%), and mononucleotide (2,929, 26.61%). These three SSR repeat types were also found to be the most highly represented in the SW lineage (mononucleotide: 4,904, 31.58%; dinucleotide: 4,500, 28.97%; trinucleotide: 4,649, 29.92%) (Additional file 3: Figure S3). The dominant EST-SSRs identified here were A/T (CSE: 2,924, 29.54%; SW: 4,819, 34.29%), AG/CT (CSE: 2,381, 24.06%; SW: 3,236, 23.03%), and AAG/CTT (CSE: 1,022, 10.33%; SW: 1,316, 9.36%) (Additional file 4: Figure S4). Very few CG/CG repeats (CSE: 2, 0.02%; SW: 2, 0.01%) were identified in the two databases (Additional file 4: Figure S4).
To maximize the universal applicability of markers, we also searched for SSRs in the 6,692 pairs of putative orthologs, and found 286 SSRs distributed among 200 pairs of orthologs (Table 4). Of the 286 SSRs loci, 78 exhibited variation in the number of specific repeat units between the two lineages and should be the best choices for future population genetic studies of this genus.
Table 4.
Test results of polymorphism and neutrality for each SCNG screened in representative individuals of T. hemsleyanum
| Locus ID | S | N | Haplotype diversity (hT) | Nucleotide diversity (πT) | Test of neutrality | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Tajima’s D | P | Fu and Li’s D | P | Fu and Li’s F | P | |||||
| Th-41 | 22 | 19 | 0.960 | 0.00944 | -0.586 | P > 0.01 | -0.457 | P > 0.01 | -0.590 | P > 0.01 |
| ThR-3 | 41 | 32 | 0.991 | 0.01185 | -0.684 | P > 0.10 | 0.932 | P > 0.10 | 0.451 | P > 0.10 |
| ThR-6 | 41 | 31 | 0.989 | 0.01527 | -0.014 | P > 0.10 | 0.980 | P > 0.10 | 0.759 | P > 0.10 |
| ThR-7 | 40 | 15 | 0.923 | 0.01682 | 1.049 | P > 0.10 | -0.183 | P > 0.10 | 0.275 | P > 0.10 |
| ThR-11 | 23 | 25 | 0.979 | 0.00798 | -1.228 | P > 0.10 | -0.548 | P > 0.10 | -0.910 | P > 0.10 |
| ThR-28 | 31 | 21 | 0.960 | 0.00946 | -0.819 | P > 0.10 | -0.697 | P > 0.10 | -0.870 | P > 0.10 |
| ThR-31 | 24 | 19 | 0.919 | 0.01380 | -0.172 | P > 0.10 | -0.754 | P > 0.10 | -0.663 | P > 0.10 |
| ThR-34 | 25 | 27 | 0.981 | 0.01027 | -0.559 | P > 0.10 | 0.871 | P > 0.10 | 0.458 | P > 0.10 |
Number of polymorphic sites, N Numbers of haplotypes.
Mining of SCNGs and multilocus coalescence analyses
A total of 353 and 167 genes from transcriptome data of both the CSE and SW lineages were found to have hits against the SCNG sets of APVO and Vitaceae species, respectively. Of the 520 putative SCNGs, 34 were shared between the results of the above two approaches. In addition, 730 putative SCNGs were identified by markerminer, of which 19 were classified as ‘strictly single-copy’, and 711 as ‘mostly single-copy’ (Fig. 1, Additional file 5: Table S2). Among the 730 SCNGs identified by markerminer, 198 were detected by the first two approaches (Fig. 1). The final set consisted of 1,018 putative SCNGs (Fig. 1, Additional file 5: Table S2). These genes were most likely to be SCNGs in both lineages.
Fig. 1.

Distribution of gene ontology (GO) term categories in the transcriptomes of CSE and SW lineages. They-axis indicates the percent of unigenes (left y-axis), and the number of unigenes (right y-axis) per (sub-) category. Black bar, CSE lineage; grey bar, SW lineage.
Pairwise alignments of all the 1,018 SCNGs demonstrated that a large proportion of loci yielded medium to high levels of sequence divergence between the two lineages: approximately one third of those loci (346, 34%) were estimated to have greater than 1% (identity < 99%) sequence divergence, and nearly half (451, 44.3%) showed 0.5%–1% divergence for the inter-lineage comparisons (Additional file 6: Figure S5 and Additional file 5: Table S2). Notably, 417 of these putative SCNGs showed significant signals of purifying selection (Ka/Ks < 0.5, P < 0.05) (see blue dots in Additional file 6: Figure S5). To test the utility of the 1,018 putative SCNGs for phylogeographic and phylogenetic studies in T. hemsleyanum, 50 candidate loci were randomly selected for primer design, and 23 yielded a single band at the expected size range. Of these 23 primer pairs, 12 were confirmed to be polymorphic in the initial screening with the 15 individuals from the five populations by Sanger sequencing (primer sequences and amplification results presented in Additional file 5: Table S1 and Additional file 7: Figure S6). However, four loci (ThR-4, ThR-5, ThR-8, ThR-10 ) were found to be less informative, and were excluded from further analyses.
The eight candidate loci (Th-41, ThR-3, ThR-6, ThR-7, ThR-11, ThR-28, ThR-31, and ThR-34) that showed relatively high intraspecific variation were finally chosen to genotype all individuals listed in Additional file 5: Table S3. The aligned sequences of each of the eight amplified loci for the 30 samples from the two lineages ranged from 463 to 895 bp (see Additional file 5: Table S3 for accession numbers). We observed a total of 247 polymorphic sites, with the number for each locus varying from 20 to 41. For each locus, the number of haplotypes identified ranged from 19 to 32, with a mean of 24 haplotypes per locus. The levels of haplotype (hT) and nucleotide (πT) diversity were consistently high, with each marker varying from 0.909 to 0.993, and from 7.32 × 10-3 and 16.01 × 10-3, respectively (Table 4). At the lineage level, SW (h = 0.815; π= 5.32 × 10-3) exhibited, in general, lower levels of diversity than CSE (h = 0.992; π= 10.99 × 10-3). Tests of neutrality showed no significant departure from the neutral model for each locus (Table 4), so all the eight loci were retained for subsequent coalescence analyses. Most nuclear networks supported the split of the two lineages except for two loci (i.e. ThR11, ThR34), which were unresolved and therefore not conflicting (Additional file 8: Figure S7).
For the ima analysis, since cpDNA genealogical patterns [6] were mostly congruent with those retrieved from the nuclear loci, we employed the largest non-recombining blocks of the eight SCNG loci together with the previously obtained cpDNA sequences [6]. The maximum-likelihood estimates (MLEs) and the 90% highest probability density (HPD) intervals of the six ima-derived parameters are summarized in Table 5, and their marginal posterior probability (MPP) distributions are illustrated in Fig. 2. Based on the geometric average mutation rate calculated (V = 5.04 × 10-7 mutations per locus yr-1), these parameter estimates were converted to absolute values of years or individuals. We estimated the time of the split between the SW and CSE lineages at about 2,964,251 yr bp, with a 90% HPD interval ranging from 1,592,603 to 4,688,583 yr bp. (Table 5, Fig. 2). The current effective population size (Ne) of each descendant lineage (NSW: 6.67 × 105; NCSE: 8.45 × 105) was estimated to be much larger than that of the ancestral population (NA = 2.7× 105). Peak posterior estimates of post-divergence migrations from the CSE to SW lineage (mCSE-SW = 0.048), and vice versa (mSW-CSE = 0.073), were low (Table 5, Fig.2), corresponding to population migration rate estimates (2Nm= Θm/2) of 0.204 and 0.245 migrants per generation, respectively.
Table 5.
Maximum-likelihood estimates (MLE) and 90% highest posterior density (HPD) intervals of demographic parameters of T. hemsleyanum based on ima multi-locus analyses.
| Estimates | Θ CSE | Θ SW | Θ A | m cse-sw | m sw-cse | t | N CSE | N SW | N A | 2NCSEMCSE-SW | 2NSWMSW-CSE | T (years BP) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MLE | 8.518 | 6.718 | 2.728 | 0.048 | 0.073 | 1.494 | 845282 | 666590 | 270737 | 0.201 | 0.232 | 2,964,251 |
| HPD90HPDLO | 4.963 | 1.888 | 1.188 | 0.000 | 0.000 | 0.803 | 492425 | 187295 | 117835 | 0.000 | 0.000 | 1,592,630 |
| HPD90HPDHi | 12.69 | 12.96 | 4.412 | 0.133 | 0.226 | 2.362 | 1258971 | 1286259 | 437849 | 0.562 | 0.738 | 4,688,583 |
ΘCSE, ΘSW, ΘA represent the scaled effective population sizes (Ne) of CSE lineage, SW lineage of T. hemsleyanum, and the ancestral population, respectively. mcse-sw and msw-cse refer to the scaled migration rates forward in time from CSE to SW lineage and vice versa. t is the time since ancestral population splitting in mutational units. 2NCSEMCSE-SW and 2NSWMSW-CSE are the effective migration rates (number of migrants per generation). All estimates include the per gene mutation rate V (geometric mean of the mutation rates of all the loci). Parameters in the first six columns are scaled by the mutation rate, while the rest are scaled by years or individuals.
Fig. 2.
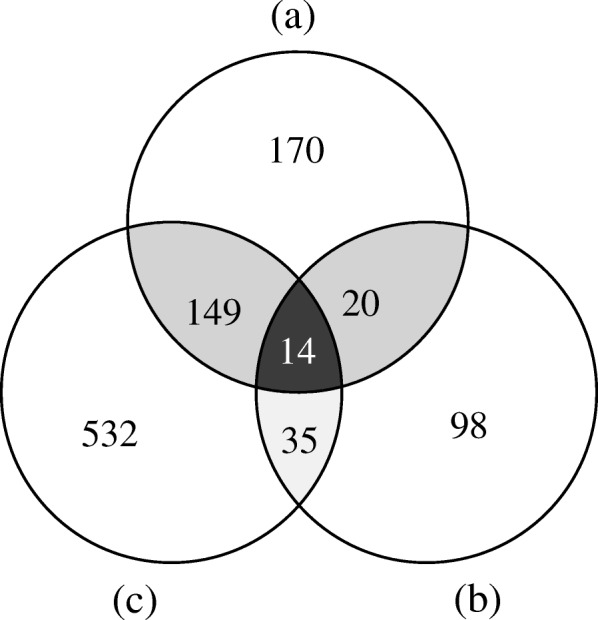
Each of the circles represents the number of SCNG loci homologous to the gene datasets of (a) APVO or (b) Vitaceae species, or detected by (c) the software markerminer. The grey and black overlapping area of the three circles correspond to the number of SCNGs shared between the results of the two, and three approaches, respectively.
Discussion
Assembly and annotation
We sequenced transcriptomes of geographically separated and genetically differentiated lineages (CSE vs. SW) of the perennial herb T. hemsleyanum. Our results are similar to the transcriptome assembly outcome of close relatives of T. hemsleyanum in Vitaceae using similar technologies (number of unigenes: 70,977–154,609; N50: 1,098–1,566; [27]). However, the number of protein-coding genes assembled for each lineage of T. hemsleyanum (CSE: 49,915 genes; SW: 54,373) is higher than that obtained in the Vitis vinifera genome (30,434 genes; [28]), indicating that our sequencing and assembly captured a considerable fraction of T. hemsleyanum protein-coding genes but that our assembly includes multiple unigenes that correspond to a single gene. Moreover, the tripartite nature of the Vitis genome (and of other Vitaceae as well as the common ancestor of all core eudicots) means that, in general, there are three copies of each gene relative to a gene from Arabidopsis thaliana, which has undergone extensive reduction in gene number relative to Vitis (e.g. [28]; Amborella Genome Project 2013). Thus, assembly of transcriptomes into unigenes likely produces an overestimate of the true gene copy number as cDNA reads are not all assembled into full-length genes.
A very high proportion of assembled unigenes matched blast searches to the known proteins in public databases (Nr, Swiss-Prot, KEGG, and COG) (CSE: 92.16%; SW: 89.92%) (Table 2). Functional annotations of these unigenes were highly similar between the two lineages (Fig. 3), in terms of both the types and relative frequencies of GO categories expressed, suggesting their overall similarity in gene expression profiles. A substantial portion of unigenes with blastx hits to Nr databases shared the highest sequence similarity with the closely related model species V. vinifera, which accounted for 92.44% and 89.62% in the CSE and SW lineages, repectively, with a very number hitting other plant groups. Only a small fraction of unigenes from both the CSE (7.84%) and SW (10.38%) lineages were not annotated or had no blast matches to protein databases. As expected, these sequences had a much smaller average sequence length (CSE: 342 bp; SW: 316 bp) and thus were less likely to obtain significant blastx matches [29, 30]. Nevertheless, some of them may also represent novel proteins unique to T. hemsleyanum, fast-evolving genes, or untranslated regions (UTRs) [23, 31].
Fig. 3.
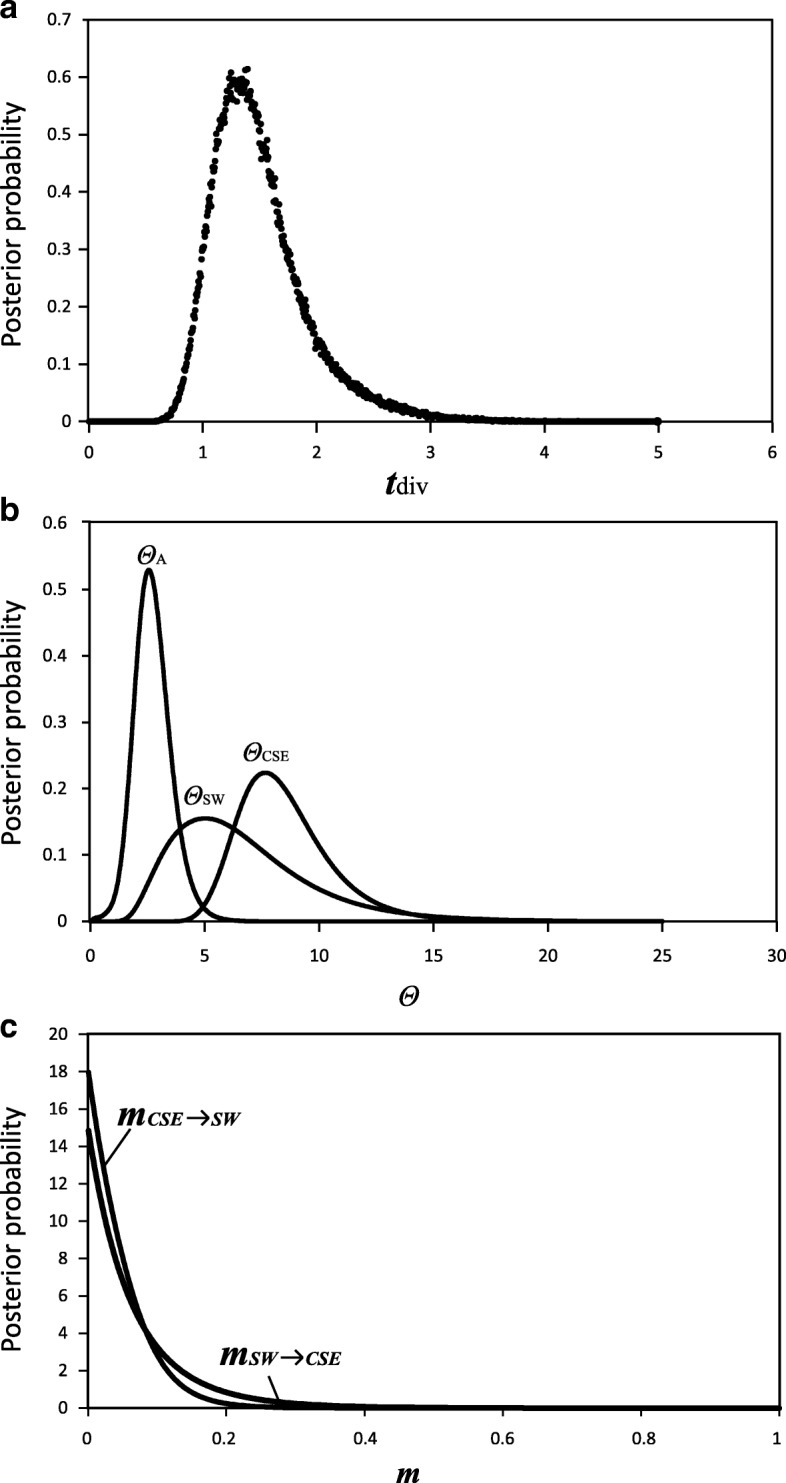
Marginal posterior probability (MPP) distributions of ima model parameters between the Central-south-east (CSE) and Southwest (SW) lineages identified in T. hemsleyanum: (a) the time (t) since ancestral population splitting in mutational units (b) the scaled effective population sizes of both lineages (ΘCSE, ΘSW), and the ancestral population (ΘA) (c) the scaled migration rates from CSE to SW lineage (mCSE-SW), and vice versa (mSW-CSE).
Nucleotide substitution effect on CDS of the two lineages
To describe genome-wide levels of coding sequence evolution and to estimate the effects of selection on lineage divergence, we calculated the ratio of Ka/Ks for orthologs of the CSE and SW lineages. This ratio has frequently been used as an indicator of frequency and mode of selection under which a protein-coding gene is evolving [32, 33]. Of the 4,906 orthologs shared between lineages that permit the calculation of Ka and Ks, only two gene pairs exhibited Ka/Ks ratios significantly >1, and an additional eight putative orthologs were above the less stringent threshold of 0.5 (Table 3). These genes were involved in several biological functions (e.g. disease resistance, metabolic process; Table 3) and may constitute candidates that are under the effect of positive selection or relaxed purifying selection and that are thus potentially associated with lineage divergence. Nearly half of the remaining orthologous pairs (2,099, 42.9%) had Ka/Ks ratios significantly < 0.5 (P < 0.05). Moreover, the average Ka/Ks ratio across all pairs is much lower than 1 (0.214), suggesting that purifying selection has a general influence on the evolution of most protein-coding regions of the two T. hemsleyanum lineages, as has been observed in other plants [34]. In fact, these genes most under the influence of purifying selection (i.e., with Ka/Ks near zero) contained primarily structural or “housekeeping” genes, e.g. tyrosyl-tRNA synthetase, transmembrane protein. As these genes are involved in processes that are crucial for organisms, purifying selection has acted to eliminate deleterious, nonsynonymous mutations [35, 36].
Overall, although we detected only a few genes under positive selection between the two lineages of T. hemsleyanum, selection might have played a role in shaping the lineage divergence of this species, given that the CSE and SW lineages are exposed to the Pacific monsoon (with cold winters and warm/humid summers) and the Indian monsoon (with rainy summers and autumns), respectively [37]. In this study, a peak Ks distribution between SW and CSE was observed at 0.051 ± 0.259. This value is higher than that found for two Primula species (0.027 ± 0.017; [38]) and falls within a mean Ks value of 0.03 – 0.10 between congeneric species [39], suggesting that the CSE and SW lineages may be at an incipient stage of allopatric speciation and may each merit species status (see below).
SCNG discovery and the estimation of divergence between lineages
In this study, we developed two sets of genetic markers, including nuclear microsatellites (EST-SSR) and single-copy nuclear genes, from 6,692 pairs of orthologous unigenes of the two T. hemsleyanum lineages. Compared with untranslated regions (UTRs), EST-SSRs located in protein-coding regions are proved to have higher transferability and equivalent levels of polymorphism [40]. Thus, the targeting of exonic repeat motifs might be the best strategy for developing portable sets of polymorphic EST-SSR markers [41]. The advantages of single-copy nuclear genes for phylogeographic and phylogenetic studies have long been recognized because of their high evolutionary rates and clear avoidance of paralogy [42, 43]. As a result, SCNGs have been shown to provide a valid alternative to nrDNA and chloroplast regions for resolving shallow-scale phylogenetics, intraspecific divergence, and population dynamics [6, 44]. SCNG databases have been reported from model plant species in many clades; these genes are expected to evolve in concordance with species evolution, thus providing invaluable resources for mining SCNGs from transcriptomes of non-model species. In this study, using the curated lists of SCNGs reported in Arabidopsis-Populus-Vitis-Oryza (APVO, [45]), Vitaceae [27], and 17 genomes broadly distributed across angiosperm phylogeny ([46]; implemented in markerminer, [20]) as the blast query, we identified 353, 167, and 730 putative SCNGs (1,018 in total) in T. hemsleyanum, respectively.
The pairwise overlapping SCNG numbers among the three gene sets were 34, 49 and 163, respectively, and only 14 genes were shared by all databases (Fig. 1). Such a low overlapping proportion of genes indicated that the SCNGs identified in different studies vary substantially with the reference databases employed for blast searches. As a result, we processed multiple queries against our putative orthologs, with SCNG datasets reported across different plant group or phylogenetic frameworks, in order to obtain a more comprehensive candidate list of SCNGs that we can use for further validation for both T. hemsleyanum and closely related species. Many of the SCNGs identified here, especially those orthologous to loci from Duarte et al. [45] and De Smet et al. [46], had general “housekeeping” functions, and are supposed to be conserved across species or even higher taxonomic levels. Nevertheless, based solely on exonic regions of these SCNGs, we estimated that 34% of the loci are > 1% divergent between intraspecific lineages of T. hemsleyanum, and over 78% showed levels of divergence > 0.5% (Additional file 6: Figure S5). These SCNG loci may represent more desirable choices for shallow-scale phylogeographic surveys when partial or complete intronic regions with faster evolutionary rates are captured along with their exonic counterparts [47].
Inference of genetic structure and demography based on multiple nuclear loci
We developed an efficient strategy for mining SCNG markers by comparative transcriptome analysis. By testing eight of the candidate markers, we illustrated the usefulness of these markers in population genetic and phylogeographic studies of T. hemsleyanum. These eight loci screened in a subset of individuals of T. hemsleyanum were informative and revealed ample polymorphism at the species level (hT: 0.919–0.991, πT: 0.008–0.017; Table 4). Concordant with the previous cpDNA data [6], haplotype networks reconstructed for each locus revealed that each lineage region mostly harboured a distinct set of haplotypes, and significant divergences were observed at most of the loci between haplotypes found in Southwest China and those restricted to Central or South-east China (Additional file 8: Figure S7). Thus, the general genealogical structure of haplotypes based on the nuclear genes combined revealed the same phylogeographic pattern as did the cpDNA genealogical structure. However, we believe the lack of geographic structure shown by a few loci (ThR11, ThR28, ThR34) is due to their longer coalescence times. The pattern of genetic structure we observed across cytoplasmic–nuclear data sets suggests long-term lineage/regional population isolation and differentiation over multiple glacial/interglacial cycles with little admixture.
Divergence dates estimated from gene trees, including phylogenies estimated from concatenated sequence data, can lead to overestimates of divergence times because gene divergence necessarily predates speciation [48]. In contrast, methods using multiple unlinked loci in conjunction with a coalescent framework, such as models of ‘isolation with migration’ (IM) implemented herein, have been proposed to provide more biologically realistic estimates because multiple unlinked genetic loci provide independent realizations of divergence history, accounting for mutational and coalescent stochasticity [48, 49]. In the present study, our ima analysis of combined multi-locus data (cpDNA and SCNGs) revealed that the separation between the SW and CSE lineages most likely occurred during the mid-Pliocene, c. 2.96 Ma (90% HPD: 1.59–4.69 Ma) (Fig. 2, Table 5). The estimate of lineage divergence time is more recent than that inferred from the previous fossil-calibrated phylogenetic dating of Vitaceae based on cpDNA sequences (c. 5.07 Ma). In fact, the new estimate provided here coincides with a long transition from a warm and stable global climate towards a cooler regime during the Piacenzian (3.60–2.58 Ma) [50, 51]. It seems likely, therefore, that the cooling and aridification at the Pliocene-Pleistocene boundary may have induced an ancient vicariant event, which promoted lineage divergence in T. hemsleyanum.
In terms of the demographic history, the previous results inferred from mismatch analyses of cpDNA alone [6] suggest long-term population stability of the SW lineage and latitudinal range shifts of the CSE lineage. In contrast, the current ima analysis suggested a somewhat larger effective population size (Ne) in both lineages compared to their ancestral population (Table 3, Table 5). The coalescent analyses using more rapidly evolving nuclear genes can trace more recent demographic events [52, 53]. Therefore, based on multiple nuclear genes, our ima analysis may reflect the recent demographic expansion signal in each lineage. Considering the pronounced geographic structure observed in T. hemsleyanum across two genetic data sets (SCNGs and cpDNA), the recent expansion dynamics, even for the CSE lineage, in which NCSE is larger than ancestral NA by 2–3 orders of magnitude, likely occur at localized scales. Our coalescent analyses show that despite their ancient divergence, both lineages have continued to exchange genes at a low rate (mCSE-SW = 0.048; mSW-CSE = 0.073). This effective rate of gene exchange between the two lineages corresponds to less than one immigrant per generation (2Nm < 0.25). Thus, this climate-induced range expansion at regional scales during inter-/postglacial periods presumably provided recurring opportunities for gene exchange between diverging lineages through secondary contact zones, for example, possibly at the southeastern Yungui Plateau. This hypothesis is also supported by previous ENM results [6]. In addition, a few genes involved in resistance to biotic and abiotic stresses were found to be under positive selection between the two lineages, suggesting that ecological forces may be important in the divergence and potentially in the maintenance of lineage boundaries of T. hemsleyanum. Thus, the two lineages might have undergone divergent evolution in physiological and/or life history traits resulting from adaptation to different eco-climatic conditions. We consider this as a viable hypothesis because the CSE lineage is exposed to the Pacific monsoon, whereas the SW lineage is affected by the Indian monsoon [54]. Future work that includes codon-level analyses, genome-wide association analyses, studies of genomic patterns of introgression along secondary contact zones, and larger numbers of samples from additional localities will shed more light on the specific loci that are most important in speciation, species integrity, and adaptation [55]. However, in view of their differences in some morphological traits and phytochemical composition, together with their deep genetic divergence at nuclear gene loci and cpDNA regions, the two lineages may represent distinct phylogenetic species, irrespective of whether they are reproductively isolated. The results of future studies may therefore have taxonomic implications as well.
Conclusions
The results illustrate the utility of transcriptome sequencing as a basis for single-copy nuclear gene development in non-model species. Analyses of the eight SCNGs combined revealed the same two groups as did the cpDNA. Our coalescent analyses of the combined data sets (SCNGs and cpDNA) suggested that T. hemsleyanum experienced a dichotomous split at c. 2.96 Ma and that, although the two groups have increased in population size, the lineages have remained isolated with limited gene flow. Accordingly, the present study demonstrated that multilocus coalescence analyses can improve estimates of process parameters such as divergence time and population expansion and thus offer a powerful complementary data set to cpDNA markers alone in tracing the evolutionary history of recently diverged lineages or species. In addition, comparative transcriptome analysis offers preliminary support for the hypothesis that ecological forces may be important in the divergence and potentially in the maintenance of lineage boundaries of T. hemsleyanum. Overall, our transcriptome analysis provides a solid foundation for future studies of gene expression, natural selection, and speciation in Tetrastigma.
Methods
Plant materials, total RNA extraction, and Illumina sequencing
Fresh juvenile leaf samples of T. hemsleyanum were harvested from the two major chloroplast lineages identified in our previous phylogeographic study [6]. As the CSE lineage contained two cpDNA sublineages (South/East and Central) [6], leaf materials of the CSE lineage were obtained in July 2014 from one individual in South/East sublineages (Ningbo, Zhejiang Province; 30.35°N, 122.32°E, alt. 162 m) and one individual in Central sublineages (Malipo, Yunnan Province; 23.12°N, 104.84°E, alt. 1700 m), while those of the SW lineage were collected in April 2015 from one individual in Guilin Botanical Garden, Guangxi Province (transplanted from Quanzhou, Guangxi Province; 25.08°N, 110.30°E, alt. 170 m). No specific collecting permits required for the collection of plant materials. Leaf tissue samples were frozen in liquid nitrogen and stored at -80°C immediately until total RNA extraction. For each individual, total RNA was extracted using a modified CTAB method. Equal amounts of high-quality RNA from two individuals of the CSE lineage were subsequently pooled into a single lineage sample. After assessing RNA quality using an Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA), the quantified total RNAs were sent to Beijing Genome Institute (BGI, Shenzhen, China) for further processing. The cDNA libraries were constructed using a cDNA Synthesis Kit (Illumina, Inc., San Diego, CA, USA) following the manufacturer’s instructions and evaluated with an Agilent 2100 Bioanalyzer and ABI StepOnePlus real-time PCR system prior to Illumina sequencing. Paired-end sequencing (2 x 90 bp) was then performed using a HiSeq3000 (Illumina, Inc, San Diego, CA, USA). Raw sequence reads were deposited in the NCBI Sequence Read Archive (SRA) with accession numbers SAMN07520791 (CSE lineage) and SAMN07502792 (SW lineage).
Sequence cleaning, de novo assembly, and gene annotation
Raw sequence reads were processed using the filter_fq program (BGI, Shenzhen, China) to remove sequencing adaptors, low-quality reads with percentage of unknown nucleotides ‘N’ higher than 5%, and percentage of low-quality bases (quality scores < 10) higher than 20%. Sequences that were retained following these filtering steps were de novo assembled using trinityv20131110 [56] with the default parameters. For each lineage sample, the assembled unigenes were further processed by sequence splicing and redundancy removal using tgicl v2.1 [57] to acquire the final non-redundant unigene dataset.
All unigenes were queried against the National Center for Biotechnology Information (NCBI) non-redundant (Nr) protein database, the SwissProt protein database (http://www.expasy.ch/sprot), the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways database, and the Cluster of Orthologous Groups (COG) database (http://www.ncbi.nlm.nih.gov/COG/) using blastx [58] with an E-value cut-off of 1e-5. If the aligning results from different databases were not in accordance with each other, a priority order of Nr, Swiss-Prot, KEGG, and COG was followed. Gene ontology (GO) terms [59] of the unigenes were obtained using blast2go v2.6.0 [60] based on the best blastx hits from the NCBI Nr database with an E-value cut-off of 1e-5. The distributions of level-2 GO terms were plotted with functional classification using wego [61].
Prediction of protein-coding sequence (CDS) regions
The coding region sequences (CDS) of unigenes were predicted according to the blast results against the Nr, Swiss-Prot, KEGG, and COG protein databases in that order (E-value < 1e-5). Unigenes with hits against the high-priority database were not aligned to those of lower priority. The CDS regions of unigenes that could be aligned to the databases were defined based on blastx results and were translated into peptides using the standard codon table. Unigenes without hits against any of the above four databases were screened by estscan v2.1 [62] to predict CDS region, determine the nucleotide sequence direction (5'–3'), and translate into peptide sequences. Both nucleotide and protein sequences of the unigene coding regions were obtained for further analysis.
Identification of orthologous contigs between lineage-specific transcriptomes and estimation of substitution rates of putative orthologous pairs
In principle, explicit phylogenetic analysis is the most appropriate method for disentangling orthologous and paralogous genes, but they are computationally expensive to construct for large numbers of genes. Previous studies comparing tree-based analysis and heuristic algorithms indicated that, despite conceptual differences, they produce similar sets of orthologs, especially at short evolutionary distances [63]. As a result, in this study, ortholog detection was conducted by similarity methods. Firstly, the predicted CDS regions of both transcriptomes were used as queries and targets respectively to search against those of the other species using all-versus-all blastp method [58]. The protein-coding sequences with unexpected stop codons in the Blast hit region and/or shorter than 150 bp in length were removed. The best hits of the longest isoforms with E-value < 1e-5 were retrieved. Orthologous pairs with identity < 60% were excluded and only 1:1 orthologous pairs in both lineages were retained. Next, based on the blast results, Markov clustering was conducted using orthomcl v2.0.9 [64] with default settings. Finally, we compared the pairs of sequences against the Swiss-Prot database (E-value < 1e-5), and only those gene pairs that mapped unambiguously to the same protein were retained as orthologous genes. The software kaks_calculator v1.2 [65] was employed to estimate nonsynonymous (Ka) and synonymous (Ks) substitution rates, and Ka/Ks ratios of each putative orthologous pair using the YN [66] algorithm.
Identification of EST-SSRs and mining of SCNGs
A Perl script known as MIcroSAtellite (MISA, http://pgrc.ipkgatersleben.de/misa) [67] was employed to identify and localize the mono-to-hexanuclecotide SSR motifs from each of the two non-redundant unigene datasets and also from the putative orthologous pairs identified between lineages. Primers with a repeat-unit length of at least 16 bp were designed for each SSR-containing sequence using primer premier v6.0 (Premier Biosoft International, Palo Alto, CA, USA). The putative function of SSR-containing sequences was obtained from the results of blastx searches of the Cluster of Orthologous Groups (COG) database (http://www.ncbi.nlm.nih.gov/COG/).
In this study, three methods were used for mining single-copy nuclear genes (SCNGs). The first two approaches employed data from the results of two previous studies that used algorithms to identify putative SCNGs at wider taxonomic scales: [58] the 959 SCNGs shared by four model plants (APVO, Arabidopsis thaliana, Populus trichocarpa, Vitis vinifera, and Oryza sativa) from the TAIR10 database [45]; and [59] the 417 SCNGs extracted from the transcriptomes of 15 species of Vitaceae [27]. The protein sequences encoded by the above published SCNGs were then queried against the orthologous genes between the lineages of T. hemsleyanum using orthomcl v2.0.9 [64] with default settings. All of the queries with top reciprocal blast hits were considered to be putative SCNGs in T. hemsleyanum. In a third approach, we used markerminer v1.0 [20] to infer the SCNGs between lineages via the iPlant Collaborative Atmosphere cloud-computing infrastructure (https://www.cyverse.org/; [68]). In this analysis, we chose the proteome of V. vinifera from the plaza v2.5 database [69] as a reference to filter the putative orthologous pairs. Then a user-specified SCNG reference [46] implemented in markerminer v1.0, which included 177 ‘strictly SCNGs’ (single-copy in all 17 angiosperm reference genomes) and 2,809 ‘mostly SCNGs’ (with duplicates detected in at least one to as many as three other genomes), was chosen as a final data filter for SCNGs. Putative orthologous pairs between lineages whose transcripts have top reciprocal BLAST hits against the reference proteins were retained and classified as putative SCNGs. The sequence difference for each pair of SCNG (identified by all three approaches) of the two T. hemsleyanum lineages was observed by sequence alignments and the calculations of pairwise identities performed in muscle v3.8.31 [70]. To predict the intron-exon boundaries of all SCN loci and approximate the intron size, the reference CDS of V. vinifera containing introns were aligned to their respective SCNG alignments of the focal species using mafft v7 [71].
SCNGs validation and multilocus coalescence analyses
To assess the utility of SCNG markers in phylogeographic inference, we designed primers and tested their amplifications in a subset of T. hemsleyanum individuals. Specifically, we selected SCNG loci for primer design at random using primer premier v6.0 (Premier Biosoft International, Palo Alto, CA) and verified whether the following three criteria were met for each locus. The locus: [58] was identified as SCNG by at least two bioinformatic approaches; [59] possessed 20%–60% intron content to increase phylogenetic resolution among recently diverged lineages; and [17] ranged in length from approximately 600 to 1200 bp to facilitate the subsequent PCR and sequencing steps.
We selected 50 loci for primer design following standard primer design guidelines and aimed for exon-anchored primers with a length of 18–24 bp, a GC content of 40–60%, melting temperature (Tm) = 55–62°C, and without repeats, runs, secondary structures such as hairpins, dimers, and cross-dimers. Priming sites containing ambiguous bases due to intraspecific polymorphism were not allowed.
Between one and three pairs of primers for each selected locus were synthesized by Eurofins Genomics (Huntsville, Alabama, USA). To assess the primer performance, we tested the amplifications in a set of T. hemsleyanum individuals (see Additional file 5: Table S3) representing all of the chloroplast haplotypes and populations reported in the previous study [6] via PCR. Our PCR recipe (25-μL reactions) was as follows: 5 μL of 5×Buffer (Mg+2 free), 1.25 μL MgCl2 (50 mM), 2 μL dNTP (5 mM), 1.25 μL of each primer (10 μM), 0.25 μL (5 units/μL) of Taq polymerase, and 60 ng template DNA (1.5 μL). We adopted the following PCR cycling conditions in a Biometra T3 Thermocycler (Whatman Biometra, Goettingen, Germany): a denaturing step at 94°C for 5 min, followed by 35 cycles of 30 s at 94°C, annealing at a specific temperature (optimized Tm for each locus presented in Additional file 5: Table S1) for 30 s and extension for 60 s, and a final extension for 10 min at 72°C.
For primer pairs that consistently yielded a single band in at least 90% of the individuals tested, we performed bidirectional sequencing for the PCR products at the University of Florida Interdisciplinary Center for Biotechnology Research. When more than one band amplified, we isolated bands, reamplified, and sequenced directly. Individual alleles (haplotypes) were determined from diploid nuclear loci using the software phase v2.1.1 [72], considering a threshold of 60% (p = q = 0.6) to reduce the number of genotype uncertainties with little or no increase in false positives [73]. The input files for software phase were created in seqphase [74]. If cloning was necessary, PCR products were purified, ligated into PMD19-T vector (Takara), and transformed into DHB-5α–competent cells (Invitrogen, Carlsbad, California, USA), reamplified, and sequenced. All of the generated sequences were edited, assembled, and aligned in geneious v7.1.7 [75].
We estimated the numbers of polymorphic sites, numbers of haplotypes, levels of nuclear sequence diversity (πT) for these SCNGs, and carried out tests of departure from the neutral model based on Tajima’s D [76] and Fu & Li’s D* and F* statistics [77] using dnasp v5.1 [78]. Genealogical relationships of the haplotypes identified at each nuclear locus were constructed from a 95% statistical parsimony network using tcs v1.21 [79]. To infer a more robust divergence and demographic history of T. hemsleyanum, we used the ‘isolation with migration’ model (IM) [80, 81] as implemented in ima2 [82] to estimate population rate parameters (Θ) and effective population sizes (Ne) of the SW lineage in Southwest China (ΘSW), the CSE lineage in Central-South-East China (ΘCSE), and their common ancestral population (ΘA; Θ = 4Neu), as well as bidirectional migration rates (mSW–CSE and mCSE–SW; M = m/u) and divergence times (τ = tu) between the two lineages. All parameters in the IM model are scaled by the neutral mutation rate (u). In our coalescence analyses, we jointly employed the eight nuclear loci identified here and the previously obtained sequences of three cpDNA regions (petL–psbE, trnK–matK, rbcL), because the cpDNA genealogical patterns [6] were mostly congruent with those retrieved from the nuclear loci (see Results). IM model involves several simplifying assumptions, e.g. no recombination within loci, free recombination among loci, no population structure within each species or populations, no genetic contribution from unsampled populations or species, and selective neutrality [80, 81]. However, recent simulation studies have revealed that ima parameter estimates are robust to small to moderate violations of IM model assumptions, and to significant levels of recombination when data sets are pared down to apparently nonrecombining blocks [83]. In this study, we determined the longest non-recombining block for each locus using the program imgc [84].
We applied the infinite sites (IS) model of nucleotide substitution to nuclear loci and the Hasegawa, Kishino and Yano (HKY) model to chloroplast loci as suggested in the ima2 software manual ([80]; https://bio.cst.temple.edu/~hey/software). We used locus-specific inheritance scalars to account for autosomal (nuclear loci = 1) and maternal (chloroplast loci = 0.5) inheritance. Based on the estimated divergence time and the average divergence of sequences between SW and CSE lineages (c. 5.07 Ma) [6], the substitution rate (substitution /site/year) for each locus was calculated as between 10-10 and 10-9 s/s/y. For the combined cpDNA sequences, an average substitution rate of 5×10-10 s/s/y, as estimated from our previous clock-calibrated beast tree of Tetrastigma, was adopted [74].
The geometric average mutation rate of the two marker sets was used to rescale the ima parameter estimates from the combined analysis. After deleting the first 105 generations as burn-in, five independent Markov Chain Monte Carlo (MCMC) runs of 10 million generations were conducted under a geometric heating scheme (hn = 40; ha = 0.979, hb = 0.60), and trees were sampled every 100 steps. The convergence of parameter estimates was assessed by monitoring the effective sample size (ESS > 300), the swapping rates between successive chains of MCMC, and the trend-line plots of the parameters.
Additional files
Figure S1. Size frequency distribution of contigs (a, CSE lineage; b, SW lineage) and unigenes (c, CSE lineage; d, SW lineage). (XLS 1093 kb)
Figure S2. Distribution of gene ontology (GO) classifications for the 3737 orthologous pairs with Ka/Ks ratios significantly < 0.5. The y-axis indicates the percent (left y-axis) or number of unigenes (right y-axis) per (sub-) category. (PDF 436 kb)
Figure S3. Frequency distribution of EST-SSR unit size in the transcriptomes of CSE and SW lineages. (PDF 348 kb)
Figure S4. Frequency distribution of EST-SSR repeat motifs (mono- to tri-nucleotide motifs) between CSE and SW lineages. (PDF 337 kb)
Table S1. Characteristics of the twelve nuclear primer pairs newly developed in this study based on SCNG loci identified by different approaches. Table S2. Full list of 1018 SCNG loci identified from the transcriptomes of CSE and SW lineage, with their basic information including identification approaches, substitution rates between lineages and gene annotations. '+' denotes the approaches that supported the single-copy status of the locus. Table S3. List of individuals included in this study, with corresponding sampling localities and GenBank accession numbers. (PDF 415 kb)
Figure S5. The pairwise identity of each SCNG pair between the two lineages. Each dot in the scatter diagram denotes a SCNG locus, and the blue colored ones represent those genes under purifying selection). (PDF 177 kb)
Figure S6. Agarose-gel electrophoresis patterns of a subset of SCNG primer pairs for T. hemsleyanum (PDF 148 kb)
Figure S7. TCS-derived network of genealogical relationships among the identified haplotypes for each of the SCNG locus (PDF 198 kb)
Acknowledgements
All data analysis and PCR experiments were performed at the University of Florida; we thank Clayton Visger and Xiaoxian Liu for their help in data analysis and Dr. Evgeny Mavrodiev for great support with laboratory work.
Funding
This research was supported by the National Natural Science Foundation of China (grant Nos. 31700193, 31570214), Zhejiang Provincial Natural Science Foundation (LY14C020002), and Biodiversity International Collaborative Project of NSFC-NSF (No. 31461123001). In all cases, the funding bodies played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Availability of data and materials
Raw sequence reads of the transcriptomes of CSE and SW lineage were deposited in the NCBI Sequence Read Archive (SRA) with accession numbers SAMN07520791 and SAMN07502792, respectively. All SCNG sequences obtained in this study are deposited in GenBank (Accession nos. listed in Additional file 5: Table S3, Supporting information).
Abbreviations
- APVO
Arabidopsis-Populus-Vitis-Oryza
- BGI
Beijing Genome Institute
- CDS
coding region sequences
- COG
Cluster of Orthologous Groups
- cpDNA
Chloroplast DNA
- CSE
Central-South-East
- EST-SSR
Expressed sequence tag–simple sequence repeat
- GO
Gene ontology
- HKY model
Kishino and Yano model
- HPD
Highest probability density
- IM
Isolation with migration
- Ka
Nonsynonymous
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- Ks
Synonymous
- MCMC
Markov Chain Monte Carlo
- MISA
MIcroSAtellite
- MLEs
maximum-likelihood estimates
- MPP
marginal posterior probability
- NCBI
National Center for Biotechnology Information
- NGS
Next-generation sequencing
- Nr
Non-redundant
- RNA-Seq
Sequencing of RNA
- SCNGs
Southwest (SW) single-copy nuclear genes
- SRA
Sequence Read Archive
- WTE
Warm-temperate evergreen
Authors’ contributions
YHW, CXF and YXQ conceived and designed the project. WMJ collected the samples. YHW and MAG performed the experiments and analyzed dataset. YHW drafted the manuscript. WQY, PSS, DES, and YXQ revised the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable. Our study did not involve endangered or protected species. There is no need to get a permit for collection of plant material from natural habitats. And Professor Yingxiong Qiu should be contacted for future permissions.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Yihan Wang, Email: wyiris6258@126.com.
Weimei Jiang, Email: wmjiang@zju.edu.cn.
Wenqing Ye, Email: yewenqing@zju.edu.cn.
Chengxin Fu, Email: cxfu@zju.edu.cn.
Matthew A Gitzendanner, Email: magitz@ufl.edu.
Pamela S Soltis, Email: psoltis@flmnh.ufl.edu.
Douglas E Soltis, Email: dsoltis@ufl.edu.
Yingxiong Qiu, Email: qyxhero@zju.edu.cn.
References
- 1.Chen P, Chen L, Wen J. The first phylogenetic analysis of Tetrastigma (Miq.) Planch. the host of Rafflesiaceae. Taxon. 2011;60:499–512. [Google Scholar]
- 2.Moran C, Catterall CP, Kanowski J. Reduced dispersal of native plant species as a consequence of the reduced abundance of frugivore species in fragmented rainforest. Biological Conservation. 2009;142:541–552. doi: 10.1016/j.biocon.2008.11.006. [DOI] [Google Scholar]
- 3.Tiffney BH, Barghoorn ES. Flora of the Brandon lignite. IV. Illiciaceae. Am J Botany. 1979;66:321–329. doi: 10.1002/j.1537-2197.1979.tb06230.x. [DOI] [Google Scholar]
- 4.Wen J. Vitaceae. In: Kubitzki K, editor. Flowering Plants-Eudicots. Berlin: Springer-Verlag; 2007. pp. 467–479. [Google Scholar]
- 5.Dai Y, Shen Z, Ying L, Wang LL, Hannaway D, Lu HF. Effects of shade treatments on the photosynthetic capacity, chlorophyll fluorescence, and chlorophyll content of Tetrastigma hemsleyanum Diels et Gilg. Environmental and Experimental Botany. 2009;65:177–182. doi: 10.1016/j.envexpbot.2008.12.008. [DOI] [Google Scholar]
- 6.Wang YH, Jiang WM, Comes HP, Hu FS, Qiu YX, Fu CX. Molecular phylogeography and ecological niche modelling of a widespread herbaceous climber, Tetrastigma hemsleyanum (Vitaceae): insights into Plio–Pleistocene range dynamics of evergreen forest in subtropical China. New Phytol. 2015;206:852–867. doi: 10.1111/nph.13261. [DOI] [PubMed] [Google Scholar]
- 7.Shao Q, Deng Y, Fang H, Zhao X. Optimization of polysaccharides extraction from Tetrastigma hemsleyanum Diels et Gilg using response surface methodology. Int J Biol Macromol. 2011;49:958–962. doi: 10.1016/j.ijbiomac.2011.08.015. [DOI] [PubMed] [Google Scholar]
- 8.Feng Z, Hao W, Lin X, Fan D, Zhou J. Antitumor activity of total flavonoids from Tetrastigma hemsleyanum Diels et Gilg is associated with the inhibition of regulatory T cells in mice. Onco Targets Ther. 2014;7:947–956. doi: 10.2147/OTT.S61794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peng X, Zhang YY, Wang J, Ji Q. Ethylacetate extract from Tetrastigma hemsleyanuminduces apoptosis via the mitochondrial caspase-dependent intrinsic pathway in HepG2 cells. Tumor Biol. 2016;37:865–876. doi: 10.1007/s13277-015-3579-8. [DOI] [PubMed] [Google Scholar]
- 10.Xu CJ, Ding GQ, Fu JY Meng J, Zhang RH, Lou XM. Immunoregulatory effects of ethyl-acetate fraction of extracts from Tetrastigma Hemsleyanum Diels et. Gilg on immune functions of ICR mice. Biomed Environ Sci. 2008;21:325–331. doi: 10.1016/S0895-3988(08)60050-1. [DOI] [PubMed] [Google Scholar]
- 11.Jiang WM. Studies on germplasm evaluation and quality controltechnology of Tetrastigma hemsleyanum. PhD Dissertation. Hangzhou: Zhejiang University; 2015.
- 12.Chen P, Wen J, Chen L. Spatial and temporal diversification of Tetrastigma (Vitaceae) Gardens Bulletin Singapore. 2011;63:307–327. [Google Scholar]
- 13.Ollerton J, Stott A, Allnutt E, Shove S, Taylor C, Lamborn E. Pollination niche overlap between a parasitic plant and its host. Oecologia. 2007;151:473–485. doi: 10.1007/s00442-006-0605-y. [DOI] [PubMed] [Google Scholar]
- 14.Davis MB, Shaw RG. Range shifts and adaptive responses to Quaternary climate change. Science. 2001;292:673–679. doi: 10.1126/science.292.5517.673. [DOI] [PubMed] [Google Scholar]
- 15.Gavrilets S, Vose A. Case studies and mathematical models of ecological speciation. 2. Palms on an oceanic island. Mol Ecol. 2007;16:2910–2921. doi: 10.1111/j.1365-294X.2007.03304.x. [DOI] [PubMed] [Google Scholar]
- 16.Zhou L, Bawa R, Holliday J. Exome resequencing reveals signatures of demographic and adaptive processes across the genome and range of black cottonwood (Populus trichocarpa) Mol Ecol. 2014;23:2486–2499. doi: 10.1111/mec.12752. [DOI] [PubMed] [Google Scholar]
- 17.Bonasio R, Zhang G, Ye C, Mutti NS, Fang X, Qin N, et al. Genomic comparison of the ants Camponotus floridanus and Harpegnathos saltator. Science. 2010;329:1068–1071. doi: 10.1126/science.1192428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li R, Li Y, Fang X, Yang H, Wang J, Kristiansen K, et al. SNP detection for massively parallel whole-genome resequencing. Genome Res. 2009;19:1124–1132. doi: 10.1101/gr.088013.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang F, Gao Q, Khan G, Luo K, Chen S. Comparative transcriptome analysis of aboveground and underground tissues of Rhodiola algida, an important ethno-medicinal herb endemic to the Qinghai-Tibetan Plateau. Gene. 2014;553:90–97. doi: 10.1016/j.gene.2014.09.063. [DOI] [PubMed] [Google Scholar]
- 20.Chamala S, García N, Godden GT, Krishnakumar V, Jordonthaden IE, Smet RD, et al. MarkerMiner 1.0: A new application for phylogenetic marker development using angiosperm transcriptomes. Appl Plant Sci. 2015;3:1400115. doi: 10.3732/apps.1400115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ekblom R, Galindo J. Applications of next generation sequencing in molecular ecology of non-model organisms. Heredity. 2011;107:1–15. doi: 10.1038/hdy.2010.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kawakami T, Backström N, Burri R, Husby A, Olason P, Rice AM, et al. Estimation of linkage disequilibrium and interspecific gene flow in Ficedula flycatchers by a newly developed 50k single-nucleotide polymorphism array. Mol Ecol Res. 2014;14:1248–1260. doi: 10.1111/1755-0998.12270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mao YR, Zhang YH, Xu C, Qiu YX. Comparative transcriptome resources of two Dysosma species (Berberidaceae) and molecular evolution of the CYP719A gene in Podophylloideae. Mol Ecol Res. 2016;16:228–241. doi: 10.1111/1755-0998.12415. [DOI] [PubMed] [Google Scholar]
- 24.Lemmon AR, Lemmon EM. High-throughput identification of informative nuclear loci for shallow-scale phylogenetics and phylogeography. Syst Biol. 2012;61:745–761. doi: 10.1093/sysbio/sys051. [DOI] [PubMed] [Google Scholar]
- 25.Reginato M, Michelangeli FA. Primers for low-copy nuclear genes in the Melastomataceae. Appl Plant Sci. 2016;4:1500092. doi: 10.3732/apps.1500092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Swanson WJ, Wong A, Wolfner MF, Aquadro CF. Evolutionary expressed sequence tag analysis of Drosophila female reproductive tracts identifies genes subjected to positive selection. Genetics. 2004;168:1457–1465. doi: 10.1534/genetics.104.030478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wen J, Xiong Z, Nie ZL, Mao L, Zhu Y, Kan XZ, et al. Transcriptome sequences resolve deep relationships of the grape family. PloS One. 2013;8:e74394. doi: 10.1371/journal.pone.0074394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jaillon O, Aury J-M, Noel B, Policriti A, Clepet C, Casagrande A, et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature. 2007;449:463–467. doi: 10.1038/nature06148. [DOI] [PubMed] [Google Scholar]
- 29.Li D, Deng Z, Qin B, Liu X, Men Z. De novo assembly and characterization of bark transcriptome using Illumina sequencing and development of EST-SSR markers in rubber tree (Hevea brasiliensis Muell. Arg.) BMC Genomics. 2012;13:192. doi: 10.1186/1471-2164-13-192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parchman TL, Geist KS, Grahnen JA, Benkman CW, Buerkle CA. Transcriptome sequencing in an ecologically important tree species: assembly, annotation, and marker discovery. BMC Genomics. 2010;11:180. doi: 10.1186/1471-2164-11-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhu W, Wang L, Dong Z, Chen X, Song F, Liu N, et al. Comparative transcriptome analysis identifies candidate genes related to skin color differentiation in red tilapia. Sci Rep. 2016;6:31347. doi: 10.1038/srep31347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miyata T, Yasunaga T. Molecular evolution of mRNA: a method for estimating evolutionary rates of synonymous and amino acid substitutions from homologous nucleotide sequences and its application. J Mol Evol. 1980;16:23–36. doi: 10.1007/BF01732067. [DOI] [PubMed] [Google Scholar]
- 33.Nei M, Kumar S. Molecular Evolution and Phylogenetics. Oxford: Oxford university press; 2000.
- 34.Buschiazzo E, Ritland C, Bohlmann J, Ritland K. Slow but not low: genomic comparisons reveal slower evolutionary rate and higher dN/dS in conifers compared to angiosperms. BMC Evol Biol. 2012;12:8. doi: 10.1186/1471-2148-12-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hughes AL, Packer B, Welch R, Bergen AW, Chanock SJ, Yeager M. Widespread purifying selection at polymorphic sites in human protein-coding loci. Proc Natl Acad Sci USA. 2003;100:15754–15757. doi: 10.1073/pnas.2536718100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Renaut S, Nolte AW, Bernatchez L. Mining transcriptome sequences towards identifying adaptive single nucleotide polymorphisms in lake whitefish species pairs (Coregonus spp. Salmonidae) Mol Ecol. 2010;19:115–131. doi: 10.1111/j.1365-294X.2009.04477.x. [DOI] [PubMed] [Google Scholar]
- 37.Liu H, Xing Q, Ji Z, Xu L, Tian Y. An outline of Quaternary development of Fagus forest in China: palynological and ecological perspectives. Flora. 2003;198:249–259. doi: 10.1078/0367-2530-00098. [DOI] [Google Scholar]
- 38.Zhang L, Yan HF, Wu W, Yu H, Ge XJ. Comparative transcriptome analysis and marker development of two closely related Primrose species (Primula poissonii and Primula wilsonii) BMC Genomics. 2013;14:329. doi: 10.1186/1471-2164-14-329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kane NC, Barker MS, Zhan SH, Rieseberg LH. Molecular evolution across the Asteraceae: micro-and macroevolutionary processes. Mol Biol Evol. 2011;28:3225–3235. doi: 10.1093/molbev/msr166. [DOI] [PubMed] [Google Scholar]
- 40.Pashley CH, Ellis JR, McCauley DE, Burke JM. EST databases as a source for molecular markers: lessons from Helianthus. J Hered. 2006;97:381–388. doi: 10.1093/jhered/esl013. [DOI] [PubMed] [Google Scholar]
- 41.Ellis JR, Burke JM. EST-SSRs as a resource for population genetic analyses. Heredity. 2007;99:125–132. doi: 10.1038/sj.hdy.6801001. [DOI] [PubMed] [Google Scholar]
- 42.Mort ME, Crawford DJ. The continuing search: low-copy nuclear sequences for lower-level plant molecular phylogenetic studies. Taxon. 2004;53:257–261. doi: 10.2307/4135604. [DOI] [Google Scholar]
- 43.Sang T. Utility of low-copy nuclear gene sequences in plant phylogenetics. Crit Rev Biochem Mol Biol. 2002;37:121–147. doi: 10.1080/10409230290771474. [DOI] [PubMed] [Google Scholar]
- 44.Potter S, Bragg JG, Peter BM, Bi K, Moritz C. Phylogenomics at the tips: inferring lineages and their demographic history in a tropical lizard, Carlia amax. Mol Ecol. 2016;25:1367–1380. doi: 10.1111/mec.13546. [DOI] [PubMed] [Google Scholar]
- 45.Duarte JM, Wall PK, Edger PP, Landherr LL, Hong M, Pires JC, et al. Identification of shared single copy nuclear genes in Arabidopsis, Populus, Vitis and Oryza and their phylogenetic utility across various taxonomic levels. BMC Evol Biol. 2010;10:61. doi: 10.1186/1471-2148-10-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.De Smet R, Adams KL, Vandepoele K, Van Montagu MCE, Maere S, Peer YVD. Convergent gene loss following gene and genome duplications creates single-copy families in flowering plants. Proc Natl Acad Sci. 2013;110:2898–2903. doi: 10.1073/pnas.1300127110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Godden GT, Jordon-Thaden IE, Chamala S, Crowl AA, García N, Germain-Aubrey CC, Heaney JM, Latvis M, Qi XS, Gitzendanner MA. Making next-generation sequencing work for you: approaches and practical considerations for marker development and phylogenetics. Plant Ecology & Diversity. 2012;5:427–450. doi: 10.1080/17550874.2012.745909. [DOI] [Google Scholar]
- 48.McCormack JE, Heled J, Delaney KS, Peterson AT, Knowles LL. Calibrating divergence times on species trees versus gene trees: implications for speciation history of Aphelocoma jays. Evolution. 2011;65:184–202. doi: 10.1111/j.1558-5646.2010.01097.x. [DOI] [PubMed] [Google Scholar]
- 49.Leavitt SD, Esslinger TL, Divakar PK, Lumbsch HT. Miocene and Pliocene dominated diversification of the lichen-forming fungal genus Melanohalea (Parmeliaceae, Ascomycota) and Pleistocene population expansions. BMC Evol Biol. 2012;12:176. doi: 10.1186/1471-2148-12-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mudelsee M, Raymo ME. Slow dynamics of the Northern Hemisphere glaciation. Paleoceanography. 2005;20:4022–4035. doi: 10.1029/2005PA001153. [DOI] [Google Scholar]
- 51.Ravelo AC, Andreasen DH, Mitchell L, Lyle AO, Wara MW. Regional climate shifts caused by gradual global cooling in the Pliocene epoch. Nature. 2004;429:263. doi: 10.1038/nature02567. [DOI] [PubMed] [Google Scholar]
- 52.Hu Y, Qi D, Wang H, Wei F. Genetic evidence of recent population contraction in the southernmost population of giant pandas. Genetica. 2010;138:1297–1306. doi: 10.1007/s10709-010-9532-2. [DOI] [PubMed] [Google Scholar]
- 53.Nance HA, Klimley P, Galván-Magaña F, Martínez-Ortíz J, Marko PB. Demographic processes underlying subtle patterns of population structure in the scalloped hammerhead shark, Sphyrna lewini. PloS One. 2011;6:e21459. doi: 10.1371/journal.pone.0021459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Qiu YX, Guan BC, Fu CX, Comes HP. Did glacials and/or interglacials promote allopatric incipient speciation in East Asian temperate plants? Phylogeographic and coalescent analyses on refugial isolation and divergence in Dysosmaversipellis. Mol Phylogenet Evol. 2009;51:281–293. doi: 10.1016/j.ympev.2009.01.016. [DOI] [PubMed] [Google Scholar]
- 55.Cokus SJ, Gugger PF, Sork VL. Evolutionary insights from de novo transcriptome assembly and SNP discovery in California white oaks. BMC Genomics. 2015;16:552. doi: 10.1186/s12864-015-1761-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pertea G, Huang X, Liang F, Antonescu V, Sultana R, Karamycheva S, et al. TIGR Gene Indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics. 2003;19:651–652. doi: 10.1093/bioinformatics/btg034. [DOI] [PubMed] [Google Scholar]
- 58.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene Ontology: tool for the unification of biology. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 61.Ye J, Fang L, Zheng H, Zhang Y, Chen J, Zhang Z, et al. WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 2006;34:W293–W297. doi: 10.1093/nar/gkl031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Iseli C, Jongeneel CV, Bucher P. ESTScan: a program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc Seventh Int Conf Intell Syst Mol Biol. 1999;99:138–148. [PubMed] [Google Scholar]
- 63.Kristensen D. M., Wolf Y. I., Mushegian A. R., Koonin E. V. Computational methods for Gene Orthology inference. Briefings in Bioinformatics. 2011;12(5):379–391. doi: 10.1093/bib/bbr030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fischer S, Brunk BP, Chen F, Gao X, Harb OS, Iodice JB, et al. Using OrthoMCL to assign proteins to OrthoMCL-DB groups or to cluster proteomes into new Ortholog groups. Curr Protoc Bioinformatics. 2011;35:6.12.1–6.12.19. doi: 10.1002/0471250953.bi0612s35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang Z, Li J, Zhao XQ, Wang J, Wong GK, Yu JM. Ka/Ks_Calculator: calculating Ka and Ks through model selection and model averaging. Genomics, Proteomics Bioinformatics. 2006;4:259–263. doi: 10.1016/S1672-0229(07)60007-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yang Z, Nielsen R. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol Biol Evol. 2000;17:32–43. doi: 10.1093/oxfordjournals.molbev.a026236. [DOI] [PubMed] [Google Scholar]
- 67.Dieringer D, Schlötterer C. Microsatellite analyser (MSA): a platform independent analysis tool for large microsatellite data sets. Mol Ecol Res. 2003;3:167–169. [Google Scholar]
- 68.Goff SA, Vaughn M, McKay S, Lyons E, Stapleton AE, Gessler D, et al. The iPlant collaborative: cyberinfrastructure for plant biology. Front Plant Sci. 2011;2:34. doi: 10.3389/fpls.2011.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Van Bel M, Proost S, Wischnitzki E, Movahedi S, Scheerlinck C, Van DPY, et al. Dissecting plant genomes with the PLAZA comparative genomics platform. Plant Physiol. 2011;158:590–600. doi: 10.1104/pp.111.189514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Katoh K, Frith MC. Adding unaligned sequences into an existing alignment using MAFFT and LAST. Bioinformatics. 2012;28:3144. doi: 10.1093/bioinformatics/bts578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001;68:978–989. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Garrick RC, Sunnucks P, Dyer RJ. Nuclear gene phylogeography using PHASE: dealing with unresolved genotypes, lost alleles, and systematic bias in parameter estimation. BMC Evol Biol. 2010;10:118. doi: 10.1186/1471-2148-10-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Flot JF. SeqPHASE: a web tool for interconverting PHASE input/output files and FASTA sequence alignments. Mol Ecol Res. 2010;10:162–166. doi: 10.1111/j.1755-0998.2009.02732.x. [DOI] [PubMed] [Google Scholar]
- 75.Drummond A, Ashton B, Buxton S, Cheung M, Cooper A, Kearse M, et al. Geneious v4. 8. 2009. DOI: http://www. geneious. com. Accessed 21 Sept 2015.
- 76.Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fu YX, Li WH. Statistical tests of neutrality of mutations. Genetics. 1993;133:693–709. doi: 10.1093/genetics/133.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- 79.Clement M, Posada D, Crandall KA. TCS: a computer program to estimate gene genealogies. Mol Ecol. 2000;9:1657–1659. doi: 10.1046/j.1365-294x.2000.01020.x. [DOI] [PubMed] [Google Scholar]
- 80.Hey J, Nielsen R. Multilocus methods for estimating population sizes, migration rates and divergence time, with applications to the divergence of Drosophila pseudoobscura and D. persimilis. Genetics. 2004;167:747–760. doi: 10.1534/genetics.103.024182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nielsen R, Wakeley J. Distinguishing migration from isolation: a Markov chain Monte Carlo approach. Genetics. 2001;158:885. doi: 10.1093/genetics/158.2.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hey J. Isolation with migration models for more than two populations. Mol Biol Evol. 2009;27:905–920. doi: 10.1093/molbev/msp296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Strasburg JL, Rieseberg LH. How robust are ‘Isolation with Migration’ analyses to violations of the IM model? A simulation study. Mol Biol Evol. 2010;27:297–310. doi: 10.1093/molbev/msp233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Woerner AE, Cox MP, Hammer MF. Recombination-filtered genomic datasets by information maximization. Bioinformatics. 2007;23:1851–1853. doi: 10.1093/bioinformatics/btm253. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Size frequency distribution of contigs (a, CSE lineage; b, SW lineage) and unigenes (c, CSE lineage; d, SW lineage). (XLS 1093 kb)
Figure S2. Distribution of gene ontology (GO) classifications for the 3737 orthologous pairs with Ka/Ks ratios significantly < 0.5. The y-axis indicates the percent (left y-axis) or number of unigenes (right y-axis) per (sub-) category. (PDF 436 kb)
Figure S3. Frequency distribution of EST-SSR unit size in the transcriptomes of CSE and SW lineages. (PDF 348 kb)
Figure S4. Frequency distribution of EST-SSR repeat motifs (mono- to tri-nucleotide motifs) between CSE and SW lineages. (PDF 337 kb)
Table S1. Characteristics of the twelve nuclear primer pairs newly developed in this study based on SCNG loci identified by different approaches. Table S2. Full list of 1018 SCNG loci identified from the transcriptomes of CSE and SW lineage, with their basic information including identification approaches, substitution rates between lineages and gene annotations. '+' denotes the approaches that supported the single-copy status of the locus. Table S3. List of individuals included in this study, with corresponding sampling localities and GenBank accession numbers. (PDF 415 kb)
Figure S5. The pairwise identity of each SCNG pair between the two lineages. Each dot in the scatter diagram denotes a SCNG locus, and the blue colored ones represent those genes under purifying selection). (PDF 177 kb)
Figure S6. Agarose-gel electrophoresis patterns of a subset of SCNG primer pairs for T. hemsleyanum (PDF 148 kb)
Figure S7. TCS-derived network of genealogical relationships among the identified haplotypes for each of the SCNG locus (PDF 198 kb)
Data Availability Statement
Raw sequence reads of the transcriptomes of CSE and SW lineage were deposited in the NCBI Sequence Read Archive (SRA) with accession numbers SAMN07520791 and SAMN07502792, respectively. All SCNG sequences obtained in this study are deposited in GenBank (Accession nos. listed in Additional file 5: Table S3, Supporting information).