

Abstract
For conditions with inflammatory flare-ups, fast drug-release from a depot is crucial to reduce cell infiltration and prevent long-term tissue destruction. While this concept has been explored for chronic diseases, preventing acute inflammatory flares has not been explored. To address this issue, we developed a preventative inflammation-sensitive system and applied it to acute gout, a condition where millions of inflammatory cells are recruited rapidly, causing excruciating and debilitating pain.
Rapid drug release was first demonstrated from a pH-responsive acetalated dextran particle loaded with dexamethasone (AcDex-DXM), reducing proinflammatory cytokines in vitro as efficiently as free drug. Then, using the air pouch model of gout, mice were pre-treated 24 hours before inducing inflammation. AcDex-DXM reduced overall cell infiltration with decreased neutrophils, increased monocytes, and diminished cytokines and chemokines. In a more extended prophylaxis model, murine joints were pre-treated eight days before initiating inflammation. After quantifying cell infiltration, only AcDex-DXM reduced the overall joint inflammation, where neither free drug nor a conventional drug-depot achieved adequate anti-inflammatory effects.
Here, the superior efficacy of disease-triggered drug-delivery to prevent acute inflammation was demonstrated over free drug and slow-release depots. This approach and results promise exciting treatment opportunities for multiple inflammatory conditions suffering from acute flares.
Keywords: Disease-triggered, prophylaxis, inflammatory flare-ups, gout, stimuli-responsive particles
ToC figure
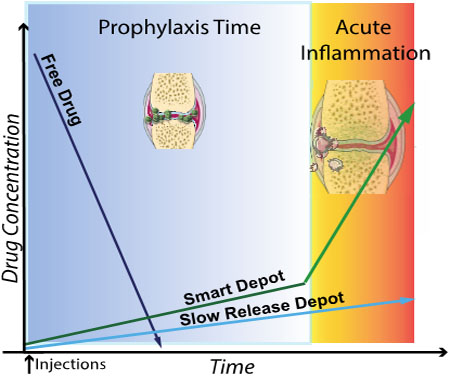
1. Introduction
Emerging technologies in drug-delivery such as disease-triggered particles and constituent polymers offer enabling tools to create tight control drug-delivery systems. While free drug enable high drug availability at time of administration, multiple injections may be needed to reach the therapeutic window, and the concentration variability can be high. To circumvent such issues, conventional drug depots slowly dispense drug over time, however this occurs at non-controllable rates, potentially sub-treating the affected area or demanding higher doses over longer time to reach efficiency.[1] Instead, controlled drug-release from a depot can offer advantages such as reduced dosing frequency, lower doses, and reduced side effects. Several different drug-delivery vehicles, both slow release and bioresponsive, have been developed to optimize treatment of inflammatory diseases and cancer.[2–7] The bioresponsive drug-delivery vehicles enhanced drug efficacy compared to non-responsive controls, delivering drugs to established chronic inflammatory disease models such as multiple sclerosis (MS).[8] However, many chronic inflammatory diseases suffer from low-grade inflammation in between disease symptoms– such as in MS or rheumatoid arthritis or non-existing inflammation with no symptoms during times of recession such as in gouty arthritis. Preventing acute inflammatory flares that can arise in such diseases has not yet been explored, where early resolution is critical to avoid inflammatory cell infiltration and long-term tissue destruction.[9] Ideally, a pre-administered depot to prevent acute flares should only release drug if a flare arises, thereby automating drug-delivery and reducing the required dose to achieve inflammation control. Administering a preventative drug-depot to a joint at risk of developing an inflammatory flare, but at time of administration is considered healthy or asymptomatic, would be greatly advantageous in diseases such as gout. Preventing an inflammatory reaction before or close to its onset demands smart drug-delivery vehicles. A preventative concept for inflammatory flares has previously not been explored, as the technology has not been available.
To achieve on-demand drug delivery and explore the concept of a prophylaxis for acute flares, we chose to utilize the biochemistry of the inflammatory process. Increased cellular metabolism decreases pH locally,[10–14] which we exploit to demonstrate our concept in a straightforward manner. We chose the naturally occurring and FDA approved dextran as our polysaccharide base, as it possesses a number of appealing characteristics: biocompatibility, biodegradatibilty and affordability. Non-modified dextran has clinically been used as a plasma expander for decades due to its water solubility.[15] Acetalated dextran (AcDex) has been studied as a bioresponsive polymer for many medical applications, exhibiting high versatility in delivery functions and very low toxicity.[8, 15–28] AcDex is readily synthesized, easily tunable and its degradation products are pH neutral and water-soluble.[25] We used the single step electrospray process to make the particles, enabling a high loading efficiency and reduced amount of potentially interfering surfactants used in emulsion.[6, 29] During electrospray, a high voltage is applied to an organic solution containing the polymer and payload to break the liquid into a jet of very fine aerosol droplets. As the solvent evaporates in flight, solid polymer particles are formed.[30] These properties make drug-loaded AcDex an excellent candidate for the concept of preventing inflammatory flares.
Flare-ups occur in inflammatory conditions such as gout, a disease with low treatment adherence and sub-optimal management.[31–33] Gout is the most common inflammatory arthritis (4% prevalence in the USA),[31–33] and impacts patient morbidity and premature mortality.[34–37] Flares are thought to occur due to increased uric acid in circulation, depositing crystals in joints. The resulting inflammatory flare consists mainly of inflammatory neutrophils being rapidly recruited, causing an excruciating acute pain.[9, 31] Increased gout incidence (29% from 1997–2012)[32] and co-morbidities on the rise such as cardiovascular disease, kidney disease and the metabolic syndrome challenge drug therapy research to increase specificity towards the affected joint and spare other tissues from toxicity. In particular, gout flares have increased due to hospitalization or medical illness.[38–41] Here, recommended treatments such as anti-inflammatory NSAIDs and steroids can pose a risk due to side effects such as impaired wound healing.[38–41] Such unwanted effects are directly dependent on the dose and timing of glucocorticoid administration.[42] Local administration of corticosteroids leads to improved outcomes,[43] and long-lasting corticosteroid suspensions are clinically available to treat joint inflammations.[44] However, a slow drug release often requires higher doses for sufficient efficacy. More importantly, they dispense drug when potentially not needed, increasing the risk of side effects.[45] Gout patients undergoing surgery, for instance due to a co-morbidity, develop flares due to the procedure itself.[46] Flares can involve previously affected joints and develop within eight days after surgery.[46] These features with rapid onset of inflammation in joints make gout an ideal model disease for investigating the concept of a local, prophylaxis option to be administered before an intervention.
In this work, we examined the efficacy of AcDex-particles carrying dexamethasone (DXM) as a prophylactic in murine models of gout, and show enhanced efficacy of DXM on protection from inflammatory cell infiltration and tissue destruction. Our results suggest that a significant suppression of an activated immune system is only reached by using a disease-triggered prophylaxis system.
2. Results and Discussion
2.1. DXM was efficiently encapsulated by AcDex using electrospray and was well tolerated
The synthesized AcDex polymer (Supplementary Figure S1) was formulated into disease-triggered particles (mean diameter 3.36 ± 0.35 μm, Figure 1a). The micron size was chosen to enhance joint retention time.[47] The control particles were formulated from poly(lactic-co-glycolic acid) (PLGA), a Food and Drug Administration (FDA) approved non-bioresponsive control polymer (mean diameter 3.48 ± 0.27 μm, Supplementary Figure S4a). DXM loading was determined to be 1.5–6.7% (15–67% encapsulation efficiency) for AcDex, and 3–8% for PLGA (30–80% encapsulation efficiency). The payload distribution within the particles was modeled using Rhodamine 6G at 1 % to avoid a potential quenching phenomenon.[48] Imaging using confocal microscopy (Figure 1c) revealed a clustered distribution pattern throughout the particles. Such distribution pattern is both facilitated by blending the payload directly in the polymer solution, as well as the single-phase fabrication method.[30, 49] Homogenous payload distribution for electrosprayed particles has previously been shown using fluorescence, where a coaxial (two-phase) setup is needed to provide distinct layers for payload distributions and core-shell structures.[50–52] As the drug is loaded at a higher percentage it is possible that drug distribution is even more homogenously distributed than the dye.
Figure 1. AcDex-DXM Microparticles were engineered to 3μm and were stable over eight days.
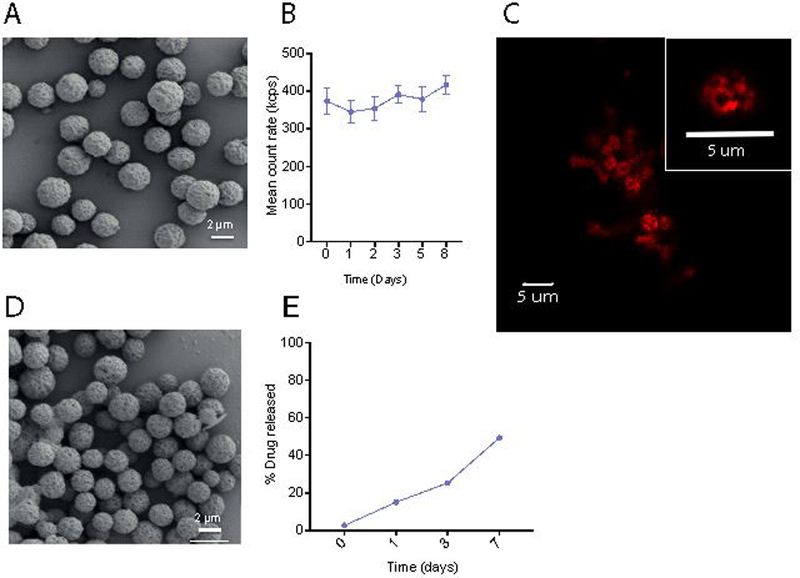
(A) Intact AcDex particles containing DXM, scale bar represents 2 μm. AcDex-DXM particles were incubated for eight days in 0.02 % Tween 80/ phosphate buffered saline (PBS) pH 7.4, and stability was measured using DLS (B), imaged using confocal microscopy, scale bar represents 5 μm (C) with insert showing zoomed in single particle. The particles were also visualized after the eight day incubation by SEM (D). (E) Drug-release over 7 days was quantified using LC-MS.
AcDex particle stability was then determined both by DLS (Figure 1b) as well as by scanning electron microscopy (SEM, Figure 1d) over eight days (PLGA stability shown in Supplementary Figure S4b–c). Release was quantified by liquid chromatography -mass spectrometry (LC-MS, AcDex Figure 1e, PLGA in Supplementary Figure S4d).
Nonspecific adsorption of proteins can cause particle aggregation and limit their performance. Compared to the density values of two saturated model proteins, bovine serum albumin (BSA, (290 ng/cm2) and ovalbumin (OVA, 313 ng/cm2),[53] minimal amount of these proteins were found to be adsorbed to AcDex particles, i.e., 0.56 ± 0.25 ng of BSA/cm2 and 0.45 ± 0.05 ng of OVA/cm2. In vitro, empty AcDex particles were added to isolated bone marrow derived macrophages (BMDM) for 24–48 hours to test for tolerability. When tested at equivalent doses of 0.66–28 μM DXM cells showed similar viability as controls (Supplementary Figure S5). When empty particles were tested in vitro, AcDex itself did not contribute to an additive anti-inflammatory effect (Supplementary Figure S6). In vivo, empty AcDex particles were injected into murine joints, where haemotoxylin and eosin (H&E) staining after eight days revealed no tissue destruction or cell infiltration compared to PBS control (Supplementary Figure S7). Taken together, these results demonstrated good stability and high viability of AcDex-DXM as a potential treatment in the relevant regimes; duration time as well as tolerability in vitro and in vivo. Where traditional fabrication methods may suffer from inhomogeneous release profiles and lack batch-to-batch reproducibility, our particles manufactured by electrospray resulted in highly reproducible particles with excellent control of size and monodispersity. This is in accordance with other reports using electrospray, showing that electrospray provides a versatile method for particle manufacturing over a wide size range and being applicable to several different polymers, including AcDex.[19, 30, 54, 55] In addition, the particles showed high stability at pH 7.4 over eight days (Figure 1 A-C), indicating that the particles could be used within the suggested eight-day timeframe.
2.2. AcDex-DXM reduces IL-1β to the same extent as free drug
We first investigated the ability of the particles loaded with anti-inflammatory drug to reduce the gout inflammation-driving cytokine interleukin (IL)-1β. We chose macrophages as they are the first line of defense to initiate an inflammation.[56] We differentiated macrophages from murine bone marrow and added either free drug, AcDex or PLGA simultaneously with stimulation. After overnight stimulation, AcDex-DXM had reduced IL-1β levels similar to free drug, demonstrating a fast drug release (Figure 2a), reducing levels more than PLGA-DXM. Cells were confirmed to be alive by a rezasurin viability assay (Supplementary Figure S8a). To investigate the potential pH reduction during cell stimulation, we measured the pH in the media reduced by stimulated cells during a 24h incubation. In general, the activated cells changed the media from initial pH 7.4 before stimulation, to a final range of 6.71 to 7.29 after the incubation (Figure 2b), which was enough to trigger drug release from AcDex over a 24-hour period.
Figure 2. AcDex-DXM reduces proinflammatory cytokines from macrophages similar to free drug and releases drug already at pH 6.
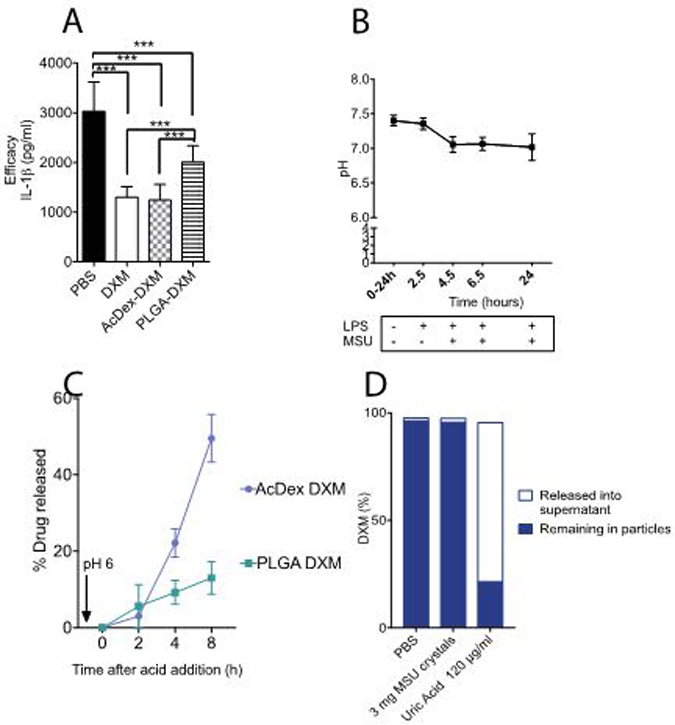
(A) BMDMs were stimulated to produce cytokines and treated with 40 nM DXM. Stimulation consisted of lipopolysaccharide (LPS, 0.1 μg/ml) for 2–3 hours, and then MSU crystals were added (0.2 mg/ml) overnight, and supernatants were saved at −80°C and then analyzed for cytokine levels by enzyme-linked immunosorbent assay (ELISA). Immortalized macrophages were stimulated during 24h and pH was measured at the indicated timepoints (B). (C) Triggered drug-release by pH 6 after 24 hours pre-incubation was measured by LC-MS over 8 hours. (D) AcDex-DXM particles were incubated for eight hours with PBS, MSU crystals or uric acid (pH 4.5) and DXM was measured in the supernatant or remaining in particles. Data is presented as pg/ml (A), mean ± SEM (A,B), ***p < 0.001
At physiological pH of 7.4 and 37 °C, the urate ion forms monosodium urate (MSU) crystals in humans.[57] To establish that MSU crystals themselves did not induce DXM release, AcDex-DXM particles were incubated with crystals for eight hours. DXM was mainly found retained in the particles after incubation with MSU crystals, whereas DXM was found primarily in the supernatant when incubated with uric acid at pH 4.5 (Figure 2b).
These results demonstrate that AcDex-DXM can be used as anti-inflammatory therapy and confirmed that AcDex-DXM display similar anti-inflammatory effects as free drug in reducing proinflammatory cytokine production from macrophages in vitro, making AcDex microparticles a good option for investigating the concept of a prophylaxis for acute inflammatory flares.
2.3. AcDex-DXM particles efficiently inhibits inflammation as a 24-hour prophylaxis in vivo
We used the air pouch gout model to investigate the in vivo anti-inflammatory prophylaxis potential.[58, 59] Treatments were injected 24 hours before MSUs into the pouch. Eight hours later, cells were collected, counted and analyzed by fluorescence-activated cell sorting (FACS). When prophylaxis was used at the equivalent doses of 120 μM DXM 24h before initiating inflammation, cell infiltration was reduced by AcDex-DXM as well as PLGA-DXM compared to control (Table 1).
Table 1.
Recovered leukocytes (106 cells/lavage, mean ± SD) after a 24h gout prophylaxis model (n= 5 mice per group, analysis of variance (ANOVA) comparing all treatments).
| Treatment (120 μM) | PBS | DXM | AcDex-DXM | PLGA-DXM |
|---|---|---|---|---|
| 106 cells/lavage (mean ± SD) | 13±6 | 8.7±3.7 | 1.2±0.8 **,♦ | 2.4.7±0.7 * |
p < 0.05,
p < 0.01 compared to PBS control,
p < 0.05 compared to DXM treatment.
At lower doses (15 μg), AcDex-DXM maintained an efficiently reduced cell infiltration as a 24 hours prophylaxis (Figure 3a). At this dose, PLGA-DXM did not reduce cell infiltration to the same extend (Figure 3a). Percentage of neutrophils decreased by AcDex-DXM (Figure 3b, d), while recruitment of monocytes/macrophages had increased (Figure 3c, d). These results show that triggered fast release kinetics enables reduced dosing, being delivered at the right time and right place.
Figure 3. AcDex-DXM reduces inflammatory flares and decreases pro-inflammatory neutrophils while increasing monocytes/macrophages as a 24h prophylaxis in vivo.
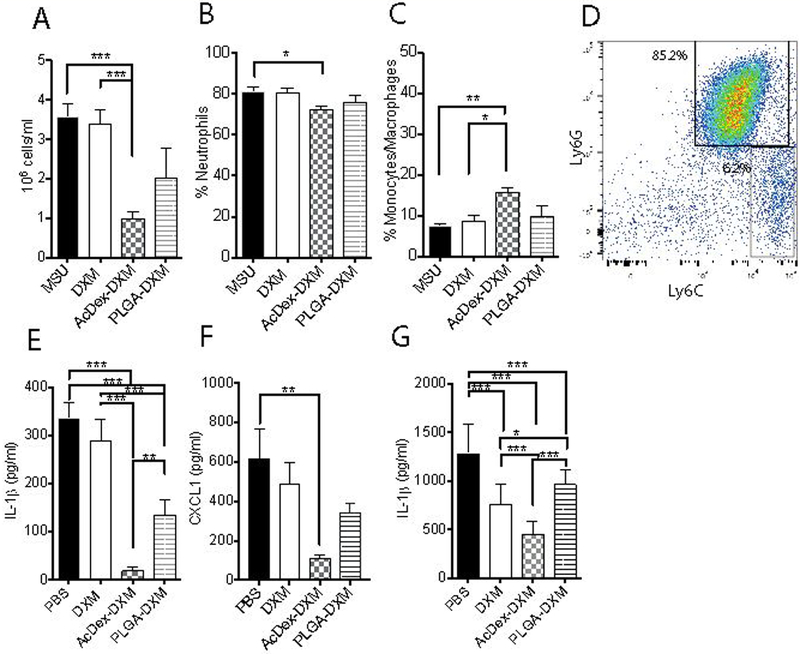
Mice were injected with prophylactic treatments (PBS control, DXM, AcDex-DXM or PLGA-DXM, 15 μg DXM/mouse) 24 hours before MSU crystals injections (3 mg in 0.5 ml PBS) into subcutaneous air pouches. After eight hours of stimulation, leukocytes from the exudates were counted (A), and FACS was used to analyze the percentage of CD11b+Ly6G+ neutrophils (B) and CD11b+Ly6C+ monocytes/macrophages (C) populations. A representative FACS plot is shown in D. IL-1β (E) and CXCL1 (F) were measured from the exudates by ELISA. In similar assays to Figure 1a, treatments were added 24h before first stimulations in BMDM (G). Data is presented as pg/ml. ***p < 0.001, ** p < 0.01, *p < 0.05.
Further, both AcDex-DXM and PLGA-DXM reduced IL-1β in the exudate (Figure 3e). However, only AcDex-DXM reduced the neutrophil attracting chemokine (C-X-C motif) ligand 1 (CXCL1, Figure 3f). These results together with a potential cell shift suggested a pro-healing environment,[60] however we were not able to confirm this by anti-inflammatory IL-10 levels as they were below detection limits (results not shown).
We further examined the direct effect on cells in vitro by the prophylaxis, adding treatments 24h before stimulation. The resulting release from AcDex-DXM proved superior in reducing IL-1β levels from stimulated macrophages, compared to both free drug and non-bioresponsive drug-delivery (Figure 3g), although all prophylaxis suppressed the cytokine. Since an inflammatory flare occurs rapidly, triggered fast release kinetics to inhibit or reduce inflammation is crucial for minimizing pain, reducing symptoms and long-term tissue destruction.
By utilizing local depot delivery, particles potentially integrated into the tissue lining for sustained prophylaxis potential,[47] being advantageously localized to the right place for rapid on-time release. The H&E staining of the skin revealed high cell-infiltration in the MSU control (Figure 4a), and no difference in infiltration was seen by free drug prophylaxis (Figure 4b). Much less infiltration was seen with AcDex-DXM (Figure 4c), whereas PLGA-DXM showed high cell infiltration (Figure 4d). Cell infiltration was quantified, revealing that AcDex-DXM was again superior to all other treatments (Figure 4e).
Figure 4. Reduced tissue infiltration after MSU crystals by AcDex-DXM as a 24-hour prophylaxis.
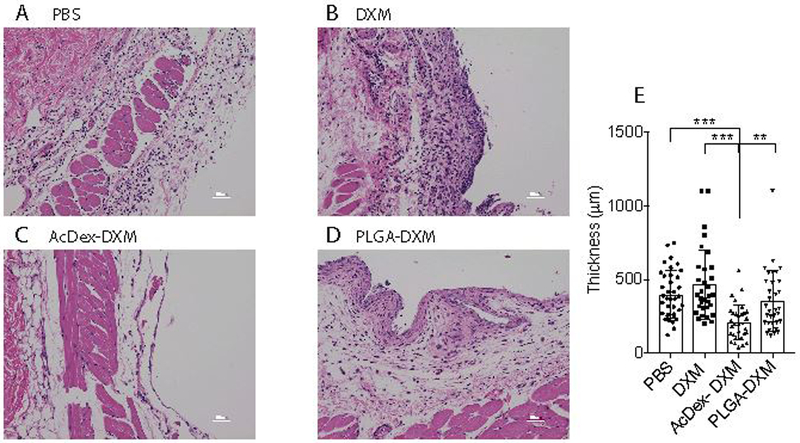
Mice were injected with prophylactic treatments (PBS control, DXM, AcDex-DXM or PLGA-DXM, 15 μg DXM/mouse) 24 hours before MSU crystal injections (3 mg in 0.5 ml PBS) into subcutaneous air pouches. After eight hours of stimulation, skin lining the pouch was preserved in 4% paraformaldehyde and subsequently stained by H&E. Representative images are shown in (A) MSU control, (B) DXM, (C) AcDex-DXM, and (D) PLGA-DXM. Scale bar represents 50 μm. The infiltrated cell area was quantified by Image J (E). The data is displayed as individual measurement points of 3 mice/group, with 6–16 sections per individual, and 32–37 distances per individual counted in total, ***p < 0.001, ** p < 0.01.
To confirm that the particles integrate in the tissue layer, we synthesized rhodamine B-labeled AcDex. These fluorescent particles were injected in the air pouch model and imaged after 32h to mimic the 24h prophylaxis study. We detected the expression of rhodamine B in the skin sections (Supplementary Figure S9a), indicating that the particles integrated in the tissues within 24h. We further investigated if these particles were uptaken by macrophages, by comparing free dye to particles loaded with dye. After either 4 or 24h incubation, cells were washed, fixed and fluorescence was subsequently measured. No fluorescence was measureable in macrophages after AcDex particle incubation after either 4 hours (results not shown) or after 24h, whereas in comparison, free dye was clearly measureable (Supplementary Figure S9b). Combining the cell study together with the in vivo data suggests that the particles integrate into the extracellular matrix rather than being uptaken by cells when injected locally, potentially due to their large size.[61] The particles are assumed to degrade in to benign biproducts over time, as cyclic acetalated dextran degrades to water-soluble dextran and acetone through hydrolysis, and acyclic acetals degrade rapidly to methanol and acetone. This has been shown by multiple other studies.[15, 16, 25] Low tissue pH in inflammation has long been regarded as an important mechanism for the failure of local anesthesia of inflamed tissues. Lactic acid and acidic by-products are concentrated near inflamed tissues, where earlier reported studies suggest a lower tissue pH at the order of 0.5–1.0 pH units.[13, 14] This depends on inflammation initiation and duration, and highly dependent on amount of recruited cells. Older studies have measured high hydrogen ion concentrations in inflamed tissue (down to pH 5.4), fracture related hematomas (down to pH 4.7) and in caridac ischemia (down to pH 5.7).[62–64] Indeed, our group has previously used AcDex for treatingtreat models of cardiac ischemia.[23, 24] Due to the airpouch model cell and tissue recovery process where a buffer is injected to collect the cells, the measured pH ranging from pH 7.2–7.4 was most likely not representative values of the pH at the time of inflammation and drug release, and satisfactory conclusions could not be drawn. Measuring pH-fluctuations non-invasively would certainly aid in the design of disease-triggered drug release, and highly sensitive probes are being developed.[65–67] Expanding the versatility of the particles and tailor them to the desired disease could include tuning the degree of acetalation and particle size to better suit the desired pH-range, where the tuned release kinetics and stability could extend the prophylactic capacity of the particles.[15, 16, 23, 24, 27, 68]
2.4. AcDex-DXM was superior as prophylaxis up to eight days, reducing proinflammatory cytokines in vitro and inhibits cell infiltration in murine joints
We continued with longer prophylaxis models (up to eight days) with triggered release in cuvette systems as well as in cells. The particles were incubated for eight days before pH was reduced to 6, and drug-release was quantified over 8 hours (Figure 5a). These results confirmed a triggered release by reduced pH was still possible in AcDex-DXM after eight days, maintaining its fast release kinetics. Drug release was reflected in the ability to decrease IL-1β levels from stimulated macrophages after 6 days (Figure 5b), and the chemokine CXCL1 from endothelial cells after eight days, important for recruiting more inflammatory cells (Figure 5c).[69] Cell viability of the primary macrophages was confirmed after the six-day prophylaxis (Supplementary Figure S8b), their maximum viability length, as was evident by the slight decrease in viability after free DXM and PLGA-DXM. The decreased viability for free DXM could be attributed to the apoptosis-inducing effects of DXM itself after longer exposure.[70, 71] PLGA has inherent vaccine adjuvant-properties, indicating that PLGA itself may have elicited a premature inflammatory response and reduced viability of the cells.[72–74] In these settings, AcDex-DXM was the only treatment that did not affect viability and the only option to reduce both pro-inflammatory cytokines (IL-1β) and the recruiting chemokine (CXCL1).
Figure 5. AcDex-DXM release drug for eight hours and reduces cytokine production as a long-term prophylaxis.
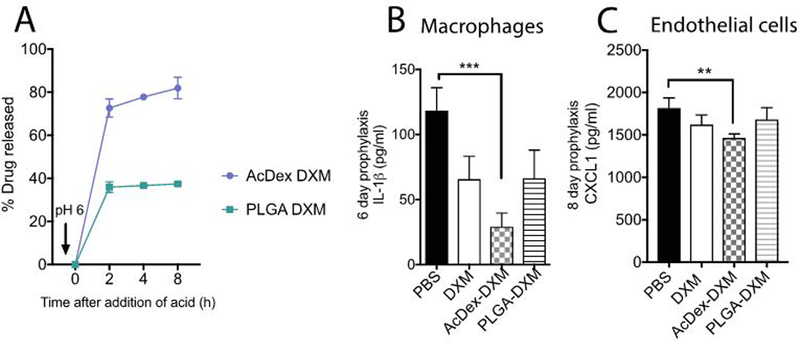
Drug was released by pH 6 after an 8-day pre-incubation as measured by LC-MS over 8 hours (A). In BMDMs (B) or C166 endothelial cells (C), treatments (equivalent doses of 40 nM DXM) were added six (B) or eight (C) days before first stimulations. Stimulation consisted of LPS (0.1 μg/ml) for 2–3 hours, and then MSU crystals were added (0.2 mg/ml) overnight, and supernatants were saved at −80°C and then analyzed for IL-1β (B) or CXCL1 (C) levels by ELISA. Data is presented as pg/ml, (***p < 0.001, *p < 0.05).
For a longer in vivo prophylaxis model we injected 10 μg DXM intra-articularly eight days before stimulation. The joints were injected with MSU crystals (100 μg) to induce inflammation, and collected after eight hours of stimulation. The joints were stained with H&E (representative histological images in Figure 6b–e) and cell infiltration was quantified, revealing that only AcDex-DXM particles reduced cell infiltration, showing superiority compared to both free drug and non-bioresponsive PLGA-DXM (Figure 6a). These results demonstrate that using a simple disease-triggered drug-delivery option enables and achieves prophylaxis for inflammatory flares. The individual data points indicate that there may be residual effects from administering free drug. The effects of steroids in human can last longer than just immediate relief, however, the result is highly unpredictable and believed to depend on factors such as genetic background, overall patient health, as well as the extent of inflammation. This thus prompts the administration of higher doses more frequently, increasing the risk of side effects.[42, 44] Similarly, some PLGA-treated individuals showed a reduced inflammatory reaction. Again, factors such as necessary dose would be hard to predict. Indeed, as with the 24h prophylaxis model (Table 1 and Figure 3), it may be that the dose could be further reduced with AcDex and retain an adequate anti-inflammatory effect. As is evident from the data, an option that reliably releases an anti-inflammatory dose exactly when needed circumvents both the need for higher doses initially, as well as repeated injections, and achieves better anti-inflammatory effect by automating the drug dispensing based on the inflammatory reaction.
Figure 6. AcDex-DXM as an eight-day prophylaxis reduces inflammation in murine joints.
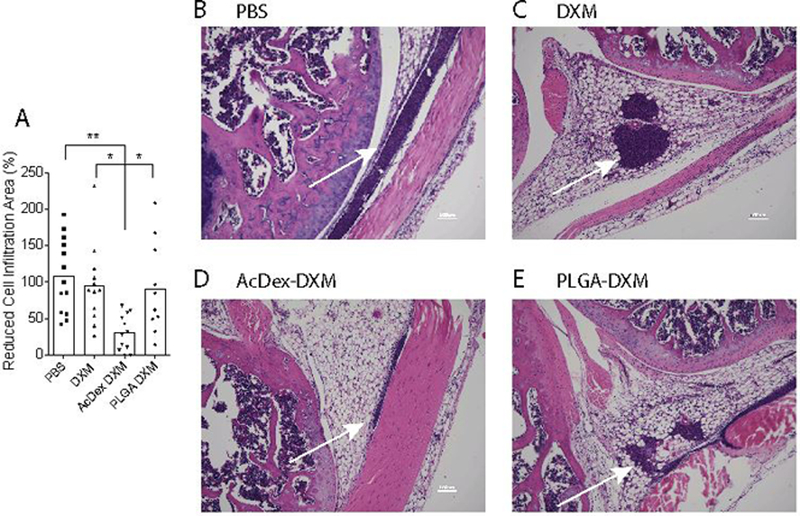
Mice were injected with prophylactic treatments (DXM, AcDex-DXM or PLGA-DXM, 10 μg DXM/mouse) eight days before MSU crystal injections (100 μg in 10 μl PBS) into murine knee joints. Eight hours after stimulation, knees were harvested, H&E stained and cell infiltration area was measured by Image J (A). (B-D) shows representative H&E stains of joints, scale bar is 100 μm. Arrows indicate cell infiltrations. In (A), dots represents individual data points pooled from two independent experiments (mean ± SEM), **p < 0.01,*p < 0.05.
3. Conclusion
In summary, we have for the first time harnessed the bioresponsive sensitivity of the AcDex polymer particle for prophylactic drug-delivery with the intention to prevent and reduce an inflammatory reaction as close as possible to the onset of the damaging acute inflammatory flare. Since an inflammatory flare occurs rapidly, triggered fast release kinetics to inhibit or reduce inflammation is crucial for minimizing pain, reducing symptoms and long-term tissue destruction. We demonstrate that sufficient anti-inflammatory effects can be achieved with reduced drug amounts if administered in a disease-triggered vehicle, and was the only option that reduced inflammatory cell infiltration as an eight-day prophylaxis. Pre-administered bioresponsive particles would be ready to release drugs to diminish the inflammatory response or preventing the flare altogether and increase recovery. The concept is widely applicable, extendable to failed drugs due to solubility issues, and has potential future applications for numerous other diseases suffering from flare-ups such as exercise-induced asthma, inflammatory bowel disease, and surgically induced systemic inflammatory response syndrome.
4. Experimental
4.1. Reagents
All reagents were from Sigma Aldrich (St. Louis, Missouri, USA), unless otherwise stated.
4.2. Responsive modified dextran
AcDex was synthesized as previously described.[15] Briefly, AcDex was synthesized by reacting dextran (1 g, 100 μmol) in anhydrous DMSO (10 mL) with 2-methoxypropene (3.4 mL, 37 mmol) and pyridinium p-toluenesulfonate (6 mg, 24 μmol) and the reaction mixture was quenched with triethylamine after 70 min (Supplementary Figure S1). The acetal content and ratio of cyclic/acyclic were determined by 1HNMR after acid degradation of the purified polymer. The methanol (3.34 ppm) and acetone (2.08 ppm) peaks in D2O/DCl were used to calculate the cyclic acetal percentage to 48.6% (Supplementary Figure S1), as previously described.[8] AcDex-labeled rhodamine B (Ex 570/ Em 590) was synthesized as described above, using rhodamine B labeled dextran (100 mg, Thermo Fisher).
4.3. Microparticle formulation with dexamethasone palmitate
Microparticles were prepared by an electrospray ionization method.[30, 55] In general, 60 mg of AcDex or the non-bioresponsive control polymer poly(lactic-co-glycolic acid); PLGA, 6 mg of Lutrol F127 (10 % wt/wt), and with or without 6 mg (10 % wt/wt) of dexamethasone palmitate (DXM; Toronto Research Chemicals, Toronto, Canada) were dissolved in either 400 μL of 4:1 de-acidified CHCl3 (EMD Millipore, Billerica MA, USA):DMSO, or acetonitrile (PLGA). The solutions were briefly sonicated to dissolve all constituents and loaded into a 4 mm diameter tube to prepare for electrospray. A voltage of 14 kV was applied with a flow rate of 0.24 mL/hr and sprayed out of a 31 gauge needle for 1.5 on to glass microscope slides (Thermo-Fisher Scientific). Microparticles were then collected by submerging the slides in 50 mL of 0.02 % Tween 80/PBS (pH 7.4) as an optimal medium to allow for passive diffusion of the hydrophobic DXM. Slides were sonicated briefly to remove the particles, and weighed before and after washing to calculate the amount of particles. The particle suspension was centrifuged at 4500 RPM for 15 min, and resuspended in Tween/PBS solution at 1 mg/mL and hydrated for 20–24 hours at 37°C on a an incubator-shaker at 60 RPM (New Brunswick Scientific). Particles were then again spun and resuspended in appropriate media for the following application.
4.4. Particle characterization
Microparticles were characterized by scanning electron microscopy (SEM) using an Agilent 8500 Fe-SEM (Agilent Technologies Inc, Santa Clara CA, USA). Particles were suspended at a concentration of 1 mg/mL and the resulting dispersions were dripped onto a silicon wafer and allowed to air dry. The particles were then sputter coated with a 2 nm layer of a palladium/gold alloy and imaged. Particle diameter distributions were extracted from recorded SEM photographs and dark-field images using NIS Elements (Nikon) and ImageJ software (NIH, Bethesda, USA). The hydrodynamic diameter and particles count rate were obtained by DLS (Malvern, Zetasizer).
Payload distribution was visualized by confocal scanning microscopy using a Zeiss LSM 880 Confocal with FAST Airyscan and loading the fluorescent probe Rhodamine 6G at 1%.
4.4.1. Particle loading and release
For loading and release studies, microparticle suspensions (at 1 mg/ml 0.02 % Tween 80/PBS pH 7.4) were divided evenly into 0.5 mL aliquots and placed inside an incubator shaker. At predetermined time points, up to eight days, individual samples were removed and centrifuged at 13,400 RPM for 10 minutes. 400 μL of supernatant was carefully removed. 100 μL of the supernatant was then combined with 900 μL of MeOH, filtered, and analyzed using Liquid Chromatography-Mass Spectroscopy (LC-MS, Agilent). The remaining particle pellet was resuspended in 100 μL of the residual supernatant. 900 μL of MeOH was added to dissolve the particles. In the case of PLGA microparticles, the solution was incubated at 60 °C for one hour in order to completely dissolve the particles. Dissolved polymer was removed using centrifugal filters (3 kDa MWCO, MilliPore Amicon Ultra; 13,400 RPM, 10 min). The eluent was filtered through a 0.2 μm pore-sized filter and analyzed by LC-MS. Particle loading studies were performed similarly. Due to a clear burst release by PLGA within the first 24 hours, (Supplementary Figure S2) particles were washed for 20–24 hours before use in subsequent experiments.
For acid-triggered release studies, particles were incubated for either one or eight days. 400 μL of supernatant was removed after either 24h or eight days. The pH was lowered to 6.0–6.5 by adding 900 μL of acidified phosphate buffered saline with 0.02% Tween 80, and placed back into the incubator shaker. Individual samples were removed from the shaker at 2, 4, and 8 hours after the addition of acid and processed as described above to analyze both the amount of DXM release into the supernatant and the amount of residual DXM in the polymer microparticles.
4.5. MSU crystals
Monosodium urate (MSU) crystals were prepared as previously described[75, 76] and confirmed to be the correct crystalline structure by X-ray diffractometer analysis (XRD, Supplementary Figure S3). Crystals were kept dry at room temperature or −20°C until resuspended in endotoxin-free PBS and confirmed to be free of detectable pyrogen by the limulus lysate assay (Lonza, Walkersville, Maryland, USA).
4.6. Particle release by MSU crystals and uric acid
Particles were incubated for eight hours at 37°C with MSU crystals (3 mg/ml PBS/tween,) or uric acid (120 μM in PBS/tween, pH 4.5). LC-MS analysis determined DXM concentrations retained in the particles or released in the supernatant.
4.7. Quantification of protein adsorption
Aliquots of microparticles (0.1 mg) were incubated overnight with BSA-AF488 (0.1 mg) and OVA-AF594 (0.1 mg) in 1 mL of PBS. After incubation, the samples were collected and washed by centrifugation to remove any unbound proteins and dispersed in 1 mL of PBS. The fluorescence of the particles was measured and the protein concentration quantified by linear calibration. The surface density coverage in ng/cm2 was obtained by dividing the amount of bound protein to the total surface area of the particles. Fluorescence measurements were performed using a fluorometer (FL-1065; Horiba Jobin Yvon).
4.8. In vitro efficacy and prophylaxis studies
4.8.1. Bone marrow derived macrophages
To generate bone marrow derived macrophages (BMDMs), bone marrow cells from femurs and tibias from C57Bl/6 mice (Jackson laboratories) were incubated in Dulbecco’s Modified Eagle Media (DMEM, Mediatech, Herndon, VA) supplemented with 10% heat-inactivated FBS, 1% penicillin/streptomycin, 1% glutamine, and 20% L929 cell supernatant (containing macrophage colony stimulating factor). On day 7 in culture the cells were washed, counted and re-plated in DMEM media (without L929 supernatant but with 10 μg/ml M-CSF, R&D systems, Minneapolis, MN, USA) at a density of 1–2×105 cells/well (96-well plate, Falcon).
Treatments were added at a final concentration of 40 nM DXM/well: dexamethasone (DXM), AcDex dexamethasone-palmitate loaded particles (AcDex-DXM), or PLGA dexamethasone-palmitate loaded particles (PLGA-DXM) at 0 h, −24 hours or -six days. For the six-day assay, half of the medium (no added treatment) was changed every other day to improve cell viability, and to mimic in vivo settings of sink conditions and single dosing. After 24 hours or six-day incubations, cells were stimulated 2–3 hours with LPS (100 ng/ml), and then MSU crystals (0.2 mg/ml) were added and cells were incubated over night before supernatants were collected. Assays were performed in triplicates and repeated three times.
4.8.2. Endothelial cell line C166
Murine C166 endothelial cells (ATCC) were plated at 1×105 cells/well (96-well plate) in full DMEM media. Treatments were added at a final concentration of 40 nM DXM/well: DXM, AcDex-DXM), or PLGA-DXM at -eight days. Half of the medium (no added treatment) was changed every other day. Cells were then stimulated o.n with LPS (100 ng/ml), and then MSU crystals (0.2 mg/ml) were added for 3 hours before supernatants were collected.
IL-1β and CXCL-1 were measured in the supernatants using Mouse DuoSet ELISAs (R&D systems).
4.9. Animal models.
All animal experiments were performed in agreement with the Institutional Animal Care and Use Committee (IACUC). C57Bl/6 male mice (Jackson laboratories, Sacramento, CA, USA) were housed in a temperature-controlled room with a 06.00–18.00 hour light cycle and consumed regular chow and tap water ad libitum. Mice were 7–9 weeks at start of the experiments, and randomized before starting treatment to reduce bias.
4.9.1. Air pouch model of inflammation.
Dermal air pouches were established by injecting mice dorsally with 3 mL filtered (0.20 μm) air on day 0 and 3. On day 6, 120 μM (all treatments) or 15μg (47 AcDex-DXM and PLGA-DXM, or 76 μM free DXM) was injected as DXM, AcDex-DXM, PLGA-DXM, or PBS control (n=3–5 mice/ group, 3 trials). 24 hours later, MSU crystals were injected (3 mg in 0.5 ml PBS). After eight hours of stimulation, pouch exudate was collected by rinsing with 1 × 3 mL endotoxin-free PBS, followed by 30 seconds of gentle massage. The collection was centrifuged (5 minutes, 450 × g) and supernatants were saved at −80°C. Supernatants were subsequently tested for IL-1β, CXCL1 and IL-10 using Mouse DuoSet ELISAs (R&D systems). The collected cells were resuspended in FACS buffer and counted by an automated cell counter (BioRad, Hercules CA, USA). Cell surface markers were detected using fluorochrome-conjugated antibodies following Fc-blocking (anti-mouse CD16/CD32; clone 2.4G2, BD Biosciences, San Jose, CA, USA); anti-Mouse Ly-6G (Gr-1) APC (clone 1A8-Ly6G, Brilliant Violet 421, Thermo Fisher) and from Biolegend, San Diego, CA USA: anti-mouse Ly-6C PE/Cy7 (clone HK1.4) and anti-mouse CD11b (clone M1/70). Samples were analyzed by a FacsCanto II (BD Biosciences). Skin tissue covering the pouch was excised and fixed in 4% paraformaldehyde (Thermo Fisher) for subsequent hematoxylin and eosin (H&E) staining. Slides were imaged using a Keyence (Itasca, IL, USA) BZ-X700 widefield microscope. Infiltrated lining was measured using Image J at 33–37 measurements per group.[51]
4.9.2. Intra-articular MSU injections.
Murine knee joints were shaved and injected with 10 μg DXM (DXM, AcDex-DXM, PLGA-DXM, or PBS control; n=4–7 joints per treatment per experiment). Eight days later, MSU crystals (100 μg/knee) were injected into the same joints. After eight hours of stimulation, the joints were collected, fixed and decalcified (Calex II, Thermo Fisher Scientific). Slides were stained with H&E, and imaged as above. Infiltrated cell area was measured by Image J and quantification was performed in a blinded manner. Three cutting depths were used per knee and the largest infiltrated area per treatment was used. The experiment was repeated two times (total number of 10–14 joints per group) with similar results and subsequently pooled.
4.10. Statistical analysis
Data is presented as mean values ± SEM unless otherwise indicated. Statistical analysis was performed using SPSS software 20.0.0 (IBM Corp) and GraphPad Prism (GraphPad Software) version 7.0b. Data was examined for normal distribution. One-way analysis of variance (ANOVA) was used to compare independent groups; all groups were statistically compared followed by Tukey’s post hoc multiple comparisons test. Whenever Levene’s test revealed unequal variances between the groups, Dunnett’s T3 post hoc test was used instead. An analysis of covariance (ANCOVA) followed by a Tukey’s post hoc test was used when adjustments for covariates were needed, i.e. when two experiments were pooled. P values <0.05 were considered statistically significant.
Supplementary Material
The use of a disease-triggered depot for acute inflammation specifically releases drug upon a flare, eliminating repeated injections for the desired effect, as well as reducing the overall amount of drug necessary for adequate effect. This system represents an automated, controlled drug delivery system to increase patient compliance and a step towards personalized medicine.
Acknowledgements
The authors gratefully acknowledge Dr. Curtis Moore at the UCSD X-ray Crystallography Facility for confirming the MSU crystal structure. NMR data was acquired at the UCSD Skaggs School of Pharmacy and Pharmaceutical Sciences NMR Facility. The Moores Cancer Center Histology Core performed histological services and UCSD School of Medicine Microscopy Core (NS047101) was used for imaging histology
The work was supported by the Air Force Office of Scientific Research (AFOSR) FA9550-15-1-0273, National Institutes of Health R01EY024134, National Institutes of Arthritis and Musculoskeletal and Skin (MG: 1K08AR064834) and Swedish Research Council’s International Postdoc Grant (AS: 2015–06470).
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
References
- [1].Sahu SK, Ram A. Curr Drug Deliv. 2013, 10, 601. [DOI] [PubMed] [Google Scholar]
- [2].Crucho CI. ChemMedChem. 2015, 10, 24. [DOI] [PubMed] [Google Scholar]
- [3].Joshi-Barr S, de Gracia Lux C, Mahmoud E, Almutairi A. Antioxid Redox Signal. 2014, 21, 730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Alshememry AK, El-Tokhy SS, Unsworth LD. Curr Pharm Des. 2017, 23, 5358. [DOI] [PubMed] [Google Scholar]
- [5].Arunkumar P, Indulekha S, Vijayalakshmi S, Srivastava R. Mater Sci Eng C Mater Biol Appl. 2016, 61, 534. [DOI] [PubMed] [Google Scholar]
- [6].Chen W, Meng F, Cheng R, Deng C, Feijen J, Zhong Z. J Control Release. 2015, 210, 125. [DOI] [PubMed] [Google Scholar]
- [7].Reum Son A, Kim DY, Hun Park S, Yong Jang J, Kim K, Ju Kim B, Yun Yin X, Ho Kim J, Hyun Min B, Keun Han D, Suk Kim M. Sci Rep. 2015, 5, 14713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Peine KJ, Guerau-de-Arellano M, Lee P, Kanthamneni N, Severin M, Probst GD, Peng H, Yang Y, Vangundy Z, Papenfuss TL, Lovett-Racke AE, Bachelder EM, Ainslie KM. Mol Pharm. 2014, 11, 828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Terkeltaub R. Nat Rev Rheumatol. 2010, 6, 30. [DOI] [PubMed] [Google Scholar]
- [10].Edlow DW, Sheldon WH. Proc Soc Exp Biol Med. 1971, 137, 1328. [DOI] [PubMed] [Google Scholar]
- [11].Hutchins GM, Sheldon WH. Proc Soc Exp Biol Med. 1973, 143, 1014. [DOI] [PubMed] [Google Scholar]
- [12].Kominsky DJ, Campbell EL, Colgan SP. J Immunol. 2010, 184, 4062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].De Backer D. Intensive Care Med. 2003, 29, 699. [DOI] [PubMed] [Google Scholar]
- [14].Punnia-Moorthy A. J Oral Pathol. 1987, 16, 36. [DOI] [PubMed] [Google Scholar]
- [15].Bachelder EM, Pino EN, Ainslie KM. Chem Rev. 2017, 117, 1915. [DOI] [PubMed] [Google Scholar]
- [16].Broaders KE, Cohen JA, Beaudette TT, Bachelder EM, Frechet JM. Proc Natl Acad Sci U S A. 2009, 106, 5497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Collier MA, Gallovic MD, Bachelder EM, Sykes CD, Kashuba A, Ainslie KM. Pharm Res. 2016, 33, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Fontana F, Shahbazi MA, Liu D, Zhang H, Makila E, Salonen J, Hirvonen JT, Santos HA. Adv Mater. 2017, 29, 1603239. [DOI] [PubMed] [Google Scholar]
- [19].Gallovic MD, Schully KL, Bell MG, Elberson MA, Palmer JR, Darko CA, Bachelder EM, Wyslouzil BE, Keane-Myers AM, Ainslie KM. Adv Healthc Mater. 2016, 5, 2617. [DOI] [PubMed] [Google Scholar]
- [20].Hoang KV, Borteh HM, Rajaram MV, Peine KJ, Curry H, Collier MA, Homsy ML, Bachelder EM, Gunn JS, Schlesinger LS, Ainslie KM. Int J Pharm. 2014, 477, 334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ornelas-Megiatto C, Shah PN, Wich PR, Cohen JL, Tagaev JA, Smolen JA, Wright BD, Panzner MJ, Youngs WJ, Frechet JM, Cannon CL. Mol Pharm. 2012, 9, 3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Schully KL, Bell MG, Prouty AM, Gallovic MD, Gautam S, Peine KJ, Sharma S, Bachelder EM, Pesce JT, Elberson MA, Ainslie KM, Keane-Myers A. Int J Pharm. 2015, 495, 849. [DOI] [PubMed] [Google Scholar]
- [23].Suarez S, Grover GN, Braden RL, Christman KL, A. Almutairi. Biomacromolecules. 2013, 14, 3927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Suarez SL, Munoz A, Mitchell A, Braden RL, Luo C, Cochran JR, Almutairi A, Christman KL. ACS Biomater Sci Eng. 2016, 2, 197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Bachelder EM, Beaudette TT, Broaders KE, Dashe J, Frechet JM. J Am Chem Soc. 2008, 130, 10494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Liu D, Chen J, Jiang T, Li W, Huang Y, Lu X, Liu Z, Zhang W, Zhou Z, Ding Q, Santos HA, Yin G, Fan J. Adv Mater. 2018, 30, e1706032. [DOI] [PubMed] [Google Scholar]
- [27].Liu D, Cito S, Zhang Y, Wang CF, Sikanen TM, Santos HA. Adv Mater. 2015, 27, 2298. [DOI] [PubMed] [Google Scholar]
- [28].Liu D, Zhang H, Makila E, Fan J, Herranz-Blanco B, Wang CF, Rosa R, Ribeiro AJ, Salonen J, Hirvonen J, Santos HA. Biomaterials. 2015, 39, 249. [DOI] [PubMed] [Google Scholar]
- [29].Allouche J. Synthesis of Organic and Bioorganic Nanoparticles: An Overview of the Preparation Methods In: Brayner R, Fiévet F, Coradin T, editors. Nanomaterials: A Danger or a Promise? A Chemical and Biological Perspective. London: Springer London; 2013. p. 27. [Google Scholar]
- [30].Almeria B, Deng WW, Fahmy TM, Gomez A. Journal of Colloid and Interface Science. 2010, 343, 125. [DOI] [PubMed] [Google Scholar]
- [31].Dalbeth N, Merriman TR, Stamp LK. Lancet. 2016, 388, 2039. [DOI] [PubMed] [Google Scholar]
- [32].Kuo CF, Grainge MJ, Mallen C, Zhang W, Doherty M. Ann Rheum Dis. 2015, 74, 661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zhu Y, Pandya BJ, Choi HK. Arthritis Rheum. 2011, 63, 3136. [DOI] [PubMed] [Google Scholar]
- [34].Abbott RD, Brand FN, Kannel WB, Castelli WP. J Clin Epidemiol. 1988, 41, 237. [DOI] [PubMed] [Google Scholar]
- [35].De Vera MA, Rahman MM, Bhole V, Kopec JA, Choi HK. Ann Rheum Dis. 2010, 69, 1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Krishnan E, Baker JF, Furst DE, Schumacher HR. Arthritis Rheum. 2006, 54, 2688. [DOI] [PubMed] [Google Scholar]
- [37].Sheane BJ, Cunnane G. J Clin Rheumatol. 2007, 13, 293. [DOI] [PubMed] [Google Scholar]
- [38].Cain TM. Pediatr Radiol. 2015, 45, 481. [DOI] [PubMed] [Google Scholar]
- [39].Chen CL, Shao H, Block JL, Chen AF. J Arthroplasty. 2016, 31, 1431. [DOI] [PubMed] [Google Scholar]
- [40].Dubreuil M, Neogi T, Chen CA, Choi HK, Chaisson CE, Hunter DJ, Zhang Y. Am J Med. 2013, 126, 1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Roper RB, Mozena JD, Boyce-Smith G. J Am Podiatry Assoc. 1984, 74, 168. [DOI] [PubMed] [Google Scholar]
- [42].Rosen J, Miner JN. Endocr Rev. 2005, 26, 452. [DOI] [PubMed] [Google Scholar]
- [43].Schumacher HR, Chen LX. Am J Med. 2005, 118, 1208. [DOI] [PubMed] [Google Scholar]
- [44].Hollander JL. Ann N Y Acad Sci. 1955, 61, 511. [DOI] [PubMed] [Google Scholar]
- [45].Rull M, Clayburne G, Sieck M, Schumacher HR. Rheumatology (Oxford). 2003, 42, 1093. [DOI] [PubMed] [Google Scholar]
- [46].Kang EH, Lee EY, Lee YJ, Song YW, Lee EB. Ann Rheum Dis. 2008, 67, 1271. [DOI] [PubMed] [Google Scholar]
- [47].Pradal J, Maudens P, Gabay C, Seemayer CA, Jordan O, Allemann E. Int J Pharm. 2016, 498, 119. [DOI] [PubMed] [Google Scholar]
- [48].Viger ML, Live LS, Therrien OD, D. Boudreau. Plasmonics. 2008, 3, 33. [Google Scholar]
- [49].Bock N, Dargaville TR, Woodruff MA. Eur J Pharm Biopharm. 2014, 87, 366. [DOI] [PubMed] [Google Scholar]
- [50].Makadia HK, Siegel SJ. Polymers (Basel). 2011, 3, 1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Loscertales IG, Barrero A, Guerrero I, Cortijo R, Marquez M, Ganan-Calvo AM. Science. 2002, 295, 1695. [DOI] [PubMed] [Google Scholar]
- [52].Zhang L, Huang J, Si T, Xu RX. Expert Rev Med Devices. 2012, 9, 595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Cantarero LA, Butler JE, Osborne JW. Analytical Biochemistry. 1980, 105, 375. [DOI] [PubMed] [Google Scholar]
- [54].Malik SA, Ng WH, Bowen J, Tang J, Gomez A, Kenyon AJ, Day RM. J Colloid Interface Sci. 2016, 467, 220. [DOI] [PubMed] [Google Scholar]
- [55].Viger ML, Collet G, Lux J, Nguyen Huu VA, Guma M, Foucault-Collet A, Olejniczak J, Joshi-Barr S, Firestein GS, A. Almutairi. Biomaterials. 2017, 133, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Soehnlein O, Lindbom L. Nat Rev Immunol. 2010, 10, 427. [DOI] [PubMed] [Google Scholar]
- [57].Martillo MA, Nazzal L, Crittenden DB. Curr Rheumatol Rep. 2014, 16, 400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Edwards JC, Sedgwick AD, Willoughby DA. J Pathol. 1981, 134, 147. [DOI] [PubMed] [Google Scholar]
- [59].Terkeltaub R, Baird S, Sears P, Santiago R, Boisvert W. Arthritis Rheum. 1998, 41, 900. [DOI] [PubMed] [Google Scholar]
- [60].Landis RC, Yagnik DR, Florey O, Philippidis P, Emons V, Mason JC, Haskard DO. Arthritis Rheum. 2002, 46, 3026. [DOI] [PubMed] [Google Scholar]
- [61].Manolova V, Flace A, Bauer M, Schwarz K, Saudan P, Bachmann MF. Eur J Immunol. 2008, 38, 1404. [DOI] [PubMed] [Google Scholar]
- [62].Steen KH, Steen AE, Reeh PW. J Neurosci. 1995, 15, 3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Rajamaki K, Nordstrom T, Nurmi K, Akerman KE, Kovanen PT, Oorni K, Eklund KK. J Biol Chem. 2013, 288, 13410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Ueno T, Tsuchiya H, Mizogami M, Takakura K. J Inflamm Res. 2008, 1, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Chen LQ, Pagel MD. Adv Radiol. 2015, 2015, [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Duwel S, Hundshammer C, Gersch M, Feuerecker B, Steiger K, Buck A, Walch A, Haase A, Glaser SJ, Schwaiger M, Schilling F. Nat Commun. 2017, 8, 15126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Anderson M, Moshnikova A, Engelman DM, Reshetnyak YK, Andreev OA. Proc Natl Acad Sci U S A. 2016, 113, 8177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Chen N, Johnson MM, Collier MA, Gallovic MD, Bachelder EM, Ainslie KM. J Control Release. 2018, 273, 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Haskard DO, Landis RC. Arthritis Res. 2002, 4 Suppl 3, S91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Schmidt M, Pauels HG, Lugering N, Lugering A, Domschke W, Kucharzik T. J Immunol. 1999, 163, 3484. [PubMed] [Google Scholar]
- [71].Haim YO, Unger ND, Souroujon MC, Mittelman M, Neumann D. Sci Rep. 2014, 4, 4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Salvador A, Sandgren KJ, Liang F, Thompson EA, Koup RA, Pedraz JL, Hernandez RM, Lore K, Igartua M. Int J Pharm. 2015, 496, 371. [DOI] [PubMed] [Google Scholar]
- [73].Demento SL, Eisenbarth SC, Foellmer HG, Platt C, Caplan MJ, Mark Saltzman W, Mellman I, Ledizet M, Fikrig E, Flavell RA, Fahmy TM. Vaccine. 2009, 27, 3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Sharp FA, Ruane D, Claass B, Creagh E, Harris J, Malyala P, Singh M, O’Hagan DT, Petrilli V, Tschopp J, O’Neill LA, Lavelle EC. Proc Natl Acad Sci U S A. 2009, 106, 870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Schiltz C, Liote F, Prudhommeaux F, Meunier A, Champy R, Callebert J, Bardin T. Arthritis Rheum. 2002, 46, 1643. [DOI] [PubMed] [Google Scholar]
- [76].Wilcox WR, Khalaf AA. Ann Rheum Dis. 1975, 34, 332. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.