

SUMMARY
Gram-positive bacteria deploy type IV secretion systems (T4SSs) to facilitate horizontal gene transfer. The T4SSs of Gram-positive bacteria rely on surface adhesins as opposed to conjugative pili to facilitate mating. Enterococcus faecalis PrgB is a surface adhesin that promotes mating pair formation and robust biofilm development in an extracellular DNA (eDNA) dependent manner. Here we report the structure of the adhesin domain of PrgB. The adhesin domain binds and compacts DNA in vitro. In vivo PrgB deleted of its adhesin domain does not support cellular aggregation, biofilm development and conjugative DNA transfer. PrgB also binds lipoteichoic acid (LTA), which competes with DNA binding. We propose that PrgB binding and compaction of eDNA facilitates cell aggregation and plays an important role in establishment of early biofilms in mono- or polyspecies settings. Within these biofilms, PrgB mediates formation and stabilization of direct cell-cell contacts through alternative binding of cell-bound LTA, which in turn promotes establishment of productive mating junctions and efficient intra- or interspecies T4SS-mediated gene transfer.
Keywords: Conjugation, DNA transfer, Mobile genetic elements, antibiotic resistance, biofilms, adhesins
The enterococcal adhesin protein PrgB promotes cellular aggregation by a mechanism dependent on binding of eDNA. We report the structure of PrgB’s adhesin domain and its capacity to both bind and condense DNA. These functions are required for cellular aggregation, formation of robust biofilms, and for PrgB-stimulated conjugative DNA transfer. PrgB alternatively binds surface-exposed lipoteichoic acids presumably to stabilize intercellular contacts for efficient type IV-mediated conjugative DNA transfer.
INTRODUCTION
Enterococci are among the leading causes of multidrug-resistant hospital acquired infections, mainly of the bloodstream, urinary tract and surgical wounds (Hidron et al., 2008; Gilmore et al., 2013; Lebreton et al., 2013). The two species of greatest medical concern, Enterococcus faecalis and Enterococcus faecium, are particularly problematic because of the presence of highly transmissible mobile genetic elements (MGEs) in their genomes. These elements code for virulence and fitness traits, as well as resistance to many antibiotics including those currently considered ‘last-resort’, e.g., vancomycin, daptomycin and linezolid. MGE-encoded type IV secretion systems (T4SSs) mediate transfer of the cognate DNA elements among enterococci and also to other medically-important Gram-positive (G+) species, e.g., staphylococci and streptococci (Paoletti et al., 2007; Arias et al., 2009; Laverde Gomez et al., 2011; Arias and Murray, 2012). Consequently, enterococci are considered important reservoirs for resistance mechanisms associated with infections by various G+ species. The enterococcal T4SSs are of considerable clinical importance due to their high efficiency and broad host range, yet at this time little is known about the mechanisms underlying this promiscuity.
In Enterococcus faecalis, the conjugative plasmid pCF10 has served as an important model for defining regulatory, structural, and functional features of type IV secretion in G+ species (Dunny, 2013; Dunny and Berntsson, 2016). Early studies showed that pCF10 transfers from donor cells at high frequencies upon sensing small peptide sex pheromones released from plasmid-free recipient cells in their vicinity (Dunny et al., 1978). Pheromone sensing activates transcription of the large prgQ operon on pCF10, which codes for three surface proteins; PrgA, PrgB and PrgC, as well as the Prg/Pcf T4SS (Dunny, 2013; Bhatty et al., 2017). Of the surface proteins, PrgB (also known as Aggregation Substance 10, Asc10) has been most extensively characterized. PrgB promotes extensive cellular aggregation and efficient pCF10 plasmid transfer (Olmsted et al., 1991; Bhatty et al., 2015), and is also an important virulence factor via its capacity to promote attachment and biofilm development of E. faecalis cells on biotic (e.g., mammalian heart tissue) and abiotic (e.g., prosthetic devices, catheters) surfaces (Schlievert et al., 1998; Rakita et al., 1999; Bhatty et al., 2015).
PrgB is a large adhesin (1,305 amino acids) exported to the E. faecalis cell surface by its N-terminal signal sequence and anchored to the cell wall by a C-proximal LPXTG anchoring motif. The central region carries an adhesin domain responsible for aggregation and two Arg-Gly-Asp (RGD) motifs implicated in binding host cell integrins (Fig. 1) (Rakita et al., 1999; Süssmuth et al., 2000; Waters et al., 2004; Chuang et al., 2009). The adhesin domain is also implicated in binding potential recipient cells to stimulate formation of mating junction formation (Olmsted et al., 1991). Recently, we reported that PrgB exerts its aggregative properties and enhances pCF10 transfer frequencies by a mechanism dependent on the presence of extracellular DNA (eDNA) (Bhatty et al., 2015). This finding is of interest in view of an increasing body of evidence establishing the general importance of eDNA for cellular aggregation and biofilm formation (Liu et al., 2008; Das et al., 2014).
Figure 1.
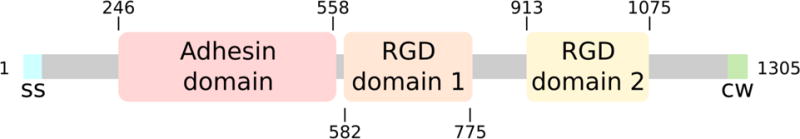
Schematic overview of the different domains of PrgB. PrgB contains a signal sequence (ss, cyan) at the N-terminus that targets it to the outside of the cell. It contains a C-proximal LPXTG motif that allows PrgB to be anchored to the cell wall (cw, green). Gray areas indicate areas of the protein predicted to be unstructured. Amino acid numbers are shown.
In this study, we sought to define a structural basis for how the PrgB – eDNA interaction promotes adherence to biotic and abiotic surfaces. We report the crystal structure of PrgB’s adhesin domain, and confirm structural similarity to the variable-region domains of the multimodal Streptococcus spp. AgI/II, SspB, and SpaP adhesins. We further show that this domain is responsible for PrgB’s ability to bind DNA, plausibly via its large positively charged surface areas. PrgB binds and compacts DNA in a cooperative fashion in vitro. A prgB mutant deleted of the adhesin domain fails to support eDNA-dependent aggregation, biofilm formation or conjugation in vivo. The PrgB adhesin domain thus contains a structural fold common to other G+ adhesins, but utilizes this fold to mediate aggregation in a completely different way through binding and compaction of eDNA.
RESULTS
Protein expression and purification
We were unable to recover full-length PrgB at detectable levels in E. coli, and therefore produced PrgB truncation mutants and individual domains identified by previous studies coupled to new analyses using the PsiPred algorithm (Waters et al., 2003; Buchan et al., 2013) (Fig. 1). The largest PrgB variant we were able to express and purify in large amounts was PrgB188-1233 (118 kDa), which lacks the N-terminal signal sequence and an adjacent disordered domain as well as the C-proximal LPXTG-anchor sequence. PrgB188-1233 purified as two peaks by size-exclusion chromatography (SEC), both species were stable and could readily be isolated. These species had molecular weights of 260 and 120 kDa, respectively, as determined by gas-phase electrophoretic mobility macromolecule analysis (GEMMA, also termed Macroion mobility spectrometer) (Kaufman et al., 1996; Bacher et al., 2001) (Fig. S1). The two peaks thus correspond to the dimeric and monomeric forms of PrgB188-1233, respectively. Although GEMMA tends to give accurate molecular masses, the method is dependent on the shape of the molecule and how it flies in the column under atmospheric pressure, likely explaining the slightly larger than expected mass of the dimeric peak. Rerunning the dimer fraction 24h after initial purification yielded only a dimer peak in SEC, and vice versa for the monomer fraction, suggesting that both oligomeric species were stable over time (Fig. S2a). We also purified the predicted adhesin domain PrgB246-558, which displays sequence homology to the variable regions of Streptococcal adhesins implicated in glucan binding, (Brady et al., 2010). Based on the eluction volume from SEC, purified PrgB246-558 was recovered only as a monomer in solution (Fig. S2b).
Overall structure of the PrgB adhesin domain
We performed crystallization trials with purified PrgB246-558. The crystals grew in spacegroup P43212 and contained one molecule in the asymmetric unit. The crystallographic phase problem was solved by single-wavelength anomalous dispersion (SAD) experiments using selenomethionine derivatized crystals, and the crystal structure was refined at a resolution of 1.6 Å (Table 1). The electron density accounted for all of the PrgB246-558 sequence. Overall, the core of the protein contains a central β-sandwich formed by two antiparallel β-sheets consisting of 16 individual β-strands (Fig. 2a). At the N-terminal end, between residues 291 and 341, a three-helix bundle is inserted. This bundle packs against one side of the β-sandwich, while an additional insertion between strand 13 and 14 protrudes out of the beta-sheet and flanks the upper portion of the sandwich. These two insertions form individual lobes, which are located on the same side of the beta-sandwich. In between the lobes, a deep and highly-negative charged cleft is formed which has a Tris molecule (from the crystallization condition) bound in its center. The opposite side of the beta-sandwich, consisting of beta-strands 1, 3, 5, 8, 10 and 15, has a large accumulation of positive surface charges (Fig. 2b). The first 40 amino acids of the domain, from residue 246 to 287, is present as a long loop wrapping around the whole crystallized domain. A DALI search using PrgB246-558 clearly demonstrated that the overall fold shows similarity to the lectin-like fold in general, and high similarity to the variable-region domains of Streptococcal Antigen I/II (AgI/II) and Streptococcus gordonii SspB (Brady et al., 2010; Holm and Rosenström, 2010). Superimposing PrgB246-558 with the variable domain of S. gordonii SspB (PDB code: 2WD6), one of the closest hits in the DALI search, gives an r.m.s.d. of 1.6 Å over 213 Cα atoms between the two structures (Fig S3).
Table 1.
Data collection and refinement statistics.
| Data collection summary | PrgB246-558 native | PrgB246-558 SeMet | PrgB246-558-DNA |
|---|---|---|---|
| Space group | P43212 | P43212 | P212121 |
| Cell dimensions | |||
| a, b, c (Å) | 60.55, 60.55, 153.32 | 60.51, 60.51, 153.98 | 32.02, 93.03, 99.10 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 |
| Resolution (Å) | 47.52-1.60 (1.70-1.60) | 47.57-1.86 (1.98-1.86) | 43.73-1.79 (1.90-1.79) |
| Completeness (%) | 99.9 (99.6) | 99.8 (99.1) | 99.3 (98.6) |
| Rmeas (%) | 5.2 (162.7) | 13.1 (242.5) | 17.2 (167.1) |
| I/σ (I) | 28.98 (1.47) | 20.40 (1.65) | 14.22 (1.63) |
| CC(1/2)* | 100.0 (64.3) | 100.0 (47.9) | 99.9 (71.0) |
| Redundancy | 18.83 | 27.6 | 13.36 |
| No. unique reflections | 38693 | 28379 | |
| Refinement summary | |||
| Resolution (Å) | 47.52 – 1.60 (1.64 – 1.60) | 43.73-1.79 (1.86-1.79) | |
| Rwork (%) | 19.00 | 17.4 | |
| Rfree (%) | 22.69 | 21.2 | |
| Number of atoms | |||
| protein | 2406 | 2244 | |
| water | 199 | 162 | |
| other ligands | 13 | 415 | |
| B-factors | |||
| protein | 44.74 | 33.16 | |
| water | 44.89 | 39.86 | |
| r.m.s. deviations | |||
| Bond lengths (Å) | 0.014 | 0.012 | |
| Bond angles (°) | 1.303 | 1.152 | |
| Ramachandran statistics | |||
| outliers (%) | 0.32 | 0.34 | |
| allowed (%) | 3.22 | 3.75 | |
| favored (%) | 96.46 | 95.90 |
Figure 2.
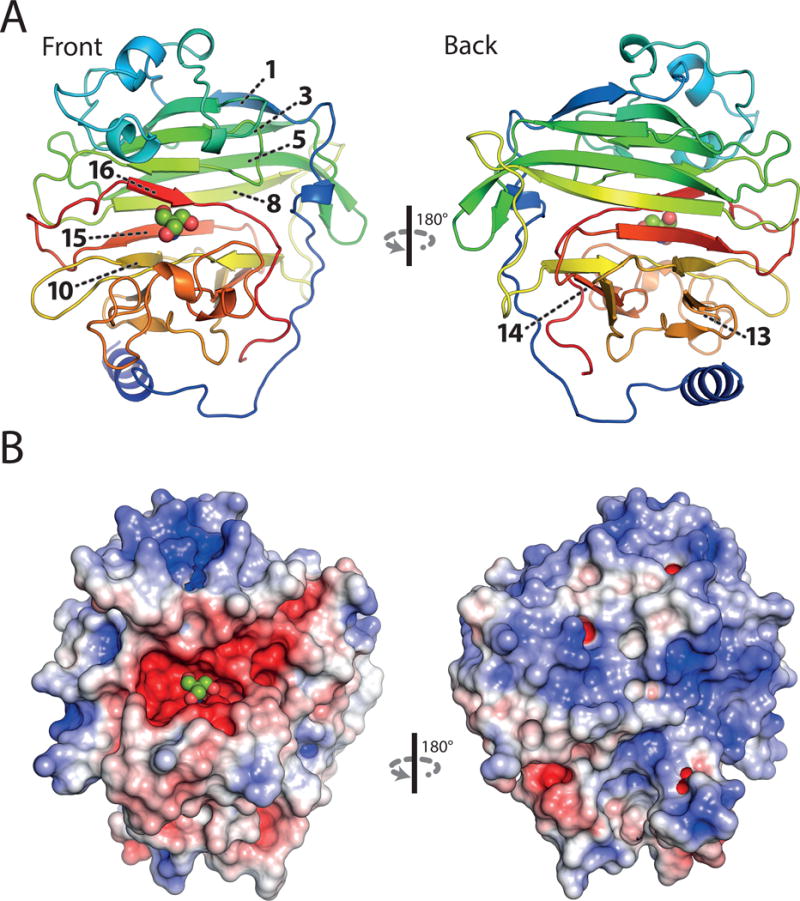
Crystal structure of the adhesion domain PrgB246-558 at 1.6 Å resolution. A: Ribbon representation, coloured in rainbow. The numbers of individual β-strands important for protein function are indicated. B: Surface charge distribution calculated by APBS, scaled from -5 (red) to 5 (blue) kbT. A bound Tris-molecule within the negatively charged cleft is shown in spheres. The top and back side of the protein is dominated by positively charged surface areas.
DNA binding properties
In view of the evidence that PrgB promotes cell aggregation and biofilm formation in an eDNA-dependent manner (Bhatty et al., 2015), we performed DNA-binding assays with PrgB188-1233 (monomer and dimer) and PrgB246-558. Using a combination of electrophoretic mobility shift assay (EMSA), surface plasmon resonance (SPR) and isothermal titration calorimetry (ITC) approaches, we first confirmed that PrgB188-1233 directly binds DNA and then established that the binding affinity of PrgB for DNA is strongly dependent on the DNA length. The dissociation constants range from 364 ± 74 μM for PrgB188-1233 monomer binding to a 19-bp long DNA substrate, as determined by ITC, to 0.2 ± 0.01 μM for PrgB188-1233 dimer binding to a 100-bp substrate as determined by EMSA (Fig. 3 & S4, Table 2, Table S1 & S2). The dimeric form of PrgB188-1233 has an affinity for DNA about twice that of the monomeric form, suggestive of additive binding (Table 2 & Fig. S4). We could not, however, detect a preference for single- or double stranded DNA, as the determined affinities were almost identical (Table 2, Fig. S4). PrgB bound DNA without sequence specificity, as deduced from SPR experiments showing that the PrgB188-1233 monomer bound two unrelated DNA sequences, designated DNA-1 and DNA-2 (220 and 240 bp, respectively), with no significant differences in affinity of 3.4 ± 0.3 μM and 3.2 ± 0.3 μM, respectively (Fig. 3a, Table 2). PrgB246-558 also directly bound both the 19-bp and 250-bp DNA substrates, although with greater affinity for the 19-bp substrate and a small decrease towards the 250-bp substrate relative to the affinity of the PrgB188-1233 variant (Table 2).
Figure 3.
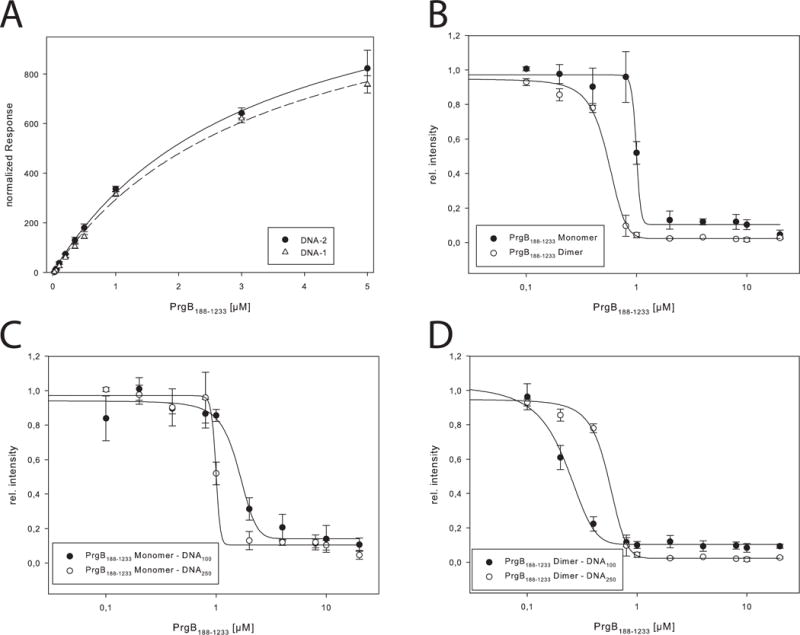
DNA interaction studies. A: SPR measurements of PrgB188-1233 with DNA-1 and DNA-2, the normalized response is plotted against the concentration of PrgB. The determined Kd values are 3.4 ± 0.3 μM and 3.2 ± 0.3 μM for DNA-1 and DNA-2, respectively. B: Binding of 250 bp long DNA to PrgB188-1233 monomer and dimer determined by EMSAs. Determined KD values are 1.0 ± 0.02 μM and 0.55 ± 0.02 μM, respectively. C: Binding of 100bp and 250bp long DNA to PrgB188-1233 monomer determined by EMSAs. Determined KD values are 1.6 ± 0.1 μM and 1.0 ± 0.01 μM, respectively. D: Binding of 100bp and 250bp long DNA to PrgB188-1233 dimer determined by EMSAs. Determined KD values are 0.2 ± 0.01 μM and 0.05 ± 0.02 μM, respectively.
Table 2.
KD values of PrgB188-1233 (monomer & dimer) and PrgB246-558 binding to different single and double stranded DNA, competition experiments with LTA as well as binding to glycerol phosphate, determined via ITC, EMSA or SPR experiments.
| DNA affinity | ||
|---|---|---|
| Protein | DNA | Kd [μM] |
| PrgB188-1233 Monomer | 19 | 364.0 ± 73.8 (ITC) |
| 100 | 1.6 ± 0.1 (EMSA) | |
| ss100 | 1.5 ± 0.09 (EMSA) | |
| DNA-1 | 3.4 ± 0.3 (SPR) | |
| DNA-2 | 3.2 ± 0.3 (SPR) | |
| 250 | 1.0 ± 0.01 (EMSA) | |
| PrgB188-1233 Dimer | 19 | 163.6 ± 42.4 (ITC) |
| 100 | 0.2 ± 0.01 (EMSA) | |
| ss100 | 0.4 ± 0.08 (EMSA) | |
| 250 | 0.55 ± 0.02 (EMSA) | |
| PrgB246-558 | 19 | 46.2 ± 14.5 (ITC) |
| 250 | 3.04 ± 0.32 (EMSA) | |
| LTA affinity | IC50 [mg/ml] | |
| PrgB188-1233 Monomer | 0.39 ± 0.09 (EMSA) | |
| PrgB188-1233 Dimer | 0.69 ± 0.03 (EMSA) | |
| PrgB246-558 | 0.19 ± 0.10 (EMSA) | |
| Glycerol phosphate affinity | Kd [μM] | |
| PrgB188-1233 | 309.2 ± 55.3 | |
| PrgB246-558 | 276.1 ± 71.2 | |
To further investigate the mode of DNA binding on a structural level we performed co-crystallization experiments with PrgB246-558 and a 10bp long dsDNA, which led to the structure of a PrgB-DNA complex at 1.9 Å (Fig. 4a, Fig. S5). Although the electron density is weak and ambiguous in places, we were able to model in the 10 bp dsDNA in the structure. The density indicated that the DNA was weakly bound, and the occupancy was set to 0.5. From a simulated annealing omit map it is however clear that the density is real (Fig. S5). In the asymmetric unit of the crystal, one DNA molecule is bound to a positive charged patch of PrgB, mediated by electrostatic interactions between K368, K371 as well as K426 and the phosphate groups of the DNA backbone as well as a possible hydrogen bond between K282 and a guanidine base of the DNA. Further DNA strands from symmetry related molecules in the crystal interact with K296 and K450. The major part of the positively charged protein surface is, however, unaccessible to the DNA due to crystal packing contacts with other PrgB molecules (Fig. 4b).
Figure 4.
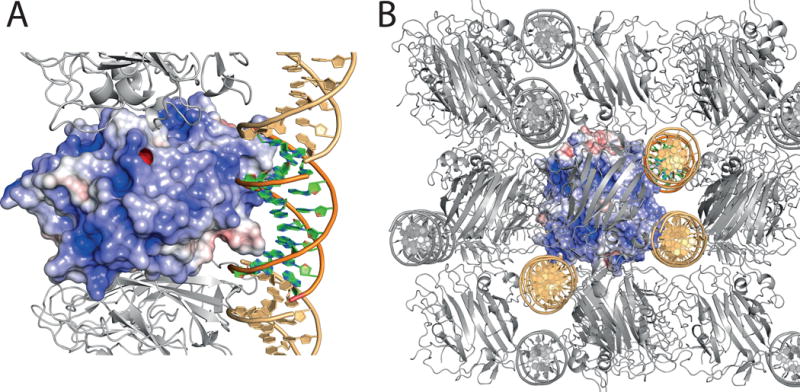
Crystal structure of the adhesion domain PrgB246-558 with bound DNA. A: Surface charge distribution calculated by APBS, scaled from -5 (red) to 5 (blue) kbT. The bound DNA molecule is shown in cartoon representation. Crystal symmetry related molecules are shown in grey for protein and light orange for DNA B: Crystal packing of the PrgB-DNA complex. Contents of the asymmetric unit are shown as colored surface and cartoon, symmetry related molecules are represented in grey for protein and light orange for DNA.
PrgB condenses long DNA
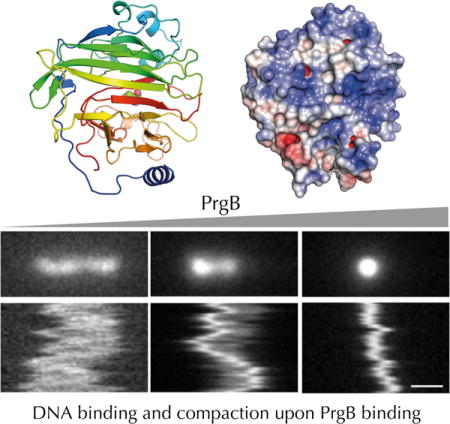
PrgB exhibits strong binding to long fragments of DNA, and we further analyzed this property by use of nanofluidic channels. Nanochannels have been extensively used for studies of single DNA molecules and have recently been adopted to studies of DNA-protein interactions (Frykholm et al., 2017). They are particularly well suited for studies of proteins that cause large conformational changes in the DNA, and hence changes in the extension in the channels (Fig 5a). We used relatively wide nanochannels (800×150 nm2) to minimize the effect of confinement on the properties of the DNA-protein complexes. Fig. 5b shows representative snapshots and kymographs of single, 48.5 kb long DNA molecules with added PrgB188-1233. Depending on the PrgB:DNA ratio, we observed three different behaviors: i) non-compacted, ii) partly compacted, and iii) fully compacted DNA. Interestingly, for the partly compacted DNA molecules, a majority initiated compaction from the end of the molecule, suggesting a higher affinity of PrgB for ends of DNA substrates or, alternatively, for the 12 bp of ssDNA comprising the ends of these DNA substrates. In Fig. 5d, we quantify the extension of DNA at different PrgB188-1233 concentrations by plotting the extension of the DNA as a function of the DNA:protein ratio. At over-threshold concentrations of PrgB188-1233, we observed full compaction of DNA (complexes smaller than 1 μm). At lower concentrations of 1:100 PrgB:DNA (DNA concentration in base pairs), we detect both fully- and partly-compacted DNA molecules in the same sample. This large heterogeneity indicates that the binding of PrgB to DNA is cooperative, and compares well with previous studies on the DNA binding protein Cox from bacteriophage P2 in nanochannels (Frykholm et al., 2016). At even lower PrgB concentrations (1:1000 PrgB:bpDNA) we observe both partly compacted and non-compacted DNA molecules. Close inspection of the kymographs reveals two additional categories of molecules at intermediate PrgB concentrations. A very small fraction of the molecules have either two condensed ends or three condensed points (Fig. 5c, left and right panels, respectively). The extension of these molecules (~3 m) is larger than detected with the naked DNA substrates, so it is reasonable to assume that these DNA molecules are two partially-compacted DNAs joined together. These findings suggest that in addition to binding and compacting single DNA molecules, PrgB potentially also has the capacity to bridge two different DNA substrates.
Figure 5.
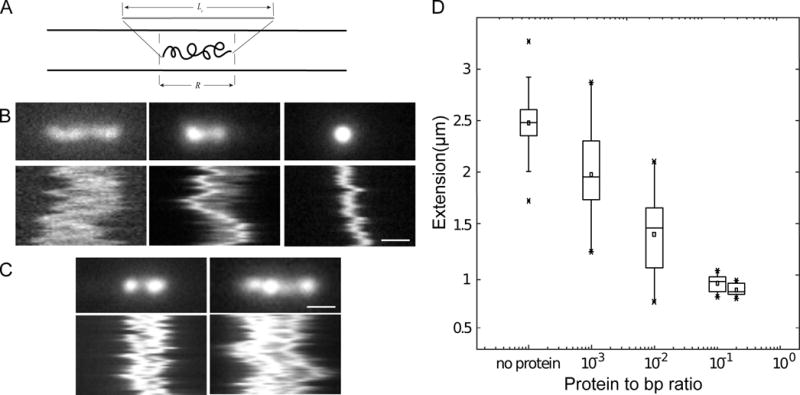
Nanochannel experiments with DNA and dimeric PrgB. A: A sketch showing a DNA molecule confined inside a nanochannel. DNA will be partially stretched out in the nanochannel, with an extension R, shorter than its contour length, Lc. B: Snapshots and kymographs of λ-DNA with PrgB bound confined in 800×150 nm2 channels. i) non-compacted DNA. ii) partly compacted DNA. iii) fully compacted DNA. C: Snapshots and kymographs of λ-DNA molecules confined in 800×150 nm2 channels. Left: Partially compacted DNA with two ends condensed. Right: DNA dimer with three condensed points. D: Boxplot of extension of λ-DNA inside 800×150 nm2 channels. The box is determined by the 25th and 75th percentiles and the whiskers are determined by the 5th and 95th percentiles. The line in the box is the median value and the square symbol in the box is the mean value. The cross symbols are the maximum and minimum values, respectively. The DNA concentration is 5 μM base pairs. The scale bar is 1μm in all figures.
Lipoteichoic acid competes with DNA for binding to PrgB
In early studies, E. faecalis mutants with altered production of lipoteichoic acid (LTA) were shown to be defective as recipients with pCF10-carrying donors (Trotter and Dunny, 1990). Further work established that PrgB binds LTA in vitro and that addition of free LTA blocks PrgB-mediated cellular aggregation in vivo (Ehrenfeld et al., 1986). Finally, an N-terminal fragment of PrgB (PrgB44-331) bound purified LTA whereas a PrgB mutant deleted of residues 156-358 showed attenuated binding (Waters et al., 2004). Results of these studies suggested that PrgB ligand – LTA receptor contacts promote formation of intercellular aggregates as a prerequisite for high-frequency plasmid transfer (Waters et al., 2004). In view of these findings, we explored the interaction network between PrgB, DNA and LTA. Interestingly, addition of LTA altered the DNA binding affinities of both PrgB188-1233 and PrgB246-558 fragments, as shown by EMSAs (Fig. 6). While PrgB fragments bound substrate DNAs in the absence of LTA, complex formation was diminished in the presence of increasing concentrations of LTA. The monomeric and dimeric forms of PrgB188-1233 had IC50 values of 0.39 and 0.69 mg/mL LTA, respectively, and PrgB246-558 had an IC50 of 0.19 mg/mL LTA (Fig. 6, Table 2). Glycerol phosphate is a main building block of LTA, so we also experimentally tested its binding to PrgB. However, the number of binding sites of glycerol phosphate to PrgB is unknown We observed an average of 8 binding sites for glycerol phosphate when we allowed the stoichiometry to float during fitting of the ITC data. We therefore set the number of binding sites to 8, a plausible value as we expect there to be multiple binding sites for glycerol phosphate, based on the surface charge of PrgB. These ITC experiments showed that PrgB246-558 and PrgB188-1233 bound glycerol phosphate equally well, with KD’s of ~0.3 mM (Table 2, Fig. S6).
Figure 6.
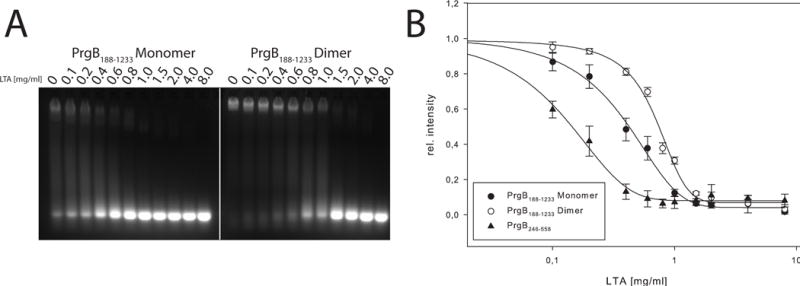
LTA competition to PrgB bound DNA, demonstrated by EMSA. A: LTA binding to either dimeric or monomeric PrgB188-1233 pre-incubated with 250 bp long DNA. B: Relative intensity of PrgB-DNA bands plotted against LTA concentration for PrgB188-1233 monomer and dimer. The determined IC50 values are 0.39 ± 0.09 and 0.69 ± 0.03 mg/mL, respectively.
PrgB246-558 mediates aggregation and biofilm formation
The above findings show that the PrgB246-558 adhesin domain binds DNA and LTA in vitro, and that LTA binding competitively interferes with DNA binding. Next, we analysed the in vivo effects of eDNA and LTA on PrgB-mediated aggregation, biofilm development, and pCF10 transfer. To this end, E. faecalis strains were engineered to produce native PrgB or PrgB deleted of its adhesin domain (Δ246-558) from the constitutive P23 promoter. Initial studies confirmed that PrgB and PrgBΔ246-558 accumulated at comparable levels when produced in OG1RF and OG1RF(pCF10) cells, and at slightly lower but detectable levels in strains with pCF10ΔprgB (denoted as ΔprgB) or pCF10 deleted of the entire surface adhesin cassette (ΔprgA-C) (Fig. 7a). As expected, production of native PrgB induced extensive clumping of cells both in the presence and absence of pCF10 or its variants (Fig. 7b). Clumping was observed regardless whether the sex pheromone cCF10 was added to cell cultures to activate expression of the prgQ operon encoding the prgA-C adhesin and prg/pcf T4SS gene modules. Production of PrgBΔ246-558 in OG1RF or isogenic strains with the pCF10ΔprgB or pCF10ΔprgA-C mutant plasmids did not induce aggregation. Production of the PrgB adhesion domain mutant in OG1RF(pCF10) resulted in a slightly reduced clumping phenotype, possibly as a result of dominant negative effects accompanying complex formation between the native and mutant forms of PrgB. Addition of DNase to pheromone-induced E. faecalis cells producing PrgB from the PQ promoter abolishes the clumping phenotype (Bhatty et al., 2015). DNase similarly blocked clumping of strains producing PrgB in the absence or presence of other pCF10-encoded proteins (Fig. 7b). Purified LTA similarly inhibited PrgB-mediated aggregation, which is consistent with our findings that LTA blocks PrgB-DNA complex formation in vitro. OG1RF(pCF10) develops significantly higher biofilm biomass than plasmid-free OG1RF on a polystyrene surface, and PrgB is a major contributor to robust biofilm development (Bhatty et al., 2015). Consistently, production of PrgB in plasmid-free OG1RF or OG1RF with pCF10 or the ΔprgB or ΔprgA-C variant plasmids, similarly resulted in significantly higher levels of biofilm biomass compared with OG1RF or even OG1RF(pCF10) (Fig. 7c). DNase completely blocked biofilm production by OG1RF(pCF10) as well as the PrgB-overproducing strains. Purified LTA also strongly inhibited biofilm development (Fig. 7c).
Figure 7.
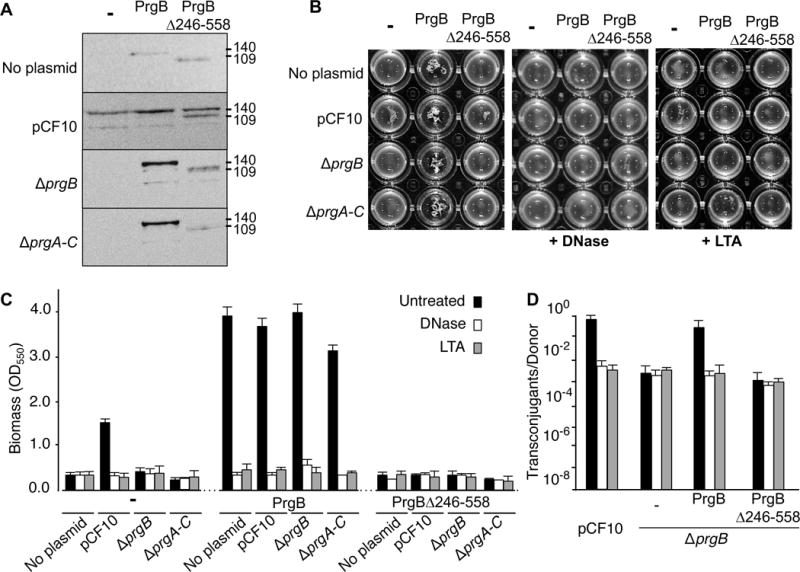
Contributions of the PrgB adhesin domain to aggregation, biofilm formation, and conjugation. In all panels, strains analysed were OG1RF alone (denoted ‘no plasmid’) or with pCF10 or pCF10ΔprgB (ΔprgB); these strains also were engineered to produce native PrgB or PrgBΔ246-558 from the constitutive P23 promoter. A: Steady-state levels of Prg proteins in strains induced for 1 h with cCF10 pheromone (10 ng ml−1). Immunoblots were developed with anti-PrgB antibodies. Protein sizes (in kilodaltons, kDa) are listed at the right. Protein extracts were loaded on a per-cell equivalent basis. B: Photographs of strains grown in microtiter plates showing the effects of DNase and LTA on PrgB-mediated clumping. C: Biofilm formation on polystyrene microtiterplates. The biofilm biomass was assayed as a function of crystal violet stain retained. DNase or LTA treatment was initiated at the onset of cCF10 induction and biofilm formation and continued for the full incubation time of 24 h. Results are expressed as OD550 values and values represent the average of at least three independent experiments; the error bars represent standard deviations. D: Effect of the PrgB adhesin domain on plasmid transfer. Transfer frequencies are presented as the number of transconjugants per donor cell. Experiments were repeated at least three times in duplicate, and results from a representative experiment are shown.
In contrast to the observed effects of PrgB, the PrgBΔ246-558 variant failed to support robust biofilm development. Furthermore, production of this variant even blocked biofilm development in OG1RF(pCF10), despite the fact that this strain produces native PrgB. Purified PrgB188-1233 multimerizes in solution, suggesting that native PrgB functions as a dimer or higher-order multimer in vivo. PrgBΔ246-558 production therefore might poison PrgB function through formation of non-productive dimers or higher-order complexes. Finally, in line with previous findings (Ehrenfeld et al., 1986; Bhatty et al., 2015), exposure of liquid mating mixtures to DNase or LTA resulted in decreased plasmid transfer by donors producing native PrgB to levels observed by donors lacking PrgB. Donors producing PrgBΔ246-558 transferred the plasmid at frequencies comparable to ΔprgB mutant donors, and plasmid transfer was unaffected by exposure to DNase or LTA (Fig. 7d).
DISCUSSION
In the present study, we determined the structure and characterized the biological functions of the N-proximal adhesin domain of PrgB (PrgB246-558). The overall fold of this domain is similar to the variable domains of Streptococcal Antigen I/II (AgI/II) and SspB adhesins. Members of the AgI/II superfamily have been shown to bind various eukaryotic cellular factors including salivary glycoproteins, fibrinogen, and the mucin-like protein gp-340 (Brady et al., 2010; Maddocks et al., 2011). The variable domains possess an overall negatively-charged lectin-like trench for carbohydrate binding, which among the oral streptococci is thought to mediate interactions with fucosylated carbohydrates displayed on the mineral matrix of teeth (Jenkinson and Lamont, 1997; Troffer-Charlier et al., 2002; Nobbs et al., 2009). However, S. gordonii SspB does not appear to bind carbohydrates, establishing that this is not a universal mechanism for attachment by this adhesin superfamily (Forsgren et al., 2009). Here, we found that the PrgB246-558 domain also possesses a negatively-charged cleft located between two lobes on one side of the beta-sandwich. Furthermore, PrgB’s cleft is lined with side-chains of S442, N444, E455, D489 and D503, which are also conserved residues among the AgI/II proteins. Although the cleft is conserved, the surface of the protein is not, as the AgI/II and SspB proteins do not have such positively charged surface areas (Fig. S3). Previous work on AgI/II proteins suggested that this binding cleft is a carbohydrate binding site. Reminiscent of SspB, however, we and others have not been able to demonstrate carbohydrate binding by PrgB, raising the possibility that PrgB evolved another mechanism(s) for promoting aggregation.
We discovered that the surface of PrgB246-558 is both covered with positively-charged lysines and arginines whose side-chains point away from the domain surface (Fig. 2b, S7) and are capable of binding DNA substrates. We also gained structural evidence from co-crystallization of PrgB246-558 and DNA for charge-based interactions. However, due to extensive crystal contacts with other PrgB molecules, the major part of the positively charged surface area is not available for DNA interactions in the crystal, which likely explains why only a short stretch of DNA is observed to be bound to PrgB in the crystal structure. Using SPR, EMSA and ITC with different DNA sequences, we further confirmed that both PrgB188-1233 and PrgB246-558 bind DNA, and that they do so in a sequence nonspecific manner (Fig. 3). These studies, together with single-molecule analyses using nanochannels, identified three biochemical properties of possible biological importance. First, the affinity of PrgB for DNA was strongly dependent on the length of the DNA, such that PrgB bound longer stretches of DNA with higher affinities (KDs approaching 0.2 μM) and shorter DNA molecules with comparatively lower affinities (KDs several orders of magnitude larger) (Fig. 3, Table 2). One explanation for these findings is that PrgB binds DNA along its entire positively-charged surface area such that short DNA substrates (e.g., 19 bp) do not cover enough of the surface to bind with a high affinity. Second, dimeric PrgB also exhibited higher affinities than monomeric PrgB towards the same DNA substrates. Conceivably, both monomers in the PrgB dimer form an overall charged-surface requiring DNA of a defined minimum length to span the entire binding site. Finally, in our nanochannel studies, the dimeric form of PrgB188-1233 strongly compacted linear DNA molecules in a cooperative fashion. Both monomers of the PrgB188-1233 dimer either bind a single DNA molecule, or potentially coordinate through binding of two different molecules to build a DNA network or mesh.
The DNA binding activity of PrgB246-558 clearly is important for PrgB function in vivo, as shown by the findings that i) DNase blocks aggregation and biofilm development of WT OG1RF(pCF10) cells, and also leads to a reduction in conjugative DNA transfer frequencies to levels observed for ΔprgB mutant donors (Bhatty et al., 2017), ii) DNase attenuates PrgB-mediated activities even when the adhesin is produced in OG1RF cells lacking other pCF10-encoded factors (Fig. 7), and ii) DNase treatment essentially phenocopies effects accompanying deletions of either full-length prgB or the 246-558 adhesin domain (Fig. 7). Thus, our data strongly suggest that PrgB246-558 exerts its aggregative properties through electrostatic binding of eDNA, which raises the mechanistic question of how a DNA binding activity confers cell-cell aggregation? It is noteworthy that growing cultures of E. faecalis accumulate eDNA either through a dedicated release/secretion pathway or by cell lysis (Thomas et al., 2008; Barnes et al., 2012). Indeed, one mechanism for eDNA release involves PrgB, which is toxic to cells when overproduced. Upon pheromone induction, pCF10-carrying cells express the plasmid-borne prgQ operon, but a subpopulation of cells appears to stochastically overexpress this operon leading to enhanced production of the Prg adhesins and PrgB overproduction toxicity. This cell subpopulation lyses and supplies the remaining viable cells with a source of eDNA and other biofilm matrix components (Bhatty et al., 2017). The eDNA accumulates as thick mats on E. faecalis cell surfaces, and also forms long, intercellular filamentous structures that bind nonspecifically to biotic or abiotic surfaces (Barnes et al., 2012). Binding of PrgB to these long eDNA filaments can be predicted to mediate a network of connections between pCF10-carrying donors with other cells in the vicinity, which in a polymicrobial growth setting would consist of plasmid-free E. faecalis as well as other bacterial species.
Our finding that PrgB compacts DNA suggests a model in which PrgB condensation of eDNA promotes formation of close cell-cell contacts and establishment of stable mating junctions. In E. coli, the F pilus undergoes dynamic rounds of extension and retraction to bring F plasmid-carrying donors into direct contact with potential recipients for mating junction formation (Clarke et al., 2008). In E. faecalis, which lacks such retractile conjugative pili, PrgB might function analogously by binding eDNA filaments and, through compaction, drawing neighboring cells into close apposition to stimulate intra- and interspecies gene transfer.
PrgB also binds LTA (Waters et al., 2004), and we here demonstrated that PrgB246-558 is responsible for this binding activity. We also found that LTA competitively interfered with DNA binding in vitro (Fig. 6) and abolished PrgB-mediated aggregation in vivo (Fig. 7), strongly suggesting that LTA and DNA bind the same positively-charged surface residues on the PrgB246-558 adhesin domain. LTA is anchored on the E. faecalis cell surface, and early genetic studies implicated LTA as a binding receptor for PrgB (Ehrenfeld et al., 1986; Trotter and Dunny, 1990). Since both PrgB and LTA are both located at the surface of the cell, it is possible that PrgB could interact with LTA on the same donor cell. Importantly, however, the domain architecture of PrgB (Fig. 1) suggests that the N-terminal adhesin domain is spatially distant and inaccessible to LTA produced by the same cell but potentially accessible to LTA displayed on the surface of neighboring cells (Fig. 1).
In a unifying model, therefore, we propose that pheromone induction of pCF10-carrying donor cells leads to production of the Prg adhesins and assembly of the Prg/Pcf T4SS. A subset of pheromone-induced cells stochastically overproduce PrgB, resulting in toxicity, lysis, and release of eDNA, which forms part of the biofilm matrix. pCF10-carrying donor cells and other potential recipient cells, be they E. faecalis or other species, are incorporated into this biofilm. PrgB-producing donors bind eDNA filaments, and then PrgB compacts eDNA filaments to form closer cell-cell interactions through a histone-like DNA condensation mechanism. Among closely juxtaposed cells, PrgB alternatively binds LTA, which stabilizes direct cell-cell contacts and promotes formation of mating junctions. Again, an analogy can be drawn to the E. coli F plasmid system. Once the F pilus retracts, the surface-exposed protein TraN forms a stabilizing bridge through specific contacts with receptors on recipient cells. This essentially ‘staples’ donors and recipients together and renders mating pairs extremely refractory to physical disruption (Arutyunov and Frost, 2013). We envision that the PrgB-LTA interaction supplies this stabilizing function for the pCF10 transfer system.
Although we do not yet have a structure of the entire PrgB protein, the structural homology of the N-proximal adhesin domain with AgI/II and SspB and a predicted elongated structure of the C-proximal region, suggest an overall head-stalk architecture reminiscent of the AgI/II adhesin family. Indeed, by Phyre2 modeling, the C-proximal third of PrgB structurally resembles the corresponding regions of the streptococcal AgI/II and SspB adhesins (PDB codes 3QE5 & 2WOY, 100 % confidence). PrgB, however, has two features that distinguish it from the streptococcal adhesins. First, our study indicates that PrgB assembles minimally as a dimer and that dimerization enhances its DNA-binding activities. Second, pCF10 codes for two other surface proteins besides PrgB, denoted as PrgA and PrgC (Bhatty et al., 2015). While PrgC’s functions in vivo do not seem to depend on the other surface proteins, several lines of evidence suggest that PrgA and PrgB physically and functionally interact: i) the two genes overlap suggestive of translational coupling, which can facilitate cosynthesis of interacting partners, ii) co-expression of prgA and prgB is necessary for full complementation of individual ΔprgA and ΔprgB mutations and for accumulation of abundant levels of both proteins, iii) PrgA production is correlated with accumulation of a 73-kDa PrgB species and iv) although synthesis of PrgB alone suffices for eDNA-dependent cell-cell aggregation, both PrgA and PrgB are necessary for efficient attachment of enterococcal cells to abiotic surfaces and robust biofilm formation (Bhatty et al., 2015). Taken together, these findings suggest that PrgB self-assembles and also forms higher-order complexes with PrgA on the cell surface. Elucidating the architecture of PrgA-PrgB complexes and defining the role of PrgA in modulating PrgB’s adhesin functions remain exciting areas for future investigations.
EXPERIMENTAL PROCEDURES
prgB and prgBΔ246-558 expression plasmids
pMC002 constitutively expresses prgB from the P23 promotor. For this construction, prgB was amplified using pCF10 as a template and gene-specific F/R primers with BamHI and SphI restriction sites respectively. The PCR product was digested using the specified restriction enzymes and introduced into similarly digested pDLP278p23 (Bhatty et al., 2017). The P23::PrgBΔ246-558 plasmid was constructed by inverse PCR to delete the adhesion domain of PrgB using the primers F: 5´-ATTTTCAATTATGGGAATCCAAAAGAACC-3´ and R: 5´-CTCTTTTTCCGCTTTGTTTTTTGCC-3´ and the pMC002 plasmid as template. For preparation of vectors for the biochemical and structural characterization of PrgB, the DNAs encoding for PrgB246-558 and PrgB188-1233 from Enterococcus faecalis pCF10 were cloned into the expression vector (pET28) by the Protein Science Facility at Karolinska Institutet/SciLifeLab (http://psf.ki.se). For sequence information of the DNAs used in the experiments, see Table S2.
Protein expression and purification for biophysical and structural characterization
Both PrgB truncations were expressed as N-terminal hexa-histidine tagged proteins in Escherichia coli BL21(DE3). The cells were grown at 37 °C in TB medium until they reached an OD600 of 1.5, at which time the temperature was lowered to 18 °C and protein expression was induced by addition of 0.5 mM IPTG. Cells were grown overnight before harvesting. Cells were disrupted using a Constant cell disruptor (Constant Systems) in 20 mM HEPES/NaOH (pH 7.0), 300 mM NaCl and 30 mM Imidazole. The lysate was clarified by centrifugation for 30 minutes at 30,000 × g. The proteins were purified at 4 °C on Ni-NTA-Sepharose (Macherey-Nagel). The column was washed with 10 column volumes (CV) of 20 mM HEPES/NaOH (pH 7.0), 300 mM NaCl, 30-50 mM imidazole and bound proteins were eluted from the column with wash buffer supplemented with 500 mM Imidazole. For PrgB246-558, the histidine affinity tag was cleaved by incubation with TEV protease (ratio 1:100) for 20 h at 4 °C. Further purification was achieved by a Superdex-200 Increase 10/300 GL gelfiltration column (GE Healthcare) in buffer containing 20 mM HEPES/NaOH (pH 7.0) and 150 mM NaCl. For PrgB188-1233 two peaks corresponding to the expected molecular weight of monomeric and dimeric protein were observed on the gel filtration, both peaks were handled separately in the following steps. Purified PrgB188-1233 and PrgB246-558 were concentrated using an Amicon Ultra Centrifugal Filter (Merck-Millipore).
Expression of selenomethionine derivatized PrgB246-558 was performed in Escherichia coli BL21(DE3) grown in M9 minimal medium supplemented with 50 mg/L L-Selenomethionine by inhibition of the methionine biosynthesis pathway as described earlier (Van Duyne et al., 1993). The purification was performed as described for native PrgB246-558 with the exception of adding 1 mM TCEP (tris(2-carboxyethyl)phosphine) to all buffers after affinity purification.
Gas-phase electrophoretic mobility macromolecule analysis (GEMMA)
GEMMA analysis was performed as previously described (Rofougaran et al., 2008). Briefly, the two peaks of purified PrgB188-1233 from size exclusion chromatography were analysed by GEMMA. Prior to GEMMA analysis, the protein samples were diluted to a concentration of 0.05–0.1 mg/ml in a buffer containing 20 mM ammonium acetate, pH 7.8. If needed to increase the signal-to-noise ratio, the samples were scanned several times (3–6 times).
Crystallization and structure determination
Crystals of native as well as of selenomethionine derivatized PrgB246-558 were grown at 20 °C by sitting drop vapor diffusion in a condition containing 0.12 M ethylene glycol, 0.1 M Bicine/Tris pH 8.5, 20% (v/v) glycerol, 10 % (w/v) PEG 4000 with a protein concentration of 30 mg/ml and a protein to reservoir ratio of 1:1 in the drop. For co-crystallization of PrgB246-558 with DNA, the protein solution was mixed with a 2-fold molar excess of 10nt double-stranded DNA prior to crystallization, and crystals appeared in the same condition as apo PrgB246-558. Crystals were flash cooled in liquid nitrogen.
X-ray diffraction data of native and selenomethionine derivatized crystals of apo PrgB246-558 was collected on BL14.1 operated by the Helmholtz-Zentrum Berlin (HZB) at the BESSY II electron storage ring (Berlin-Adlershof, Germany) (Mueller et al., 2015). Diffraction data of PrgB246-558 co-crystallized with DNA was recorded at beamline ID30A-3 at the European Synchrotron Radiation Facility (ESRF) (Grenoble, France). The data was processed using XDS and XSCALE, respectively (Kabsch, 2010). The PrgB246-558 crystals belong to the monoclinic space group P43212 and contain one molecule in the asymmetric unit. The crystallographic phase problem was solved by means of single-wavelength anomalous dispersion (SAD), the methionine sites were found and refined by the Auto-Rickshaw pipeline and an initial model was built by ARP/wARP (Panjikar et al., 2005; Langer et al., 2008). The crystals of PrgB246-558 with DNA belong to the orthorhombic space group P212121 with one molecule in the asymmetric unit. The structures of the high resolution native data, as well as that of the co-crystallized protein, were solved by molecular replacement with PHASER using the initial PrgB structure as a search model. Building of the models were conducted in COOT (Emsley et al., 2010). The structures were both refined at 1.6 and 1.8 Å resolutions, respectively, and with crystallographic R and Rfree values of 19.0/22.7 and 17.4/21.2, respectively, using Refmac5 and PHENIX refine (Winn et al., 2001; Adams et al., 2002). Although the electron density for the bound DNA was ambiguous in places, it is clear that there was DNA bound. Rwork/Rfree decreased by 1.0/1.6% when including the DNA in the refinement. For complete data collection and refinement statistics, see Table 1. The final PrgB246-558 model consists of residues 246 – 558, and was validated using MolProbity (Chen et al., 2010). Atomic coordinates and structure factors of the PrgB246-558 crystal structures, apo and co-crystallised with DNA, have been deposited in the Protein Data Bank (PDB id 6evu and 6ged, respectively).
Electrophoretic mobility shift assay (EMSA)
For Lipoteichoic acid (LTA) competition experiments, 120 μM mono- as well as dimeric PrgB was mixed with 175 μM oriT DNA and LTA from Enterococcus hirae (Sigma) in varying concentrations from 0 to 8 mg/ml. DNA interaction studies were performed by mixing 50 nM of 100bp or 250bp DNA with PrgB188-1233 in varying concentrations between 0.1 and 20 μM. All reactions were performed in a buffer solution containing 20 mM HEPES-NaOH pH 7.0, 150 mM NaCl. The reaction mixtures were incubated for one hour at RT and subsequently run on a 10% PAGE at 4°C. Detection of the nucleic acids was achieved by staining with GelRed and the gel was imaged in a Chemidoc Touch Imaging System (Biorad). The bands corresponding to the unbound DNA were quantified in ImageLab (Biorad) and the curves from the resulting values were fit to a sigmoidal dose-response equation using SigmaPlot (Systat Software).
Isothermal Titration Calorimetry (ITC)
The ITC experiments were performed on a MicroCal iTC200 (Malvern). For the DNA binding experiments, 1 mM DNA solution was titrated into 0.05 mM PrgB188-1233 or PrgB246-558. The dsDNA used had the sequence GGGGGCGGGGCGGGCGGCG). For interaction studies with glycerol phosphate (GPO4), a 5 mM GPO4 solution was titrated into 0.05 mM PrgB246-558. All solutions were prepared with identical buffer solution; 20 mM HEPES-NaOH pH 7.0, 150 mM NaCl. The injection sequence consisted of an initial injection of 0.4 μl to prevent artifacts arising from the filling of the syringe (not used in data fitting), followed by injections of 2 μl aliquots with a 270 s interval between each injection. To correct for heats of dilution and mixing, blank titrations of DNA or GPO4 into buffer were subtracted from the DNA- or GPO4-protein titration. The resulting titration curves were fit to a one-site model using the Origin ITC software package supplied by Malvern to numerically obtain the apparent association constant (K), the number of binding sites (n) and binding enthalpy ΔH.
Surface plasmon resonance (SPR)
The interaction study between PrgB188-1233 and DNA was performed using a BIAcore® 3000 biosensor (GE Healthcare®, Uppsala, Sweden) equipped with a CM5 sensor chip (GE Healthcare®). First, Streptavidin was immobilized on the dextran layer of the chip using standard amino coupling reagents at pH 4.0. In a second step, biotinylated DNA-1 and DNA-2 was coupled to the Streptavidin coating at a density of 3500 response units (RUs). For the measurements, PrgB was flushed over the chip surface in concentrations varying between 0.025 and 5 μM. All SPR experiments were performed at 25 °C at a flow rate of 20 μL·min−1 in 20 mM HEPES buffer (pH 7.0) and in the presence of 150 mm NaCl. All sensograms were corrected for nonspecific interactions using a reference surface according to standard procedures (Myszka, 1999).
Nanofluidics
The single DNA molecule experiments were performed in nanochannels with a width of 800 nm and a depth of 150 nm. The devices were fabricated using advanced nanofabrication described elsewhere (Persson and Tegenfeldt, 2010). The channel system consists of a pair of feeding channels (micro-size), spanned by a set of parallel nanochannels. The sample is loaded into the channel system from one of the four reservoirs that are connected to the feeding channels, and moved into the nanochannels by flow. The flow is induced by applying nitrogen pressure onto the reservoirs. To avoid non-specific binding of protein to the negatively charged channel walls, the channels were precoated with a lipid bilayer comprising 99% 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC, Avanti) and 1% N-(fluorescein-5-thiocarbamoyl)-1,2-dihexadecanoyl-sn-Glycero-3-phosphoethanolamine, triethylammonium salt (fluorescein-DHPE, Invitrogen). The coating procedure is described elsewhere (Persson et al., 2012). DNA from phage lambda ( -DNA, Roche) was prestained with YOYO-1 (YOYO, Invitrogen) at a ratio of 1 dye molecule per 25 basepairs. Pre-stained DNA was then mixed with the PrgB protein (dimeric PrgB188-1233), incubated at 4°C for 12 h and then introduced into the nanofluidic system and equilibrated for 60s before image capture. The DNA concentration was 5 M (basepairs) in all samples and 3% (v/v) -mercaptoethanol (Sigma-Aldrich) was used as oxygen scavenger to suppress oxygen radical induced photodamage of the DNA. The buffer is 30mM NaCl with 20mM Hepes-NaOH (pH 7). The DNA and DNA–protein complexes were imaged using an epifluorescence microscope (Zeiss AxioObserver.Z1) equipped with a Hamamatsu digital CMOS C11440-22CU camera, a 63× oil immersion TIRF objective (NA = 1.46) and a 1.6× optovar from Zeiss. Using the microscopy imaging software ZEN, 50 subsequent images were recorded with an exposure time of 200 ms. Data analysis was performed using a custom-written MATLAB-based software. The microscopy image stacks were used as input to the software and converted into kymographs (time traces). Sizes were extracted from the kymographs as the length of each row and averaged for each molecule.
PrgB protein detection by immunoblotting
Exponential-phase cultures (10 ml) of E. faecalis OG1RF strains carrying and pCF10 or pCF10 variants normalized to an OD600 of 0.3 were induced with 10 ng ml−1 of peptide cCF10 for 2 h at 37 °C. The cells were pelleted by centrifugation at 13,200 × g for 15 min at 4 °C and washed once with cold 1X physiological buffer saline (PBS). The pellet was resuspended in 125 μl of SMM buffer (0.5M sucrose, 0.02M MgCl2, 0.02M maleate, pH 6.5) containing 60 μl ml−1 of mutanolysin (Sigma-Aldrich) and 10 mg ml−1 of lysozyme (Sigma-Aldrich), and the resulting mix was incubated for 1 h at 37 °C with shaking. Material released from the digested cell wall was separated from cell-bound material by centrifugation at 13.200 × g for 15 min at 4 °C. Equal amounts of the cell-wall (supernatant) fractions were electrophoresed through sodium dodecyl sulfate (SDS)-polyacrylamide gels, and the surface protein PrgB was detected by Western transfer and immunostaining with the anti-PrgB antibodies (Christie et al., 1988; Chen et al., 2007; Chen et al., 2008).
Aggregation assays
The aggregation assay was performed as previously described (Bhatty et al., 2015). Overnight cultures of E. faecalis strains were diluted 1:10 in BHI and grown at 37°C for 1 h. This culture was further diluted 1:100 with BHI containing 10 ng ml−1 of peptide cCF10; for LTA competition aggregation assays, 6 mg/ml of Lipoteichoic acid (LTA) from E. hirae (Sigma) was added to one set of cultures. The effect of LTA treatment on PrgB-induced aggregation was monitored by visual inspection after 24 h.
Crystal violet biofilm formation assay
A biofilm formation assay was performed in a 96-well polystyrene flat-bottomed microtiter plate (Greiner Bio-One), as previously described (Bhatty et al., 2015) with modifications. Briefly, overnight cultures were diluted 1:100 in 200 μl of M9-YEG (Kristich et al., 2007) containing 10 ng/ml of cCF10 with or without 6 mg/ml of LTA and incubated for 24 h at 37 °C under static conditions. At least six wells were inoculated per strains. To quantify biofilm formation, the wells were washed 3 times with 1 X PBS to remove the non-adherent cells. The plates were dried at room temperature (RT) for 3 h and then stained with 0.1% crystal violet (CV) for 15 min at RT. The wells were washed three times with 1 X PBS to remove unbound CV. Ethanol was used to solubilize CV and absorbances were measured at 550 nm. The results were the averages of at least twelve replicate wells.
Conjugation assays
E. faecalis donor and recipient cultures grown overnight were diluted 1:10 in BHI and incubated for 1 h at 37°C without shaking. Donor and recipient cells were mixed in a ratio of 1:10 and allowed to mate in liquid without shaking for 3 h at 37°C. To evaluate the effect of LTA (Sigma) on conjugative transfer, 6 mg/ml of LTA was added at the onset of mating. Mating mixtures were serially diluted in BHI, and the numbers of donors and transconjugants were obtained by plating on selective BHI agar plates. The plasmid transfer frequencies were calculated as the number of transconjugants per donor cell (Chen et al., 2008). The results are reported as an average of three replicates of each experiment.
Supplementary Material
Acknowledgments
The authors thank Prof. Gary Dunny for fruitful discussions and critical reading of the manuscript. We thank the beam-line scientists at BESSY, Berlin and ESRF, Grenoble for their support in data collection. The group of Assoc. Prof. Tobias Ambjörnsson at Lund University is acknowledged for developing the data analysis software for extracting lengths of DNA-protein complexes in the nanochannel experiments. Dr. Sriram KK is acknowledged for fabricating the nanofluidic devices. This work was supported by grants from the Swedish Research Council (2016-03599) and Kempestiftelserna (JCK-1524) to R.P-A.B., and the Swedish Research Council (2015-5062) and Olle Engkvist Byggmästare Foundation to F.W. Work in the Christie laboratory was supported by the NIH grants R01GM48476 and R21AI105454. We thank the Protein Science Facility at Karolinska Institutet/SciLifeLab (http://psf.ki.se) for their help in cloning.
Footnotes
Author contributions
A.S. and R.P-A.B. performed protein purification, structure determination, DNA and LTA binding assays. K.J. performed and analyzed the DNA condensation studies. M.I.C. performed in vivo functional assays. V.R.J. and A.H. performed and analyzed the GEMMA experiments. A.S., F.W., P.J.C. and R.P-A.B. planned the experiments, performed the data analysis and wrote the manuscript with input from all authors.
References
- Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, et al. PHENIX : building new software for automated crystallographic structure determination. Acta Crystallogr Sect D Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- Arias CA, Murray BE. The rise of the Enterococcus: beyond vancomycin resistance. Nat Rev Microbiol. 2012;10:266–78. doi: 10.1038/nrmicro2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias CA, Panesso D, Singh KV, Rice LB, Murray BE. Cotransfer of antibiotic resistance genes and a hylEfm-containing virulence plasmid in Enterococcus faecium. Antimicrob Agents Chemother. 2009;53:4240–6. doi: 10.1128/AAC.00242-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arutyunov D, Frost LS. F conjugation: Back to the beginning. Plasmid. 2013;70:18–32. doi: 10.1016/j.plasmid.2013.03.010. [DOI] [PubMed] [Google Scholar]
- Bacher G, Szymanski WW, Kaufman SL, Zöllner P, Blaas D, Allmaier G. Charge-reduced nano electrospray ionization combined with differential mobility analysis of peptides, proteins, glycoproteins, noncovalent protein complexes and viruses. J Mass Spectrom. 2001;36:1038–52. doi: 10.1002/jms.208. [DOI] [PubMed] [Google Scholar]
- Barnes AMT, Ballering KS, Leibman RS, Wells CL, Dunny GM. Enterococcus faecalis produces abundant extracellular structures containing DNA in the absence of cell lysis during early biofilm formation. MBio. 2012;3:e00193–12. doi: 10.1128/mBio.00193-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatty M, Camacho MI, Gonzalez-Rivera C, Frank KL, Dale JL, Manias DA, et al. PrgU: a suppressor of sex pheromone toxicity in Enterococcus faecalis. Mol Microbiol. 2017;103:398–412. doi: 10.1111/mmi.13563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatty M, Cruz MR, Frank KL, Laverde Gomez Ja, Andrade F, Garsin Da, et al. Enterococcus faecalis pCF10-encoded surface proteins PrgA, PrgB (aggregation substance) and PrgC contribute to plasmid transfer, biofilm formation and virulence. Mol Microbiol. 2015;95:660–677. doi: 10.1111/mmi.12893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady LJ, Maddocks SE, Larson MR, Forsgren N, Persson K, Deivanayagam CC, Jenkinson HF. The changing faces of Streptococcus antigen I/II polypeptide family adhesins. Mol Microbiol. 2010;77:276–86. doi: 10.1111/j.1365-2958.2010.07212.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchan DWA, Minneci F, Nugent TCO, Bryson K, Jones DT. Scalable web services for the PSIPRED Protein Analysis Workbench. Nucleic Acids Res. 2013;41:W349–57. doi: 10.1093/nar/gkt381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VB, Arendall WB, Headd JJ, Keedy Da, Immormino RM, Kapral GJ, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Staddon JH, Dunny GM. Specificity determinants of conjugative DNA processing in the Enterococcus faecalis plasmid pCF10 and the Lactococcus lactis plasmid pRS01. Mol Microbiol. 2007;63:1549–64. doi: 10.1111/j.1365-2958.2007.05610.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Zhang X, Manias D, Yeo HJ, Dunny GM, Christie PJ. Enterococcus faecalis PcfC, a Spatially Localized Substrate Receptor for Type IV Secretion of the pCF10 Transfer Intermediate. J Bacteriol. 2008;190:3632–3645. doi: 10.1128/JB.01999-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie PJ, Kao SM, Adsit JC, Dunny GM. Cloning and expression of genes encoding pheromone-inducible antigens of Enterococcus (Streptococcus) faecalis. J Bacteriol. 1988;170:5161–8. doi: 10.1128/jb.170.11.5161-5168.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang ON, Schlievert PM, Wells CL, Manias DA, Tripp TJ, Dunny GM. Multiple functional domains of Enterococcus faecalis aggregation substance Asc10 contribute to endocarditis virulence. Infect Immun. 2009;77:539–48. doi: 10.1128/IAI.01034-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke M, Maddera L, Harris RL, Silverman PM. F-pili dynamics by live-cell imaging. Proc Natl Acad Sci U S A. 2008;105:17978–81. doi: 10.1073/pnas.0806786105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das T, Sehar S, Koop L, Wong YK, Ahmed S, Siddiqui KS, Manefield M. Influence of calcium in extracellular DNA mediated bacterial aggregation and biofilm formation. PLoS One. 2014;9:e91935. doi: 10.1371/journal.pone.0091935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunny GM. Enterococcal sex pheromones: signaling, social behavior, and evolution. Annu Rev Genet. 2013;47:457–82. doi: 10.1146/annurev-genet-111212-133449. [DOI] [PubMed] [Google Scholar]
- Dunny GM, Berntsson RPA. Enterococcal sex pheromones. J Bacteriol. 2016;198:1556–1562. doi: 10.1128/JB.00128-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunny GM, Brown BL, Clewell DB. Induced cell aggregation and mating in Streptococcus faecalis: evidence for a bacterial sex pheromone. Proc Natl Acad Sci U S A. 1978;75:3479–83. doi: 10.1073/pnas.75.7.3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunny GM, Hancock LE, Shankar N. Enterococcal Biofilm Structure and Role in Colonization and Disease. 2014 [PubMed] [Google Scholar]
- Van Duyne GD, Standaert RF, Karplus PA, Schreiber SL, Clardy J. Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J Mol Biol. 1993;229:105–24. doi: 10.1006/jmbi.1993.1012. [DOI] [PubMed] [Google Scholar]
- Ehrenfeld EE, Kessler RE, Clewell DB. Identification of pheromone-induced surface proteins in Streptococcus faecalis and evidence of a role for lipoteichoic acid in formation of mating aggregates. J Bacteriol. 1986;168:6–12. doi: 10.1128/jb.168.1.6-12.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsgren N, Lamont RJ, Persson K. Crystal structure of the variable domain of the Streptococcus gordonii surface protein SspB. Protein Sci. 2009;18:1896–905. doi: 10.1002/pro.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frykholm K, Berntsson RPA, Claesson M, De Battice L, Odegrip R, Stenmark P, Westerlund F. DNA compaction by the bacteriophage protein Cox studied on the single DNA molecule level using nanofluidic channels. Nucleic Acids Res. 2016;44:7219–7227. doi: 10.1093/nar/gkw352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frykholm K, Nyberg LK, Westerlund F. Exploring DNA-protein interactions on the single DNA molecule level using nanofluidic tools. Integr Biol (Camb) 2017;9:650–661. doi: 10.1039/c7ib00085e. [DOI] [PubMed] [Google Scholar]
- Gilmore MS, Lebreton F, van Schaik W. Genomic transition of enterococci from gut commensals to leading causes of multidrug-resistant hospital infection in the antibiotic era. Curr Opin Microbiol. 2013;16:10–6. doi: 10.1016/j.mib.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidron AI, Edwards JR, Patel J, Horan TC, Sievert DM, Pollock DA, et al. NHSN annual update: antimicrobial-resistant pathogens associated with healthcare-associated infections: annual summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2006-2007. Infect Control Hosp Epidemiol. 2008;29:996–1011. doi: 10.1086/591861. [DOI] [PubMed] [Google Scholar]
- Holm L, Rosenström P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010;38:W545–9. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkinson HF, Lamont RJ. Streptococcal adhesion and colonization. Crit Rev Oral Biol Med. 1997;8:175–200. doi: 10.1177/10454411970080020601. [DOI] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66:125–32. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman SL, Skogen JW, Dorman FD, Zarrin F, Lewis KC. Macromolecule analysis based on electrophoretic mobility in air: globular proteins. Anal Chem. 1996;68:1895–904. doi: 10.1021/ac951128f. [DOI] [PubMed] [Google Scholar]
- Kristich CJ, Chandler JR, Dunny GM. Development of a host-genotype-independent counterselectable marker and a high-frequency conjugative delivery system and their use in genetic analysis of Enterococcus faecalis. Plasmid. 2007;57:131–44. doi: 10.1016/j.plasmid.2006.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer G, Cohen SX, Lamzin VS, Perrakis A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat Protoc. 2008;3:1171–9. doi: 10.1038/nprot.2008.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laverde Gomez JA, Hendrickx APA, Willems RJ, Top J, Sava I, Huebner J, et al. Intra- and interspecies genomic transfer of the Enterococcus faecalis pathogenicity island. PLoS One. 2011;6:e16720. doi: 10.1371/journal.pone.0016720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebreton F, van Schaik W, McGuire AM, Godfrey P, Griggs A, Mazumdar V, et al. Emergence of epidemic multidrug-resistant Enterococcus faecium from animal and commensal strains. MBio. 2013;4:1–10. doi: 10.1128/mBio.00534-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HH, Yang YR, Shen XC, Zhang ZL, Shen P, Xie ZX. Role of DNA in bacterial aggregation. Curr Microbiol. 2008;57:139–44. doi: 10.1007/s00284-008-9166-0. [DOI] [PubMed] [Google Scholar]
- Maddocks SE, Wright CJ, Nobbs AH, Brittan JL, Franklin L, Strömberg N, et al. Streptococcus pyogenes antigen I/II-family polypeptide AspA shows differential ligand-binding properties and mediates biofilm formation. Mol Microbiol. 2011;81:1034–49. doi: 10.1111/j.1365-2958.2011.07749.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller U, Förster R, Hellmig M, Huschmann FU, Kastner A, Malecki P, et al. The macromolecular crystallography beamlines at BESSY II of the Helmholtz-Zentrum Berlin: Current status and perspectives. Eur Phys J Plus. 2015;130 [Google Scholar]
- Myszka DG. Improving biosensor analysis. J Mol Recognit. 1999;12:279–84. doi: 10.1002/(SICI)1099-1352(199909/10)12:5<279::AID-JMR473>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Nobbs AH, Lamont RJ, Jenkinson HF. Streptococcus adherence and colonization. Microbiol Mol Biol Rev. 2009;73:407–50. doi: 10.1128/MMBR.00014-09. Table of Contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olmsted SB, Kao SM, van Putte LJ, Gallo JC, Dunny GM. Role of the pheromone-inducible surface protein Asc10 in mating aggregate formation and conjugal transfer of the Enterococcus faecalis plasmid pCF10. J Bacteriol. 1991;173:7665–72. doi: 10.1128/jb.173.23.7665-7672.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panjikar S, Parthasarathy V, Lamzin VS, Weiss MS, Tucker PA. Auto-rickshaw: an automated crystal structure determination platform as an efficient tool for the validation of an X-ray diffraction experiment. Acta Crystallogr D Biol Crystallogr. 2005;61:449–57. doi: 10.1107/S0907444905001307. [DOI] [PubMed] [Google Scholar]
- Paoletti C, Foglia G, Princivalli MS, Magi G, Guaglianone E, Donelli G, et al. Co-transfer of vanA and aggregation substance genes from Enterococcus faecalis isolates in intra- and interspecies matings. J Antimicrob Chemother. 2007;59:1005–9. doi: 10.1093/jac/dkm057. [DOI] [PubMed] [Google Scholar]
- Persson F, Fritzsche J, Mir KU, Modesti M, Westerlund F, Tegenfeldt JO. Lipid-based passivation in nanofluidics. Nano Lett. 2012;12:2260–5. doi: 10.1021/nl204535h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson F, Tegenfeldt JO. DNA in nanochannels-directly visualizing genomic information. Chem Soc Rev. 2010;39:985–999. doi: 10.1039/b912918a. [DOI] [PubMed] [Google Scholar]
- Rakita RM, Vanek NN, Jacques-Palaz K, Mee M, Mariscalco MM, Dunny GM, et al. Enterococcus faecalis bearing aggregation substance is resistant to killing by human neutrophils despite phagocytosis and neutrophil activation. Infect Immun. 1999;67:6067–75. doi: 10.1128/iai.67.11.6067-6075.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rofougaran R, Crona M, Vodnala M, Sjöberg BM, Hofer A. Oligomerization status directs overall activity regulation of the Escherichia coli class Ia ribonucleotide reductase. J Biol Chem. 2008;283:35310–8. doi: 10.1074/jbc.M806738200. [DOI] [PubMed] [Google Scholar]
- Schlievert PM, Gahr PJ, Assimacopoulos AP, Dinges MM, Stoehr JA, Harmala JW, et al. Aggregation and binding substances enhance pathogenicity in rabbit models of Enterococcus faecalis endocarditis. Infect Immun. 1998;66:218–23. doi: 10.1128/iai.66.1.218-223.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Süssmuth SD, Muscholl-Silberhorn A, Wirth R, Susa M, Marre R, Rozdzinski E. Aggregation substance promotes adherence, phagocytosis, and intracellular survival of Enterococcus faecalis within human macrophages and suppresses respiratory burst. Infect Immun. 2000;68:4900–6. doi: 10.1128/iai.68.9.4900-4906.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas VC, Thurlow LR, Boyle D, Hancock LE. Regulation of autolysis-dependent extracellular DNA release by Enterococcus faecalis extracellular proteases influences biofilm development. J Bacteriol. 2008;190:5690–8. doi: 10.1128/JB.00314-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troffer-Charlier N, Ogier J, Moras D, Cavarelli J. Crystal structure of the V-region of Streptococcus mutans antigen I/II at 2.4 A resolution suggests a sugar preformed binding site. J Mol Biol. 2002;318:179–88. doi: 10.1016/S0022-2836(02)00025-6. [DOI] [PubMed] [Google Scholar]
- Trotter KM, Dunny GM. Mutants of Enterococcus faecalis deficient as recipients in mating with donors carrying pheromone-inducible plasmids. Plasmid. 1990;24:57–67. doi: 10.1016/0147-619x(90)90025-8. [DOI] [PubMed] [Google Scholar]
- Turnbull WB, Daranas AH. On the value of c: can low affinity systems be studied by isothermal titration calorimetry? J Am Chem Soc. 2003;125:14859–66. doi: 10.1021/ja036166s. [DOI] [PubMed] [Google Scholar]
- Waters CM, Hirt H, McCormick JK, Schlievert PM, Wells CL, Dunny GM. An amino-terminal domain of Enterococcus faecalis aggregation substance is required for aggregation, bacterial internalization by epithelial cells and binding to lipoteichoic acid. Mol Microbiol. 2004;52:1159–71. doi: 10.1111/j.1365-2958.2004.04045.x. [DOI] [PubMed] [Google Scholar]
- Waters CM, Wells CL, Dunny GM. The aggregation domain of aggregation substance, not the RGD motifs, is critical for efficient internalization by HT-29 enterocytes. Infect Immun. 2003;71:5682–9. doi: 10.1128/IAI.71.10.5682-5689.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn MD, Isupov MN, Murshudov GN. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr D Biol Crystallogr. 2001;57:122–33. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.