

Abstract
The obesity epidemic is a leading cause of health problems in the United States, increasing the risk of cardiovascular, endocrine, and psychiatric diseases. Though many people lose weight through changes in diet and lifestyle, keeping the weight off remains a challenge. Here, we discuss a hypothesis that seeks to explain why obesity is so persistent. There is a great degree of overlap in the circuits implicated in substance use disorder and obesity, and neural plasticity of these circuits in response to drugs of abuse is well documented. We hypothesize that obesity is also associated with neural plasticity in these circuits, and this may underlie persistent changes in behavior, energy balance, and body weight. Here, we discuss how obesity-associated reductions in motivation and physical activity may be rooted in neurophysiological alterations in these circuits. Such plasticity may alter how humans and animals use, expend, and store energy, even after weight loss.
Keywords: obesity, plasticity, synaptic, glutamate, high-fat diet
Graphical abstract
The obesity epidemic is a leading cause of health problems in the United States, increasing the risk of cardiovascular, endocrine, and psychiatric diseases. Here, we discuss how obesity-associated reductions in motivation and physical activity may be rooted in neurophysiological alterations in these circuits. Such plasticity may alter how humans and animals use, expend, and store energy, even after weight loss.
Introduction
Despite widespread knowledge of the health risks of obesity, there has been little progress in reversing obesity in America. The obesity rate in the United States has increased approximately 30% since 1980, including a rise of about 10% since obesity was declared a national epidemic in 1999.1 Studies show that obesity is a leading cause of death in the United States, second only to tobacco-related deaths.2,3 Although dozens of weight loss theories and solutions have been put forward, effective approaches for combatting obesity still elude most Americans. A common perception is that reducing exposure to factors that contribute to weight gain will lead to weight loss. However, evidence suggests that this premise is flawed. While reducing caloric intake often results in immediate weight loss, it is difficult to maintain over the long term.4 This also occurs in animals. For example, it has been known since the 1980s that obese rodents maintain a higher average body weight than those that were never obese, even when maintained on identical diets.5,6 Similar associations are seen in humans, supporting the concept of obesity changing a body weight “set point.”7 Here, we propose that obesity engages persistent changes in neural circuits that cause animals to defend an elevated body weight, even after weight loss.
The biological mechanisms that underlie such a shift in set point remain unclear and likely involve more than one system. Hypothalamic pro-opiomelanocortin (POMC)-expressing neurons in the arcuate nucleus control energy homeostasis and food intake and may contribute to the determination of body-weight set point.8 Basal metabolic rates can slow after weight loss, a phenomenon known as metabolic adaptation.9 Behavioral changes, such as reductions in physical activity, can persist following weight loss,10 contributing to a reduction in energy expenditure via physical activity. At the same time, craving for high-fat foods can increase after weight loss in animals11, which may engage similar mechanisms in people to drive weight re-gain.12 To the extent that the mechanisms underlying these changes persist, they may contribute to the persistence of obesity. Here, we discuss the hypothesis that altered plasticity in basal ganglia circuitry may underlie persistent changes in energy balance and weight.13,14 We believe that identifying and reversing such plasticity will be necessary to achieve long-term weight loss and meaningful progress in reducing obesity rates.
Common circuits, behaviors, and underlying mechanisms between obesity and addictive disorders
Despite differences, obesity shares some similarities with substance use disorder. Obesity is associated with difficulty in controlling food intake, repeated desires and cravings for food, unsuccessful attempts to curb excessive eating, and continued overeating despite negative consequences.15,16 Attempts to lose weight through dieting can also be reminiscent of attempts to quit drugs of abuse; both share a pattern wherein individuals abstain from the substance for a period of time followed by relapse.17 And while there is disagreement over whether obesity should be classified as an addictive state,18 there are common neural features of both substance use disorders and obesity.19 Consumption of high-fat food activates the mesolimbic dopaminergic system,20,21 the same circuit that is activated by drugs of abuse.22,23 Animals exposed to palatable foods can also become resistant to outcome devaluation, suggesting that palatable foods can facilitate habitual behaviors.24–27 Here, we seek to outline a framework of circuit-based changes in substance use disorders and discuss how similar circuit adaptations may underlie the persistent behavioral changes seen in obesity.
Glutamatergic plasticity in substance use disorders
Substance abuse disorders have been linked to synaptic plasticity in the striatum, a subcortical brain region that serves as the primary input nucleus of the basal ganglia. Medium spiny neurons (MSNs) are the principal striatal cells, accounting for over 95% of rodent striatal neurons.28,29 The striatum receives excitatory projections from the cortex, thalamus, and amygdala, as well as dopaminergic innervation from the midbrain.30,31 The dorsal striatum is involved in regulation of motor functions, decision making, and the development of habits.32,33 In contrast, the ventral striatum contains the nucleus accumbens (NAc) and mediates goal-directed reward and motivation, particularly under conditions that require the animal to exert effort.34–36
At the cellular level, connections between brain regions are mediated by billions of synapses, and plasticity in these synapses regulates the flow of information. Drugs of abuse alter synaptic plasticity in mesolimbic nuclei, including the NAc and ventral tegmental area (VTA),37 and dopaminergic and glutamatergic changes in the VTA underlie behavioral adaptations in response to drugs of abuse. Cocaine modifies VTA plasticity––a single in vivo cocaine exposure is sufficient to increase α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)/N-methyl-D-aspartic acid (NMDA) ratios at VTA synapses, and synapses in the VTA of animals treated repeatedly with cocaine appear to be potentiated to the point of saturation38 or exhibit aberrant susceptibility to long-term potentiation (LTP).39 Additional work elucidated a form of cocaine-induced plasticity dependent on a distinct mechanism that appeared to involve the insertion of calcium-permeable, GluA2-lacking AMPA receptors.40 Such cocaine-evoked plasticity in the VTA is gated by metabotropic glutamate receptors, which also control plasticity in the NAc.41 Together, this shows that synaptic plasticity in the mesolimbic circuits can be altered by acute and repeated cocaine exposure.
Interestingly, these plasticity mechanisms may interact with stress. This may be relevant to the development of substance use disorder, given the strong associations between stress and drug use in both humans and animals.42–44 Accumbal MSNs in mice susceptible to stress-induced depressive-like phenotypes incorporated more synaptic calcium-permeable, glutamate ionotropic receptor AMPA type subunit 2 (GluA2)-lacking AMPARs relative to MSNs from control or resilient mice.45,46 This suggests that related plasticity mechanisms in mesolimbic circuitry may underlie stress susceptibility and drug abuse. However, recent work examining the effects of cocaine versus chronic social defeat on evoked currents at established mushroom spines in the NAc suggests that shared mechanisms may be nuanced and cell type or stress-paradigm dependent; a week-long cocaine exposure and a chronic social-defeat paradigm produced opposite changes in corticostriatal plasticity.47
The effects of cocaine on synaptic plasticity can be long lasting. Exposure to and withdrawal from cocaine increased accumbal expression of functionally silent synapses and promoted silent-to-functional synaptic conversion involving insertion of calcium-permeable AMPARs, respectively.48,49 Silent synapses are important for metaplastic priming of brain regions, including the NAc, rendering them susceptible to more permanent changes in plasticity.48,50,51 Cocaine has been shown to increase extracellular glutamate in the NAc52,53 and potentiate accumbal glutamatergic synapses.54 Surprisingly, in light of these synaptic changes that predict enhancements in glutamatergic synapses in the NAc, in vivo firing rates of NAc neurons are depressed following long-term cocaine exposure.55,56 This indicates that changes in excitatory transmission may not translate directly into enhancements in accumbal firing, but may alter how accumbal neurons translate glutamatergic input into spiking. Intriguingly, reversing glutamatergic plasticity onto NAc neurons can reverse locomotor sensitization to cocaine, suggesting that this plasticity is a necessary feature of this behavioral change.57,58 Thus, the modification of glutamatergic corticostriatal synapses by drugs of abuse can alter both striatal function and drug-adapted changes in behavior.
Cocaine can also alter LTP in the hippocampus, one of the glutamatergic inputs to the NAc. Self-administration of cocaine in rats enhanced in vitro hippocampal LTP in cornu ammonis area 1 (CA1),59 which was sustained across 1 week of withdrawal.59,60 In contrast, LTP was suppressed in CA1 following 3 months of withdrawal.60 Enhanced synaptic transmission (as measured by input–output curves in CA1) was seen in rats 3–5 weeks following abstinence from cocaine self-administration.61 This suggests that cocaine alters hippocampal synaptic plasticity and that these changes can persist over long periods of time, although the specifics of these synaptic changes may depend on the dose and time course of cocaine exposure.
Finally, cocaine and other drugs of abuse are associated with dysfunction of frontal brain regions,62 which also send projections to the dorsal striatum and NAc. Prefrontal activity in chronic cocaine users is reduced during withdrawal, leading to the hypofrontality hypothesis of substance use disorders.63 Men with alcohol use disorder exhibited lower prefrontal cortical activity (measured by functional magnetic resonance imaging (fMRI)) during a visuospatial task relative to healthy controls.64 Furthermore, hypofrontality may persist after drug use ceases––relative glucose metabolism in frontal brain regions, as measured by positron emission tomography (PET) with [18F]-fluorodeoxyglucose, was lower in cocaine abusers 3–4 months after drug use than in healthy controls.65 Similar effects were seen in people with alcohol use disorder over a week after alcohol withdrawal.65 Together with alterations in corticostriatal plasticity, a persistent decrease in frontal activity may contribute to long-lasting behavioral changes in people with substance use disorder.66 Specifically, these include cognitive changes and impulsivity that can persist after cessation of drug use.67,68
Glutamatergic plasticity in obesity
Compared with the vast literature linking drugs of abuse to corticostriatal plasticity, relatively little is known about how these synapses change in obesity. Despite a relatively small number of studies, high-fat diet (HFD) exposure has been linked to disruptions in synaptic plasticity in the NAc (Fig. 1A). An in vitro study found that MSNs from obesity-prone rats fed HFD exhibited a mild synaptic potentiation in response to a stimulation protocol that normally would induce a synaptic depression, while MSNs from obesity-resistant rats fed HFD exhibited only a modest depression.69 This suggests that exposure to a diet with higher fat content can alter NAc plasticity.69 Accumbal MSNs from obesity-prone rats fed a junk food diet also exhibited an increased prevalence of calcium-permeable AMPA receptors in the membrane,70 similar to what is seen in the NAc after exposure to cocaine.
Figure 1.
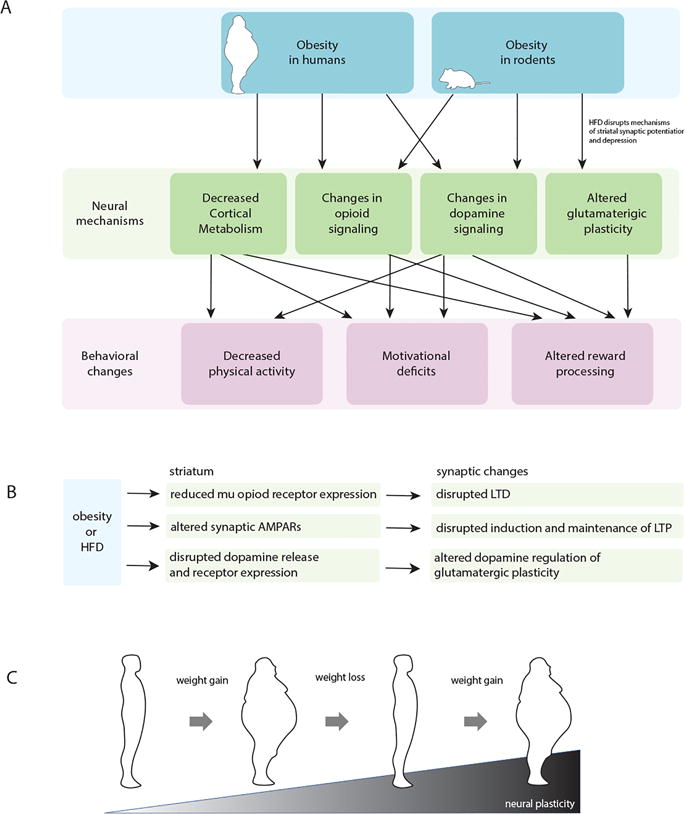
(A) Schematic showing how obesity induces neural mechanisms that lead to behavioral changes in both humans and rodents. (B) Flowchart of proposed biomolecular changes in the striatum during obesity or after HFD that lead to altered synaptic function. (C) Cartoon illustrating the cyclic nature of obesity. Obese states lead to changes in synaptic plasticity that can persist despite weight loss. This contributes to physiological and behavioral factors contributing to relapse and ultimately the cyclic nature of weight gain and loss.
Electrophysiological studies that suggest HFD affects synaptic plasticity in the NAc are bolstered by investigations of biomolecular changes in response to HFD. For instance, brain-derived neurotrophic factor (BDNF) plays an important role in structural synaptic changes72 and susceptibility to compulsive drug seeking.73,74 Consumption of HFD increased BDNF in NAc tissue lysates as measured via western blotting.11 This suggests that biochemical aspects of reward signaling pathways are also altered after HFD consumption. Rats fed saturated fat had increased protein levels of phosphorylated accumbal AMPA receptors, as measured by western blot, relative to rats fed a diet rich in monounsaturated fat or low-fat controls.75 The insertion and phosphorylation of calcium-permeable AMPARs are thought to contribute to the induction of LTP,76,77 further suggesting that HFD can engage aberrant plasticity mechanisms in the NAc (Fig. 1C). Interestingly, the rats in this study did not become obese on these diets, demonstrating that some effects of HFD can occur independent of obesity. These studies demonstrate that NAc adaptations can occur independent of weight gain.
There is also evidence of alterations in hippocampal plasticity following HFD exposure. In vivo78 and slice79 electrophysiological recordings from rodents show that high-frequency stimulation (HFS) of perforant pathway induces significantly lower field excitatory postsynaptic potentials (EPSPs) in dentate gyrus granular cells in overweight animals fed HFD relative to controls. Recordings in CA1 in response to Schaeffer collateral stimulation showed similar disruptions; field EPSP slopes from overweight mice fed HFD were decreased relative to controls in response to Schaeffer collateral theta burst stimulation80 or HFS.81 However, these effects may be transient; one study that showed decreased hippocampal LTP in obese mice fed HFD saw that this phenotype was reversed after switching to a diet lower in fat.79 In addition, it is difficult to reconcile why experiments with cocaine reported an enhanced ability to induce synaptic plasticity in the hippocampus,59–61 while studies with HFD found the opposite.
Finally, obesity has been linked to alterations in prefrontal cortical (PFC) activity. In general, there appears to be a reduction of activity in frontal cortical areas in people with obesity, consistent with the hypofrontality hypothesis of substance use disorders.63,82 In humans, PET scans showed women with obesity had lower cerebral blood flow in dorsolateral PFC in response to a meal (following a 36-h fast), compared with lean or formerly obese women.83 This suggests that obesity may blunt frontal PFC activity in response to salient food stimuli. In the absence of a food stimulus, PET and single-photon emission-computed tomography (sPECT) scans revealed an inverse relationship between glucose metabolism and body mass index (BMI)84 or cerebral blood flow and BMI85 in prefrontal cortical areas, respectively. However, these studies were conducted with relatively few subjects (n = 8–12 people per lean, obese, or formerly obese group,83, n = 21 subjects,84 n = 36 subjects total85) or only found differences in PFC activation in females.83 Furthermore, other imaging studies in humans with obesity have reported increased prefrontal cortical activity relative to controls after consuming a satiating meal86 or in response to images of highly satiating food.87 PFC activity in children or adolescents with obesity has also been inconsistent. One fMRI study reported decreased prefrontal cortical blood flow in response to consumption of a glucose-rich beverage in adolescents with obesity relative to lean adolescent subjects.88 Another reported higher PFC activation in response to pre-meal pictures of food in adolescents with obesity relative to controls.89 Together, these studies suggest alterations in PFC metabolism in obesity, but it may be heterogeneous and not present in all individuals.
Dopamergic changes in substance use disorders
Dopaminergic circuits encode or incentivize reward or reward-learning,90 and a rich body of literature has shown that substance abuse alters dopaminergic neurotransmission.91–94 Exposure to drugs of abuse, including morphine, ethanol, cocaine, or methamphetamine induces morphological,95–98 electrophysiological,38,99 biomolecular,100,101 and plasticity98,102–104 changes in midbrain VTA dopaminergic neurons and their projections to the ventral striatum.
Drugs of abuse have many effects on the dopaminergic system and the accumbens.105–107 Cocaine and other drugs of abuse increase extracellular dopamine in the NAc,108–111 which is central addictive behavior in animal models.109,112 Cocaine inhibits accumbal dopamine reuptake, the mechanism by which it increases extracellular dopamine levels.22,113 Cocaine withdrawal is associated with decreased dopamine receptor availability.82 A seminal PET study showed decreased striatal dopamine receptor type 2 (D2R) availability in cocaine abusers after 1 week of detoxification,114 which returned to baseline after a month. A larger cohort of cocaine users showed that decreased D2R binding persisted as long as 4 months.115 Decreased D2R availability was also observed in individuals with alcohol use disorder.116 Animal work largely supports these human imaging studies. After cocaine self-administration, rodents displayed decreased D2R messenger ribonucleic acid (mRNA) levels, as measured by in situ hybridization117 and decreased D2R protein in the NAc.118 Similar results were observed via autoradiography in monkeys.119 Together, there is evidence across multiple species that cocaine use reduces the availability of striatal D2Rs.
In addition to the acute effects of dopamine on circuit function, dopamine regulates plasticity mechanisms of glutamatergic synapses.120 Therefore, these drug-induced changes in dopamine transmission can affect subsequent glutamatergic plasticity. Some of the earliest work studying the effects of cocaine on midbrain synapses showed that a single in vivo exposure of cocaine was sufficient to alter AMPA channels at excitatory synapses in the VTA.38 Furthermore, cocaine increases dopaminergic terminal density and striatal spinogenesis,121,122 and cocaine withdrawal increases surface-level AMPA receptors in the ventral striatum.123 Related changes in dopamine transmission have also been reported following obesity and HFD exposure.
Dopaminergic changes in obesity and following HFD
It is less clear how dopamine neurotransmission is disrupted in humans with obesity. The DRD2 Taq1A allele has been linked to reduced D2R availability in humans and increased likelihood of obesity.124–126 Consistent with these findings, early evidence linked obesity to decreased D2 receptor availability.127–129 Wang et al.27 explored the striatum using PET scans for [C-11]raclopride. Later, it was reported that individuals with severe obesity (average BMI of 51.5) also exhibited lower D2R binding in prefrontal cortical regions.128 Additionally, when D2R binding was examined in 33 subjects (BMIs ranging from 19 to 35) weak negative correlations between D2R and BMI were seen in the left caudate nucleus, right ventral striatum, and amygdala.129 This suggested that the severity of decreased D2R may depend on the degree of obesity. A reduction in striatal D2R availability was also reported in a sample of women with obesity, relative to controls,130,131 yet here without evidence of negative correlations between BMI and D2R availability. However, more recently, this point has been disputed. While Guo et al.132 reported a negative correlation between D2R binding and BMI in the ventromedial striatum, they found significant positive correlations between BMI and D2R binding in other striatal areas. This is supported by several other studies reporting positive or nonexistent correlations between BMI and striatal D2R availability.133–136 PET scans measuring D2R binding with [18F]fallypride in women showed a positive correlation between BMI and D2R binding in the caudate.133 Similarly, combined D2R and D3R binding was positively correlated with BMI in the right dorsal caudate of 12 individuals as measured by PET scans with [11C]propyl-hexahydro-naphtho-oxazin ([11C]PHNO).134 D2R binding as measured by [11C]n-methyl-benperidol ([11C]NMB) PET scans did not correlate with the BMI of 30 individuals,135 nor did D2R binding seen in the PET scans of 27 women using the radioligand [11C]raclopride.136 Together, this indicates that the link between D2Rs and obesity is complex and may occur differently across varying striatal compartments or different subject populations. Of interest is the idea that the relationship between D2Rs and obesity may be nonlinear, such that reductions in D2R are only seen in people with severe obesity.137 Alternatively, the relationship may depend on subsets of people with obesity, such as people with different levels of emotional138 or opportunistic eating.132 These subsets may not have been equally represented across studies.
Other changes in dopamine neurotransmission have been linked to obesity in humans. Postmortem studies of human striatum revealed an inverse correlation between BMI and dopamine transporter (DAT) binding, as well as decreased DAT and tyrosine hydroxylase (TH) mRNA in the substantia nigra of people with obesity.139 Another study of 50 subjects supported this point, reporting highly significant negative correlations between striatal DAT binding and BMI.140 However, these findings have been contradicted by others, who showed no correlation between BMI and striatal DAT binding.141–143 Thus, while the specifics of dopamine receptor availability and dopamine transporter surface expression differs between studies, a large body of evidence supports the conclusion that dopaminergic circuits are altered in humans with obesity (Fig. 1).
Experiments conducted using animal models also suggest that diet-induced obesity alters striatal dopamine signaling.14 Compared to the complexity seen in the human literature, animal studies have consistently reported decreases in dopamine D2Rs in obesity. For instance, D2R binding144–147 and expression25 were reduced in obese rodents fed HFD144,147 and other rodent models of obesity.145,146 Adolescent rats maintained on a high-sugar diet also had lower dopamine receptor type 1 (D1R) and D2R expression in the NAc.148 Dopaminergic receptor gene expression levels (D1R, D2R) were reported to be decreased in the VTA, NAc, and prefrontal cortex of overweight mice fed HFD from adolescence to adulthood,149 an effect that recovered in VTA and prefrontal cortex, but not NAc, following 4 weeks of withdrawal from HFD.149 Further work that examined D2R binding at an earlier time point (3 weeks after HFD) showed an increase in D2R striatal binding in overweight mice fed HFD,150 suggesting a time-dependent effect of HFD on dopamine receptor expression. Protein homogenates of the NAc of rats fed a diet high in saturated fat for 8 weeks during adulthood showed increased D1R levels via western blot quantification, despite no difference in weight gain relative to controls,75 suggesting that D1R translation may be differentially regulated depending on specific fat components of the diet, the age of exposure to HFD, or both. Intriguingly, some changes in ventral striatal dopamine transmission appear to occur even in the absence of weight gain. For example, in the absence of weight gain, rats exhibited decreased D2R protein levels and dopamine reuptake in the ventral striatum after HFD.151,152 This indicates that these changes may not be secondary to the increased storage of fat. Furthermore, this highlights an important issue in the literature, namely that studies rarely parse the effect of HFD exposure versus. the effects from accumulated fat and changes in circulating factors. This would require studies to pair-feed control groups limited amounts of HFD, such that they receive all of their calories from HFD but do not gain weight.
Reports of extracellular dopamine concentrations in rodents maintained on HFD vary. Dopamine levels in the VTA and NAc were reported to be reduced in obese mice maintained on an HFD.153,154 Using high-performance liquid chromatography (HPLC) assays, dopamine levels were also reduced in accumbal homogenates of obese mice maintained on an HFD.153 Obesity-prone rats fed HFD for 5 days exhibited lower dopamine levels in the NAc as measured by in vivo microdialysis and subsequent HPLC.154 In obese rats fed a cafeteria diet, NAc extracellular dopamine levels and amplitude of evoked dopamine responses decreased relative to chow controls.155 However, a different group reported no difference in extracellular dopamine levels between rats fed HFD and controls, yet dopamine turnover––assessed by normalizing concentrations of 3,4-dihydroxyphenylacetic acid (DOPAC), a dopamine metabolite, to extracellular dopamine levels–– decreased in the striatum.156 This effect was not observed in mice maintained on an HFD for 18 weeks, after which extracellular striatal dopamine, DOPAC, and the ratio between them were no different from control mice.147 Finally, other studies reported that HFD increased extracellular dopamine in the accumbens of obesity-prone rats as measured by microdialysis157 and that peri-adolescent exposure to HFD increased burst firing in the VTA and subsequent extracellular dopamine levels in the NAc.148 On the basis of the variance in this animal work, conclusions regarding the effect of HFD on extracellular dopamine levels in rodents remain difficult to draw.
In contrast to the variance seen in studies of extracellular dopamine, HFD appears to negatively regulate surface expression of ventral striatal dopamine transporters. Fast-scan cyclic voltammetry experiments in the NAc showed that obese mice on an HFD did not show normal insulin-enhanced dopamine clearance and thus exhibited inhibited DAT function,158 while high-speed chrono-amperometry experiments showed delayed clearance of injected dopamine in the striatum of obese rats fed HFD.159 Inhibited dopamine transporter insertion into the membrane appears to occur after HFD even in the absence of weight gain,152 supported by decreased DAT protein levels seen in NAc lysates following exposure to saturated fat in the absence of weight gain.75 The apparent inhibition of DAT function after HFD is similar to what has been documented following the effects of cocaine; cocaine causes delayed presynaptic dopamine transport, and repeated exposure over time reduces the ability of cocaine to inhibit accumbal synaptic DATs.106 This highlights further potential parallel mechanisms spanning drug abuse and obesity and brings to light the question of what mechanistic deficiencies underlie both states.
These effects of HFD on dopamine transmission may alter dopamine-dependent striatal glutamatergic plasticity. As discussed above, drugs of abuse, including cocaine and alcohol, increase dopamine release into the striatum, and over time result in diminished dopamine receptor availability and inhibited dopamine reuptake.114–116 This disrupts normal synaptic plasticity in projections from the prefrontal cortex to the NAc.160 The disruptions in dopamine release, binding, or reuptake seen in rodents fed HFD144–147,149,151 suggest a role for HFD in altering D2R-dependent plasticity mechanisms29 (Fig. 1B). Such changes may underlie the changes in physical inactivity and food choices that accompany obesity (Fig. 1A).
Opioid changes in substance use disorders
Opioid receptors have been implicated in regulating intake of multiple drugs of abuse. For example, alcohol interacts with the opioid system,161,162 and alcohol drinking in rats was bidirectionally gated by injection of μ-opioid receptor agonists or antagonists into the ventral pallidum.163 Furthermore, intraventricular administration of a κ-opioid receptor antagonist attenuates anxiety-like phenotypes exhibited by mice following alcohol withdrawal, while κ-opioid receptor agonist administration induces anxiety-like behaviors similar to those seen in alcohol withdrawal.164 Similarly, stress-induced cocaine seeking in rats was precluded with treatment with a κ-opioid receptor antagonist.165 These studies suggest that opioid receptors may gate certain behavioral aspects of substance abuse.
Opioid receptors are also involved in the regulation of glutamatergic synaptic plasticity. Administration of μ-opioid receptor antagonists hindered the induction of LTP at mossy fiber–CA3 terminals in the hippocampus.166. In vitro recordings in the dorsal striatum showed that endogenous opioids, including met-enkephalin and dynorphin-A, as well as μ- and δ-opioid agonists, induced long-term depression (LTD),167 suggesting that opioid receptors modulate corticostriatal plasticity per se by facilitating synaptic depression. Recent work suggests a specific role for dynorphin in the control of LTP; optogenetic-facilitated release of dynorphin in striatal slices following theta-burst stimulation of synaptic potentiation impaired LTP in the dorsal medial striatum.168 Studies examining the effects of μ-opioid receptor agonists in striatum suggested that μ-opioid receptors gate excitatory and inhibitory inputs to NAc MSNs in a cell type–specific manner.169,170 κ-Opioid receptors enhance excitatory inputs from the amygdala to D1R-expressing MSNs in the NAc and inhibit excitatory input from amygdala and ventral hippocampus to accumbal D2R-expressing MSNs,171 suggesting that κ-opioid receptors control the balance of excitation and inhibition to the NAc in a cell-specific and projection-specific manner. These studies suggest that opioid receptors regulate glutamatergic synaptic plasticity. Furthermore, opioid receptors may control both the onset and persistence of obesity through their regulation of food intake as well as synaptic plasticity.
Opioid receptors can also regulate drug-induced synaptic changes. Alcohol-induced LTD between fast-spiking interneurons and MSNs in the dorsolateral striatum is dependent on δ-opioid receptors and was attenuated in the presence of the antagonist naloxone.172 κ-Opioid receptor antagonists reversed the effects of stress on LTP of γ-aminobutyric acid (GABA)-ergic synapses in the VTA and prevented reinstatement of cocaine seeking.173 Finally, stress-induced changes in hippocampal metaplasticity were prevented by administration of a κ-opioid receptor antagonist.174 Synaptic plasticity and various drug-seeking behaviors are differentially regulated by opioid receptors, raising the question as to whether these receptors also gate circuit and behavioral changes associated with obesity.
Opioid changes in obesity
Opioids have a complex interaction with food intake, and specifically the intake of palatable diets. It has been known for decades that stimulation of endogenous μ-opioid receptors in the NAc promotes feeding.175,176 A genome-wide association study also revealed that adolescent humans carrying the minor OPRM1 allele were more likely to eat less fat and have a lower body fat percentage.177 Supporting this, systemic administration of naltrexone, a nonselective opiate receptor antagonist, reduced the consumption of palatable foods in rats without altering intake of chow.178 Consistently, gene knockout mice lacking μ-opioid receptors exhibited normal feeding when presented with a low-fat diet, yet were resistant to obesity when given access to an HFD.179 Together, these results suggest that μ-opioid receptors are necessary for fat appetite. However, men180 and women136 with obesity have reportedly lower levels of accumbal μ-opioid receptors, which is difficult to reconcile with this point. In addition, others have reported that μ-opioid receptor gene knockout mice gained weight on a chow diet, relative to control mice,181,182 indicating that μ-opioid receptors may have effects on food intake independent from fat content. There has been less work investigating how the other classes of opioid receptor relate to feeding, but evidence suggests that κ-opiates can also drive intake of fat. Injecting a κ-opioid receptor agonist into the lateral ventricle of rats caused them to increase intake of high-fat but not low-fat food.183 Consistently, κ-opioid receptor gene knockout mice had lower body weights than controls after being maintained on an HFD.184
While the role of opioid receptors on feeding and obesity has been well documented, the effects of HFD on opioid receptors are not well understood. Obese rodents fed HFD or cafeteria diet exhibited reduced μ-opioid mRNA in the accumbens and in the VTA and prefrontal cortex,185–188 possibly due to increased methylation or heterochromatic location of the μ-opioid receptor.185,188 A study in rats showed that one week of HFD exposure increased the binding of naloxone, an opioid receptor antagonist, in tissue homogenates of the cortex and midbrain.189 Interestingly, Alsio et al.190 showed that access to a palatable, high-fat/high-sugar diet in the absence of weight gain did not alter NAc μ-opioid receptor mRNA, but that ad libitum access to such a diet (which caused weight gain) decreased NAc μ-opioid receptor mRNA. Other studies reported no difference in μ-opioid receptor binding in the NAc of rats who gained weight on a diet including 33% condensed milk.191 Further work aimed at elucidating the mechanisms underlying opioid receptor control of feeding and HFD, and importantly of the role of HFD in modulating opioid receptors, is needed to understand potential opioid contributions to obesity.
Changes in dopamine persist after weight loss
Multiple studies suggest that an HFD has persistent actions on dopamine release and transmission in striatum, even after weight loss. Rodent exposure to HFD increased body weight and depleted striatal dopamine levels, which persisted despite weight loss192 or decreased further11,153 after the rodents were switched back to chow. HFD-induced obesity decreased dopaminergic gene expression in the NAc.149 This effect appeared to persist partially, with D1R expression levels recovering to baseline after diet was switched back to chow, and D2R gene expression remaining low.149 Mice on an HFD had lower TH protein levels in the NAc relative to mice fed a low-fat control diet; when these HFD mice were switched to a normal chow diet, the low accumbal TH protein levels persisted.11 While more studies are needed to understand the time course and persistence of these changes, there are several indications that dopaminergic changes can persist following withdrawal from HFD and subsequent weight loss. To the extent that these changes drive behavior and energetic regulation, they may contribute to the persistence of obesity.
Many researchers have also looked at the persistence of behavioral adaptations following withdrawal from obesogenic diets. Female rats given a “junk -food” high-carbohydrate, low-protein diet had lower break points in an operant task for sucrose reward that persisted for 9 days after the diet was withdrawn.193 Mice maintained on an HFD spent less time in the open arm of an elevated plus maze (interpreted as a high anxiety score), and this was exacerbated after withdrawal from HFD.11 However, rats put on a long-term monounsaturated or saturated fat diet did not show increased anxiety-like behavior,194 and these rats did not gain weight. Together, this suggests that HFD-induced obese states contribute to anxiety-like behavior, but the consumption of fat alone is not anxiogenic.
Human studies also support a role for persistent changes in dopamine function and physical activity after weight loss. Two PET studies showed lack of recovery of D2R binding after gastric bypass and associated weight loss.195,196 However, a report from another group demonstrated a partial recovery of D2R binding 6 weeks after bariatric surgery.197 Similarly, objective assessments of vigorous physical activity 6–12 months after bariatric surgery show that, despite weight loss, physical activity levels remained low in these individuals.10,198,199 Animal studies have supported these observations. Loss of adiposity correlated with further decreases physical activity in nonhuman primates200 and dogs.201,202 Further work is needed to investigate the circuit changes that may underlie these persistent behavioral effects, but it is possible they reflect persistent changes in dopamine function.
Lastly, persistent effects seen in offspring exposed to HFD in utero or in those born to mothers with obesity also support a lasting role for these––at times transient––conditions. Experiments in nonhuman primates showed Japanese macaques exposed to HFD in utero had lower D1R and D2R protein levels and a decrease in TH positive terminal fibers in the prefrontal cortex, as measured by immunohistochemistry,203 and that maternal obesity (but not maternal HFD) resulted in increased offspring body weight 7 months after weaning.203 Rats exposed to HFD in utero exhibited impaired adaptive dopamine signals in the NAc in response to repeated pinch stress204 and decreased levels of NAc dopamine in response to a palatable food reward compared with rats born to lean mothers.205 Rats exposed to a high-fat, high-sugar diet in utero exhibited increased lever presses to achieve a palatable reward relative to those born to chow-controlled mothers,206 Together, these studies suggest that exposure to HFD during development can alter dopamine transmission later in life, which may increase the vulnerability of the offspring to obesity. While exposure to HFD during fetal development is much different than exposure during adolescence or adulthood, it underscores how changes caused by short-term exposure to HFD or obesity may have long-lasting impacts on neural circuits and behavior.
Potential mechanisms of persistent effects of obesity
There are multiple possible contributing factors involved in the sustained effects of obesity or obesogenic diets. Factors such as triglycerides and free fatty acids are elevated following HFD exposure, and fat itself can alter synaptic plasticity.207 Evidence of this is found in multiple brain regions. The presence of acutely applied triglycerides disrupted the maintenance of in vitro hippocampal LTP,208 suggesting that acute exposure to fat can disrupt synaptic potentiation. Recordings in rodent hippocampus after exposure to HFD and subsequent obesity showed disruptions in LTD209 and reductions in synaptic efficiency.80,209 Recent work showed that bath application of monounsaturated fatty acid decreased both neuronal firing of VTA dopaminergic neurons in slice recordings and action potential–independent excitatory release onto those neurons,210 suggesting that fat itself may affect VTA projections to the NAc. Together, this supports the hypothesis that fat, or components of fat including fatty acids, can alter synaptic plasticity. Subcellular mechanisms underlying changes in plasticity may include disruptions in pathways involving the holoenzyme protein kinase A (PKA), which is important for LTP211,212 and LTD regulation.213 During LTP, AMPA channels are driven into the membrane,214 in part due to activity-dependent PKA phosphorylation of glutamate ionotropic receptor AMPA subunit 1 (GluA1) at serine 845.215 Rats fed saturated fat diets in the absence of weight gain showed increases in accumbal protein markers known to be targets of PKA: phospho-dopamine and cAMP–regulated phosphoprotein 32 (DARPP32) and phospho-serine845-GluA1.75 Mice fed HFD became obese and exhibited increased PKA activity in the hypothalamus.216 Together, these studies suggest that HFD and obese states alter PKA-based molecular pathways in the brain, which may be particularly important for synaptic plasticity in the striatum.
Persistent effects of HFD may also include epigenetic or gene expression changes, which have been implicated in altered synaptic plasticity following exposure to drugs of abuse.217 Mice fed HFD exhibited altered gene expression in the cortex,218 and RNA-sequencing data from an Alzheimer’s mouse model following exposure to HFD showed an upregulation of immune-linked genes and a downregulation of genes ontologically linked to synaptic transmission in the cortex.219 A study examining DNA methylation after HFD in mice showed that the μ-opioid receptor (MOR) gene had enriched methylation in tissue from the VTA, NAc, and PFC of mice fed HFD and was more likely to be located within inactive chromatin, suggesting that HFD acted to epigenetically suppress MOR transcription.185 This may relate to human findings of reductions in MOR binding in striatum of subjects with obesity.136,180 Furthermore, protein levels of the canonical transcription factors phosphorylated-cAMP response element binding protein (CREB) and δ-FBJ murine osteosarcoma viral oncogene homolog B (deltaFosB) were increased in the amygdala and NAc of mice fed HFD, respectively.11 CREB has also been implicated in the influence of drugs of abuse over cellular plasticity.105 Overexpression of CREB in the NAc induced aversion to cocaine220 and reduced preference for morphine221 in rats in a conditioned place- preference task, while expression of a dominant-negative mutant CREB enhanced preference for both cocaine and morphine.220,221 This suggests that regulation of transcription factors, such as CREB, may underlie subcellular changes common to exposure to drugs of abuse and obesogenic diets. Recent work showed that, in the absence of weight gain, rats fed monounsaturated or saturated fats exhibited altered transcription profiles of stress-related genes, including corticotropin releasing hormone (CRH) and its associated receptor CRH receptor 1 (CRH-R1), in the paraventricular nucleus (PVN) of the hypothalamus as well as amygdalar nuclei.194 PVN CRH transcription levels positively correlate with susceptibility to social stress.222 Together, these findings suggest a mechanism for persistent changes in gene expression of multiple brain circuits following HFD.
Additionally, obesity-linked alterations in insulin and leptin resistance may underlie molecular mechanisms contributing to sustained changes in the dopaminergic system. Obesity causes prolonged increased secretion and deficient clearance of insulin, which ultimately leads to insulin resistance.223,224 Through phosphoinositide 3-kinase (PI3K) and renin–angiotensin system (Ras) pathway activation, insulin binding in the brain is involved in DAT surface expression,225 suggesting that sustained insulin exposure due to insulin resistance may disrupt DAT expression. Reduced DAT expression has been linked to obesity in humans139,140 and HFD in rodents,75 though there have been conflicting reports on this point.141–143 Indeed, obese rats fed HFD for a month exhibited elevated plasma insulin levels indicative of insulin resistance and also showed reduced striatal DAT expression,159 similar to the observed insulin resistance, impaired blood glucose clearance, and reduced dopamine clearance prevalent in mice fed HFD for 6 weeks.158 Inhibition of protein tyrosine phosphtase 1B––and subsequent promotion of phosphorylation of insulin receptor tyrosine residues––promoted the insulin receptor signaling cascade and effectively rescued dopamine reuptake,158 supporting the link between insulin resistance and changes observed in the dopamine system. Even in the absence of obesity, mice fed high-fructose corn syrup (HFCS) for several months had impaired blood glucose clearance and impaired dopamine release in the dorsal striatum.226
Finally, a large literature indicates that obesity is associated with inflammation in the hippocampus227 and impaired cognition or hippocampal-dependent memory.228,229 Broadly, sustained inflammation alters synaptic plasticity and disrupts neural homeostasis,230 and recent work suggests that HFD-induced obesity caused blood–brain barrier leakage231 and may increase markers of immune response in the brain,219 even acutely in the absence of weight gain.232 Elevated levels of the inflammatory molecules interleukin 6 (IL-6) or tumor necrosis factor α (TNF-α) inhibit LTP in rodent hippocampus.233,234 Persistent inflammation is associated with depression,235 a comorbidity of obesity that involves the ventral striatum and prefrontal cortex. Obesity is also associated with inflammation in the hypothalamus,236,237 which projects to the VTA, further implicating the circuitry that is involved in reward and depression. Though hypothalamic inflammation appears to recede with weight loss,238 increased inflammation during obesity may affect hypothalamic to VTA plasticity and in turn alter VTA to NAc projections.237 Persistent effects of inflammation may be another mechanism whereby obesity disrupts neural circuits, even after weight loss.
Conclusions and ongoing questions
Understanding how synaptic plasticity is linked to obesity may reveal new insights into the challenges of both weight loss and weight maintenance. However, many outstanding questions remain. For instance, given the heterogeneity of human food consumption, it is difficult to attribute changes in weight to the presence or absence of any particular food substance. A similar problem exists in rodent studies, where experiments are often conducted using different formulations of HFD. Thus, the question of what elements of a diet contribute most strongly to weight gain or loss remains controversial. With regard to plasticity, it is possible that certain types of fat are more likely to alter synaptic plasticity mechanisms than others. Finally, fat itself is an endocrine organ, and the accumulation of fat during obesity alters the concentration of many circulating factors in the blood. Therefore, there is a need to decouple the effects of HFD and obesity to identify whether specific components of the diet are likely to alter synaptic plasticity mechanisms. This can be done with pair feeding that allows animals to gain all of their calories from an HFD without becoming obese.
While there is widespread acknowledgement that obesity is associated with reductions in physical activity, the mechanism underlying the link between the two remains unclear. We have reviewed evidence that HFD alters dopamine binding, which in turn controls synaptic potentiation and depression. This leads to the question of whether overconsumption of fat disrupts striatal dopamine binding or transmission and whether this is a main contributor to decreases in physical activity that accompany obesity. Physical activity is one of the best predictors of human health, and unraveling the mechanisms that cause inactivity may be necessary to increase activity in people with obesity.
Perhaps the most pertinent question on this topic is whether persistent changes observed in people with obesity can be reversed. There is some evidence of D2R recovery after food restriction in rats,146 as well as recovery of D2R binding in human striatum following gastric bypass.197 This suggests that dramatic changes in diet may be one option for treating persistent effects of obesity. Additionally, a study in mice demonstrated increased D2R availability in striatum after chronic wheel running,239 suggesting that high-intensity physical activity may aid in the recovery of striatal dopamine dysfunction after obesity. However, this has not yet been examined by other researchers or in humans.
Changes in basal ganglia circuitry in obesity may be like the circuit alterations caused by drugs of abuse and may lead to some behavioral adaptations that resemble substance use disorders. Such changes may persist in people who have lost weight and may underlie susceptibility to relapse for compulsive eating and weight gain. Understanding these circuit alterations may lead to new treatments for obesity and to better understanding of why obesity is so difficult to reverse.
Acknowledgments
This work was funded by the National Institutes of Health Intramural Research Program (NIDDK).
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Mokdad AH, et al. The spread of the obesity epidemic in the United States, 1991–1998. Jama. 1999;282:1519–1522. doi: 10.1001/jama.282.16.1519. [DOI] [PubMed] [Google Scholar]
- 2.National Center for Health, S. Health, United States, 2015: With Special Feature on Racial and Ethnic Health Disparities. National Center for Health Statistics (US); 2016. [PubMed] [Google Scholar]
- 3.McGinnis JM, Foege WH. Actual causes of death in the United States. Jama. 1993;270:2207–2212. [PubMed] [Google Scholar]
- 4.Hall KD, Guo J. Obesity Energetics: Body Weight Regulation and the Effects of Diet Composition. Gastroenterology. 2017;152:1718–1727.e1713. doi: 10.1053/j.gastro.2017.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guo J, Jou W, Gavrilova O, Hall KD. Persistent diet-induced obesity in male C57BL/6 mice resulting from temporary obesigenic diets. PLoS One. 2009;4:e5370. doi: 10.1371/journal.pone.0005370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rolls BJ, Rowe EA, Turner RC. Persistent obesity in rats following a period of consumption of a mixed, high energy diet. J Physiol. 1980;298:415–427. doi: 10.1113/jphysiol.1980.sp013091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harris RB. Role of set-point theory in regulation of body weight. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 1990;4:3310–3318. doi: 10.1096/fasebj.4.15.2253845. [DOI] [PubMed] [Google Scholar]
- 8.Chhabra KH, et al. Reprogramming the body weight set point by a reciprocal interaction of hypothalamic leptin sensitivity and Pomc gene expression reverts extreme obesity. Molecular metabolism. 2016;5:869–881. doi: 10.1016/j.molmet.2016.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hall KD. Predicting metabolic adaptation, body weight change, and energy intake in humans. American journal of physiology. Endocrinology and metabolism. 2010;298:E449–466. doi: 10.1152/ajpendo.00559.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berglind D, et al. Women undergoing Roux-en-Y Gastric Bypass surgery: Family resemblance in pre- to postsurgery physical activity and sedentary behavior in children and spouses. Surgery for obesity and related diseases: official journal of the American Society for Bariatric Surgery. 2015;11:690–696. doi: 10.1016/j.soard.2014.10.018. [DOI] [PubMed] [Google Scholar]
- 11.Sharma S, Fernandes MF, Fulton S. Adaptations in brain reward circuitry underlie palatable food cravings and anxiety induced by high-fat diet withdrawal. International journal of obesity (2005) 2013;37:1183–1191. doi: 10.1038/ijo.2012.197. [DOI] [PubMed] [Google Scholar]
- 12.Nair SG, Adams-Deutsch T, Epstein DH, Shaham Y. The neuropharmacology of relapse to food seeking: methodology, main findings, and comparison with relapse to drug seeking. Progress in neurobiology. 2009;89:18–45. doi: 10.1016/j.pneurobio.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beeler JA, Frazier CR, Zhuang X. Putting desire on a budget: dopamine and energy expenditure, reconciling reward and resources. Frontiers in integrative neuroscience. 2012;6:49. doi: 10.3389/fnint.2012.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kravitz AV, O’Neal TJ, Friend DM. Do Dopaminergic Impairments Underlie Physical Inactivity in People with Obesity? Frontiers in human neuroscience. 2016;10:514. doi: 10.3389/fnhum.2016.00514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ifland JR, et al. Refined food addiction: a classic substance use disorder. Medical hypotheses. 2009;72:518–526. doi: 10.1016/j.mehy.2008.11.035. [DOI] [PubMed] [Google Scholar]
- 16.Rogers PJ. Food and drug addictions: Similarities and differences. Pharmacology, biochemistry, and behavior. 2017;153:182–190. doi: 10.1016/j.pbb.2017.01.001. [DOI] [PubMed] [Google Scholar]
- 17.Davis C, et al. Evidence that ‘food addiction’ is a valid phenotype of obesity. Appetite. 2011;57:711–717. doi: 10.1016/j.appet.2011.08.017. [DOI] [PubMed] [Google Scholar]
- 18.Finlayson G. Food addiction and obesity: unnecessary medicalization of hedonic overeating. Nature reviews Endocrinology. 2017;13:493–498. doi: 10.1038/nrendo.2017.61. [DOI] [PubMed] [Google Scholar]
- 19.Kenny PJ, Voren G, Johnson PM. Dopamine D2 receptors and striatopallidal transmission in addiction and obesity. Curr Opin Neurobiol. 2013;23:535–538. doi: 10.1016/j.conb.2013.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Valdivia S, Patrone A, Reynaldo M, Perello M. Acute high fat diet consumption activates the mesolimbic circuit and requires orexin signaling in a mouse model. PLoS One. 2014;9:e87478. doi: 10.1371/journal.pone.0087478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu S, et al. Consumption of palatable food primes food approach behavior by rapidly increasing synaptic density in the VTA. Proc Natl Acad Sci U S A. 2016;113:2520–2525. doi: 10.1073/pnas.1515724113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sulzer D. How addictive drugs disrupt presynaptic dopamine neurotransmission. Neuron. 2011;69:628–649. doi: 10.1016/j.neuron.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Avena NM, Bocarsly ME, Hoebel BG, Gold MS. Overlaps in the nosology of substance abuse and overeating: the translational implications of “food addiction”. Current drug abuse reviews. 2011;4:133–139. doi: 10.2174/1874473711104030133. [DOI] [PubMed] [Google Scholar]
- 24.Furlong TM, Jayaweera HK, Balleine BW, Corbit LH. Binge-like consumption of a palatable food accelerates habitual control of behavior and is dependent on activation of the dorsolateral striatum. J Neurosci. 2014;34:5012–5022. doi: 10.1523/jneurosci.3707-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnson PM, Kenny PJ. Dopamine D2 receptors in addiction-like reward dysfunction and compulsive eating in obese rats. Nat Neurosci. 2010;13:635–641. doi: 10.1038/nn.2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tantot F, et al. The effect of high-fat diet consumption on appetitive instrumental behavior in rats. Appetite. 2017;108:203–211. doi: 10.1016/j.appet.2016.10.001. [DOI] [PubMed] [Google Scholar]
- 27.Pitman KA, Borgland SL. Changes in mu-opioid receptor expression and function in the mesolimbic system after long-term access to a palatable diet. Pharmacology & therapeutics. 2015;154:110–119. doi: 10.1016/j.pharmthera.2015.07.005. [DOI] [PubMed] [Google Scholar]
- 28.Tepper JM, Abercrombie ED, Bolam JP. Basal ganglia macrocircuits. Progress in brain research. 2007;160:3–7. doi: 10.1016/s0079-6123(06)60001-0. [DOI] [PubMed] [Google Scholar]
- 29.Kreitzer AC, Malenka RC. Striatal plasticity and basal ganglia circuit function. Neuron. 2008;60:543–554. doi: 10.1016/j.neuron.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alexander GE, DeLong MR, Strick PL. Parallel organization of functionally segregated circuits linking basal ganglia and cortex. Annu Rev Neurosci. 1986;9:357–381. doi: 10.1146/annurev.ne.09.030186.002041. [DOI] [PubMed] [Google Scholar]
- 31.Bolam JP, Hanley JJ, Booth PA, Bevan MD. Synaptic organisation of the basal ganglia. Journal of anatomy. 2000;196(Pt 4):527–542. doi: 10.1046/j.1469-7580.2000.19640527.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.DeLong MR. Primate models of movement disorders of basal ganglia origin. Trends in neurosciences. 1990;13:281–285. doi: 10.1016/0166-2236(90)90110-v. [DOI] [PubMed] [Google Scholar]
- 33.Balleine BW, Delgado MR, Hikosaka O. The role of the dorsal striatum in reward and decision-making. J Neurosci. 2007;27:8161–8165. doi: 10.1523/jneurosci.1554-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Salamone JD, Correa M, Farrar A, Mingote SM. Effort-related functions of nucleus accumbens dopamine and associated forebrain circuits. Psychopharmacology. 2007;191:461–482. doi: 10.1007/s00213-006-0668-9. [DOI] [PubMed] [Google Scholar]
- 35.Schmidt L, Lebreton M, Clery-Melin ML, Daunizeau J, Pessiglione M. Neural mechanisms underlying motivation of mental versus physical effort. PLoS Biol. 2012;10:e1001266. doi: 10.1371/journal.pbio.1001266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lerma-Cabrera JM, et al. Control of food intake by MC4-R signaling in the lateral hypothalamus, nucleus accumbens shell and ventral tegmental area: interactions with ethanol. Behavioural brain research. 2012;234:51–60. doi: 10.1016/j.bbr.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van Huijstee AN, Mansvelder HD. Glutamatergic synaptic plasticity in the mesocorticolimbic system in addiction. Front Cell Neurosci. 2014;8:466. doi: 10.3389/fncel.2014.00466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ungless MA, Whistler JL, Malenka RC, Bonci A. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature. 2001;411:583–587. doi: 10.1038/35079077. [DOI] [PubMed] [Google Scholar]
- 39.Liu QS, Pu L, Poo MM. Repeated cocaine exposure in vivo facilitates LTP induction in midbrain dopamine neurons. Nature. 2005;437:1027–1031. doi: 10.1038/nature04050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bellone C, Luscher C. Cocaine triggered AMPA receptor redistribution is reversed in vivo by mGluR-dependent long-term depression. Nat Neurosci. 2006;9:636–641. doi: 10.1038/nn1682. [DOI] [PubMed] [Google Scholar]
- 41.Mameli M, et al. Cocaine-evoked synaptic plasticity: persistence in the VTA triggers adaptations in the NAc. Nat Neurosci. 2009;12:1036–1041. doi: 10.1038/nn.2367. [DOI] [PubMed] [Google Scholar]
- 42.Farrell MR, Schoch H, Mahler SV. Modeling cocaine relapse in rodents: Behavioral considerations and circuit mechanisms. Progress in neuropsychopharmacology & biological psychiatry. 2018 doi: 10.1016/j.pnpbp.2018.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koob GF, Volkow ND. Neurobiology of addiction: a neurocircuitry analysis. The lancet Psychiatry. 2016;3:760–773. doi: 10.1016/s2215-0366(16)00104-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Belujon P, Grace AA. Hippocampus, amygdala, and stress: interacting systems that affect susceptibility to addiction. Ann N Y Acad Sci. 2011;1216:114–121. doi: 10.1111/j.1749-6632.2010.05896.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vialou V, et al. DeltaFosB in brain reward circuits mediates resilience to stress and antidepressant responses. Nat Neurosci. 2010;13:745–752. doi: 10.1038/nn.2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lim BK, Huang KW, Grueter BA, Rothwell PE, Malenka RC. Anhedonia requires MC4R-mediated synaptic adaptations in nucleus accumbens. Nature. 2012;487:183–189. doi: 10.1038/nature11160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Khibnik LA, et al. Stress and Cocaine Trigger Divergent and Cell Type-Specific Regulation of Synaptic Transmission at Single Spines in Nucleus Accumbens. Biological psychiatry. 2016;79:898–905. doi: 10.1016/j.biopsych.2015.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang YH, et al. In vivo cocaine experience generates silent synapses. Neuron. 2009;63:40–47. doi: 10.1016/j.neuron.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee BR, et al. Maturation of silent synapses in amygdala-accumbens projection contributes to incubation of cocaine craving. Nat Neurosci. 2013;16:1644–1651. doi: 10.1038/nn.3533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yao J, Qi J, Chen G. Actin-dependent activation of presynaptic silent synapses contributes to long-term synaptic plasticity in developing hippocampal neurons. J Neurosci. 2006;26:8137–8147. doi: 10.1523/jneurosci.1183-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee BR, Dong Y. Cocaine-induced metaplasticity in the nucleus accumbens: silent synapse and beyond. Neuropharmacology. 2011;61:1060–1069. doi: 10.1016/j.neuropharm.2010.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reid MS, Berger SP. Evidence for sensitization of cocaine-induced nucleus accumbens glutamate release. Neuroreport. 1996;7:1325–1329. doi: 10.1097/00001756-199605170-00022. [DOI] [PubMed] [Google Scholar]
- 53.Churchill L, Swanson CJ, Urbina M, Kalivas PW. Repeated cocaine alters glutamate receptor subunit levels in the nucleus accumbens and ventral tegmental area of rats that develop behavioral sensitization. Journal of neurochemistry. 1999;72:2397–2403. doi: 10.1046/j.1471-4159.1999.0722397.x. [DOI] [PubMed] [Google Scholar]
- 54.Pascoli V, Turiault M, Luscher C. Reversal of cocaine-evoked synaptic potentiation resets drug-induced adaptive behaviour. Nature. 2011;481:71–75. doi: 10.1038/nature10709. [DOI] [PubMed] [Google Scholar]
- 55.Peoples LL, Kravitz AV, Guillem K. The role of accumbal hypoactivity in cocaine addiction. TheScientificWorldJournal. 2007;7:22–45. doi: 10.1100/tsw.2007.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Peoples LL, Uzwiak AJ, Gee F, West MO. Tonic firing of rat nucleus accumbens neurons: changes during the first 2 weeks of daily cocaine self-administration sessions. Brain research. 1999;822:231–236. doi: 10.1016/s0006-8993(98)01271-2. [DOI] [PubMed] [Google Scholar]
- 57.Terrier J, Luscher C, Pascoli V. Cell-Type Specific Insertion of GluA2-Lacking AMPARs with Cocaine Exposure Leading to Sensitization, Cue-Induced Seeking, and Incubation of Craving. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2016;41:1779–1789. doi: 10.1038/npp.2015.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Creed M, Ntamati NR, Chandra R, Lobo MK, Luscher C. Convergence of Reinforcing and Anhedonic Cocaine Effects in the Ventral Pallidum. Neuron. 2016;92:214–226. doi: 10.1016/j.neuron.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Del Olmo N, et al. Hippocampal synaptic plasticity and water maze learning in cocaine self-administered rats. Ann N Y Acad Sci. 2006;1074:427–437. doi: 10.1196/annals.1369.043. [DOI] [PubMed] [Google Scholar]
- 60.Thompson AM, Swant J, Gosnell BA, Wagner JJ. Modulation of long-term potentiation in the rat hippocampus following cocaine self-administration. Neuroscience. 2004;127:177–185. doi: 10.1016/j.neuroscience.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 61.Keralapurath MM, Briggs SB, Wagner JJ. Cocaine self-administration induces changes in synaptic transmission and plasticity in ventral hippocampus. Addiction biology. 2017;22:446–456. doi: 10.1111/adb.12345. [DOI] [PubMed] [Google Scholar]
- 62.Goldstein RZ, Volkow ND. Dysfunction of the prefrontal cortex in addiction: neuroimaging findings and clinical implications. Nat Rev Neurosci. 2011;12:652–669. doi: 10.1038/nrn3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kalivas PW. Addiction as a pathology in prefrontal cortical regulation of corticostriatal habit circuitry. Neurotoxicity research. 2008;14:185–189. doi: 10.1007/bf03033809. [DOI] [PubMed] [Google Scholar]
- 64.Pfefferbaum A, et al. Reorganization of frontal systems used by alcoholics for spatial working memory: an fMRI study. Neuroimage. 2001;14:7–20. doi: 10.1006/nimg.2001.0785. [DOI] [PubMed] [Google Scholar]
- 65.Volkow ND, et al. Decreased brain metabolism in neurologically intact healthy alcoholics. The American journal of psychiatry. 1992;149:1016–1022. doi: 10.1176/ajp.149.8.1016. [DOI] [PubMed] [Google Scholar]
- 66.Kalivas PW, Volkow ND. New medications for drug addiction hiding in glutamatergic neuroplasticity. Mol Psychiatry. 2011;16:974–986. doi: 10.1038/mp.2011.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Volkow ND, Fowler JS. Addiction, a disease of compulsion and drive: involvement of the orbitofrontal cortex. Cerebral cortex (New York, N.Y. : 1991) 2000;10:318–325. doi: 10.1093/cercor/10.3.318. [DOI] [PubMed] [Google Scholar]
- 68.Winstanley CA. The orbitofrontal cortex, impulsivity, and addiction: probing orbitofrontal dysfunction at the neural, neurochemical, and molecular level. Ann N Y Acad Sci. 2007;1121:639–655. doi: 10.1196/annals.1401.024. [DOI] [PubMed] [Google Scholar]
- 69.Brown RM, et al. Addiction-like Synaptic Impairments in Diet-Induced Obesity. Biological psychiatry. 2017;81:797–806. doi: 10.1016/j.biopsych.2015.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Oginsky MF, Goforth PB, Nobile CW, Lopez-Santiago LF, Ferrario CR. Eating ‘Junk-Food’ Produces Rapid and Long-Lasting Increases in NAc CP-AMPA Receptors: Implications for Enhanced Cue-Induced Motivation and Food Addiction. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2016;41:2977–2986. doi: 10.1038/npp.2016.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Verdoorn TA, Burnashev N, Monyer H, Seeburg PH, Sakmann B. Structural determinants of ion flow through recombinant glutamate receptor channels. Science. 1991;252:1715–1718. doi: 10.1126/science.1710829. [DOI] [PubMed] [Google Scholar]
- 72.Russo SJ, et al. IRS2-Akt pathway in midbrain dopamine neurons regulates behavioral and cellular responses to opiates. Nat Neurosci. 2007;10:93–99. doi: 10.1038/nn1812. [DOI] [PubMed] [Google Scholar]
- 73.Hall FS, Drgonova J, Goeb M, Uhl GR. Reduced behavioral effects of cocaine in heterozygous brain-derived neurotrophic factor (BDNF) knockout mice. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2003;28:1485–1490. doi: 10.1038/sj.npp.1300192. [DOI] [PubMed] [Google Scholar]
- 74.St Laurent R, Helm SR, Glenn MJ. Reduced cocaine-seeking behavior in heterozygous BDNF knockout rats. Neurosci Lett. 2013;544:94–99. doi: 10.1016/j.neulet.2013.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hryhorczuk C, et al. Dampened Mesolimbic Dopamine Function and Signaling by Saturated but not Monounsaturated Dietary Lipids. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2016;41:811–821. doi: 10.1038/npp.2015.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- 77.Plant K, et al. Transient incorporation of native GluR2-lacking AMPA receptors during hippocampal long-term potentiation. Nat Neurosci. 2006;9:602–604. doi: 10.1038/nn1678. [DOI] [PubMed] [Google Scholar]
- 78.Karimi SA, et al. Effect of high-fat diet and antioxidants on hippocampal long-term potentiation in rats: an in vivo study. Brain research. 2013;1539:1–6. doi: 10.1016/j.brainres.2013.09.029. [DOI] [PubMed] [Google Scholar]
- 79.Hao S, Dey A, Yu X, Stranahan AM. Dietary obesity reversibly induces synaptic stripping by microglia and impairs hippocampal plasticity. Brain, behavior, and immunity. 2016;51:230–239. doi: 10.1016/j.bbi.2015.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liu Z, et al. High-fat diet induces hepatic insulin resistance and impairment of synaptic plasticity. PLoS One. 2015;10:e0128274. doi: 10.1371/journal.pone.0128274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Krishna S, et al. Neurochemical and electrophysiological deficits in the ventral hippocampus and selective behavioral alterations caused by high-fat diet in female C57BL/6 mice. Neuroscience. 2015;297:170–181. doi: 10.1016/j.neuroscience.2015.03.068. [DOI] [PubMed] [Google Scholar]
- 82.Goldstein RZ, Volkow ND. Drug addiction and its underlying neurobiological basis: neuroimaging evidence for the involvement of the frontal cortex. The American journal of psychiatry. 2002;159:1642–1652. doi: 10.1176/appi.ajp.159.10.1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Le DS, et al. Less activation in the left dorsolateral prefrontal cortex in the reanalysis of the response to a meal in obese than in lean women and its association with successful weight loss. The American journal of clinical nutrition. 2007;86:573–579. doi: 10.1093/ajcn/86.3.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Volkow ND, et al. Inverse association between BMI and prefrontal metabolic activity in healthy adults. Obesity (Silver Spring, Md.) 2009;17:60–65. doi: 10.1038/oby.2008.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Willeumier KC, Taylor DV, Amen DG. Elevated BMI is associated with decreased blood flow in the prefrontal cortex using SPECT imaging in healthy adults. Obesity (Silver Spring, Md.) 2011;19:1095–1097. doi: 10.1038/oby.2011.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Del Parigi A, et al. Neuroimaging and obesity: mapping the brain responses to hunger and satiation in humans using positron emission tomography. Ann N Y Acad Sci. 2002;967:389–397. [PubMed] [Google Scholar]
- 87.Stoeckel LE, et al. Widespread reward-system activation in obese women in response to pictures of high-calorie foods. Neuroimage. 2008;41:636–647. doi: 10.1016/j.neuroimage.2008.02.031. [DOI] [PubMed] [Google Scholar]
- 88.Jastreboff AM, et al. Altered Brain Response to Drinking Glucose and Fructose in Obese Adolescents. Diabetes. 2016;65:1929–1939. doi: 10.2337/db15-1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bruce AS, et al. Obese children show hyperactivation to food pictures in brain networks linked to motivation, reward and cognitive control. International journal of obesity (2005) 2010;34:1494–1500. doi: 10.1038/ijo.2010.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Schultz W. Getting formal with dopamine and reward. Neuron. 2002;36:241–263. doi: 10.1016/s0896-6273(02)00967-4. [DOI] [PubMed] [Google Scholar]
- 91.Gonzales RA, Job MO, Doyon WM. The role of mesolimbic dopamine in the development and maintenance of ethanol reinforcement. Pharmacology & therapeutics. 2004;103:121–146. doi: 10.1016/j.pharmthera.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 92.Salamone JD, Correa M, Mingote SM, Weber SM. Beyond the reward hypothesis: alternative functions of nucleus accumbens dopamine. Current opinion in pharmacology. 2005;5:34–41. doi: 10.1016/j.coph.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 93.Koob GF. The neurobiology of addiction: a neuroadaptational view relevant for diagnosis. Addiction (Abingdon, England) 2006;101(Suppl 1):23–30. doi: 10.1111/j.1360-0443.2006.01586.x. [DOI] [PubMed] [Google Scholar]
- 94.Robinson TE, Berridge KC. The psychology and neurobiology of addiction: an incentive-sensitization view. Addiction (Abingdon, England) 2000;95(Suppl 2):S91–117. doi: 10.1080/09652140050111681. [DOI] [PubMed] [Google Scholar]
- 95.Enrico P, et al. Morphofunctional alterations in ventral tegmental area dopamine neurons in acute and prolonged opiates withdrawal. A computational perspective. Neuroscience. 2016;322:195–207. doi: 10.1016/j.neuroscience.2016.02.006. [DOI] [PubMed] [Google Scholar]
- 96.Sklair-Tavron L, et al. Chronic morphine induces visible changes in the morphology of mesolimbic dopamine neurons. Proc Natl Acad Sci U S A. 1996;93:11202–11207. doi: 10.1073/pnas.93.20.11202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Spiga S, Serra GP, Puddu MC, Foddai M, Diana M. Morphine withdrawal-induced abnormalities in the VTA: confocal laser scanning microscopy. The European journal of neuroscience. 2003;17:605–612. doi: 10.1046/j.1460-9568.2003.02435.x. [DOI] [PubMed] [Google Scholar]
- 98.Sarti F, Borgland SL, Kharazia VN, Bonci A. Acute cocaine exposure alters spine density and long-term potentiation in the ventral tegmental area. The European journal of neuroscience. 2007;26:749–756. doi: 10.1111/j.1460-9568.2007.05689.x. [DOI] [PubMed] [Google Scholar]
- 99.Mejias-Aponte CA, Ye C, Bonci A, Kiyatkin EA, Morales M. A subpopulation of neurochemically-identified ventral tegmental area dopamine neurons is excited by intravenous cocaine. J Neurosci. 2015;35:1965–1978. doi: 10.1523/jneurosci.3422-13.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Heller EA, et al. Morphine and cocaine increase serum- and glucocorticoid-inducible kinase 1 activity in the ventral tegmental area. Journal of neurochemistry. 2015;132:243–253. doi: 10.1111/jnc.12925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Self DW, McClenahan AW, Beitner-Johnson D, Terwilliger RZ, Nestler EJ. Biochemical adaptations in the mesolimbic dopamine system in response to heroin self-administration. Synapse. 1995;21:312–318. doi: 10.1002/syn.890210405. [DOI] [PubMed] [Google Scholar]
- 102.Hausknecht K, et al. Excitatory synaptic function and plasticity is persistently altered in ventral tegmental area dopamine neurons after prenatal ethanol exposure. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2015;40:893–905. doi: 10.1038/npp.2014.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hausknecht K, Shen YL, Wang RX, Haj-Dahmane S, Shen RY. Prenatal Ethanol Exposure Persistently Alters Endocannabinoid Signaling and Endocannabinoid-Mediated Excitatory Synaptic Plasticity in Ventral Tegmental Area Dopamine Neurons. J Neurosci. 2017;37:5798–5808. doi: 10.1523/jneurosci.3894-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Saal D, Dong Y, Bonci A, Malenka RC. Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron. 2003;37:577–582. doi: 10.1016/s0896-6273(03)00021-7. [DOI] [PubMed] [Google Scholar]
- 105.Nestler EJ. Cellular basis of memory for addiction. Dialogues in clinical neuroscience. 2013;15:431–443. doi: 10.31887/DCNS.2013.15.4/enestler. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Siciliano CA, Calipari ES, Ferris MJ, Jones SR. Adaptations of presynaptic dopamine terminals induced by psychostimulant self-administration. ACS chemical neuroscience. 2015;6:27–36. doi: 10.1021/cn5002705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Volkow ND, Fowler JS, Wang GJ, Baler R, Telang F. Imaging dopamine’s role in drug abuse and addiction. Neuropharmacology. 2009;56(Suppl 1):3–8. doi: 10.1016/j.neuropharm.2008.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Di Chiara G, Imperato A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci U S A. 1988;85:5274–5278. doi: 10.1073/pnas.85.14.5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Nestler EJ. The neurobiology of cocaine addiction. Science & practice perspectives. 2005;3:4–10. doi: 10.1151/spp05314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Willuhn I, Wanat MJ, Clark JJ, Phillips PE. Dopamine signaling in the nucleus accumbens of animals self-administering drugs of abuse. Current topics in behavioral neurosciences. 2010;3:29–71. doi: 10.1007/7854_2009_27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Volkow ND, Morales M. The Brain on Drugs: From Reward to Addiction. Cell. 2015;162:712–725. doi: 10.1016/j.cell.2015.07.046. [DOI] [PubMed] [Google Scholar]
- 112.Kuhar MJ, Ritz MC, Boja JW. The dopamine hypothesis of the reinforcing properties of cocaine. Trends in neurosciences. 1991;14:299–302. doi: 10.1016/0166-2236(91)90141-g. [DOI] [PubMed] [Google Scholar]
- 113.Juarez B, Han MH. Diversity of Dopaminergic Neural Circuits in Response to Drug Exposure. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2016;41:2424–2446. doi: 10.1038/npp.2016.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Volkow ND, et al. Effects of chronic cocaine abuse on postsynaptic dopamine receptors. The American journal of psychiatry. 1990;147:719–724. doi: 10.1176/ajp.147.6.719. [DOI] [PubMed] [Google Scholar]
- 115.Volkow ND, et al. Decreased dopamine D2 receptor availability is associated with reduced frontal metabolism in cocaine abusers. Synapse. 1993;14:169–177. doi: 10.1002/syn.890140210. [DOI] [PubMed] [Google Scholar]
- 116.Volkow ND, et al. Decreases in dopamine receptors but not in dopamine transporters in alcoholics. Alcoholism, clinical and experimental research. 1996;20:1594–1598. doi: 10.1111/j.1530-0277.1996.tb05936.x. [DOI] [PubMed] [Google Scholar]
- 117.Besson M, et al. Cocaine modulation of frontostriatal expression of Zif268, D2, and 5-HT2c receptors in high and low impulsive rats. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2013;38:1963–1973. doi: 10.1038/npp.2013.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Conrad KL, Ford K, Marinelli M, Wolf ME. Dopamine receptor expression and distribution dynamically change in the rat nucleus accumbens after withdrawal from cocaine self-administration. Neuroscience. 2010;169:182–194. doi: 10.1016/j.neuroscience.2010.04.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Nader MA, et al. Effects of cocaine self-administration on striatal dopamine systems in rhesus monkeys: initial and chronic exposure. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2002;27:35–46. doi: 10.1016/s0893-133x(01)00427-4. [DOI] [PubMed] [Google Scholar]
- 120.Gardoni F, Bellone C. Modulation of the glutamatergic transmission by Dopamine: a focus on Parkinson, Huntington and Addiction diseases. Front Cell Neurosci. 2015;9:25. doi: 10.3389/fncel.2015.00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Dos Santos M, et al. Cocaine increases dopaminergic connectivity in the nucleus accumbens. Brain structure & function. 2017 doi: 10.1007/s00429-017-1532-x. [DOI] [PubMed] [Google Scholar]
- 122.Shen HW, et al. Altered dendritic spine plasticity in cocaine-withdrawn rats. J Neurosci. 2009;29:2876–2884. doi: 10.1523/jneurosci.5638-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Conrad KL, et al. Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature. 2008;454:118–121. doi: 10.1038/nature06995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Thompson J, et al. D2 dopamine receptor gene (DRD2) Taq1 A polymorphism: reduced dopamine D2 receptor binding in the human striatum associated with the A1 allele. Pharmacogenetics. 1997;7:479–484. doi: 10.1097/00008571-199712000-00006. [DOI] [PubMed] [Google Scholar]
- 125.Blum K, et al. Increased prevalence of the Taq I A1 allele of the dopamine receptor gene (DRD2) in obesity with comorbid substance use disorder: a preliminary report. Pharmacogenetics. 1996;6:297–305. doi: 10.1097/00008571-199608000-00003. [DOI] [PubMed] [Google Scholar]
- 126.Stice E, Spoor S, Bohon C, Small DM. Relation between obesity and blunted striatal response to food is moderated by TaqIA A1 allele. Science. 2008;322:449–452. doi: 10.1126/science.1161550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wang GJ, et al. Brain dopamine and obesity. Lancet. 2001;357:354–357. doi: 10.1016/s0140-6736(00)03643-6. [DOI] [PubMed] [Google Scholar]
- 128.Volkow ND, et al. Low dopamine striatal D2 receptors are associated with prefrontal metabolism in obese subjects: possible contributing factors. Neuroimage. 2008;42:1537–1543. doi: 10.1016/j.neuroimage.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kessler RM, Zald DH, Ansari MS, Li R, Cowan RL. Changes in dopamine release and dopamine D2/3 receptor levels with the development of mild obesity. Synapse. 2014;68:317–320. doi: 10.1002/syn.21738. [DOI] [PubMed] [Google Scholar]
- 130.de Weijer BA, et al. Lower striatal dopamine D2/3 receptor availability in obese compared with non-obese subjects. EJNMMI research. 2011;1:37. doi: 10.1186/2191-219x-1-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.van de Giessen E, Celik F, Schweitzer DH, van den Brink W, Booij J. Dopamine D2/3 receptor availability and amphetamine-induced dopamine release in obesity. Journal of psychopharmacology (Oxford, England) 2014;28:866–873. doi: 10.1177/0269881114531664. [DOI] [PubMed] [Google Scholar]
- 132.Guo J, Simmons WK, Herscovitch P, Martin A, Hall KD. Striatal dopamine D2-like receptor correlation patterns with human obesity and opportunistic eating behavior. Mol Psychiatry. 2014;19:1078–1084. doi: 10.1038/mp.2014.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Dunn JP, et al. Relationship of dopamine type 2 receptor binding potential with fasting neuroendocrine hormones and insulin sensitivity in human obesity. Diabetes care. 2012;35:1105–1111. doi: 10.2337/dc11-2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Cosgrove KP, Veldhuizen MG, Sandiego CM, Morris ED, Small DM. Opposing relationships of BMI with BOLD and dopamine D2/3 receptor binding potential in the dorsal striatum. Synapse. 2015;69:195–202. doi: 10.1002/syn.21809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Eisenstein SA, et al. A comparison of D2 receptor specific binding in obese and normal-weight individuals using PET with (N-[(11)C]methyl)benperidol. Synapse. 2013;67:748–756. doi: 10.1002/syn.21680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Karlsson HK, et al. Obesity is associated with decreased mu-opioid but unaltered dopamine D2 receptor availability in the brain. J Neurosci. 2015;35:3959–3965. doi: 10.1523/jneurosci.4744-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Horstmann A, Fenske WK, Hankir MK. Argument for a non-linear relationship between severity of human obesity and dopaminergic tone. Obesity reviews: an official journal of the International Association for the Study of Obesity. 2015;16:821–830. doi: 10.1111/obr.12303. [DOI] [PubMed] [Google Scholar]
- 138.Eisenstein SA, et al. Emotional Eating Phenotype is Associated with Central Dopamine D2 Receptor Binding Independent of Body Mass Index. Scientific reports. 2015;5:11283. doi: 10.1038/srep11283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wu C, Garamszegi SP, Xie X, Mash DC. Altered Dopamine Synaptic Markers in Postmortem Brain of Obese Subjects. Frontiers in human neuroscience. 2017;11:386. doi: 10.3389/fnhum.2017.00386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Chen PS, et al. Correlation between body mass index and striatal dopamine transporter availability in healthy volunteers--a SPECT study. Neuroimage. 2008;40:275–279. doi: 10.1016/j.neuroimage.2007.11.007. [DOI] [PubMed] [Google Scholar]
- 141.van de Giessen E, et al. No association between striatal dopamine transporter binding and body mass index: a multi-center European study in healthy volunteers. Neuroimage. 2013;64:61–67. doi: 10.1016/j.neuroimage.2012.09.011. [DOI] [PubMed] [Google Scholar]
- 142.Thomsen G, et al. No correlation between body mass index and striatal dopamine transporter availability in healthy volunteers using SPECT and [123I]PE2I. Obesity (Silver Spring Md.) 2013;21:1803–1806. doi: 10.1002/oby.20225. [DOI] [PubMed] [Google Scholar]
- 143.Versteeg RI, et al. Serotonin Transporter Binding in the Diencephalon Is Reduced in Insulin-Resistant Obese Humans. Neuroendocrinology. 2017;105:141–149. doi: 10.1159/000450549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Huang XF, et al. Dopamine transporter and D2 receptor binding densities in mice prone or resistant to chronic high fat diet-induced obesity. Behavioural brain research. 2006;175:415–419. doi: 10.1016/j.bbr.2006.08.034. [DOI] [PubMed] [Google Scholar]
- 145.Hajnal A, Margas WM, Covasa M. Altered dopamine D2 receptor function and binding in obese OLETF rat. Brain research bulletin. 2008;75:70–76. doi: 10.1016/j.brainresbull.2007.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Thanos PK, Michaelides M, Piyis YK, Wang GJ, Volkow ND. Food restriction markedly increases dopamine D2 receptor (D2R) in a rat model of obesity as assessed with in-vivo muPET imaging ([11C] raclopride) and in-vitro ([3H] spiperone) autoradiography. Synapse. 2008;62:50–61. doi: 10.1002/syn.20468. [DOI] [PubMed] [Google Scholar]
- 147.Friend DM, et al. Basal Ganglia Dysfunction Contributes to Physical Inactivity in Obesity. Cell metabolism. 2017;25:312–321. doi: 10.1016/j.cmet.2016.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Naneix F, et al. Protracted motivational dopamine-related deficits following adolescence sugar overconsumption. Neuropharmacology. 2017 doi: 10.1016/j.neuropharm.2017.11.021. [DOI] [PubMed] [Google Scholar]
- 149.Carlin JL, et al. Removal of high-fat diet after chronic exposure drives binge behavior and dopaminergic dysregulation in female mice. Neuroscience. 2016;326:170–179. doi: 10.1016/j.neuroscience.2016.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.South T, Huang XF. High-fat diet exposure increases dopamine D2 receptor and decreases dopamine transporter receptor binding density in the nucleus accumbens and caudate putamen of mice. Neurochemical research. 2008;33:598–605. doi: 10.1007/s11064-007-9483-x. [DOI] [PubMed] [Google Scholar]
- 151.Adams WK, et al. Long-term, calorie-restricted intake of a high-fat diet in rats reduces impulse control and ventral striatal D2 receptor signalling – two markers of addiction vulnerability. The European journal of neuroscience. 2015;42:3095–3104. doi: 10.1111/ejn.13117. [DOI] [PubMed] [Google Scholar]
- 152.Cone JJ, Chartoff EH, Potter DN, Ebner SR, Roitman MF. Prolonged high fat diet reduces dopamine reuptake without altering DAT gene expression. PLoS One. 2013;8:e58251. doi: 10.1371/journal.pone.0058251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Carlin J, Hill-Smith TE, Lucki I, Reyes TM. Reversal of dopamine system dysfunction in response to high-fat diet. Obesity (Silver Spring, Md.) 2013;21:2513–2521. doi: 10.1002/oby.20374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Rada P, Bocarsly ME, Barson JR, Hoebel BG, Leibowitz SF. Reduced accumbens dopamine in Sprague-Dawley rats prone to overeating a fat-rich diet. Physiology & behavior. 2010;101:394–400. doi: 10.1016/j.physbeh.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Geiger BM, et al. Deficits of mesolimbic dopamine neurotransmission in rat dietary obesity. Neuroscience. 2009;159:1193–1199. doi: 10.1016/j.neuroscience.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Davis JF, et al. Exposure to elevated levels of dietary fat attenuates psychostimulant reward and mesolimbic dopamine turnover in the rat. Behavioral neuroscience. 2008;122:1257–1263. doi: 10.1037/a0013111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Narayanaswami V, Thompson AC, Cassis LA, Bardo MT, Dwoskin LP. Diet-induced obesity: dopamine transporter function, impulsivity and motivation. International journal of obesity (2005) 2013;37:1095–1103. doi: 10.1038/ijo.2012.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Fordahl SC, Jones SR. High-Fat-Diet-Induced Deficits in Dopamine Terminal Function Are Reversed by Restoring Insulin Signaling. ACS chemical neuroscience. 2017;8:290–299. doi: 10.1021/acschemneuro.6b00308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Speed N, et al. Impaired striatal Akt signaling disrupts dopamine homeostasis and increases feeding. PLoS One. 2011;6:e25169. doi: 10.1371/journal.pone.0025169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Moussawi K, et al. N-Acetylcysteine reverses cocaine-induced metaplasticity. Nat Neurosci. 2009;12:182–189. doi: 10.1038/nn.2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Volpicelli JR, Alterman AI, Hayashida M, O’Brien CP. Naltrexone in the treatment of alcohol dependence. Archives of general psychiatry. 1992;49:876–880. doi: 10.1001/archpsyc.1992.01820110040006. [DOI] [PubMed] [Google Scholar]
- 162.Drews E, Zimmer A. Modulation of alcohol and nicotine responses through the endogenous opioid system. Progress in neurobiology. 2010;90:1–15. doi: 10.1016/j.pneurobio.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 163.Kemppainen H, Raivio N, Suo-Yrjo V, Kiianmaa K. Opioidergic modulation of ethanol self-administration in the ventral pallidum. Alcoholism, clinical and experimental research. 2012;36:286–293. doi: 10.1111/j.1530-0277.2011.01611.x. [DOI] [PubMed] [Google Scholar]
- 164.Berger AL, Williams AM, McGinnis MM, Walker BM. Affective cue-induced escalation of alcohol self-administration and increased 22-kHz ultrasonic vocalizations during alcohol withdrawal: role of kappa-opioid receptors. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2013;38:647–654. doi: 10.1038/npp.2012.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Groblewski PA, Zietz C, Willuhn I, Phillips PE, Chavkin C. Repeated stress exposure causes strain-dependent shifts in the behavioral economics of cocaine in rats. Addiction biology. 2015;20:297–301. doi: 10.1111/adb.12123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Derrick BE, Rodriguez SB, Lieberman DN, Martinez JL., Jr Mu opioid receptors are associated with the induction of hippocampal mossy fiber long-term potentiation. The Journal of pharmacology and experimental therapeutics. 1992;263:725–733. [PubMed] [Google Scholar]
- 167.Atwood BK, Kupferschmidt DA, Lovinger DM. Opioids induce dissociable forms of long-term depression of excitatory inputs to the dorsal striatum. Nat Neurosci. 2014;17:540–548. doi: 10.1038/nn.3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Hawes SL, Salinas AG, Lovinger DM, Blackwell KT. Long-term plasticity of corticostriatal synapses is modulated by pathway-specific co-release of opioids through kappa-opioid receptors. J Physiol. 2017;595:5637–5652. doi: 10.1113/jp274190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Ma YY, et al. Regional and cell-type-specific effects of DAMGO on striatal D1 and D2 dopamine receptor-expressing medium-sized spiny neurons. ASN neuro. 2012;4 doi: 10.1042/an20110063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.James AS, et al. Opioid self-administration results in cell-type specific adaptations of striatal medium spiny neurons. Behavioural brain research. 2013;256:279–283. doi: 10.1016/j.bbr.2013.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Tejeda HA, et al. Pathway- and Cell-Specific Kappa-Opioid Receptor Modulation of Excitation-Inhibition Balance Differentially Gates D1 and D2 Accumbens Neuron Activity. Neuron. 2017;93:147–163. doi: 10.1016/j.neuron.2016.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Patton MH, Roberts BM, Lovinger DM, Mathur BN. Ethanol Disinhibits Dorsolateral Striatal Medium Spiny Neurons Through Activation of A Presynaptic Delta Opioid Receptor. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2016;41:1831–1840. doi: 10.1038/npp.2015.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Polter AM, et al. Poststress block of kappa opioid receptors rescues long-term potentiation of inhibitory synapses and prevents reinstatement of cocaine seeking. Biological psychiatry. 2014;76:785–793. doi: 10.1016/j.biopsych.2014.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Keralapurath MM, Clark JK, Hammond S, Wagner JJ. Cocaine- or stress-induced metaplasticity of LTP in the dorsal and ventral hippocampus. Hippocampus. 2014;24:577–590. doi: 10.1002/hipo.22250. [DOI] [PubMed] [Google Scholar]
- 175.Pecina S, Berridge KC. Hedonic hot spot in nucleus accumbens shell: where do mu-opioids cause increased hedonic impact of sweetness? J Neurosci. 2005;25:11777–11786. doi: 10.1523/jneurosci.2329-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Zhang M, Kelley AE. Enhanced intake of high-fat food following striatal mu-opioid stimulation: microinjection mapping and fos expression. Neuroscience. 2000;99:267–277. doi: 10.1016/s0306-4522(00)00198-6. [DOI] [PubMed] [Google Scholar]
- 177.Haghighi A, et al. Opioid receptor mu 1 gene, fat intake and obesity in adolescence. Mol Psychiatry. 2014;19:63–68. doi: 10.1038/mp.2012.179. [DOI] [PubMed] [Google Scholar]
- 178.Apfelbaum M, Mandenoff A. Naltrexone suppresses hyperphagia induced in the rat by a highly palatable diet. Pharmacology, biochemistry, and behavior. 1981;15:89–91. doi: 10.1016/0091-3057(81)90344-0. [DOI] [PubMed] [Google Scholar]
- 179.Tabarin A, et al. Resistance to diet-induced obesity in mu-opioid receptor-deficient mice: evidence for a “thrifty gene”. Diabetes. 2005;54:3510–3516. doi: 10.2337/diabetes.54.12.3510. [DOI] [PubMed] [Google Scholar]
- 180.Burghardt PR, Rothberg AE, Dykhuis KE, Burant CF, Zubieta JK. Endogenous Opioid Mechanisms Are Implicated in Obesity and Weight Loss in Humans. The Journal of clinical endocrinology and metabolism. 2015;100:3193–3201. doi: 10.1210/jc.2015-1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Zuberi AR, Townsend L, Patterson L, Zheng H, Berthoud HR. Increased adiposity on normal diet, but decreased susceptibility to diet-induced obesity in mu-opioid receptor-deficient mice. European journal of pharmacology. 2008;585:14–23. doi: 10.1016/j.ejphar.2008.01.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Han W, et al. Increased body weight in mice lacking mu-opioid receptors. Neuroreport. 2006;17:941–944. doi: 10.1097/01.wnr.0000221829.87974.ad. [DOI] [PubMed] [Google Scholar]
- 183.Ookuma K, Barton C, York DA, Bray GA. Effect of enterostatin and kappa-opioids on macronutrient selection and consumption. Peptides. 1997;18:785–791. doi: 10.1016/s0196-9781(97)00029-6. [DOI] [PubMed] [Google Scholar]
- 184.Czyzyk TA, et al. kappa-Opioid receptors control the metabolic response to a high-energy diet in mice. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2010;24:1151–1159. doi: 10.1096/fj.09-143610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Vucetic Z, Kimmel J, Reyes TM. Chronic high-fat diet drives postnatal epigenetic regulation of mu-opioid receptor in the brain. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2011;36:1199–1206. doi: 10.1038/npp.2011.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Ong ZY, Wanasuria AF, Lin MZ, Hiscock J, Muhlhausler BS. Chronic intake of a cafeteria diet and subsequent abstinence. Sex-specific effects on gene expression in the mesolimbic reward system. Appetite. 2013;65:189–199. doi: 10.1016/j.appet.2013.01.014. [DOI] [PubMed] [Google Scholar]
- 187.Martire SI, et al. Extended exposure to a palatable cafeteria diet alters gene expression in brain regions implicated in reward, and withdrawal from this diet alters gene expression in brain regions associated with stress. Behavioural brain research. 2014;265:132–141. doi: 10.1016/j.bbr.2014.02.027. [DOI] [PubMed] [Google Scholar]
- 188.Blendy JA, et al. Reduced nicotine reward in obesity: cross-comparison in human and mouse. Psychopharmacology. 2005;180:306–315. doi: 10.1007/s00213-005-2167-9. [DOI] [PubMed] [Google Scholar]
- 189.Tsujii S, et al. Effects of food deprivation and high fat diet on opioid receptor binding in rat brain. Neurosci Lett. 1986;72:169–173. doi: 10.1016/0304-3940(86)90074-1. [DOI] [PubMed] [Google Scholar]
- 190.Alsio J, et al. Dopamine D1 receptor gene expression decreases in the nucleus accumbens upon long-term exposure to palatable food and differs depending on diet-induced obesity phenotype in rats. Neuroscience. 2010;171:779–787. doi: 10.1016/j.neuroscience.2010.09.046. [DOI] [PubMed] [Google Scholar]
- 191.Smith SL, Harrold JA, Williams G. Diet-induced obesity increases mu opioid receptor binding in specific regions of the rat brain. Brain research. 2002;953:215–222. doi: 10.1016/s0006-8993(02)03291-2. [DOI] [PubMed] [Google Scholar]
- 192.Ma D, et al. Effects of discontinuing a high-fat diet on mitochondrial proteins and 6-hydroxydopamine-induced dopamine depletion in rats. Brain research. 2015;1613:49–58. doi: 10.1016/j.brainres.2015.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Blaisdell AP, et al. Food quality and motivation: a refined low-fat diet induces obesity and impairs performance on a progressive ratio schedule of instrumental lever pressing in rats. Physiology & behavior. 2014;128:220–225. doi: 10.1016/j.physbeh.2014.02.025. [DOI] [PubMed] [Google Scholar]
- 194.Hryhorczuk C, et al. Saturated high-fat feeding independent of obesity alters hypothalamus-pituitary-adrenal axis function but not anxiety-like behaviour. Psychoneuroendocrinology. 2017;83:142–149. doi: 10.1016/j.psyneuen.2017.06.002. [DOI] [PubMed] [Google Scholar]
- 195.Dunn JP, et al. Decreased dopamine type 2 receptor availability after bariatric surgery: preliminary findings. Brain research. 2010;1350:123–130. doi: 10.1016/j.brainres.2010.03.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.de Weijer BA, et al. Striatal dopamine receptor binding in morbidly obese women before and after gastric bypass surgery and its relationship with insulin sensitivity. Diabetologia. 2014;57:1078–1080. doi: 10.1007/s00125-014-3178-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Steele KE, et al. Alterations of central dopamine receptors before and after gastric bypass surgery. Obesity surgery. 2010;20:369–374. doi: 10.1007/s11695-009-0015-4. [DOI] [PubMed] [Google Scholar]
- 198.Bond DS, et al. Pre- to postoperative physical activity changes in bariatric surgery patients: self report vs. objective measures. Obesity (Silver Spring, Md.) 2010;18:2395–2397. doi: 10.1038/oby.2010.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Ramirez-Marrero FA, Miles J, Joyner MJ, Curry TB. Self-reported and objective physical activity in postgastric bypass surgery, obese and lean adults: association with body composition and cardiorespiratory fitness. Journal of physical activity & health. 2014;11:145–151. doi: 10.1123/jpah.2012-0048. [DOI] [PubMed] [Google Scholar]
- 200.Sullivan EL, et al. Chronic consumption of a high-fat diet during pregnancy causes perturbations in the serotonergic system and increased anxiety-like behavior in nonhuman primate offspring. J Neurosci. 2010;30:3826–3830. doi: 10.1523/jneurosci.5560-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Morrison R, Reilly JJ, Penpraze V, Pendlebury E, Yam PS. A 6-month observational study of changes in objectively measured physical activity during weight loss in dogs. The Journal of small animal practice. 2014;55:566–570. doi: 10.1111/jsap.12273. [DOI] [PubMed] [Google Scholar]
- 202.Vitger AD, Stallknecht BM, Nielsen DH, Bjornvad CR. Integration of a physical training program in a weight loss plan for overweight pet dogs. Journal of the American Veterinary Medical Association. 2016;248:174–182. doi: 10.2460/javma.248.2.174. [DOI] [PubMed] [Google Scholar]
- 203.Rivera HM, et al. Maternal high-fat diet and obesity impact palatable food intake and dopamine signaling in nonhuman primate offspring. Obesity (Silver Spring, Md.) 2015;23:2157–2164. doi: 10.1002/oby.21306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Naef L, Gratton A, Walker CD. Exposure to high fat during early development impairs adaptations in dopamine and neuroendocrine responses to repeated stress. Stress (Amsterdam, Netherlands) 2013;16:540–548. doi: 10.3109/10253890.2013.805321. [DOI] [PubMed] [Google Scholar]
- 205.Romani-Perez M, et al. Impact of perinatal exposure to high-fat diet and stress on responses to nutritional challenges, food-motivated behaviour and mesolimbic dopamine function. International journal of obesity (2005) 2017;41:502–509. doi: 10.1038/ijo.2016.236. [DOI] [PubMed] [Google Scholar]
- 206.Camacho A, Montalvo-Martinez L, Cardenas-Perez RE, Fuentes-Mera L, Garza-Ocanas L. Obesogenic diet intake during pregnancy programs aberrant synaptic plasticity and addiction-like behavior to a palatable food in offspring. Behavioural brain research. 2017;330:46–55. doi: 10.1016/j.bbr.2017.05.014. [DOI] [PubMed] [Google Scholar]
- 207.Liu J, Han L, Zhu L, Yu Y. Free fatty acids, not triglycerides, are associated with non-alcoholic liver injury progression in high fat diet induced obese rats. Lipids in health and disease. 2016;15:27. doi: 10.1186/s12944-016-0194-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Farr SA, et al. Obesity and hypertriglyceridemia produce cognitive impairment. Endocrinology. 2008;149:2628–2636. doi: 10.1210/en.2007-1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Valladolid-Acebes I, et al. High-fat diets induce changes in hippocampal glutamate metabolism and neurotransmission. American journal of physiology Endocrinology and metabolism. 2012;302:E396–402. doi: 10.1152/ajpendo.00343.2011. [DOI] [PubMed] [Google Scholar]
- 210.Hryhorczuk C, et al. Oleic Acid in the Ventral Tegmental Area Inhibits Feeding, Food Reward, and Dopamine Tone. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2018;43:607–616. doi: 10.1038/npp.2017.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Esteban JA, et al. PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat Neurosci. 2003;6:136–143. doi: 10.1038/nn997. [DOI] [PubMed] [Google Scholar]
- 212.Fourcaudot E, et al. cAMP/PKA signaling and RIM1alpha mediate presynaptic LTP in the lateral amygdala. Proc Natl Acad Sci U S A. 2008;105:15130–15135. doi: 10.1073/pnas.0806938105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Liu S, Rao Y, Daw N. Roles of protein kinase A and protein kinase G in synaptic plasticity in the visual cortex. Cerebral cortex (New York, N.Y.: 1991) 2003;13:864–869. doi: 10.1093/cercor/13.8.864. [DOI] [PubMed] [Google Scholar]
- 214.Hayashi Y, et al. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science. 2000;287:2262–2267. doi: 10.1126/science.287.5461.2262. [DOI] [PubMed] [Google Scholar]
- 215.Roche KW, O’Brien RJ, Mammen AL, Bernhardt J, Huganir RL. Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron. 1996;16:1179–1188. doi: 10.1016/s0896-6273(00)80144-0. [DOI] [PubMed] [Google Scholar]
- 216.London E, Nesterova M, Stratakis CA. Acute vs chronic exposure to high fat diet leads to distinct regulation of PKA. Journal of molecular endocrinology. 2017;59:1–12. doi: 10.1530/jme-16-0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Robison AJ, Nestler EJ. Transcriptional and epigenetic mechanisms of addiction. Nat Rev Neurosci. 2011;12:623–637. doi: 10.1038/nrn3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Hellsten SV, et al. The gene expression of the neuronal protein, SLC38A9, changes in mouse brain after in vivo starvation and high-fat diet. PLoS One. 2017;12:e0172917. doi: 10.1371/journal.pone.0172917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Nam KN, et al. Effect of high fat diet on phenotype, brain transcriptome and lipidome in Alzheimer’s model mice. Scientific reports. 2017;7:4307. doi: 10.1038/s41598-017-04412-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Carlezon WA, Jr, et al. Regulation of cocaine reward by CREB. Science. 1998;282:2272–2275. doi: 10.1126/science.282.5397.2272. [DOI] [PubMed] [Google Scholar]
- 221.Barrot M, et al. CREB activity in the nucleus accumbens shell controls gating of behavioral responses to emotional stimuli. Proc Natl Acad Sci U S A. 2002;99:11435–11440. doi: 10.1073/pnas.172091899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Elliott E, Ezra-Nevo G, Regev L, Neufeld-Cohen A, Chen A. Resilience to social stress coincides with functional DNA methylation of the Crf gene in adult mice. Nat Neurosci. 2010;13:1351–1353. doi: 10.1038/nn.2642. [DOI] [PubMed] [Google Scholar]
- 223.Ye J. Mechanisms of insulin resistance in obesity. Frontiers of medicine. 2013;7:14–24. doi: 10.1007/s11684-013-0262-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Gupta N, Jensen MD. Clinical effects of high-fat meals and weight gain due to high-fat feeding. International journal of obesity supplements. 2012;2:S51–55. doi: 10.1038/ijosup.2012.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Nash AI. Crosstalk between insulin and dopamine signaling: A basis for the metabolic effects of antipsychotic drugs. Journal of chemical neuroanatomy. 2017;83–84:59–68. doi: 10.1016/j.jchemneu.2016.07.010. [DOI] [PubMed] [Google Scholar]
- 226.Meyers AM, Mourra D, Beeler JA. High fructose corn syrup induces metabolic dysregulation and altered dopamine signaling in the absence of obesity. PLoS One. 2017;12:e0190206. doi: 10.1371/journal.pone.0190206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Lu J, et al. Ursolic acid improves high fat diet-induced cognitive impairments by blocking endoplasmic reticulum stress and IkappaB kinase beta/nuclear factor-kappaB-mediated inflammatory pathways in mice. Brain, behavior, and immunity. 2011;25:1658–1667. doi: 10.1016/j.bbi.2011.06.009. [DOI] [PubMed] [Google Scholar]
- 228.Miller AA, Spencer SJ. Obesity and neuroinflammation: a pathway to cognitive impairment. Brain, behavior, and immunity. 2014;42:10–21. doi: 10.1016/j.bbi.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 229.Kohman RA, Rhodes JS. Neurogenesis, inflammation and behavior. Brain, behavior, and immunity. 2013;27:22–32. doi: 10.1016/j.bbi.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Yirmiya R, Goshen I. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain, behavior, and immunity. 2011;25:181–213. doi: 10.1016/j.bbi.2010.10.015. [DOI] [PubMed] [Google Scholar]
- 231.Hargrave SL, Davidson TL, Zheng W, Kinzig KP. Western diets induce blood-brain barrier leakage and alter spatial strategies in rats. Behavioral neuroscience. 2016;130:123–135. doi: 10.1037/bne0000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Bilbo SD. Early-life infection is a vulnerability factor for aging-related glial alterations and cognitive decline. Neurobiology of learning and memory. 2010;94:57–64. doi: 10.1016/j.nlm.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Bellinger FP, Madamba SG, Campbell IL, Siggins GR. Reduced long-term potentiation in the dentate gyrus of transgenic mice with cerebral overexpression of interleukin-6. Neurosci Lett. 1995;198:95–98. doi: 10.1016/0304-3940(95)11976-4. [DOI] [PubMed] [Google Scholar]
- 234.Cunningham AJ, Murray CA, O’Neill LA, Lynch MA, O’Connor JJ. Interleukin-1 beta (IL-1 beta) and tumour necrosis factor (TNF) inhibit long-term potentiation in the rat dentate gyrus in vitro. Neurosci Lett. 1996;203:17–20. doi: 10.1016/0304-3940(95)12252-4. [DOI] [PubMed] [Google Scholar]
- 235.Hodes GE, Kana V, Menard C, Merad M, Russo SJ. Neuroimmune mechanisms of depression. Nat Neurosci. 2015;18:1386–1393. doi: 10.1038/nn.4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Dorfman MD, Thaler JP. Hypothalamic inflammation and gliosis in obesity. Current opinion in endocrinology, diabetes, and obesity. 2015;22:325–330. doi: 10.1097/med.0000000000000182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Thaler JP, Guyenet SJ, Dorfman MD, Wisse BE, Schwartz MW. Hypothalamic inflammation: marker or mechanism of obesity pathogenesis? Diabetes. 2013;62:2629–2634. doi: 10.2337/db12-1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Fischer IP, et al. A history of obesity leaves an inflammatory fingerprint in liver and adipose tissue. International journal of obesity (2005) 2017 doi: 10.1038/ijo.2017.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Clark PJ, et al. Wheel running alters patterns of uncontrollable stress-induced cfos mRNA expression in rat dorsal striatum direct and indirect pathways: A possible role for plasticity in adenosine receptors. Behavioural brain research. 2014;272:252–263. doi: 10.1016/j.bbr.2014.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]