

Viral inhibition of innate immunity contributes to herpes simplex virus pathogenesis. Although this complex process involves multiple factors, the underlying events remain unclear. We demonstrate that an HSV virulence factor γ134.5 precludes the activation of STING, a central adaptor in the intracellular DNA sensing pathway. Upon HSV infection, this viral protein engages with and inactivates STING. Consequently, it compromises host immunity and facilitates HSV replication. These observations uncover an HSV mechanism that is likely to mediate viral virulence.
KEYWORDS: herpes simplex virus, STING, interferons, viral replication, virus-host interactions
ABSTRACT
The γ134.5 gene of herpes simplex virus 1 (HSV-1) encodes a virulence factor that promotes viral pathogenesis. Although it perturbs TANK-binding kinase 1 (TBK1) in the complex network of innate immune pathways, the underlying mechanism is obscure. Here we report that HSV-1 γ134.5 targets stimulator of interferon genes (STING) in the intracellular DNA recognition pathway that regulates TBK1 activation. In virus-infected cells the γ134.5 protein associates with and inactivates STING, which leads to downregulation of interferon regulatory factor 3 (IRF3) and IFN responses. Importantly, HSV-1 γ134.5 disrupts translocation of STING from the endoplasmic reticulum to Golgi apparatus, a process necessary to prime cellular immunity. Deletion of γ134.5 or its amino-terminal domain from HSV-1 abolishes the observed inhibitory activities. Consistently, an HSV mutant that lacks functional γ134.5 replicated less efficiently in STING+/+ than in STING−/− mouse embryonic fibroblasts. Moreover, reconstituted expression of human STING in the STING−/− cells activated IRF3 and reduced viral growth. These results suggest that control of the DNA sensing pathway by γ134.5 is advantageous to HSV infection.
IMPORTANCE Viral inhibition of innate immunity contributes to herpes simplex virus pathogenesis. Although this complex process involves multiple factors, the underlying events remain unclear. We demonstrate that an HSV virulence factor γ134.5 precludes the activation of STING, a central adaptor in the intracellular DNA sensing pathway. Upon HSV infection, this viral protein engages with and inactivates STING. Consequently, it compromises host immunity and facilitates HSV replication. These observations uncover an HSV mechanism that is likely to mediate viral virulence.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) is a large DNA virus that switches between latent and lytic cycles, which can lead to genital ulcers, keratitis, and encephalitis (1). In this process, an HSV virulence γ134.5 is believed to play a role (2, 3). Published work has established that HSV-1 γ134.5 promotes viral replication in the peripheral tissues and penetration to the peripheral nervous systems in experimental models (4–6). In addition, it facilitates HSV infection and replication in the central nervous system (2, 3). Likewise, the γ134.5 gene product encoded by HSV-2 is crucial to mediate viral pathogenesis in vivo, although its precise function is incompletely characterized (7, 8).
Emerging evidence suggests that upon HSV infection, intracellular DNA sensors initiate IFN responses (9). Among them are cyclic GAMP synthase (cGAS), interferon-inducible protein 16 (IFI16), and DEAD box helicase 41 (DDX41) (10–12). In the cytoplasm, cGAS synthesizes cyclic GMP-AMP, a second messenger that activates stimulator of interferon genes (STING), which recruits TANK-binding kinase 1 (TBK1) (10). As a result, TBK1 phosphorylates interferon regulatory factor 3 (IRF3), resulting in the production of alpha/beta interferon (IFN-α/β), chemokines, and interferon-stimulated genes. Moreover, DDX41 detects cytosolic DNA to activate TBK1 in a STING-dependent manner (12). Remarkably, IFI16 recognizes nuclear DNA and subsequently interacts with STING in the cytoplasm to activate TBK1 (11, 13, 14). These DNA receptors work cooperatively and relay danger signals to STING, subsequently activating host immunity (15–17).
HSV-1 γ134.5 is present in two copies per viral genome within the inverted repeat regions (18). Upon infection, viral DNA replication activates double-stranded RNA-dependent protein kinase PKR, which phosphorylates the α subunit of translation initiation factor 2 (eIF-2α) and shuts off protein synthesis (19, 20). To evade this, HSV-1 γ134.5 recruits cellular phosphatase 1 (PP1), forming a high-molecular-weight complex that dephosphorylates eIF-2α (21–23). Accordingly, mutations in the PP1 binding site abrogate viral virulence (24, 25). The γ134.5 protein also inhibits autophagy, IFN induction, and dendritic cell maturation and facilitates glycoprotein processing as well as nuclear egress (6, 26–34). Of note, in HSV-infected cells the γ134.5 protein overcomes a blockade in viral growth by TBK1 (28, 29), a host factor implicated in the control of human HSV encephalitis (35). However, as a complex TBK1 network exists (9, 36), the precise way by which γ134.5 functions is to be deciphered.
Here we report that HSV-1 γ134.5 targets STING in the intracellular DNA recognition pathway upon infection. We provide evidence that HSV-1 γ134.5 binds to STING, prevents its translocation from the endoplasmic reticulum (ER) to the Golgi apparatus, and thereby inhibits antiviral immunity. Our work identifies a molecular mechanism of γ134.5 that likely contributes to HSV virulence.
RESULTS
Ectopic expression of γ134.5 suppresses IFN promoter activation by STING and cGAS.
To understand the mechanism of γ134.5 actions, we asked whether it modulates the intracellular DNA sensing pathway. For this purpose, we examined the cGAS-STING axis that limits HSV-1 infection (10, 37, 38). As shown in Fig. 1B, when coexpressed in 293T cells, cGAS and STING stimulated IFN-β promoter activation in a reporter assay. Ectopic expression of intact γ134.5 inhibited IFN-β promoter activation in a dose-dependent manner. To dissect the functional domain, we analyzed the Δ146 mutant, with a deletion of amino acids (aa) 1 to 146 from γ134.5, and the N159 mutant, with a deletion of amino acids 159 to 263 (Fig. 1A). Similar to intact γ134.5, the N159 mutant suppressed IFN-β promoter activation. In contrast, the Δ146 mutant had little inhibitory activity (Fig. 1C). These activities coincided with the ability of γ134.5 to interact with STING, as measured by immunoprecipitation (Fig. 1D and E). Thus, ectopic expression of γ134.5 inhibits gene activation potentiated by STING and cGAS via its amino-terminal domain.
FIG 1.

(A) Schematic diagram of HSV-1 genome organization. The γ134.5 gene is located in the inverted repeats, with two copies per genome. The expanded section shows intact γ134.5, the N-terminal deletion mutant (Δ146), and the C-terminal deletion mutant (N159). Numbers indicate amino acid positions. (B and C) The N terminus of γ134.5 inhibits IFN-β promoter activation by STING and cGAS. 293T cells were cotransfected with pIFN-β-luc (50 ng) and pRL-TK (10 ng) along with the indicated plasmid (cGAS [50 ng], STING [5 ng], γ134.5 [in panel B, 10 to 500 ng, and in panel C, 300 ng], Δ146 [300 ng], or N159 [300 ng]). At 36 h after transfection, cells were harvested for luciferase assays. Fold activation was determined compared to the empty vector. Data represent the mean values from three independent experiments, with SDs. The data were statistically analyzed by a two-tailed Student t test (*, P < 0.05; **, P < 0.01; ns, not significant). (D and E) The N terminus of γ134.5 interacts with STING. 293T cells were transfected with Myc-STING along with a vector (Vec) or a plasmid expressing the wild-type γ134.5 or the Δ146 or N159 mutant. At 36 h after transfection, cells were processed for immunoprecipitation (IP) with anti-Myc (D) or anti-Flag (E) antibody. Precipitated proteins and whole-cell lysates (WCL) were probed with antibodies against Flag, Myc, and β-actin. The data are representative of results from three independent experiments.
The γ134.5 protein inhibits STING pathway activation and IFN expression in virus-infected cells.
To assess the relevance of the γ134.5-STING interaction in HSV infection, we first examined the impact of γ134.5 on STING phosphorylation, a hallmark of activation (39). Human foreskin fibroblasts (HFF), mock infected or infected with viruses, were processed for immunoblot analysis. As illustrated in Fig. 2A, STING was expressed in all cells. Like the mock control, wild-type HSV-1(F) did not induce STING phosphorylation at serine 366. However, recombinant R3616, a γ134.5 null mutant (2), induced phosphorylated STING species. Moreover, H1001, in which the amino-terminal domain (aa 1 to 146) was deleted (29), also caused STING phosphorylation. As expected, H1002 (29), a repair virus with intact γ134.5, prevented the appearance of phosphorylated STING species. These results were not due to differences in viral infectivity, as determined by ICP27 expression. Intriguingly, ICP0 was present in cells infected with all viruses except the γ134.5 null mutant. However, deletion of the N-terminal domain from γ134.5 had no effect on ICP0 expression but induced STING phosphorylation, indicative of an ICP0-independent cellular response.
FIG 2.

The γ134.5 protein inhibits STING pathway activation via its amino-terminal domain. (A) Effects of γ134.5 variants on STING phosphorylation. HFF-1 cells were mock infected or infected with wild-type HSV-1, R3616, H1001, and H1002 (5 PFU/cell). At 5 h postinfection, cell lysates were prepared and processed for Western blot analysis with antibodies against p-STING, STING, ICP0, ICP27, and β-actin. (B) Effects of γ134.5 variants on IRF3 phosphorylation. HFF-1 cells were infected with viruses as for panel A and then processed for Western blot analysis with antibodies against p-IRF3, IRF3, ICP0, ICP27, and β-actin. (C) Effects of γ134.5 variants on gene expression. HFF-1 cells were infected with viruses as for panel A. At 5 h postinfection, total RNA extracted from cells was subjected to quantitative real-time PCR amplification for IFN-β, ISG54, ISG56, and RANTES. The results were normalized to 18S rRNA and expressed as fold activation with SDs among triplicate samples. The data were statistically analyzed by a two-tailed Student t test (**, P < 0.01).
Next, we determined the phosphorylation of IRF3, which mediates the induction of the type I IFN response. HFF were subjected to virus infection and Western blot analysis. Figure 2B shows that IRF3 expression remained virtually unchanged in mock- or virus-infected cells. Infection with R3616 or H1001 resulted in a phosphorylated IRF3 species. This band was not seen with wild-type HSV-1 or H1002 infection. Further, ICP27 was detectable in all virus-infected cells, although ICP0 was absent in cells infected with R3616. These results, in line with STING phosphorylation analysis, suggest that HSV-1 γ134.5 has a distinct activity independent of ICP0 expression.
In addition, we tested the gene expression by quantitative real-time PCR. As indicated in Fig. 2C, wild-type HSV-1 triggered little expression of IFN-β, ISG54, ISG56, and RANTES. However, the γ134.5 null mutant R3616 caused a sharp increase in the expression of IFN-β, ISG54, ISG56, and RANTES. Although with a different magnitude, H1001 induced a similar gene expression pattern. H1002 behaved like wild-type virus. We conclude that the γ134.5 protein blocks STING pathway activation, which dampens the IFN response.
The ability of γ134.5 to bind STING is coupled with increased virus growth.
To determine whether γ134.5 interacts with endogenous STING in HSV-infected cells, we performed immunoprecipitation analysis. As shown in Fig. 3A, in cells infected with wild-type HSV-1, STING was precipitated with the γ134.5 protein. This was not detectable in cells infected with R3616 or H1001. However, STING appeared in the precipitates in cells infected with H1002. Therefore, the γ134.5 protein forms a complex with STING, which is dependent on its amino-terminal domain in HSV-infected cells.
FIG 3.
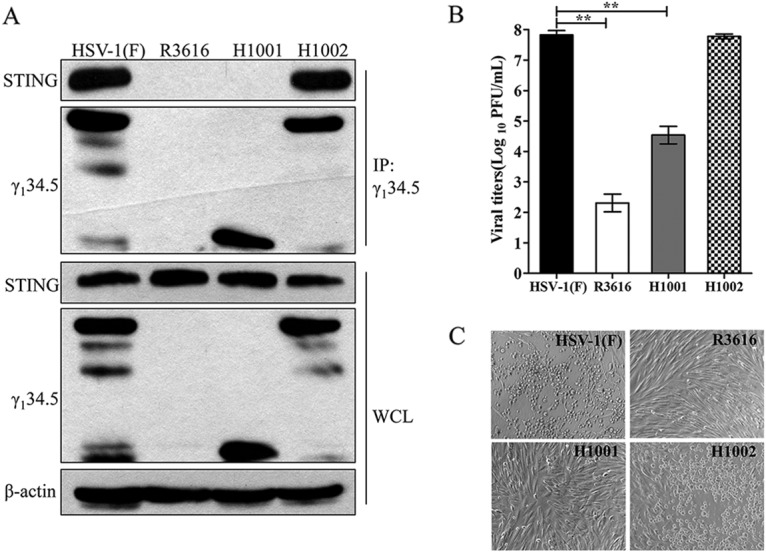
(A) The γ134.5 protein interacts with endogenous STING. HFF-1 cells were infected with wild-type HSV-1(F), R3616 (which lacks γ134.5), H1001 (in which the amino-terminal domain [aa 146 to 263] was deleted), or H1002 (in which intact γ134.5 was restored [5 PFU/cell]). At 15 h postinfection, cells were processed for immunoprecipitation with anti-γ134.5 antibody. Whole-cell lysates and precipitated proteins were probed with antibodies against STING, γ134.5, and β-actin. (B) Viral replication on HFF-1 cells. HFF-1 cells were infected with the indicated virus (0.01 PFU/cell). At 48 h postinfection, the total virus yields were titrated on Vero cells. (C) Cytopathic effects in HFF-1 cells. Viral infections were carried out as for panel B, and images were taken under the microscope. The data are representative of those from three independent experiments. Viral growth results are for triplicate samples and assessed by a two-tailed Student t test (**, P < 0.01).
To investigate viral replication, we compared growth of γ134.5 variants. As illustrated in Fig. 3B, wild-type HSV-1replicated efficiently in human foreskin fibroblasts, reaching a titer of 7.8 × 107 PFU/ml at 48 h postinfection. Under this condition, R3616 replicated poorly, with a titer of 2.3 × 102 PFU/ml. H1001 replicated modestly, reaching a titer of 4.5 × 104 PFU/ml, which is an intermediate level between those of wild-type virus and the γ134.5 null mutant. H1002 replicated to 7.8 × 107 PFU/ml. Consistently, cytopathic effects in infected cells paralleled viral growth properties (Fig. 3C). These results argue that the amino-terminal domain of γ134.5, required to bind STING, promotes HSV replication.
STING expression restricts HSV replication in the absence of functional γ134.5.
To evaluate the impact of STING on HSV infection, we examined viral growth in STING+/+ and STING−/− mouse embryo fibroblasts (MEFs). Figure 4A shows that in STING+/+ cells, both wild-type HSV-1 and H1002 replicated efficiently, with a titer of 6.9 × 107 PFU/ml. R3616 replicated to 2 × 102 PFU/ml, whereas H1001 grew to 3.7 × 103 PFU/ml. In STING−/− cells, replication of wild-type HSV-1 as well as H1002 remained unchanged. However, replication of R3616 and H1001 increased about 10-fold, with titers reaching 3.2 × 103 PFU/ml and 5.3 × 105 PFU/ml, respectively.
FIG 4.

The γ134.5-STING interaction influences HSV replication. STING+/+ (A) and STING−/− (B) MEFs were infected with the indicated viruses (0.01 PFU/cell). At 48 h postinfection, virus yields were titrated on Vero cells. (C) Expression of STING. STING+/+, STING−/−, and reconstituted STING−/− MEFs expressing Flag-tagged human STING were grown and processed for Western blot analysis with antibodies against STING and β-actin. (D) Reconstituted expression of human STING reduces viral replication. STING−/− MEFs stably expressing human STING were infected with viruses as for panel A, and viral yields were measured. (E) The γ134.5 protein interacts with STING in reconstituted cells. STING−/− MEFs expressing h-STING were infected with the indicated viruses (5 PFU/cell). At 15 h postinfection, cells were processed for immunoprecipitation with anti-γ134.5 antibody. Whole-cell lysates and precipitated proteins and were probed with antibodies against Flag, γ134.5, and β-actin. The data are representative of results from three independent experiments.
To further assess STING, we stably expressed human STING (h-STING) in the STING−/− MEFs (Fig. 4C). With this reconstituted Flag-tagged h-STING-expressing cell line, we examined viral replication (Fig. 4D). Wild-type HSV-1 and H1002 replicated efficiently to a titer of 6.7 × 106 PFU/ml. R3616 replicated to 2.1 × 102 PFU/ml, whereas H1001 replicated to 3.8 × 103 PFU/ml. While STING expression had little effect on wild-type HSV-1 and H1002, it reduced replication of R3616 and H1001 10-fold. These phenotypes correlated with the γ134.5-STING interaction as determined by immunoprecipitation assay (Fig. 4E). Therefore, expression of STING imposes a restriction on HSV replication, particularly in the absence of functional γ134.5.
Reconstituted STING activates cytokine expression in response to γ134.5 mutants.
We further determined cytokine expression upon virus infection. The stable h-STING-expressing MEFs were mock infected or infected with viruses. At 5 h postinfection, samples were analyzed by quantitative real-time PCR. As summarized in Fig. 5A, like R3616, H1001 induced robust IFN-β expression. A similar response was seen with expression of RANTES, ISG54, and ISG56. In all experiments, wild-type HSV-1 as well as H1002 consistently inhibited the host antiviral responses. Furthermore, R3616 or H1001 stimulated IRF3 phosphorylation, whereas wild-type HSV-1 inhibited this process (Fig. 5B). Western blot analysis showed that wild-type HSV-1 and H1001 expressed ICP27 and ICP0. Nevertheless, R3616 expressed ICP27 but not ICP0. These results suggest that the γ134.5 protein inhibits IRF3 phosphorylation and the IFN response, a process that is dependent on its N-terminal domain.
FIG 5.

(A) The γ134.5 protein inhibits the IFN response in STING reconstituted MEFs. STING−/− MEFs expressing h-STING were mock infected or infected with HSV-1, R3616, H1001, and H1002 (5 PFU/cell). At 5 h postinfection, total RNA extracted from cells was subjected to quantitative real-time PCR amplification for IFN-β, ISG54, ISG56, and RANTES. The results were normalized to 18S rRNA and expressed as fold activation with SDs among triplicate samples. The data were statistically analyzed by a two-tailed Student t test (**, P < 0.01). (B) Phosphorylation of IRF3. STING−/− MEFs expressing h-STING were mock infected or infected with viruses as for panel A. Cell lysates were prepared and processed for Western blot analysis with antibodies against p-IRF3, IRF3, ICP0, ICP27, and β-actin. The data are representative of results from three independent experiments.
The γ134.5 protein precludes STING translocation to the Golgi apparatus in viral infection.
STING is an ER resident protein which, upon activation, moves to the perinuclear region (37, 40). Therefore, we asked whether HSV-1 γ134.5 interferes with this step. The h-STING-expressing MEFs were mock infected or infected with viruses. Cells were then processed for immune-staining of STING. As illustrated in Fig. 6, in mock-infected cells STING diffused throughout the cytoplasm, with <5% of cells exhibiting distinct foci. A similar distribution pattern was observed in cells infected with wild-type virus, although STING foci were detectable in 20% of cells. However, larger puncta appeared in the perinuclear regions of cells infected with R3616 or H1001. Approximately 60 to 70% of the cells were positive for the STING foci. A sharp increase in the STING puncta inversely correlated with the presence of functional γ134.5.
FIG 6.

(A) HSV-1 γ134.5 inhibits punctum formation of STING. STING−/− MEFs expressing h-STING were mock infected or infected with the indicated viruses (5 PFU/cell). At 5 h postinfection, cells were fixed, stained with 4′,6-diamidino-2-phenylindole (DAPI) and antibody specific for Flag, and examined by confocal microscopy. Scale bars, 50 μm. (B) Quantitative analysis. Cells with puncta and diffused signals were enumerated. The relative abundance was calculated among 170 to 200 cells in each treatment group.
To define the subcellular localization of STING (41), we further examined the ER and Golgi apparatus. Mock- or virus-infected cells were stained with calreticulin (ER marker), GM130 (Golgi marker), and Flag-STING, respectively. As illustrated in Fig. 7A, STING colocalized with calreticulin in mock- or wild-type-virus-infected cells. Following infection with R3616 or H1001, STING was no longer retained in the ER, suggesting its transit away from the ER. Consistent with this, STING was colocalized with GM130 in cells infected with R3616 or H1001 (Fig. 7B), suggesting its move to the Golgi apparatus. This colocalization pattern was barely seen cells mock infected or infected with wild-type HSV-1. Thus, upon HSV infection, HSV γ134.5 precludes STING translocation from the ER to the Golgi apparatus via its N-terminal domain.
FIG 7.

HSV-1 γ134.5 inhibits the trafficking of STING from the ER to Golgi apparatus. (A and B) STING−/− MEFs expressing h-STING were infected with viruses as for Fig. 6A. Cells were stained with DAPI and incubated with antibodies specific for Flag, GM130 (Golgi marker), and calreticulin (ER marker). Samples were examined under a confocal microscope. Scale bars, 10 μm.
DISCUSSION
HSV-1 γ134.5 is a leaky late protein (42), but it is also expressed early in virus infection (28, 43). The early expression of γ134.5 is believed to dampen the host response (28). In line with this model, we found that unlike wild-type virus, an HSV mutant that lacks functional γ134.5 stimulated the phosphorylation of STING, with subsequent activation of IRF3 and the IFN response. Indeed, HSV-1 γ134.5 physically associated with endogenous STING in virus-infected cells. This interaction was also detectable in transfected cells in the absence of any other HSV proteins. Consistently, γ134.5 inhibited IFN-β promoter activation mediated by cGAS and STING. These results demonstrate that HSV-1 γ134.5 possesses a distinct function to restrict STING pathway activation.
Our work suggests that inhibition of STING by γ134.5 facilitates viral replication. Although incompletely defined, this process is linked to the γ134.5-STING interaction. Deletion of γ134.5 or its N terminus severely impaired viral growth. On the other hand, depletion of STING restored growth of the γ134.5 mutant approximately 10-fold. This effect was partial but significant. Rescue of STING expression reversed the observed phenotype. These results lend support to the notion that the γ134.5-STING interplay may represent a regulatory point in HSV replication. Furthermore, given that STING critically primes adaptive immunity (37, 38), it is reasonable to postulate that control over STING is advantageous to HSV, likely at multiple levels in vivo. This is consistent with published work showing that HSV-1 is more virulent in STING−/− than in STING+/+ mice (37, 44, 45).
As a key innate immune component, STING usually resides in the ER (40). Upon stimulation, it travels to the Golgi compartment to recruit IRF3 and TBK1, forming a complex that mediates gene expression (41, 46). It has been suggested that the trafficking of STING relies on iRhom2 and translocon-associated protein (TRAPβ), where iRhom2 serves as a bridge (47). A relevant question then arises as to how the γ134.5 protein exerts its inhibitory activity. We noted that wild-type virus caused ER retention of STING, whereas the γ134.5 mutant induced STING accumulation in the Golgi network. A simple explanation would be that γ134.5 acts to perturb the trafficking of STING. At least two scenarios exist. An attractive possibility is that upon interacting with STING, γ134.5 may block its access to iRhom2 or TRAPβ and disrupt subsequent IRF3 activation. Alternatively, the γ134.5 protein may preclude binding of cGAMP or recruitment of TBK1 and IRF3 to STING in response to HSV infection.
The γ134.5 protein negatively regulates STING via its N-terminal domain. When expressed, this domain bound to and inhibited STING. Its deletion relieved the inhibitory effect of HSV-1 γ134.5. Consistent with our previous finding that the N-terminal domain of γ134.5 is required to block IFN-β expression in PKR−/− cells (29), these results argue that this region constitutes a separate module to cope with host DNA sensing. The way through which the N terminus of γ134.5 interacts with STING remains unknown. This domain, consisting of 146 amino acids, is predicted to form four α-helices separated by a stretch of turn or coil regions. We suspect that one or more of these α-helices may form a contact site(s) for STING. Crystal structure coupled with mutational analysis is necessary to clarify this issue.
The γ134.5 mutants analyzed stimulate the phosphorylation of STING and IRF3, but they differ in the expression of ICP0, a viral inhibitor of IFI16 (13). As the γ134.5 mutant that lacks the N-terminal domain expressed ICP0 normally, a separate function of γ134.5 must have been affected. This is supported by a γ134.5-STING connection. However, the γ134.5 null mutant was unable to express ICP0, a defect that is also reported by others (48). Thus, IRF3 phosphorylation induced by the γ134.5null mutant may reflect disruption of a function specifically associated with γ134.5, ICP0, or both. We postulate that besides ICP0 expression, the γ134.5 protein has evolved a different function to evade DNA sensing. Our work adds to a growing number of HSV-1 proteins, such as ICP0, ICP27, UL46, UL41, and UL49 (13, 49–53), which manipulate the intracellular DNA recognition pathway. As HSV-1 interacts with host cells in a complex way, these viral proteins may function coordinately to establish successful HSV infection.
MATERIALS AND METHODS
Cells and viruses.
Vero, HFF-1, and 293T cells were obtained from the American Type Culture Collection. STING+/+ and STING−/− mouse embryonic fibroblasts (MEFs) have been described previously (40). STING−/− MEFs reconstituted with h-STING were established by lentiviral transduction and puromycin (Santa Cruz) selection. All cells were propagated in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum. HSV-1(F) is a prototype HSV-1 strain (54). In recombinant virus R3616, a 1-kb fragment from the coding region of the γ134.5 gene was deleted (2). In H1001, the sequences of the γ134.5 gene encoding amino acids 1 to 146 were deleted, and in H1002 the deleted region was repaired with wild-type γ134.5 (29).
Viral infections were done at appropriate multiplicities of infection (28). Cells were then harvested at the desired time points and processed for immunoblot, immunoprecipitation, or real-time PCR analysis (29, 34). To measure viral growth, cells were harvested and viral yields were determined by plaque assay on Vero cells (28).
Plasmids and reporter assays.
To construct pMyc-STING and pFlag-STING, a PCR fragment of human STING was cloned into the BamHI and EcoRI sites of pCDNA3-Myc and pCDH-Flag, respectively. To construct pcGAS-HA, a human cGAS cDNA fragment was cloned into the HindIII and XbaI sites of pcDNA3. The plasmids Flag-γ134.5, Flag-Δ146, Flag-N159, pIFN-β-Luc, and pTK-Luc were described elsewhere (24). Reporter assays were carried out using a dual-luciferase reporter assay system (Promega) as described previously (29).
Antibodies.
Anti-Myc antibody (sc-40), ant-HSV-1 ICP0 antibody (sc-53070), and m-IgGκ BP-fluorescein isothiocyanate (FITC) (sc-516140) were purchased from Santa Cruz Biotechnology. Anti-Myc-horseradish peroxidase (HRP) antibody (number 2040), anti-Flag antibody (number 14793), anti-STING antibody (number 13647), anti-p-STING antibody (Ser366; number 85735), anti-IRF3 antibody (number 4302), and anti-p-IRF3 antibody (Ser396; number 4947) were purchased from Cell Signaling. Anti-Flag antibody (F3165), anti-Flag-HRP antibody (A8582), anti-Flag M2 affinity gel (A2220), and anti-β-actin antibody (A5316) were purchased from Sigma. Anti-STING antibody (564835), anti-GM130 antibody (610822), and anti-calreticulin antibody (612136) were purchased from BD Biosciences. Anti-HSV ICP27 antibody (P1113) was purchased from Virusys. Goat anti-rabbit IgG(H+L) cross-absorbed secondary antibody tetramethyl rhodamine isocyanate (TRITC) was purchases from Thermo Fisher. Anti-γ134.5 antibody was generated as described previously (55).
Quantitative real-time PCR assay.
Total RNA was harvested from cells using an RNeasy kit (Qiagen) and subjected to DNase I digestion (New England BioLabs). cDNA was synthesized using a high-capacity cDNA reverse transcription kit (Applied Biosystems). Quantitative real-time PCR was performed using an Applied Biosystems ABI Prism 7900HT instrument with SYBR green master mix (Applied Biosystems). Gene expression levels were normalized to that of endogenous control 18S rRNA. Relative gene expression was determined as described previously (56). Primer sequences were as follows: human IFN-β, CCT GAA GGC CAA GGA GTA CA and AAG CAA TTG TCC AGT CCC AG; human ISG54, AAG AGT GCA GCT GCC TGA AC and CCT CCA TCA AGT TCC AGG TG; human ISG56, CCT CCT TGG GTT CGT CTA CA and AGT GGC TGA TAT CTG GGT GC; human RANTES, CCT GCT GCT TTG CCT ACA TT and ACA CAC TTG GCG GTT CTT TC; mouse IFN-β, AAT TTC TCC AGC ACT GGG TG and AGT TGA GGA CAT TC CCA CG; mouse ISG54, GCAAGA TGC ACC AAG ATG AG and CAC TCT CCA GGC AAC CTC TT; mouse ISG56, CAA GGC AGG TTT CTG AGG AG and AAG CAG ATT CTC CAT GAC CTG; mouse RANTES, CTG CTG CTT TGC CTA CCT CT and CAC TTC TTC TCT GGG TTG GC; and 18S rRNA, CCT GCG GCT TAA TTT GAC TC and AAC CAG ACA AAT CGC TCC AC.
Confocal microscopy.
Cells were grown on coverslips in a 12-well plate. After viral infection, cells were fixed with 4% paraformaldehyde (Thermo Fisher), permeabilized in 0.5% Triton X-100, and then incubated with 3% bovine serum albumin (BSA) in phosphate-buffered saline (PBS). The cells were stained with primary antibodies at 4°C overnight and then reacted with fluorescently conjugated secondary antibodies. After being washed with PBS, the cells were mounted, and images were acquired under Zeiss LSM710 confocal microscope and processed with Zen 2010 (Zeiss).
ACKNOWLEDGMENTS
This work was supported by grants from the National Institute of Allergy and Infectious Diseases (AI119618 to B.H.) and the National Natural Science Foundation of China (81672010 to Y.C.). Shuang Pan was supported in part by a fellowship from the China Scholarship Council (CSC NO.201606200021).
We thank Glen Barber for providing STING+/+ and STING−/− cells.
REFERENCES
- 1.Whitley RJ, Roizman B. 2001. Herpes simplex virus infections. Lancet 357:1513–1518. doi: 10.1016/S0140-6736(00)04638-9. [DOI] [PubMed] [Google Scholar]
- 2.Chou J, Kern ER, Whitley RJ, Roizman B. 1990. Mapping of herpes simplex virus-1 neurovirulence to γ134.5, a gene nonessential for growth in culture. Science 250:1262–1266. doi: 10.1126/science.2173860. [DOI] [PubMed] [Google Scholar]
- 3.MacLean AR, ul-Fareed M, Robertson L, Harland J, Brown SM. 1991. Herpes simplex virus type 1 deletion variants 1714 and 1716 pinpoint neurovirulence-related sequences in Glasgow strain 17+ between immediate early gene 1 and the ‘a’ sequence. J Gen Virol 72:631–639. doi: 10.1099/0022-1317-72-3-631. [DOI] [PubMed] [Google Scholar]
- 4.Whitley RJ, Kern ER, Chatterjee S, Chou J, Roizman B. 1993. Replication, establishment of latency, and induced reactivation of herpes simplex virus γ134.5 deletion mutants in rodent models. J Clin Invest 91:2837–2843. doi: 10.1172/JCI116527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perng GC, Ghiasi H, Slanina SM, Nesburn AB, Wechsler SL. 1996. High-dose ocular infection with a herpes simplex virus type 1 ICP34.5 deletion mutant produces no corneal disease or neurovirulence yet results in wild-type levels of spontaneous reactivation. J Virol 70:2883–2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mao H, Rosenthal KS. 2003. Strain-dependent structural variants of herpes simplex virus type 1 ICP34.5 determine viral plaque size, efficiency of glycoprotein processing, and viral release and neuroinvasive disease potential. J Virol 77:3409–3417. doi: 10.1128/JVI.77.6.3409-3417.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.MacLean A, Robertson L, McKay E, Brown SM. 1991. The RL neurovirulence locus in herpes simplex virus type 2 strain HG52 plays no role in latency. J Gen Virol 72:2305–2310. doi: 10.1099/0022-1317-72-9-2305. [DOI] [PubMed] [Google Scholar]
- 8.Davis KL, Korom M, Morrison LA. 2014. Herpes simplex virus 2 ICP34.5 confers neurovirulence by regulating the type I interferon response. Virology 468-470:330–339. [DOI] [PubMed] [Google Scholar]
- 9.Ma Y, He B. 2014. Recognition of herpes simplex viruses: Toll-like receptors and beyond. J Mol Biol 426:1133–1147. doi: 10.1016/j.jmb.2013.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun L, Wu J, Du F, Chen X, Chen ZJ. 2013. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339:786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, Sirois CM, Jin T, Latz E, Xiao TS, Fitzgerald KA, Paludan SR, Bowie AG. 2010. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol 11:997–1004. doi: 10.1038/ni.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Z, Yuan B, Bao M, Lu N, Kim T, Liu YJ. 2011. The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat Immunol 12:959–965. doi: 10.1038/ni.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Orzalli MH, DeLuca NA, Knipe DM. 2012. Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc Natl Acad Sci U S A 109:E3008–E3017. doi: 10.1073/pnas.1211302109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li T, Diner BA, Chen J, Cristea IM. 2012. Acetylation modulates cellular distribution and DNA sensing ability of interferon-inducible protein IFI16. Proc Natl Acad Sci U S A 109:10558–10563. doi: 10.1073/pnas.1203447109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Orzalli MH, Broekema NM, Diner BA, Hancks DC, Elde NC, Cristea IM, Knipe DM. 2015. cGAS-mediated stabilization of IFI16 promotes innate signaling during herpes simplex virus infection. Proc Natl Acad Sci U S A 112:E1773–E1781. doi: 10.1073/pnas.1424637112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Almine JF, O'Hare CA, Dunphy G, Haga IR, Naik RJ, Atrih A, Connolly DJ, Taylor J, Kelsall IR, Bowie AG, Beard PM, Unterholzner L. 2017. IFI16 and cGAS cooperate in the activation of STING during DNA sensing in human keratinocytes. Nat Commun 8:14392. doi: 10.1038/ncomms14392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jønsson KL, Laustsen A, Krapp C, Skipper KA, Thavachelvam K, Hotter D, Egedal JH, Kjolby M, Mohammadi P, Prabakaran T, Sorensen LK, Sun C, Jensen SB, Holm CK, Lebbink RJ, Johannsen M, Nyegaard M, Mikkelsen JG, Kirchhoff F, Paludan SR, Jakobsen MR. 2017. IFI16 is required for DNA sensing in human macrophages by promoting production and function of cGAMP. Nat Commun 8:14391. doi: 10.1038/ncomms14391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ackermann M, Chou J, Sarmiento M, Lerner RA, Roizman B. 1986. Identification by antibody to a synthetic peptide of a protein specified by a diploid gene located in the terminal repeats of the L component of herpes simplex virus genome. J Virol 58:843–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chou J, Roizman B. 1992. The γ134.5 gene of herpes simplex virus 1 precludes neuroblastoma cells from triggering total shutoff of protein synthesis characteristic of programed cell death in neuronal cells. Proc Natl Acad Sci U S A 89:3266–3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chou J, Chen JJ, Gross M, Roizman B. 1995. Association of a M(r) 90,000 phosphoprotein with protein kinase PKR in cells exhibiting enhanced phosphorylation of translation initiation factor eIF-2α and premature shutoff of protein synthesis after infection with γ134.5- mutants of herpes simplex virus 1. Proc Natl Acad Sci U S A 92:10516–10520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.He B, Gross M, Roizman B. 1997. The γ134.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1α to dephosphorylate the α subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc Natl Acad Sci U S A 94:843–848. doi: 10.1073/pnas.94.3.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.He B, Gross M, Roizman B. 1998. The γ134.5 protein of herpes simplex virus 1 has the structural and functional attributes of a protein phosphatase 1 regulatory subunit and is present in a high molecular weight complex with the enzyme in infected cells. J Biol Chem 273:20737–20743. doi: 10.1074/jbc.273.33.20737. [DOI] [PubMed] [Google Scholar]
- 23.He B, Chou J, Liebermann DA, Hoffman B, Roizman B. 1996. The carboxyl terminus of the murine MyD116 gene substitutes for the corresponding domain of the γ134.5 gene of herpes simplex virus to preclude the premature shutoff of total protein synthesis in infected human cells. J Virol 70:84–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Verpooten D, Feng Z, Valyi-Nagy T, Ma Y, Jin H, Yan Z, Zhang C, Cao Y, He B. 2009. Dephosphorylation of eIF2α mediated by the γ134.5 protein of herpes simplex virus 1 facilitates viral neuroinvasion. J Virol 83:12626–12630. doi: 10.1128/JVI.01431-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilcox DR, Muller WJ, Longnecker R. 2015. HSV targeting of the host phosphatase PP1alpha is required for disseminated disease in the neonate and contributes to pathogenesis in the brain. Proc Natl Acad Sci U S A 112:E6937–E6944. doi: 10.1073/pnas.1513045112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tallóczy Z, Jiang W, Virgin HW IV, Leib DA, Scheuner D, Kaufman RJ, Eskelinen EL, Levine B. 2002. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc Natl Acad Sci U S A 99:190–195. doi: 10.1073/pnas.012485299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Orvedahl A, Alexander D, Talloczy Z, Sun Q, Wei Y, Zhang W, Burns D, Leib DA, Levine B. 2007. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 1:23–35. doi: 10.1016/j.chom.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 28.Verpooten D, Ma Y, Hou S, Yan Z, He B. 2009. Control of TANK-binding kinase 1-mediated signaling by the γ134.5 protein of herpes simplex virus 1. J Biol Chem 284:1097–1105. doi: 10.1074/jbc.M805905200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma Y, Jin H, Valyi-Nagy T, Cao Y, Yan Z, He B. 2012. Inhibition of TANK binding kinase 1 by herpes simplex virus 1 facilitates productive infection. J Virol 86:2188–2196. doi: 10.1128/JVI.05376-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jin H, Ma Y, Prabhakar BS, Feng Z, Valyi-Nagy T, Yan Z, Verpooten D, Zhang C, Cao Y, He B. 2009. The γ134.5 protein of herpes simplex virus 1 is required to interfere with dendritic cell maturation during productive infection. J Virol 83:4984–4994. doi: 10.1128/JVI.02535-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jin H, Yan Z, Ma Y, Cao Y, He B. 2011. A herpesvirus virulence factor inhibits dendritic cell maturation through protein phosphatase 1 and Ikappa B kinase. J Virol 85:3397–3407. doi: 10.1128/JVI.02373-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brown SM, MacLean AR, Aitken JD, Harland J. 1994. ICP34.5 influences herpes simplex virus type 1 maturation and egress from infected cells in vitro. J Gen Virol 75:3679–3686. doi: 10.1099/0022-1317-75-12-3679. [DOI] [PubMed] [Google Scholar]
- 33.Wang Y, Yang Y, Wu S, Pan S, Zhou C, Ma Y, Ru Y, Dong S, He B, Zhang C, Cao Y. 2014. p32 is a novel target for viral protein ICP34.5 of herpes simplex virus type 1 and facilitates viral nuclear egress. J Biol Chem 289:35795–35805. doi: 10.1074/jbc.M114.603845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu S, Pan S, Zhang L, Baines J, Roller R, Ames J, Yang M, Wang J, Chen D, Liu Y, Zhang C, Cao Y, He B. 2016. Herpes simplex virus 1 induces phosphorylation and reorganization of lamin A/C through the γ134.5 protein that facilitates nuclear egress. J Virol 90:10414–10422. doi: 10.1128/JVI.01392-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Herman M, Ciancanelli M, Ou YH, Lorenzo L, Klaudel-Dreszler M, Pauwels E, Sancho-Shimizu V, Perez de Diego R, Abhyankar A, Israelsson E, Guo Y, Cardon A, Rozenberg F, Lebon P, Tardieu M, Heropolitanska-Pliszka E, Chaussabel D, White MA, Abel L, Zhang SY, Casanova JL. 2012. Heterozygous TBK1 mutations impair TLR3 immunity and underlie herpes simplex encephalitis of childhood. J Exp Med 209:1567–1582. doi: 10.1084/jem.20111316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ahmad L, Zhang SY, Casanova JL, Sancho-Shimizu V. 2016. Human TBK1: a gatekeeper of neuroinflammation. Trends Mol Med 22:511–527. doi: 10.1016/j.molmed.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ishikawa H, Ma Z, Barber GN. 2009. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461:788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li XD, Wu J, Gao D, Wang H, Sun L, Chen ZJ. 2013. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science 341:1390–1394. doi: 10.1126/science.1244040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, Du F, Ren J, Wu YT, Grishin NV, Chen ZJ. 2015. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 347:aaa2630. doi: 10.1126/science.aaa2630. [DOI] [PubMed] [Google Scholar]
- 40.Ishikawa H, Barber GN. 2008. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455:674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dobbs N, Burnaevskiy N, Chen D, Gonugunta VK, Alto NM, Yan N. 2015. STING Activation by translocation from the ER is associated with infection and autoinflammatory disease. Cell Host Microbe 18:157–168. doi: 10.1016/j.chom.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chou J, Roizman B. 1986. The terminal a sequence of the herpes simplex virus genome contains the promoter of a gene located in the repeat sequences of the L component. J Virol 57:629–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McKay EM, McVey B, Marsden HS, Brown SM, MacLean AR. 1993. The herpes simplex virus type 1 strain 17 open reading frame RL1 encodes a polypeptide of apparent M(r) 37K equivalent to ICP34.5 of herpes simplex virus type 1 strain F. J Gen Virol 74:2493–2497. doi: 10.1099/0022-1317-74-11-2493. [DOI] [PubMed] [Google Scholar]
- 44.Royer DJ, Carr DJ. 2016. A STING-dependent innate-sensing pathway mediates resistance to corneal HSV-1 infection via upregulation of the antiviral effector tetherin. Mucosal Immunol 9:1065–1075. doi: 10.1038/mi.2015.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Parker ZM, Murphy AA, Leib DA. 2015. Role of the DNA sensor STING in protection from lethal infection following corneal and intracerebral challenge with herpes simplex virus 1. J Virol 89:11080–11091. doi: 10.1128/JVI.00954-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mukai K, Konno H, Akiba T, Uemura T, Waguri S, Kobayashi T, Barber GN, Arai H, Taguchi T. 2016. Activation of STING requires palmitoylation at the Golgi. Nat Commun 7:11932. doi: 10.1038/ncomms11932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luo WW, Li S, Li C, Lian H, Yang Q, Zhong B, Shu HB. 2016. iRhom2 is essential for innate immunity to DNA viruses by mediating trafficking and stability of the adaptor STING. Nat Immunol 17:1057–1066. doi: 10.1038/ni.3510. [DOI] [PubMed] [Google Scholar]
- 48.Manivanh R, Mehrbach J, Knipe DM, Leib DA. 2017. Role of herpes simplex virus 1 γ34.5 in the regulation of IRF3 signaling. J Virol 91:e01156-. doi: 10.1128/JVI.01156-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Christensen MH, Jensen SB, Miettinen JJ, Luecke S, Prabakaran T, Reinert LS, Mettenleiter T, Chen ZJ, Knipe DM, Sandri-Goldin RM, Enquist LW, Hartmann R, Mogensen TH, Rice SA, Nyman TA, Matikainen S, Paludan SR. 2016. HSV-1 ICP27 targets the TBK1-activated STING signalsome to inhibit virus-induced type I IFN expression. EMBO J 35:1385–1399. doi: 10.15252/embj.201593458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Deschamps T, Kalamvoki M. 2017. Evasion of the STING DNA-sensing pathway by VP11/12 of herpes simplex virus 1. J Virol 91:e00535-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Su C, Zheng C. 2017. Herpes simplex virus 1 abrogates the cGAS/STING-mediated cytosolic DNA-sensing pathway via its virion host shutoff protein, UL41. J Virol 91:e02414-16. doi: 10.1128/JVI.02414-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Orzalli MH, Broekema NM, Knipe DM. 2016. Relative contributions of herpes simplex virus 1 ICP0 and vhs to loss of cellular IFI16 vary in different human cell types. J Virol 90:8351–8359. doi: 10.1128/JVI.00939-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huang J, You H, Su C, Li Y, Chen S, Zheng C. 23 May 2018. Herpes simplex virus 1 tegument protein VP22 abrogates cGAS/STING-mediated antiviral innate immunity. J Virol doi: 10.1128/JVI.00841-18. [DOI] [PMC free article] [PubMed]
- 54.Ejercito PM, Kieff ED, Roizman B. 1968. Characterization of herpes simplex virus strains differing in their effects on social behaviour of infected cells. J Gen Virol 2:357–364. doi: 10.1099/0022-1317-2-3-357. [DOI] [PubMed] [Google Scholar]
- 55.Cheng G, Brett ME, He B. 2002. Signals that dictate nuclear, nucleolar, and cytoplasmic shuttling of the γ134.5 protein of herpes simplex virus type 1. J Virol 76:9434–9445. doi: 10.1128/JVI.76.18.9434-9445.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]