

Abstract
Streptococcus agalactiae (Group B Streptococcus or GBS) is a frequent cause of serious disease in newborns and adults. Epidemiological evidence indicates a strong association between GBS strains belonging to the hypervirulent CC17 clonal complex and the occurrence of meningitis in neonates. We investigate here the role of PbsP, a cell wall plasminogen binding protein, in colonization of the central nervous system by CC17 GBS. Deletion of pbsP selectively impaired the ability of the CC17 strain BM110 to colonize the mouse brain after intravenous challenge, despite its unchanged capacity to persist at high levels in the blood and to invade the kidneys. Moreover, immunization with a recombinant form of PbsP considerably reduced brain infection and lethality. In vitro, pbsP deletion markedly decreased plasmin-dependent transmigration of BM110 through brain microvascular endothelial cells. Although PbsP was modestly expressed in bacteria grown under standard laboratory conditions, pbsP expression was markedly upregulated during in vivo infection or upon contact with cultured brain endothelial cells. Collectively, our studies indicate that PbsP is a highly conserved Plg binding adhesin, which is functionally important for invasion of the central nervous system by the hypervirulent CC17 GBS. Moreover, this antigen is a promising candidate for inclusion in a universal GBS vaccine.
Introduction
Streptococcus agalactiae (also referred to as Group B Streptococcus or GBS), is a Gram-positive encapsulated bacterium that is frequently found as a commensal in the human gastrointestinal and genital tracts1,2. In addition, GBS can cause invasive infections particularly in neonates, pregnant women and elderly adults. Neonatal disease occurring during the first 6 days of life (referred to as early-onset disease or EOD) is likely due to transmission of the bacterium from the pregnant mother to the neonate secondary to aspiration of infected amniotic fluid or vaginal secretions during labor. Most cases of EOD are characterized by pneumonia followed by septicemia3. Late-onset disease (LOD), occurring from age 7 days to 3 months, also consists of septicemia, but displays a higher rate of meningitis, compared with EOD4–6.
Of the 10 GBS capsular types, the majority of invasive neonatal diseases are associated with the serotype III7–9. Among this serotype, one highly homogenous clone belonging to the clonal complex (CC) 17 is isolated in the majority of LODs and is referred to as the “hypervirulent” GBS clone7,8,10,11. Comparative genome analyses suggest an adaptation of the CC17 clones to humans, with an enhanced ability to colonize the gastro-intestinal tract and to cause invasive diseases, especially meningitis12,13. Several studies have attempted to explain the mechanisms underlying brain invasion by GBS and the striking meningeal tropism of the CC17 clones14. The first step in brain invasion involves adhesion of blood-born GBS to the microvascular endothelium, which is mediated by multiple interactions between bacterial surface proteins and cognate receptors, such as fibrinogen, laminin and fibronectin present on the endothelial lining15–22. After adhesion, GBS are internalized by endothelial cells and transmigrate within phagosomes23,24. In this process, GBS exploit the physiological process of transcytosis by which endothelial cells move macromolecular cargo from the circulation into the brain interstitium within membrane-bound vescicles25. In addition, bacterial toxins and proteolytic or inflammatory host factors can increase the permeability of the endothelial layer, thereby further promoting transversal of the brain blood barrier (BBB) by bacterial pathogens26–28. The surface proteins of CC17 strains are distinct from those belonging to other clonal complexes and might confer selective advantages during colonization or invasive infections29. For examples, the CC17-specific HvgA and Srr2 cell-wall proteins contribute to the increased virulence and to the meningeal tropism of CC17 strains30,31.
Recently, the ability of GBS to hijack the host fibrinolytic system and to invade the brain by plasminogen-dependent mechanisms has attracted considerable attention. Plasminogen (Plg) is an inactive proenzyme that is present in plasma at concentrations of approximately 2 μM and can be converted to plasmin (Pln) by host activators, such as tissue type (tPA) or urokinase type (uPA) activators. Contrary to Group A Streptococcus, which uses streptokinase to convert Plg to plasmin (Pln), GBS does not express endogenous Plg activators. Instead, after binding to the GBS surface, Plg is converted to Pln by host tPA or uPA during infection32,33. Pln, in turn, contributes to a number of amplification loops leading to increased Plg activation, generation of active metalloproteases, degradation of the extracellular matrix (ECM) and severe weakening of the BBB28,32,33.
We recently characterized PbsP (Plasminogen binding surface Protein), a cell wall-anchored protein that is required for acquisition of surface-associated Pln activity by GBS and for its dissemination from the blood to the brain34. This protein is a valuable vaccine candidate since PbsP is highly conserved (>99.3% identity at the protein level) and the pbsP gene is present in all sequenced human GBS strains. However, expression of PbsP at the GBS cell surface was shown to be strain-dependent34. Immunization with PbsP is protective against infection with the CC23 NEM316 strain34, but the role of PbsP in the virulence and meningeal tropism of the CC17 strains is unclear. In contrast to CC23 strains, the protein is expressed at very low levels on the surface of CC17 strains grown under standard laboratory conditions34, which may greatly limit the potential usefulness of PbsP in an anti-GBS vaccine34.
The present study was undertaken to investigate whether PbsP has a role in the context of infections caused by CC17 GBS and whether immunization with PbsP is effective against this major strain lineage causing meningitis in neonates. Using a mouse model of meningitis, we demonstrate upregulation of PbsP in CC17 GBS specifically during infection and its role in the invasion of the central nervous system. Accordingly, PbsP expression increases in vitro upon contact with cultured brain endothelial cells and PbsP has a non-redundant role in plasmin-dependent transmigration through monolayers of endothelial cells. In addition, we demonstrate that immunization with PbsP prevents the development of meningo-encephalitis in mice infected with CC17 GBS.
Results
PbsP is required for virulence of CC17 GBS
To investigate the role of PbsP in the pathogenesis of invasive GBS infection, the pbsP gene was deleted in-frame in the chromosome of the CC17 strain BM110. The viability, morphology and growth in Todd-Hewitt broth (THB) of the ∆pbsP mutant were similar to those of the wild type (WT) parental strain (Figs S1 and S2). Western-blot analysis of GBS cell wall extracts using a polyclonal mouse serum (pAb) raised against recombinant PbsP showed a reactive band in the WT strain that was absent in the ∆pbsP mutant (Fig. 1A). We next compared the virulence of BM110 WT with that of the mutant using a mouse model of invasive GBS infection involving hematogenous spreading of the bacteria to the brain after intravenous challenge. Under these conditions, survival was increased in mice infected with the ∆pbsP mutant and, at the end of the experiment, only 2 out of 16 (12%) of the mice infected with the mutant strain died, while 10 out of 16 (62%) of those challenged with the WT strain succumbed to infection (Fig. 1B).
Figure 1.
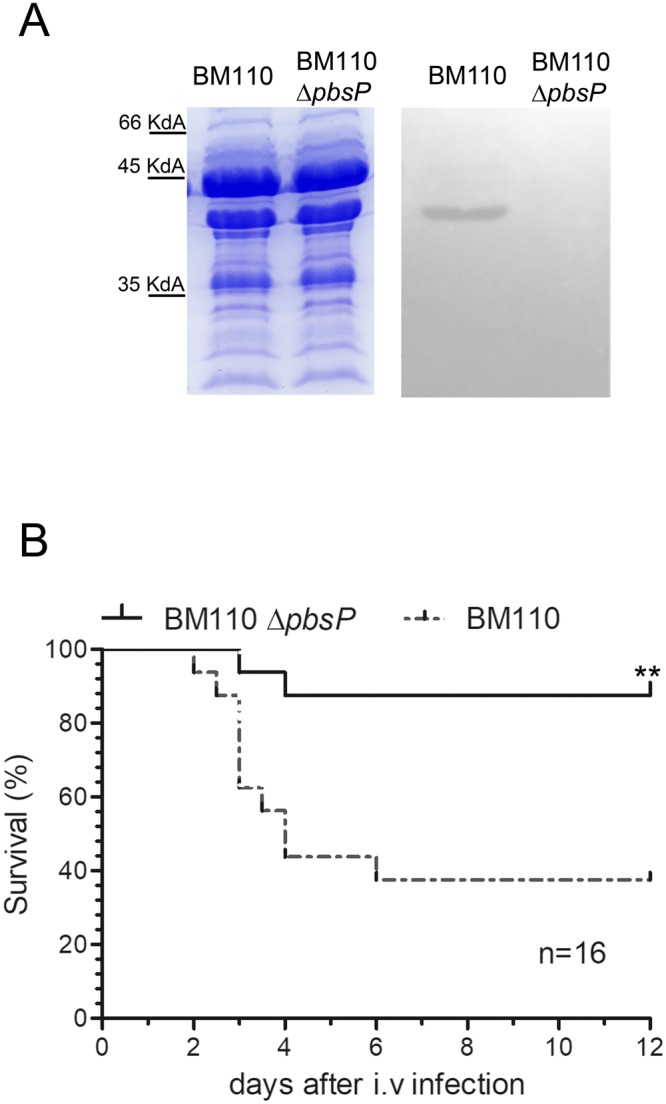
PbsP is required for virulence of CC17 GBS. (A) Western blot analysis: 10 µg of cell wall extracts from the wild-type CC17 BM110 strain (BM110) and its pbsP deletion mutant (BM110 ΔpbsP) were run on SDS-PAGE gels. Duplicate gels were stained with Coomassie (left) or blotted on nitrocellulose membranes for western blot analysis (right) using a polyclonal anti-PbsP mouse serum followed by peroxidase-conjugated goat anti-mouse IgG. (B) Effect of pbsP deletion on survival of BM110-infected mice. Adult CD1 mice were infected intravenously (i.v.) with 1 × 108 CFU of wild-type BM110 or its pbsP deletion mutant (BM110 ΔpbsP). Survival was monitored every 12 h. **p < 0.01 by Kaplan-Meier analysis. Shown are the cumulative results of two experiments, each involving 8 mice per group.
To determine whether the decreased lethality induced by the ∆pbsP mutant was linked to an impaired ability to invade the central nervous system, we compared the ability of BM110 WT and ∆pbsP mutant to persist in the blood and to colonize the kidneys and brains at 48 h after infection. Mice challenged with the WT strain had significantly higher bacterial counts than those infected with the ΔpbsP mutant in the brain, but not in the blood or kidneys (Fig. 2A–C).
Figure 2.
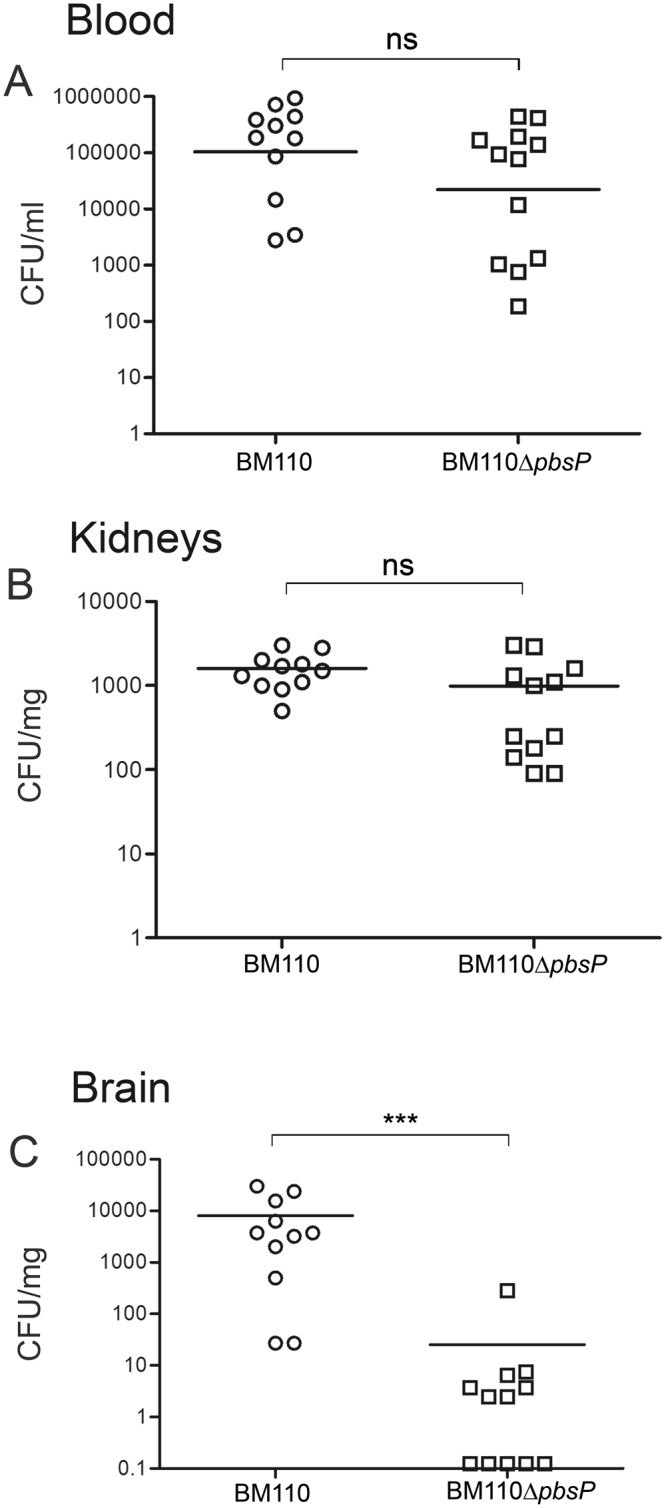
PbsP is required for brain invasion by CC17 GBS. Bacterial burden in the blood (A) kidneys (B) and brains (C) at 48 h after i.v. challenge with 1 × 108 CFU of wild-type BM110 GBS (BM110) or its pbsP deletion mutant (BM110 ΔpbsP). ***p < 0.001 by the Mann-Whitney U test; ns, non-significant. Shown are cumulative results from 2 experiments, each involving 6 mice per group.
Next, we determined whether infection with the ΔpbsP mutant was associated with decreased brain inflammation, which is a major determinant of lethality in this animal model. To this end, we measured the levels of pro-inflammatory cytokines and of myeloperoxidase (MPO), an indicator of neutrophil infiltration, in brain homogenates of infected mice. MPO, interleukin-1 beta (IL-1β) and tumor necrosis factor-alpha (TNF-α) levels in the brains of mice infected with the ΔpbsP mutant were not significantly different from those measured in uninfected mice and were markedly lower than those measured in mice infected with WT BM110 (p < 0.001; Fig. 3). Therefore, these data indicate that PbsP selectively promotes hematogenous spreading of bacteria to the brain and the subsequent development of encephalitis.
Figure 3.
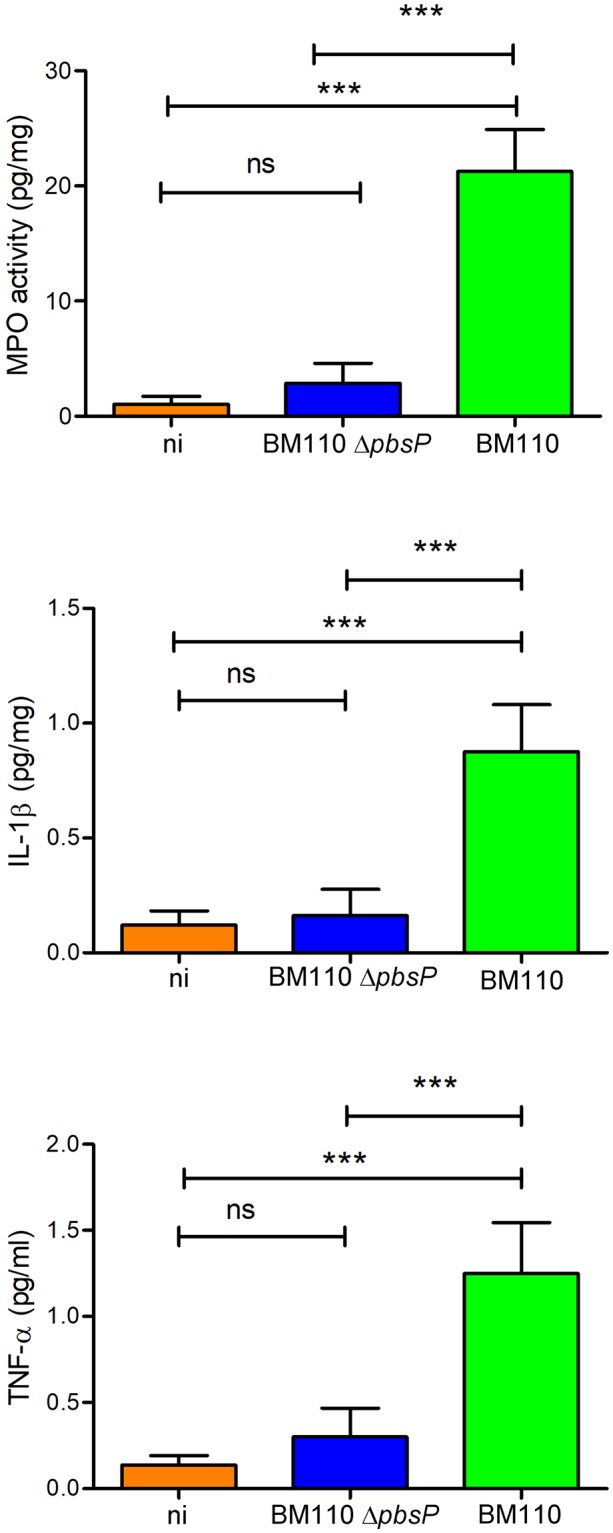
PbsP is required for the development of GBS-induced encephalitis. Myeloperoxidase (MPO), interleukin beta (IL-1β) and tumor necrosis factor-alpha (TNF-α) protein levels were measured in organ homogenates from wild-type BM110 GBS (BM110) or its pbsP deletion mutant (BM110 ΔpbsP). ni, non-infected. Data are expressed as means ± standard deviations of three observations, each conducted with a different animal. ***p < 0.001 by one-way ANOVA; ns, non-significant.
Since this antigen is an interesting vaccine candidate due to its high degree of conservation among GBS isolates34, we next investigated if immunization with PbsP prevents invasive disease by CC17 GBS. Adult mice were immunized with GST-PbsP or GST, used as a negative control, and challenged intravenously three weeks later with WT BM110. Under these conditions, immunization with PbsP resulted in increased survival (75% versus 37% in immunized and control animals, respectively; P < 0.05 by log-rank Kaplan-Meyer analysis) and in decreased bacterial colony counts in the brain but not in the blood or kidneys at 48 h post challenge (Fig. 4). In addition, lower levels of inflammation markers were measured in brain homogenates from PbsP-immunized animals relative to those from mice immunized with the GST control protein (Fig. S3).These data indicate that PbsP immunization can induce significant protection against encephalitis and lethality produced by CC17 GBS.
Figure 4.

PbsP-based immunization protects against lethal CC17 GBS infection in mice. (A) Mice were immunized with recombinant PbsP fused to glutathione-S-transferase (GST-PbsP) or with GST as a control and challenged i.v. with 1 × 108 CFU of wild-type CC17 strain BM110. *p < 0.05 by Kaplan-Meier analysis. Shown are the cumulative results of two experiments, each involving 8 mice per group. (B–D) Effects of PbsP immunization on organ bacterial burden after i.v. challenge with 1 × 108 CFU of BM110. **p < 0.01 by the Mann-Whitney U test; ns, non-significant. Shown are cumulative results from 2 experiments, each involving 6 mice per group.
PbsP is highly expressed in vivo
The protective activity of PbsP after immunization was unexpected since this protein is expressed at low levels on the surface of CC17 GBS strains, at least following growth under standard laboratory conditions34. We thus investigated whether PbsP is upregulated in vivo by performing quantitative RT-PCR analysis of bacterial RNA extracted from the kidneys and brains of mice infected with the BM110 strain. Figure 5A shows that PbsP mRNA levels were approximately 20–30 fold higher in bacteria harvested from the organs of infected mice, as compared with those grown in Todd-Hewitt broth, indicating that pbsP transcription is markedly upregulated in vivo.
Figure 5.
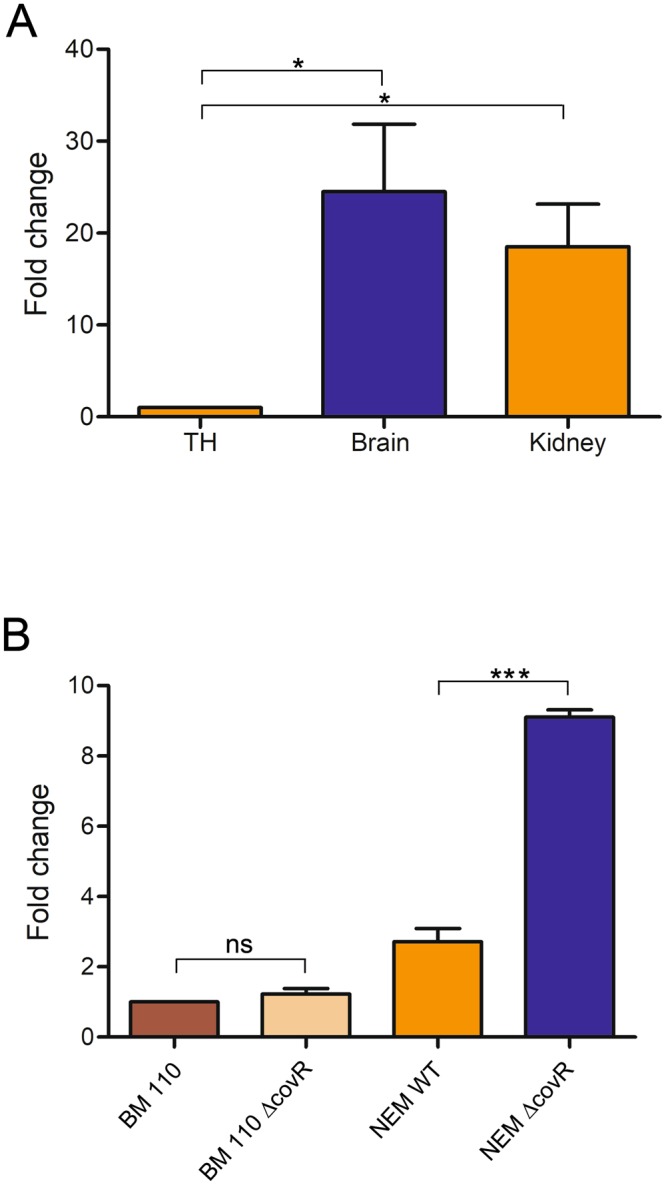
Expression of pbsP is upregulated in vivo. (A) RT-PCR analysis of pbsP mRNA levels in bacteria grown in Todd-Hewitt broth (TH) or isolated from the kidneys and brains of BM110-infected mice. Values are presented as ratios relative to the values observed in bacteria grown in TH broth. Results are means ± SD from four independent experiments performed in triplicate. (B) RT-PCR analysis of pbsP mRNA levels in wild-type CC17 BM110 strain (BM110), wild-type CC23 NEM316 strain (NEM316) and their respective CovR deletion mutants (BM110 ΔCovR and NEM316 ΔCovR). Values were normalized for those observed in wild-type BM110. Results are means ± SD from three independent experiments performed in triplicate. *p < 0.05; ***p < 0.001 by Student’s t test; ns, non-significant.
In the CC23 NEM316 strain, PbsP expression is increased in the absence of the CovRS two-component system (TCS), the master regulator of GBS virulence34,35. However, this was not the case for the CC17 strain BM110 where only a slight, non-significant, increase in pbsP mRNA levels was observed in the ∆covR mutant compared to the WT BM110 (Fig. 5B). To ascertain whether the differential effects of CovR on pbsP expression in the two clonal complexes could be explained by differences in regulatory sequences, we compared the promoter regions of PbsP in NEM316 and BM110 genomes36. However, these sequences were identical in both strains (data not shown), suggesting that the regulatory role of CovR on pbsP expression in NEM316 is not direct, in agreement with our previous data showing that CovR do not bind to the promoter region of PbsP34.
PbsP promotes interactions of BM110 GBS with brain endothelial cells
Since invasion of the endothelial barrier is a critical step in brain infection by GBS37,38, we compared the adhesion and invasion properties of the ΔpbsP mutant with those of the parental BM110 strain using the brain endothelial cell line hCMEC/D3. The deletion mutant was significantly impaired in its ability to both adhere to and invade hCMEC/D3 cells compared to the WT and to the complemented strain (Fig. 6A,B). We next investigated whether PbsP is upregulated under the conditions used in the adhesion and invasion assays. We observed a 12- to 15-fold increase of pbsP mRNA expression in BM110 GBS incubated with hCMEC/D3 monolayers, relative to the values observed with bacteria incubated in cell-free cell culture medium (Fig. 6C). Increased expression of PbsP in bacteria exposed to hCMEC/D3 was also confirmed at the protein level by immunofluorescence flow cytometry analysis using anti-PbsP antibodies (Fig. 6E,F). These data indicate that PbsP expression is upregulated in BM110 after contact with brain vascular endothelial cells and that this surface protein is required for adhesion and invasion.
Figure 6.

PbsP is required for adhesion to and transmigration across brain endothelial cells by CC17 GBS. (A,B and D) GBS strains were compared for their ability to interact with endothelial cells. WT, BM110 wild-type strain; ΔpbsP, isogenic BM110 pbsP deletion mutant; ΔpbsP + pbsP, ΔpbsP strain carrying a complementing vector with constitutive pbsP expression. Results are means ± SD from four independent experiments performed in triplicate. *p < 0.05 and **p < 0.01 by Bonferroni test and one way ANOVA. Adhesion (A) invasion (B) and traslocation (D) were assessed using the brain endothelial cell line hCMEC/D3. Plg, plasminogen; (tPA), tissue Plg activator; (6-ACA), 6-amino-n-caproic acid (100 nM). (C) RT-PCR analysis of pbsP mRNA levels in BM110 GBS incubated for 3 h in the presence of hCMEC/D3 cells (hCMEC/D3) or fresh, cell-free hCMEC/D3 culture medium (cell culture medium). Values were normalized for those observed in bacteria grown in Todd-Hewitt broth (TH). (E, F) Immunofluorescence flow cytometry analysis using mouse polyclonal anti-PbsP serum (blue line) or control anti-GST serum (red line) of PbsP surface expression in BM110 GBS exposed to cultured brain micro-vascular endothelial cells as detailed in (C).
Previous studies using the CC23 NEM316 strain indicated that, after Plg binding to the bacterial surface, conversion of the zymogen to Pln by host tissue tPA is important for migration across brain endothelial cells29,34. Therefore, we investigated whether transmigration across the brain endothelium is Pln-dependent also in the context of CC17 strains and whether PbsP plays a role in this process. We first observed that the ability of WT CC17 BM110 to migrate through hCMEC/D3 monolayers was largely Pln-dependent, as it was markedly decreased in the absence of either Plg or tPA (Fig. 6D). We have previously shown that lysine and the lysine analog 6-aminocaproic acid (6-ACA) selectively inhibit PbsP binding to Plg, suggesting that PbsP might engage lysine binding sites on the kringle domains of the zymogen molecule34. Accordingly, bacterial migration through hCMEC/D3 monolayers was significantly impaired by 6-ACA (Fig. 6D). Moreover, the BM110 ∆pbsP mutant displayed significantly decreased Pln-dependent transmigration across the monolayers, a defect which was reversed by genetic complementation (Fig. 6D). Collectively, these data indicate that PbsP plays a crucial role in Pln-dependent transmigration of the CC17 strain BM110 across brain endothelial cells.
Discussion
The present study demonstrates that the cell wall-anchored PbsP protein is critical in the context of GBS infection caused by a representative strain of the hypervirulent CC17 clone. Hallmarks of CC17 strains are their tropism for the central nervous system and their specific set of adhesins. A crucial step in the pathogenesis of GBS disease is penetration of the BBB, a process dependent on the ability of these bacteria to exploit the Plg-Pln fibrinolytic system29,34. Such a system has a dual role in the interactions between microbial pathogens and the endothelium39–42. This is the case for GBS where Plg bound to endothelial cell membranes can function as a receptor for GBS adhesins, while soluble Plg can be activated to Pln, after binding to the bacterial surface, by the action of Plg activators produced by endothelial cells and other cell types. Surface-associated Pln, in turn, can degrade extracellular matrix and basement membrane proteins either directly or indirectly (e.g. through activation of matrix metalloproteases) and initiate the release of fibrinogen and bradykininogen fragments, promoting inflammation and blood vessel permeability42–46.
We previously showed that the CC23 strain NEM316 uses PbsP to bind human Plg, which is then activated into plasmin by tPA34. Moreover, PbsP-dependent Plg activation was required for hematogenous spreading of NEM316 GBS to the brain during experimental infection. In this study, we extend the role of PbsP to the virulence of the CC17 BM110 strain. First we found that, in the absence of PbsP, BM110 displayed a selective impairment in its ability to colonize the brain, despite an unchanged capacity to persist at high levels in the blood and to invade the kidney. Second, immunization with a recombinant form of the antigen largely prevented brain infection and lethality, and this also occurred by a highly selective mechanism. Accordingly, in vitro experiments indicated that pbsP deletion impaired the ability of BM110 GBS to adhere to brain endothelial cells and to transmigrate through them using plasmin-dependent mechanisms. These findings were unexpected since very low PbsP levels were originally detected on the CC17 surface34, in contrast to the high expression of the CC17-specific Srr2 protein, which plays a major role in the binding of both Plg and fibrinogen30,31.
By analyzing PbsP expression in the context of infection, we demonstrate here that pbsP transcription is markedly up-regulated in CC17 bacteria during mouse infection or upon contact with cultured brain endothelial cells. Our data extend those of Mereghetti and coworkers who reported profound changes in the transcriptome of the CC23 strain NEM316 when grown in the presence of human blood47. Strikingly, in that study, only a few virulence genes were up-regulated, and among these, the highest levels of transcription were displayed by pbsP (referred to as gbs0428) and by other genes encoding proteins implicated in binding or activation of Plg, such as enolase, glyceraldeyde 3-phosphate dehydrogenase and Skizzle. While this manuscript was under review, Cook et al. reported that PbsP expression is increased in CC7 GBS (strain A909) during colonization of the mouse vagina or upon contact with vaginal lavage fluid48. Therefore, GBS belonging to different clonal complexes selectively upregulate PbsP during interaction with host cells or host fluids, both in vivo and in vitro, suggesting a conserved role of PbsP during colonization and infection.
In contrast to our previous observations done with the CC23 strain NEM316, in which pbsP deletion impaired the ability to invade several organs, including the brain and the kidney, the deletion of pbsP in the BM110 strain impairs brain invasion only. This difference might be related to the different set of adhesins expressed at the surface of CC17 strains. For example, expression of the adhesins HgvA and Srr2 is restricted to CC17 GBS, while Srr1 is expressed by non-CC17 strains only31. Therefore, it is possible that CC17-specific adhesins can compensate for absence of PbsP in the context of colonization of the kidney, but not of the brain. In addition, strain-specific regulation of adhesin expression might explain this difference.
PbsP expression was shown to be repressed by the CovRS TCS49–51 in CC23 GBS35, but we observe in this study that CovRS-dependent regulation is not conserved in the BM110 strain. Instead, Cook et al. recently reported that PbsP expression is directly regulated by the SaeRS two-component system in CC7 GBS48. Therefore, further studies will be needed to analyze the role of SaeRS and other regulation systems in controlling PbsP expression in GBS strains belonging to different clonal complexes and the external environment signal triggering in vivo PbsP expression.
In conclusion, our studies indicate that PbsP is a conserved Plg binding adhesin, is expressed during infection, and is necessary for brain invasion by GBS belonging to the major CC responsible for neonatal meningitis. As such, this protein might represent a promising candidate for inclusion in a universal GBS vaccine.
Materials and Methods
Bacterial strains and reagents
The GBS strain BM110, a human clinical isolate and a prototype strain belonging to the hypervirulent CC17 clonal complex7, was used throughout this study. The BM110 ΔpbsP and BM110 ΔcovR GBS mutants were obtained as previously described, using pG1 shuttle plasmids containing gene-specific deletion cassettes10,34. A ΔpbsP strain carrying a complementing vector with constitutive pbsP expression (ΔpbsP + pbsP) was obtained as previously described34. Recombinant PbsP was produced as a fusion with glutathione-S-transferase (GST) from Escherichia coli BL21(DE3) after transformation with the pGEX-SN_PbsP plasmid, and purified as previously described34,52. Mouse anti PbsP sera were raised in CD1 mice (5 weeks old, Charles River Labs) as previously described34. Briefly, the mice were bled at 2 weeks after the last immunization and the sera were tested for reactivity to the purified antigen using ELISA15,52.
Cell wall extracts and immunoblots
Cell wall extracts were obtained by mutanolysin extraction in a hypertonic sucrose buffer and used for Western blot analyses as described previously52,53. Briefly, 10 µg of cell wall proteins were run on polyacrylamide gels (SDS-PAGE), transferred to a nitrocellulose membrane and PbsP was visualized by chemi-luminescence after incubation with mouse anti-PbsP serum and horseradish peroxidase conjugated goat anti mouse IgG (Abcam ab6789).
Mouse meningitis model
The CC17 BM110 strain and the deletion mutants were used to induce infection in 8-week-old CD1 female mice, as previously described34,52. Mice were infected i.v with 1 × 108 CFU and monitored every 12 hours for clinical signs and lethality. Animals with signs of sepsis or neurological manifestations were humanely euthanized and GBS invasion of organs confirmed as the cause of disease, as described34. In a second set of experiments GBS-infected mice were sacrificed at 48 h after infection to collect blood, brains and kidneys. The number of CFU was measured in organ homogenates using standard methods, as described53. The levels of MPO, IL-1β and TNF-α were measured in brain homogenates by using commercially available kits purchased from R&D, as previously described54. The protective effect of PbsP immunization was evaluated as previously described34. Briefly, CD1 female mice were immunized by intraperitoneal injection with 30 µg of recombinant PbsP or GST in complete (first injection) or incomplete (second and third injection) Freund’s adjuvant emulsions (in a total volume of 0.2 ml) on day 0, 14 and 28. Three week after the last immunization, mice were challenged i.v. with 1 × 108 CFUs of BM110 WT bacteria. All studies involving mice were performed in accordance with the European Union guidelines for the use of laboratory animals. The procedures were approved by the animal welfare committee of University of Messina (OPBA permit n. 18052010) and by the Ministero della Salute of Italy (permit n° 665/2015). Data were analyzed using GraphPad Prism 5.0 (GraphPad Software, San Diego, California).
Quantitative RT-PCR
To measure pbsP mRNA, total RNA was extracted from the organs of infected mice or from bacteria exposed in vitro to different conditions, retro-transcribed and analyzed by RT-PCR. The first step in isolation of bacteria from infected brains or kidneys (obtained from CD1 mice at 24 h after i.v. challenge with 1 × 108 CFUs of BM110 WT bacteria) consisted in homogenizing the organs with the gentle MACS Dissociator system (Mylteni), according to manufacturer’s instructions. Next, tissue debris and residual intact cells were sedimented by centrifugation at 200 g for 10 min and bacteria were recovered from supernatants by high speed centrifugation (12,000 g for 10 min). In in vitro experiments, 5 × 106 GBS grown to the exponential phase in TH broth were added to 0.5 ml of brain microvascular endothelial cell cultures or to fresh, cell-free, medium in 24 well plates. After centrifugation at 500 g for 2 min, the plates were incubated at 37 °C for 2 h. After scraping the contents of the wells were collected and bacteria were recovered by differential centrifugation, as described above.
To extract RNA, bacterial pellets were suspended in 350 µl of Tris-HCl (pH 8, 10 mM) in 1.5 ml microcentrifuge tubes, to which 25 µg of glass beads (106 µm, Sigma-Aldrich) were added. The tubes were placed in a RETSH MM30 homogenizer and shaken at 30 Hz for 20 minutes. RNA in the homogenized samples was purified using Qiagen RNA purification columns and stored at −80 °C after quantification by Nanodrop 2000 (Thermo Fischer) readings and gel electrophoresis analysis. Reverse transcription was performed with the M-MLV reverse transcriptase (Invitrogen) and random primers (Promega). Specific primer pairs were designed to obtain amplicons of 68 (pbsP) and 74 bp (gyrA), as shown in Table S1, Supplementary Methods. Quantitative PCR (qPCR) was carried out with the Taqman Gene Expression Master MIX (Applied Biosystem) in a 7500 Real-Time PCR Detection System (Applied Biosystem). Relative gene expression levels were calculated with the ΔΔCT method55,56 where expression values were normalized with the expression of the housekeeping gyrA gene. Each assay was performed in triplicate. Expression of the PbsP protein on the bacterial surface was analyzed, in selected experiments, by immuno-fluorescence flow cytometry using anti-PbsP mouse sera, as described34.
Adhesion, invasion and transmigration assays
The human brain endothelial cell line hCMEC/D3 was provided by P.O. Couraud (INSERM, Paris, France) and the adherence and invasion assays were performed as described15,57. Briefly, bacteria were grown to the mid-log phase and added to confluent monolayers at a multiplicity of infection (MOI) of 20 bacteria/cell. After a one-hour incubation, monolayers were washed with PBS to remove non-adherent bacteria, lysed, and plated to enumerate cell-associated bacteria. For the invasion assay, after washing, the monolayers were further incubated for 1 h with medium supplemented with penicillin and streptomycin (200 units/ml 100 µg/ml, respectively) to kill extracellular bacteria. Percentages of bacterial adhesion and invasion were calculated as 100× (recovered cfu/initial inoculum cfu). To test the transmigration ability of BM110, an endothelial blood-brain barrier in vitro model was used by cultivating hCMEC/D3 cells in collagen coated-polycarbonate transwell membrane inserts with a pore size of 3 µm, as previously described15,57. This model allows access to the upper (“blood” side) and lower (“brain side”) chambers and mimics GBS penetration into the brain. The hCMEC/D3 cells were grown for 5–7 days at 37 °C in a humidified chamber containing 5% CO2 to reach confluence. Prior to the assay, the integrity of the monolayer was verified by adding the Evans blue stain to the upper chamber. The hCMEC/D3 cells were then washed and resuspended in serum-free culture medium without antibiotics. Log-phase GBS were added to the upper chamber together with Plg and/or tPA. At 2 h post-infection, the lower chamber medium was entirely removed and plated onto TH agar to measure transmigration.
Electronic supplementary material
Acknowledgements
Work described here was supported in part by funds granted to Scylla Biotech Srl and Charybdis Vaccines Srl by the Ministero dell’Università e della Ricerca Scientifica of Italy (Project n. 4/13 ex art. 11 D.M. n. 593 and Cluster Medintech, respectively).
Author Contributions
G.L., A.M. and L.R. performed the experiments and analyzed the data. E.M., A.P. and G.M. analyzed data. R.G. designed experiments and analyzed data. C.Bi., C.Be., A.F., P.T. and G.T. designed experiments, analyzed data and wrote the manuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Germana Lentini and Angelina Midiri contributed equally.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-32774-8.
References
- 1.Tam T, Bilinski E, Lombard E. Recolonization of group B Streptococcus (GBS) in women with prior GBS genital colonization in pregnancy. J Matern-Fetal Neo M. 2012;25:1987–1989. doi: 10.3109/14767058.2012.670331. [DOI] [PubMed] [Google Scholar]
- 2.Najmi N, Jehan I, Sikandar R, Zuberi NF. Maternal genital tract colonisation by Group-B Streptococcus: A hospital based study. Journal of the Pakistan Medical Association. 2013;63:1103–1107. [PubMed] [Google Scholar]
- 3.Lin FYC, Troendle JF. Neonotol respiratory distress may be related to asymptomatic colonization with group B streptococci. Pediatric Infectious Disease Journal. 2006;25:884–888. doi: 10.1097/01.inf.0000239322.58890.94. [DOI] [PubMed] [Google Scholar]
- 4.Luan SL, et al. Multilocus sequence typing of Swedish invasive group B streptococcus isolates indicates a neonatally associated genetic lineage and capsule switching. Journal of Clinical Microbiology. 2005;43:3727–3733. doi: 10.1128/JCM.43.8.3727-3733.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bohnsack JF, et al. Population structure of invasive and colonizing strains of Streptococcus agalactiae from Neonates of six US academic Centers from 1995 to 1999. Journal of Clinical Microbiology. 2008;46:1285–1291. doi: 10.1128/JCM.02105-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Manning SD, et al. Multilocus Sequence Types Associated with Neonatal Group B Streptococcal Sepsis and Meningitis in Canada. Journal of Clinical Microbiology. 2009;47:1143–1148. doi: 10.1128/JCM.01424-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Musser JM, Mattingly SJ, Quentin R, Goudeau A, Selander RK. Identification of a High-Virulence Clone of Type-Iii Streptococcus-Agalactiae (Group-B Streptococcus) Causing Invasive Neonatal Disease. Proceedings of the National Academy of Sciences of the United States of America. 1989;86:4731–4735. doi: 10.1073/pnas.86.12.4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Poyart C, et al. Invasive group B streptococcal infections in infants, France. Emerging Infectious Diseases. 2008;14:1647–1649. doi: 10.3201/eid1410.080185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Edmond KM, et al. Group B streptococcal disease in infants aged younger than 3 months: systematic review and meta-analysis. Lancet. 2012;379:547–556. doi: 10.1016/S0140-6736(11)61651-6. [DOI] [PubMed] [Google Scholar]
- 10.Lamy MC, et al. Rapid detection of the “highly virulent” group B streptococcus ST-17 clone. Microbes and Infection. 2006;8:1714–1722. doi: 10.1016/j.micinf.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 11.Phares CR, et al. Epidemiology of invasive group B streptococcal disease in the United States, 1999–2005. Jama-Journal of the American Medical Association. 2008;299:2056–2065. doi: 10.1001/jama.299.17.2056. [DOI] [PubMed] [Google Scholar]
- 12.Jones N, et al. Enhanced invasiveness of bovine-derived neonatal sequence type 17 group B Streptococcus is independent of capsular serotype. Clinical Infectious Diseases. 2006;42:915–924. doi: 10.1086/500324. [DOI] [PubMed] [Google Scholar]
- 13.Bisharat N, et al. Hyperinvasive neonatal group B streptococcus has arisen from a bovine ancestor. Journal of Clinical Microbiology. 2004;42:2161–2167. doi: 10.1128/JCM.42.5.2161-2167.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tazi A, et al. Group B Streptococcus surface proteins as major determinants for meningeal tropism. Curr Opin Microbiol. 2012;15:44–49. doi: 10.1016/j.mib.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 15.Buscetta M, et al. FbsC, a Novel Fibrinogen-binding Protein, Promotes Streptococcus agalactiae-Host Cell Interactions. Journal of Biological Chemistry. 2014;289:21003–21015. doi: 10.1074/jbc.M114.553073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hull JR, Tamura GS, Castner DG. Interactions of the streptococcal C5a peptidase with human fibronectin. Acta Biomaterialia. 2008;4:504–513. doi: 10.1016/j.actbio.2008.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ragunathan P, Spellerberg B, Ponnuraj K. Structure of laminin-binding adhesin (Lmb) from Streptococcus agalactiae. Acta Crystallographica Section D-Biological Crystallography. 2009;65:1262–1269. doi: 10.1107/S0907444909038359. [DOI] [PubMed] [Google Scholar]
- 18.Boone TJ, Burnham CAD, Tyrrell GJ. Binding of group B streptococcal phosphoglycerate kinase to plasminogen and actin. Microbial Pathogenesis. 2011;51:255–261. doi: 10.1016/j.micpath.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 19.Margarit I, et al. Capturing host-pathogen interactions by protein microarrays: identification of novel streptococcal proteins binding to human fibronectin, fibrinogen, and C4BP. Faseb Journal. 2009;23:3100–3112. doi: 10.1096/fj.09-131458. [DOI] [PubMed] [Google Scholar]
- 20.Adderson EE, Wang Y, Armstrong JL, Bohnsack JF. Fibronectin-binding proteins of group B Streptococcus agalactiae (GBS) Pediatric Research. 2003;53:315a–315a. [Google Scholar]
- 21.Devi AS, Ponnuraj K. Cloning, expression, purification and ligand binding studies of novel fibrinogen-binding protein FbsB of Streptococcus agalactiae. Protein Expression and Purification. 2010;74:148–155. doi: 10.1016/j.pep.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 22.Papasergi Salvatore, Lanza Cariccio Veronica, Pietrocola Giampiero, Domina Maria, D’Aliberti Deborah, Trunfio Maria Grazia, Signorino Giacomo, Peppoloni Samuele, Biondo Carmelo, Mancuso Giuseppe, Midiri Angelina, Rindi Simonetta, Teti Giuseppe, Speziale Pietro, Felici Franco, Beninati Concetta. Immunogenic Properties of Streptococcus agalactiae FbsA Fragments. PLoS ONE. 2013;8(9):e75266. doi: 10.1371/journal.pone.0075266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rubin LL, Staddon JM. The cell biology of the blood-brain barrier. Annual Review of Neuroscience. 1999;22:11–28. doi: 10.1146/annurev.neuro.22.1.11. [DOI] [PubMed] [Google Scholar]
- 24.Mucke L, Eddleston M. Astrocytes in Infectious and Immune-Mediated Diseases of the Central-Nervous-System. Faseb Journal. 1993;7:1226–1232. doi: 10.1096/fasebj.7.13.8405808. [DOI] [PubMed] [Google Scholar]
- 25.Tuma PL, Nyasae LK, Hubbard AL. Dynamin-2 regulates the basolateral internalization of transcytosing glycolipid-anchored and single transmembrane proteins in polarized hepatocytes. Molecular Biology of the Cell. 2001;12:468a–469a. [Google Scholar]
- 26.Mook-Kanamori BB, Geldhoff M, van der Poll T, van de Beek D. Pathogenesis and Pathophysiology of Pneumococcal Meningitis. Clinical Microbiology Reviews. 2011;24:557–591. doi: 10.1128/CMR.00008-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Combes V, Guillemin GJ, Chan-Ling TL, Hunt NH, Grau GER. The crossroads of neuroinflammation in infectious diseases: endothelial cells and astrocytes. Trends in Parasitology. 2012;28:311–319. doi: 10.1016/j.pt.2012.05.008. [DOI] [PubMed] [Google Scholar]
- 28.Iovino F, Seinen J, Henriques-Normark B, van Diji JM. How Does Streptococcus pneumoniae Invade the Brain. Trends in Microbiology. 2016;24:307–315. doi: 10.1016/j.tim.2015.12.012. [DOI] [PubMed] [Google Scholar]
- 29.Almeida, A. et al. Parallel Evolution of Group B Streptococcus Hypervirulent Clonal Complex 17 Unveils New Pathoadaptive Mutations. mSystems2 (2017). [DOI] [PMC free article] [PubMed]
- 30.Six A, et al. Srr2, a multifaceted adhesin expressed by ST-17 hypervirulent Group B Streptococcus involved in binding to both fibrinogen and plasminogen. Molecular Microbiology. 2015;97:1209–1222. doi: 10.1111/mmi.13097. [DOI] [PubMed] [Google Scholar]
- 31.Seo HS, et al. Characterization of Fibrinogen Binding by Glycoproteins Srr1 and Srr2 of Streptococcus agalactiae. Journal of Biological Chemistry. 2013;288:35982–35996. doi: 10.1074/jbc.M113.513358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Magalhães Vanessa, Andrade Elva Bonifácio, Alves Joana, Ribeiro Adilia, Kim Kwang Sik, Lima Margarida, Trieu-Cuot Patrick, Ferreira Paula. Group B Streptococcus Hijacks the Host Plasminogen System to Promote Brain Endothelial Cell Invasion. PLoS ONE. 2013;8(5):e63244. doi: 10.1371/journal.pone.0063244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Magalhaes V, et al. Interaction with human plasminogen system turns on proteolytic activity in Streptococcus agalactiae and enhances its virulence in a mouse model. Microbes and Infection. 2007;9:1276–1284. doi: 10.1016/j.micinf.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 34.Buscetta M, et al. PbsP, a cell wall-anchored protein that binds plasminogen to promote hematogenous dissemination of group B Streptococcus. Molecular Microbiology. 2016;101:27–41. doi: 10.1111/mmi.13357. [DOI] [PubMed] [Google Scholar]
- 35.Papasergi S, et al. Analysis of the Streptococcus agalactiae exoproteome. Journal of proteomics. 2013;89:154–164. doi: 10.1016/j.jprot.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 36.Rosinski-Chupin I, et al. Single nucleotide resolution RNA-seq uncovers new regulatory mechanisms in the opportunistic pathogen Streptococcus agalactiae. BMC genomics. 2015;16:419. doi: 10.1186/s12864-015-1583-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nizet V, et al. Invasion of brain microvascular endothelial cells by group B streptococci. Infect Immun. 1997;65:5074–5081. doi: 10.1128/iai.65.12.5074-5081.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Doran KS, et al. Blood-brain barrier invasion by group B Streptococcus depends upon proper cell-surface anchoring of lipoteichoic acid. Journal of Clinical Investigation. 2005;115:2499–2507. doi: 10.1172/JCI23829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Behnsen J, et al. The opportunistic human pathogenic fungus Aspergillus fumigatus evades the host complement system. Infect Immun. 2008;76:820–827. doi: 10.1128/IAI.01037-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fox D, Smulian AG. Plasminogen-binding, activity of enolase in the opportunistic pathogen Pneumocystis carinii. Medical Mycology. 2001;39:495–507. doi: 10.1080/mmy.39.6.495.507. [DOI] [PubMed] [Google Scholar]
- 41.Jong AY, et al. Binding of Candida albicans enolase to plasmin(ogen) results in enhanced invasion of human brain microvascular endothelial cells. Journal of Medical Microbiology. 2003;52:615–622. doi: 10.1099/jmm.0.05060-0. [DOI] [PubMed] [Google Scholar]
- 42.Stie J, Fox D. Blood-brain barrier invasion by Cryptococcus neoformans is enhanced by functional interactions with plasmin. Microbiology-Sgm. 2012;158:240–258. doi: 10.1099/mic.0.051524-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eberhard T, Kronvall G, Ullberg M. Surface bound plasmin promotes migration of Streptococcus pneumoniae through reconstituted basement membranes. Microbial Pathogenesis. 1999;26:175–181. doi: 10.1006/mpat.1998.0262. [DOI] [PubMed] [Google Scholar]
- 44.Dano K, et al. Plasminogen Activators, Tissue Degradation, and Cancer. Advances in Cancer Research. 1985;44:139–266. doi: 10.1016/S0065-230X(08)60028-7. [DOI] [PubMed] [Google Scholar]
- 45.Castellino FJ, Ploplis VA. Structure and function of the plasminogen/plasmin system. Thrombosis and Haemostasis. 2005;93:647–654. doi: 10.1160/TH04-12-0842. [DOI] [PubMed] [Google Scholar]
- 46.Lahteenmaki K, et al. Bacterial Plasminogen Receptors - in-Vitro Evidence for a Role in Degradation of the Mammalian Extracellular-Matrix. Infect Immun. 1995;63:3659–3664. doi: 10.1128/iai.63.9.3659-3664.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mereghetti Laurent, Sitkiewicz Izabela, Green Nicole M., Musser James M. Extensive Adaptive Changes Occur in the Transcriptome of Streptococcus agalactiae (Group B Streptococcus) in Response to Incubation with Human Blood. PLoS ONE. 2008;3(9):e3143. doi: 10.1371/journal.pone.0003143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cook, L. C. C., Hu, H., Maienschein-Cline, M. & Federle, M. J. A Vaginal Tract Signal Detected by the Group B Streptococcus SaeRS System Elicits Transcriptomic Changes and Enhances Murine Colonization. Infect Immun86 (2018). [DOI] [PMC free article] [PubMed]
- 49.Lembo A, et al. Regulation of CovR expression in Group B Streptococcus impacts blood-brain barrier penetration. Molecular Microbiology. 2010;77:431–443. doi: 10.1111/j.1365-2958.2010.07215.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Park SE, Jiang SM, Wessels MR. CsrRS and Environmental pH Regulate Group B Streptococcus Adherence to Human Epithelial Cells and Extracellular Matrix. Infect Immun. 2012;80:3975–3984. doi: 10.1128/IAI.00699-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Patras KA, et al. Group B Streptococcus CovR regulation modulates host immune signalling pathways to promote vaginal colonization. Cellular Microbiology. 2013;15:1154–1167. doi: 10.1111/cmi.12105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Papasergi S, et al. Plasminogen- and Fibronectin-binding Protein B Is Involved in the Adherence of Streptococcus pneumoniae to Human Epithelial Cells. Journal of Biological Chemistry. 2010;285:7517–7524. doi: 10.1074/jbc.M109.062075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cardaci Angela, Papasergi Salvatore, Midiri Angelina, Mancuso Giuseppe, Domina Maria, Cariccio Veronica Lanza, Mandanici Francesca, Galbo Roberta, Passo Carla Lo, Pernice Ida, Donato Paolo, Ricci Susanna, Biondo Carmelo, Teti Giuseppe, Felici Franco, Beninati Concetta. Protective Activity of Streptococcus pneumoniae Spr1875 Protein Fragments Identified Using a Phage Displayed Genomic Library. PLoS ONE. 2012;7(5):e36588. doi: 10.1371/journal.pone.0036588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Biondo, C. et al. Essential Role of Interleukin-1 Signaling in Host Defenses Against Group B Streptococcus. Mbio5 (2014). [DOI] [PMC free article] [PubMed]
- 55.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(T) (−Delta Delta C) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 56.Biondo C, et al. IFN-alpha/beta signaling is required for polarization of cytokine responses toward a protective type 1 pattern during experimental cryptococcosis. J Immunol. 2008;181:566–573. doi: 10.4049/jimmunol.181.1.566. [DOI] [PubMed] [Google Scholar]
- 57.Weksler BB, et al. Blood-brain barrier-specific properties of a human adult brain endothelial cell line. Faseb Journal. 2005;19:1872–+. doi: 10.1096/fj.04-3458fje. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.