

Previous studies demonstrated the experimental evolution of vaccinia virus (VACV) following deletion of a host range gene important for evasion of host immune defenses. We have extended experimental evolution to essential genes that cannot be deleted but could be replaced by a divergent orthologous gene from another poxvirus. Replacement of a VACV transcription factor gene with one from a distantly related poxvirus led to decreased fitness as evidenced by diminished replication. Serially passaging the hybrid virus at a low multiplicity of infection provided conditions for selection of adaptive mutations that improved replication. Notably, these included five independent mutations of the largest and second largest RNA polymerase subunits. This approach should be generally applicable for investigating adaptation to swapping of orthologous genes encoding additional essential proteins of poxviruses as well as other viruses.
KEYWORDS: evolution, homologous recombination, myxoma virus, poxvirus, transcription factors, vaccinia virus
ABSTRACT
High-throughput DNA sequencing enables the study of experimental evolution in near real time. Until now, mutants with deletions of nonessential host range genes were used in experimental evolution of vaccinia virus (VACV). Here, we guided the selection of adaptive mutations that enhanced the fitness of a hybrid virus in which an essential gene had been replaced with an ortholog from another poxvirus genus. Poxviruses encode a complete system for transcription, including RNA polymerase and stage-specific transcription factors. The abilities of orthologous intermediate transcription factors from other poxviruses to substitute for those of VACV, as determined by transfection assays, corresponded with the degree of amino acid identity. VACV in which the A8 or A23 intermediate transcription factor subunit gene was replaced by the myxoma (MYX) virus ortholog exhibited decreased replication. During three parallel serial passages of the hybrid virus with the MYXA8 gene, plaque sizes and virus yields increased. DNA sequencing of virus populations at passage 10 revealed high frequencies of five different single nucleotide mutations in the two largest RNA polymerase subunits, RPO147 and RPO132, and two different Kozak consensus sequence mutations predicted to increase translation of the MYXA8 mRNA. Surprisingly, there were no mutations within either intermediate transcription factor subunit. Based on homology with Saccharomyces cerevisiae RNA polymerase, the VACV mutations were predicted to be buried within the internal structure of the enzyme. By directly introducing single nucleotide substitutions into the genome of the original hybrid virus, we demonstrated that both RNA polymerase and translation-enhancing mutations increased virus replication independently.
IMPORTANCE Previous studies demonstrated the experimental evolution of vaccinia virus (VACV) following deletion of a host range gene important for evasion of host immune defenses. We have extended experimental evolution to essential genes that cannot be deleted but could be replaced by a divergent orthologous gene from another poxvirus. Replacement of a VACV transcription factor gene with one from a distantly related poxvirus led to decreased fitness as evidenced by diminished replication. Serially passaging the hybrid virus at a low multiplicity of infection provided conditions for selection of adaptive mutations that improved replication. Notably, these included five independent mutations of the largest and second largest RNA polymerase subunits. This approach should be generally applicable for investigating adaptation to swapping of orthologous genes encoding additional essential proteins of poxviruses as well as other viruses.
INTRODUCTION
The Poxviridae comprise a large family of viruses that infect vertebrates and invertebrates (1). During their evolution, chordopoxviruses segregated into 11 recognized genera as well as additional unassigned species. Analysis of the genomes of representatives of the various genera showed that approximately 90 genes encoding proteins for essential functions, including entry, transcription, genome replication, disulfide bond formation, and virion assembly have been preserved (2). A similar number of less well conserved genes are unnecessary for replication in cell culture; many of these genes are involved in host interactions and are present in only a subset of poxvirus genera (3). The diversity of the latter genes is likely related to their acquisition and adaptive modification during the long period of poxvirus evolution and speciation in various hosts (4). In some cases, these viral proteins counteract cellular innate immune responses, whereas others have roles that are not yet understood (5).
In contrast to the relatively low rate of natural evolution within a single host species, propagation of vaccinia virus (VACV) as the smallpox vaccine in unnatural hosts such as calf skin and cell culture over the past 200 years has promoted rapid changes (6). A striking example of this is modified VACV Ankara (MVA), which lost approximately 15% of the genome and suffered a severe host restriction during >500 passages in chicken embryo fibroblasts (7). Presently, high-throughput sequencing methods enable investigation of the experimental evolution of poxviruses in near real time. Thus far, such studies have been limited to VACV host range mutants with deletions of genes involved in evasion of the PKR/eukaryotic initiation factor 2α (eIF2α) antiviral pathway (8–11). The genetic changes comprised both copy number amplification and point mutation of individual viral genes. We are seeking to extend experimental evolution to essential genes encoding proteins that have coadapted with other viral proteins during natural selection. Our approach is to swap orthologous genes from distantly related poxviruses, thereby encouraging the selection of adaptive mutations that enhance replication. In principle, the results could provide insights into protein interactions and pathways. In this first effort to apply orthologous gene swapping for experimental evolution of poxviruses, we targeted the transcription system.
Poxviruses have three stages of gene expression: early, intermediate, and late (12). An eight-subunit DNA-dependent RNA polymerase (Pol) that is homologous to the polymerases of archaea and eukaryotes (13, 14) acts in conjunction with stage-specific transcription factors and promoters. The VACV early transcription factor is a heterodimer of an 82-kDa and a 70-kDa subunit with ATPase activity that binds to the core region of early promoters and DNA downstream of the RNA start site, thereby altering the DNA conformation (15–17), and to the RNA Pol-associated protein RAP94 (18, 19). The intermediate factor is a heterodimer of 34- and 45-kDa proteins (20), and the late factor is comprised of 17-, 26-, and 30-kDa proteins (21). The intermediate and late transcription factors have no known catalytic activities, nor is there evidence of direct association with DNA or RNA Pol.
The likelihood that chordopoxviruses use a common mechanism for mRNA synthesis can be inferred from the conservation of their transcription apparatus and from the sequence similarity and functional interchangeability of their promoters (22–25). However, the compatibility of orthologous transcription factors has yet to be investigated. Transient expression experiments described here indicated that orthologous intermediate transcription factors exhibit a range of compatibilities with VACV. Using this information, we constructed hybrid viruses in which the genes encoding the VACV 34-kDa (A8) or 45-kDa (A23) intermediate transcription factor subunit were individually replaced with the myxoma virus (MYXV) ortholog (abbreviated MYXA8 and MYXA23, respectively) and then carried out serial passaging of the poorly replicating viruses to allow experimental evolution and adaptive selection. Substantial increases in replicative abilities of the MYXA8 VACV were associated with mutations in RNA Pol subunits and sequence changes around the translation initiation site of the MYXA8 gene.
RESULTS
Transient expression assays.
To initiate this study, we compared the A8 and A23 intermediate transcription factor orthologs of chordopoxvirus genera. For diversity, we selected the genes from myxoma virus (MYXV), canarypox virus (CNPV), Nile crocodilepox virus (CRV), and salmon gill poxvirus (SGPV), which encode proteins that vary in amino acid identity from 65% to 23% relative to VACV sequences (Table 1). The order of similarity to VACV was MYXV > CNPV > CRV > SGPV, consistent with their overall genome sequence similarities.
TABLE 1.
Comparison of intermediate transcription factor orthologs
| Virus | % amino acid identity |
|
|---|---|---|
| A8 | A23 | |
| Vaccinia | 100 | 100 |
| Myxoma | 65 | 58 |
| Canarypox | 38 | 51 |
| Crocodilepox | 31 | 40 |
| Salmon gill pox | 29 | 23 |
Our plan was to analyze intermediate gene expression after infecting cells with a recombinant VACV lacking the gene encoding A8, A23, or both (ΔA8, ΔA23, and ΔA8/ΔA23 strains, respectively) and transfecting DNA encoding corresponding orthologs. Because the A8 and A23 genes are essential, deletion viruses were previously constructed with the aid of a complementing cell line that expressed both intermediate transcription factors (26). We made new versions of these viruses that constitutively express the bacteriophage T7 RNA polymerase so that a T7 promoter could be used for expression of A8 and A23 genes from transfected plasmids. The starting virus vTF7-3 (27) contains the A8 and A23 genes from the VACV Western Reserve (WR) strain and will be referred to here as the wild type (WT). The VACV A8 or A23 gene or both genes were deleted and replaced by the green fluorescent protein (GFP) or Discosoma sp. red fluorescent protein (DsRed) open reading frame (ORF) regulated by a VACV promoter to facilitate isolation in the complementing cell line (Fig. 1A). Loss of the A8 and A23 ORFs was confirmed by PCR, and the inability of the viruses to express viral intermediate proteins was demonstrated by Western blotting (data not shown).
FIG 1.
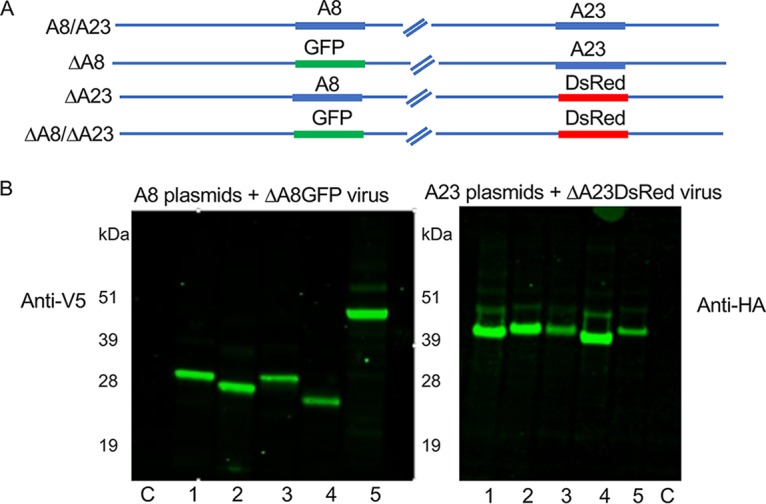
Transient expression of intermediate transcription factor orthologs. (A) Recombinant VACV that express T7 RNA Pol and contains the VACV A8 and A23 intermediate transcription factors (A8/A23), only A23 (ΔA8), only A8 (ΔA23), or neither A8 or A23 (ΔA8/ΔA23) were constructed by homologous recombination using GFP and/or DsRed as a reporter. In the scheme shown, for example, A8/A23 virus expresses both A8 and A23, while ΔA8 virus expresses a GFP reporter and A23. (B) Expression of A8 and A23 orthologs. BS-C-1 cells were infected with ΔA8 or ΔA23 virus and transfected with plasmids that encode A8 orthologs with V5 epitope tags or A23 orthologs with HA epitope tags regulated by T7 promoters. Following cell lysis, proteins were resolved on SDS-polyacrylamide gels, transferred to membranes, and detected with anti-V5 and anti-HA antibodies. The poxviruses from which the A8 or A23 orthologs were derived are as follows: lanes 1, VACV; lanes 2, MYXV; lanes 3, CNPV; lanes 4, CRV; lanes 5, SGPV. C represents a control plasmid not expressing a protein. The masses in kilodaltons to the left of each blot indicate the mobilities of marker proteins.
For construction of the A8 and A23 expression plasmids, the orthologous ORFs were mammalian-codon optimized to minimize differences in G/C content, which might affect expression. Each plasmid of one set contained an A8 ORF with a V5 epitope tag, while each plasmid of the other set had an A23 ORF with a hemagglutinin (HA) epitope tag. Expression of the proteins following transfection of the plasmids into cells infected with recombinant viruses is shown in Fig. 1B. Some differences in the intensities of the bands were noted, but the weaker bands were not enhanced by altering the amount of plasmid transfected.
A plasmid with the firefly luciferase (Fluc) gene regulated by the VACV G8 intermediate/late promoter (12, 28) was used to monitor the activities of the intermediate transcription factors. The scheme in which cells are infected with one recombinant VACV of the group expressing T7 RNA polymerase (ΔA8, ΔA23, and ΔA8/ΔA23) and transfected with one or two plasmids of the group containing the T7 promoter (A8, A23, and both A8 and A23) in which A8 and A23 are derived from VACV, MYXV, CNPV, CRV, or SGPV and a second reporter plasmid, G8-Fluc, is outlined in Fig. 2A. Only the VACV and MYXV A8 genes allowed substantial expression of Fluc (Fig. 2B, left). In contrast, all of the orthologous A23 genes except the SGPV A23 gene enhanced Fluc in the order VACV > MYXV > CNPV > CRV (Fig. 2B, middle). In the experiments described so far, one of the intermediate transcription factor subunits was from VACV, while the other was one of the orthologs. We considered that the activities of the transcription factor orthologs might be relatively low because they had to work in concert with the VACV factor. Therefore, in the next experiment we infected cells with a VACV lacking both A8 and A23 and transfected two plasmids expressing both transfection factors from the same virus. However, the ranking was similar to that obtained by transfecting A8 or A23 alone (Fig. 2B, right).
FIG 2.

Complementation of intermediate gene expression by orthologous transcription factors. (A) Experimental plan. Cells were infected with ΔA8, ΔA23, or ΔA8/ΔA23 VACV encoding T7 RNA polymerase and transfected with plasmids encoding A8, A23, or both A8 and A23 (A8+A23) regulated by the T7 promoter and with Fluc regulated by an intermediate/late promoter. Fluc activity depended on expression of A8 and A23 proteins. (B) The plan depicted in panel A was carried out by transfecting plasmids into HeLa cells infected with ΔA8, ΔA23, and ΔA8/ΔA23 viruses. Relative luminescence values are shown. Experiments were carried out in triplicate, and the bars indicate the standard errors of the means.
Construction of hybrid viruses.
We made hybrid viruses in order to further investigate the effects of orthologous transcription factors. Our strategy was to start with viruses that had A8 or A23 replaced by GFP or DsRed and then exchange the fluorescent reporter gene with MYXV or CNPV A8 with an N-terminal V5 tag or with A23 with an N-terminal HA tag by homologous recombination. The recombinant viruses with MYXV genes were detected by formation of nonfluorescent plaques in BS-C-1 cells, and three separate clones with MYXA8 or MYXA23 designated A, B, or C were isolated (Fig. 3A). The presence of the MYXV genes was confirmed by DNA sequencing. The MYXA8 hybrid made predominantly tiny plaques, whereas the MYXA23 hybrid made intermediate-sized plaques (Fig. 3B). The different plaque sizes of the hybrid viruses were consistent with the relative activities of MYXA8 and MYXA23 in transfection experiments (Fig. 2B). A similar attempt to make CNPV and CRV A23 hybrids in BS-C-1 cells failed, evidently because of poor replication.
FIG 3.
Plaque formation by hybrid viruses expressing VACV and MYXV intermediate transcription factors. (A) Diagram of viruses expressing the VACV WRA8 and WRA23 (WT), MYXA8 and WRA23 (MYXA8), and WRA8 and MYXA23 (MYXA23). (B) Plaques were stained with crystal violet at 48 h after infection of BS-C-1 cells with WT or three independent clones of the hybrid viruses labeled A, B, and C.
Experimental evolution.
The inefficient replication of the hybrid viruses containing MYXV transcription factors suggested that beneficial mutations might arise, perhaps in the transcription factors themselves, during serial passaging. The scheme shown in Fig. 4 was carried out starting with stocks of the three independent plaque-purified clones (A, B, and C) of MYXA8 and MYXA23, which we refer to as passage 0 (P0). Ten passages were carried out, and a portion of each was frozen as the “fossil record.” Plaques formed with the MYXA8 virus at passage 10 (P10) exhibited a range of sizes, including many plaques larger than those of P0 (data not shown). In contrast, the plaques formed with MYXA23 did not significantly increase in size after passaging (data not shown). The 24-h yields of the MYXA8 P10 viruses were 3 to 5 times higher than those of the P0 viruses, whereas the yields of MYXA23 did not appreciably change during passage (data not shown) and will not be discussed further.
FIG 4.
Experimental evolution of MYXA8 virus. (A) Scheme for serial passaging in BS-C-1 cells of MYXA8 virus containing the MYXVA8 and VACVA23 genes. After each passage, a sample was saved to serve as the fossil record. (B) Three clones of MYXA8 virus designated A, B, and C in Fig. 3 were independently passaged at a multiplicity of infection of approximately 0.1 PFU/cell. Plaques formed in 48 h on BS-C-1 cells by WT virus containing the VACV A8 and A23 genes and by hybrid viruses at passage 0 (MYXA8 P0) and passage 10 (MYXA8 P10) plaque-purified clones from serial passages A, B, and C are shown.
Large plaques were picked from each of the three independent A, B, and C P0 and P10 MYXA8 passages, and the viruses were plaque purified several times. The plaque-purified viruses derived from the series A, B, and C passage P10 populations are referred to as P10 clones A, B, and C, respectively, to indicate their origin. The plaque sizes of the P10 clones, relative to those of the P0 and the WT virus with VACV A8, are shown in Fig. 4B. The plaque-purified P10 clones were significantly larger than those of P0, and the P10 A clones approached the size of the WT clones. The enhanced replication of the MYXA8 A, B, and C clones from P10 compared to levels of P0 and the WT are shown in Fig. 5. At 12 h, the titers of the P10 A and B clones were significantly higher than those of the P0 viruses, and by 24 h all three were higher, with A and B approaching the level of the WT.
FIG 5.

Replication of MYXA8 P0 and P10 clones. Triplicate BS-C-1 cell monolayers were infected for 6, 12, and 24 h with 5 PFU/cell of WT virus, and MYXA8 P0 and P10 clones from passages A, B, and C. Virus titers were determined in duplicate by plaque assay in BS-C-1 cells. Bars represent standard errors of the means. *, P < 0.05; **, P < 0.01.
Genome sequencing.
Whole-genome sequencing was performed on the population of viruses in P10 of series A, B, and C and on plaque-purified clones from each series after amplification in BS-C-1 cells. Both synonymous and nonsynonymous changes in the viral genome were found, and the complete sequences have been archived. The genetic changes considered most significant are listed in Table 2. Single nucleotide nonsynonymous mutations of RNA Pol were detected in each of the passage populations. Two of the mutations are in RPO147 (ORF J6), the largest subunit, and three are in RPO132 (ORF A24), the second-largest subunit. Two of the RPO132 mutations were found in relatively high frequency in the passage B population but were not in the plaque-purified clone from this passage (Table 2). The five mutations are all different, indicating that they arose independently in the three separate passages. Sequence changes that potentially increase expression of MYXA8 were also found (Table 2) and will be discussed below. The mutations in the clones were present at 100%, indicating their purity.
TABLE 2.
Mutation abundance in MYXA8 P10 passage population and clones
| Passage series | Amino acid change | ORF | Gene | Mutation frequency (%) |
|
|---|---|---|---|---|---|
| Passage populationa | Clone populationb | ||||
| A | M424I | J6R | RPO147e | 30 | 100 |
| L013M | MYXA8c | MYXA8 | 20 | 100 | |
| B | A1212V | J6R | RPO147 | 6 | 100 |
| None | MYXA8d | MYXA8 | 18 | 100 | |
| A1026V | A24R | RPO132 | 14 | 0 | |
| A1133T | A24R | RPO132 | 30 | 0 | |
| C | K1046Q | A24R | RPO132 | 69 | 100 |
Passage 10 virus was amplified prior to DNA purification.
Virus from large plaques was clonally purified and amplified.
New translation initiation codon within N-terminal V5 tag preceding MYXA8 ORF.
Improved Kozak consensus sequence preceding N-terminal V5 tag; carries a single nucleotide change at the −3 position relative to the start codon.
RPO, RNA polymerase subunit.
In addition to whole-genome sequencing of the cloned viruses and the P10 population, we performed amplicon sequencing of genomes from sequential passages to determine when changes in the RPO147 and RPO132 sequences occurred (Table 3). The mutation K1046Q in RPO132 was detected in P2 of the passage series C population and steadily increased in frequency, reaching 55% of the population at P10. The mutation M4241 in RPO147 was detected at P4 of the passage series A population and increased to 24% of the population by P10. The mutation A1212V in RPO147 was detected at P8 of the passage series B population and was only 1.9% of the total population at P10. Note that the RNA Pol mutations of passage series A, B, and C increased to 30%, 6%, and 69% of the population, respectively, with the additional passages used to purify DNA for whole-genome sequencing, indicating that selection was still occurring (Table 2).
TABLE 3.
Changes in abundance of RNA Pol mutations during passages
| Passage no. | Frequency of mutation (%)a |
||
|---|---|---|---|
| RPO147 M424I | RPO147 A1212V | RPO132 K1046Q | |
| 0 | ND | ND | ND |
| 2 | ND | ND | 1.7 |
| 4 | 3.0 | ND | 10.1 |
| 6 | 6.1 | ND | 17.6 |
| 8 | 11.2 | 1.2 | 38.1 |
| 9 | 16.8 | 1.5 | 50.1 |
| 10 | 24.2 | 1.9 | 55.3 |
ND, not detected.
RNA Pol mutations increase replication of the MYXA8 hybrid VACV.
The RNA Pol mutations were mapped to the homologous Saccharomyces cerevisiae RNA Pol II (RNAP II) subunits (Fig. 6A). The yeast amino acids corresponding to the mutated ones are located internally, rather than in the solvent-exposed surface where interactions with transcription factors might be expected. To determine the significance of the RNA Pol mutations in the absence of other sequence changes in the hybrid virus genomes, homologous recombination with PCR products containing the mutations was used to directly modify the P0 viruses. DNA segments of approximately 1,000 bp that encompassed the single nucleotide RNA Pol mutations were derived by PCR from the DNA of the cloned P10 viruses and transfected into BS-C-1 cells that were infected with the original MYXA8 P0 viruses. After 20 h, virus was collected, and individual plaques were screened by PCR and Sanger sequencing. Approximately 20 to 30% of the plaques had the desired mutations, attesting to the high rate of recombination, and no additional mutations were found by whole-genome sequencing. Following plaque purification and amplification, the plaque sizes of the mutated and cloned viruses were compared with those of the MYXA8 P0 virus containing the WT RNA Pol sequences. The plaques of the viruses with the RPO147 M424I mutation and the RPO132 K1046Q mutation were larger than those of the original MYXA8 virus (Fig. 6B), whereas the difference was small in the case of the virus with the RPO147 A1212V mutation (data not shown). Plaque size measurements of all three recombinant viruses are presented in a subsequent figure.
FIG 6.

RNA Pol mutations. (A) Mutations in RPO147 and RPO132 that occurred during passages of MYXA8 were mapped onto the S. cerevisiae RNAP II crystal structure. Green, RPB1; blue, RPB2. (B) Single nucleotide mutations were inserted by homologous recombination into RPO147 and RPO132 of MYXA8 P0 viruses. Plaques formed by the cloned recombinant viruses and a MYXA8 P0 virus are shown. (C) Virus yields at 2, 24, 48, and 72 h after infection with 0.01 PFU/cell of WT, MYXA8 P0 clone A (MYX-A8-A), MYXA8 P0 clone C (MYX-A8-C), and the recombinant viruses with single nucleotide mutations in RPO147 (M424I) or RPO132 (K1046Q). (D) Replication competition between the MYXA8 P0 clone A virus and the recombinant virus with mutation in RPO147. BS-C-1 cells were infected in triplicate with 0.01 PFU/cell of a mixture comprised of 90% MYXA8 P0 and 10% MYXA8 with the M424I mutation in RPO147. After each of three rounds of passaging, an aliquot was removed for Sanger sequencing of the region including the M424I mutation. Graphs of the sequence are shown with a dotted oval around the mutated nucleotide. The approximate percentages of M424I after the third passages are indicated at the right.
We also determined the virus yields following infection with 0.01 PFU/cell of the MYXA8 P0 clones, recombinant viruses with mutated RNA Pol, and WT virus. Notably, the yields of viruses with the RPO147 M424I mutation from P10 clone A and the RPO132 K1046Q mutation from P10 clone C were higher than those of the P0 MYXA8 A and C clones, which had unmutated RPO147 and RPO132 (Fig. 6C), indicating that these mutations were adaptive.
A competition experiment was carried out to confirm the ability of the recombinant virus with the RPO147 M424I mutation to enhance replication relative to the level of the original MYXA8 P0 virus. Three BS-C-1 cell monolayers were each infected with 0.01 PFU cell of a mixture comprised of 90% MYXA8 P0 virus with the WT RPO147 sequence and 10% RPO147 M424I virus for 48 h. The cells were then harvested and titrated, and the virus was passaged two more times. After each passage, PCR and Sanger sequencing were performed to estimate the ratios of the WT RPO147 sequence and the mutated sequence. In the three independent experiments, the percentages of mutated sequence after passage 3 were ∼90%, 70%, and 100% (Fig. 6D), demonstrating the selective advantage of the mutation. In a parallel competition experiment with WT VACV (90%) and WT VACV that had the RPO147 mutation (10%), the relative amounts of the two remained constant during the three passages (data not shown). Thus, the advantage of the RPO147 M424I mutation was specific for the MYXA8 hybrid virus.
Mutations of translation initiation sequences of MYXA8 gene.
The P10 clones of passage A and B had a mutation that introduced a new translation initiation codon with an optimal Kozak sequence within the N-terminal V5 tag and a mutation that was predicted to improve the original translation initiation sequence of the MYXA8 ORF, respectively (Fig. 7A). The nucleotides around the translation initiation codon of the V5-tagged MYXA8 in the original P0 clones are CTTATGG, with the ones at −3 and +4 underlined. Although the G at +4 is consistent with good translation initiation, a purine rather than a C is optimal at −3 (29). The single nucleotide mutation in P10 clone A introduced a new translation initiation codon with preferred nucleotides at both −3 and +4 (GGCATGG) within the V5 tag of clone A, while the mutation in P10 clone B changed the C at −3 to a preferred A. Homologous recombination with mutated PCR product was used to introduce these mutations into the MYXA8 P0 clones, similar to the protocol used to make the RNA Pol mutations. Random plaques were selected for Sanger sequencing, and those with the expected mutation were cloned. Comparison of the plaques formed by a MYXA8 P0 clone and the recombinant viruses revealed that the latter produced larger plaques (Fig. 7B). As anticipated, the V5-tagged MYXA8 protein was expressed more highly in cells infected with the virus with the clone B mutation than with the MYXA8 P0 clone, whereas E3, another early viral protein, was expressed in similar amounts (Fig. 7C). A comparable Western blot analysis of the P10 clone A mutant was not possible because of the interruption of the V5 tag.
FIG 7.

Mutations at translation initiation sites of the MYXA8 gene. (A) The nucleotide and amino acid sequences around the N-terminal V5 tag of the MYXA8 gene in the original construct and the nucleotide mutations in the P10 A and B clones are shown. The ATG start codon at the start of the tag sequence is shaded green, and the nucleotides at the −3 and +4 positions are underlined. The new ATG within the V5 tag of the clone A mutant is shaded yellow. (B) Single nucleotide mutations were inserted by homologous recombination into the MYXA8 P0 virus genome to introduce a new start codon within the V5 tag (clone A mutation) or at the −3 position relative to the original start codon (clone B mutation). Plaques formed in BS-C-1 cells after 48 h by the cloned recombinant viruses and the MYXA8 P0 virus are shown. (C) Expression of the MYXA8 protein. BS-C-1 cells were infected with MYXA8 P0 and the virus with the clone B mutation at a multiplicity of infection of 5 PFU/cell. At 8 and 12 h after infection (hpi), the cells were harvested, and the proteins were analyzed by Western blotting with antibody to V5 to visualize the MYXA8 protein and to E3, another early protein.
Comparison of plaque sizes of clonal isolates and recombinants with single nucleotide changes.
The P10 clone A and B viruses had both RNA Pol and translation initiation site mutations. In order to determine the relative contributions of the individual mutations, we compared the plaque sizes of the P10 clones with those of mutants with single nucleotide changes in either an RNA Pol subunit or MYXA8 translation initiation sequence. The measurements in Fig. 8 are presented as box-and-whisker plots: the top and bottom of the box show the upper and lower quartiles, the horizontal line within the box is the median, and the vertical lines are the maximum and minimum values, with the dots representing outliers that are more or less than 3/2 of the upper and lower quartiles, respectively. The plaque sizes of the viruses with individual RNA Pol mutations were each larger than those of the MYXA8 P0 virus. The plaques formed by the K1046Q mutant virus were similar in size to those of the P10 clone C virus, confirming that the latter had no other significant adaptive mutations, as indicated in Table 2. In contrast, the plaques formed by the M424I and A212V RNA Pol mutant viruses were smaller than the clone A and B P10 viruses, which also had mutations in the MYXA8 Kozak consensus sequences (Table 2). Furthermore, the plaques of the mutants with changes in the Kozak consensus sequences were also larger than those of the MYXA8 P0 virus, indicating that the latter as well as the RNA Pol mutations were adaptive. However, none of the P10 clones made plaques as large as those of the WT VACV (Fig. 8). The color coding in Fig. 8 groups the mutations present in the cloned viruses as detailed in the legend.
FIG 8.

Comparison of plaque sizes of recombinant and passaged virus clones. The plaque sizes of recombinant viruses with RNA Pol or Kozak sequence mutations, of MYXA8 P10 clones, of MYXA8 P0 clone A, and of WT virus with the VACV A8 gene are shown. Three independent wells, for a total of 150 to 300 plaques, were analyzed for each virus. Box-and-whisker plots are shown. Color coding: red, clone A and recombinant viruses with individual mutations in that clone; violet, clone B and recombinant viruses with individual mutations in that clone; yellow, clone C and the recombinant virus with the individual mutation in that clone.
DISCUSSION
Experimental evolution studies are usually carried out with RNA viruses, which are facilitated by high rates of mutation compared to those of DNA viruses. The low mutation rate of VACV has been attributed to intrinsic 3′ → 5′ exonuclease activity of the viral DNA polymerase (30–32). Nevertheless, recent studies have shown that adaptation of VACV host range mutants with deletions of genes involved in evasion of the PKR/eIF2α antiviral pathway can occur both by gene amplification and point mutations during serial passaging (8–11). We initiated the current project to investigate experimental evolution of essential genes and chose the A8 and A23 intermediate transcription factor genes for this purpose. Since there were no mutagenesis studies of these essential proteins, we replaced the VACV factors with orthologs from other chordopoxvirus genera. Transfection experiments indicated that the ability of orthologs to functionally substitute for their VACV counterparts corresponded directly with amino acid sequence identity values. We were able to individually substitute the genes encoding the MYXA8 and MYXA23 proteins, which had ∼60% amino acid identity with the VACV proteins, but not those of CNPV and CRV, which had 51% and 40% identity, respectively. Both MYXA8 and MYXA23 hybrid viruses made smaller plaques than the WT virus, but MYXA8 produced the smallest, providing a better model virus for experimental evolution.
Viruses that made larger plaques and replicated to higher titers than the original VACV MYXA8 virus arose during each of three parallel serial passages. Since the hybrid viruses had an MYXV intermediate transcription factor, we had anticipated that adaptive mutations might occur in either the MYXA8 or VACVA23 intermediate transcription factor subunit or in the RNA Pol. Although we found no mutations in either transcription factor subunit, two different single nucleotide mutations in two independent passage series predicted more efficient translation of MYXA8 mRNA. More interesting was the occurrence of RNA Pol mutations in each of the three parallel passages: there were two independent mutations in RPO147 and three in RPO132, the largest and second largest of the eight subunits in VACV RNA Pol, respectively. The RNA Pol mutations were not detected in P0 but varied from 6 to 69% of the entire population after P10, suggesting strong selection. Amplicon sequencing of specific RNA Pol mutations confirmed progressive increases in mutation frequency in successive passages. Although we did not detect gene duplications in the P10 populations, such changes could have occurred transiently in earlier passages that were not examined by whole-genome sequencing.
Three MYXA8 viruses were clonally purified from the P10 populations, each of which formed larger plaques than the MYXA8 P0 clones. The P10 A and B clones each had mutations in RNA Pol and the translation site of the MYXA8 gene, while P10 clone C had only an RNA Pol mutation. To attribute phenotypic changes to single nucleotides, we mutated the RNA Pol of MYXA8 P0 viruses by transfection of ∼1,000-bp PCR products. The high rate of recombination, which allowed this simple procedure without a selection marker, is not generally appreciated and for this purpose is far simpler than the transient dominant selection method (33). The absence of any other changes in the mutant viruses was verified by whole-genome sequencing. These recombinant viruses allowed us to prove that RPO147 M424I and RPO132 K1046Q increased plaque size and virus yield substantially although the RPO147 A1212V mutation had a smaller effect. Furthermore, a competition experiment carried out between the RPO147 M424I mutant and the MYXA8 P0 virus with WT RPO147 demonstrated that the mutant rapidly became dominant. The plaque size and virus yield of the RPO132 K1046Q mutant were similar to those of the P10 clone C virus, confirming the absence of other adaptive mutations in the latter. While the RPO147 M424I and RPO147 A1212V mutant viruses made larger plaques than the MYXA8 P0 clones, they were smaller than those of the P10 A and B clones, suggesting that the MYXA8 translation site mutations in the P10 clones were responsible. The MYXA8 gene in the starting virus has a C at the −3 position and a G in the +4 position relative to the first nucleotide of the translation codon. According to the Kozak model, G at +4 is a good context for high translation initiation, but a purine is preferred to the C at −3 (29). Interestingly, a C → A mutation in the −3 position relative to the ATG start codon of MYXA8 occurred in one P10 clone. In the other P10 clone, the point mutation created a new ATG with a good Kozak consensus sequence within the N-terminal V5 epitope tag. Genome sequencing of the passage 10 populations revealed a frequency of 18% for the C → A mutation and a frequency of 20% for the new ATG, suggesting that selection had occurred. Homologous recombination was used to make these exact mutations upstream of the MYXA8 open reading frame of the original virus. In each case, the mutants enhanced virus replication, as shown by the presence of increased plaque size. In addition, Western blotting with anti-V5 antibody showed that the C → A mutation increased expression of MYXA8. Although the mutation within the V5 tag likely increased expression as well, this could not be confirmed because of the inability of anti-V5 antibody to detect the protein. Curiously, the Kozak mutation within the V5 tag conferred higher replication than the upstream Kozak mutation, suggesting the possibility that the V5 tag is deleterious and that the disruption is beneficial. However, previous studies showed that addition of a V5 tag to the VACV A8 protein did not have a noticeable effect (26).
Based on homology with the yeast RNA Pol, the mutations in RPO147 and RPO132 were predicted to be located internally near the catalytic site rather than in a solvent-accessible region where interaction with other proteins might occur. This would suggest that the mutations do not directly enhance binding of the transcription factors. Some mutations found by other investigators in RPO147 (A535V and S288Y) and RPO132 (Y462H) confer resistance to isatin-β-thiosemicarbazone (IBT) by reducing transcription elongation (34, 35). In addition, mutants in RPO132 were found during experimental evolution with E3 deletion mutants that have defects related to double-stranded RNA with activation of the oligoadenylate synthetase (OAS)/RNase L and PKR/eIF2α pathways. In one report, an RPO132 T1120M mutation partially reduced PKR phosphorylation and prevented eIF2α phosphorylation, resulting in increased viral protein synthesis and resistance to IBT (10), whereas in another report, an RPO132 Leu18Phe mutation increased levels of PKR and eIF2α phosphorylation; an RPO132 Lys452Asn mutation elicited no change in PKR and eIF2α phosphorylation compared to levels in the E3 deletion mutant (11). Thus, the previously described RNA Pol mutations appear to have diverse effects.
We plan to investigate the role of RNA Pol mutations in enhancing replication of MYXA8 in a follow-up study. We are considering several possibilities: (i) that the mutated RNA Pol is more promiscuous and less dependent on intermediate transcription factors for RNA synthesis, (ii) that the mutations slow down transcription, allowing more time for the heterologous transcription factors to act, and (iii) that diminished processivity may reduce double-stranded RNA and indirectly enhance replication. With regard to the last idea, it will be interesting to determine the effects on replication of MYXA8 of previously described mutations in RPO147 and RPO132. To conclude, we have shown that accelerated experimental evolution can be achieved by swapping orthologous poxvirus transcription factor genes and suggest that this approach is generally applicable to interrogating additional gene functions.
MATERIALS AND METHODS
Construction of deletion and hybrid viruses.
RK13 A8-23 cells (26) in 24-well dishes were infected with 0.05 PFU per cell of vTF7-3, a WR strain of VACV that encodes the bacteriophage T7 RNA Pol regulated by a VACV early/late promoter (27). After 1 h, the cells were washed with fresh medium and transfected with 2 μl of Lipofectamine 2000 (Invitrogen) and 500 ng of a PCR product encoding GFP or DsRed regulated by the VACV p11 late promoter and sequences flanking the A8 or A23 gene, respectively. In the case of the A8 deletion, all of the N-terminal nucleotides of the ORF as well as three nucleotides in the preceding intergenic region were deleted, but 28 nucleotides of the C terminus were retained because of overlap of the A9 gene. In the case of the A23 gene, all N-terminal nucleotides were deleted, but 47 nucleotides at the C terminus were retained because of overlap of the A24 gene. The medium was replaced after 24 h, and the incubation continued for 48 h. Diluted cell extracts were used to infect fresh RK13 A8-23 cell monolayers, which were then overlaid with 0.5% methylcellulose. After 48 h, fluorescent plaques were picked with sterile toothpicks. The viruses were clonally purified by repeated plaquing on RK13 A8-23 cells, and the purity was assessed by PCR. Deletion of both A8 and A23 from the same virus was carried out by successive deletion of the individual genes.
Hybrid viruses containing orthologous mammalian codon-optimized A8 or A23 genes were constructed using a similar recombination protocol in HeLa cells, except that the mammalian-optimized ortholog replaced GFP or DsRed and identified by formation of nonfluorescent plaques in BS-C-1 cells.
Construction of expression plasmids.
Nucleotide sequences for the A8 and A23 orthologs were obtained from GenBank and included VACV WR (accession number NC_006998), MYXV-6918 (EU552530), CNPV-VR111 (NC_005309), CRV (NC_008030), and SGPV (NC_027707). The genes were codon optimized for expression in mammalian cells and synthesized by Life Technologies GeneArt Gene Synthesis service. Primers were designed to amplify genes from the plasmids supplied by the company using Phusion High-Fidelity (HF) master mix with HF buffer (M0531L; New England Biolabs [NEB]). The PCR products were purified using a QiaQuick PCR purification kit (catalog no. 28104) and inserted into a Blunt TOPO vector (45-0245; Invitrogen). A8 and A23 ORFs were tagged at their N termini with V5 and HA, respectively. Plasmid DNA was extracted using a QiaPrep Spin Miniprep kit (catalog no. 27106). Colonies of transformed Escherichia coli were picked and grown, and the DNA was sequenced.
Western blotting for analysis of plasmid expression.
HeLa cells were infected with 3 PFU per cell of vTF7-3 for 1 h and then transfected with 500 ng of plasmid in Lipofectamine 2000. Samples were incubated overnight at 37°C and harvested. Anti-V5 (ab27671; Abcam) and anti-HA (H3663; Sigma) antibodies were used to detect expression of the viral proteins. Secondary antibodies were fluorescently labeled (1:10,000; Li-Cor). Bands were visualized using a Li-Cor Odyssey CLx.
Luciferase assays.
HeLa cells were infected in triplicate with 3 PFU per cell of the deletion viruses and then transfected with 500 ng of one or both A8 and A23 plasmids and the luciferase plasmid. Cells were incubated at 37°C for 12 h, and luciferase activity was measured (catalog no. E1501; Promega) with a luminometer (Berthold Detection Systems) and normalized to the activity of the WR orthologs. Samples were diluted to keep readings in the linear range.
Serial passaging of hybrid viruses.
BS-C-1 cells in 10-cm-diameter dishes were infected with 0.1 PFU per cell at 37°C, harvested at 48 h, and then resuspended in 1 ml of medium. The cells were frozen and thawed three times and then sonicated before each passage.
Whole-genome sequencing.
DNA was purified as described by Esposito et al. (36) with minor modifications. T75 flasks of BS-C-1 cells (∼1 × 107 cells) were infected and 2 to 3 days later collected by scraping. Cell pellets were washed with phosphate-buffered saline (PBS) and resuspended in 400 μl of 20 mM Tris, pH 8.0, 5 mM EDTA, and 1% Triton X-100; 10 min later, NaCl was added to a final concentration of 0.2 M. The cell extract was centrifuged at 800 × g for 3 min, and the nuclear pellet was discarded. The supernatant was treated with proteinase K in 0.5% SDS for 1 h at 50°C, and the DNA was extracted with phenol. Two volumes of ethanol was added to the aqueous phase; the visible threads of DNA were collected by brief centrifugation and washed twice with 80% ethanol, and the pellet was dissolved in 100 μl of water. Genome sequencing was done with an Illumina Miseq 2- by 150-bp configuration by the Genewiz company or the NIAID core facility.
Amplicon sequencing.
Amplicon sequencing was carried out by either Illumina or Sanger sequencing. For the latter, the PCR primers were designed to produce a product of about 400 bp with the mutation in the middle. Cells were lysed by three freeze/thaw cycles and sonication, and 4 μl of the lysate was used for PCR in 50-μl reaction volumes (Phusion High-Fidelity PCR Mix; NEB). PCR products were purified by a Qiagen PCR kit, and Sanger sequencing was performed with the original primers used for PCR. The percentage of a mutation was estimated based on the heights of the peaks corresponding to specific nucleotides. For better quantitation, primers were designed to produce products of about 150 bp with the mutation in the middle, and the PCR products were analyzed by ultradeep sequencing on the Illumina Miseq platform by Genewiz.
Introduction of single nucleotide mutations into the VACV genome.
BS-C-1 cells were infected with 3 PFU/cell of VACV and after 1.5 h transfected with a PCR product of approximately 1,000 bp with the desired nucleotide change in the middle. At 18 to 20 h after infection, the cells were lysed, and diluted virus was used to infect BS-C-1 cells. At 48 h, the virus from individual plaques was placed in 250 μl of medium and subjected to three freeze-thaw cycles, and 4 μl of the lysate was used for PCR with the original primers in a 50-μl reaction volume (Phusion High-Fidelity PCR Mix; NEB). PCR products were purified with a Qiagen PCR kit, and clones with mutations were identified by Sanger sequencing. The clones with the desired mutations were plaque purified once more; 100 μl of the virus from the plaque was used to infect wells of a 24-well plate of BS-C-1 cells, and virus was collected after 2 to 3 days when the cytopathic effect was complete. For the second passage, six wells of a six-well plate were each infected with aliquots from the first passage.
Plaque size determination.
BS-C-1 cells were infected and covered with 0.5% methylcellulose. After 48 h at 37°C, the plates were stained with 0.1% crystal violet and scanned using a Canon 9000F Mark II color image scanner. Plaque sizes were determined using ImageJ, version 1.51w, software. Three independent wells, for a total of 150 to 300 plaques, were analyzed for each virus.
Statistical analysis.
Statistical analysis was carried out using GraphPad Prism (version 7.0c). Unpaired two-tailed t tests were performed and P values of <0.05 were deemed statistically significant.
Competition experiment.
Approximately 2.5 × 105 BS-C-1 cells were infected in triplicate with 0.01 PFU/cell of a virus mixture comprised of 90% MYXA8 P0 clone and 10% MYXA8 with the M424I mutation in the RPO147 gene. After 48 h, the cells were resuspended in 250 μl of medium. Virus was released by three freeze-thaw cycles and sonication, which was followed by two additional passages in BS-C-1 cells at 0.01 PFU/cell for 48 h.
Mapping of VACV RNA Pol mutations on the eukaryotic RNA Pol structure.
Alignments between the VACV J6 (RPO147) and A24 (RPO132) gene products and S. cerevisiae RPB1 and RBP2 subunits of RNAP II were generated using the PRALINE web server (37) (http://www.ibi.vu.nl/programs/pralinewww/). Corresponding J6 and A24 variant residues were then mapped onto the S. cerevisiae RNAP II crystal structure (PDB accession number 5U5Q) by use of Chimera software (38) (http://www.cgl.ucsf.edu/chimera/).
Accession number(s).
Data are available at the Sequence Read Archive under BioProject accession number PRJNA481995.
ACKNOWLEDGMENTS
We thank Catherine Cotter for help with cell culture and Wei Xiao for preparation of plasmids.
The research was supported by the Division of Intramural Research, NIAID, NIH.
REFERENCES
- 1.Moss B. 2013. Poxviridae, p 2129–2159. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Rancaniello VR, Roizman B (ed), Fields virology, 6th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Upton C, Slack S, Hunter AL, Ehlers A, Roper RL. 2003. Poxvirus orthologous clusters: toward defining the minimum essential poxvirus genome. J Virol 77:7590–7600. doi: 10.1128/JVI.77.13.7590-7600.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bratke KA, McLysaght A, Rothenburg S. 2013. A survey of host range genes in poxvirus genomes. Infect Genet Evol 14:406–425. doi: 10.1016/j.meegid.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haller SL, Peng C, McFadden G, Rothenburg S. 2014. Poxviruses and the evolution of host range and virulence. Infect Genet Evol 21:15–40. doi: 10.1016/j.meegid.2013.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith GL, Benfield CT, Maluquer de Motes C, Mazzon M, Ember SWJ, Ferguson BJ, Sumner RP. 2013. Vaccinia virus immune evasion: mechanisms, virulence and immunogenicity. J Gen Virol 94:2367–2392. doi: 10.1099/vir.0.055921-0. [DOI] [PubMed] [Google Scholar]
- 6.Qin L, Favis N, Famulski J, Evans DH. 2015. Evolution of and evolutionary relationships between extant vaccinia virus strains. J Virol 89:1809–1824. doi: 10.1128/JVI.02797-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mayr A, Hochstein-Mintzel V, Stickl H. 1975. Abstammung, eigenschaften und verwendung des attenuierten vaccinia-stammes MVA. Infection 3:6–14. doi: 10.1007/BF01641272. [DOI] [Google Scholar]
- 8.Elde NC, Child SJ, Eickbush MT, Kitzman JO, Rogers KS, Shendure J, Geballe AP, Malik HS. 2012. Poxviruses deploy genomic accordions to adapt rapidly against host antiviral defenses. Cell 150:831–841. doi: 10.1016/j.cell.2012.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brennan G, Kitzman JO, Rothenburg S, Shendure J, Geballe AP. 2014. Adaptive gene amplification as an intermediate step in the expansion of virus host range. PLoS Pathog 10:e1004002. doi: 10.1371/journal.ppat.1004002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brennan G, Kitzman JO, Shendure J, Geballe AP. 2015. Experimental evolution identifies vaccinia virus mutations in A24R and A35R that antagonize the protein kinase R pathway and accompany collapse of an extragenic gene amplification. J Virol 89:9986–9997. doi: 10.1128/JVI.01233-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cone KR, Kronenberg ZN, Yandell M, Elde NC. 2017. Emergence of a viral RNA polymerase variant during gene copy number amplification promotes rapid evolution of vaccinia virus. J Virol 91:e01428-16. doi: 10.1128/JVI.01428-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baldick CJ Jr, Moss B. 1993. Characterization and temporal regulation of mRNAs encoded by vaccinia virus intermediate stage genes. J Virol 67:3515–3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Broyles SS, Moss B. 1986. Homology between RNA polymerases of poxviruses, prokaryotes, and eukaryotes: nucleotide sequence and transcriptional analysis of vaccinia virus genes encoding 147-kDa and 22-kDa subunits. Proc Natl Acad Sci U S A 83:3141–3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Knutson BA, Broyles SS. 2008. Expansion of poxvirus RNA polymerase subunits sharing homology with corresponding subunits of RNA polymerase II. Virus Genes 36:307–311. doi: 10.1007/s11262-008-0207-3. [DOI] [PubMed] [Google Scholar]
- 15.Broyles SS, Yuen L, Shuman S, Moss B. 1988. Purification of a factor required for transcription of vaccinia virus early genes. J Biol Chem 263:10754–10760. [PubMed] [Google Scholar]
- 16.Luo Y, Hagler J, Shuman S. 1991. Discrete functional stages of vaccinia virus early transcription during a single round of RNA synthesis in vitro. J Biol Chem 266:13303–13310. [PubMed] [Google Scholar]
- 17.Cassetti MC, Moss B. 1996. Interaction of the 82-kDa subunit of the vaccinia virus early transcription factor heterodimer with the promoter core sequence directs downstream DNA binding of the 70-kDa subunit. Proc Natl Acad Sci U S A 93:7540–7545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ahn B-Y, Moss B. 1992. RNA polymerase-associated transcription specificity factor encoded by vaccinia virus. Proc Natl Acad Sci U S A 89:3536–3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang Z, Moss B. 2009. Interaction of the vaccinia virus RNA polymerase-associated 94-kilodalton protein with the early transcription factor. J Virol 83:12018–12026. doi: 10.1128/JVI.01653-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sanz P, Moss B. 1999. Identification of a transcription factor, encoded by two vaccinia virus early genes, that regulates the intermediate stage of viral gene expression. Proc Natl Acad Sci U S A 96:2692–2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Keck JG, Baldick CJ, Moss B. 1990. Role of DNA replication in vaccinia virus gene expression: A naked template is required for transcription of three late transactivator genes. Cell 61:801–809. doi: 10.1016/0092-8674(90)90190-P. [DOI] [PubMed] [Google Scholar]
- 22.Senkevich TG, Bugert JJ, Sisler JR, Koonin EV, Darai G, Moss B. 1996. Genome sequence of a human tumorigenic poxvirus: prediction of specific host response-evasion genes. Science 273:813–816. doi: 10.1126/science.273.5276.813. [DOI] [PubMed] [Google Scholar]
- 23.Senkevich TG, Koonin EV, Bugert JJ, Darai G, Moss B. 1997. The genome of molluscum contagiosum virus: analysis and comparison with other poxviruses. Virology 233:19–42. doi: 10.1006/viro.1997.8607. [DOI] [PubMed] [Google Scholar]
- 24.Boyle DB. 1992. Quantitative assessment of poxvirus promoters in fowlpox and vaccinia virus recombinants. Virus Genes 6:281–290. doi: 10.1007/BF01702566. [DOI] [PubMed] [Google Scholar]
- 25.Mendez-Rios JD, Yang Z, Erlandson KJ, Cohen JI, Martens CA, Bruno DP, Porcella SF, Moss B. 2016. Molluscum contagiosum virus transcriptome in abortively infected cultured cells and human skin lesion. J Virol 90:4469–4480. doi: 10.1128/JVI.02911-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Warren RD, Cotter C, Moss B. 2012. Reverse genetic analysis of poxvirus intermediate transcription factors. J Virol 86:9514–9519. doi: 10.1128/JVI.06902-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fuerst TR, Niles EG, Studier FW, Moss B. 1986. Eukaryotic transient-expression system based on recombinant vaccinia virus that synthesizes bacteriophage T7 RNA polymerase. Proc Natl Acad Sci U S A 83:8122–8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang Z, Reynolds SE, Martens CA, Bruno DP, Porcella SF, Moss B. 2011. Expression profiling of the intermediate and late stages of poxvirus replication. J Virol 85:9899–9908. doi: 10.1128/JVI.05446-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kozak M. 1992. Regulation of translation in eukaryotic systems. Annu Rev Cell Biol 8:197–225. doi: 10.1146/annurev.cb.08.110192.001213. [DOI] [PubMed] [Google Scholar]
- 30.Challberg MD, Englund PT. 1979. Purification and properties of the deoxyribonucleic acid polymerase induced by vaccinia virus. J Biol Chem 254:7812–7819. [PubMed] [Google Scholar]
- 31.Hamilton MD, Evans DH. 2005. Enzymatic processing of replication and recombination intermediates by the vaccinia virus DNA polymerase. Nucleic Acids Res 33:2259–2268. doi: 10.1093/nar/gki525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Czarnecki MW, Traktman P. 2017. The vaccinia virus DNA polymerase and its processivity factor. Virus Res 234:193–206. doi: 10.1016/j.virusres.2017.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Falkner FG, Moss B. 1990. Transient dominant selection of recombinant vaccinia viruses. J Virol 64:3108–3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Prins C, Cresawn SG, Condit RC. 2004. An isatin-beta-thiosemicarbazone-resistant vaccinia virus containing a mutation in the second largest subunit of the viral RNA polymerase is defective in transcription elongation. J Biol Chem 279:44858–44871. doi: 10.1074/jbc.M408167200. [DOI] [PubMed] [Google Scholar]
- 35.Cresawn SG, Prins C, Latner DR, Condit RC. 2007. Mapping and phenotypic analysis of spontaneous isatin-beta-thiosemicarbazone resistant mutants of vaccinia virus. Virology 363:319–332. doi: 10.1016/j.virol.2007.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Esposito J, Condit R, Obijeski J. 1981. The preparation of orthopoxvirus DNA. J Virol Methods 2:175–179. doi: 10.1016/0166-0934(81)90036-7. [DOI] [PubMed] [Google Scholar]
- 37.Simossis VA, Heringa J. 2005. PRALINE: a multiple sequence alignment toolbox that integrates homology-extended and secondary structure information. Nucleic Acids Res 33(Suppl 2):W289–294. doi: 10.1093/nar/gki390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera—a visualization system for exploratory research analysis. J Comput Chem 25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]