

SUMMARY
Transporting epithelial cells like those that line the gut, build large arrays of actin-supported protrusions called microvilli, which extend from the apical surface into luminal spaces to increase functional surface area. Although critical for maintaining physiological homeostasis, mechanisms controlling the formation of microvilli remain poorly understood. Here we report that the I-BAR domain containing protein insulin receptor tyrosine kinase substrate (IRTKS, also known as BAIAP2L1) promotes the growth of epithelial microvilli. Super-resolution microscopy and live imaging of differentiating epithelial cells revealed that IRTKS localizes to the distal tips of actively growing microvilli via a mechanism that requires its N-terminal I-BAR domain. At microvillar tips, IRTKS promotes elongation through a mechanism involving its C-terminal actin binding WH2 domain. IRTKS can also drive microvillar elongation using its SH3 domain to recruit the bundling protein EPS8 to microvillar tips. These results provide new insight on mechanisms that control microvillar growth during the differentiation of transporting epithelial cells, and help explain why IRTKS is targeted by enteric pathogens that disrupt microvillar structure during infection of the intestinal epithelium.
Keywords: actin, protrusion, cytoskeleton, brush border, enterocyte, differentiation
Graphical Abstract
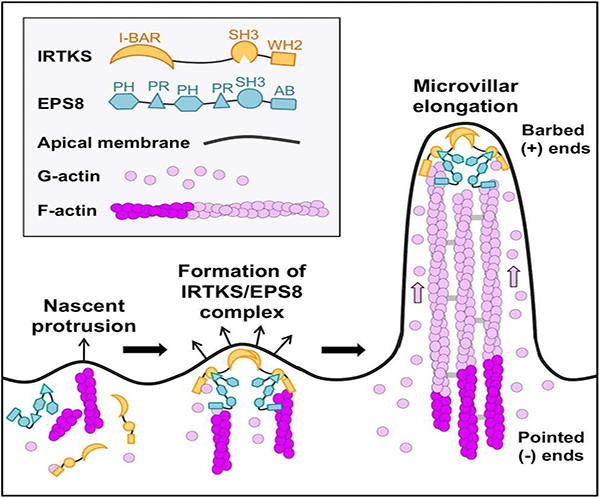
eTOC Blurb
Postema et al. show that IRTKS (BAIAP2L1) localizes to the distal tips of actively growing epithelial microvilli through its N-terminal I-BAR domain. At these sites, IRTKS drives microvillar elongation directly using its C-terminal actin binding WH2 domain, and indirectly using its SH3 domain to recruit the actin regulatory protein EPS8.
INTRODUCTION
Transporting epithelial cells line the luminal surface of many hollow organs where they promote solute uptake from the external environment. Transport is aided by actin-supported membrane protrusions called microvilli, which increase apical membrane surface area [1]. A single microvillus contains a core bundle of parallel actin filaments with barbed ends oriented away from the cell and pointed ends embedded in an underlying terminal web [2]. Certain epithelial cell types, such as intestinal enterocytes, build hundreds of densely packed microvilli in an array known as the ‘brush border’ (BB) [3, 4]. In addition to promoting nutrient absorption, intestinal microvilli provide a barrier against luminal pathogens and toxins [5]. Interestingly, a subset of pathogenic microbes, including enterohemorrhagic Escherichia coli (EHEC), have evolved mechanisms to destroy microvilli, which leads to nutrient malabsorption and osmotic imbalances that can prove life threatening [6]. Despite the critical physiological role of the BB, the molecules and mechanisms controlling microvillar growth remain poorly understood.
Microvillar growth occurs during enterocyte differentiation, which takes place in pit-like “crypts”, sites that harbor intestinal stem cells [7]. Although crypt epithelial cells exhibit short, disorganized microvilli [8, 9], the apical domain undergoes a striking transition as nascent enterocytes migrate out of crypts and onto the villus [8, 10]. Defining features of this transition include an increase in microvillar packing density (number of microvilli/cell) and an increase in length. Both of these changes increase apical membrane surface area and contribute to maximizing the absorptive capacity of mature enterocytes.
Although mechanisms that drive tight microvillar packing are beginning to emerge [3, 11–15], molecules responsible for elongation of microvilli during differentiation remain poorly understood. Previous studies implicated actin filament bundling proteins, including villin and espin, in elongation [16, 17]. Bundling proteins also play a role in the growth, elongation, and maintenance of other actin-supported protrusions including stereocilia (espin-1, plastin-1 and fascin-2) [18–20] and filopodia (fascin-1) [21], respectively. Remarkably, studies on KO mice lacking the three major microvillar actin bundlers (villin, espin, and plastin) revealed that BBs are still present [22], suggesting the existence of alternate assembly pathways. Factors that target to the ends of core actin bundles would also be well positioned to exert control over bundle length. Indeed, cordon bleu targets near the pointed ends of microvillar actin cores and has been shown to promote their growth [23–25].
Given the high outward membrane curvature generated during protrusion, another group of molecules likely to play important roles in microvillar elongation are I-BAR (Inverse-Bin-Amphiphysin-Rvs) proteins [26, 27]. I-BAR domains are small membrane-binding, three helix bundles that dimerize and exhibit a structural curvature that is well-matched to outward bending [28, 29]. Members of the I-BAR family (including IRSp53, IRTKS, BAIAP2L2, MIM, and ABBA) are comprised of an N-terminal BAR domain that interacts with acidic phospholipids, a central SH3 domain, and an actin-binding Wiskott-Aldrich syndrome protein homology 2 (WH2) motif at the C-terminus [26, 27]. I-BAR proteins have been implicated in generating filopodia on the surface of motile cells [30, 31] and dendritic spines in neurons [32], physiological scenarios that both require membrane protrusion.
A previous proteomic study by our laboratory identified insulin receptor tyrosine kinase substrate (IRTKS or brain-specific angiogenesis inhibitor 1-associated protein like 1 or BAIAP2L1) as the only I-BAR domain containing protein in the intestinal BB [33]. IRTKS was first identified as a tyrosine phosphorylated insulin receptor substrate [34] and subsequently predicted to be an I-BAR domain protein [31]. Later work showed that over-expression of IRTKS induced dramatic effects on the actin cytoskeleton in a WH2 domain-dependent manner [35]. Studies on IRTKS KO mouse models implicate this molecule in insulin signaling and glucose homeostasis [36], as well as the formation of dorsal filopodia in embryonic fibroblasts [37]. IRTKS is also hijacked by EHEC during its infection of the intestinal tract. Under these conditions, the IRTKS SH3 domain binds tightly to virulence factors that ultimately stimulate actin polymerization and adherent pedestal formation, which are required for survival of the microbe and continued infection [38–41]. The dependence of EHEC pathogenesis on IRTKS strongly implicates this molecule as a resident of the enterocyte apical domain, the initial site of EHEC contact with the epithelium, yet how IRTKS contributes to normal BB function remains unknown.
Here we report that IRTKS plays an important role in elongating microvilli. Using super resolution microscopy, we discovered that IRTKS exhibits striking localization to the distal tips of microvilli, where the growing ends of actin filaments are found. IRTKS targets to these sites of outward membrane curvature using its N-terminal I-BAR domain. Once at the tips, IRTKS serves to elongate microvilli via distinct mechanisms that require functional WH2 and SH3 domains. A role for the SH3 domain in elongation is in part explained by binding to EPS8, an established F-actin capping and bundling protein. We show that IRTKS controls the localization of EPS8 and promotes its targeting to microvillar tips. Thus, IRTKS functions in microvillar elongation directly using its WH2 domain, and indirectly using its SH3 domain to recruit EPS8, which also harbors its own elongation activity. Together these studies identify the first I-BAR protein in the epithelial apical domain, illuminate molecular mechanisms that promote microvillar growth during enterocyte differentiation, and provide an evolutionary rationale on the targeting of IRTKS during EHEC infection.
RESULTS
IRTKS localizes to the tips of microvilli in differentiating epithelial cells.
Using 2D-LC-MS/MS to define the mouse BB proteome, we previously identified IRTKS as one of 646 proteins enriched in BB fractions [33]. To examine the expression and localization of IRTKS, we stained mouse small intestinal organoids with an IRTKS specific antibody. Intestinal organoids are derived from isolated stem cell-containing crypts; in culture, these structures reseal and differentiate through formation of new crypt domains budding from a shared villus domain that encapsulates a closed lumen (Figure 1A) [42, 43]. The resulting primary cultures are highly amenable to whole mount imaging, which makes it easier to orient and capture full crypt-villus axes. Importantly, the accumulation of apical F-actin, which defines the crypt-villus transition, is recapitulated in this system (Figure 1A). In these cultures, IRTKS is enriched at the apical surface in both crypt and villus domain cells. However, expression is much higher in crypt domains (Figure 1B, S1A, Video S1). Interestingly, line scans orthogonal to the apex of crypt cells revealed peak IRTKS signal close to microvillar tips (0.78 ± 0.21, base = 0, tips = 1) (Figure 1C). To define IRTKS localization at higher resolution, we examined Ls174T-W4 (W4) cells, an intestinal epithelial cell line that can be induced to form microvilli [44]. W4 cells mimic the partially differentiated state of crypt cells and provide a model for studying IRTKS function in cells that have actively growing microvilli (Videos S2 and S3). Consistent with organoid staining, super-resolution structured illumination microscopy (SIM) of endogenous IRTKS in W4 cells revealed puncta throughout the cytoplasm, as well as enrichment at microvillar tips (Figure 1D,E). Thus, IRTKS is highly expressed in differentiating epithelial cells of the crypt and localizes to the tips of nascent microvilli.
Figure 1: IRTKS localizes to the distal tips of epithelial microvilli.
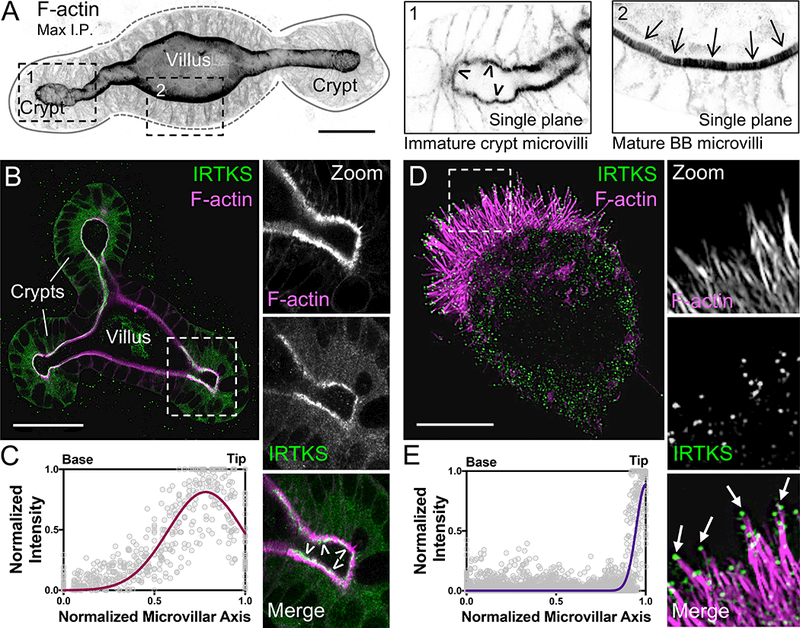
(A) Confocal maximum intensity projection of a mouse small intestinal organoid stained with phalloidin to label F-actin. Zooms indicate single plane images: 1) crypt cells, arrowheads highlight immature microvilli and 2) villar cells, arrows highlight mature microvilli. Scale bar, 25 μm. (B) Endogenous IRTKS (green) and phalloidin (magenta) labeling of an intestinal organoid. Dashed box indicates zoom of the crypt; arrowheads highlight IRTKS tip localization. Scale bar, 40 μm. (C) Line scans (n = 36 microvilli) of normalized endogenous IRTKS intensity parallel to the microvillar axis in organoid crypt domains. Length values are normalized such that 0 = base and 1 = tip. (D) SIM projection of a Ls174T-W4 (W4) cell showing endogenous IRTKS (green) and stained with phalloidin (magenta). Dashed box indicates zoom of the BB, arrows point to IRTKS puncta at the microvillar distal tips. Scale bar, 5 μm. (E) Line scans (n = 42 microvilli) of normalized endogenous IRTKS intensity parallel to the microvillar axis in W4 cells. Length values are normalized such that 0 = base and 1 = tip. See also Figure S1 and Videos S1–S3.
IRTKS tracks the distal tips of growing microvilli.
We next examined IRTKS dynamics in live W4 cells expressing EGFP-IRTKS and mCherry-Utrophin (UtrCH) [45] to label F-actin (Figure 2A, Video S3). In kymographs created with lines along the microvillar axis, IRTKS tracked the tips of growing bundles, producing diagonal features with the slope indicating an elongation rate of ~0.75 μm/min (Figure 2B). Montages of individual microvilli also showed IRTKS puncta persistently tracking the tips of newly emerging protrusions (Figure 2C, Video S4). Of note, we were unable to detect clear examples of IRTKS tip tracking during microvillar retraction. This might suggest that IRTKS prefers the tips of growing bundles. However, we cannot rule out the possibility that retraction events may be obscured by the dynamic movements of microvilli during these time-lapse.
Figure 2: IRTKS tracks the tips of growing F-actin protrusions.
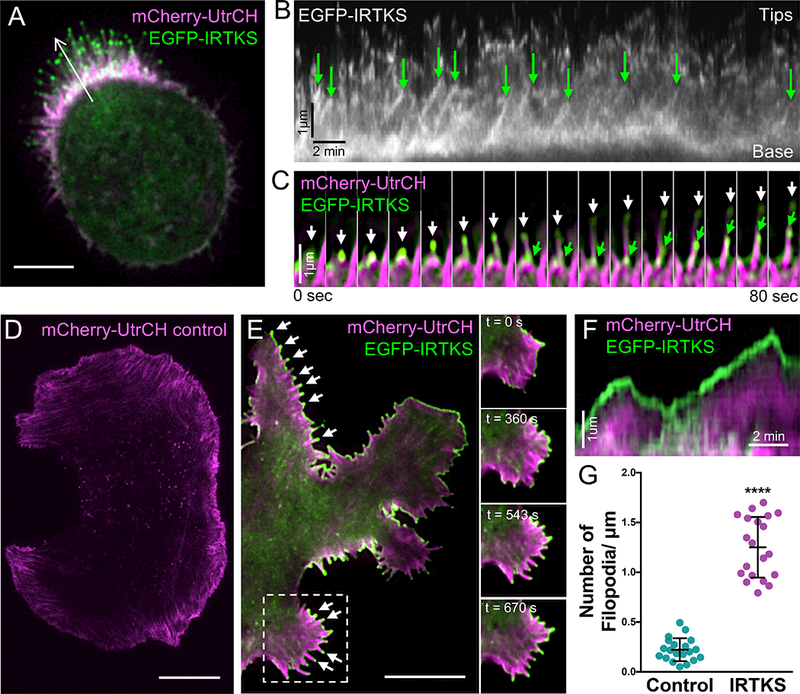
(A) EGFP-IRTKS (green) targets to the tips of mCherry-Utrophin (UtrCH, magenta) labeled microvilli in live W4 cells. Scale bar, 5 μm. (B) Kymograph from the white arrow in A; EGFP-IRTKS puncta track the tips of protruding microvilli as indicated with green arrows. (C) Montage of EGFP-IRTKS (green) puncta tracking the tips of growing microvillar actin bundles in a W4 cell expressing mCherry-UtrCH (magenta). White and green arrows indicate IRTKS at the tips of two distinct elongating microvillar bundles. (D) TIRF live-cell imaging of a control B16F1 melanoma cell expressing mCherry-UtrCH. Scale bar, 10 μm. (I) TIRF live-cell imaging of a B16F1 melanoma cell expressing EGFP-IRTKS (green) and mCherry-UtrCH (magenta); arrows point to individual filopodial protrusions with IRTKS enrichment at the distal tips. Dashed box indicates single Video frames with time in seconds. Scale bar, 10 μm. (J) Kymograph of a B16F1 cell filopodia. IRTKS (green) persists at the tips of dynamic filopodial actin bundles (magenta). (K) Quantitation of the number of filopodia per μm of cell perimeter in control and IRTKS expressing B16F1 melanoma cells; 20 cells/condition. Error bars indicate mean ± SD; p value calculated using a t test (****p<0.0001). See also Videos S3–S6.
To determine if tip targeting was specific to the parallel actin bundles in epithelial microvilli, we examined the localization of EGFP-tagged IRTKS in B16F1 melanoma cells. Time-lapse imaging showed strong localization to the tips of dynamic filopodia, as well as an increase in the total number of filopodia over control cells (Figure 2D-2G, Videos S5 & S6). Together, these results suggest that the IRTKS tip targeting mechanism senses general features of the distal tip compartment, such as outward membrane curvature and/or the enrichment of actin filament barbed ends.
IRTKS tip targeting is driven by the N-terminal I-BAR domain.
To define the mechanism of IRTKS tip targeting, we examined the impact of eliminating or mutating specific structural motifs (Figure 3A). A full-length EGFP-IRTKS construct enriched at the tips of W4 microvilli similar to endogenous IRTKS (Figure 3B, S2A). A construct containing loss-of-function mutations (W378K/W391K) in the SH3 domain (SH3*)[46] still displayed some tip targeting, but also accumulated near the base of microvilli (Figure 3C, S2B). Deletion of the C-terminal WH2 domain ΔWH2) had no impact on tip targeting (Figure 3D, S2C), whereas deletion of the N-terminal I-BAR domain ΔI-BAR) completely eliminated all membrane localization and tip targeting (Figure 3E, S2D). The IRTKS I-BAR domain alone displayed targeting to the membrane and the tips of microvilli (Figure 3F, S2E). Thus, IRTKS tip targeting is controlled primarily by the I-BAR domain, but is refined by the SH3 domain, likely through its interactions with binding partners.
Figure 3: IRTKS promotes microvillar elongation.
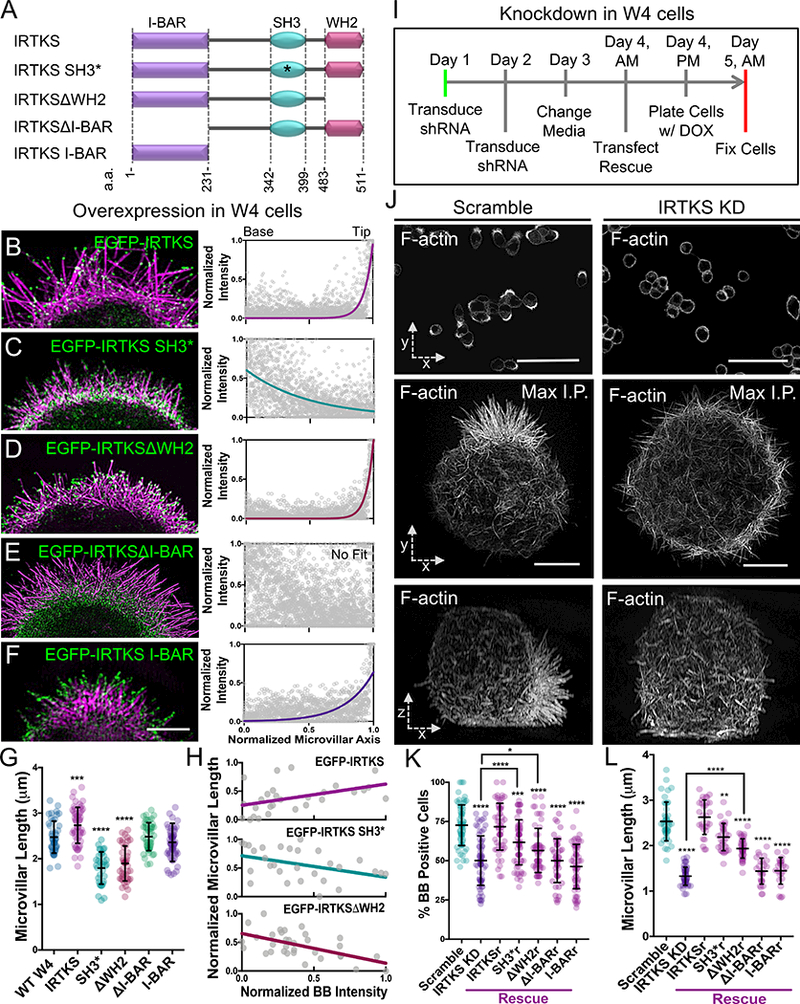
(A) Construct cartoons for the IRTKS variants used in this study. *refers to W378K/W391K point mutations within the SH3 domain; a.a. refers to the amino acid numbers of the indicated domain. (B-F) SIM projections of W4 cells expressing EGFP-IRTKS constructs (green) and stained with phalloidin (magenta). Line scans (n ≥ 32 microvilli for each construct) parallel to the microvillar axis show the intensity distribution of each IRTKS construct; 0 = base, 1 = tip. Scale bar, 3.5 μm. (G) Quantitation of microvillar length from cells expressing IRTKS constructs; >34 cells/condition, >10 microvilli/cell. (H) Quantitation of total IRTKS fluorescent intensity within the BB versus averaged microvillar length for individual cells; >28 cells per condition, >10 microvilli averaged per cell. The slope of EGFP-IRTKS is significantly different from the slopes of EGFP-IRTKS SH3* and EGFP-IRTKSΔWH2 (**p<0.0021 and ****p<0.0001, respectively). The slopes of EGFP-IRTKS SH3* and EGFP-IRTKSΔWH2 are not significantly different (p<0.4957). (I) Schematic of KD/rescue experimental design. W4 cells are plated with doxycycline (DOX) to induce the formation of a BB (Baas et al., 2004). (J) Images of scramble or IRTKS KD W4 cells stained with phalloidin. Top panels: low magnification confocal images that were scored to generate “% BB positive cells” plot; scale bars, 40 μm. Middle panels: SIM whole cell maximum intensity projections (en face, x-y); scale bars, 5 μm. Bottom panels: SIM whole cell maximum intensity projections of the same cell as above (lateral, x-z). (K,L) Quantitation of IRTKS KD and rescue experiments in W4 cells. Percentage of BB positive cells; >200 cells/condition, individual points correspond to the percent of cells containing a BB in an individual 40× confocal image field. Microvillar length >28 cells/condition, >10 microvilli averaged/cell. All error bars indicate mean ± SD; all p values calculated using a t test (*p<0.033, **p<0.002, ***p<0.0002, ****p<0.0001). See also Figure S2.
IRTKS promotes microvillar elongation using its SH3 and WH2 domains.
Our analysis of IRTKS targeting mechanisms revealed that overexpression of variants significantly impacted microvillar length (Figure 3G). W4 cells overexpressing full-length IRTKS exhibited a minor increase in length over WT cells (2.7 ± 0.4 μm vs. 2.5 ± 0.4 μm, respectively). However, SH3* and ΔWH2 constructs significantly decreased length (1.8 ± 0.4 μm and 1.9 ± 0.4 μm, respectively), likely due to a dominant negative effect. Effects on length also scaled with expression level (Figure 3H). Constructs lacking the I-BAR or consisting of the I-BAR domain alone did not impact microvillar length (Figure 3G).
Based on these data, we hypothesized that IRTKS plays a role in controlling microvillar length. To determine if IRTKS is required for microvillar growth, we used shRNA to knockdown (KD) IRTKS in W4 cells (Figs. 3J, S2G), and then examined the impact using confocal microscopy and SIM (Figure 3J). IRTKS KD significantly reduced the fraction of cells able to grow a BB (72.7 ± 12.9% in scramble control vs. 50.1 ± 15.7% in KD) (Figure 3K) and significantly decreased microvillar length (2.5 ± 0.5 μm control vs. 1.3 ± 0.2 μm KD) (Figure 3L). These perturbations were specific to loss of IRTKS, as a construct refractory to KD (IRTKSr) rescued the fraction of cells able to grow microvilli (71.7 ± 14.9%) and microvillar length (2.7 ± 0.3 μm) (Figure 3K, 3L, & S2J). Interestingly, expression of refractory SH3* and ΔWH2 constructs only partially rescued microvillar growth parameters, while refractory ΔI-BAR and I-BAR constructs had no effect on microvillar length (Figure 3K, 3L, & S2K-N). These overexpression and KD/rescue assays reveal that IRTKS is needed for normal microvillar elongation, which requires functional SH3 and actin-binding WH2 domains.
EPS8 colocalizes with IRTKS and interacts with its SH3 domain.
Involvement of the IRTKS WH2 domain in microvillar elongation makes biological sense as this motif is known to bind actin directly [35]. How the IRTKS SH3 domain contributes to microvillar length control is less clear. To develop our understanding of these observations, we sought to identify a binding partner for this domain. Previous studies identified the IRTKS SH3 domain as a high affinity-binding partner of the proline rich (PR) motifs in the EHEC virulence factor, EspFu [40]. Sequence analysis of EspFu PR motifs implicates several mammalian proteins as potential IRTKS binding partners under normal conditions. One candidate that stands out based on high sequence conservation is EPS8 (Figure 4A), which exhibits actin bundling and capping activities through its C-terminal region (Figure 4B)[47, 48]. Previous studies suggest that EPS8 targets to the tips of intestinal microvilli [47, 49], hair cell stereocilia [50–52], and filopodia [53] where it controls the length of these protrusions, although significant questions remain regarding its mechanism of targeting and action in these different contexts. In W4 cells, endogenous EPS8 was highly enriched at the tips of microvilli (Figure 4C, 4D) and colocalized with endogenous IRTKS puncta at these sites (Figure 4E-4G). Whereas the majority of tip puncta contained both EPS8 and IRTKS, the stoichiometry of colocalization in the tip compartment was not fixed; this likely reflects structural variability inherent to the tips of individual microvilli. Finally, using in vitro pull-downs, we found that FLAG-tagged EPS8 interacted with full length EGFP-tagged IRTKS; we also confirmed that the SH3 domain was sufficient for binding (Figure 4H). Thus, using its SH3 domain, IRTKS holds the potential to interact with EPS8 at the tips of microvilli.
Figure 4: EPS8 colocalizes and interacts with IRTKS.
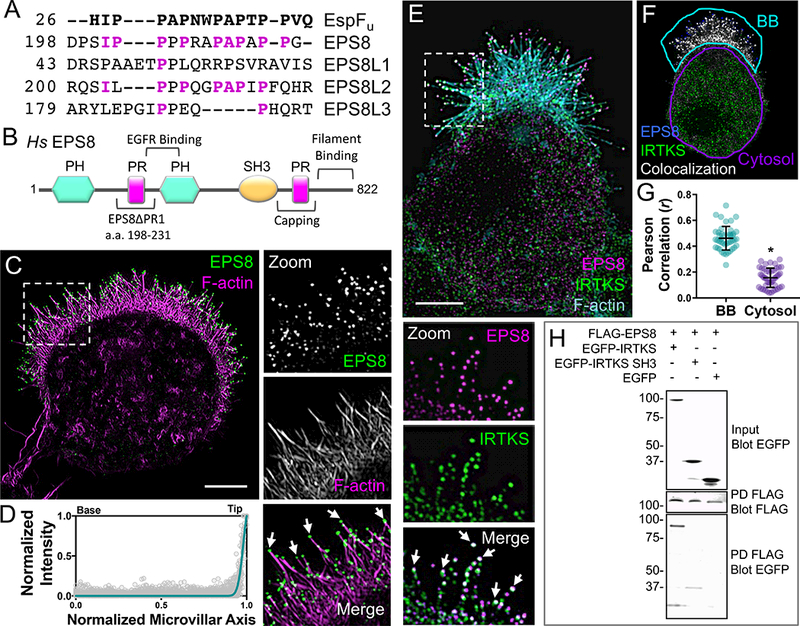
(A) Sequence alignment of the proline rich (PR) motifs of EspFu and EPS8 isoforms. Magenta letters indicate EPS8 amino acids that align with the EspFu PR. (B) Domain organization of human EPS8. EPS8ΔPR1 refers to the construct used in Figure 5D,E; capping and filament binding refer to F-actin interactions. (C) SIM projection of a W4 cell stained for endogenous EPS8 (green) and phalloidin (magenta). Dashed box indicates zoom of the BB, arrows highlight EPS8 puncta at microvillar distal tips. Scale bar, 5 μm. (D) Line scans (n = 42) of endogenous EPS8 intensity parallel to the microvillar axis in W4 cells; 0 = base, 1 = tip. (E) SIM projection of a W4 cell stained for endogenous IRTKS (green), EPS8 (magenta), and phalloidin (cyan). Dashed box indicates zoom of the BB (rotated 90° clockwise); arrows point to colocalized IRTKS and EPS8 puncta (white spots). Scale bar, 5 μm. (F) Colocalization of EPS8 and IRTKS; colocalized pixels (white), EPS8 pixels (blue), and IRTKS pixels (green). Teal outline designates the BB and purple outline designates the cytosol. (G) Pearson’s Correlation analysis of IRTKS and EPS8 colocalization in the BB vs. cytosol (n = 44 cells). Error bars indicate mean ± SD; p value calculated using a t test (*p<0.0001). (H) Pulldown of FLAG-tagged EPS8 co-expressed with EGFP-tagged IRTKS variants reveals binding between EPS8 and the IRTKS SH3 domain. See also Figure S3 and Video S9.
Interestingly, co-expressing IRTKS and EPS8 in W4 cells did not significantly elongate microvilli (Figure S3B). This observation might be due to the fact that, even under normal conditions, a major fraction of cellular G-actin is incorporated into microvillar actin bundles upon induction with doxycycline. We tested this by co-expressing IRTKS and EPS8 in cells that do not normally build BBs: B16F1 and HeLa cells. In both cell types, dual expression dramatically increased the number of both dorsal and substrate-attached filopodia over control cells (Figure S3C-S3F & Video S9), indicating that IRTKS and EPS8 can work together to stimulate finger-like protrusion growth.
IRTKS promotes EPS8 enrichment at the tips of microvilli.
To further elucidate the functional relationship between EPS8 and IRTKS at microvillar tips, we examined the impact of IRTKS KD on EPS8 localization. Although some EPS8 was still present in the shortened microvilli on the surface of IRTKS KD cells (Figure 5A,B), we also found that cytosolic EPS8 levels were significantly increased relative to controls (Figure 5A-C), suggesting that IRTKS promotes the tip targeting of EPS8 under normal conditions. To test this concept, we generated a variant of EPS8 lacking the predicted IRTKS interacting motif (EPS8ΔPR1, Figure 4A,B). Strikingly, this variant localized in puncta throughout the cytoplasm (Figure 5D), with significantly reduced tip targeting relative to EGFP-EPS8 (Figure 5E). Using pulldowns, we also found that less EGFP-EPS8ΔPR1 bound to a FLAG-tagged IRTKS SH3 bait relative to EGFP-EPS8 (Figure S4A). To further determine if IRTKS is sufficient to control EPS8 localization, we fused its SH3 domain to TOM20, thus directing it to the surface of mitochondria. In cells expressing TOM20-mCherry-IRTKS-SH3, but not TOM20-mCherry alone, a large fraction of endogenous EPS8 was sequestered to mitochondria (Figure 5F-5G). Microvillar length was also slightly decreased in these cells, likely due to the loss of EPS8 from the BB (Figure 5I). Together, these results indicate that IRTKS can use its SH3 domain to promote the localization of EPS8 to microvillar tips.
Figure 5: IRTKS recruits EPS8 using its SH3 domain.
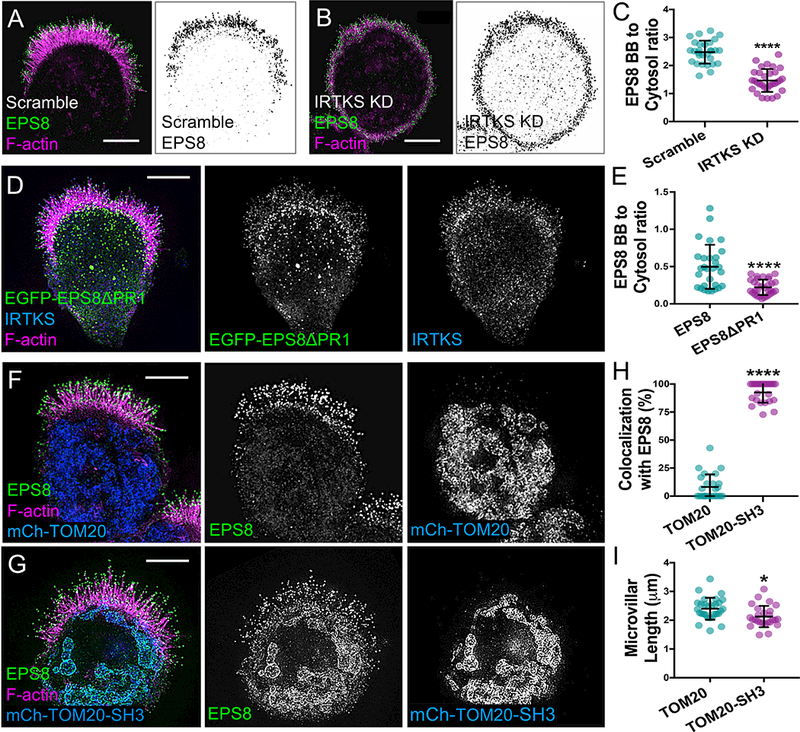
(A,B) SIM projections of scramble control and IRTKS KD W4 cells stained for EPS8 (green) and phalloidin (magenta). Inverted black and white images highlight the increased cytosolic localization of EPS8 in the IRTKS KD cells. Scale bars, 5 μm. (C) Quantitation of the ratio of endogenous EPS8 in the BB vs. cytosol for scramble and IRTKS KD W4 cells; scramble n = 29, IRTKS KD n = 31. (D) SIM projection of a W4 cell expressing EGFP-EPS8ΔPR1 (green) and stained with IRTKS (blue) and phalloidin (magenta). Scale bar, 5 μm. (E) Quantitation of the ratio of expressed EGFP-EPS8 (n = 29) and EGFP-EPS8ΔPR1 (n = 33) constructs in the BB vs. cytosol in W4 cells. (F) SIM projection of a W4 cell expressing mCherry-TOM20 (blue), stained for EPS8 (green) and with phalloidin (magenta) in control conditions. Scale bar, 5 μm. (G) SIM projection of an W4 cell expressing mCherry-TOM20 fused to the SH3 domain of IRTKS (blue) and stained with EPS8 (green) and phalloidin (magenta). The SH3 domain of IRTKS recruits EPS8 to the mitochondria. Scale bar, 5 μm. (H) Quantitation of the percentage of cells with EPS8 colocalized with mitochondria (n = 24). (I) Quantitation of microvillar length from cells expressing mCherry-TOM20 and mCherry-TOM20-SH3; >26 cells/condition, >10 microvilli averaged/cell. All error bars indicate mean ± SD; all p values calculated using a t test (*p<0.033, **p<0.002, ***p<0.0002, ****p<0.0001). See also Figure S4.
EPS8 promotes microvillar elongation.
Previous studies implicated EPS8 in controlling the length of actin bundle-supported protrusions [47, 52], and we confirmed this in EPS8 depleted W4 cells (Figures 6A, 6B & S4B). Interestingly, EPS8 KD phenocopied IRTKS KD (Figure 3J-3L), by reducing the fraction of cells able to assemble BBs (71.9 ± 11.7% scramble vs. 26.5 ± 10.7% EPS8 KD) (Figure 6C) and decreasing microvillar length (2.5 ± 0.4 μm scramble vs. 1.6 ± 0.3 μm EPS8 KD) (Figure 6D). Both microvillar growth parameters were rescued by expression of a refractory variant of EGFP-EPS8, but only partially rescued with a refractory EGFP-EPS8ΔPR1 (Figure 6C, 6D, S4B-S4D). Thus, EPS8 is also required for normal microvillar elongation though a mechanism that requires the PR1 motif.
Figure 6: IRTKS elongates microvilli using EPS8 dependent and independent mechanisms.
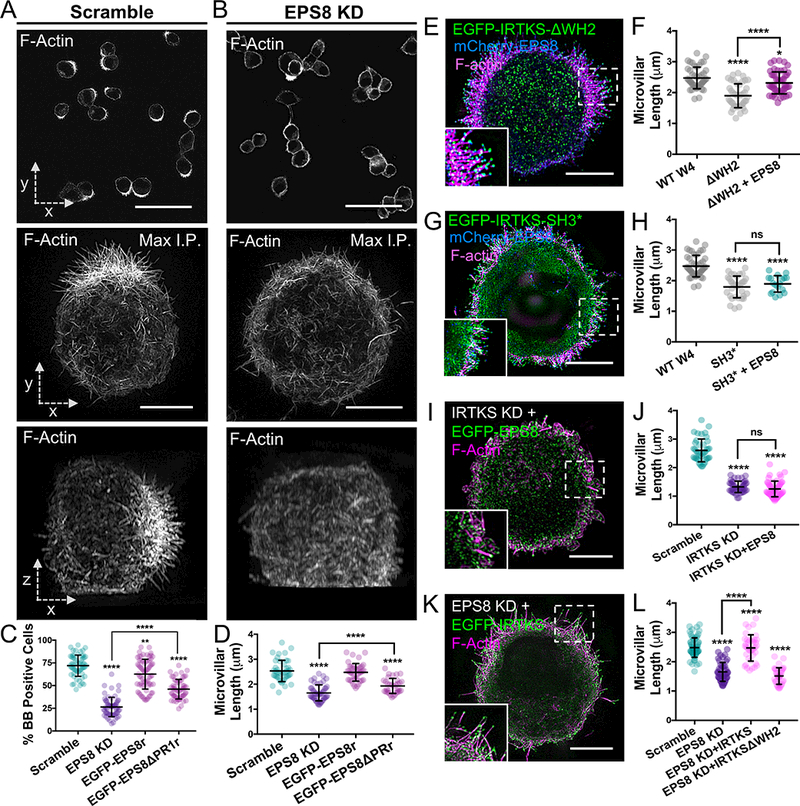
(A,B) Images of scramble and EPS8 KD W4 cells stained with phalloidin. Top panels: low magnification confocal images that were scored for “% BB positive cells” plot; scale bars, 40 μm. Middle panels: SIM whole cell maximum intensity projections (en face, x-y); scale bars, 5 μm. Bottom panels: SIM whole cell maximum intensity projections of the same cells as above (lateral, x-z). (C) Quantitation of the percentage of BB positive cells in EPS8 KD and rescue experiments; >200 cells/condition. Individual points correspond to the percent of cells containing a BB as scored in a 40× confocal image field. (D) Quantitation of microvillar length in EPS8 KD and rescue experiments; >41 cells/ condition, >10 microvilli averaged/cell. (E,F) SIM projection and quantitation of microvillar length in W4 cells expressing EGFP-IRTKSΔWH2 (green) and mCherry-EPS8 (blue) and stained with phalloidin (magenta). The WT W4 and ΔWH2 data (shaded gray tones) in this graph are replicated from Figure 2G for a direct comparison. Scale bar, 5 μm; >30 cells, >10 microvilli/cell. (G,H) SIM projection and quantitation of microvillar length in W4 cells expressing EGFP-IRTKS SH3* (green) and mCherry-EPS8 (blue), and stained with phalloidin (magenta). The WT W4 and SH3* data (shaded gray tones) in this graph are replicated from Figure 2G for a direct comparison. Scale bar, 5 μm; 18 cells, >10 microvilli/cell. (I,J) SIM projection and quantitation of microvillar length in IRTKS KD W4 cells expressing EGFP-EPS8. Scale bar, 5 μm; 45 cells/condition, >10 microvilli averaged/cell. (K,L) SIM projection and quantitation of microvillar length in EPS8 KD W4 cells expressing EGFP-IRTKS. Overexpressing IRTKS rescues microvillar length, whereas overexpressing IRTKSΔWH2 fails to rescue. Scale bar, 5 μm; ≥33 cells/condition, >10 microvilli averaged/cell. All error bars are mean ± SD; all p values calculated using a t test (*p<0.033, **p<0.002, ***p<0.0002, ****p<0.0001). See also Figure S4 and Figure S5.
IRTKS uses EPS8-dependent and independent mechanisms to elongate microvilli.
Our results up to this point suggest a model where IRTKS promotes microvillar elongation using two distinct mechanisms: (1) a direct mechanism that involves its actin-binding WH2 domain, and (2) an indirect mechanism that involves SH3 domain-dependent recruitment of EPS8, which in turn exerts its own elongation activity. As proposed, both mechanisms require functional IRTKS. If this model is accurate, EPS8 co-expression should rescue microvillar shortening observed in response to overexpression of IRTKSΔWH2, but not IRTKS SH3* (Figure 3D, 3G). Indeed, EPS8 expression rescued microvillar length in cells expressing IRTKSΔWH2, but had no impact on the length reduction caused by IRTKS SH3* (Figure 6E-6H). A second prediction is that a functional SH3/PR1 interaction should be required for EPS8 to increase filopodia numbers when co-expressed with IRTKS in B16 cells. Consistent with this proposal, co-expressing EPS8 with IRTKS SH3* or expressing EPS8ΔPR1 on its own did not impact filopodia number over controls (Figure S5A, S5D, S5E). However, co-expressing EPS8 with IRTKSΔWH2 significantly increased protrusion numbers (Fig S5B, S5E), likely because EPS8 can still localize using the intact IRTKS SH3 domain. Similarly, co-expressing IRTKS with EPS8ΔPR1 significantly increased protrusion numbers (Fig S5C, S5E), likely because IRTKS is able to target independent of EPS8. A third prediction based on our proposed model is that EPS8 overexpression should be unable to rescue the shortened microvilli observed with IRTKS KD (Figure 3L). We confirmed this experimentally; EPS8 overexpression had no impact on microvillar length in cells lacking IRTKS (Figure 6I, 6J). A fourth and final prediction states that IRTKS overexpression should rescue the shortening of microvilli observed with EPS8 KD (Figure 6D), but this rescue would require the IRTKS WH2 domain. We found that microvillar shortening in EPS8 KD cells was in fact rescued by overexpression of IRTKS, but not IRTKSΔWH2 (Figure 6K, 6L). Taken together, these findings indicate that IRTKS functions to elongate microvilli using EPS8-dependent and independent mechanisms, which represent structurally distinct and thus experimentally separable activities.
DISCUSSION
IRTKS has been studied extensively as a target of EHEC virulence factors [38, 39, 54, 55]. EHEC is a Shiga toxin-secreting attaching/effacing (A/E) pathogen [56] that colonizes distal small intestine or proximal colon epithelium by effacing microvilli and building adherent ‘pedestals’ on the apical surface [57]. To build pedestals, EHEC injects host cells with virulence factors that remodel the actin cytoskeleton [58]. IRTKS is targeted by the virulence factor EspFu [59], which uses tandem polyproline motifs to bind tightly to the SH3 domain [40]. This interaction is notable due to its high affinity (KD ~500 nM) [40]. The apically localized IRTKS-EspFu complex recruits N-WASP and Arp2/3, leading to a massive induction of actin assembly and pedestal formation beneath EHEC [41, 59]. The current study may help explain why EHEC evolved to target IRTKS using EspFu in its pathogenic mechanism. Our findings reveal that IRTKS is well-positioned and contains the appropriate domains and activities to influence apical actin assembly in response to microbial contact.
The studies presented here focused on elucidating the normal function of IRTKS in the apical domain. We found that IRTKS targets to and tracks the distal tips of growing microvilli (Figure 2B, 2C), where the barbed ends of core actin filaments interface with highly curved plasma membrane. Combined with results from our overexpression and loss-of-function experiments, and the demonstrated physical association with the established protrusion elongation factor EPS8 [47, 49], our results indicate IRTKS plays an important role in elongating microvilli. Indeed, IRTKS KD eliminated BB formation in ~50% of the W4 cells assayed (Figure 3K). In cells that were still able to form a BB, IRTKS KD significantly shortened microvilli compared to controls (Figure 3L). Because short microvilli still form in KD cells, IRTKS-driven elongation may involve detection of outwardly curved regions of plasma membrane at some point after initiation of microvillar growth.
Our molecular dissection of IRTKS function revealed that the N-terminal I-BAR domain is the primary driver of tip targeting. Deletion of the I-BAR domain completely eliminated targeting to microvillar tips (Figure 3E, S3D). The I-BAR domain alone was able to tip target, but also exhibited general plasma membrane labeling (Figure 3F, S3E). This suggests that IRTKS probably requires additional motifs or interactions with other factors to focus its enrichment at microvillar tips. Indeed, mutation of the SH3 domain ligand-binding pocket also disrupted localization, although some distal tip labeling persisted (Figure 3C, S3B). Although it is tempting to speculate that SH3 domain targeting is related to EPS8 binding, IRTKS still localized to microvillar tips in cells lacking EPS8 (Figure 6K).
Structure/function studies revealed that the IRTKS WH2 domain is critical for microvillar elongation. Whereas WH2 domain deletion had no impact on IRTKS localization, overexpression of IRTKSΔWH2 exerted a dominant negative effect on protrusion length (Figure 3D, 3G, 3H), likely mediated by interactions with endogenous IRTKS. In addition, IRTKS KD phenotypes were only partially rescued by IRTKSΔWH2 (Figure 3K, 3L). Exactly how the IRTKS WH2 domain contributes to elongation remains unclear, but WH2 domain containing proteins in general have been implicated in actin filament nucleation and elongation [60]. IRTKS is different from many of these other factors in two ways. Whereas most WH2 domain molecules contain multiple copies of this 17–20 amino acid motif arranged in tandem, IRTKS contains only a single WH2 motif. Moreover, the IRTKS WH2 domain is divergent, containing ‘LRPT’ rather than the conserved ‘LKKV’ motif that typically contributes to monomer binding; IRTKS is also missing canonical hydrophobic residues upstream of this motif [60]. Despite these distinctions, the IRTKS WH2 domain does bind actin and may prefer filaments over monomers [35]. Interestingly, the WH2 domain of closely related IRSp53 binds strongly to G-actin despite containing an IRTKS-like ‘LKPT’ motif [29]. Espin, which functions in the elongation of microvilli and stereocilia [16, 19, 61, 62], also contains a WH2 domain characterized by ‘LKPT’ [63]. The espin WH2 domain binds to G-actin and may function to increase local monomer concentration at the distal tips, which could promote elongation. Notably, espin-2B exerts its effects on microvillar length without significantly altering F-actin treadmilling rates in microvilli [16]. This is consistent with our FRAP measurements on W4 cells expressing EGFP-β-actin, which revealed IRTKS KD had minimal impact on BB actin turnover (Figure S2H,I).
Our findings suggest that the IRTKS SH3 domain also contributes to microvillar length control (Figure 3C, 3G, & 3H), likely through its interaction with EPS8, an established elongation factor that enriches at the tips of actin protrusions [47, 52, 64]. Interaction with EPS8 was predicted in previous structural studies as its proline rich motifs are nearly identical to those found in EspFu peptides that bind IRTKS with high affinity [40]. Interestingly, higher cytoplasmic levels of EPS8 were seen with both the IRTKS KD and in cells expressing a variant of EPS8 lacking PR1, a predicted IRTKS binding motif (Figure 5A-5E). Forced targeting of the IRTKS SH3 domain to the mitochondrial surface also recruited EPS8 to these sites (Figure 5F-5G). Thus, we propose that IRTKS and EPS8 form a complex at the tips of microvilli, which stabilizes EPS8 in this compartment (Figure 7). Once at the tips, both IRTKS and EPS8 hold the potential to contribute to core bundle elongation: IRTKS through its WH2 domain and EPS8 through its actin bundling and capping activities [47, 48]. Our findings indicate these represent separable, parallel activities that both depend on IRTKS (Figure 7).
Figure 7: Models of IRTKS function in microvillar elongation.
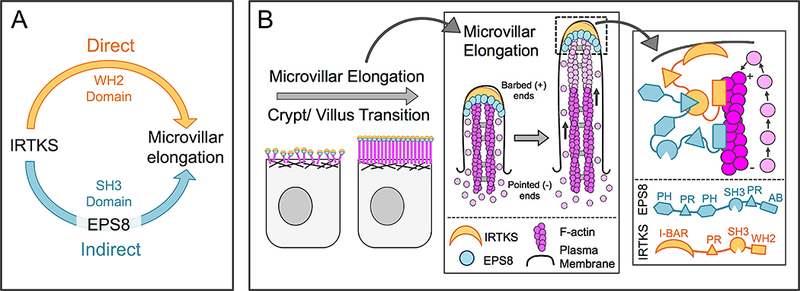
(A) These studies reveal two distinct pathways for microvillar elongation: a direct pathway that requires the IRTKS WH2 domain, and a second indirect pathway that uses the IRTKS SH3 domain to recruit to EPS8. (B) IRTKS and EPS8 form a microvillar elongation complex at the tips of actively growing microvilli. AB = actin binding domain. The EPS8 PR1 domain drawn binding to the IRTKS SH3 domain is representative, as other PR domains may also exhibit the ability to bind.
Of note, overexpression of the I-BAR domain alone did not impact microvillar length (Figure 3G). This might seem to contradict early reports suggesting I-BAR domains exhibit actin bundling activity [31, 35, 65]. Yet our data are consistent with studies on IRSp53 and MIM, which indicate that membrane binding rather than actin filament bundling is the primary function of this domain in cells [66]. We note here that our results do not rule out the possibility that the IRTKS I-BAR domain exerts direct effects on membrane mechanical properties that facilitate microvillar elongation [67].
Early models of microvillar growth emphasized the role of F-actin bundling proteins in protrusion assembly [17, 68]. However, more recent studies on a mouse model lacking all three established bundling proteins, fimbrin, villin, and espin, revealed that microvilli still form [22]. Our studies highlight new mechanisms that also contribute to this biologically robust process. Future studies must focus on understanding how the IRTKS/EPS8 complex interfaces with known polarity pathways during enterocyte differentiation, and on defining the temporal sequence of molecular recruitment that drives microvillar growth.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Matthew J. Tyska (matthew.tyska@vanderbilt.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS Cell Culture
Cell Culture
Ls174T-W4 cells (female Hs colon epithelial cells) were cultured in DMEM with high glucose and 2 mM L-glutamine supplemented with 10% tetracycline-free fetal bovine serum (FBS), G418 (1 mg/ml), blasticidin (10 μg/ml), and phleomycin (20 μg/ml). The cell line was obtained directly from Dr. Hans Clevers (Utrecht University, Netherlands) and has not been additionally authenticated beyond confirmation of polarization with the addition of DOX. B16F1 (Male Mm melanoma cells) and HeLa cells (female Hs cervix epithelial cells) were cultured in DMEM with high glucose and 2 mM L-glutamine supplemented with 10% FBS. Intestinal organoids were generated from P42–56 mice (C57BL/6, both genders). Organoids were suspended in 50 μl matrigel (BD Biosciences) and cultured in advanced DMEM/F12 (Invitrogen) supplemented with the following growth factors: 500 ng/ml EGF (Invitrogen), 100 ng/ml Noggin (R&D Systems), 500 ng/ml R-spondin 1 (R&D Systems), and 1mM N-Acetylcysteine (Sigma). All cells were grown at 37°C and 5% CO2.
METHOD DETAILS
Transfections and lentivirus production:
All transfections were performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions and the cells were allowed to recover overnight (ON). For KD experiments, lentivirus was generated by cotransfecting HEK293FT cells (Fetal Hs embryonic epithelial cells; T75 flasks at 80% confluency) with 6 μg of pLKO.1 shRNA KD plasmids (Open Biosystems; IRTKS, TRCN0000005350; EPS8, TRCN0000061545; scramble control [69]), 4 μg of psPAX2 packaging plasmid, and 0.8 μg of pMD2.G envelope plasmid using FuGENE 6 (Promega). For efficient lentiviral production, cells were incubated for 48 hr, then lentivirus-containing media was collected and concentrated with Lenti-X concentrator (Clontech). To transduce W4 cells with lentivirus, the media was supplemented with 6 ug/ml polybrene (Sigma) and the lentiviral shRNAs. After a 24-hour incubation, the media was changed and resupplemented with 6 ug/ml polybrene and lentiviral shRNAs for an additional 24 hours. The cells were then seeded into plates or flasks and incubated ON in the absence or presence of 1 μg/ml doxycycline, and then prepared for immunofluorescence or SDS-PAGE. For rescue experiments, cells were transiently transfected 48 hours after the second lentiviral infection using Lipofectamine 2000, induced with 1 ug/ml doxycycline ON, and fixed the following morning for immunofluorescence.
Immunofluorescence:
For SIM imaging, cells were plated on glass coverslips and allowed to adhere for at least 6 hrs (W4 cells) or 3 hrs (HeLa cells). Cells were washed with pre-warmed phosphate-buffered saline (PBS) and fixed with warm 4% paraformaldehyde/PBS for 15 min at 37°C. Cells were then washed three times with PBS and permeablized with 0.1% Triton X-100/PBS for 15 min at room temperature. Cells were once again washed three times with PBS and blocked for 1 hr at 37°C in 5% bovine serum alb umin (BSA)/PBS. Primary antibodies anti-IRTKS (2 μg/ml, HPA021257; Sigma-Aldrich), anti-EPS8 (1 μg/ml Cat# 610143; BD Biosciences), anti-GFP (50 μg/ml, GFP-1020; Aves Labs), or anti-mCherry (1 μg/ml, Cat# M11217; Invitrogen) were diluted in PBS and incubated with cells at 37°C for 1 hr, followed by four washes with PBS. Cells were then incubated for 1 hr with goat anti-rabbit Alexa Fluor 488 F(ab’)2 Fragment (2 μg/ml, A11070; Molecular Probes), goat anti-mouse Alexa Fluor 488 F(ab’)2 Fragment (2 μg/ml, A11017; Molecular Probes), or Alexa Fluor 568-phalloidin or Alexa Flour 647-phalloidin (1:200, A12380 and A22287 respectively; Invitrogen) at room temperature. Coverslips were then washed four times with PBS and mounted on glass slides in ProLong Gold (P36930; Invitrogen). Organoids were processed in a similar manner as above with some minor modifications: 1) Organoids were fixed with warm 4% paraformaldehyde/PBS for 40 min at 37°C, 2) permeab lized with 0.1% Triton X-100/PBS for 30 min, and 3) primary antibodies were incubated with organoids ON at 4°C. For live-cell TIRF imaging, transfected B16F1 cells were seeded onto 35 mm glass bottom dishes (Invitro Scientific, D35–20-1.5-N) coated with 25 μg/ml laminin (Sigma, L2020) in PBS and allowed to adhere for 2 to 3 hours. Images of single cells were acquired every 5 seconds for 15 minutes. For live-cell confocal imaging of W4 cells, transfected cells were plated on glass-bottom dishes with 1 μg/ml of doxycycline and allowed to adhere for 6 hours. Images of single W4 cells were acquired every 5 seconds for 30 minutes or continuously for 4 minutes. For FRAP of scramble and IRTKS KD, images were acquired every 5 seconds with an initial acquisition of 15 sec followed by bleaching and an additional 10 minutes. Bleaching was performed within a 20 μm2 ROI using 30% 405 laser power for a duration of 100 ms. All live cells were maintained in a humid environment at 37°C and 5% CO2 using a stage-top incubation system. Image acquisition was controlled with Nikon Elements software.
Cloning and Constructs:
pTOB7-IRTKS (Open Biosystems) containing full-length human IRTKS was purchased and shuttled into a pEGFP-C1 vector (Clontech) adapted for Gateway cloning using the Gateway conversion kit (Invitrogen) (herein referred to as “Gateway-adapted pEGFP-C1”). The resulting clone, pEGFP-C1-IRTKS (aa 1–511), was verified by sequencing. IRTKSΔWH2 (aa 1–482), I-BAR alone (aa 1–249), ΔI-BAR (aa 250–511), WH2 alone (aa 473–511) constructs were generated by PCR and TOPO cloned into the pCR8 Gateway entry vector (Invitrogen). To generate the IRTKS-SH3* construct, mutations W378K and W391K were introduced into pEGFP-IRTKS using site-directed mutagenesis. All entry vectors were verified by DNA sequencing and then shuttled into the destination vector Gateway-adapted pEGFP-C1—pDONR221 EPS8 (Open Biosystems) was also purchased and shuttled into Gateway-adapted pEGFP-C1 to generate pEGFP-C1-EPS8 (aa 1–822) and verified by sequencing. EPS8ΔPR1 (deleted aa 198–231) construct was generated by reverse PCR and TOPO cloned into the pCR8 Gateway entry vector. The mCherry-tagged UtrCH used for B16F1, HeLa, and W4 live-cell imaging was purchased from Addgene (26740, deposited by W. Bement). The TOM20-mCherry construct was generously provided by the Kaverina Lab (Vanderbilt), and the SH3 domain of IRTKS was fused to the C-terminus of mCherry. A nontargeting scramble control shRNA (Addgene; plasmid 1864), IRTKS KD shRNA, and EPS8 KD shRNA clones were expressed in pLKO.1, corresponding to TRC clones TRCN0000005350 and TRCN0000061545 (Sigma) respectively. To generate an IRTKS construct refractory to KD, three silent mutations were introduced into full-length IRTKS using site-directed mutagenesis. For the IRTKS shRNA, which targets nucleotides (nts) 1190–1211, nt 1190 (c→ a), nt 1192 (t → a), nt 1196 (a → t), nt 1197 (g→ c), and nt 1198 (t → g) were mutated. These silent mutations were also introduced into pEGFP-IRTKS SH3* and pEGFP-ΔWH2. For the EPS8 shRNA, which targets EPS8 nts 1712–1732, nt 1718 (c→ t), nt 1720 (g→ a), nt 1724 (t → a), nt 1725 (c→ g), and nt 1726 (t → c) were mutated. The same nts were mutated in the EGFP-EPS8ΔPR1 refractory construct.
Western Blot Analysis:
IRTKS and EPS8 knockdown and scramble cells were seeded into T25 flasks and allowed to grow for 24 hr. Cells were harvested with a cell scraper, pelleted at low speed, and lysed using ice-cold RIPA buffer containing 2 mM ATP, cOmplete ULTRA tablets (Roche), and 1 mM Pefabloc (Roche). The resulting cell lysates were then centrifuged at 15,000 RPM for 20 min and the soluble material was diluted with Laemmli sample buffer and heated at 95°C for 5 min. Equal sample volumes were loaded on a 4–12% Nu-Page gradient gel (Invitrogen) and the proteins were transferred to nitrocellulose at 25V for 16 hr. Membranes were blocked for 1 h in 10% milk-PBS and incubated with primary antibodies diluted in PBS containing 0.1% Tween-20 (PBS-T) ON at 4.°C The pri mary antibodies used were anti-IRTKS (0.08 μg/ml, HPA021257; Sigma-Aldrich), anti-EPS8 (0.1 μg/ml, Cat# 610143; BD Biosciences) and anti-glyceraldehyde 3-phosphate dehydrogenase (0.5 μg/ml, Cat# 3907; Cell Signaling). Membranes were then washed four times with PBS-T and incubated with donkey anti-rabbit 800 IRdye (0.01 ug/ml, 926–32213; Li-Cor) or donkey anti-mouse 800 IRdye (0.01 μg/ml, 926–32212; Li-Cor) for 30 min. Membranes were washed with PBS-T an additional four times and imaged using a Li-Cor Odyssey infrared imaging system. The same protocol was used to generate EGFP-tagged IRTKS constructs for expression levels. Images of membranes were cropped and adjusted using ImageJ (NIH) and relative protein expression levels were quantified using the signal from GAPDH to normalize the samples from loading.
Pulldown Assays:
COS7 cells were grown in T75 flasks to 80% confluency and transfected with pull-down constructs using Lipofectamine 2000 according to the manufacturer’s protocol. After 48 hr, cells were lysed using 1 ml of ice-cold Cellytic M buffer (Sigma) containing 2 mM ATP, 1mM Pefabloc (Roche), and complete ULTRA tablets (Roche) and centrifuged at 15,000 RPM. The soluble material was recovered after centrifugation and incubated with a 30μl bed volume pre-equilibrated anti-FLAG M2 resin (Sigma) for 2 hr with continuous rocking at 4°C. Resin and bound material were pelleted at 300 RPM, washed four times using RIPA buffer supplemented with 2 mM ATP, 1 mM Pefabloc (Roche), and complete ULTRA tablets (Roche), and eluted by boiling in SDS buffer to recover bound material. Resin-bound material was detected by western analysis with the following antibody dilutions: mouse anti-FLAG M2 (10 μg/ml; Sigma cat. #F3165) and chicken anti-EGFP (2 μg/ml; Aves Labs cat. #GFP-1020). The pull-down assays were repeated three times, and the results shown are representative.
Microscopy
SIM of W4 and HeLa cells was performed using an Applied Precision DeltaVision OMX equipped with a 60X Plan-Apochromat N/1.42 NA oil immersion objective (Olympus) using softWorx software (GE Healthcare). Laser scanning confocal microscopy was performed using a Nikon A1R laser-scanning confocal microscope. Live cell B16F1 imaging was performed on a Nikon TiE inverted light microscope equipped with 488 and 561 nm excitation LASERs, a 100x/1.49 NA TIRF objective, and an Andor Neo sCMOS detector. Imaging was performed using near-TIRF illumination, where the incident angle of the LASER was adjusted to increase the depth of penetration of the excitation field. Live-cell imaging of W4 cells was performed on a Nikon Ti2 equipped with a Yokogawa CSU-X1 spinning disk confocal unit, 488 and 561 nm LASERs, a 100x/1.49 NA objective, and a Photometrics PRIME 95B sCMOS detector. Images were contrast enhanced and cropped using FIJI/ImageJ software (NIH). Further details regarding the preparation of cells for imaging and data analysis and quantification can be found in Supplemental Experimental Procedures.
QUANTIFICATION AND STATISTICAL ANALYSIS
All image analysis was performed using FIJI or Nikon Elements software and all quantitative data are from at least three independent experiments. To perform line-scan analysis, a line was drawn along the axis of microvilli that were entirely in plane with a distinct tip and base visible. The intensity of the IRTKS or EPS8 signal was recorded and normalized with the lowest intensity set to 0 and the maximum set to 1. The microvillar length axis from individual scans was also normalized with the base set to 0 and the tip set to 1. Normalized line-scans were then plotted together and fit to a single Gaussian using nonlinear regression (Prism v.7, GraphPad). For quantification of percentage of cells with BB, cells were scored as BB positive if they displayed polarized F-actin accumulation as visualized using a 40X objective on a Nikon A1R laser-scanning confocal microscope. The same microscope and objective was used to quantify the percent of EPS8 and mitochondrial localization, and cells were scored as localized when they a majority of the signal colocalized with the mitochondria. Microvillar length measurements were performed on projected SIM images by tracing individual microvillar actin bundles using FIJI. For analyses in which individual microvilli were measured, at least 10 microvillar actin bundles were scored per cell and at least 25 cells measured per experiment. In B16F1 melanoma cells, filopodia number was quantified in ImageJ by counting the number of actin protrusions on the outside of a cell. The BB:cytosol ratio measured in W4 cells was defined as the ratio of the mean intensities from these two regions. Percent BB and microvillar length data were analyzed with a D’Agostino and Pearson omnibus normality test to determine normal distribution. Normally distributed data were statistically analyzed to determine significance using the unpaired Student’s t test. Statistical analyses performed are stated in the figure legends. All graphs were generated and statistical analyses performed using Prism (v.7, GraphPad).
Supplementary Material
Video S8, Related to Figure 3. FRAP of IRTKS shRNA Ls174T-W4 cells.
Spinning Disk confocal imaging of an induced, IRTKS KD Ls174T-W4 cell expressing mCherry-β-actin. Bleaching within a 10 μm2 ROI was performed using 30% laser power for a duration of 100 ms. Video was acquired every 5 seconds for 4 minutes and is played at 6.25 FPS. Scale bar, 5 μm.
Video S9, Related to Figure 4. EPS8 and IRTKS co-expression induces both substrate-attached and dorsal filopodia.
TIRF imaging of a B16F1 melanoma cell expressing EGFP-EPS8 (green) and mCherry-IRTKS (magenta). Video was acquired every 5 seconds for 15 minutes and is played at 20 FPS. Scale bar, 10 μm.
Video S1, Related to Figure 1. IRTKS is expressed at the tips of microvilli in the organoid crypt domain.
Slices through a confocal Z stack of IRTKS (green) and phalloidin (magenta) staining in a mouse intestinal organoid. Video is played at 8 frames per second (FPS). Scale bar, 40 μm.
Video S2, Related to Figure 2. Live imaging of Ls174T-W4 cell microvilli.
Spinning disk confocal imaging of an induced Ls174T-W4 cell expressing EGFP-Utrophin (UtrCH) to label F-actin. Video was acquired every 10 seconds for 30 minutes and is played at 12.5 FPS. Scale bar, 5 μm.
Video S3, Related to Figure 2. IRTKS localizes to the distal tips of growing microvilli.
Spinning disk confocal imaging of an induced Ls174T-W4 cell expressing mCherry-UtrCH (magenta) and EGFP-IRTKS (green). Video was acquired every 10 seconds for 30 minutes and is played at 12.5 FPS. Scale bar, 5 μm.
Video S4, Related to Figure 2. IRTKS tracks the tips of growing microvillar protrusions.
Spinning disk confocal imaging of an induced Ls174T-W4 cell expressing mCherry-UtrCH (magenta) and EGFP-IRTKS (green). Video was acquired every 5 seconds for 80 seconds and is played at 5 FPS.
Video S5, Related to Figure 2. Representative filopodial protrusions in a control B16F1 melanoma cell.
TIRF imaging of a B16F1 melanoma cell expressing mCherry-UtrCH (magenta); indicates the representative number of filopodia in a control cell. Video was acquired every 5 seconds for 12 minutes and is played at 20 FPS. Scale bar, 10 μm.
Video S6, Related to Figure 2. IRTKS expression induces filopodial protrusions in a B16F1 melanoma cell.
TIRF imaging of a B16F1 melanoma cell expressing EGFP-IRTKS (green) and mCherry-UtrCH (magenta). Video was acquired every 5 seconds for 15 minutes and is played at 20 FPS. Scale bar, 10 μm.
Video S7, Related to Figure 3. FRAP of scramble control shRNA Ls174T-W4 cells.
Spinning Disk confocal imaging of an induced, scramble control Ls174T-W4 cell expressing mCherry-β-actin. Bleaching within a 20 μm2 ROI was performed using 30% laser power for a duration of 100 ms. Video was acquired every 5 seconds for 4 minutes and is played at 6.25 FPS. Scale bar, 5 μm.
Highlights.
IRTKS uses its N-terminal I-BAR domain to target the tips of growing microvilli.
IRTKS promotes microvilli elongation using its C-terminal WH2 domain.
IRTKS also drives elongation using its SH3 domain to recruit EPS8.
An IRTKS/EPS8 complex is required for normal microvillar elongation.
ACKNOWLEDGMENTS
We thank all members of the Tyska laboratory, Vanderbilt Microtubule and Motors Club, and the Vanderbilt Epithelial Biology Center for feedback and advice. Super-resolution imaging was performed through the Vanderbilt University Cell Imaging Shared Resource. This work was supported by an American Heart Association Predoctoral Fellowship (M.M.P.), American Heart Association Predoctoral Fellowship (N.G.L.) and NIH grants R01DK075555, R01DK111949 and R01DK095811 (M.J.T.).
Footnotes
AUTHOR CONTRIBUTIONS
M.M.P. and M.J.T. designed experiments, analyzed data, and wrote the manuscript. M.M.P. performed experiments, and M.J.T. supervised the study. N.G.L. and A.C.N. helped with data collection. All authors contributed to editing the manuscript.
DECLARATION OF INTEREST
Nothing to declare
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Helander HF, and Fandriks L (2014). Surface area of the digestive tract - revisited. Scand J Gastroenterol 49, 681–689. [DOI] [PubMed] [Google Scholar]
- 2.Mooseker MS, and Tilney LG (1975). Organization of an actin filament-membrane complex. Filament polarity and membrane attachment in the microvilli of intestinal epithelial cells. J Cell Biol 67, 725–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Crawley SW, Shifrin DA Jr., Grega-Larson NE, McConnell RE, Benesh AE, Mao S, Zheng Y, Zheng QY, Nam KT, Millis BA, et al. (2014). Intestinal brush border assembly driven by protocadherin-based intermicrovillar adhesion. Cell 157, 433–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Delacour D, Salomon J, Robine S, and Louvard D (2016). Plasticity of the brush border - the yin and yang of intestinal homeostasis. Nat Rev Gastroenterol Hepatol 13, 161–174. [DOI] [PubMed] [Google Scholar]
- 5.Shifrin DA Jr., McConnell RE, Nambiar R, Higginbotham JN, Coffey RJ, and Tyska MJ (2012). Enterocyte microvillus-derived vesicles detoxify bacterial products and regulate epithelial-microbial interactions. Curr Biol 22, 627–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vallance BA, Chan C, Robertson ML, and Finlay BB (2002). Enteropathogenic and enterohemorrhagic Escherichia coli infections: emerging themes in pathogenesis and prevention. Can J Gastroenterol 16, 771–778. [DOI] [PubMed] [Google Scholar]
- 7.Friedman NB (1945). Cellular Dynamics in the Intestinal Mucosa: The Effect of Irradiation on Epithelial Maturation and Migration. J Exp Med 81, 553–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fath KR, Obenauf SD, and Burgess DR (1990). Cytoskeletal protein and mRNA accumulation during brush border formation in adult chicken enterocytes. Development 109, 449–459. [DOI] [PubMed] [Google Scholar]
- 9.Specian RD, and Neutra MR (1981). The surface topography of the colonic crypt in rabbit and monkey. The American journal of anatomy 160, 461–472. [DOI] [PubMed] [Google Scholar]
- 10.van Dongen JM, Visser WJ, Daems WT, and Galjaard H (1976). The relation between cell proliferation, differentiation and ultrastructural development in rat intestinal epithelium. Cell and tissue research 174, 183–199. [DOI] [PubMed] [Google Scholar]
- 11.Crawley SW, Weck ML, Grega-Larson NE, Shifrin DA Jr., and Tyska MJ (2016). ANKS4B Is Essential for Intermicrovillar Adhesion Complex Formation. Dev Cell 36, 190–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weck ML, Crawley SW, Stone CR, and Tyska MJ (2016). Myosin-7b Promotes Distal Tip Localization of the Intermicrovillar Adhesion Complex. Curr Biol 26, 2717–2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li J, He Y, Lu Q, and Zhang M (2016). Mechanistic Basis of Organization of the Harmonin/USH1C-Mediated Brush Border Microvilli Tip-Link Complex. Dev Cell 36, 179–189. [DOI] [PubMed] [Google Scholar]
- 14.Li J, He Y, Weck ML, Lu Q, Tyska MJ, and Zhang M (2017). Structure of Myo7b/USH1C complex suggests a general PDZ domain binding mode by MyTH4-FERM myosins. Proc Natl Acad Sci U S A 114, E3776–E3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu IM, Planelles-Herrero VJ, Sourigues Y, Moussaoui D, Sirkia H, Kikuti C, Stroebel D, Titus MA, and Houdusse A (2017). Myosin 7 and its adaptors link cadherins to actin. Nat Commun 8, 15864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loomis PA, Zheng L, Sekerkova G, Changyaleket B, Mugnaini E, and Bartles JR (2003). Espin cross-links cause the elongation of microvillus-type parallel actin bundles in vivo. J Cell Biol 163, 1045–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Friederich E, Huet C, Arpin M, and Louvard D (1989). Villin induces microvilli growth and actin redistribution in transfected fibroblasts. Cell 59, 461–475. [DOI] [PubMed] [Google Scholar]
- 18.Krey JF, Krystofiak ES, Dumont RA, Vijayakumar S, Choi D, Rivero F, Kachar B, Jones SM, and Barr-Gillespie PG (2016). Plastin 1 widens stereocilia by transforming actin filament packing from hexagonal to liquid. J Cell Biol 215, 467–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sekerkova G, Richter CP, and Bartles JR (2011). Roles of the espin actin-bundling proteins in the morphogenesis and stabilization of hair cell stereocilia revealed in CBA/CaJ congenic jerker mice. PLoS Genet 7, e1002032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shin JB, Longo-Guess CM, Gagnon LH, Saylor KW, Dumont RA, Spinelli KJ, Pagana JM, Wilmarth PA, David LL, Gillespie PG, et al. (2010). The R109H variant of fascin-2, a developmentally regulated actin crosslinker in hair-cell stereocilia, underlies early-onset hearing loss of DBA/2J mice. J Neurosci 30, 9683–9694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vignjevic D, Kojima S, Aratyn Y, Danciu O, Svitkina T, and Borisy GG (2006). Role of fascin in filopodial protrusion. J Cell Biol 174, 863–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Revenu C, Ubelmann F, Hurbain I, El-Marjou F, Dingli F, Loew D, Delacour D, Gilet J, Brot-Laroche E, Rivero F, et al. (2012). A new role for the architecture of microvillar actin bundles in apical retention of membrane proteins. Mol Biol Cell 23, 324–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grega-Larson NE, Crawley SW, Erwin AL, and Tyska MJ (2015). Cordon bleu promotes the assembly of brush border microvilli. Mol Biol Cell 26, 3803–3815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grega-Larson NE, Crawley SW, and Tyska MJ (2016). Impact of cordon-bleu expression on actin cytoskeleton architecture and dynamics. Cytoskeleton (Hoboken) 73, 670–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wayt J, and Bretscher A (2014). Cordon Bleu serves as a platform at the basal region of microvilli, where it regulates microvillar length through its WH2 domains. Mol Biol Cell 25, 2817–2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ahmed S, Goh WI, and Bu W (2010). I-BAR domains, IRSp53 and filopodium formation. Semin Cell Dev Biol 21, 350–356. [DOI] [PubMed] [Google Scholar]
- 27.Zhao H, Pykalainen A, and Lappalainen P (2011). I-BAR domain proteins: linking actin and plasma membrane dynamics. Curr Opin Cell Biol 23, 14–21. [DOI] [PubMed] [Google Scholar]
- 28.Millard TH, Bompard G, Heung MY, Dafforn TR, Scott DJ, Machesky LM, and Futterer K (2005). Structural basis of filopodia formation induced by the IRSp53/MIM homology domain of human IRSp53. EMBO J 24, 240–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee SH, Kerff F, Chereau D, Ferron F, Klug A, and Dominguez R (2007). Structural basis for the actin-binding function of missing-in-metastasis. Structure 15, 145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lim KB, Bu W, Goh WI, Koh E, Ong SH, Pawson T, Sudhaharan T, and Ahmed S (2008). The Cdc42 effector IRSp53 generates filopodia by coupling membrane protrusion with actin dynamics. J Biol Chem 283, 20454–20472. [DOI] [PubMed] [Google Scholar]
- 31.Yamagishi A, Masuda M, Ohki T, Onishi H, and Mochizuki N (2004). A novel actin bundling/filopodium-forming domain conserved in insulin receptor tyrosine kinase substrate p53 and missing in metastasis protein. J Biol Chem 279, 14929–14936. [DOI] [PubMed] [Google Scholar]
- 32.Saarikangas J, Kourdougli N, Senju Y, Chazal G, Segerstrale M, Minkeviciene R, Kuurne J, Mattila PK, Garrett L, Holter SM, et al. (2015). MIM-Induced Membrane Bending Promotes Dendritic Spine Initiation. Dev Cell 33, 644–659. [DOI] [PubMed] [Google Scholar]
- 33.McConnell RE, Benesh AE, Mao S, Tabb DL, and Tyska MJ (2011). Proteomic analysis of the enterocyte brush border. American journal of physiology. Gastrointestinal and liver physiology 300, G914–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yeh TC, Ogawa W, Danielsen AG, and Roth RA (1996). Characterization and cloning of a 58/53-kDa substrate of the insulin receptor tyrosine kinase. J Biol Chem 271, 2921–2928. [DOI] [PubMed] [Google Scholar]
- 35.Millard TH, Dawson J, and Machesky LM (2007). Characterisation of IRTKS, a novel IRSp53/MIM family actin regulator with distinct filament bundling properties. Journal of cell science 120, 1663–1672. [DOI] [PubMed] [Google Scholar]
- 36.Huang LY, Wang YP, Wei BF, Yang J, Wang JQ, Wu BH, Zhang ZZ, Hou YY, Sun WM, Hu RM, et al. (2013). Deficiency of IRTKS as an adaptor of insulin receptor leads to insulin resistance. Cell Res 23, 1310–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sudhaharan T, Sem KP, Liew HF, Yu YH, Goh WI, Chou AM, and Ahmed S (2016). The Rho GTPase Rif signals through IRTKS, Eps8 and WAVE2 to generate dorsal membrane ruffles and filopodia. Journal of cell science 129, 2829–2840. [DOI] [PubMed] [Google Scholar]
- 38.Aitio O, Hellman M, Skehan B, Kesti T, Leong JM, Saksela K, and Permi P (2012). Enterohaemorrhagic Escherichia coli exploits a tryptophan switch to hijack host f-actin assembly. Structure 20, 1692–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Crepin VF, Girard F, Schuller S, Phillips AD, Mousnier A, and Frankel G (2010). Dissecting the role of the Tir:Nck and Tir:IRTKS/IRSp53 signalling pathways in vivo. Mol Microbiol 75, 308–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aitio O, Hellman M, Kazlauskas A, Vingadassalom DF, Leong JM, Saksela K, and Permi P (2010). Recognition of tandem PxxP motifs as a unique Src homology 3-binding mode triggers pathogen-driven actin assembly. Proc Natl Acad Sci U S A 107, 21743–21748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vingadassalom D, Kazlauskas A, Skehan B, Cheng HC, Magoun L, Robbins D, Rosen MK, Saksela K, and Leong JM (2009). Insulin receptor tyrosine kinase substrate links the E. coli O157:H7 actin assembly effectors Tir and EspF(U) during pedestal formation. Proc Natl Acad Sci U S A 106, 6754–6759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sato T, and Clevers H (2013). Growing self-organizing mini-guts from a single intestinal stem cell: mechanism and applications. Science 340, 1190–1194. [DOI] [PubMed] [Google Scholar]
- 43.Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ, et al. (2009). Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459, 262–265. [DOI] [PubMed] [Google Scholar]
- 44.Baas AF, Kuipers J, van der Wel NN, Batlle E, Koerten HK, Peters PJ, and Clevers HC (2004). Complete polarization of single intestinal epithelial cells upon activation of LKB1 by STRAD. Cell 116, 457–466. [DOI] [PubMed] [Google Scholar]
- 45.Burkel BM, von Dassow G, and Bement WM (2007). Versatile fluorescent probes for actin filaments based on the actin-binding domain of utrophin. Cell Motil Cytoskeleton 64, 822–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tu Y, Liang L, Frank SJ, and Wu C (2001). Src homology 3 domain-dependent interaction of Nck-2 with insulin receptor substrate-1. Biochem J 354, 315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Croce A, Cassata G, Disanza A, Gagliani MC, Tacchetti C, Malabarba MG, Carlier MF, Scita G, Baumeister R, and Di Fiore PP (2004). A novel actin barbed-end-capping activity in EPS-8 regulates apical morphogenesis in intestinal cells of Caenorhabditis elegans. Nat Cell Biol 6, 1173–1179. [DOI] [PubMed] [Google Scholar]
- 48.Hertzog M, Milanesi F, Hazelwood L, Disanza A, Liu H, Perlade E, Malabarba MG, Pasqualato S, Maiolica A, Confalonieri S, et al. (2010). Molecular basis for the dual function of Eps8 on actin dynamics: bundling and capping. PLoS Biol 8, e1000387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tocchetti A, Soppo CB, Zani F, Bianchi F, Gagliani MC, Pozzi B, Rozman J, Elvert R, Ehrhardt N, Rathkolb B, et al. (2010). Loss of the actin remodeler Eps8 causes intestinal defects and improved metabolic status in mice. PLoS One 5, e9468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zampini V, Ruttiger L, Johnson SL, Franz C, Furness DN, Waldhaus J, Xiong H, Hackney CM, Holley MC, Offenhauser N, et al. (2011). Eps8 regulates hair bundle length and functional maturation of mammalian auditory hair cells. PLoS Biol 9, e1001048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Behlouli A, Bonnet C, Abdi S, Bouaita A, Lelli A, Hardelin JP, Schietroma C, Rous Y, Louha M, Cheknane A, et al. (2014). EPS8, encoding an actin-binding protein of cochlear hair cell stereocilia, is a new causal gene for autosomal recessive profound deafness. Orphanet J Rare Dis 9, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Manor U, Disanza A, Grati M, Andrade L, Lin H, Di Fiore PP, Scita G, and Kachar B (2011). Regulation of stereocilia length by myosin XVa and whirlin depends on the actin-regulatory protein Eps8. Curr Biol 21, 167–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Disanza A, Mantoani S, Hertzog M, Gerboth S, Frittoli E, Steffen A, Berhoerster K, Kreienkamp HJ, Milanesi F, Di Fiore PP, et al. (2006). Regulation of cell shape by Cdc42 is mediated by the synergic actin-bundling activity of the Eps8-IRSp53 complex. Nat Cell Biol 8, 1337–1347. [DOI] [PubMed] [Google Scholar]
- 54.de Groot JC, Schluter K, Carius Y, Quedenau C, Vingadassalom D, Faix J, Weiss SM, Reichelt J, Standfuss-Gabisch C, Lesser CF, et al. (2011). Structural basis for complex formation between human IRSp53 and the translocated intimin receptor Tir of enterohemorrhagic E. coli. Structure 19, 1294–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yi CR, and Goldberg MB (2009). Enterohemorrhagic Escherichia coli raises the I-BAR. Proc Natl Acad Sci U S A 106, 6431–6432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ho NK, Henry AC, Johnson-Henry K, and Sherman PM (2013). Pathogenicity, host responses and implications for management of enterohemorrhagic Escherichia coli O157:H7 infection. Can J Gastroenterol 27, 281–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Licois D, Reynaud A, Federighi M, Gaillard-Martinie B, Guillot JF, and Joly B (1991). Scanning and transmission electron microscopic study of adherence of Escherichia coli O103 enteropathogenic and/or enterohemorrhagic strain GV in enteric infection in rabbits. Infect Immun 59, 3796–3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jarvis KG, and Kaper JB (1996). Secretion of extracellular proteins by enterohemorrhagic Escherichia coli via a putative type III secretion system. Infect Immun 64, 4826–4829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Campellone KG, Robbins D, and Leong JM (2004). EspFU is a translocated EHEC effector that interacts with Tir and N-WASP and promotes Nck-independent actin assembly. Dev Cell 7, 217–228. [DOI] [PubMed] [Google Scholar]
- 60.Dominguez R (2016). The WH2 Domain and Actin Nucleation: Necessary but Insufficient. Trends Biochem Sci 41, 478–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zheng L, Sekerkova G, Vranich K, Tilney LG, Mugnaini E, and Bartles JR (2000). The deaf jerker mouse has a mutation in the gene encoding the espin actin-bundling proteins of hair cell stereocilia and lacks espins. Cell 102, 377–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sekerkova G, Zheng L, Loomis PA, Changyaleket B, Whitlon DS, Mugnaini E, and Bartles JR (2004). Espins are multifunctional actin cytoskeletal regulatory proteins in the microvilli of chemosensory and mechanosensory cells. J Neurosci 24, 5445–5456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Loomis PA, Kelly AE, Zheng L, Changyaleket B, Sekerkova G, Mugnaini E, Ferreira A, Mullins RD, and Bartles JR (2006). Targeted wild-type and jerker espins reveal a novel, WH2-domain-dependent way to make actin bundles in cells. Journal of cell science 119, 1655–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Roffers-Agarwal J, Xanthos JB, and Miller JR (2005). Regulation of actin cytoskeleton architecture by Eps8 and Abi1. BMC Cell Biol 6, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bompard G, Sharp SJ, Freiss G, and Machesky LM (2005). Involvement of Rac in actin cytoskeleton rearrangements induced by MIM-B. Journal of cell science 118, 5393–5403. [DOI] [PubMed] [Google Scholar]
- 66.Mattila PK, Pykalainen A, Saarikangas J, Paavilainen VO, Vihinen H, Jokitalo E, and Lappalainen P (2007). Missing-in-metastasis and IRSp53 deform PI(4,5)P2-rich membranes by an inverse BAR domain-like mechanism. J Cell Biol 176, 953–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Prevost C, Zhao H, Manzi J, Lemichez E, Lappalainen P, Callan-Jones A, and Bassereau P (2015). IRSp53 senses negative membrane curvature and phase separates along membrane tubules. Nat Commun 6, 8529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Franck Z, Footer M, and Bretscher A (1990). Microinjection of villin into cultured cells induces rapid and long-lasting changes in cell morphology but does not inhibit cytokinesis, cell motility, or membrane ruffling. J Cell Biol 111, 2475–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sarbassov DD, Guertin DA, Ali SM, and Sabatini DM (2005). Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S8, Related to Figure 3. FRAP of IRTKS shRNA Ls174T-W4 cells.
Spinning Disk confocal imaging of an induced, IRTKS KD Ls174T-W4 cell expressing mCherry-β-actin. Bleaching within a 10 μm2 ROI was performed using 30% laser power for a duration of 100 ms. Video was acquired every 5 seconds for 4 minutes and is played at 6.25 FPS. Scale bar, 5 μm.
Video S9, Related to Figure 4. EPS8 and IRTKS co-expression induces both substrate-attached and dorsal filopodia.
TIRF imaging of a B16F1 melanoma cell expressing EGFP-EPS8 (green) and mCherry-IRTKS (magenta). Video was acquired every 5 seconds for 15 minutes and is played at 20 FPS. Scale bar, 10 μm.
Video S1, Related to Figure 1. IRTKS is expressed at the tips of microvilli in the organoid crypt domain.
Slices through a confocal Z stack of IRTKS (green) and phalloidin (magenta) staining in a mouse intestinal organoid. Video is played at 8 frames per second (FPS). Scale bar, 40 μm.
Video S2, Related to Figure 2. Live imaging of Ls174T-W4 cell microvilli.
Spinning disk confocal imaging of an induced Ls174T-W4 cell expressing EGFP-Utrophin (UtrCH) to label F-actin. Video was acquired every 10 seconds for 30 minutes and is played at 12.5 FPS. Scale bar, 5 μm.
Video S3, Related to Figure 2. IRTKS localizes to the distal tips of growing microvilli.
Spinning disk confocal imaging of an induced Ls174T-W4 cell expressing mCherry-UtrCH (magenta) and EGFP-IRTKS (green). Video was acquired every 10 seconds for 30 minutes and is played at 12.5 FPS. Scale bar, 5 μm.
Video S4, Related to Figure 2. IRTKS tracks the tips of growing microvillar protrusions.
Spinning disk confocal imaging of an induced Ls174T-W4 cell expressing mCherry-UtrCH (magenta) and EGFP-IRTKS (green). Video was acquired every 5 seconds for 80 seconds and is played at 5 FPS.
Video S5, Related to Figure 2. Representative filopodial protrusions in a control B16F1 melanoma cell.
TIRF imaging of a B16F1 melanoma cell expressing mCherry-UtrCH (magenta); indicates the representative number of filopodia in a control cell. Video was acquired every 5 seconds for 12 minutes and is played at 20 FPS. Scale bar, 10 μm.
Video S6, Related to Figure 2. IRTKS expression induces filopodial protrusions in a B16F1 melanoma cell.
TIRF imaging of a B16F1 melanoma cell expressing EGFP-IRTKS (green) and mCherry-UtrCH (magenta). Video was acquired every 5 seconds for 15 minutes and is played at 20 FPS. Scale bar, 10 μm.
Video S7, Related to Figure 3. FRAP of scramble control shRNA Ls174T-W4 cells.
Spinning Disk confocal imaging of an induced, scramble control Ls174T-W4 cell expressing mCherry-β-actin. Bleaching within a 20 μm2 ROI was performed using 30% laser power for a duration of 100 ms. Video was acquired every 5 seconds for 4 minutes and is played at 6.25 FPS. Scale bar, 5 μm.