

Abstract
A comprehensive survey of pathways leading to the generation of α-trifluoromethyl monocyclic piperidinic derivatives is provided (65 references). These compounds have been synthesized either from 6-membered rings e.g., pipecolic acid or lactam derivatives by introduction a trifluoromethyl group, from pyridine or pyridinone derivatives by reduction, and from 5-membered rings e.g., prolinol derivatives by ring expansion, from linear amines by cyclization or from dienes/dienophiles by [4 + 2]-cycloaddition.
Keywords: nitrogen heterocycles, fluorine, piperidine, trifluoromethyl group, ring expansion, cyclization, cycloaddition
1. Introduction
Functionalized piperidinic derivatives are among the most ubiquitous heterocyclic cores in natural products and bioactive compounds, therefore a huge number of methods has been developed to prepare piperidinic derivatives [1,2,3,4,5,6,7]. With respect to the biologically active targets, there is great interest in introducing substituents that can increase their biological activity which can be related to an increase of the lipophilicity, the bioavailability and the metabolic stability. In this context, the trifluoromethyl group is often used as a bioisostere of a chloride or a methyl group to modulate the steric and electronic properties of a lead compound or to protect a reactive methyl group from metabolic oxidation. This substituent can also increase the lipophilicity of molecules [8].
Herein, we report the synthesis of α-trifluoromethylpiperidinic derivatives of type A. These compounds have been synthesized either from 6-membered rings B and C by introduction of a CF3 group, from pyridines D or pyridinones E by reduction, from 5-membered rings F by ring expansion, from linear amines G by cyclization, from dienes/dienophiles H/I by cycloaddition (Scheme 1). We will only report the formation of monocyclic piperidinic derivatives, and of bicyclic derivatives only when they were transformed into the monocyclic derivatives.
Scheme 1.

Precursors of α-trifluoromethyl piperidinic derivatives.
2. From Cyclic Substrates
2.1. From 6-Membered Rings
The synthesis of α-trifluoromethylpiperidines A has been achieved from pipecolic acid, from δ-lactams, from pyridines and from pyridinones.
2.1.1. From Pipecolic Acid
The first synthesis of 2-(trifluoromethyl)piperidine (2) was realized in 1962 by Raash [9], from the sodium salt of pipecolic acid (1) that was treated with sulfur tetrafluoride (SF4) in the presence of HF at 120 °C. However, 2 was isolated in a very low yield of 9.6% (Scheme 2).
Scheme 2.

Synthesis of 2-trifluoromethyl piperidine from pipecolic acid.
2.1.2. From 2-Trifluoromethylpyridine
One easy access to 2-trifluoromethylpiperidine (2) was realized by hydrogenation of the commercially available 2-trifluoromethylpyridine (3) in the presence of Pd, Pt or Rh catalysts (Scheme 3) [10,11,12].
Scheme 3.

Hydrogenation of α-trifluoromethylpyridine.
2.1.3. From Trifluoromethyl Pyridinones
Access to trifluoromethylpiperidinones 5 from trifluoromethylpyridinones 4 has been developed by hydrogenation in the presence of Pd/C. Lactams 5 were isolated in 45%–88% yield and subsequent reduction should lead to the 5-amino 2-trifluoromethylpiperidines 6 (Scheme 4) [13].
Scheme 4.

Hydrogenation of trifluoromethylpyridinones.
2.1.4. From δ-Lactams
Since the first access to 2-trifluoromethylpiperidine (2) different methods have been developed to produce α-trifluoromethylpiperidines of type A from δ-lactams C via either enamines C1, or imines C2 or C3 (Scheme 5).
Scheme 5.

Synthesis of α-trifluoromethylpiperidines from δ-lactams.
From Enamines C1
α-Trifluoromethyl cyclic enamines C1 can be good precursors of A as they are easily obtained from δ-lactams. When lactam 7 was treated with trichloroborane in the presence of an excess of CF3Br and HEPT, enamine 8 was formed in 30% yield via the formation of iminium species I which can be attacked by the CF3– anion, generated from CF3Br. Intermediate II was thus produced and after a β-elimination, enamine 8 was formed (Scheme 6) [14]. A reduction of this enamine should lead to 2-trifluoromethylpiperidine 9. It is worth noting that CF3Br is not easy to handle as it is a gas and alternatives to synthesize 2-trifluoromethylpiperidines from δ-lactams from imines C2 and C3 have been devised.
Scheme 6.

Synthesis of α-trifluoromethylpiperidine from δ-lactam via enamine.
From Imines C2
Imines of type C2 were synthesized in four steps from N-(diethoxymethyl)piperidin-2-one (10). First, 10 was subjected to a Claisen condensation with ethyl trifluoroacetate (NaH, CF3CO2Et) and, after deprotection under acidicic conditions (HCl), the corresponding fluorinated acyl lactam 11 was isolated as the hydrate, due to the ability of the electron-withdrawing CF3 group to stabilize the tetrahedral adducts of such type of compounds [15].
The hydrolysis and the decarboxylation of perfluoro acyl lactam 11 under acidic conditions (6 M HCl) and heating at reflux led to III. When the decarboxylation was complete, the reaction was carefully made alkaline with KOH and hemiaminal 12 was obtained. After azeotropic removal of water or distillation, 12 was transformed into imine 13 (Scheme 7) [15].
Scheme 7.

Synthesis of α-trifluoromethylpiperidine from δ-lactam via imine.
Imine 13 can be involved in a variety of reactions such as reduction, phosphorylation, alkylation under Friedel-Craft conditions, and Ugi-type reactions, producing a diversity of α-trifluoromethyl piperidinic derivatives. Imine 13 was transformed to 2-trifluoromethylpiperidine (2) in a yield of 71% by reduction with NaBH(OAc)3 in MeOH (Scheme 8) [15].
Scheme 8.

Synthesis of α-trifluoromethylpiperidine from an imine by reduction.
When 13 was treated with diethyl phosphite in the presence of boron trifluoride etherate (BF3.OEt2) as the catalyst, 2-(trifluoromethyl)-2-ethylphosphonate piperidine (14) was obtained (92%) and easily transformed to the corresponding α-trifluoromethyl substituted cyclic α-aminophosphonic acid 15 by treatment with trimethylbromosilane in CHCl3 followed by the addition of aqueous MeOH. It is worth pointing out that to avoid the formation of the ammonium salt, due to the liberation of HBr during the hydrolysis of the ester, the addition of propylene oxide was necessary and the free amino acid 15 was isolated (Scheme 9) [16].
Scheme 9.

Synthesis of 2-(trifluoromethyl)-2-ethylphosphonate piperidine from imine.
Imine 13 was transformed to the piperidinic derivatives 16 when subjected to an Ugi-type reaction using isocyanides. Thus, when 13 was treated with an isocyanide (RNC) in the presence of CF3CO2H, piperidine 16 was formed via intermediate V (Scheme 10, eq. 1) [17]. It is worth mentioning that an azido-Ugi reaction was performed using isocyanate (RNC) in the presence of TMSN3 in MeOH. Under these conditions, tetrazole 17 was obtained via intermediate VI. In addition, when benzylisocyanate (BnNC) was used, the corresponding piperidyltetrazole (17, R = Bn) was isolated and debenzylated by hydrogenolysis (H2, Pd/C, MeOH) to produce 18 in good yield (96%) (Scheme 10, eq. 2) [18].
Scheme 10.

Synthesis of α-trifluoromethylpiperidines by Ugi-type reactions.
If tetrazole can be introduced at the α-position of α-trifluoromethylpiperidyl derivatives, using an Ugi-type reaction, other heterocycles such as pyrroles, indoles can also be introduced in the α-position by using a Friedel-Craft reaction. When imine 13 was treated with the non-protected pyrrole 19 in the presence of BF3.OEt2, the unexpected piperidine 20, possessing a C2-substituted pyrrole was obtained (67%) (Scheme 11, eq. 1). On the contrary, when the N-methylpyrrole 21 was involved in the Friedel-Craft reaction, piperidine 22, substituted in the α-position by the C3-substituted pyrrole, was isolated (Scheme 11, eq. 2). The same regioselectivity at C3 was observed with pyrrole 23. It is worth mentioning that no diastereoselectivity was observed with pyrrole 23 as a ratio of 1 to 1 was observed for 24 (Scheme 11, eq. 3). As noticed, the reaction with non-protected and protected pyrroles is very regioselective. This regioselectivity can be the result of steric interactions between the N-substituent of the pyrrole and the cyclic imine which can be quite significant due to the bulkiness of the trifluoromethyl group and its specific nature and, the C3 isomers were formed. When less steric interactions were present, e.g., with unprotected pyrroles, the C2-isomers were formed (Scheme 11, eq. 1) [19,20].
Scheme 11.

Synthesis of α-trifluoromethylpiperidines by Friedel-Craft reactions with pyrroles.
Indoles such 25 can also be involved in a Friedel-Craft reaction with imine 13 leading to the piperidines 26 in good yield (Scheme 12) [21].
Scheme 12.

Synthesis of α-trifluoromethylpiperidines by a Friedel-Craft reaction with indoles.
From Imines C3
In imines 13, the CF3 group is initially located in the α-position and then different R substituents can also be introduced in the α-position. From an imine intermediate, the reverse strategy can be used to access compound A, e.g., imines are synthesized from a δ-lactam and then the trifluoromethyl group is introduced. Thus, when imines 27, prepared from the corresponding δ-lactams, were treated with the Ruppert-Prakash reagent (TMSCF3) in the presence of TFA and KHF2, in MeCN, the α,α-disubstituted piperidines 28 were isolated in good yields. The activation of imines 27 under acidic conditions was necessary to obtained piperidines 28 (Scheme 13) [22].
Scheme 13.

Synthesis of α,α-disubstituted piperidines.
It is worth mentioning that all the 2-trifluoromethylpiperidines were obtained in a racemic form, however, recently, we have reported a method that allows the synthesis of optically active substituted 2-trifluoromethylpiperidines by using a stereospecific ring expansion applied to 2′-trifluoromethylprolinols of type F.
2.2. From 5-Membered Rings
The ring expansion of pyrrolidines to piperidines via an aziridinium intermediate under kinetic and thermodynamic conditions is well-established [23,24,25,26,27,28]. This method was used to access piperidines 30, 30′ from prolinols 29, 29′.
Prolinols 29 and 29′ were prepared from l-proline in a seven-step sequence [29]. When prolinols 29 and 29′ were treated with DAST, I2/PPh3 or by Tf2O, followed by the addition of different nucleophiles (alcohols, amines, thiols, cuprates, cyanides), a diversity of 2-(trifluoromethyl)- 3-substituted piperidines 30 and 30′ were obtained with good diastereo- and enantioselectivities (de > 94% and ee > 99%). The regioselective attack of the nucleophiles on the aziridinium intermediates VII/VII′ is due to the presence of the CF3 group (Scheme 14) [29,30].
Scheme 14.

Synthesis of C3-substituted α-trifluoromethylpiperidines from l-proline.
3. From Non-cyclic Substrates
To synthesize 2-trifluoromethyl-substituted piperidines from non-cyclic substrates, a cyclisation step or a cycloaddition is necessary to access the piperidine skeleton.
3.1. Cyclization
3.1.1. By Metathesis
Metathesis reactions have changed the way molecules are constructed due to the commercialization of robust catalysts such as the Grubbs first and second generation catalysts, G-I and G-II, and the Grubbs-Hoveyda catalysts GH-II (Figure 1). Metathesis is a powerful method to access both carbo- and heterocyclic ring systems [31,32,33,34,35] and the reaction is useful to synthesize piperidines and particularly to synthesize 2-trifluoromethylpiperidinic derivatives.
Figure 1.

Catalysts for olefin metathesis.
From Dienes
The ammonium salt of the 2-trifluoromethylpiperidine 2.HCl was formed from 36, which was obtained from the α-trifluoroaminodiene 35 when treated with G-I. This diene has been synthesized in six steps from the hemiacetal 31 which was transformed into imine 32 by treatment with the imine of benzophenone followed by a silylation step (ImSiMe3). After an allylation under acidic conditions (allylTMS, BF3.OEt2), followed by a deprotection/protection sequence, the allyltrifluoroamine 34 was isolated and allylated to produce the piperidinic core precursor 35. Thus, after treatment of 35 with G-I catalyst (1 mol %) in dichloromethane at rt, the unsaturated piperidine 36 was isolated in 91% yield. After hydrogenation (Pd/C, EtOH, rt, 24 h) and treatment with HCl, 2.HCl was isolated quantitatively (Scheme 15) [36,37]. Compared to the synthesis of 2 from pipecolic acid the synthesis of 2 from 31 is more efficient in terms of yield (47.9% versus 9.6%).
Scheme 15.

Synthesis of α-trifluoromethylpiperidine from trifluoromethylhemiacetal.
A shorter synthesis of unsaturated piperidines from imines 37 using a Barbier-type reaction (allylBr, Zn, DMF, rt) followed by a N-allylation has been reported. The resulting dienic amino compound 38 was isolated in good yied. After treatment with G-I (5–10 mol %) the unsaturated piperidines 39 were isolated (Scheme 16) [38].
Scheme 16.

Synthesis of α-trifluoromethylpiperidines from trifluoromethylimines.
Starting from imines, the strategy is very versatile to access a variety of unsaturated piperidines and, particularly, 2-(trifluoromethyl)-2-substituted tetrahydropyridines. Thus, when imines 40 were treated with allylmagnesium bromide (THF, –78 °C) then allylated (NaH, allylbromide) dienes 42 were isolated in good yields and then involved in a ring-closing metathesis (RCM) using different catalysts such as G-I or the ruthenium catalyst J [39]. It is worth noting that the yields obtained with G-I are better than with catalyst J (Scheme 17) [40,41,42,43].
Scheme 17.

Synthesis of α,α-disubstituted unsaturated piperidines from trifluoromethylimines.
It is worth noting that the dienes, precursors of 2-(trifluoromethyl)-unsaturated piperidines, could be synthesized from trifluorodiazo derivatives 44. Treatment of N,N-diallyl-N-methylamine by the diazo compound 44 in the presence of the copper catalyst [Cu(F3-acac)2], produced ylide VIII which, after a [2,3]-sigmatropic rearrangement, led to the dienic derivatives 45, precursor of the unsaturated piperidines 46 (Scheme 18) [43].
Scheme 18.

Synthesis of α,α-disubstituted piperidines from trifluorodiazo derivatives.
When the ring-opening metathesis (ROM)/ ring-closing metathesis (RCM) was applied to 47, using the G-I catalyst (10 mol %), the unsaturated piperidines 48, substituted at C4, were isolated in good yields (74%–86%) (Scheme 19) [44].
Scheme 19.

Synthesis of α-trifluoromethylpiperidines using a ROM/RCM sequence.
From Enynes
It is worth noting that a substituent at C4 could not be introduced on the piperidinic core by using a RCM applied to enyne 49 (Scheme 20, eq. 1). On the contrary, 2-(trifluoromethyl) unsaturated piperidines substituted at C5 can be produced by applying a RCM to enyne 51. When this latter was treated with G-I (5 mol %) the resulting unsaturated piperidine 52 was obtained with an excellent yield (95%) (Scheme 20, eq. 2).
Scheme 20.


Synthesis of α-trifluoromethylpiperidines by a RCM to enynes.
The alkyne can be substituted by different alkyl groups (compound 53), aryl groups (compound 55) and ketone (compound 57). In all cases, the corresponding unsaturated piperidinic compounds substituted at C5, were isolated in good yields (Scheme 20, eqs. 3, 4 and 5) [37,38,45].
It is worth mentioning that in the case of 55, when the aryl group is substituted in the para position the yields in 56 are good (60%–74%). On the contrary, when the aryl group is substituted in the ortho position, 55 is not transformed into 56 whatever the catalyst used, either the G-II or the GH-II catalysts [45].
3.1.2. By Cycloaddition
Only the cycloadditions leading to the monocyclic piperidinic core will be reported thus, the [2 + 2]-cycloaddition of allenynes [46] and the Paulson-Khan reaction [38,43,47] will be excluded.
[4 + 2]-Cycloaddition: Aza Diels-Alder
If imines are used as dienophiles, activation of the imine moiety either by electron-withdrawing substituents or by a Lewis acid is required for a successful [4 + 2]-cycloaddition. Imines 59, possessing three electron-withdrawing groups, are very reactive and the aza Diels-Alder reaction was achieved at low temperature in the presence of different dienes 60, to produce the corresponding unsaturated piperidines 61 in good yields. It is worth mentioning that phosphonylimines were less reactive than sulfonylimines as the reaction has to be performed at higher temperature and longer reaction times were required (120 h for phosphonylimines versus 14 h for sulfonylimines) (Table 1) [48].
Table 1.
Aza Diels-Alder cycloadditions.
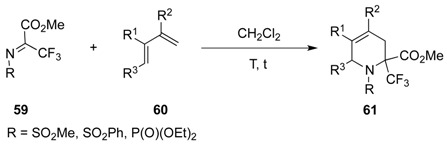
| Entry | R | R1 | R2 | R3 | T (°C) | t (h) | Yield |
|---|---|---|---|---|---|---|---|
| 1 | SO2Ph | Me | Me | H | –30 to 20 | 13 | 71% |
| 2 | SO2Me | Me | Me | H | –30 to 20 | 14 | 91% |
| 3 | P(O)(OEt)2 | Me | Me | H | 20 | 120 | 95% |
| 4 | SO2Ph | H | Me | H | –30 to 20 | 13 | 81% |
| 5 | SO2Me | H | Me | H | –30 to 20 | 13 | 97% |
| 6 | P(O)(OEt)2 | H | Me | H | 20 | 150 | 68% |
| 7 | SO2Ph | H | H | Me | 4 | 120 | 75% |
| 8 | P(O)(OEt)2 | H | H | Me | 20 | 720 | 89% |
When the silyloxydiene 62 was involved in the aza Diels-Alder reaction, piperidinone 63 was isolated after acidic treatment (Scheme 21) [48]
Scheme 21.

Synthesis of piperidinones by an aza Diels-Alder reaction.
Aza Diels-Alder Reaction with 1-Azadiene
The unsaturated piperidine 67 was obtained from an aza Diels-Alder reaction between the iso-butyl vinyl ether 66 and the sulfinimine intermediates IX generated in situ from the trifluoromethyl α,β-unsaturated ketone 64 and the sulfinamine 65. In this aza Diels-Alder reaction, the HOMO orbital of 66 reacts with the LUMO orbital of the azadienes IX. The trifluoromethyl-piperidines 67 were obtained with a good diastereoselectivity in favor of the endo product. In addition, when the reaction was performed with an optically active sulfinamine, the cycloadduct was obtained with a good enantioselectivity (ee > 99%) (Scheme 22) [49].
Scheme 22.

Aza Diels-Alder reaction with 1-azadienes.
3.1.3. 1,4-Addition: Aza-Michael
A Michael acceptor, tethered to a nucleophilic amine, can undergo an intramolecular 1,4-addition with concomitant formation of a piperidinic core. After the preparation of the optically active ω-amino α,β-unsaturated ketone 70 in three steps from the trifluoromethylhemiacetal 68, the aminoketone 70 was treated under basic conditions to produce the 2-trifluoromethyl-substituted piperidine 71 in 80% yield and with an excellent diastereoselectivity of 94:6 in favor of the cis-isomer. This piperidine was transformed to an analog of pinidinol (Scheme 23) [50].
Scheme 23.

Synthesis of α-trifluoromethylpiperidine by aza-Michael addition.
2-Trifluoromethylpiperidines with a quaternary center at C2 were synthesized from hemiaminal 72 and an α,β-unsaturated ketone 73 in the presence of a chiral amine ((2S,3S)-2-amino-N-((S)-1-hydroxy-3-phenyl-propan-2-yl)-3-methyl-pentanamide) under acidic conditions [para-nitrobenzoic acid (PNBA)]. Hemiaminal 72 reacts with the chiral amine to form intermediate 74 which then reacts with the enolized form of 73 to produce intermediate 75. An intramolecular aza-Michael reaction then occurs and the tricyclic compounds 76 were isolated in good yields and with excellent dr(s) and ee(s). After reduction of the ketone (NaBH4) and cleavage of the C–S and N–S bonds (sodium naphthalide), piperidine 77 (R1 = Ph) was isolated with a good enantiomeric excess (ee = 92%) (Scheme 24) [51].
Scheme 24.

Synthesis of chiral α-trifluoromethylpiperidines.
3.1.4. Electrophilic-Induced Cyclization
Amino-iodo cyclization has been used to prepare 2-trifluoromethylpipecolic acid derivatives starting from the ω-unsaturated imine 78. The transformation of 79 to 80 was realized in a one-pot process with an excellent yield of 80%. After treatment of imine 78 with iodine in the presence of NaH, the bicyclic product 79 was formed and, when reacted with I2, a migration of the CF3 group took place leading to the diiodo derivative 80 via an iminium alcoholate intermediate XI. The bicyclic compound 80 was then transformed into a variety of 2-trifluoromethylpipecolic acid derivatives 81 in a few steps (Scheme 25) [52,53].
Scheme 25.

Synthesis of α-trifluoromethylpipecolic acid derivatives.
If in the electrophilic-induced cyclization of ω-aminoalkenes the CF3 group was present in the starting material, on the contrary, in the electrophilic-induced cyclization of ω-amino alkynes the CF3 was introduced after the cyclization.
When the ω-amino alkynes 82 were treated with AgF in the presence of H2O, a hydroamination takes place to produce the enamine intermediate XII, which was reacted with TMSCF3 to produce 2-trifluoromethylpiperidines 83 with excellent yields (88%–92%) (Scheme 26) [54].
Scheme 26.

Synthesis of α-trifluoromethylpiperidines by electrophilic-induced cyclization.
3.1.5. Nucleophilic Attack of Iminium Ions
In reaction where iminium ions are involved, the iminium ion is attacked by a nucleophile such as in a Mannich type reaction and a Prins cyclization. A diastereoselective synthesis of 2-trifluoromethylpiperidines was realized by using an intramolecular Mannich reaction starting from the trifluoromethyl amine 84. After condensation of the latter with aldehydes, the formed imines 85 were transformed to the iminium intermediates XIII under acidic conditions (p-TsOH, refluxing toluene). A cyclization is taking place to produce the 2-trifluoromethylpiperidinic derivatives 86. The diastereoselectivity can be explained by the six-membered ring chair transition states XIV, in which the steric interactions are minimized (Scheme 27) [55,56,57].
Scheme 27.

Synthesis of α-trifluoromethylpiperidines by an intramolecular Mannich reaction.
A silyl-aza-Prins reaction was also used to prepare highly functionalized 2-trifluoromethyl- piperidines. Treatment of the vinyl silyl trifluoromethyl amine 87 with ethyl glyoxylate in the presence of InCl3 led to 88 via the iminium intermediate XV. After an intramolecular attack of the iminium XV by the vinyl silane, intermediate XVI was formed, a desilylation took place and piperidine 88 was isolated in 65% yield. This piperidine can then be transformed to the highly functionalized 2-trifluoromethylpiperidine 89 (Scheme 28) [58].
Scheme 28.

Synthesis of an α-trifluoromethylpiperidine by a silyl-aza-Prins reaction.
3.1.6. Intramolecular Condensation of Aminoketones and Aminoesters
Intramolecular lactamization and hemiaminalization were used to build up the C–N bond of the 2-trifluoromethylpiperidine derivatives. 2-Trifluoromethyllactam 93, which can led to the corresponding piperidine by reduction, was synthesized in three steps from ketoester 90. After treatment of 90 with aniline (p-TsOH, toluene, reflux), imine 91 was reduced to the δ-aminoester 92 which was cyclized to the corresponding lactam 93 under basic conditions (NaH, THF) (Scheme 29) [59].
Scheme 29.

Synthesis of a trifluoromethyllactam from a ketoester.
The α-substituted trifluoromethyl-δ-lactam 96 was prepared from the α-trifluoromethyl nitrosulfinamine 94, prepared in two steps from trifluoromethylhemiacetal 68. After a 1,4-addition of 94 to ethyl acrylate under basic conditions, the resulting aminoester 95 was treated with TFA to produce, after deprotection (NH3, MeOH), the trifluoromethyl-δ-lactam 96 (Scheme 30) [60].
Scheme 30.

Synthesis of a trifluoromethyllactam from a trifluoromethylhemiacetal.
A lactamization was also utilized to access 2-trifluoromethyl-3-hydroxypiperidine 102. The latter was synthesized from aldimine 97 and the 2-trimethylsilyloxyfuran 98, and transformed into the unsaturated lactone 99 after treatment with TBSOTf. After hydrogenation, the obtained lactone 100 was transformed to the δ-lactam 101 under acidic conditions (H2SO4, 80%), and then to the 2,3-disubstituted piperidine 102 by reduction (LiAlH4, AlCl3) (Scheme 31) [61].
Scheme 31.

Synthesis of an α-trifluoromethylpiperidine by lactamization.
Due to the high reactivity of trifluoromethyl ketones they can be attacked by weak nucleophiles such as amines to form a hemiaminal. When the carboxylic trifluoromethyl ketones 103 were treated with ammonium carbonate in refluxing toluene, the six-membered ring hemiaminal intermediates 104 were formed and dehydrated under acidic conditions (p-TsOH, reflux) to furnish the unsaturated lactams 105 (Scheme 32) [62].
Scheme 32.

Synthesis of unsaturated lactams from carboxylic trifluoromethyl ketones.
Highly substituted 2-trifluoromethylpiperidines 112 were obtained from isoxazoles 106, trifluoromethylketoester 107, aldehydes 108 and ammonium acetate in ethanol. In this multicomponent reaction, isoxazoles 106 react with the enolized β-ketoester 107, to form intermediates 109 which can react with imines 110 (obtained by condensation of aldehydes 108 with NH4OAc) to produce the aminotrifluoromethylketones 111. The intramolecular attack of the amino group to the highly reactive trifluoromethylketones led to the trifluoromethylpiperidines 112 (Scheme 33) [63].
Scheme 33.

Synthesis of highly substituted α-trifluoromethylpiperidines.
3.1.7. Intramolecular Nucleophilic Substitution
The intramolecular nucleophilic displacement of a good leaving group by an amine is one of the must common methods to prepare six-membered nitrogen heterocycles. This method was used to synthesize 2-trifluoromethylpiperidines 114 from the highly reactive aziridines 113. The high reactivity of 113 is due to the presence of the trifluoromethyl group and the N-tosyl group. The attack of aziridine 113 by a variety of nucleophiles was very regioselective and produced intermediates XVII that cyclized according to an intramolecular nucleophilic substitution of the chloride to produce 114 (Scheme 34) [64].
Scheme 34.

Synthesis of α-trifluoromethylpiperidines from trifluoromethylaziridines.
3.1.8. Intramolecular Nucleophilic Attack of Aldehydes
The annulation of aminoaldehyde 115 was realized by an intramolecular attack of a diphenyl sulfonium species on an aldehyde. When aminoaldehyde 115 was reacted with the trifluoromethyl- vinyl diphenylsulfonium 116, the ylide intermediate XVIII was formed and a cyclization took place to produce XIX which led to the piperidino-epoxide 117. It is worth noting that 117 was isolated with a good yield (72%) and a good diastereoselectivity (dr > 20:1) (Scheme 35) [65].
Scheme 35.

Synthesis of α-trifluoromethylpiperidino-epoxides.
4. Conclusions
Despite the considerable synthetic efforts spent to synthesize 2-trifluoromethylpiperidinic derivatives, most of these compounds have been obtained in a racemic form. When they are optically active, the methods involve a chiral auxiliary or the starting material comes from the chiral pool. Thus, there is still a strong demand for catalytic enantioselective methods to efficiently access 2-trifluoromethylpiperidine derivatives in high diastereo- and enantioselectivity.
Acknowledgments
One of us, S. R. thanks GSK for a grant.
Author Contributions
All authors were involved in the preparation of the manuscript: The articles were collected by S. R., the organization and the writing of the manuscript were realized by J. C. and the typing by D. G. P.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Laschat S., Dickner T. Stereoselective synthesis of piperidines. Synthesis. 2000:1781–1813. doi: 10.1055/s-2000-8218. [DOI] [Google Scholar]
- 2.Toyooka N., Nemoto H. Application of chiral building blocks to the synthesis of drugs. Drugs Fut. 2002;27:143–158. doi: 10.1358/dof.2002.027.02.654810. [DOI] [Google Scholar]
- 3.Felpin F.-X., Lebreton J. Recent advances in the total synthesis of piperidine and pyrrolidine natural alkaloids with ring-closing metathesis as a key step. Eur. J. Org. Chem. 2003:3693–3712. doi: 10.1002/ejoc.200300193. [DOI] [Google Scholar]
- 4.Basra S., Blechert S. Ring rearrangement metathesis—A new concept in piperidine and pyrrolidine synthesis. Strategies Tactics Org. Synt. 2004;4:315–346. [Google Scholar]
- 5.Pearson M.S.M., Mathe-Allainmat M., Fargeas V., Lebreton J. Recent advances in the total synthesis of piperidine aza-sugars. Eur. J. Org. Chem. 2005:2159–2191. doi: 10.1002/ejoc.200400823. [DOI] [Google Scholar]
- 6.Vicario J.L., Badia D., Carrillo L., Ruiz N., Reyes E. New methods for the asymmetric synthesis of piperidines and pyrrolidines: Chiral auxiliaries and asymmetric organocatalysis. Targets Heterocycl. Syst. 2008;12:302–327. doi: 10.1002/chin.201016249. [DOI] [Google Scholar]
- 7.Amat M., Perez M., Bosch J. Stereoselective conjugate addition reactions to phenylglycinol-derived, unsaturated oxazolopiperidone lactams. Chem. Eur. J. 2011;17:7724–7732. doi: 10.1002/chem.201100471. [DOI] [PubMed] [Google Scholar]
- 8.Meyer F. Trifluoromethyl nitrogen heterocycles: Synthetic aspects and potential biological targets. Chem. Commun. 2016;52:3077–3094. doi: 10.1039/C5CC09414C. [DOI] [PubMed] [Google Scholar]
- 9.Raasch M.S. Chemistry of sulfur tetrafluoride. IX. Reaction with amino acids in hydrogen fluoride. J. Org. Chem. 1962;27:1406–1409. doi: 10.1021/jo01051a068. [DOI] [Google Scholar]
- 10.Albrecht B.K., Gehling V.C., Harmange J.-C., Vaswani R.G. Preparation of Substituted Indolecarboxamides as Modulators of Methyl Modifying Enzymes. WO 2016130396. 2016 Aug 18;
- 11.Rice K. Preparation of Benzoxazepines as PI3K/mTOR Inhibitors Useful in the Treatment of Cancer. WO 2012071509. 2012 May 31;
- 12.Rice K., Markby D. Benzoxazepines as Inhibitors of PI3K/mTOR and Methods of their Use as Antitumor Agents and Manufacture. WO 2012068106. 2012 May 24;
- 13.Tolmachova N.A., Dolovanyuk V.G., Gerus I.I., Kondratov I.S., Polovinko V.V., Bergander K., Haufe G. Catalytic hydrogenation of 3-amino-6-(trifluoromethyl)-5,6-dihydropyridin-2(1H)-ones and its use in the synthesis of trifluoromethyl-containing mimetics of ornithine and thalidomide. Synthesis. 2011:1149–1156. doi: 10.1055/s-0030-1258448. [DOI] [Google Scholar]
- 14.Bürger H., Dittmar T., Pawelke G. A simple one-pot synthesis of α-trifluoromethyl-substituted enamines by C-trifluoromethylation of dialkylamides with P(NEt2)3/CF3Br. J. Fluor. Chem. 1995;70:89–93. doi: 10.1016/0022-1139(94)03090-M. [DOI] [Google Scholar]
- 15.Shevchenko N.E., Balenkova E.S., Röschenthaler G.-V., Nenajdenko V.G. Practical synthesis of α-perfluoroalkyl cyclic imines and amines. Synthesis. 2010:120–126. doi: 10.1055/s-0029-1217104. [DOI] [Google Scholar]
- 16.Odinets I.L., Artyushin O.I., Lyssenko K.A., Shevchenko N.E., Nenajdenko V.G., Röschenthaler G.-V. Facile synthesis of cyclic α-perfluoroalkyl-α-aminophosphonates. J. Fluor. Chem. 2009;130:662–666. doi: 10.1016/j.jfluchem.2009.05.002. [DOI] [Google Scholar]
- 17.Gulevich A.V., Shevchenko N.E., Balenkova E.S., Röschenthaler G.-V., Nenajdenko V.G. Efficient multicomponent synthesis of α-trifluoromethyl proline, homoproline, and azepan carboxylic acid dipeptides. Synlett. 2009:403–406. doi: 10.1055/s-0028-1087529. [DOI] [Google Scholar]
- 18.Shmatova O.I., Nenajdenko V.G. Tetrazole-substituted five-, six-, and seven-membered cyclic amines bearing perfluoroalkyl groups—efficient synthesis by azido-Ugi reaction. Eur. J. Org. Chem. 2013:6397–6403. doi: 10.1002/ejoc.201300861. [DOI] [Google Scholar]
- 19.Shmatova O.I., Shevchenko N.E., Balenkova E.S., Röschenthaler G.-V., Nenajdenko V.G. Highly β-regioselective Friedel-Crafts aminoalkylation of pyrroles with cyclic perfluoroalkylated imines. Eur. J. Org. Chem. 2013:3049–3058. doi: 10.1002/ejoc.201201725. [DOI] [Google Scholar]
- 20.Shmatova O.I., Shevchenko N.E., Balenkova E.S., Röschenthaler G.-V., Nenajdenko V.G. Friedel-Crafts alkylation of natural amino acid-derived pyrroles with CF3-substituted cyclic imines. Mendeleev Commun. 2013;23:92–93. doi: 10.1016/j.mencom.2013.03.013. [DOI] [Google Scholar]
- 21.Shevchenko N.E., Shmatova O.I., Balenkova E.S., Röschenthaler G.-V., Nenajdenko V.G. Aminoalkylation of indoles with α-polyfluoroalkylated cyclic imines. Eur. J. Org. Chem. 2013:2237–2245. doi: 10.1002/ejoc.201201661. [DOI] [Google Scholar]
- 22.Shevchenko N.E., Vlasov K., Nenajdenko V.G., Röschenthaler G.-V. The reaction of cyclic imines with the Ruppert-Prakash reagent. Facile approach to α-trifluoromethylated nornicotine, anabasine, and homoanabasine. Tetrahedron. 2011;67:69–74. doi: 10.1016/j.tet.2010.11.032. [DOI] [Google Scholar]
- 23.Cossy J., Gomez Pardo D. Enantioselective ring expansion via aziridinium intermediates. Synthesis of substituted piperidines from substituted pyrrolidines. Synthetic applications. Targets Heterocycl. Syst. 2002;6:1–26. doi: 10.1002/chin.200429273. [DOI] [Google Scholar]
- 24.Cossy J., Gomez Pardo D., Dumas C., Mirguet O., Dechamps I., Métro T.-X., Burger B., Roudeau R., Appenzeller J., Cochi A. Rearrangement of β-amino alcohols and application to the synthesis of biologically active compounds. Chirality. 2009;21:850–856. doi: 10.1002/chir.20716. [DOI] [PubMed] [Google Scholar]
- 25.Tynoshenko D.D. Ring expansions of 1-azabicyclo[n.1.0]alkanes. Recent developments. Arkivoc. 2011:329–345. doi: 10.3998/ark.5550190.0012.105. [DOI] [Google Scholar]
- 26.Cochi A., Gomez Pardo D., Cossy J. Access to optically active 3-aminopiperidines by ring expansion of prolinols: Thermodynamic versus kinetic control. Eur. J. Org. Chem. 2012:2023–2040. doi: 10.1002/ejoc.201101829. [DOI] [Google Scholar]
- 27.Gomez Pardo D., Cossy J. Access to optically active 3-substituted piperidines by ring expansion of prolinols and derivatives. Chem. Eur. J. 2014;20:4516–4525. doi: 10.1002/chem.201304924. [DOI] [PubMed] [Google Scholar]
- 28.Dolfen J., Yadav N.N., De Kimpe N., D'hooghe M., Ha H.-J. Bicyclic aziridinium ions in azaheterocyclic chemistry—Preparation and synthetic application of 1-azoniabicyclo[n.1.0]alkanes. Adv. Synth. Catal. 2016;358:3485–3511. doi: 10.1002/adsc.201600750. [DOI] [Google Scholar]
- 29.Rioton S., Orliac A., Antoun Z., Bidault R., Gomez Pardo D., Cossy J. Stereoselective rearrangement of (trifluoromethyl)prolinols to enantioenriched 3-substituted 2-(trifluoromethyl)piperidines. Org. Lett. 2015;17:2916–2919. doi: 10.1021/acs.orglett.5b01084. [DOI] [PubMed] [Google Scholar]
- 30.Orliac A. Ph.D. Thesis. Université Pierre et Marie Curie; Paris, France: Nov 13, 2013. Synthèse et propriétés physico‑chimiques des fluoro-pipéridines. XtalFluor-E et synthèse d′amides. [Google Scholar]
- 31.Schmalz H.-G. Catalytic ring-closing metathesis: a new, powerful technique for carbon-carbon coupling in organic synthesis. Angew. Chem. Int. Ed. 1995;34:1833–1836. doi: 10.1002/anie.199518331. [DOI] [Google Scholar]
- 32.Grubbs R.H., Miller S.J., Fu G.C. Ring-closing metathesis and related processes in organic synthesis. Acc. Chem. Res. 1995;28:446–452. doi: 10.1021/ar00059a002. [DOI] [Google Scholar]
- 33.Schuster M., Blechert S. Olefin metathesis in organic chemistry. Angew. Chem. Int. Ed. 1997;36:2037–2056. doi: 10.1002/anie.199720361. [DOI] [Google Scholar]
- 34.Armstrong S.K. Ring-closing diene metathesis in organic synthesis. J. Chem. Soc., Perkin Trans. 1: Org. Bio-Org. Chem. 1998:371–388. doi: 10.1039/a703881j. [DOI] [Google Scholar]
- 35.Wright D.L. Application of olefin metathesis to organic synthesis. Curr. Org. Chem. 1999;3:211–240. [Google Scholar]
- 36.Billard T., Langlois B.R. Reactivity of stable trifluoroacetaldehyde hemiaminals. 2. Generation and synthetic potentialities of fluorinated iminiums. J. Org. Chem. 2002;67:997–1000. doi: 10.1021/jo016265t. [DOI] [PubMed] [Google Scholar]
- 37.Gille S., Ferry A., Billard T., Langlois B.R. Synthesis of α-trifluoromethylated nitrogen heterocycles. J. Org. Chem. 2003;68:8932–8935. doi: 10.1021/jo035097x. [DOI] [PubMed] [Google Scholar]
- 38.Magueur G., Legros J., Meyer F., Ourévitch M., Crousse B., Bonnet-Delpon D. A one-pot synthesis of doubly unsaturated trifluoromethyl amines: Easy access to CF3-substituted piperidines. Eur. J. Org. Chem. 2005:1258–1265. doi: 10.1002/ejoc.200400719. [DOI] [Google Scholar]
- 39.Fürstner A., Picquet M., Bruneau C., Dixneuf P.H. Cationic ruthenium allenylidene complexes as a new class of performing catalysts for ring-closing metathesis. Chem. Commun. 1998:1315–1316. doi: 10.1039/a803286f. [DOI] [PubMed] [Google Scholar]
- 40.Osipov S.N., Bruneau C., Picquet M., Kolomiets A.F., Dixneuf P.H. Synthesis of fluorine-containing cyclic amino acid derivatives via ring-closing olefin metathesis. Chem. Commun. 1998:2053–2054. doi: 10.1039/a804792h. [DOI] [Google Scholar]
- 41.Osipov S.N., Artyushin O.I., Kolomiets A.F., Bruneau C., Dixneuf P.H. α-CF3-substituted phosphorus containing analogs of α-amino acids. Novel six- and seven-membered α-amino phosphonates via ring-closing metathesis with LnRu=C=C=CR2 precatalyst. Synlett. 2000:1031–1033. doi: 10.1055/s-2000-6650. [DOI] [Google Scholar]
- 42.Osipov S.N., Artyushin O.I., Kolomiets A.F., Bruneau C., Picquet M., Dixneuf P.H. Synthesis of fluorine-containing cyclic α-amino acid and α-amino phosphonate derivatives by alkene metathesis. Eur. J. Org. Chem. 2001:3891–3897. doi: 10.1002/1099-0690(200110)2001:20<3891::AID-EJOC3891>3.0.CO;2-R. [DOI] [Google Scholar]
- 43.Vorobyeva D.V., Mailyan A.K., Peregudov A.S., Karimova N.M., Vasilyeva T.P., Bushmarinov I.S., Bruneau C., Dixneuf P.H., Osipov S.N. Synthesis of functionalized CF3-containing heterocycles via [2,3]-sigmatropic rearrangement and sequential catalytic carbocyclization. Tetrahedron. 2011;67:3524–3532. doi: 10.1016/j.tet.2011.03.031. [DOI] [Google Scholar]
- 44.Osipov S.N., Kobelikova N.M., Shchetnikov G.T., Kolomiets A.F., Bruneau C., Dixneuf P.H. Novel synthesis of cyclic α-amino acid esters via ene reaction and ruthenium-catalyzed ring rearrangement. Synlett. 2001:621–622. doi: 10.1055/s-2001-13362. [DOI] [Google Scholar]
- 45.Mailyan A.K., Krylov I.M., Bruneau C., Dixneuf P.H., Osipov S.N. Access to cyclic α-CF3-substituted α-amino acid derivatives by ring-closing metathesis of functionalized 1,7-enynes. Eur. J. Org. Chem. 2013:5353–5363. doi: 10.1002/ejoc.201300619. [DOI] [Google Scholar]
- 46.Mailyan A.K., Krylov I.M., Bruneau C., Dixneuf P.H., Osipov S.N. Thermal [2 + 2]-cycloaddition of CF3-substituted allenynes: Access to novel cyclobutene-containing α-amino acids. Synlett. 2011:2321–2324. doi: 10.1055/s-0030-1261217. [DOI] [Google Scholar]
- 47.Ferry A., Billard T., Langlois B.R. Synthesis of α-trifluoromethylated nitrogen bicycles. Synlett. 2005:1027–1029. doi: 10.1055/s-2005-864812. [DOI] [Google Scholar]
- 48.Kobel′kova N.M., Osipov S.N., Kolomiets A.F. Aza Diels-Alder reactions of methyl trifluoropyruvate sulfonyl- and phosphorylimines with 1,3-dienes. Russ. Chem. Bull. Int. Ed. 2002;51:1298–1302. doi: 10.1023/A:1020964916405. [DOI] [Google Scholar]
- 49.Li P., Liu L.-J., Liu J.-T. Per(poly)fluoroalkanesulfinamides assisted diastereoselective three-component inverse-electron-demand aza Diels-Alder reaction. Org. Biomol. Chem. 2011;9:74–77. doi: 10.1039/C0OB00699H. [DOI] [PubMed] [Google Scholar]
- 50.Fustero S., Monteagudo S., Sanchez-Rosello M., Flores S., Barrio P., Del Pozo C. N-Sulfinyl amines as a nitrogen source in the asymmetric intramolecular aza-Michael reaction: Total synthesis of (−)-pinidinol. Chem. Eur. J. 2010;16:9835–9845. doi: 10.1002/chem.201000615. [DOI] [PubMed] [Google Scholar]
- 51.Zhang S., Cha L., Li L., Hu Y., Li Y., Zha Z., Wang Z. Asymmetric formal aza-Diels-Alder reaction of trifluoromethyl hemiaminals with enones catalyzed by primary amines. J. Org. Chem. 2016;81:3177–3187. doi: 10.1021/acs.joc.6b00087. [DOI] [PubMed] [Google Scholar]
- 52.Fustero S., Albert L., Aceña J.L., Sanz-Cervera J.F., Asensio A. An Efficient entry to optically active anti- and syn-β-amino-α-trifluoromethyl alcohols. Org. Lett. 2008;10:605–608. doi: 10.1021/ol702947n. [DOI] [PubMed] [Google Scholar]
- 53.Fustero S., Albert L., Mateu N., Chiva G., Miro J., Gonzalez J., Aceña J.L. Stereoselective access to fluorinated and non-fluorinated quaternary piperidines: Synthesis of pipecolic acid and imino-sugar derivatives. Chem. Eur. J. 2012;18:3753–3764. doi: 10.1002/chem.201102351. [DOI] [PubMed] [Google Scholar]
- 54.Han J., Xu B., Hammond G.B. Synthesis of α-CN and α-CF3 N-heterocycles through tandem nucleophilic additions. Org. Lett. 2011;13:3450–3453. doi: 10.1021/ol2011902. [DOI] [PubMed] [Google Scholar]
- 55.Bariau A., Roblin J.-P., Troin Y., Canet J.-L. A simple stereoselective access to α-trifluoromethylated piperidines. Synlett. 2005:1731–1733. doi: 10.1055/s-2005-871552. [DOI] [Google Scholar]
- 56.Bariau A., Jatoi W.B., Calinaud P., Troin Y., Canet J.-L. A simple stereoselective route to α-trifluoromethyl analogues of piperidine alkaloids. Eur. J. Org. Chem. 2006:3421–3433. doi: 10.1002/ejoc.200600157. [DOI] [Google Scholar]
- 57.Ciblat S., Besse P., Canet J.-L., Troin Y., Veschambre H., Gelas J. A practical asymmetric synthesis of 2,6-cis-disubstituted piperidines. Tetrahedron: Asymmetry. 1999;10:2225–2235. doi: 10.1016/S0957-4166(99)00226-8. [DOI] [Google Scholar]
- 58.Dobbs A.P., Parker R.J., Skidmore J. Rapid access to CF3-containing heterocycles. Tetrahedron Lett. 2008;49:827–831. doi: 10.1016/j.tetlet.2007.11.178. [DOI] [Google Scholar]
- 59.Wan W., Hou J., Jiang H., Yuan Z., Ma G., Zhao G., Hao J. Reductive amination/cyclization of ω-trifluoromethyl keto esters to trifluoromethylated δ-amino alcohols and lactams. Eur. J. Org. Chem. 2010:1778–1786. doi: 10.1002/ejoc.200901424. [DOI] [Google Scholar]
- 60.García Ruano J.L., De Haro T., Singh R., Belén Cid M. An efficient method for the synthesis of nitropiperidones. J. Org. Chem. 2008;73:1150–1153. doi: 10.1021/jo702130a. [DOI] [PubMed] [Google Scholar]
- 61.Spanedda M.V., Ourévitch M., Crousse B., Bégué J.-P., Bonnet-Delpon D. Vinylogous Mannich reactions. additions of trimethylsilyloxyfuran to fluorinated aldimines. Tetrahedron Lett. 2004;45:5023–5025. doi: 10.1016/j.tetlet.2004.05.003. [DOI] [Google Scholar]
- 62.Okano T., Fumoto M., Kusukawa T., Fujita M. Synthesis of optically active trifluoromethylated indolizidine derivatives via stereoselective radical cyclization. Org. Lett. 2002;4:1571–1573. doi: 10.1021/ol025792b. [DOI] [PubMed] [Google Scholar]
- 63.Shi W., Wang Y., Zhu Y., Zhang M., Song L., Deng H. A one-pot, multicomponent synthesis of trifluoromethylated spiropiperidines under catalyst-free conditions. Synthesis. 2016;48:3527–3536. [Google Scholar]
- 64.Dolfen J., Kenis S., Van Hecke K., De Kimpe N., D′hooghe M. Selective synthesis of functionalized trifluoromethylated pyrrolidines, piperidines, and azepanes starting from 1-tosyl-2-(trifluoromethyl)aziridine. Chem. Eur. J. 2014;20:10650–10653. doi: 10.1002/chem.201304759. [DOI] [PubMed] [Google Scholar]
- 65.Fritz S.P., West T.H., McGarrigle E.M., Aggarwal V.K. Diastereoselective synthesis of CF3-substituted, epoxide-fused heterocycles with β-(trifluoromethyl)vinylsulfonium salts. Org. Lett. 2012;14:6370–6373. doi: 10.1021/ol303200n. [DOI] [PubMed] [Google Scholar]