

Abstract
We have reported that short-term stimulation of primary human monocyte-derived macrophages (MDM) with interferon-γ (IFN-γ) and tumor necrosis factor-α (TNF-α), i.e. M1 polarization, leads to a significant containment of virus replication. Here we show that M1-MDM restimulation with these cytokines 7 days after infection (M12 MDM) promoted an increased restriction of HIV-1 replication characterized by very low levels of virus production near to undetectable levels. In comparison to control and M1-MDM that were not restimulated, M12 MDM showed a stronger reduction of both total and integrated HIV DNA as well as of viral mRNA expression. M12 MDM were characterized by an upregulated expression of restriction factors acting at the level of reverse transcription (RT), including apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 3A (APOBEC3A) and APOBEC3G, but not SAM domain and HD domain-containing protein 1 (SAMHD1). M12 MDM also showed an increased expression of Class II Transactivator (CIITA) and Tripartite Motif22 (TRIM22), two negative regulators of proviral transcription, whereas expression and phosphorylation of transcriptional inducers of HIV-1, such as nuclear factor kB (NF-kB) and signal transducer and activator of transcription 1 (STAT1), were not impaired in these cells. The almost quiescent state of the infection in M12 MDM was promptly reversed by coculture with mitogen-stimulated leukocytes or cell incubation with their filtered culture supernatant. M12 MDM harbored replication-competent HIV-1 as virus spreading following cell stimulation was fully prevented by the RT inhibitor lamivudine/3TC. Selective reactivation of proviral expression in M12 MDM, but not in control or in M1-MDM that were not restimulated, was confirmed in cells infected with single round Vesicular Stomatitis Virus-G-pseudotyped HIV-1. Thus, M12 MDM represent an in vitro model of reversible, almost quiescent HIV-1 infection of primary human macrophages that could be further exploited for “Cure” related investigations.
Introduction
The human immunodeficiency virus type-1 (HIV-1) infects cells expressing the primary entry receptor CD4 and either CCR5 or CXCR4 as mandatory entry coreceptors. This entry requirements restrict the number of cells susceptible to HIV-1 infection mostly to a major subset of T lymphocytes and to myeloid cells, with the exception of the astrocytes of the central nervous system (CNS) that are infected in vivo and infectable in vitro via a CD4/chemokine receptor independent route1.
In addition to entry requirements, infection of T cells and myeloid cells share several features, such as the capacity to counteract active virus replication by means of constitutively expressed restriction factors, including members of the apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like (APOBEC) family, usually promoting G-to-A hypermutations in the viral sequence during reverse transcription process2,3, or SAM domain and HD domain-containing protein 1 (SAMHD1), a phosphohydrolase that curtails the reverse transcription step in the virus life cycle by depleting the pool of dideoxynucleoeside triphosphates4. Once integrated as a provirus, HIV-1 can undergo active transcription leading to translation and assembly of its viral proteins with genomic RNA at the plasma membrane in order to release new progeny virions or it can be silenced by either epigenetic mechanisms, lack of positive transcription factors and/or dominance of negative transcription factors5. In this regard, a crucial role is played by the provirus-encoded protein Tat that acts as anti-terminator of viral transcripts by promoting the assembly of the Positive Transcriptional Elongation Factor b (P-TEFb) complex, composed of cyclin T1 and cyclin-dependent kinase 9 (CDK9)5,6.
HIV-1 replication in vivo leads to a progressive and selective depletion of CD4+ T lymphocytes leading to the acquired immunodeficiency syndrome (AIDS) in the absence of combination antiretroviral therapy (cART). Although the precise mechanism(s) of CD4+ T cell depletion in vivo are still debated7, in vitro infection of activated primary CD4+ T cells is associated with virus-induced cytopathicity and cell death. In contrast, no depletion of circulating monocytes or tissue-associated macrophages is usually seen in infected individuals whereas in vitro infection of primary human monocyte-derived macrophages (MDM) does not lead to a significant cytopathicity and cell death8,9. Furthermore, both in vivo and in vitro macrophage infections are characterized by the active accumulation of mature virions in subcellular compartments generated as invaginations of the plasma membrane nowadays referred to as virus-containing compartments (VCC)9,10, a feature not observed in CD4+ T cells. These distinctive features of macrophage infection render them potential candidates for contributing to viral persistence in individuals profoundly depleted of CD4+ T cells as well as in those receiving cART1.
The potential role of infected macrophages as long-lived reservoirs of silent infectious provirus is challenged by several observations, including the uncertainty of the lifespan of infected tissue-resident macrophages, potential “false positive” findings in tissue preparations due to the presence of residual infected CD4+ T cells11, phagocytosis of infected CD4+ T cells (with unclear consequences in terms of macrophage infection or virus elimination)12,13, and the lack of a molecular signature that would unequivocally demonstrate the non-CD4+ T cell origin of virus rebounding after cART suspension, as reviewed9,14–16. In support of a relevant role of infected macrophages in HIV-1 persistency there are different observations in macaques experimentally infected with the simian immunodeficiency virus (SIV)17,18, a close relative of HIV-1, as well as clinical evidence of active HIV-1 replication and/or HIV-related inflammation in the CNS occurring in a fraction of individuals with suppressed viremia by cART19. The hypothesis that, in addition to CD4+ T cells, macrophages could be prominent contributors to the cART-resistant reservoir of infectious proviruses has been further supported by the recent evidence that mice with severe combined immunodeficiency (SCID) reconstituted with human myeloid cells in the absence of CD4+ T cells can undergo both acute and chronic HIV-1 infection with establishment of an inducible HIV-1 reservoir, at least in some animals20.
An additional element fostering the perplexity on the role of tissue-associated macrophages in persistent HIV-1 infection is the lack of robust in vitro models based on primary cells rather than on immortalized myeloid cell lines. In this regard, we have previously shown that stimulation of either primary human MDM21 or monocytes10 with potent pro-inflammatory cytokines, namely interferon-γ (IFN-γ) and tumor necrosis factor-α (TNF-α), before infection leads to a significant containment of virus replication. This HIV-restrictive profile was characterized by several features, including downregulation of the CD4 molecule from the cell surface, upregulated secretion of CCR5-binding chemokines and induction of the expression of the putative restriction factor APOBEC3A22. In the present study, we have investigated the potential consequences of restimulating infected M1-MDM with IFN-γ and TNF-α, as these cytokines are credited with latency-reversal capacity driven by activation of STAT1 and NF-kB transcription factors, respectively23–26. Quite surprisingly, we observed that a second stimulation of infected M1-MDM with these pro-inflammatory cytokines several days after infection, a condition here defined as “M1 squared (M12) MDM”, further inhibited HIV-1 replication down to very low levels for several weeks. However, this quasi-silent infection was fully reversed leading to rapid viral spreading by either cocultivation with activated peripheral blood mononuclear cell (PBMC) or by incubation with their culture supernatants, an effect that was prevented by the reverse transcriptase (RT) inhibitor lamivudine/3TC. As both the selective repression of virus expression and its reactivation in M12 MDM upon PHA blast coculture were reproduced in cells infected with single round Vesicular Stomatitis Virus-G (VSV-G) pseudotyped HIV-1, M12 MDM represent an in vitro model that could be further exploited to decipher the molecular and immuno-pharmacological features of reversible latency/low levels HIV-1 production in primary human macrophages.
Results
Restimulation of infected M1-MDM with pro-inflammatory cytokines leads to a stronger containment of HIV-1 replication
Having described that M1 polarization of primary human MDM leads to a reduction of HIV-1 replicative capacity21,22, we have investigated the consequences of reexposing M1-MDM that were infected 7 days earlier by the R5 HIV-1Bal strain to the same cytokines (Fig. 1A), an experimental condition further referred to as “M12 MDM”. We confirmed that M1 stimulation before infection lead to a significant reduction of virus replication vs. unstimulated, control (CTR) MDM (Fig. 1B). However, a much greater inhibition of virus propagation, measured both in terms of kinetics and area under the curve (AUC), was consistently observed in M12 MDM vs. M1-MDM obtained from multiple seronegative donors (Fig. 1B). The strong inhibitory profile associated with M12 MDM was not caused by cytotoxic effects of cytokine stimulation. Freeze-fracture experiments were performed in order to quantify the levels of cell-associated vs. released virions, as reported27, in these experimental conditions. However, no detectable cell-associated RT activity was observed in both M1-MDM and M12 MDM (Fig. 1S). Furthermore, if infected CTR MDM were stimulated 7 days post-infection with M1 cytokines only a moderate inhibition of virus replication was observed (Fig. 2S).
Figure 1.

M1-MDM stimulation with polarizing cytokines after infection further inhibits HIV-1 replication. (A) Experimental scheme. Seven days-old MDM were either left unstimulated (CTR) or were stimulated with IFN-γ plus TNF-α (M1-MDM) 18 h before HIV-1 infection (m.o.i = 0.1). A second stimulation of M1-MDM with the same polarizing cytokines was performed 7 days after infection (M12 MDM); as for the first stimulation, cytokine-enriched medium was removed 18 h later and replaced with complete medium. (B) M12 MDM show a superior capacity of curtailing HIV-1 replication than M1-MDM. Left panel: kinetics of supernatant-associated RT activity; right panel: Area under curve (AUC); statistical analysis by One-way ANOVA; **p < 0.01, ****p < 0.0001 (n = 8).
Thus, a second stimulation of infected M1-MDM with the same pro-inflammatory cytokines several days after infection further strengthened their capacity to contain virus replication.
M12 MDM are characterized by persistently low levels of total and integrated HIV-1 DNA together with a silent transcriptional profile
The profound containment of HIV-1 replication in M12 MDM observed by quantification of RT activity was consistent with a significant reduction of the levels of total HIV-1 DNA in comparison to both CTR, infected MDM and also to M1-MDM that were not restimulated (Fig. 3S). M1-MDM showed an initial containment of viral DNA accumulation, as reported22; however, as early as 12 days after infection, their HIV DNA levels became superimposable to those of CTR MDM. In contrast, M12 MDM maintained significantly lower levels of HIV-1 DNA (although with some fluctuations) at least up to 21 days after infection (Fig. 3S). Reduced levels of proviral DNA (however not reaching statistical significance) were detected by Alu-PCR in M12 MDM vs. CTR and M1-MDM 15 days post-infection (i.e. 8 days after cytokine restimulation in the case of M12 MDM; Fig. 2A).
Figure 2.
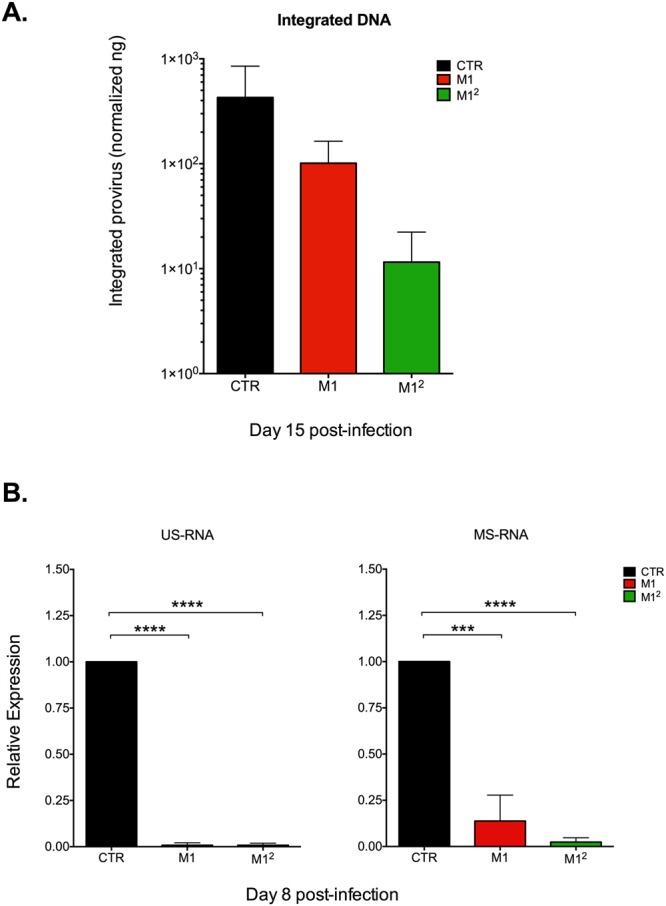
M12 MDM show reduced levels of integrated (proviral) HIV DNA than CTR and M1-MDM together with a silent profile of proviral transcription. (A) Alu-PCR was evaluated by semi-quantitative determination of HIV integrated provirus normalized to mitochondrial DNA (ng/ng) in extracts from MDM obtained 15 days after infection. Reduced levels of integrated provirus were observed in M1-MDM and, particularly, in M12 MDM that, however, were not statistically significant by paired ANOVA; the results show the means ± SD of infections of cells obtained from 5 independent donors. (B) Silent transcriptional profile of M12 MDM after cytokine restimulation. Cytokine restimulation of M1-MDM 8 days after infection did not result in increased levels of either US or MS HIV-1 RNA. Bar graphs represent the fold difference of M1 and M12 macrophages in respect with unpolarized CTR cells Days post-infection: 8 (n = 2), 12 (n = 8), 18 and 21 (n = 4); statistical analysis by One-way ANOVA *p < 0.05, **p < 0.01; ***p < 0.001.
When the levels of both unspliced (US) and multiply-spliced (MS) viral RNAs were quantified in the different experimental conditions 8 days post-infection (i.e. one day after restimulation with pro-inflammatory cytokines in the case of M12 MDM) both M1 and M12 MDM showed significantly lower levels of viral transcripts in comparison to those of CTR infected MDM (Fig. 2B). This quasi-silent transcriptional profile of M12 MDM was confirmed also at later time points both in the presence or absence of the RT inhibitor lamivudine/3TC (Fig. 4S).
Altogether, these results support the hypothesis that M12 MDM may represent an in vitro model of latent HIV-1 infection of primary human macrophages.
Upregulation of HIV-1 restriction factors in M12 MDM
We next investigated whether the strong containment of virus replication observed in M12 MDM was associated with an increased expression of restriction factors known to counteract HIV-1 infection by acting at the level of reverse transcription, before proviral integration, namely APOBEC3A, APOBEC3G and SAMHD128. As we22 and others29 have originally reported, APOBEC3A was not expressed in uninfected control MDM whereas APOBEC3G was visible by Western blotting as a faint band (Fig. 5S). M1 polarization induced a modest upregulation of APOBEC3G, but a clear-cut expression of APOBEC3A (Figs 3A and 5S), as published22. Eight days after infection, the levels of expression of these two restriction factors in M1-MDM returned to levels comparable to those observed at baseline; conversely, M12 MDM showed significantly higher levels of both restriction factors than both CTR and M1-MDM (Figs 3A and 5S). In contrast, SAMHD1 was clearly expressed by MDM, but it was not significantly modulated in these experimental conditions (Fig. 6S).
Figure 3.

Upregulation of HIV-1 restriction factors in M12 MDM. (A) Expression of APOBEC3A (A3A) and APOBEC3G (A3G). Cytokine stimulation induced the expression of A3A and upregulated that of A3G both before infection and 8 days after. The quantification of the original Western blot shown as Fig. 5S was obtained with the use of the ImageJ software (version 1.47 v, WS Rasband ImageJ, NIH; http://rsb.info.nih.gov). (B) Upregulation of TRIM22 and CIITA mRNA expression. Cytokine stimulation induced the expression of both negative regulators of proviral transcription before and 8 days after infection; the levels of mRNA expression of TRIM22, but not those of CIITA, were also increased in CTR MDM 7 days after infection. Bar graphs represent the fold difference as compared to CTR cells at day 0 prior to infection. Statistical analysis by One-way ANOVA; *p < 0.05, **p < 0.01; ***p < 0.001 (n = 4).
It is well known that macrophage stimulation with IFN-γ and TNF-α triggers multiple signaling pathways resulting in cell activation according to a pro-inflammatory mode30. Of interest, some factors either induced or post-translationally modified by M1 polarization have been previously reported to modulate HIV-1 proviral transcription either in a positive or negative fashion. In particular, IFN-γ was reported to upregulate the expression of TRIM22 and CIITA, two inducible restriction factors acting as repressors of proviral transcription by independent mechanisms31–34. The expression of both TRIM22 and CIITA mRNAs was indeed promptly upregulated after 18 h of cell stimulation of MDM with M1 cytokines before infection (Fig. 3B). Of interest, CTR MDM showed a significant increase in TRIM22 mRNA expression in comparison to their basal levels 8 days after infection. Infected M12 MDM expressed significantly higher levels of TRIM22 mRNA vs. M1-MDM that were not restimulated (Fig. 3B, left panel). CIITA mRNA expression showed a partially different pattern in that, like TRIM22, it was strongly upregulated in uninfected MDM by stimulation with M1 cytokines; however, unlike TRIM22, its levels returned to baseline in both CTR and M1-MDM 8 days after infection (Fig. 3B, right panel) whereas they were strongly induced by cytokine restimulation in M12 MDM (Fig. 3B, right panel). In contrast, no significant changes in the low levels of either cyclin T1 or CDK9 (forming the P-TEFb complex) were observed in these different experimental conditions (Fig. 7S).
Thus, the quasi-silent profile of HIV expression in M12 MDM was associated with an increased expression of APOBEC3A and APOBEC3G as well as of TRIM22 and CIITA, two IFN-inducible factors exerting a negative effect on proviral transcription.
The lack of HIV-1 expression in M12 MDM is not explained by an impaired activation of STAT1 and NF-kB
Both IFN-γ and TNF-α have been earlier described as potent inducers of HIV-1 transcription and expression acting via activation of STAT1 and of the canonical NF-kB pathway, respectively, in different cell models, including interleukin-2 (IL-2) activated PBMC35 and myeloid cells23–26. Therefore, we have investigated whether the silent transcriptional and expression profiles observed in M12 MDM could be accounted for by an impaired activation of these two transcription factors.
When uninfected MDM were stimulated with IFN-γ and TNF-α, a prompt activation of STAT1, measured in terms of phosphorylation of this transcription factor, was observed 5 min after cytokine stimulation and remained clearly visible after 10 additional min (Fig. 4, left panel). This finding was consistent with the observation that the upregulation of CIITA expression upon cell stimulation with IFN-γ requires STAT1 activation36. Furthermore, the phosphorylated form of NF-kB p65 became promptly visible upon TNF-α stimulation of these cells. After infection and 7 additional days of culture, no evidence of residual activation of either STAT1 or NF-kB was observed in both control and M1-MDM. However, restimulation of these latter cells with IFN-γ and TNF-α resulted in the rapid and robust activation of both STAT1 and NF-kB (Fig. 4, right panel). As previously reported37, an increased expression of STAT1 was observed in M1-MDM 7 days after infection (Fig. 4, right panel).
Figure 4.

Activation of STAT1 and NF-kB in M1 and M12 MDM. Cytokine stimulation induced activation of STAT-1 and NF-kB in both uninfected (left panel) and 7-days infected (right panel) MDM. Increased expression of STAT-1 was also observed in M1-MDM 7 days after infection (right panel). These results were obtained with the cells of a single donor representative of 4 independently tested.
Thus, the silent HIV-1 transcriptional profile observed in M12 MDM was not consequent to absent or impaired activation of STAT1 and NF-kB, two transcription factors endowed with the capacity of triggering or potentiating proviral transcription.
Allogeneic mitogen-stimulated PBMC or their culture supernatant reactivate HIV-1 replication in M12 MDM, but not in CTR or M1-MDM
In order to verify whether the almost silent HIV-1 replicative profile of infected M12 MDM could be reversed by cell stimulation, we incubated both CTR and polarized infected MDM with allogeneic PBMC that were activated 72 h before with the mitogen phytohemagglutinin (PHA blasts). Of note, PHA blasts were not resuspended in medium containing IL-2 in order to curtail cell proliferation and viral spreading in these cells. In this regard, we observed that IL-2-stimulated PHA blasts rapidly overcame adherent M12 MDM leading to their elimination likely by cell fusion, release of cytotoxic cytokines, or activation-induced cell death (data not shown).
Incubation of infected CTR or M1-MDM with PHA blasts did not modify their levels of RT activity production (Fig. 5A); a similar observation was made by analyzing the levels of HIV DNA in the same experimental conditions (Fig. 8S). In sharp contrast, incubation of infected M12 MDM with PHA blasts resulted in a prompt and robust release of RT activity in culture supernatants in association with a clear-cut rise in the levels of cell-associated HIV DNA (Figs 5A and 8S, respectively).
Figure 5.

Selective induction of HIV-1 replication (or expression of VSV-G pseudotyped virus) in M12 MDM stimulated by allogeneic PHA blasts or their supernatant and its selective prevention by lamivudine/3TC. (A) Kinetics of RT activity. PHA blasts upregulated virus replication in M12 MDM, but not in CTR or M1-MDM; the arrowheads indicate the timing of PHA blast addition to the infected MDM cultures. Mean ± SEM of 7 independent donors is reported. Comparison of different time points between M1 vs. M12 plus PHA blasts was performed using One-way ANOVA. ***p < 0.001. The addition of lamivudine/3TC (1 µM - black arrowheads) did not substantially affect virus production in CTR and M1-MDM, but fully prevented HIV-1 replication in M12 MDM. (B) Left panel: kinetics of RT activity; right panel: RT activity ratios relative to unstimulated M12 MDM calculated at peak virus replication in the different conditions (n = 2). The arrowhead indicates the timing of stimuli addition to the infected MDM cultures. (C) CTR, M1-MDM and M12 MDM were infected with a VSV-G pseudotyped virus expressing eGFP. Thirteen days after infection the cells were incubated with Blast Sup for 72 h before determining the levels of eGFP expression. Both M1-MDM and M12 MDM showed reduced levels of GFP expression vs. CTR cells, but only M12 MDM were significantly enhanced in terms of virus expression by incubation with Blast Sup (p < 0,05 by T-test, n = 3; see also Fig. 9S).
In order to determine whether the released virus was produced from persistently infected MDM or by propagation of a replication-competent HIV-1, these experiments were also conducted in the presence of the RT inhibitor 3TC/lamivudine (1 µM). Of interest, a clear-cut dichotomous profile was observed in that the antiviral agent did not significantly affect virus production in unstimulated or PHA blast-stimulated CTR and M1-MDM, but abrogated virus replication in M12 MDM both in unstimulated and stimulated conditions (Fig. 5A).
Next, we investigated whether cell-to-cell contact with PHA blasts was required for virus reactivation in infected M12 MDM. To this purpose, M12 MDM cells were incubated with different dilutions of filtered PHA blast supernatants (Blast Sup). Blast Sup upregulated virus production from infected M12 MDM in a dilution-dependent manner, although, as expected, with a lower potency than that observed with PHA blast coculture with M12 MDM (Fig. 5B).
PHA blast supernatant selectively reactivates VSV-G pseudotyped HIV-1 expression in M12 MDM, but not in CTR or M1-MDM
In order to consolidate the profile observed with replication-competent HIV-1 in terms of restricted expression and proviral reactivation by cell stimulation with allogeneic PHA blasts culture supernatants, these experiments were also conducted after infection of CTR, M1 and M12 MDM with VSV-G pseudotyped virus expressing enhanced Green Fluorescent Protein (eGFP) that bypasses the CD4 and CCR5 requirement for viral entry and establishes a highly efficient single round infection of these cells.
As reported22, the M1-dependent restriction was observed also using this experimental approach; indeed, almost 50% of the CTR MDM expressed eGFP 13 days post-infection without any influence of 72 h incubation with Blast Sup (Fig. 9S). A similar observation was made for M1-MDM that were not restimulated and whose lower levels of eGFP expression vs. those of CTR cells were not enhanced by Blast Sup (Figs 5C and 9S), thus recapitulating what observed with replication-competent HIV-1 (Fig. 5A). In contrast, consistently with the results obtained with replication-competent HIV-1 (Fig. 5A), M12 MDM showed a moderate, but statistically significant, enhancement of virus expression upon Blast Sup stimulation (Figs 5C and 9S).
These observations together provide substantial evidence that HIV-1 infection of M12 MDM established a state of persistent low levels HIV-1 replication likely including cell harboring latent, replication-competent proviruses that could be reversed by immunologic stimulation to produce infectious virus.
Discussion
In the present study, we have further investigated our previous observation that short-term stimulation of human MDM with pro-inflammatory cytokines (IFN-γ and TNF-α), i.e. M1 polarization, before infection induces a partial restriction of HIV-1 replication. By restimulating M1-MDM with the same cytokines 7 days after infection (M12 MDM), we have consistently observed a more profound restriction of virus replication approaching the limits of RT activity detection in culture supernatants. Together with a silent profile of proviral transcription, M12 MDM were characterized by lower levels of total and integrated HIV DNA in comparison to CTR infected MDM that did not undergo M1 polarization and also of M1-MDM that were not restimulated with the pro-inflammatory cytokines after infection. Infected M12 MDM showed an upregulation of several well-known intracellular inhibitors of virus replication, acting either before (APOBEC3A, APOBEC3G) or after (TRIM22 and CIITA) proviral integration, although the activation of both of STAT1 and canonical NF-kB, known inducers of proviral transcription, was not impaired in these cells. Activation of HIV-1 expression was promptly triggered in M12 MDM, but not in control or M1-MDM, by either cocultivation with allogeneic PHA blasts or incubation with their culture supernatants (Blast Sup). This effect was fully prevented in M12 MDM, but not in CTR or M1-MDM, by the RT inhibitor lamivudine/3TC therefore indicating that they harbored replication-competent virus. Proviral reactivation by Blast Sup was also selectively observed in M12 MDM, but not in CTR or M1-MDM, infected with single-round VSV-G pseudotyped HIV-1.
The observation that cART suspension in individuals with fully controlled levels of HIV-1 viremia almost invariably results in the rapid resurgence of virus replication and disease progression demonstrates a central role of viral reservoirs insensitive to antiretroviral agents in life-long persistency of HIV-1 infection38. The demonstration of the existence of a small pool of long-lived, resting memory CD4+ T lymphocytes harboring replication-competent provirus has provided the first demonstration of a viral reservoir39–41. Several contributions have refined the composition of latently infected CD4+ T cell subsets highlighting a prominent role of follicular T helper cells42, and a more accurate estimate of the size of the replication-competent proviral reservoir43,44. In contrast, the contribution of myeloid cells, and of tissue macrophages in particular, has remained elusive with the partial exception of the central role of microglial cells, together with astrocytes, in the pathogenesis of brain infection with compelling evidence obtained in SIV-infected macaques17,18 and in individuals with viremia suppressed by cART, showing evidence of either active virus replication or HIV-related inflammation in the CNS19.
A component of the perplexity on the potential role of macrophages in HIV-1 persistency is the lack of a robust in vitro model of reversible latent infection in primary cells. This is partially explained by the observation that in vitro infection of MDM is associated with continuous virus production for several weeks in culture, likely reflecting an absent or reduced virus-dependent cytopathicity45,46. However, Brown and colleagues have provided evidence of reversible latent HIV-1 infection in primary human MDM infected with an EGFP-coding virus by purifying EGFP negative cells containing integrated provirus that could be reactivated by cell stimulation with IL-447. Additional evidence of a potential latent state of HIV-1 infection of primary human MDM was earlier associated with cell stimulation by IL-10, although this cytokine also interfered with a pre-integration step of the virus life cycle48, making difficult to dissect out the contribution of the inhibition of proviral transcription from the global inhibition of virus replication induced by IL-10 stimulation.
In this context, we have earlier reported that short-term (18 h) stimulation of primary human MDM with the pro-inflammatory cytokines IFN-γ and TNF-α, an experimental condition commonly referred to as “M1 polarization”30, before infection caused a significant containment of virus replication initially attributed to an impairment of viral entry21, consistently with earlier observations49. In a subsequent study22, we described a strong association between the HIV-restrictive profile of M1-MDM and the induction of expression of APOBEC3A that was previously described to act as a restriction factor for HIV-1 infection of monocytes freshly isolated from peripheral blood29. As this restrictive profile was induced by stimulation of MDM shortly before infection, we have here investigated the consequence of re-exposing infected M1-MDM to the same stimuli 7 days after infection, a time window sufficient to allow the establishment of a pool of cells harboring integrated proviruses. We initially hypothesized that restimulation of infected M1-MDM with pro-inflammatory cytokines known to be potent upregulators of proviral transcription23–26 would lead to a burst of virus production from these cells. In sharp contrast, we consistently observed a stronger containment of virus replication, frequently approaching the limit of detection of our RT activity assay, in comparison to infected M1-MDM that were not restimulated. As we excluded that this quasi-silent phenotype could be consequent to either cytokine-dependent cytotoxicity50 or accumulation of preformed virions in VCC27, we switched our hypothesis to consider M1 restimulation as a mean to drive primary human macrophages towards a state of proviral latency. Indeed, restimulation of infected M1-MDM with IFN-γ and TNF-α upregulated the expression of restriction factors APOBEC3A and APOBEC3G that could likely account for the decreased levels of HIV DNA observed in both M1-MDM and M12 MDM2,3. Of interest, SAMHD1, a hydroxylase inhibited by the HIV-2 associated protein Vpx, which acts by reducing the availability of the pool of deoxynucleotides required for the RT step preceding proviral integration28,51, was not significantly modified in terms of its expression in the different experimental conditions here explored. In this regard, a recent study showed that a significant fraction (up to 20%) of both microglia and monocyte-derived tissue resident macrophages, unlike MDM, is in a G1-like state of their life cycle that, although not coupled with cell proliferation, allowed these cells to bypass the restriction imposed by SAMHD152.
Restimulation of M1-MDM with M1 cytokines 8 days after infection failed to upregulate both US and MS viral transcripts that remained barely detectable also at later time points. This partially unexpected finding was not explained by the lack of STAT1 and NF-kB activation, two transcription factors previously linked to activation of proviral transcription both in T lymphocytes and myeloid cells23–26. We hypothesize that their inductive effects on proviral transcription was overcome by the simultaneous expression of TRIM22 and CIITA, two inducible restriction factors acting as negative transcriptional regulators of HIV expression. In this regard, we have earlier shown that TRIM22 interferes with Sp1 driven proviral transcription31,32 whereas other investigators have reported its negative effects also in MDM53,54. CIITA, in addition to its key role of the upregulation of MHC Class II antigens55, was demonstrated to compete with Tat for binding to the P-TEFb complex56, also in myeloid cells33, thereby resulting in the downregulation of HIV-1 gene expression. More recently, we have reported that TRIM22 and CIITA can aggregate in nuclear bodies also containing TRIM19/PML and Cyclin T1 (a component of the P-TEFb complex)34 suggesting that these factors may act in concert.
However, we suggest that STAT1 and NF-kB activation could still play a role in terms of “priming” of the selective upregulation of virus production observed in M12 MDM, but not in control or M1-MDM, cocultured with PHA blasts or incubated with their filtered supernatant. In this regard, the observation that Blast Sup induced virus production from M12 MDM indicated that, on the one hand, cell-to-cell contact between infected MDM and PHA blasts was dispensable and, on the other hand, that virus replication occurred mostly, if not only, in M12 MDM rather than in PHA blasts (that were maintained in medium devoid of IL-2 to prevent their proliferation). The observation that addition of lamivudine/3TC completely prevented the inductive effect of PHA blasts or of their supernatant on virus production from M12 MDM (as well as on the minimal levels of virus replication observed in unstimulated cells) demonstrates, on the one hand, that these cells harbored replication-competent virus and, on the other hand, that most of the released virus derived from newly infected cells rather than from persistently infected cells as observed in CTR and M1-MDM whose virus production was insensitive to the presence of the RT inhibitor. In this regard, in order to provide formal evidence that at least a fraction of M12 MDM harbored latently infected proviruses we reproduced the same profiles of virus restriction and reactivation upon infection of CTR, M1- and M12 MDM with VSV-G pseudotyped HIV-1 that undergoes a single round of infection and it is incapable of viral spreading.
Thus, M12 MDM represent a robust model of low levels virus replication and reversible HIV-1 latency in primary human macrophages that could be further exploited for testing latency-reversing and/or latency-enhancing agents in these cells. Our study also emphasizes the potential role of restriction factors of the APOBEC family and of physiological negative regulators of proviral transcription, such as TRIM22 and CIITA, in infected primary human macrophages, at least in vitro. As, at least in the case of TRIM22, allelic variants affecting its coding sequences have been linked to differential outcomes of HIV-1 disease57, it is tempting to speculate that they could also differentially affect the nature and size of the inducible viral reservoir in infected individuals receiving cART.
Methods
Reagents
Human, endotoxin-free recombinant cytokines IFN-γ and TNF-α were purchased from R&D Systems (Minneapolis, MN, USA); Ficoll-Hypaque and Percoll density gradient were bought from Amersham Biosciences Europe (Milan, Italy) and from GE Healthcare (Fairfield, CT), respectively. Dulbecco’s modified Eagle’s medium (DMEM), phosphate-buffered saline (PBS), fetal bovine serum (FBS), normal human serum (NHS), penicillin, streptomycin (pen/strep) and glutamine were purchased from Lonza (Cologne, Germany). Polyadenylic Acid polyA and oligo d(T)12–18 were purchased from Amersham Pharmacia Biotech (Uppsala, Sweden); [α-32P]-dTTP was purchased from Perkin Elmer (Waltham, MA, USA); DE81 paper was purchased from Whatman (Maidstone, UK). SSC (0.3M sodium citrate in 3 M NaCl), KCl and phorbol-12, 3TC (Lamivudine) and Phytohemagglutinin (PHA) were purchased from Sigma-Aldrich (St. Louis, MO, USA); MgCl2 was purchased from BDH lab Supplies (Poole, U.K.); Accutase was purchased from Sigma Aldrich (Merck KGaA, Darmstadt, Germany).
Isolation of human monocytes from peripheral blood mononuclear cells (PBMC) and differentiation into monocyte-derived macrophages (MDM)
PBMC were isolated from the buffy coats of healthy HIV-seronegative blood donors by Ficoll-Hypaque density gradient centrifugation. Monocytes were then purified by Percoll density gradient centrifugation reaching 80–90% purity, as determined by CD14 expression and other lineage markers, as described46. The cells were then washed, resuspended in DMEM containing pen/strep (1%), glutamine (1%), heat-inactivated FBS (10%) and NHS (5%) (Complete Medium). Monocytes were seeded into 48-wells plastic plates at the concentration of 5 × 105 cells/mL and cultivated for 7 additional days at 37 °C in 5% CO2 to promote their full differentiation into MDM (≥95% CD14+), as previously described21.
M1 and M12 polarization of MDM
Fully differentiated MDM were stimulated for 18 h with IFN-γ (20 ng/ml) and TNF-α (2 ng/ml) to induce M1 polarization21. Then, the medium containing polarization cytokines was removed and replaced with complete medium before HIV-1 infection. In order to obtain M12 MDM, infected M1-MDM were restimulated in the same conditions described above with IFN-γ (20 ng/ml) and TNF-α (2 ng/ml) 7 days after infection, as indicated in Fig. 1A. The cytokine containing medium was removed 18 h after restimulation and complete medium was added to the adherent cell cultures.
CCR5-dependent (R5) HIV-1 infection of MDM
Both CTR and M1-MDM were infected with the macrophage-tropic, laboratory-adapted R5 HIV-1BaL strain at the multiplicity of infection (m.o.i.) of 0.1. Multiple aliquots of culture supernatants were collected every 3 days up to 24 days post-infection and stored at −20 °C to assess virus production. At the end of each infection experiment, supernatants were thawed and analyzed for their viral content by measuring the levels of virion-associated Mg2+-dependent RT activity present in the supernatant, with a radioactive assay. As 99% of the RT enzyme is particle-associated, this assay is a faithful indication of the production of new progeny virions58.
VSV-G enhanced green fluorescence protein (eGFP) pseudotyped HIV-1 infection
The HIV-1 vector and plasmid used in this study have been described previously59. HIV-eGFP virus (in which nef was replaced with the eGFP reporter gene) was produced by cotransfection with a ratio of 1:7 of pMD2.G together with pNL4–3_GFP_R-Env-60 (both plasmids were obtained from the NIH AIDS Research and Reference Program, Division of AIDS, NIAID, NIH, Bethesda, Maryland, USA). Vector containing supernatants were harvested 48 h after transfection in 293 T cells, cleared by centrifugation, filtered by a 0.45 mm filter (MILLEX-HV PVDF; Millipore, Carrigtwohill, County Cork, Ireland), and stored at −80 °C.
CTR, M1-MDM and M12 MDM were infected with 60 µl/well of HIV-eGFP viral stock. Infected MDM (5 × 105 cells/condition) were detached from plastic adhesion after incubation with Accutase (150 µl/well for 30 min at 37 °C), as described46, spun, and their pellet was resuspended in a fixing solution containing 4% paraformaldehyde (PFA). Flow cytometry for GFP expression was performed using a FACS Calibur instrument (Becton Dickinson Italia, SpA, Milano, Italy), and the results were analyzed with the FlowJo software version 8.4.3 (Tree Star, Ashland, Oregon, USA).
Stimulation of infected MDM with allogeneic PHA blasts or their culture supernatant
Human PBMC of different donors were stimulated for 72 h with PHA (5 μg/ml) in order to induce their blast transformation. Cells were then washed and resuspended in RPMI 1640, 10% FBS without supplementation of IL-2 in order to prevent their cellular proliferation. Culture supernatants from PHA blasts were collected after 72 h and filtered through a 0.45 µm filter before incubation with MDM.
Cytotoxicity assay
ToxiLightTM bioassay kit (Lonza, Cologne, Germany) was used to check the potential citotoxicity at different times post-infection in the different experimental conditions. This kit quantitatively measures the release of adenylate kinase (AK) activity from damaged cells, providing an accurate and sensitive determination of cytolysis61.
PCR-based quantification of HIV-1 gag DNA
MDM were washed, lysed and treated with proteinase k at 65 °C for 2 h and then at 95 °C for 15 min. An aliquot of cell lysate corresponding to 1 × 106 cells was incubated in a PCR mix containing the following HIV-1 gag gene primer pair and probe: forward 5′-acatcaagcagccatgcaaat-3′; reverse 5′-atctggcctggtgcaatagg-3′; probe 5′-(FAM)catcaatgaggaagctgcagaatgggataga(TAMRA)-3′. The number of HIV-1 DNA copies was then determined by interpolating a reference standard curve (showing a linear distribution between 101 and 107 copies, r = 0.99) after normalization for human glyceraldehyde 3-phosphate dehydrogenase (GAPDH) DNA copy number (forward primer 5′-accacagtccatgccatcact-3′, reverse primer 5′-ggccatcacgccacagtt-3′ and probe, 5′-(FAM)cccagaagactgtggatggcccc(TAMRA)-3′). The thermal cycling conditions were 50 °C for 2 min, 95 °C for 12 min, and 40 cycles of 95 °C for 15 s and 65 °C for 1 min62.
Quantification of integrated (proviral) DNA by Alu-PCR
The levels of integrated proviral DNA were estimated adopting the published Alu-PCR protocol, involving a nested PCR-based assay with two sets of Alu-LTR primers and a probe, with minor modifications63. Genomic DNA (100 ng) was first amplified with AccuPrime Taq DNA Polymerase High Fidelity (Life Technologies Italia, Monza, Italy) in a DNA thermal cycler (Perkin Elmer, Waltham, Massachusetts, USA). Then, a real-time PCR was performed with an aliquot equivalent to 1/10th of the 25th cycle PCR product with the second-round LTR primers and TaqMan probe. For control, all samples were also amplified with a primer pair and probe targeting mitochondrial DNA. Standard curves for both target and normalizer were obtained by nested PCR, as described above, using serially diluted genomic DNA (from 500 to 0.16 ng). The levels of proviral DNA, calculated from the standard curves, were expressed as ng of integrated provirus normalized to those of a mitochondrial DNA standard curve.
Quantification of HIV-1 RNA by real-time PCR
HIV-1 RNA, both unspliced (US) and multiply spliced (MS), was analyzed 8 days after infection. Total RNA was extracted by using TRIzol reagent and PureLink RNA Mini Kit (Ambion, Life Technologies) and cDNA was synthesized from 1 µg total RNA. Multiplex TaqMan qPCR (Applied Biosystems) was performed with 50 ng of cDNA in a total volume of 25 µl with the following primer pairs and probe: forward 5′-cagactcatcaagcttctctatcaaa-3′, reverse 5′-tcactaatcgaatggatctgtctc-3′, probe RNAS 5′-(FAM)acccgacaggcccgaaggaa(TAMRA)-3′ (for MS RNA); forward 5′-acatcaagcagccatgcaaat-3′, reverse 5′-atctggcctggtgcaatagg-3′; RNA gag probe 5′-(FAM)catcaatgaggaagctgcagaatgggataga(TAMRA)-3′ (for US RNA). mRNA expression was calculated by using the relative quantification method (∆∆Ct) to uninfected CTR MDM all normalized to human 18S ribosomal RNA expression using Taqman ribosomal RNA control reagent kit (Applied Biosystems). All reactions were performed with an ABI 7700 Prism instrument (Applied Biosystems) using the following thermal cycling conditions: 50 °C for 2 min, 95 °C for 15 min, and 40 cycles of 95 °C for 15 sec and 60 °C for 1 min.
Quantification of cellular transcripts by real-time PCR
Relative expression of TRIM22, CIITA, CdK9 and Cyclin T1 RNA was analyzed at day 0 before infection and 8 days post-infection. Total RNA was extracted from cells using TRIzol reagent. cDNA was synthesized from 0.5 μg total RNA; cDNA (25 ng for each reaction) was amplified by PCR by using an ABI Prism 7000 Sequence Detection System (Applied Biosystems) with IQSYBR Green PCR master mix (Bio-Rad) according to the manufacture’s protocol. All mRNA values were normalized to Ribosomal Protein S7 (RPS7) mRNA. For each gene analyzed the mRNA level in CTR MDM at day 0 was set to 1. The following primer pair sets were used: CIITA, forward 5′-ggatcctcacggcctttt-3′, reverse 5′-ccccgatcttgttctcactc-3′; TRIM22, forward 5′-ggcttggtgagtgaatctgg-3′, reverse 5′-tcacaaactcctgcagtgct-3′; CdK9, forward 5′-ttcggggaggtgttcaag-3′, reverse 5′-atctcccgcaaggctgtaat-3′; CyT1, forward 5′-ggcgtggacccagataaag-3′, reverse 5′-ctgtgtgaaggactgaatcat-3′; RPS7, forward 5′-tggagatgaactcggacctc-3′, reverse 5′-cgaccaccaccaacttcaa-3′.
APOBEC3A/G, SAMHD1, NF-kB and STAT1 detection by Western blotting
MDM were lysed in NP40 buffer [50 mM Tris HCl (pH 7.5), 150 mM NaCl, 1% NP40 and 0.5% w/v deoxycholate] containing protease and phosphatase inhibitors. Proteins were separated by 10% (for STAT1 and NF-kB detection) or 7,5% (for APOBEC3A/G and SAMHD1 detection) SDS-PAGE, transferred to a nitrocellulose membrane by electroblot analysis and probed with the following antibodies (Ab): rabbit anti-APOBEC3A/G polyclonal Ab, kindly provided by Dr. Klaus Strebel (Laboratory od Molecular Microbiology, National Institute of Allergy and Infectious disease, National Institute of Health, Bethesda, MD, USA); polyclonal rabbit anti-STAT1 and monoclonal anti-phospho-STAT1 (tyr701) Ab (mAb; Cell Signaling Technology Inc., Denvers, MA, USA); mouse anti-NF-kB p65 and anti-phospho-p65 (ser536) mAbs (Termofisher Scientific, San Diego, California, USA; polyclonal rabbit anti-SAMHD1 Ab (Sigma-Aldrich, St. Louis, MO, USA); rabbit anti-GAPDH mAb was used as control (14C10 clone; Cell Signaling Technology, Inc., Denvers, MA, USA).
Statistical analysis
Statistical analysis was performed using the Prism GraphPad software v. 6.0 (GraphPad Software, www.graphpad.com). Results are reported as means ± SD. Comparison between groups was performed using the parametric one-way ANOVA; p values < 0.05 were considered significant. To control for inter-donor variability, all assays were performed in triplicate samples per condition with MDM derived from independent donors. T-test analysis was performed when appropriate, as indicated in the text.
Ethical Approval
The use of human peripheral blood cells derived from buffy coats for experimentation has been approved by the San Raffaele Hospital Ethical Committee; volunteers blood donors (>18 years old) sign a statement that material not of medical use could be utilized for research, noncommercial purposes. All methods were carried out in accordance with relevant guidelines and regulations approved by the aforementioned Institutional Ethical Committee; experiments involving infectious material (HIV) were conducted in BSL-3 laboratories.
Electronic supplementary material
Acknowledgements
We thank Silvia Ghezzi and Filipa Blasco-Lopez (Division of Immunology, Transplantation and Infectious Diseases, San Raffaele Scientific Institute, Milano) for their technical support. This work was funded in part by the AIM-HIV (to EV and GP) and HepaVAC (to RSA) EC-FP7-HEALTH projects (n. 305938 and n. 602893, respectively) and by the University of Insubria (intramural grant FAR 2014–2016 to RSA and GF and Institutional grant 2017 to RSA). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author Contributions
F.G. and G.A. performed most of the experiments here shown, except those related to CIITA and TRIM22 expression that were performed by G.F.; F.T. participated with G.A. in the generation of the results regarding STAT1 and NF-kB expression and activation; R.A., E.V. and G.P. participated to the overall discussion and interpretation of the results and to writing the paper. G.P. conceived and supervised all the experiments and provided a primary interpretation of the results.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Francesca Graziano and Giulia Aimola contributed equally.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-32451-w.
References
- 1.Deeks SG, et al. International AIDS Society global scientific strategy: towards an HIV cure 2016. Nat Med. 2016;22:839–850. doi: 10.1038/nm.4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koning FA, Goujon C, Bauby H, Malim MH. Target cell-mediated editing of HIV-1 cDNA by APOBEC3 proteins in human macrophages. J Virol. 2011;85:13448–13452. doi: 10.1128/JVI.00775-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ran X, Ao Z, Yao X. Apobec3G-Based Strategies to Defeat HIV Infection. Curr HIV Res. 2016;14:217–224. doi: 10.2174/1570162X14999160224100541. [DOI] [PubMed] [Google Scholar]
- 4.Ahn J. Functional organization of human SAMHD1 and mechanisms of HIV-1 restriction. Biol Chem. 2016;397:373–379. doi: 10.1515/hsz-2015-0260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mbonye U, Karn J. Transcriptional control of HIV latency: cellular signaling pathways, epigenetics, happenstance and the hope for a cure. Virology. 2014;454–455:328–339. doi: 10.1016/j.virol.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wei P, Garber ME, Fang SM, Fischer WH, Jones KA. A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell. 1998;92:451–462. doi: 10.1016/S0092-8674(00)80939-3. [DOI] [PubMed] [Google Scholar]
- 7.Vidya Vijayan KK, Karthigeyan KP, Tripathi SP, Hanna LE. Pathophysiology of CD4+ T-Cell Depletion in HIV-1 and HIV-2 Infections. Front Immunol. 2017;8:580. doi: 10.3389/fimmu.2017.00580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alfano M, Graziano F, Genovese L, Poli G. Macrophage polarization at the crossroad between HIV-1 infection and cancer development. Arterioscler Thromb Vasc Biol. 2013;33:1145–1152. doi: 10.1161/ATVBAHA.112.300171. [DOI] [PubMed] [Google Scholar]
- 9.Sattentau QJ, Stevenson M. Macrophages and HIV-1: An Unhealthy Constellation. Cell Host Microbe. 2016;19:304–310. doi: 10.1016/j.chom.2016.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Graziano F, Vicenzi E, Poli G. Immuno-Pharmacological Targeting of Virus-Containing Compartments in HIV-1-Infected Macrophages. Trends Microbiol. 2016;24:558–567. doi: 10.1016/j.tim.2016.02.018. [DOI] [PubMed] [Google Scholar]
- 11.Josefsson L, et al. The HIV-1 reservoir in eight patients on long-term suppressive antiretroviral therapy is stable with few genetic changes over time. Proc Natl Acad Sci USA. 2013;110:E4987–4996. doi: 10.1073/pnas.1308313110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Calantone N, et al. Tissue myeloid cells in SIV-infected primates acquire viral DNA through phagocytosis of infected T cells. Immunity. 2014;41:493–502. doi: 10.1016/j.immuni.2014.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DiNapoli SR, et al. Tissue-resident macrophages can contain replication-competent virus in antiretroviral-naive, SIV-infected Asian macaques. JCI Insight. 2017;2:e91214. doi: 10.1172/jci.insight.91214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barton K, Winckelmann A, Palmer S. HIV-1 Reservoirs During Suppressive Therapy. Trends Microbiol. 2016;24:345–355. doi: 10.1016/j.tim.2016.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Churchill MJ, Deeks SG, Margolis DM, Siliciano RF, Swanstrom R. HIV reservoirs: what, where and how to target them. Nat Rev Microbiol. 2016;14:55–60. doi: 10.1038/nrmicro.2015.5. [DOI] [PubMed] [Google Scholar]
- 16.Stevenson M. HIV persistence in macrophages. Nat Med. 2017;23:538–539. doi: 10.1038/nm.4337. [DOI] [PubMed] [Google Scholar]
- 17.Gama L, et al. Reactivation of simian immunodeficiency virus reservoirs in the brain of virally suppressed macaques. AIDS. 2017;31:5–14. doi: 10.1097/QAD.0000000000001267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spector SA, Rappaport J. HIV cure strategists: ignore the central nervous system at your patients’ peril. AIDS. 2017;31:167–168. doi: 10.1097/QAD.0000000000001268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kugathasan R, et al. Diffuse White Matter Signal Abnormalities on Magnetic Resonance Imaging Are Associated With Human Immunodeficiency Virus Type 1 Viral Escape in the Central Nervous System Among Patients With Neurological Symptoms. Clin Infect Dis. 2017;64:1059–1065. doi: 10.1093/cid/cix035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Honeycutt JB, et al. HIV persistence in tissue macrophages of humanized myeloid-only mice during antiretroviral therapy. Nat Med. 2017;23:638–643. doi: 10.1038/nm.4319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cassol E, Cassetta L, Rizzi C, Alfano M, Poli G. M1 and M2a polarization of human monocyte-derived macrophages inhibits HIV-1 replication by distinct mechanisms. J Immunol. 2009;182:6237–6246. doi: 10.4049/jimmunol.0803447. [DOI] [PubMed] [Google Scholar]
- 22.Cassetta L, et al. M1 polarization of human monocyte-derived macrophages restricts pre and postintegration steps of HIV-1 replication. AIDS. 2013;27:1847–1856. doi: 10.1097/QAD.0b013e328361d059. [DOI] [PubMed] [Google Scholar]
- 23.Griffin GE, Leung K, Folks TM, Kunkel S, Nabel GJ. Activation of HIV gene expression during monocyte differentiation by induction of NF-kappa B. Nature. 1989;339:70–73. doi: 10.1038/339070a0. [DOI] [PubMed] [Google Scholar]
- 24.Bachelerie F, Alcami J, Arenzana-Seisdedos F, Virelizier JL. HIV enhancer activity perpetuated by NF-kappa B induction on infection of monocytes. Nature. 1991;350:709–712. doi: 10.1038/350709a0. [DOI] [PubMed] [Google Scholar]
- 25.Biswas P, et al. Interferon gamma induces the expression of human immunodeficiency virus in persistently infected promonocytic cells (U1) and redirects the production of virions to intracytoplasmic vacuoles in phorbol myristate acetate-differentiated U1 cells. J Exp Med. 1992;176:739–750. doi: 10.1084/jem.176.3.739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Appelberg KS, et al. HIV-1 Infection Primes Macrophages Through STAT Signaling to Promote Enhanced Inflammation and Viral Replication. AIDS Res Hum Retroviruses. 2017;33:690–702. doi: 10.1089/AID.2016.0273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Graziano F, et al. Extracellular ATP induces the rapid release of HIV-1 from virus containing compartments of human macrophages. Proc Natl Acad Sci USA. 2015;112:E3265–3273. doi: 10.1073/pnas.1500656112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Strebel K. HIV accessory proteins versus host restriction factors. Curr Opin Virol. 2013;3:692–699. doi: 10.1016/j.coviro.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peng G, et al. Myeloid differentiation and susceptibility to HIV-1 are linked to APOBEC3 expression. Blood. 2007;110:393–400. doi: 10.1182/blood-2006-10-051763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Locati M, Mantovani A, Sica A. Macrophage activation and polarization as an adaptive component of innate immunity. Adv Immunol. 2013;120:163–184. doi: 10.1016/B978-0-12-417028-5.00006-5. [DOI] [PubMed] [Google Scholar]
- 31.Kajaste-Rudnitski A, et al. TRIM22 inhibits HIV-1 transcription independently of its E3 ubiquitin ligase activity, Tat, and NF-kappaB-responsive long terminal repeat elements. J Virol. 2011;85:5183–5196. doi: 10.1128/JVI.02302-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Turrini F, et al. HIV-1 transcriptional silencing caused by TRIM22 inhibition of Sp1 binding to the viral promoter. Retrovirology. 2015;12:104. doi: 10.1186/s12977-015-0230-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Forlani G, et al. The MHC-II transactivator CIITA inhibits Tat function and HIV-1 replication in human myeloid cells. J Transl Med. 2016;14:94. doi: 10.1186/s12967-016-0853-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Forlani G, et al. Tripartite Motif-Containing Protein 22 Interacts with Class II Transactivator and Orchestrates Its Recruitment in Nuclear Bodies Containing TRIM19/PML and Cyclin T1. Front Immunol. 2017;8:564. doi: 10.3389/fimmu.2017.00564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kinter AL, Poli G, Fox L, Hardy E, Fauci AS. HIV replication in IL-2-stimulated peripheral blood mononuclear cells is driven in an autocrine/paracrine manner by endogenous cytokines. J Immunol. 1995;154:2448–2459. [PubMed] [Google Scholar]
- 36.Steimle V, Siegrist CA, Mottet A, Lisowska-Grospierre B, Mach B. Regulation of MHC class II expression by interferon-gamma mediated by the transactivator gene CIITA. Science. 1994;265:106–109. doi: 10.1126/science.8016643. [DOI] [PubMed] [Google Scholar]
- 37.Goropevsek A, et al. Increased Levels of STAT1 Protein in Blood CD4 T Cells from Systemic Lupus Erythematosus Patients Are Associated with Perturbed Homeostasis of Activated CD45RA-FOXP3hi Regulatory Subset and Follow-Up Disease Severity. J Interferon Cytokine Res. 2017;37:254–268. doi: 10.1089/jir.2016.0040. [DOI] [PubMed] [Google Scholar]
- 38.Henrich TJ, Deeks SG, Pillai SK. Measuring the Size of the Latent Human Immunodeficiency Virus Reservoir: The Present and Future of Evaluating Eradication Strategies. J Infect Dis. 2017;215:S134–S141. doi: 10.1093/infdis/jiw648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chun TW, et al. In vivo fate of HIV-1-infected T cells: quantitative analysis of the transition to stable latency. Nat Med. 1995;1:1284–1290. doi: 10.1038/nm1295-1284. [DOI] [PubMed] [Google Scholar]
- 40.Chun TW, et al. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature. 1997;387:183–188. doi: 10.1038/387183a0. [DOI] [PubMed] [Google Scholar]
- 41.Finzi D, et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science. 1997;278:1295–1300. doi: 10.1126/science.278.5341.1295. [DOI] [PubMed] [Google Scholar]
- 42.Perreau M, et al. Follicular helper T cells serve as the major CD4 T cell compartment for HIV-1 infection, replication, and production. J Exp Med. 2013;210:143–156. doi: 10.1084/jem.20121932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ho YC, et al. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell. 2013;155:540–551. doi: 10.1016/j.cell.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sanyal A, et al. Novel assay reveals a large, inducible, replication-competent HIV-1 reservoir in resting CD4+ T cells. Nat Med. 2017;23:885–889. doi: 10.1038/nm.4347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gendelman HE, et al. Efficient isolation and propagation of human immunodeficiency virus on recombinant colony-stimulating factor 1-treated monocytes. J Exp Med. 1988;167:1428–1441. doi: 10.1084/jem.167.4.1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gartner S. The macrophage and HIV: basic concepts and methodologies. Methods Mol Biol. 2014;1087:207–220. doi: 10.1007/978-1-62703-670-2_17. [DOI] [PubMed] [Google Scholar]
- 47.Brown A, Zhang H, Lopez P, Pardo CA, Gartner S. In vitro modeling of the HIV-macrophage reservoir. J Leukoc Biol. 2006;80:1127–1135. doi: 10.1189/jlb.0206126. [DOI] [PubMed] [Google Scholar]
- 48.Montaner LJ, Griffin P, Gordon S. Interleukin-10 inhibits initial reverse transcription of human immunodeficiency virus type 1 and mediates a virostatic latent state in primary blood-derived human macrophages in vitro. J Gen Virol. 1994;75(Pt 12):3393–3400. doi: 10.1099/0022-1317-75-12-3393. [DOI] [PubMed] [Google Scholar]
- 49.Herbein G, Montaner LJ, Gordon S. Tumor necrosis factor alpha inhibits entry of human immunodeficiency virus type 1 into primary human macrophages: a selective role for the 75-kilodalton receptor. J Virol. 1996;70:7388–7397. doi: 10.1128/jvi.70.11.7388-7397.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Biswas P, Poli G, Orenstein JM, Fauci AS. Cytokine-mediated induction of human immunodeficiency virus (HIV) expression and cell death in chronically infected U1 cells: do tumor necrosis factor alpha and gamma interferon selectively kill HIV-infected cells? J Virol. 1994;68:2598–2604. doi: 10.1128/jvi.68.4.2598-2604.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vicenzi E, Poli G. Novel factors interfering with human immunodeficiency virus-type 1 replication in vivo and in vitro. Tissue Antigens. 2013;81:61–71. doi: 10.1111/tan.12047. [DOI] [PubMed] [Google Scholar]
- 52.Mlcochova P, et al. A G1-like state allows HIV-1 to bypass SAMHD1 restriction in macrophages. EMBO J. 2017;36:604–616. doi: 10.15252/embj.201696025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tissot C, Mechti N. Molecular cloning of a new interferon-induced factor that represses human immunodeficiency virus type 1 long terminal repeat expression. J Biol Chem. 1995;270:14891–14898. doi: 10.1074/jbc.270.25.14891. [DOI] [PubMed] [Google Scholar]
- 54.Bouazzaoui A, et al. Stimulated trans-acting factor of 50 kDa (Staf50) inhibits HIV-1 replication in human monocyte-derived macrophages. Virology. 2006;356:79–94. doi: 10.1016/j.virol.2006.07.025. [DOI] [PubMed] [Google Scholar]
- 55.Tosi G, Bozzo L, Accolla RS. The dual function of the MHC class II transactivator CIITA against HTLV retroviruses. Front Biosci (Landmark Ed) 2009;14:4149–4156. doi: 10.2741/3519. [DOI] [PubMed] [Google Scholar]
- 56.Accolla RS, Mazza S. De Lerma Barbaro, A., De Maria, A. & Tosi, G. The HLA class II transcriptional activator blocks the function of HIV-1 Tat and inhibits viral replication. Eur J Immunol. 2002;32:2783–2791. doi: 10.1002/1521-4141(2002010)32:10<2783::AID-IMMU2783>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 57.Ghezzi S, et al. Identification of TRIM22 single nucleotide polymorphisms associated with loss of inhibition of HIV-1 transcription and advanced HIV-1 disease. AIDS. 2013;27:2335–2344. doi: 10.1097/01.aids.0000432474.76873.5f. [DOI] [PubMed] [Google Scholar]
- 58.Fernie BF, Poli G, Fauci AS. Alpha interferon suppresses virion but not soluble human immunodeficiency virus antigen production in chronically infected T-lymphocytic cells. J Virol. 1991;65:3968–3971. doi: 10.1128/jvi.65.7.3968-3971.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Neagu MR, et al. Potent inhibition of HIV-1 by TRIM5-cyclophilin fusion proteins engineered from human components. J Clin Invest. 2009;119:3035–3047. doi: 10.1172/JCI39354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.He J, et al. Human immunodeficiency virus type 1 viral protein R (Vpr) arrests cells in the G2 phase of the cell cycle by inhibiting p34cdc2 activity. J Virol. 1995;69:6705–6711. doi: 10.1128/jvi.69.11.6705-6711.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Crouch SP, Kozlowski R, Slater KJ, Fletcher J. The use of ATP bioluminescence as a measure of cell proliferation and cytotoxicity. J Immunol Methods. 1993;160:81–88. doi: 10.1016/0022-1759(93)90011-U. [DOI] [PubMed] [Google Scholar]
- 62.Vicenzi E, Panina-Bodignon P, Vallanti G, Di Lucia P, Poli G. Restricted replication of primary HIV-1 isolates using both CCR5 and CXCR4 in Th2 but not in Th1 CD4(+) T cells. J Leukoc Biol. 2002;72:913–920. [PubMed] [Google Scholar]
- 63.Tan W, Dong Z, Wilkinson TA, Barbas CF, 3rd, Chow SA. Human immunodeficiency virus type 1 incorporated with fusion proteins consisting of integrase and the designed polydactyl zinc finger protein E2C can bias integration of viral DNA into a predetermined chromosomal region in human cells. J Virol. 2006;80:1939–1948. doi: 10.1128/JVI.80.4.1939-1948.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.