

Abstract
Tooth agenesis (TA), the failure of development of one or more permanent teeth, is a common craniofacial abnormality observed in different world populations. The genetic etiology of TA is heterogeneous; more than a dozen genes have been associated with isolated or nonsyndromic TA, and more than 80 genes with syndromic forms. In this study, we applied whole exome sequencing (WES) to identify candidate genes contributing to TA in four Turkish families. Likely pathogenic variants with a low allele frequency in the general population were identified in four disease-associated genes, including two distinct variants in TSPEAR, associated with syndromic and isolated TA in one family each; a variant in LAMB3 associated with syndromic TA in one family; and a variant in BCOR plus a disease-associated WNT10A variant in one family with syndromic TA. With the notable exception of WNT10A (Tooth agenesis, selective, 4, MIM #150400), the genotype-phenotype relationships described in the present cohort represent an expansion of the clinical spectrum associated with these genes: TSPEAR (Deafness, autosomal recessive 98, MIM #614861), LAMB3 (Amelogenesis imperfecta, type IA, MIM #104530; Epidermolysis bullosa, junctional, MIMs #226700 and #226650), and BCOR (Microphthalmia, syndromic 2, MIM #300166). We provide evidence supporting the candidacy of these genes with TA, and propose TSPEAR as a novel nonsyndromic TA gene. Our data also suggest potential multilocus genomic variation, or mutational burden, in a single family, involving the BCOR and WNT10A loci, underscoring the complexity of the genotype-phenotype relationship in the common complex trait of TA.
Keywords: whole exome sequencing, tooth agenesis, TSPEAR, LAMB3, BCOR, WNT10A
INTRODUCTION
Tooth agenesis (TA), the developmental absence of one or more permanent teeth, is one of the most common developmental abnormalities in humans, for which at least 200 million people are affected worldwide (Yin and Bian 2015). TA is classified clinically into hypodontia, oligodontia, or anodontia based on the number of absent permanent teeth (excluding third molars). Hypodontia is a mild form of TA, characterized by the absence of five teeth or less, whereas the absence of six or more teeth is referred to as oligodontia. The rare and extreme phenotypic presentation of TA is anodontia, which has been reported only in syndromic forms, and describes the complete absence of teeth (Yin and Bian 2015). The prevalence of TA has been estimated to range from 1.6% to 9.6% in the general population (Vastardis 2000). In two Turkish cohort studies, the prevalence of TA was found to be approximately 4.7%, which included a prevalence range of 3.67% to 4.3% for hypodontia and 0.21% to 0.3% for the more severe phenotype of oligodontia (Celikoglu et al. 2010; Karadas et al. 2014).
TA can be observed both as an isolated clinical trait, as well as in association with more than 150 syndromes defined by multi-organ or multisystem involvement (Yin and Bian 2015). During the last two decades of research, at least six genes (MSX1, PAX9, AXIN2, EDA, WNT10A, LRP6) have been proposed to be associated with nonsyndromic TA, with evidence supporting the candidacy of GREM2, WNT10B, EDAR, EDARADD, DKK1, BMP2, and BMP4 associated with nonsyndromic TA (Bergendal et al. 2011; Chen et al. 2017; Dinckan et al. 2018; Gong et al. 2015; Kantaputra et al. 2015; Liu et al. 2014; Lu et al. 2016; Massink et al. 2015; Ockeloen et al. 2016; Stockton et al. 2000; Tao et al. 2006; van den Boogaard et al. 2012; Vastardis et al. 1996; Yu et al. 2016). Notably, several of these genes have been associated with both nonsyndromic and syndromic forms of TA, often underscoring their important role in ectodermal growth and maintenance. For instance, WNT10A is associated with odontoonychodermal dysplasia (OODD, MIM #257980) and Schopf-Schulz-Passarge syndrome (SSPS, MIM #224750), both syndromes encompassing TA with additional phenotypes involving the skin, hair, and nails (Adaimy et al. 2007; Bohring et al. 2009); AXIN2 with oligodontia-colorectal cancer syndrome (ODCRCS, MIM #608615) (Lammi et al. 2004), and MSX1 and EDA with different subtypes of ectodermal dysplasia (MIMs #189500 and #305100) (Jumlongras et al. 2001; Kere et al. 1996). In addition, IRF6 has been associated with van der Woude syndrome (MIM #119300) for which hypodontia is a cardinal clinical feature (Ranta and Rintala 1983). LAMA3 and COL17A1 have previously been associated with non-Herlitz type junctional epidermolysis bullosa (nH-JEB, MIM #226650) (McGrath et al. 1995a; Nakano et al. 2002), as well as with nonsyndromic TA (Dinckan et al. 2018).
To date, a significant proportion of the potential genetic etiology of TA remains unexplained by variants in known TA genes, suggesting that additional loci, additional variants in known loci, and perhaps additional genetic models for disease, remain to be elucidated. In this study, whole exome sequencing (WES) was performed on four Turkish families, one with isolated TA and three others with TA and additional clinical findings, not linked to any specific recognizable syndrome. We identified five rare potentially pathogenic variants in TSPEAR, LAMB3, BCOR and WNT10A as the likely cause of TA in four Turkish (three multiplex and one simplex) families. Our findings underscore the potential role of TSPEAR, LAMB3, and BCOR in the genetic etiology for TA, and extend what is known about genotype-phenotype correlations in these genes and WNT10A.
MATERIALS AND METHODS
Study Families
This study was approved by the Istanbul University Institutional Ethical Review Board and the University of Texas Health Science Center at Houston Committee for Protection of Human Subjects (HSC-DB-12–0255). Clinical information and DNA samples from peripheral blood were collected from four families (TA11, TA12, TA13, and TA14) who were ascertained based on radiographic records demonstrating congenital TA and recruited as part of the CRANIRARE-2 project, a European Union-funded collaborative ERA-net project on craniofacial malformations at the Istanbul University, Istanbul Medical Faculty, Medical Genetics Department. After obtaining written informed consent from the family members involved in the study, all participants underwent detailed general clinical and craniofacial examinations performed by a medical geneticist, as well as oral and dental evaluations performed by a dentist. Panoramic radiographs were obtained on all probands. A total of 18 individuals in these four families were clinically evaluated by a dentist to confirm the diagnosis of TA. An initial diagnosis of TA was made if one or more permanent teeth were missing from the oral cavity, and confirmed by clinical (all participants) and radiographic (all probands) examinations. The complete spectrum of each probands’ phenotypes are documented.
Genetic Analysis
Genomic DNA was extracted from peripheral blood samples obtained from the four probands, their parents and siblings. For families TA11, TA12, and TA14, we employed a step-wise approach, first performing WES on proband-only samples with a plan to extend analyses to trio-WES in cases for which proband-only WES did not yield candidate variants. The complex pedigree structure of TA13 (unaffected parents in the setting of two affected paternal uncles and one affected paternal aunt) prompted us to begin analysis using a trio-WES strategy.
Whole exome sequencing (WES) was performed at the Baylor College of Medicine Human Genome Sequencing Center (BCM-HGSC, https://www.hgsc.bcm.edu) through the Baylor-Hopkins Center for Mendelian Genomics (BHCMG) initiative. Using the manufacturer’s protocol (Illumina, San Diego, CA), pre-capture libraries were constructed using the in-house developed HGSC VCRome 2.1 exome capture design (Bainbridge et al. 2011). Mercury pipeline was employed to process the raw data and generate vcf files with annotation (Reid et al. 2014). Minor allele frequency data from the Atherosclerosis Risk in Communities Study (ARIC) database (The ARIC Investigators 1989), the NHLBI GO Exome Sequencing Project (ESP5400) database (http://evs.gs.washington.edu), the 1000 Genomes Project (1KGP) database (1000 Genomes Project Consortium et al. 2015), the Exome Aggregation Consortium (ExAC) database, the Genome Aggregation Database (gnomAD) (Lek et al. 2016), and the internal BHCMG database (~7,000 exomes including ~1,000 from individuals of Turkish descent) were included to parse and filter the variants.
Indels were retained if they were (1) reported in the Human Gene Mutation Database (HGMD) and have a minor allele frequency (MAF) less than 5% in the 1KGP database or (2) have a MAF less than 2% in 1KGP and pass through internal variant quality metrics. Indel variants meeting one of these two specifications must have an allele frequency less than 120 out of 10,940 in the ARIC database. Single nucleotide variants (SNVs) underwent parallel processing using the following criteria: (1) reported in HGMD or with a clinical variant value between 3 and 8 in dbSNP, and a MAF less than 5% in both 1KGP and ESP5400 African and European population databases, or (2) MAF less than 1% in both 1KGP and ESP5400 African and European population databases. SNVs meeting these specifications and passing internal variant quality metrics must also be present in fewer than 120 out of 10,940 samples studied in the ARIC database.
The filtered variants were then prioritized via comprehensive analyses considering a spectrum of biological properties including inheritance modes (e.g. biallelic variants for suspected recessive trait), the frequency of the variant (minor allele frequency [MAF] ≤ 0.005), mutation types, reported functions and involved pathway(s) of the relevant protein, associated disease(s), published studies, as well as scores for predicting deleteriousness including the CADD score calculated by a machine learning method (Kircher et al. 2014) and the GERP (Doerks et al. 2002) score utilizing the level of evolutionary conservation (Table S1). To experimentally confirm the candidate variants by an orthogonal sequencing method and evaluate their segregation within the family, conventional PCR was performed on the genomic DNA using HotStarTaq DNA polymerase (Qiagen, Valencia, CA); the amplified fragments underwent Sanger sequencing at the Baylor College of Medicine DNA sequencing core.
RESULTS
The raw data for each exome included approximately 200,000 variants, of which fewer than 1,000 rare variants were retained based on quality of variant and frequency against available public databases (Bayram et al. 2017). Further prioritization of this set of exome variants involved review of heterozygous, homozygous, and compound heterozygous variants based on autosomal dominant (AD), autosomal recessive (AR), and X-linked (XL) inheritance models for trait segregation; assessment of the expected deleteriousness of the identified variant on protein function, i.e. likelihood damaging; and comparison of identified loci with known disease genes and disease-associated pathways through interrogation of public knowledge databases, i.e. PubMed, OMIM, STRING interaction network. Between five and seven candidate variants were prioritized in each family (Table S1). Additional review of the association with known Mendelian disease, MAF in public databases, tissue expression, prediction of deleteriousness, conservation of mutation sites, and segregation analyses within each family led to the prioritization of one to two candidate variants in each family (Table 1). Rare variants in three genes, TSPEAR, LAMB3, and BCOR, and a known TA pathogenic variant in WNT10A, were identified as potentially etiologic for the observed TA phenotypes.
Table 1.
List of variants identified as pathogenic/likely pathogenic in this study.
| Gene | Nucleotide Change | Protein Change | Zygosity | MAF in ExAC/gnomAD | MAF in Turkish db. |
CADD | GERP |
|---|---|---|---|---|---|---|---|
| TSPEAR | c.1726_1728delGTCinsTT (a.k.a. c.[1726G>T; 1728delC]) | p.Val576Leufs*38 | Hom | 2.5×10−5/2.8×10−5 | 1.1×10−3 | NA | NA |
| c.1877T>C | p.Phe626Ser | Hom | 1.2×10−4/1.5×10−4 | 1.6×10−3 | 32 | 4.33 | |
| LAMB3 | c.547C>T | p.Arg183Cys | Hom | 5.1×10−4/4.7×10−4 | 4.5×10−3 | 26.4 | 4.57 |
| BCOR | c.1651G>A | p.Asp551Asn | Het | 1.4×10−4/0.95×10−4 | NA | 11.74 | 5.6 |
| WNT10A | c.682T>A | p.Phe228Ile | Hom | 0.011/0.01 | 0.022 | 6.68 | 4.46 |
Abbreviations: Het, heterozygous; Hom, homozygous; MAF, minor allele frequency; db, database.
The number of homozygotes is 14.
The number of homozygotes is 8.
In family TA11, the parents of the proband TA11-III-2 are first cousins (Supplemental Fig. 1). TA11-III-2 was evaluated as a 10 year, 10 month-old male proband diagnosed with oligodontia based on the radiographic observation of 20 permanent teeth missing, including maxillary and mandibular lateral incisors and canines, maxillary premolars and second molars bilaterally, mandibular central incisors, second premolars, and second molars bilaterally (Fig. 1a), and additional clinical features of microcephaly (FOC −2.6 SD at 12 years of age) in the setting of normal parental head circumference (mother 53 cm, 10–25th centile; father 55 cm, 50th centile). His school performance, writing and reading skills were normal. The patient also manifested dysmorphic facial features, which include a narrow forehead, increased hair growth on the forehead, a high arched palate, low set ears, and minimal striation at the border of ear lobules and antitragus. There was no evidence of anterior microdontia (conical teeth), scalp hypotrichosis, or alopecia. Although a formal audiology evaluation was not performed, profound deafness was not noted on gross neurological evaluation, nor did the proband have any history of speech delay to suggest a profound hearing impairment. No specific syndrome was clinically recognized to potentially underlie this constellation of observed clinical findings. WES analysis revealed two homozygous variants in TSPEAR (NM_144991.2) leading to a complex allele, c.1726_1728delGTCinsTT, a.k.a. c.[1726G>T; 1728delC], and a frameshift with early protein termination (p.Val576Leufs*38) (Fig. 1, b-d, Fig. S1). TSPEAR encodes a protein consisting of a laminin G-like domain and seven EAR (epilepsy-associated repeat) domains (Punta et al. 2012). The proband’s unaffected parents and sister were each found to be heterozygous carriers for the same variant allele (Fig. 1b). This complex allele was not found in any other individual in our internal database of ~1,000 Turkish exomes, and neither variant was found to occur independently of the other. The complex allele is located in exon 10 (of 12 total exons) and is predicted to impact the EAR6 domain of TSPEAR (Fig. 1c). The premature termination codon (PTC) produced by c.1726_1728delGTCinsTT is located within the last 50 base pairs in the penultimate exon, thus predicting that the mutant transcript may escape nonsense-mediated decay (NMD), and could result in the production of a nonfunctional protein or one that has a dominant negative or gain-of-function effect (Fig. 1d) (Nagy and Maquat 1998). Of note, in subject TA11-III-2, absence of heterozygosity (AOH) was deduced by BafCalculator and the calculation of B-allele frequency from exome variant data (Gambin et al. 2017; Karaca et al. 2018), and demonstrated that the complex allele c.1726_1728delGTCinsTT was located in an AOH region of approximately 442 kb (Fig. 1b) on chromosome 21. In addition, the total AOH size in the proband is significantly higher (333.472 Mb) than the average total AOH size in the non-Turkish probands presented in our internal database (130.683 Mb, N=2,379), which supports the parental consanguinity reported for this proband.
Fig. 1.
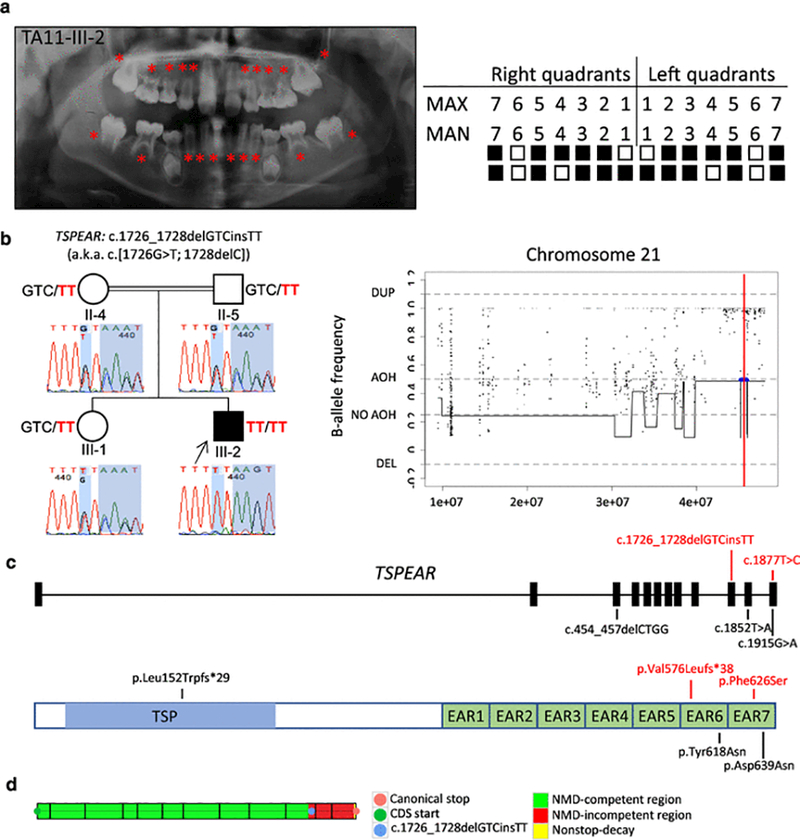
Clinical findings of TA11 family proband with tooth agenesis (TA), segregation results, absence of heterozygosity (AOH) plots, and molecular findings of patients with variants in TSPEAR. (a) TA diagnosis was confirmed through clinical and radiographic examinations for patient TA11-III-2. The panoramic radiograph was taken at 10 years 10 months. On the left, permanent teeth missing are indicated by red asterisks on the panoramic radiographs; on the right, the filled boxes represent permanent teeth missing in a schematic of the maxillary (MAX) and mandibular (MAN) arches while non-filled boxes indicate permanent teeth present excluding third molars. (b) Pedigree and genotypes of family TA11. Neighboring homozygous missense and truncating variants result in a complex allele c.1726_1728delGTCinsTT (p.Val576Leufs*38), a.k.a. c.[1726G>T; 1728delC] (red font) in TSPEAR identified in the proband (III-2, arrow), while these two variants were found in a heterozygous state in the unaffected sister (III-1) and consanguineous parents (II-4 and II-5). Sanger sequencing chromatograms demonstrate the location of identified variants (shaded in blue). AOH regions in the proband were identified by calculation of B-allele frequency from exome variant data using BafCalculator (horizontal blue line) and the position of the two variants indicated with a thin vertical red line. These two variants are located within an AOH region (~442 kb). The total calculated AOH size in the proband is 333.472 Mb. (c) Positions of the variants identified in the present study (red font) and previous studies are mapped to the 12 exons of TSPEAR. The homozygous allele c.1726_1728delGTCinsTT (in TA11) is located in exon 10, and homozygous variant c.1877T>C (in TA12) maps to exon 12; additional previously reported variants (black font) map to exons 3, 11, and 12. Below the gene structure is a schematic representation of TSPEAR protein structure. The corresponding amino acid changes are located in the thrombospondin-type laminin G (TSP) domain and two epilepsy-associated repeats (EAR) domains. (d) The complex allele c.1726_1728delGTCinsTT was predicted to escape NMD by our in-house developed tool based on the rule that PTCs within the last 50 base pairs of the penultimate exon and the last exon are hypothesized not to undergo NMD. Abbreviations: AOH, absence of heterozygosity; DEL, deletion; DUP, duplication.
In family TA12, the parents of the proband (TA12-IV-1) are first cousins once removed (Supplemental Fig. 2). The 10 year, 8 month-old female proband (ten teeth missing), together with her brother (IV-2, eight teeth missing) and father (III-7, seven teeth missing), were all affected with apparently isolated oligodontia. Radiographic examination confirmed the absence of ten permanent teeth in the proband, including maxillary lateral incisors, canines, and second molars bilaterally, and mandibular central incisors and second molars bilaterally (Fig. 2a). Clinical evaluation did not demonstrate any evidence of anterior microdontia, scalp hypotrichosis, alopecia, or severe deafness. WES analysis revealed a TSPEAR homozygous missense variant c.1877T>C (rs369010851) in the proband TA12-IV-1 (Fig. 2b, Fig. S2). This coding SNP (cSNP)/variant allele is located within exon 12 and is predicted to result in a phenylalanine to serine substitution (p.Phe626Ser) impacting the EAR7 domain of TSPEAR (Fig. 1c). Although this variant was not found in the ARIC database, it is listed in the ExAC database (MAF, 1.2×10−4), ESP database (MAF, 7.8×10−5), and our internal exome variant database (~7,000 exomes, MAF, 1.4×10−4); to date, there is no record of the homozygous form of this variant. Furthermore, the frequency of c.1877T>C in our internal Turkish cohort database (~1,000 exomes) is 1.6×10−3, which is higher than that observed in other databases. In addition, the c.1877T>C variant occurred at a highly conserved site (GERP = 4.33) and was predicted to be deleterious (CADD_phred = 32). Segregation analyses revealed that the affected brother (TA12-IV-2) also carries an identical homozygous c.1877T>C variant. The parents are both heterozygous for the variant. No additional exonic variant was found in TSPEAR in the affected father by Sanger sequencing of all exons (Fig. 2b), though disabling deep intronic alterations, and gross heterozygous deletions/duplications/inversions in or outside investigated regions could not be excluded. AOH analysis revealed that the c.1877T>C was located in a ~2.7-Mb AOH region on chromosome 21 (Fig. 2b) in the proband, who has the total AOH size of 231.807 Mb.
Fig. 2.
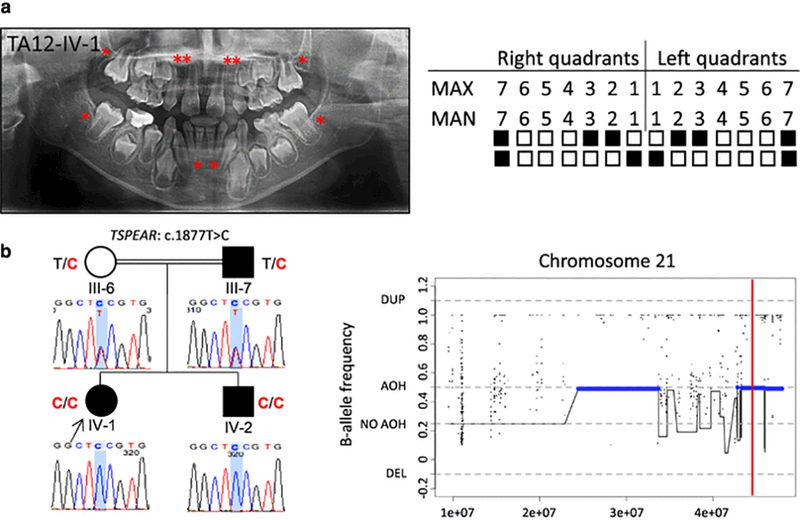
Clinical findings of TA12 family proband, segregation results, and AOH plots. (a) TA diagnosis was confirmed through clinical and radiographic examinations for patient TA12-IV-1. The panoramic radiograph of the proband was taken at 10 years 8 months. The missing permanent teeth are indicated as in Fig. 1a. (b) Pedigree and genotypes of family TA12. The same missense variant c.1877T>C (red font) in TSPEAR was identified in the proband (IV-1, arrow) and her affected brother (IV-2), while consanguineous parents (III-6 and III-7), an unaffected mother and affected father, harbor this variant in heterozygous state. This variant is located within one AOH region (~2.7 Mb). The total calculated AOH size in the proband is 231.807 Mb. Abbreviations: AOH, absence of heterozygosity; DEL, deletion; DUP, duplication.
In family TA13, the parents of the proband (TA13-V-1) are second cousins (Supplemental Fig. 3). The 12 year, 11 month-old male proband was affected with oligodontia (eleven teeth missing), pectus excavatum deformity, and mild maxillary hypoplasia. Both of his arms had hyperextensibility of 5–10º. Missing teeth included the maxillary lateral incisors, first and second premolars bilaterally, mandibular canines, first premolars, and the second left premolar (Fig. 3a, Fig. S3). No significant clinical findings were observed in his unaffected parents and two brothers on physical and dental examination. WES analysis revealed a homozygous missense nucleotide transition mutation c.547C>T (rs116124880) in exon 6 (of 23 total exons) of LAMB3 (NM_000228.2), resulting in a substitution from arginine to cysteine (p.Arg183Cys), located within the Laminin N domain of the protein (Fig. 3, b-c). The frequency of this variant is 9.0×10−4 in the 1KGP database, 5.1×10−4 in the ExAC database, and 5.8×10−4 in ESP with no homozygous individuals reported in these databases. This patient is the only individual who carries this homozygous variant among ~7,000 samples in our internal exome database, in which we observed a minor allele frequency of 1.2×10−3 overall, with a slightly higher frequency of 4.5×10−3 in our Turkish exomes cohort (~1000 exomes). This variant was not observed in the ARIC database. The p.Arg183Cys variant was predicted to be deleterious (CADD_phred = 26.4) and located at a conserved residue (GERP = 4.57); changes to cysteine can sometimes result in new disulphide linkages between thiol groups resulting in perturbed secondary structure and abnormal folding of protein (Creighton 1988). PCR amplification and Sanger sequencing were conducted on four unaffected family members (IV-11, IV-12, V-2, and V-4), who were found to be heterozygous for the c.547C>T variant (Fig. 3b). Further analysis of WES data by BafCalculator revealed this variant mapped to a ~1.5-Mb AOH genomic interval on chromosome 1 (Fig. 3b). In addition, the total AOH size in the proband is 173.918 Mb.
Fig. 3.
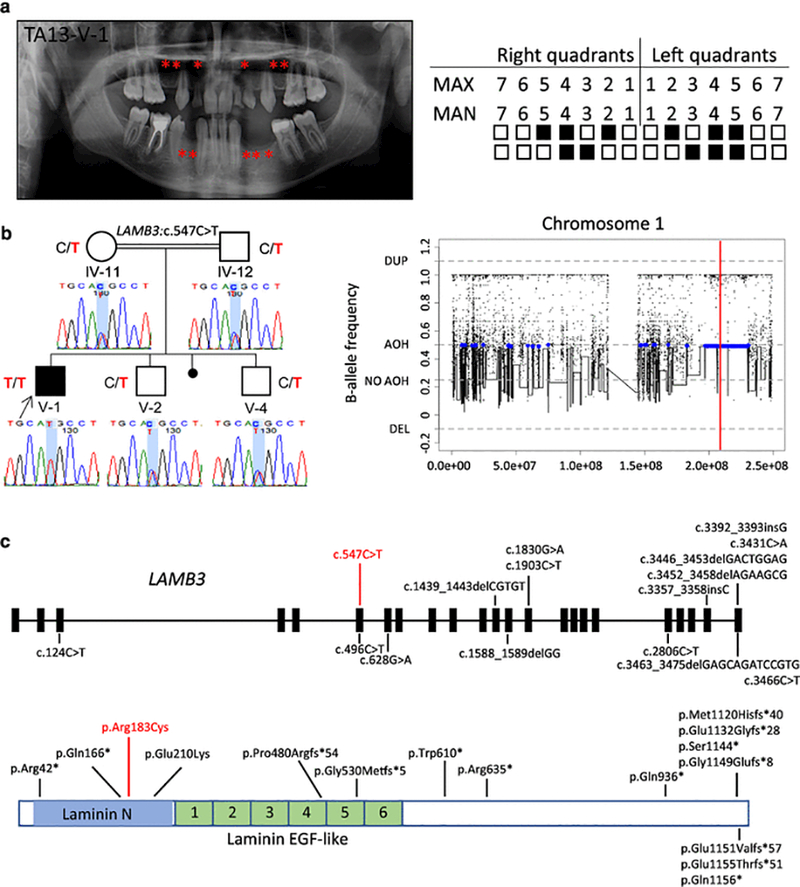
Clinical findings of TA13 family proband, segregation results, AOH plots, and molecular findings of patients with variants in LAMB3. (a) TA diagnosis was confirmed through clinical and radiographic examinations for patient TA13-V-1. The panoramic radiograph of the proband was taken at 12 years 11 months. The missing permanent teeth are indicated as in Fig. 1a. (b) Pedigree and genotypes of family TA13 and genotyping results of available family members. A homozygous missense variant c.547C>T (red font) in LAMB3 was identified in the proband (V-1, arrow), while his two unaffected brothers (V-2 and V-4) and consanguineous parents (IV-11 and IV-12) are all heterozygous for the c.547C>T variant. This variant is located within a region of AOH (~1.5 Mb). The total calculated AOH size in the proband is 173.918 Mb. (c) The variant identified in the present study and known variants previously studied map to 9 of 23 exons of LAMB3. The homozygous variant c.547C>T (in red) detected in our study is in exon 6; variants causative for dominantly inherited AI are all within the last two exons; variants contributing to recessive H-JEB and nH-JEB are primarily truncating variants. Below the gene structure is a schematic representation of LAMB3 protein structure, indicating the corresponding amino acid changes, a subset of which are located within Laminin N and six Laminin EGF-like domains. Abbreviations: AOH, absence of heterozygosity; DEL, deletion; DUP, duplication; EGF, epidermal growth factor.
In family TA14, an 8 year, 3 month-old female proband (TA-14-III-3) was diagnosed with oligodontia of eight teeth, including the maxillary second molars bilaterally, right canine and second premolar, mandibular left central incisor, right lateral incisor, and second premolars bilaterally (Fig. 4a, Fig. S4). Additional clinical features observed in this patient include high and narrow palate, and short philtrum. Her father (II-9) and sister (III-1) were also affected with hypodontia (three and four teeth were absent, respectively), and a high, narrow palate was observed in the father. Her mother and brother were not diagnosed with TA. WES analysis revealed a heterozygous variant c.1651G>A (rs372463512, p.Asp551Asn) in exon 4 (of 15 total exons) of BCOR (NM_001123383.1), shared with the affected sister (heterozygous) and father (hemizygous); and a known homozygous variant c.682T>A (rs121908120, p.Phe228Ile) in exon 3 (of 4 total exons) of WNT10A (NM_025216.2), shared with the sister (homozygous) and present in the heterozygous state in the affected father, unaffected mother, and unaffected brother (Fig. 4, b-d). Neither of these variants were found in the ARIC database. Twelve individuals with the c.1651G>A BCOR variant have been reported in ExAC: four heterozygous and eight hemizygous (MAF is 1.4×10−4), and one heterozygous variant was found in the ESP database (MAF is 1.0×10−4), and three heterozygous and two hemizygous variants were found in the 1KGP database (MAF is 1.3×10−3). Additionally, this variant occurred at a highly conserved site (GERP = 5.6), with a CADD phred-like score of 11.74. Fourteen individuals with a homozygous c.682T>A in WNT10A are reported in ExAC (MAF is 0.01), while eight homozygous variant carriers are reported in our internal Turkish database (MAF is 0.02). In addition, this variant is located at a highly conserved site (GERP = 4.46), with a CADD phred-like score of 6.68.
Fig. 4.
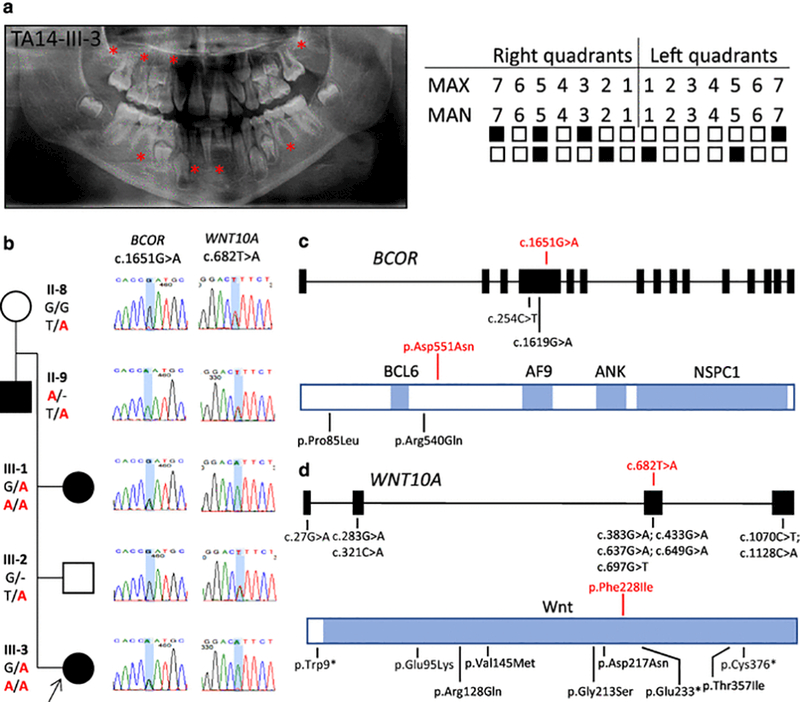
Clinical findings of TA14 family proband, segregation results, and molecular findings of patients with missense variants in BCOR and WNT10A. (a) TA diagnosis was confirmed through clinical and radiographic examinations for patient TA14-III-3. The panoramic radiograph of the proband was taken at 8 years 3 months. The missing permanent teeth are indicated as in Fig. 1a. (b) Pedigree and genotypes of family TA14 and genotyping results of available family members. A heterozygous missense variant c.1651G>A (in red) in BCOR was identified in the proband (III-3, arrow) and her sister (III-1), the variant in hemizygous state was identified in the father (II-9), but not detected in the unaffected brother (III-2) and mother (II-8). While no parental consanguinity was reported for this pedigree, the calculated total AOH for the proband was 99.983 Mb. (c) The variant identified in our study and previously reported missense variants all map to exon 4 of BCOR. The variant c.1651G>A identified in the present study is indicated in red font. Below the gene structure is a schematic representation of BCOR protein structure with known domains indicated in blue, indicating the corresponding amino acid changes, a subset of which are located upstream and downstream of the BCL6 binding site. Abbreviations: BCL6, B-cell CLL/lymphoma 6 binding cite; AF9, the common mixed lineage leukemia gene fusion partner binding site; ANK, Ankyrin repeats; NSPC1, polycomb group ring finger 1 binding site. (d) A selection of known variants map to four exons of WNT10A. The homozygous variant c.682T>A (in red) detected in the present study is in exon 3. Below the gene structure is a schematic representation of WNT10A protein structure, indicating the corresponding amino acid changes, a subset of which are located within the signal region (white) and mature protein region (blue).
DISCUSSION
In the present study, we identified rare variation in TSPEAR, LAMB3, and BCOR as likely damaging and contributing to familial TA, expanding the observed phenotypes potentially associated with alterations in these genes. We also described a known pathogenic c.682T>A variant in WNT10A, providing evidence for a possible mutational load or mutation burden involving rare variation in the BCOR and WNT10A loci in a single family. Variants in LAMB3, TSPEAR, and BCOR have been previously associated with syndromic TA including clinical diagnoses of nH-JEB (MIM #226650), ectodermal dysplasia, and syndromic microphthalmia-2 (MCOPS2, MIM #300166), respectively (McGrath et al. 1995b; Ng et al. 2004; Peled et al. 2016).
Clinical spectrum of TSPEAR variants
We report two variants in TSPEAR (the complex allele c.1726_1728delGTCinsTT (a.k.a. c.[1726G>T; 1728delC]) in TA11; and c.1877T>C in TA12) as likely pathogenic for TA (Table 2, Fig. 1c). This complex allele was first described in association with a phenotype of bilateral profound sensorineural deafness (Delmaghani et al. 2012) and shown to affect the EAR6 domain of the TSPEAR protein. Additional findings of the truncating c.1728delC variant as homozygous or compound heterozygous were reported in two patients with an ectodermal dysplasia phenotype characterized by hypodontia, anterior microdontia, scalp hypotrichosis, and facial dysmorphisms (Peled et al. 2016). All variants described above and reported by Peled et al. are either frameshift variants presumably leading to the complete abolishment of the EAR domains, or missense variants located at the conserved sites of the EAR domains (Fig. 1c), suggesting an important role of the EAR domains in normal TSPEAR function. Furthermore, functional studies demonstrated that the truncated protein was indeed synthesized and was not properly secreted to the extracellular region, consistent with predicted escape from NMD, and this was proposed to be the mechanism of disease (Delmaghani et al. 2012). Importantly, congenital sensorineural deafness was not observed by Peled et al. in the individual identified with the homozygous c.1726_1728delGTCinsTT allele (Peled et al. 2016).
Table 2.
Selected known and novel variants identified in TSPEAR, LAMB3, and WNT10A with associated phenotypes
| TSPEAR | ||||
|---|---|---|---|---|
| Nucleotide change | Protein change | Zygosity | Phenotype Spectrum | |
| c.[454_457delCTGG];[1726G>T; 1728delC] | p.[Leu152Trpfs*29];[Val576Phe;Lys577Serfs*37] | Comp het | Scalp hypotrichosis, hypodontia and conical teeth, facial dysmorphism (Peled et al. 2016) | |
| c.1726_1728delGTCinsTT (a.k.a. c.[1726G>T; 1728delC]) | p.Val576Leufs*38 | Hom | Congenital profound sensorineural deafness (Delmaghani et al. 2012); oligodontia, microcephaly, narrow forehead, increased hair growth on the forehead, high arched palate, low set ears, minimal striation at the border of ear lobules and antitragus (this study) | |
| c.[1726G > T; 1728delC] | p.[Val576Phe; Lys577Serfs*37] | Hom | Scalp hypotrichosis, hypodontia and microdontia, facial dysmorphism (Peled et al. 2016) | |
| c.[1852T>A];[1915G>A] | p.[Tyr618Asn];[Asp639Asn] | Comp het | Scalp hypotrichosis, hypodontia and conical teeth, facial dysmorphism (Peled et al. 2016) | |
| c.1877T>C | p.Phe626Ser | Hom | Oligodontia (this study) | |
| LAMB3 | ||||
| Nucleotide change | Protein change | Zygosity | Phenotype Spectrum | |
| c.124C>T | p.Arg42* | Comp w/ missense | nH-JEB (Kivirikko et al. 1996; McGrath et al. 1995b; Mellerio et al. 1998) | |
| c.124C>T | p.Arg42* | Comp w/ nonsense | H-JEB (Kivirikko et al. 1996) | |
| c.496C>T | p.Gln166* | Comp w/ nonsense | H-JEB (Takizawa et al. 1998b) | |
| c.547C>T | p.Arg183Cys | Hom | Oligodontia and mild maxillary hypoplasia (this study) | |
| c.628G>A | p.Glu210Lys | Comp w/ nonsense | nH-JEB (Kivirikko et al. 1996; McGrath et al. 1995b; Mellerio et al. 1998; Posteraro et al. 1998) | |
| c.904delT | p.Trp302Glyfs*94 | Comp w/ missense | nH-JEB (Posteraro et al. 1998) | |
| c.1439_1443delCGTGT | p.Pro480Argfs*54 | Comp w/ nonsense | nH-JEB (Pulkkinen and Uitto 1998) | |
| c.1587_1588delAG | p.Gly530Metfs*5 | Comp w/ nonsense | Uncommon/nH-JEB (Gache et al. 2001; Nakano et al. 2002) | |
| c.1830G>A | p.Trp610* | Comp w/ nonsense | H-JEB (Takizawa et al. 1998b) | |
| c.1903C>T | p.Arg635* | Comp w/ COL17A1 (digenic) | nH-JEB (Floeth and Bruckner-Tuderman 1999) | |
| c.1903C>T | p.Arg635* | Hom; Comp w/ nonsense/splicing | H-JEB (Pulkkinen et al. 1994; Pulkkinen et al. 1997) | |
| c.2806C>T | p.Gln936* | Hom | H-JEB (Takizawa et al. 1998a) | |
| c.3357_3358insC | p.Met1120Hisfs*40 | Het | AI (Lee et al. 2015) | |
| c.3392_3393insG | p.Glu1132Glyfs*28 | Het | AI (Poulter et al. 2014) | |
| c.3431C>A | p.Ser1144* | Het | AI (Kim et al. 2013) | |
| c.3446_3453delGACTGGAG | p.Gly1149Glufs*8 | Het | AI (Kim et al. 2013) | |
| c.3452_3458delAGAAGCG | p.Glu1151Valfs*57 | Het | AI (Kim et al. 2016) | |
| c.3463_3475delGAGCAGATCCGTG | p.Glu1155Thrfs*51 | Het | AI (Lee et al. 2015) | |
| c.3466C>T | p.Gln1156* | Het | AI (Wang et al. 2015) | |
| WNT10A | ||||
| Nucleotide change | Protein change | Zygosity | Phenotype Spectrum | |
| c.27G>A | p.Trp9* | Hom | OODD (Bohring et al. 2009) | |
| c.283G>A | p.Glu95Lys | Comp w/ missense | STHAG4 (van den Boogaard et al. 2012) | |
| c.321C>A | p.Cys107* | Het; Hom; Comp w/ missense |
OODD; SPSS; STHAG4 (Bohring et al. 2009) | |
| c.383G>A | p.Arg128Gln | Het; Comp w/ nonsense |
OODD; STHAG4 (Bohring et al. 2009; van den Boogaard et al. 2012) | |
| c.433G>A | p.Val145Met | Hom | STHAG4 (Dinckan et al. 2018; van den Boogaard et al. 2012) | |
| c.637G>A | p.Gly213Ser | Het; Comp w/ missense | STHAG4 (Kantaputra et al. 2014; Song et al. 2014; Yuan et al. 2017) | |
| c.649G>A | p.Asp217Asn | Comp w/ missense | STHAG4 (Kantaputra and Sripathomsawat 2011) | |
| c.682T>A | p.Phe228Ile | Het; Hom; Comp w/ nonsense |
OODD; STHAG4 (Bohring et al. 2009; Dinckan et al. 2018; Kantaputra and Sripathomsawat 2011; van den Boogaard et al. 2012); TA with high and narrow palate, and short philtrum (this study) | |
| c.697G>T | p.Glu233* | Hom | OODD (Adaimy et al. 2007; Dinckan et al. 2018) | |
| c.1070C>T | p.Thr357Ile | Het; Comp w/ missense | STHAG (Song et al. 2014; Yuan et al. 2017) | |
| c.1128C>A | p.Cys376* | Hom | OODD (Bohring et al. 2009) | |
The variants found in this study are highlighted in bold.
Abbreviations: Het, heterozygous; Hom, homozygous; Comp, compound heterozygous; AI, amelogenesis imperfecta; H-JEB, Herlitz type junctional epidermolysis dysplasia; nH-JEB, non-Herlitz type junctional epidermolysis dysplasia; OODD, odontoonychodermal dysplasia; SPSS, Schopf-Schulz-Passarge syndrome; STHAG4, tooth agenesis, selective, 4, with or without ectodermal dysplasia.
In the present study, proband TF11-III-2 was found to have the homozygous c.1726_1728delGTCinsTT allele in association with a phenotype of oligodontia, microcephaly, and additional dysmorphic facial features that included a narrow forehead and increased hair growth on the forehead, features that are distinct from those typically observed in association with ectodermal dysplasia. Microcephaly is not a common feature of ectodermal dysplasia. These findings suggest the possibility that microcephaly in association with a narrow forehead and increased hair growth on the forehead may represent an expansion of the phenotypes associated with TSPEAR. In addition, a homozygous missense variant c.1877T>C was identified in family TA12. Although this variant was observed in several control databases, all reported instances are heterozygous. Segregation of this variant in available relatives also presenting with oligodontia phenotypes demonstrated that the affected father (seven teeth missing) was heterozygous for c.1877T>C, in contrast to the affected proband (ten teeth missing) and sibling (eight teeth missing) who are both homozygous for this variant. Although no second TSPEAR variant was identified in the father through exon-by-exon Sanger sequencing, it remains possible that noncoding variation at this locus, variation at another locus, or a mutational burden may impact expression of the disease trait associated with monoallelic TSPEAR variation. Incomplete penetrance has been described in TA in individuals with WNT10A variation, and could underlie a more complex genotype-phenotype relationship (Song et al. 2014), as has been described in craniosynostosis due to a combination of a rare variant in SMAD6 and a common variant near BMP2 (Timberlake et al. 2016), or congenital scoliosis due to compound inheritance of a rare variant null allele and a common variant, noncoding, hypomorphic allele of TBX6 (Wu et al. 2015). Larger cohorts are needed to address the role of rare and common variants in TA using statistical models (Gonzaga-Jauregui et al. 2015; Timberlake et al. 2016; Wu et al. 2015).
Previous functional studies support a role for TSPEAR in normal tooth development. Functional assay of gene expression using microarray analysis revealed that down-regulation of TSPEAR leads to down-regulation of NOTCH1 together with many other Notch signaling pathway-associated genes (Peled et al. 2016). Notch signaling is a highly conserved intercellular signal transfer mechanism and plays a key role in the generation of tooth roots, formation of cusp patterns, and differentiation of odontoblasts; NOTCH1 is involved in the regulation of dental epithelial stem cell differentiation (Cai et al. 2011; Felszeghy et al. 2010).
Clinical spectrum of LAMB3 variants
In family TA13, we identified a homozygous missense variant c.547C>T in LAMB3. LAMB3 encodes a subunit of laminin 5 protein (Burgeson et al. 1994), and heterozygous pathogenic variants have been associated with AD amelogenesis imperfecta (AI, MIM #104530), while biallelic variants are associated with AR Herlitz type junctional epidermolysis bullosa (H-JEB, MIM #226700) and non-Herlitz type junctional epidermolysis bullosa (nH-JEB, MIM #226650). AI is characterized by abnormal enamel development of permanent teeth and can present as either an isolated trait or as part of a broader syndrome (Kim et al. 2013). Seven heterozygous truncating variants have been identified as pathogenic for AI in multiple families (Kim et al. 2013; Kim et al. 2016; Poulter et al. 2014; Wang et al. 2015). Escape from NMD was suggested as the molecular mechanism for the disease since these variants and their corresponding premature truncation codons are all located within the last or penultimate exons (Fig. 3c) (Kim et al. 2013; Kim et al. 2016; Poulter et al. 2014; Wang et al. 2015). The mono-allelic association with AD disease trait and a gain-of-function, GoF, versus loss-of-function, LoF, mutational mechanism is also consistent with this ‘NMD-escape’ model (Ben-Shachar et al. 2009; Coban-Akdemir et al. 2018; Khajavi et al. 2006). Lethal H-JEB and mild nH-JEB are AR disorders featuring AI associated with blistering lesions of the skin and oral mucosa with atrophic scarring and dystrophic nails (Kim et al. 2013). H-JEB is caused by biallelic LoF variants in LAMB3 (Table 2), resulting in abolishment of protein expression (Kiritsi et al. 2013; Pulkkinen et al. 1994; Takizawa et al. 1998a). Patients affected with nH-JEB can present with a spectrum of phenotypic manifestations including hypodontia, which have been reported in three siblings of Swiss origin (Bircher et al. 1993). These individuals typically have compound heterozygous alleles that include one truncating and one missense allele (Table 2, Fig. 3c), with partial functionality conferred by the hypomorphic allele resulting in a milder form of JEB (Nakano et al. 2002). In these cases, reduced laminin 5 expression was observed, consistent with partial protein functionality resulting in a less severe phenotype (Nakano et al. 2002). Although TA is not typically considered to be associated with JEB, one family of three siblings affected with nH-JEB and hypodontia has been described, however, the causative variants in these siblings were not identified (Bircher et al. 1993). Additional evidence for an association between TA and laminin proteins was demonstrated in two Turkish families with TA attributed to multilocus variation involving LAMA3 and WNT10A; affected individuals in these families demonstrated no physical evidence of JEB (Dinckan et al. 2018). In the present study, patient TA13-V-1 presented with mild maxillary hypoplasia, pectus excavatum deformity, and joint laxity, features that are not typical of AI or JEB. The milder TA phenotype observed in association with a homozygous missense variant may be yet another example of how the degree of functional impact conferred by a variant can modulate the severity and extent of the observed phenotype. Elucidation of additional families with homozygous missense variation in LAMB3 will provide further clarification of this potential genotype-phenotype correlation.
Oligogenic inheritance of BCOR and WNT10A variants
In family TA14, we demonstrate independent segregation of variants at two unlinked loci, BCOR and WNT10A, in association with a TA phenotype. BCOR has been associated with syndromic microphthalmia-2 (MCOPS2, MIM #300166), also known as oculofaciocardiodental syndrome (OFCD, an X-linked dominant condition. MCOPS2 is a rare disorder characterized by ocular, craniofacial, cardiac, and dental abnormalities including oligodontia (Gorlin et al. 1996; Ng et al. 2004). The expression of BCOR has been detected in both the dental epithelium and mesenchyme during tooth development in early embryogenesis (Cai et al. 2010). In addition, alteration in BCOR was associated with abnormal tooth root development, congenital cataracts, craniofacial defects, and congenital heart disease through epigenetic mechanisms (Fan et al. 2009). A majority of pathogenic variants described in BCOR as etiologic for MCOPS2 are nonsense, frameshift, splicing variants, and deletions, of which premature stop codons are predicted to result in NMD and a functional null mutation (Hilton et al. 2009; Ng et al. 2004). To date, two missense variants in BCOR have been reported in three male patients. The variant c.254C>T (p.Pro85Leu) in exon 4 was identified by two studies, one younger than 2-year-old male patient with microphthalmia, microcephaly, hypoplastic corpus callosum and cingulate gyrus (Ng et al. 2004); while the other 7-year-old male patient had microphthalmia, narrow forehead, simple ears, atrial septal defect, digital abnormalities, intellectual disability, and hypospadias (Hilton et al. 2009). The second missense variant c.1619G>A (p.Arg540Gln) in exon 4 was identified in a male infant with congenital heart disease, glaucoma, and cerebral white matter hypoplasia, who was deceased after cardiac surgery because of complex congenital heart disease (Zhu et al. 2015). To our knowledge, the variant c.1651G>A found in family TA14, which is located within the largest exon 4, is the third potential pathogenic missense variant in BCOR. Notably, affected individuals in TA14 do not have features of MCOPS2, though the proband and father were noted to have a narrow palate, potentially representing an expansion of the phenotype reported in association with BCOR. The three previously reported male cases have not been observed to have dental anomalies. However, the possibility of TA cannot be excluded, because of the young age of the patients and the absence of any dedicated dental evaluation.
Variations in WNT10A have been associated with both syndromic and nonsyndromic TA (Table 2). In the present study, one known biallelic variant c.682T>A (p.Phe228Ile) was identified in two affected siblings, while the affected father and two other unaffected family members are heterozygous for the variant. The protein change p.Phe228Ile of this variant is predicted to disrupt protein structure by destabilizing disulfide bridges that impact proper folding and function (Dinckan et al. 2018). The variant c.682T>A has been associated recurrently with both syndromic and nonsyndromic TA in heterozygous, homozygous, or compound heterozygous manner (Bohring et al. 2009; Dinckan et al. 2018; van den Boogaard et al. 2012). Notably, heterozygosity for the c.682T>A has been associated with reduced penetrance, and has been observed in ~2.3% of unaffected controls (van den Boogaard et al. 2012). Such reduced penetrance suggests the possibility that an additional genomic mutational burden is required for phenotypic expression. TA represents a clinically heterogeneous condition with nearly 100 associated loci, providing an excellent substrate for the identification of mutational burden. Consistent with this, previous studies have reported multilocus variation as likely etiologic for TA, often involving WNT10A and/or additional WNT genes (Arte et al. 2013). In a previous study, we found two families with suggestive evidence of multilocus variation as likely pathogenic in TA, with one family demonstrating rare variation involving WNT10A and LAMA3, and another with rare variation involving DKK1, LAMA3, and COL17A1 (Dinckan et al. 2018). Intrafamilial genotypic and phenotypic variability can enable a dissection of the phenotype (or phenotypic severity) attributed to an identified locus in a family (Karaca et al. 2018). In TA14, the unaffected male sibling has only the heterozygous WNT10A c.682T>A variant, suggesting that an additional mutational burden or inheritance of rare variants at more than one locus may be required for phenotype expression in this family. This observation favors a role for both WNT10A and BCOR in this family’s TA phenotype.
The application of WES facilitates the discovery of novel candidate genes for those TA cases that remain unsolved and enables the identification of multilocus variation. In this study, we have identified known and novel variants in TSPEAR, LAMB3, BCOR, and WNT10A as potentially pathogenic in four families with suspected syndromic and isolated TA. Our results support previous findings that underscore the role of these genes in tooth development. Furthermore, our results provide evidence for extending the phenotype associated with TSPEAR, LAMB3, and BCOR, and proposing TSPEAR as a plausible novel nonsyndromic TA gene. The complex genetic etiology and heterogeneity of TA phenotypes are further highlighted by our study.
Supplementary Material
Acknowledgements
We appreciate the participation of the patients and their families in this project. We also thank Dr. Ousheng Liu and Dr. Davut Pehlivan for helpful discussions. This work was supported in part by the Scientific and Technological Research Institution of Turkey, TUBITAK-ERA NET (CRANIRARE-2, grant number: SBAG-112S398), Istanbul University Research Fund (Project No: 48398), National Institute of Dental and Craniofacial Research (NIDCR) R03-DE024596 (to AL) and the Baylor-Hopkins Center for Mendelian Genomics, BHCMG (UM1 HG006542, to JRL). The BHCMG is jointly funded by the National Human Genome Research Institute (NHGRI) and National Heart Lung and Blood Institute (NHLBI). The GSP Coordinating Center (U24 HG008956) contributed to cross-program scientific initiatives and provided logistical and general study coordination. JEP was supported by NHGRI K08 HG008986. NW was supported by 2016 Milstein Medical Asian American Partnership Foundation Fellowship Award in Translational Medicine.
Footnotes
Compliance with ethical standards
Conflict of interest JRL has stock ownership in 23andMe and Lasergen, is a paid consultant for Regeneron, and a co-inventor on multiple United States and European patents related to molecular diagnostics for inherited neuropathies, eye diseases and bacterial genomic fingerprinting. Other authors declare that they have no conflict of interest.
References
- 1000 Genomes Project Consortium et al. (2015) A global reference for human genetic variation. Nature 526:68–74 doi: 10.1038/nature15393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adaimy L et al. (2007) Mutation in WNT10A is associated with an autosomal recessive ectodermal dysplasia: the odonto-onycho-dermal dysplasia. Am J Hum Genet 81:821–828 doi: 10.1086/520064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arte S, Parmanen S, Pirinen S, Alaluusua S, Nieminen P (2013) Candidate gene analysis of tooth agenesis identifies novel mutations in six genes and suggests significant role for WNT and EDA signaling and allele combinations. PLoS One 8:e73705 doi: 10.1371/journal.pone.0073705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bainbridge MN et al. (2011) Targeted enrichment beyond the consensus coding DNA sequence exome reveals exons with higher variant densities. Genome Biol 12:R68 doi: 10.1186/gb-2011-12-7-r68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayram Y et al. (2017) REST Final-Exon-Truncating Mutations Cause Hereditary Gingival Fibromatosis. Am J Hum Genet 101:149–156 doi: 10.1016/j.ajhg.2017.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Shachar S, Khajavi M, Withers MA, Shaw CA, van Bokhoven H, Brunner HG, Lupski JR (2009) Dominant versus recessive traits conveyed by allelic mutations - to what extent is nonsense-mediated decay involved? Clin Genet 75:394–400 doi: 10.1111/j.1399-0004.2008.01114.x [DOI] [PubMed] [Google Scholar]
- Bergendal B, Klar J, Stecksen-Blicks C, Norderyd J, Dahl N (2011) Isolated oligodontia associated with mutations in EDARADD, AXIN2, MSX1, and PAX9 genes. Am J Med Genet A 155A:1616–1622 doi: 10.1002/ajmg.a.34045 [DOI] [PubMed] [Google Scholar]
- Bircher AJ, Lang-Muritano M, Pfaltz M, Bruckner-Tuderman L (1993) Epidermolysis bullosa junctionalis progressiva in three siblings. Br J Dermatol 128:429–435 doi: 10.1111/j.1365-2133.1993.tb00204.x [DOI] [PubMed] [Google Scholar]
- Bohring A et al. (2009) WNT10A mutations are a frequent cause of a broad spectrum of ectodermal dysplasias with sex-biased manifestation pattern in heterozygotes. Am J Hum Genet 85:97–105 doi: 10.1016/j.ajhg.2009.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgeson RE et al. (1994) A new nomenclature for the laminins. Matrix Biol 14:209–211 [DOI] [PubMed] [Google Scholar]
- Cai J et al. (2010) Function analysis of mesenchymal Bcor in tooth development by using RNA interference. Cell Tissue Res 341:251–258 doi: 10.1007/s00441-010-0996-2 [DOI] [PubMed] [Google Scholar]
- Cai X, Gong P, Huang Y, Lin Y (2011) Notch signalling pathway in tooth development and adult dental cells. Cell Prolif 44:495–507 doi: 10.1111/j.1365-2184.2011.00780.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celikoglu M, Kazanci F, Miloglu O, Oztek O, Kamak H, Ceylan I (2010) Frequency and characteristics of tooth agenesis among an orthodontic patient population. Med Oral Patol Oral Cir Bucal 15:e797–801 doi: 10.4317/medoral.15.e797 [DOI] [PubMed] [Google Scholar]
- Chen YT, Liu HC, Han D, Liu Y, Feng HL (2017) Association between EDAR Polymorphisms and Non-Syndromic Tooth Agenesis in the Chinese Han Population. Chin J Dent Res 20:153–159 doi: 10.3290/j.cjdr.a38770 [DOI] [PubMed] [Google Scholar]
- Coban-Akdemir Z et al. (2018) Identifying genes whose mutant transcripts cause dominant disease traits by potential gain-of-function alleles. Am J Hum Genet. In press [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creighton TE (1988) Disulphide bonds and protein stability. Bioessays 8:57–63 doi: 10.1002/bies.950080204 [DOI] [PubMed] [Google Scholar]
- Delmaghani S, Aghaie A, Michalski N, Bonnet C, Weil D, Petit C (2012) Defect in the gene encoding the EAR/EPTP domain-containing protein TSPEAR causes DFNB98 profound deafness. Hum Mol Genet 21:3835–3844 doi: 10.1093/hmg/dds212 [DOI] [PubMed] [Google Scholar]
- Dinckan N et al. (2018) Whole-Exome Sequencing Identifies Novel Variants for Tooth Agenesis. J Dent Res 97:49–59 doi: 10.1177/0022034517724149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doerks T, Copley RR, Schultz J, Ponting CP, Bork P (2002) Systematic identification of novel protein domain families associated with nuclear functions. Genome Res 12:47–56 doi: 10.1101/gr.203201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Z et al. (2009) BCOR regulates mesenchymal stem cell function by epigenetic mechanisms. Nat Cell Biol 11:1002–1009 doi: 10.1038/ncb1913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felszeghy S, Suomalainen M, Thesleff I (2010) Notch signalling is required for the survival of epithelial stem cells in the continuously growing mouse incisor. Differentiation 80:241–248 doi: 10.1016/j.diff.2010.06.004 [DOI] [PubMed] [Google Scholar]
- Floeth M, Bruckner-Tuderman L (1999) Digenic junctional epidermolysis bullosa: mutations in COL17A1 and LAMB3 genes. Am J Hum Genet 65:1530–1537 doi: 10.1086/302672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gache Y, Allegra M, Bodemer C, Pisani-Spadafora A, de Prost Y, Ortonne JP, Meneguzzi G (2001) Genetic bases of severe junctional epidermolysis bullosa presenting spontaneous amelioration with aging. Hum Mol Genet 10:2453–2461 doi: 10.1093/hmg/10.21.2453 [DOI] [PubMed] [Google Scholar]
- Gambin T et al. (2017) Homozygous and hemizygous CNV detection from exome sequencing data in a Mendelian disease cohort. Nucleic Acids Res 45:1633–1648 doi: 10.1093/nar/gkw1237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong M et al. (2015) Association of BMP4 polymorphisms with isolated tooth agenesis in a Chinese Han population: a case-control study. Eur Rev Med Pharmacol Sci 19:2188–2194 [PubMed] [Google Scholar]
- Gonzaga-Jauregui C et al. (2015) Exome Sequence Analysis Suggests that Genetic Burden Contributes to Phenotypic Variability and Complex Neuropathy. Cell Rep 12:1169–1183 doi: 10.1016/j.celrep.2015.07.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorlin RJ, Marashi AH, Obwegeser HL (1996) Oculo-facio-cardio-dental (OFCD) syndrome. Am J Med Genet 63:290–292 doi: [DOI] [PubMed] [Google Scholar]
- Hilton E et al. (2009) BCOR analysis in patients with OFCD and Lenz microphthalmia syndromes, mental retardation with ocular anomalies, and cardiac laterality defects. Eur J Hum Genet 17:1325–1335 doi: 10.1038/ejhg.2009.52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumlongras D et al. (2001) A Nonsense Mutation in MSX1 Causes Witkop Syndrome. The American Journal of Human Genetics 69:67–74 doi: 10.1086/321271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantaputra P, Kaewgahya M, Kantaputra W (2014) WNT10A mutations also associated with agenesis of the maxillary permanent canines, a separate entity. Am J Med Genet A 164A:360–363 doi: 10.1002/ajmg.a.36280 [DOI] [PubMed] [Google Scholar]
- Kantaputra P, Sripathomsawat W (2011) WNT10A and isolated hypodontia. Am J Med Genet A 155A:1119–1122 doi: 10.1002/ajmg.a.33840 [DOI] [PubMed] [Google Scholar]
- Kantaputra PN, Kaewgahya M, Hatsadaloi A, Vogel P, Kawasaki K, Ohazama A, Ketudat Cairns JR (2015) GREMLIN 2 Mutations and Dental Anomalies. J Dent Res 94:1646–1652 doi: 10.1177/0022034515608168 [DOI] [PubMed] [Google Scholar]
- Karaca E et al. (2018) Phenotypic expansion illuminates multilocus pathogenic variation. Genet Med doi: 10.1038/gim.2018.33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karadas M, Celikoglu M, Akdag MS (2014) Evaluation of tooth number anomalies in a subpopulation of the North-East of Turkey. Eur J Dent 8:337–341 doi: 10.4103/1305-7456.137641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kere J et al. (1996) X-linked anhidrotic (hypohidrotic) ectodermal dysplasia is caused by mutation in a novel transmembrane protein. Nat Genet 13:409–416 doi: 10.1038/ng0895-409 [DOI] [PubMed] [Google Scholar]
- Khajavi M, Inoue K, Lupski JR (2006) Nonsense-mediated mRNA decay modulates clinical outcome of genetic disease. Eur J Hum Genet 14:1074–1081 doi: 10.1038/sj.ejhg.5201649 [DOI] [PubMed] [Google Scholar]
- Kim JW et al. (2013) LAMB3 mutations causing autosomal-dominant amelogenesis imperfecta. J Dent Res 92:899–904 doi: 10.1177/0022034513502054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YJ, Shin TJ, Hyun HK, Lee SH, Lee ZH, Kim JW (2016) A novel de novo mutation in LAMB3 causes localized hypoplastic enamel in the molar region. Eur J Oral Sci 124:403–405 doi: 10.1111/eos.12280 [DOI] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J (2014) A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46:310–315 doi: 10.1038/ng.2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiritsi D, Has C, Bruckner-Tuderman L (2013) Laminin 332 in junctional epidermolysis bullosa. Cell Adh Migr 7:135–141 doi: 10.4161/cam.22418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kivirikko S, McGrath JA, Pulkkinen L, Uitto J, Christiano AM (1996) Mutational hotspots in the LAMB3 gene in the lethal (Herlitz) type of junctional epidermolysis bullosa. Hum Mol Genet 5:231–237 doi: 10.1093/hmg/5.2.231 [DOI] [PubMed] [Google Scholar]
- Lammi L et al. (2004) Mutations in AXIN2 cause familial tooth agenesis and predispose to colorectal cancer. Am J Hum Genet 74:1043–1050 doi: 10.1086/386293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KE, Ko J, Le CG, Shin TJ, Hyun HK, Lee SH, Kim JW (2015) Novel LAMB3 mutations cause non-syndromic amelogenesis imperfecta with variable expressivity. Clin Genet 87:90–92 doi: 10.1111/cge.12340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M et al. (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature 536:285–291 doi: 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HC, Zhang J, Wong S, Han D, Zhao HS, Feng HL (2014) Association between rs11001553 of DKK1 and non-syndromic tooth agenesis in the Chinese Han population. Genet Mol Res 13:7133–7139 doi: 10.4238/2014.April.3.4 [DOI] [PubMed] [Google Scholar]
- Lu Y et al. (2016) Genetic Variants of BMP2 and Their Association with the Risk of Non-Syndromic Tooth Agenesis. PLoS One 11:e0158273 doi: 10.1371/journal.pone.0158273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massink MP et al. (2015) Loss-of-Function Mutations in the WNT Co-receptor LRP6 Cause Autosomal-Dominant Oligodontia. Am J Hum Genet 97:621–626 doi: 10.1016/j.ajhg.2015.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JA et al. (1995a) Mutations in the 180-kD bullous pemphigoid antigen (BPAG2), a hemidesmosomal transmembrane collagen (COL17A1), in generalized atrophic benign epidermolysis bullosa. Nat Genet 11:83–86 doi: 10.1038/ng0995-83 [DOI] [PubMed] [Google Scholar]
- McGrath JA, Pulkkinen L, Christiano AM, Leigh IM, Eady RAJ, Uitto J (1995b) Altered Laminin 5 Expression Due to Mutations in the Gene Encoding the β3 Chain (LAMB3) in Generalized Atrophic Benign Epidermolysis Bullosa. Journal of Investigative Dermatology 104:467–474 doi: 10.1111/1523-1747.ep12605904 [DOI] [PubMed] [Google Scholar]
- Mellerio JE, Eady RA, Atherton DJ, Lake BD, McGrath JA (1998) E210K mutation in the gene encoding the beta3 chain of laminin-5 (LAMB3) is predictive of a phenotype of generalized atrophic benign epidermolysis bullosa. Br J Dermatol 139:325–331 doi: 10.1046/j.1365-2133.1998.02377.x [DOI] [PubMed] [Google Scholar]
- Nagy E, Maquat LE (1998) A rule for termination-codon position within intron-containing genes: when nonsense affects RNA abundance. Trends Biochem Sci 23:198–199 doi: 10.1016/S0968-0004(98)01208-0 [DOI] [PubMed] [Google Scholar]
- Nakano A, Chao SC, Pulkkinen L, Murrell D, Bruckner-Tuderman L, Pfendner E, Uitto J (2002) Laminin 5 mutations in junctional epidermolysis bullosa: molecular basis of Herlitz vs. non-Herlitz phenotypes. Hum Genet 110:41–51 doi: 10.1007/s00439-001-0630-1 [DOI] [PubMed] [Google Scholar]
- Ng D et al. (2004) Oculofaciocardiodental and Lenz microphthalmia syndromes result from distinct classes of mutations in BCOR. Nat Genet 36:411–416 doi: 10.1038/ng1321 [DOI] [PubMed] [Google Scholar]
- Ockeloen CW et al. (2016) Novel mutations in LRP6 highlight the role of WNT signaling in tooth agenesis. Genet Med 18:1158–1162 doi: 10.1038/gim.2016.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peled A et al. (2016) Mutations in TSPEAR, Encoding a Regulator of Notch Signaling, Affect Tooth and Hair Follicle Morphogenesis. PLoS Genet 12:e1006369 doi: 10.1371/journal.pgen.1006369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posteraro P et al. (1998) Compound heterozygosity for an out-of-frame deletion and a splice site mutation in the LAMB3 gene causes nonlethal junctional epidermolysis bullosa. Biochem Biophys Res Commun 243:758–764 doi: 10.1006/bbrc.1998.8180 [DOI] [PubMed] [Google Scholar]
- Poulter JA, El-Sayed W, Shore RC, Kirkham J, Inglehearn CF, Mighell AJ (2014) Whole-exome sequencing, without prior linkage, identifies a mutation in LAMB3 as a cause of dominant hypoplastic amelogenesis imperfecta. Eur J Hum Genet 22:132–135 doi: 10.1038/ejhg.2013.76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulkkinen L, Christiano AM, Gerecke D, Wagman DW, Burgeson RE, Pittelkow MR, Uitto J (1994) A homozygous nonsense mutation in the beta 3 chain gene of laminin 5 (LAMB3) in Herlitz junctional epidermolysis bullosa. Genomics 24:357–360 doi: 10.1006/geno.1994.1627 [DOI] [PubMed] [Google Scholar]
- Pulkkinen L et al. (1997) Predominance of the recurrent mutation R635X in the LAMB3 gene in European patients with Herlitz junctional epidermolysis bullosa has implications for mutation detection strategy. J Invest Dermatol 109:232–237 doi: 10.1111/1523-1747.ep12319752 [DOI] [PubMed] [Google Scholar]
- Pulkkinen L, Uitto J (1998) Heterozygosity for premature termination codon mutations in LAMB3 in siblings with non-lethal junctional epidermolysis bullosa. J Invest Dermatol 111:1244–1246 doi: 10.1046/j.1523-1747.1998.00399.x [DOI] [PubMed] [Google Scholar]
- Punta M et al. (2012) The Pfam protein families database. Nucleic Acids Res 40:D290–301 doi: 10.1093/nar/gkr1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranta R, Rintala AE (1983) Correlations between microforms of the Van der Woude syndrome and cleft palate. Cleft Palate J 20:158–162 [PubMed] [Google Scholar]
- Reid JG et al. (2014) Launching genomics into the cloud: deployment of Mercury, a next generation sequence analysis pipeline. BMC Bioinformatics 15:30 doi: 10.1186/1471-2105-15-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song S, Zhao R, He H, Zhang J, Feng H, Lin L (2014) WNT10A variants are associated with non-syndromic tooth agenesis in the general population. Hum Genet 133:117–124 doi: 10.1007/s00439-013-1360-x [DOI] [PubMed] [Google Scholar]
- Stockton DW, Das P, Goldenberg M, D’Souza RN, Patel PI (2000) Mutation of PAX9 is associated with oligodontia. Nat Genet 24:18–19 doi: 10.1038/71634 [DOI] [PubMed] [Google Scholar]
- Takizawa Y, Pulkkinen L, Shimizu H, Lin L, Hagiwara S, Nishikawa T, Uitto J (1998a) Maternal uniparental meroisodisomy in the LAMB3 region of chromosome 1 results in lethal junctional epidermolysis bullosa. J Invest Dermatol 110:828–831 doi: 10.1046/j.1523-1747.1998.00186.x [DOI] [PubMed] [Google Scholar]
- Takizawa Y et al. (1998b) Novel mutations in the LAMB3 gene shared by two Japanese unrelated families with Herlitz junctional epidermolysis bullosa, and their application for prenatal testing. J Invest Dermatol 110:174–178 doi: 10.1046/j.1523-1747.1998.00105.x [DOI] [PubMed] [Google Scholar]
- Tao R et al. (2006) A novel missense mutation of the EDA gene in a Mongolian family with congenital hypodontia. J Hum Genet 51:498–502 doi: 10.1007/s10038-006-0389-2 [DOI] [PubMed] [Google Scholar]
- The ARIC Investigators (1989) The Atherosclerosis Risk in Communities (ARIC) Study: design and objectives. Am J Epidemiol 129:687–702 doi: 10.1093/oxfordjournals.aje.a115184 [DOI] [PubMed] [Google Scholar]
- Timberlake AT et al. (2016) Two locus inheritance of non-syndromic midline craniosynostosis via rare SMAD6 and common BMP2 alleles. Elife 5 doi: 10.7554/eLife.20125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Boogaard MJ et al. (2012) Mutations in WNT10A are present in more than half of isolated hypodontia cases. J Med Genet 49:327–331 doi: 10.1136/jmedgenet-2012-100750 [DOI] [PubMed] [Google Scholar]
- Vastardis H (2000) The genetics of human tooth agenesis: new discoveries for understanding dental anomalies. Am J Orthod Dentofacial Orthop 117:650–656 doi: 10.1067/mod.2000.103257 [DOI] [PubMed] [Google Scholar]
- Vastardis H, Karimbux N, Guthua SW, Seidman JG, Seidman CE (1996) A human MSX1 homeodomain missense mutation causes selective tooth agenesis. Nat Genet 13:417–421 doi: 10.1038/ng0896-417 [DOI] [PubMed] [Google Scholar]
- Wang X, Zhao Y, Yang Y, Qin M (2015) Novel ENAM and LAMB3 mutations in Chinese families with hypoplastic amelogenesis imperfecta. PLoS One 10:e0116514 doi: 10.1371/journal.pone.0116514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu N et al. (2015) TBX6 null variants and a common hypomorphic allele in congenital scoliosis. N Engl J Med 372:341–350 doi: 10.1056/NEJMoa1406829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin W, Bian Z (2015) The Gene Network Underlying Hypodontia. J Dent Res 94:878–885 doi: 10.1177/0022034515583999 [DOI] [PubMed] [Google Scholar]
- Yu P et al. (2016) Mutations in WNT10B Are Identified in Individuals with Oligodontia. Am J Hum Genet 99:195–201 doi: 10.1016/j.ajhg.2016.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Q et al. (2017) Role of WNT10A in failure of tooth development in humans and zebrafish. Mol Genet Genomic Med 5:730–741 doi: 10.1002/mgg3.332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Dai FR, Wang J, Zhang Y, Tan ZP, Zhang Y (2015) Novel BCOR mutation in a boy with Lenz microphthalmia/oculo-facio-cardio-dental (OFCD) syndrome. Gene 571:142–144 doi: 10.1016/j.gene.2015.07.061 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.