

Abstract
Aim
The aim of this study is to analyse the prognostic value of complement anaphylatoxin receptors in patients with non‐ischaemic cardiomyopathy undergoing endomyocardial biopsy.
Methods and results
In 102 patients (72.5% male patients, median age 54 years) with non‐ischaemic cardiomyopathy, myocardial expression of C3aR was assessed among other parameters. The primary study endpoint was a composite of death, heart transplantation, heart failure‐related re‐hospitalization, and deterioration of left ventricular ejection fraction within a mean follow‐up of 11.9 months. The number of cells, which stained positive for C3aR, was significantly increased in patients with inflammatory compared with non‐inflammatory cardiomyopathy (1.75 ± 0.31 cells in inflammatory cardiomyopathy vs. 0.94 ± 0.26 in non‐inflammatory cardiomyopathy, P = 0.049). Subsequently, positive expression for C3aR was judged based on a semi‐quantitative scoring system. Significantly, more patients with positive MHCII and CD68 expression showed an increased number of C3aR‐positive cells. C3aR expression based on this score was more pronounced in patients with human herpesvirus 6 viral genome detection. Kaplan–Meier curves illustrate that the C3aR‐negative group reached the primary endpoint significantly more often (mean follow‐up 11.9 months, log rank 5.963, P = 0.015). Lack of C3aR expression was a strong independent predictor for the primary endpoint in Cox regression analysis [hazard ratio 0.46 (0.26–0.82, P = 0.009)].
Conclusions
C3aR‐positive cells are found more often in patients with inflammatory cardiomyopathy. The relevance of C3aR‐positive cells in patients with non‐ischaemic cardiomyopathy should be further evaluated as potential predictors or modulators of adverse cardiac remodelling, the substrate of progressive heart failure.
Keywords: Anaphylatoxin receptors, Complement, Non‐ischaemic heart failure, Myocarditis, Prognosis
Introduction
Non‐ischaemic cardiomyopathy is a common cause of congestive heart failure (HF) in young patients with subsequent need for pharmacological treatment, implantation of cardioverter–defibrillator, and eventually heart transplantation.1, 2 Despite some advances in diagnosis and therapy, the prognosis in patients with non‐ischaemic cardiomyopathy remains poor. Chronic low‐grade activation of the innate immune system with increased levels of inflammatory proteins, cytokines, and chemokines has been described in patients with HF.3, 4, 5, 6 While it has been reported that activation of the immune system is associated with beneficial effects during cardiac regeneration,5, 6 long‐term immune activation is also known to promote myocardial inflammation, which causes cardiac fibrosis and, thus, progression of HF.7 Among other mechanisms, infiltration with inflammatory cells mediates an increase of pro‐inflammatory proteins in the myocardial tissue followed by myofibroblast activation, accumulation of extracellular matrix proteins, and cardiac remodelling.
The complement system is an integral part of the innate immune response, and complement receptors are of importance for the recruitment of inflammatory cells to the site of myocardial injury and for maintaining inflammatory processes.5, 8 This system of soluble factors produced by a variety of immune cells is formed by a group of plasmatic proteins, which are activated in enzymatic cascades. In particular, it is responsible for the opsonization of microbial intruders by attracted inflammatory cells and for the induction of mast cell degranulation, whereby the potent anaphylatoxins C3a and C5a are released. Different pathways of complement activation result in the cleavage of C3 into C3a and C3b, followed by cleavage of C5 to form C5a and C5b.9 The components C5b to C9 then form the membrane attack complex, which mediates osmotic lysis of targeted cells. Among its role in immune defence, the complement system is also involved in more unexpected processes of tissue homeostasis such as angiogenesis and regenerative processes.5, 10 In animal models of cardiac disease, C3a causes histamine release, tachycardia, impairment of atrioventricular conduction, coronary vasoconstriction, and left ventricular dysfunction.11 C5a induces pro‐inflammatory and cardiodepressant cytokines in human leucocytes including tumour necrosis factor‐α, interleukin (IL)‐1, IL‐6, and IL‐8.12, 13 Furthermore, the C5aR pathway regulates the expression of adhesion molecules on peripheral monocytes, as well as infiltration and cytokine production of macrophages in the heart.4, 9, 14 Thus, C3a and C5a are important inflammatory mediators but might also be of pathophysiological relevance for cardiac tissue remodelling. Despite the potential relevance of complement anaphylatoxins and their receptors for myocardial inflammation, their diagnostic and prognostic value in patients with non‐ischaemic cardiomyopathy has not been investigated, yet.
Materials and methods
Study design, patient cohort, and assessment of clinical risk factors
In this study, we analysed 102 consecutive patients with regionally or globally impaired systolic left ventricular ejection fraction (LVEF) due to non‐ischaemic cardiomyopathy who underwent endomyocardial biopsy (EMB) as part of a routine clinical evaluation for HF at our university hospital from August 2007 to November 2011. All patients were admitted to the University Hospital of Tübingen with impaired LVEF ≤60% and/or symptomatic, congestive HF with either dyspnoea New York Heart Association (NYHA) classification ≥II and/or elevated B‐type natriuretic peptide (BNP) levels (>100 ng/L) and/or cardiac arrhythmias, including recurrent palpitations with presyncope, syncope, and documented sustained and/or non‐sustained ventricular tachycardia. The indication for EMB was based on these clinical criteria or one of the following: new‐onset HF of 2 week duration with dilation of the left ventricle and haemodynamic compromise, new‐onset HF of up to 3 month duration with dilated left ventricle and malignant arrhythmias, failure to respond to usual care, or suspected cardiac involvement with impaired global or regional systolic left or right ventricular function, enlargement of the left or right ventricle, pericardial effusion, myocardial hypertrophy, or abnormal myocardial echo patterns in transthoracic echocardiography as shown in the flow chart depicted in Supporting Information, Figure S1 .15, 16
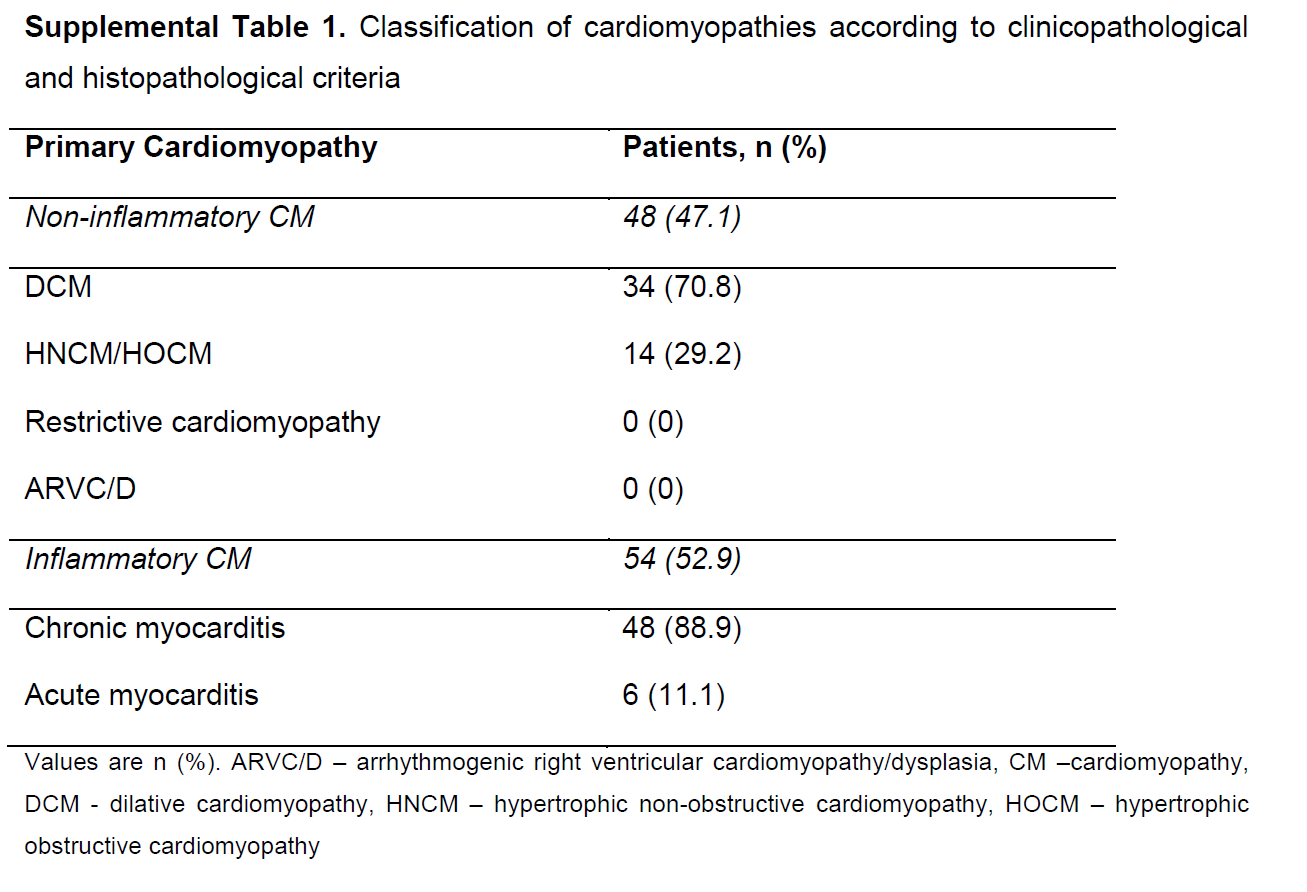
Cardiomyopathies were defined according to proposed classification criteria by the American Heart Association expert consensus panel1 and the European Society of Cardiology.17 The group of inflammatory cardiomyopathies comprised acute and chronic myocarditis (Supporting Information, Table S1 ). Non‐inflammatory cardiomyopathies comprised dilative cardiomyopathy (DCM), hypertrophic cardiomyopathy, and hypertensive cardiomyopathy. Secondary cardiomyopathies attributed to underlying diseases were excluded (Supporting Information, Table S1 ). Clinical risk factors at study entry were age, gender, body mass index, NYHA functional class, and concomitant medication. B‐type natriuretic peptide (>100 ng/L) and C‐reactive protein (CRP, >0.5 mg/dL) were classified as elevated laboratory markers. Echocardiographic parameters included LVEF and left ventricular end‐diastolic diameter (LVEDD). Left ventricular ejection fraction was estimated by echocardiography (iE33, Philips Medical Systems, Hamburg, Germany) using the modified Simpson method, with images obtained from apical four‐chamber and two‐chamber views. Left ventricular end‐diastolic diameter was analysed by two‐dimensionally guided M‐mode echocardiography in all patients. Significant coronary artery disease (>50% diameter luminal stenosis of two or more coronary vessels or left main or proximal left anterior descending coronary artery stenosis >50%) was excluded by coronary angiography in all patients. Further, cardiac function and morphology were assessed by contrast‐enhanced cardiac magnetic resonance imaging (MRI). Contrast‐enhanced cardiac MRI was performed on a 1.5 T scanner (Siemens Medical Systems, Erlangen, Germany) providing a gradient strength of 40 mT/m and maximum slew rate of 200 mT/m/ms. An advanced cardiac software package was used. Images were acquired with the subject in the supine position, by applying electrocardiographically gated breath‐hold sequences as described before.18 For late gadolinium enhancement (LGE) imaging, a two‐dimensional inversion‐recovery segmented k‐space gradient‐echo magnetic resonance sequence was performed using 0.15 mmol gadobutrol per kilogram of body weight (Gadovist, Bayer Healthcare, Berlin, Germany).18 Two experienced investigators independently reviewed the image loops of each subject in a random fashion. For LGE image analysis, both investigators visually judged the occurrence (presence vs. absence), localization, and pattern of LGE.19, 20 All patients received medication according to current European Society of Cardiology and American College of Cardiology/American Heart Association guidelines depending on their LVF and HF symptoms.16 The study conformed to the principles outlined in the Declaration of Helsinki and was approved by the Local Ethics Committee of the Eberhard Karls University of Tübingen (270/2011BO1). Written informed consent was obtained from all patients.
Study endpoints and follow‐up
All patients presented in our outpatient clinic for clinical follow‐up every 6 to 9 months. None of the patients were lost during follow‐up. The primary study endpoint was defined as a composite of all‐cause death, heart transplantation, HF‐related re‐hospitalization, and deterioration of LVEF ≥10% over time within a follow‐up period of 2 years (mean follow‐up 11.9 months, Supporting Information, Table S2 ). To determine the occurrence of an endpoint, all clinical events were reviewed by an independent endpoint committee. The clinicians, who were in charge of the decision for re‐hospitalization, were not involved in the study.
Endomyocardial biopsy, immunohistochemistry, and histopathological analysis
Biopsy samples of 1 to 3 mm were taken from the septum of the right ventricle with a bioptome (Biopsy Forceps, Cordis Corporation, Milpitas, CA) advanced through a 9 French venous sheath.21 Samples were immediately fixed under sterile conditions in 4% buffered formaldehyde for haematoxylin and eosin staining, Masson's trichrome, or Giemsa staining and for immunohistochemistry. Paraffin‐embedded, 4‐μm‐thick tissue sections were evaluated by light microscopy. Other samples were fixed in RNAlater (Ambion Inc., Foster City, CA, USA) for nested (reverse transcription) PCR detection of viral genomes [parvovirus B19, human herpesvirus types 6, 7, and 8, enteroviruses, adenoviruses, influenza A and B viruses, human cytomegalovirus (CMV), and Epstein–Barr virus].22 An avidin–biotin immunoperoxidase method (Vectastain Elite ABC Kit, Vector Laboratories, Burlingame, CA, USA) was used for immunohistochemistry according to the manufacturer's protocol, with the following monoclonal antibodies used as primary antibodies to identify the cell types in mononuclear cell infiltrates: CD3 for T cells (Novocastra Laboratories, Newcastle upon Tyne, UK), CD68 for macrophages (Dako, Glostrup, Denmark), and HLA‐DR‐α (Dako, Hamburg, Germany) to assess major histocompatibility complex class II (MHCII) expression in antigen‐presenting immune cells.
To detect C3aR and C5aR in the biopsy samples, the paraffin‐fixed tissue samples of the patients were deparaffinized with Roti‐Histol (2 × 10 min) and rehydrated through graded ethanol washes (100–70%). Antigen demasking was performed by heating the samples 3 × 5 min at 360 W in citrate buffer pH 6 in a microwave oven. Endogenous peroxidase activity was blocked by 3% H2O2 (15 min). Slides were pre‐incubated with ‘Protein Block Serum‐Free’ from Dako (X0909), then incubated with a C3aR antibody (H‐300; sc‐20138, Santa Cruz Biotechnology, Santa Cruz, CA, USA) or a C5aR antibody (anti‐C5R1, ab59390, Abcam, Cambridge, UK) followed by a biotinylated secondary antibody mix (‘Biotinylated Link Universal’, LSAB + System HRP, K0690, Dako). Rabbit IgG isotype control antibodies served as controls. For signal amplification, the slides were incubated with streptavidin–HRP (LSAB + System HRP, K0690, Dako) for 30 min. For the colour reaction, a DAB Substrate Kit (LiquidDAB + Substrate Chromogen System, K3468, Dako) was used (brown colour). After washing the slides in PBS, a second pre‐incubation with ‘Protein Block Serum‐Free’ followed. Slides were incubated with biotinylated secondary antibody mix and streptavidin–HRP again. For the colour reaction, a peroxidase substrate kit (Vector SG, Vector Laboratories) was used (grey‐blue colour). All sections were briefly counterstained with haematoxylin. Slides were covered with mounting medium and stored at room temperature until analysis.
Myocardial tissue sections stained for C3aR expression were analysed by a semi‐quantitative score scheme as described previously21, 22, 23, 24, 25, 26 and according to scores used for CD68 and MHC‐II. C3aR staining was classified as Score 0 if there were no stained cells or as Score 1 if there were one to eight stained cells. Scores for C3aR staining were obtained in a blinded manner from one or two sections for each staining by two independent investigators. The amount of cardiac fibrosis was determined as described before.27 According to the amount of fibrosis given in percentage of fibrosis in relation to the total area of the biopsy, patients were categorized using tertile distribution defined as mild (Grade 1, 0–10%), moderate (Grade 2, 11–20%), and severe fibrosis (Grade 3, >20%).27, 28 Cardiomyopathies were classified according to standardized clinical1 and histological criteria.22 Inflammatory cardiomyopathy comprised acute and chronic myocarditis (Supporting Information, Table S1 ) and was defined by lymphocytic infiltrates in association with myocyte necrosis according to the Dallas criteria as described before.15, 23, 25 Inflammation was defined by immunohistological detection of mononuclear infiltrates with >14 leucocytes/mm2 in the myocardium, in addition to enhanced expression of human leukocyte antigen Class II molecules according to the World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of Cardiomyopathies.15, 23 Endomyocardial biopsy without signs of inflammation but with myocardial damage and fibrosis was classified as non‐inflammatory cardiomyopathy; these patients had predominantly idiopathic dilative cardiomyopathy, hypertrophic cardiomyopathy, or hypertensive heart disease (shown in Supporting Information, Table S1 ).
Statistical analysis
Continuous, not normally distributed variables are expressed as median and interquartile range (IQR) and were compared using Mann–Whitney U test. Categorical data are presented as proportions and were analysed by chi‐squared test. Risk factors, including NYHA functional class, LVEF, LVEDD, LGE, BNP, CRP, C3aR, and C5aR were assessed. Continuous parameters were dichotomized at established cut‐off values. Cox proportional hazards regression analysis was carried out for multivariable analysis to assess the association of risk factors with the primary endpoint. Survival curves of patients grouped by pre‐specified variables were calculated by Kaplan–Meier analyses and compared using the log‐rank test. The risk for endpoint occurrence is presented as a hazard ratio with 95% confidence interval. Survival analysis started at the date of EMB. Values are given as mean ± standard deviation or standard error of the mean where applicable. Comparisons were considered statistically significant if the two‐sided P value was ≤0.05. Statistical analyses were performed using SPSS software version 22.0 (SPSS Inc., Chicago, IL, USA).
Results
The degree of cardiac fibrosis and inflammation detected by endomyocardial biopsy is different in patients with inflammatory cardiomyopathy compared with non‐inflammatory cardiomyopathy
Baseline characteristics and demographics of the patient cohort are given in Table 1. The classification of the underlying cardiomyopathies is described in Supporting Information, Table S1 . The median age of the population was 54 (IQR 20–84) years; 74 (72.5%) patients were men; and 86.3% of patients presented with symptomatic HF and an NYHA functional class ≥2 with a median LVEF of 40% (20–60). This study describes an ‘all‐comer collective’; the decision for EMB was based on the flow chart depicted in Supporting Information, Figure S1 . Cardiac and inflammatory biomarkers were increased. For instance, the median BNP serum concentrations were 384 (IQR 18–23211) ng/L, and the median troponin I levels were 0.05 (IQR 0–5.6) μg/L. Sixty patients (69.0%) had troponin I levels >0.03 μg/L. A median CRP of 0.53 (0–24) was found within the cohort. Fifty‐four (52.9%) patients were diagnosed with inflammatory cardiomyopathy, and the remaining 48 (47.1%) patients had non‐inflammatory cardiomyopathy. Patients with inflammatory cardiomyopathy were significantly younger than patients with non‐inflammatory cardiomyopathy (P < 0.001), showed a lower NYHA class (P = 0.009) and a better LVEF (P = 0.001), and were less often treated with diuretics (P = 0.004) and with mineralocorticoid receptor antagonists (P = 0.030).
Table 1.
Baseline characteristics of patient population
| Parameters | All patients, n = 102 | ICM, n = 54 (52.9%) | non‐ICM, n = 48 (47.1%) | P |
|---|---|---|---|---|
| Clinical characteristics | ||||
| Age (years) | 54 (20–84) | 50.5 (21–79) | 68.5 (20–84) | <0.001 |
| Male | 74 (72.5) | 40 (74.1) | 34 (70.8) | 0.825 |
| BMI (kg/m2) | 26.1 (19.9–52.0) | 26.5 (19.9–52) | 25 (19.9–31.2) | 0.603 |
| NYHA class ≥II | 88 (86.3) | 42 (77.8) | 46 (95.8) | 0.009 |
| Concomitant cardiac medication at study entry | ||||
| ß‐Blockers | 90 (88.2) | 47 (87.0) | 43 (89.6) | 0.765 |
| ACE‐I | 69 (67.6) | 34 (63.0) | 35 (72.9) | 0.299 |
| ARB | 23 (22.5) | 11 (20.4) | 12 (25.0) | 0.639 |
| Diuretics | 63 (61.8) | 26 (48.1) | 37 (77.1) | 0.004 |
| MRA | 54 (52.9) | 23 (42.6) | 31 (64.6) | 0.030 |
| Parameters of the left ventricle | ||||
| LVEF (%) | 40 (20–60) | 50 (25–60) | 35 (20–60) | 0.001 |
| LVEF <45% | 52 (51) | 20 (37) | 32 (66.7) | 0.003 |
| LVEDD (mm) | 51 (34–78) | 51 (34–78) | 52 (38–74) | 0.190 |
| LVEDD >55 mm | 40 (39.2) | 19 (35.2) | 21 (43.8) | 0.420 |
| Positive LGE | 46 (45.1%) | 26 (48.1) | 20 (41.7) | 0.687 |
| Biomarkers | ||||
| BNP (ng/L) | 384 (18–23 211) | 278 (24–23 211) | 544 (18–11 955) | 0.143 |
| TnI (μg/L) | 0.05 (0–5.6) | 0.07 (0–5.6) | 0.05 (0–0.34) | 0.184 |
| TnI >0.03 | 60 (69.0) | 33 (71.7) | 27 (65.9) | 0.645 |
| CRP (mg/dL) | 0.53 (0–24) | 0.62 (0–24) | 0.32 (0–4.6) | 0.080 |
| CRP >0.5 | 42 (41.2) | 26 (48.1) | 16 (33.3) | 0.160 |
| Virus‐positive endomyocardial biopsies | ||||
| Total | 26 (25.5) | 17 (31.5) | 9 (18.8) | 0.175 |
| EBV | 8 (7.8) | 5 (9.3) | 3 (6.3) | 0.719 |
| PVB19 | 10 (9.8) | 8 (14.8) | 2 (4.2) | 0.098 |
| HHV‐6 | 9 (8.8) | 5 (9.3) | 4 (8.3) | 1.000 |
| Influenza A/B | 2 (2.0) | 1 (1.9) | 1 (2.1) | 1.000 |
| CBV3 | 0 (0) | 0 (0) | 0 (0) | 1.000 |
| Myocardial fibrosis | ||||
| Mild | 29 (28.4%) | 21 (38.9) | 8 (16.7) | 0.024 |
| Moderate | 39 (38.2) | 20 (37.0) | 19 (39.6) | 0.675 |
| Severe | 24 (23.5) | 9 (16.7) | 15 (31.3) | 0.046 |
| Positive detection of immunohistological markers in the myocardium | ||||
| MHC II | 64 (62.7) | 50 (92.6) | 14 (29.2) | <0.001 |
| CD68 | 62 (60.8) | 50 (92.6) | 12 (25.0) | <0.001 |
| CD3 | 43 (42.2) | 41 (75.9) | 2 (4.2) | <0.001 |
ACE‐I, angiotensin‐converting enzyme inhibitor; ARB, angiotensin receptor blocker; BMI, body mass index; BNP, B‐type natriuretic peptide; CBV, Coxsackie B virus; CRP, C‐reactive protein; EBV, Epstein–Barr virus; HHV‐6, human herpesvirus 6; ICM, inflammatory cardiomyopathy; LGE, late gadolinium enhancement; LVEDD, left ventricular end‐diastolic diameter; LVEF, left ventricular ejection fraction; MRA, mineralocorticoid receptor antagonist; non‐ICM, non‐inflammatory cardiomyopathy; NYHA, New York Heart Association; PVB19, parvovirus B19; TnI, troponin I.
Values are n (%) or are given as median and interquartile range.
Significantly different values are presented in bold.
Mild myocardial fibrosis detected by histology in EMB was found significantly more often in inflammatory cardiomyopathy (38.9% in inflammatory cardiomyopathy vs. 16.7% in non‐inflammatory cardiomyopathy, P = 0.024), while severe fibrosis was increased among patients with non‐inflammatory cardiomyopathy (16.7% in inflammatory cardiomyopathy vs. 31.3% in non‐inflammatory cardiomyopathy, P = 0.046) as shown in Figure 1 A. There was no significant difference in moderate fibrosis between 37% in inflammatory cardiomyopathy vs. 39.6% in non‐inflammatory cardiomyopathy, P = 0.675. Positive LGE detected in cardiac MRI was present in 46 of the 102 patients (45.1%), but there was no difference between inflammatory and non‐inflammatory cardiomyopathy (P = 0.687, Table 1 and Figure 1 B). In immunohistochemistry, we also analysed the numbers of infiltrating inflammatory CD3‐positive T cells and CD68‐positive cells in the groups of inflammatory and non‐inflammatory cardiomyopathy. We found that numbers of CD3‐positive and CD68‐positive cells were significantly different among the two groups as shown in Figure 1 C and D. The number of CD3+ T cells was significantly increased in patients with inflammatory cardiomyopathy compared with non‐inflammatory cardiomyopathy (12.6 ± 1.84 in inflammatory cardiomyopathy vs. 2.3 ± 0.43 in non‐inflammatory cardiomyopathy, P < 0.001, Figure 1 C). CD68+ cells/macrophages were significantly increased in patients with inflammatory cardiomyopathy compared with non‐inflammatory cardiomyopathy (29.7 ± 2.23 in inflammatory cardiomyopathy vs. 11.5 ± 0.65 in non‐inflammatory cardiomyopathy, P < 0.001, Figure 1 D). Representative myocardial tissue sections illustrate myocardial architecture in histology and immunohistochemistry in the myocardium of inflammatory cardiomyopathy (Figure 1 E) and non‐inflammatory cardiomyopathy (Figure 1 F).
Figure 1.
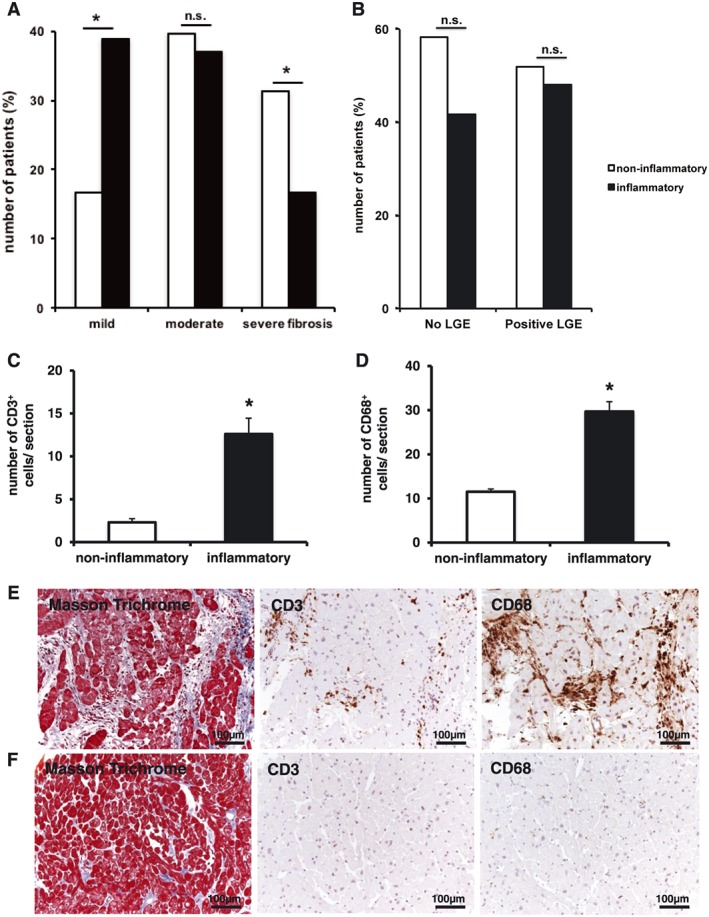
The degree of cardiac fibrosis and inflammation detected by endomyocardial biopsy is different in patients with inflammatory and non‐inflammatory cardiomyopathy, while the number of CD3‐positive T cells and CD68‐positive macrophages is increased in patients with inflammatory cardiomyopathy. According to the amount of fibrosis given in percentage (%) of fibrosis in relation to the total area of the biopsy, patients were categorized using tertile distribution defined as mild (Grade 1, 0–10%), moderate (Grade 2, 11–20%), and severe fibrosis (Grade 3, >20%). The number of CD3‐positive and CD68‐positive cells are given in cell number per section. Values are mean ± standard error of the mean; *P < 0.05. (A) Mild myocardial fibrosis was found significantly more often in patients with inflammatory cardiomyopathy (38.9% in inflammatory cardiomyopathy vs. 16.7% in non‐inflammatory cardiomyopathy, P = 0.024), while severe fibrosis was increased among patients with non‐inflammatory cardiomyopathy (16.7% in inflammatory cardiomyopathy vs. 31.3 in non‐inflammatory cardiomyopathy, P = 0.046). There was no difference in moderate fibrosis between 37% in inflammatory cardiomyopathy vs. 39.6% in non‐inflammatory cardiomyopathy, P = 0.675. (B) Positive late gadolinium enhancement (LGE) detected in cardiac MRI was present in 46 of the 102 patients (45.1%), but there was no difference between inflammatory and non‐inflammatory cardiomyopathy (P = 0.687). (C) The number of CD3‐positive T cells was significantly increased in patients with inflammatory cardiomyopathy compared with non‐inflammatory cardiomyopathy (12.6 ± 1.84 in inflammatory cardiomyopathy vs. 2.3 ± 0.43 in non‐inflammatory cardiomyopathy, P < 0.001). (D) CD68‐positive cells/macrophages were significantly increased in patients with inflammatory cardiomyopathy compared with non‐inflammatory cardiomyopathy (29.7 ± 2.23 in inflammatory cardiomyopathy vs. 11.5 ± 0.65 in non‐inflammatory cardiomyopathy, P < 0.001). (E) Representative myocardial tissue sections depict myocardial architecture in histological Masson's trichrome staining and positive expression of CD3 and CD68 on cells residing in the myocardium in inflammatory cardiomyopathy. (F) Representative myocardial tissue sections depict myocardial architecture in histological Masson's trichrome staining and positive expression of CD3 and CD68 on cells residing in the myocardium in non‐inflammatory cardiomyopathy.
Expression of anaphylatoxin receptor C3aR is increased in patients with inflammatory cardiomyopathy
C3aR staining was classified as Score 0 if there were no stained cells or as Score 1 if there were one to eight stained cells. Forty‐two out of 102 (41.2%) patients showed positive C3aR and 28/102 (27.5%) positive C5aR expression within the myocardium. The number of C3aR‐positive cells was significantly increased in patients with inflammatory cardiomyopathy compared with non‐inflammatory cardiomyopathy (C3aR‐positive cells: 1.75 ± 0.31 in inflammatory cardiomyopathy vs. 0.94 ± 0.26 in non‐inflammatory cardiomyopathy, P = 0.049, Figure 2 A). There was no difference for theexpression of C5aR (C5aR‐positive cells: 0.48 ± 0.13 in inflammatory cardiomyopathy vs. 0.62 ± 0.17 in non‐inflammatory cardiomyopathy, P = 0.513, Figure 2 B). Representative myocardial tissue sections depicting positive expression of C3aR or C5aR on cells residing in the myocardium are illustrated in Figure 2 C. The degree of myocardial fibrosis detected by EMB was not different, if staining was positive or negative for C3aR (P = 0.691, Supporting Information, Figure S2 A). Furthermore, the degree of myocardial fibrosis and the presence of late gadolinium enhancement in cardiac MRI were not associated with the expression of C3aR within the myocardium (Supporting Information, Figure S2 B).
Figure 2.
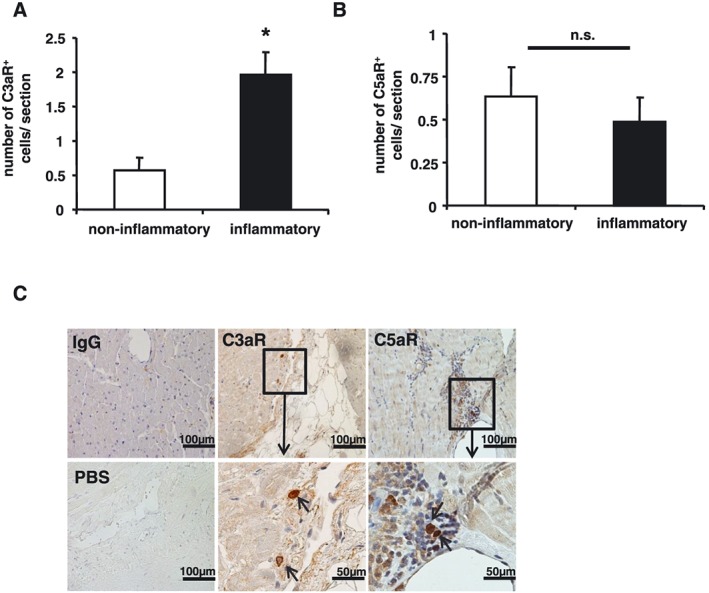
The number of anaphylatoxin receptor‐positive cells is increased in patients with inflammatory cardiomyopathy. The number of C3aR‐positive and C5aR‐positive cells is given in cell number per section. Values are mean + standard error of the mean; *P < 0.05. (A) The number of C3aR‐positive cells was significantly increased in patients with inflammatory cardiomyopathy compared with non‐inflammatory cardiomyopathy (1.75 ± 0.31 in inflammatory cardiomyopathy vs. 0.94 ± 0.26 in non‐inflammatory cardiomyopathy, P = 0.049). (B) There was no difference in the number of C5aR‐expressing cells (0.48 ± 0.13 in inflammatory cardiomyopathy vs. 0.62 ± 0.17 in non‐inflammatory cardiomyopathy, P = 0.513). (C) Representative myocardial tissue sections depict positive expression of C3aR and C5aR in the myocardium.
Expression of C3aR is increased in patients with positive expression of established inflammatory markers in endomyocardial biopsy
Up to 62.7% of patients undergoing EMB showed positive immunohistochemistry for at least one established inflammatory marker and were positive for MHCII (n = 64, 62.7%), CD68 (n = 62, 60.8%), or CD3 (n = 43, 42.2%) as depicted in Table 1. Patients with inflammatory cardiomyopathy showed significantly higher myocardial expression of MHCII (P < 0.001), CD68 (P < 0.001), and CD3 (P < 0.001) compared with patients with non‐inflammatory cardiomyopathy (Figure 3 A and B). Thirty out of 64 (46.9%) patients with positive MHCII expression were positive for C3aR (Score 1), which was higher for trend compared with 11/38 (28.9%) of MHCII‐negative patients, P = 0.076 (Figure 3 A). We found similar results in patients with positive CD68 expression. Twenty‐nine out of 62 (46.8%) patients with positive CD68 expression showed positive C3aR expression (Score 1), which was higher for trend compared with 12/40 (30.0%) CD68‐negative patients, P = 0.093 (Figure 3 B). Endomyocardial biopsy revealed 26/102 (25.5%) patients to be virus positive (Table 1). Among those patients, 10/26 (38.5%) were parvovirus B19 positive, 8/26 patients (30.8%) were Epstein–Barr virus positive, 9/26 patients (34.6%) were human herpesvirus 6 positive, and 2/26 (7.7%) were influenza A/B virus positive (Table 1).
Figure 3.
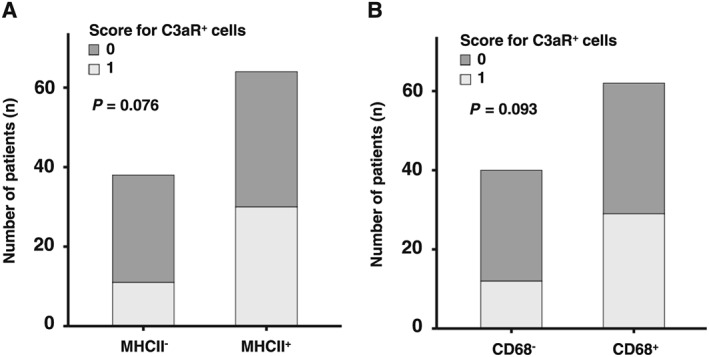
Expression of anaphylatoxin receptor C3aR is enhanced in patients with positive expression of established inflammatory markers in endomyocardial biopsy. C3aR‐positive cells were quantified by investigators blinded to the patient characteristics. Samples were categorized into two groups according to the number of cells positive for the analysed markers. Score 1 = one or more than one cells positive for the marker were counted; Score 0 = no cell positive for the marker. *P < 0.05. (A) 30/64 (46.9%) patients with positive MHCII expression showed positive C3aR expression (Score 1), which was higher for trend compared with 11/38 (28.9%) of MHCII‐negative patients, P = 0.076. (B) 29/62 (46.8%) patients with positive CD68 expression showed positive C3aR expression (Score 1), which was higher for trend compared with 12/40 (30.0%) CD68‐negative patients, P = 0.093.
Expression of C3aR is a predictor of outcome in patients with non‐ischaemic heart failure
During a mean follow‐up of 11.9 months, 36 (35.2%) patients reached the primary endpoint. The patient collective consisted of patients with non‐inflammatory cardiomyopathy (DCM 70.8% and hypertrophic non‐obstructive cardiomyopathy or hypertrophic obstructive cardiomyopathy 29.2%) and inflammatory cardiomyopathy (88.9% chronic myocarditis and 11.1% acute myocarditis, Supporting Information, Table S1 ). Supporting Information, Table S2 depicts the clinical outcome during follow‐up stratified by the underlying cardiomyopathy.
Interestingly, Kaplan–Meier curves illustrate in the overall patient cohort (n = 102, inflammatory and non‐inflammatory cardiomyopathy) that patients negative for C3aR (Score 0) reach the primary endpoint more often than patients that show positive C3aR expression (Score 1) (log rank 5.963, P = 0.015, Figure 4 A). We tested whether C3aR correlates with LVEF. As depicted in Supporting Information, Figures S3 and S4 , there was no correlation of C3aR with LVEF or LVEDD. Furthermore, in the subgroup analysis of patients with inflammatory cardiomyopathy (n = 54), the Kaplan–Meier curve demonstrates that patients negative for C3aR (Score 0) reach the primary endpoint more often than patients that show positive C3aR expression (Score 1) (log rank 3.749, P = 0.053, Figure 4 B). The primary study endpoint was defined as a composite of all‐cause death, heart transplantation, HF‐related re‐hospitalization, and deterioration of LVEF. In multivariate Cox regression analysis for the primary endpoint, among all the risk factors tested (expression of C3aR, age, gender, LVEF, CRP, and NYHA ≥II), C3aR‐negative expression status was a strong independent predictor of the occurrence of the primary endpoint (hazard ratio 0.46; 95% confidence interval 0.26–0.82, P = 0.009, Table 2).
Figure 4.
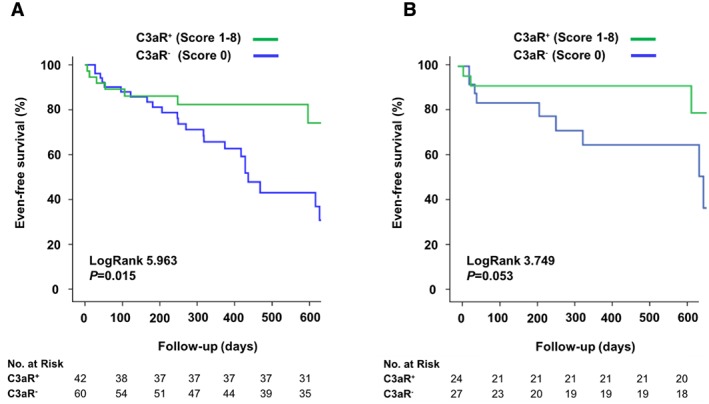
Kaplan–Meier curves illustrate the occurrence of the primary study endpoint stratified by myocardial C3aR expression. During a mean follow‐up of 11.9 months, 36 (35.2%) patients reached the primary endpoint. Out of these, 11 (30.6%) were C3aR positive (Score 1). The primary study endpoint was defined as a composite of all‐cause death, heart transplantation, HF‐related re‐hospitalization, and deterioration of LVEF. (A) Kaplan–Meier curves illustrate in the overall patient cohort (n = 102, inflammatory and non‐inflammatory cardiomyopathy) that patients negative for C3aR (Score 0) reach the primary endpoint more often than patients, who are C3aR positive (Score 1, log rank 5.963, P = 0.015). (B) In the subgroup analysis of patients with inflammatory cardiomyopathy (n = 54), Kaplan–Meier curves demonstrate that patients negative for C3aR (Score 0) reach the primary endpoint more often than patients, who are positive for C3aR with a trend for statistical significance (Score 1, log rank 3.749, P = 0.053).
Table 2.
Lack of C3aR expression in the heart tissue is an independent predictor of the primary study endpoint
| Cox regression analysis | ||
|---|---|---|
| Variable | HR (95% CI) | P |
| Gender | 2.30 (0.92–5.76) | 0.075 |
| Age | 1.04 (1.0–1.1) | 0.006 |
| CRP (mg/dL) | 1.03 (1.0–1.05) | 0.019 |
| LVEF (%) | 0.97 (0.94–1.00) | 0.052 |
| NYHA class ≥II | 0.68 (0.24–1.94) | 0.47 |
| C3aR− | 0.46 (0.26–0.82) | 0.009 |
CI, confidence interval; CRP, C‐reactive protein; HR, hazard ratio; LVEF, left ventricular ejection fraction; NYHA, New York Heart Association.
Values are given in order of decreasing HR. Significant P values are in bold.
Discussion
Inflammatory cardiomyopathy causes chronic HF associated with severe morbidity and significant mortality. Reliable predictors of persisting HF are, however, not available. Here, we describe a potential new parameter, which may add to histopathological characterization of patients with inflammatory cardiomyopathy, improve prediction of prognosis, and could open new directions of basic research in processes involving cardiac remodelling and immune activation. The major findings of our present study are (i) myocardial expression of the anaphylatoxin receptor C3aR is enhanced specifically in patients with inflammatory cardiomyopathy, (ii) C3aR expression is increased in patients positive for other markers of myocardial inflammation, and (iii) lack of C3aR expression is predictive for the occurrence of the composite endpoint consisting of all‐cause death, heart transplantation, HF‐related re‐hospitalization, and deterioration of LVEF.
The complement anaphylatoxin receptors C3aR and C5aR play a pivotal role in tissue inflammation,6 yet the impact of C3aR and C5aR expression in patients with non‐ischaemic cardiomyopathy has not been elaborated, so far.
In our study cohort, C3aR expression was found more frequently in inflammatory cardiomyopathy. In non‐inflammatory cardiomyopathy settings such as DCM, we could not detect significant differences for anaphylatoxin receptor expression. Anaphylatoxin receptors C3aR and C5aR and their active ligands C3a and C5a play a crucial role for the recruitment of inflammatory cells to the site of myocardial injury and histamine release with consequences for tissue injury, remodelling, and healing.11 We observed that over one‐third of the consecutively enrolled patients present with positive myocardial C3aR expression.
Previous data demonstrated that immune cells are important targets of myocardial enterovirus infection providing a non‐cardiac reservoir for viral RNA.24 The diagnostic and prognostic relevance of the detection of viral genome remains a matter of debate.29 Future studies will also have to address the role of anaphylatoxin receptors for viral load in the context of cardiomyopathies. In our cohort, approximately one‐third of biopsies were virus positive, but detection of virus genome was not predictive for the occurrence of clinical events during follow‐up.
Complement deposition has been described for adverse cardiac remodelling after myocardial infarction,30 and its activation was associated with leucocyte infiltration.4 We observed that significantly more patients with increased MHCII expression, typically found on antigen‐presenting cells like macrophages, had elevated levels of C3aR. Whether C3aR expression has any relevance for antigen presentation in inflammatory cardiomyopathy and adverse remodelling will have to be addressed in further studies.
To our knowledge, the present study is the first to describe a diagnostic and prognostic impact of C3aR in patients undergoing EMB for non‐ischaemic HF. If a potentially relevant biomarker for acute or chronic HF associates with myocardial markers of inflammation, it cannot be entirely ruled out that any correlation with myocardial damage and long‐term cardiovascular outcome was secondary to its association with other inflammation markers. Thus, clinical and mechanistical studies are needed to further challenge our findings. One can speculate that underlying mechanisms of positive C3aR expression and relevance for patient outcome are driven by progressive inflammation and fibrotic cardiac remodelling in chronic courses of the disease, already described in other pathologies.31 The role of complement deposition as a prognostic marker in ischaemic HF, however, has only been investigated in very few studies. Gombos et al.32 showed an association of activated complement C3a and a combined endpoint of all‐cause mortality and re‐hospitalization due to progressive HF in 182 patients. Aukrust et al.33 found systemic complement activation in 39 patients with chronic HF, who were treated with intravenous immunoglobulin, which lead to reduced complement activation and improvement of left ventricular function during a 5 months follow‐up. In contrast to previous findings,29 immunohistological detection of inflammation (CD3, CD68, and MHCII) did not allow the prediction of adverse events in our cohort of patients with inflammatory cardiomyopathy. This might reflect the patient selection criteria in our study that consecutively enrolled patients with HF not related to coronary artery disease, including inflammatory and non‐inflammatory cardiomyopathies.34
Furthermore, several prospective clinical studies have shown independently that modest elevations in baseline CRP levels, as detected by high‐sensitivity assays, predict future cardiovascular events.35 High CRP levels were furthermore described as an independent prognostic predictor for mortality in non‐ischaemic cardiomyopathy36, 37 and may contribute to myocardial damage via activation of the complement system and chemotaxis of macrophages.38 We consider elevated CRP levels at baseline rather as a marker of the systemic acute phase reaction and a characteristic finding in our patient cohort than employing it as a specific marker to identify patients with inflammatory cardiomyopathy.
Study limitations
We are aware that our study is rather observational and hypothesis generating, but our findings suggest further in‐depth evaluation of mechanistic features connecting anaphylatoxin receptors with cardiomyopathies, which has not been well characterized so far but might be of great clinical importance. There are several other limitations that should be taken into account. Here, we analysed a ‘real‐world, all‐comers’ collective with patients, for whom a clinical indication for EMB was present. Therefore, some of these patients only show a mildly impaired left ventricular function. Fifty‐one per cent of our patients revealed an LVEF <45% (37% among inflammatory and 66.7% among non‐inflammatory cardiomyopathy). We are aware that these differences regarding left ventricular impairment may be a confounder for our analysis. On the other hand, we could show in multivariate analysis that, among others, lack of C3aR remains an independent predictor for the clinical outcome in our adjusted model. Another limitation is the analysis of a combined endpoint; for future studies, it would be interesting to evaluate the predictors of ‘hard’ endpoints such as death, heart transplantation, or HF‐related re‐hospitalization as single endpoints, especially as our clinical outcome is triggered by the deterioration of LV function over time. The latter may be used to predict left ventricular reverse remodelling, an entity that influences medical treatment or other clinical decisions before other HF symptoms occur. During a 2 year follow‐up, the overall event rate of progressive myocardial remodelling defined by worsening of LVEF over time was low in our cohort of 102 patients. Therefore, larger prospective trials are needed to evaluate, whether C3aR expression remains an independent predictor for worsening of LVEF in patients with non‐ischaemic cardiomyopathy. However, we included 102 consecutive patients, who all underwent EMB and could identify an inverse association of positive myocardial C3aR expression with adverse outcome already in this hypothesis‐generating study. We also have to acknowledge that staining for the anaphylatoxin receptors was evaluated in a semi‐quantitative manner by blinded co‐investigators.1, 39, 40, 41 Even though this method has been suggested for other biomarkers,42 an exact evaluation of C5aR and C3aR expression would be desirable to standardize this quantification method.
In conclusion, expression of C3aR in the myocardial tissue is enhanced in patients with signs of myocardial damage and inflammation and is associated with adverse cardiac remodelling, the substrate of progressive HF, and clinical outcome. Thus, detection of C3aR in the myocardium could serve as a novel, additional biomarker to predict adverse outcome and, in particular, worsening of LVEF in patients with non‐ischaemic HF. Routine staining for this indicator might be useful in an intensified risk assessment of patients with non‐ischaemic cardiomyopathy. Furthermore, our observations may trigger further clinical and basic science studies challenging the pathophysiological role and underlying mechanisms of anaphylatoxin receptors contributing to inflammatory cardiomyopathy.
Conflict of interest
None declared.
Funding
This study was supported by the Volkswagen Foundation (Lichtenberg programme to H. F. L.), the Deutsche Forschungsgemeinschaft (Klinische Forschergruppe ‘Platelets‐Molecular Mechanisms and Translational Implications’ KFO 274), and the TÜFF Frauenförderungsprogramm of the University Tübingen (Karin Mueller, 2241‐0‐0).
Supporting information
Figure S1. Flow chart depicting patient selection criteria and decision for endomyocardial biopsy.
Figure S2. The degree of cardiac fibrosis detected by endomyocardial biopsy and positive late gadolinium enhancement in cardiac MRI was not associated with the expression of anaphylatoxin receptor C3aR within the myocardium. Values are presented as percentages of patients (%). (A) The degree of myocardial fibrosis detected by EMB was not different, if staining was positive or negative for C3aR (P = 0.691). (B) Similarly, presence of late gadolinium enhancement in cardiac MRI was not associated with the expression of C3aR within the myocardium (P = 0.84).
Figure S3. No correlation was observed between the number of C3aR positive cells and left ventricular ejection fraction. Values are presented as Pearson's correlation coefficient r. (A) Correlation between the number of C3aR positive cells and LVEF for the complete collective (n = 102, inflammatory and non‐inflammatory cardiomyopathy). (B) Correlation between the number of C3aR positive cells and LVEF for the subgroup with inflammatory cardiomyopathy (n = 54). (C) Correlation between the number of C3aR positive cells and LVEF for the subgroup with non‐inflammatory cardiomyopathy (n = 48).
Figure S4. There is no correlation between the number of C3aR positive cells with left ventricular end‐diastolic diameter (LVEDD). Values are presented as Pearson's correlation coefficient r. (A) Correlation between the number of C3aR positive cells and LVEDD for the complete collective (n = 102, inflammatory and non‐inflammatory cardiomyopathy). (B) Correlation between the number of C3aR positive cells and LVEDD for the subgroup with inflammatory cardiomyopathy (n = 54). (C) Correlation between the number of C3aR positive cells and LVEDD for the subgroup with non‐inflammatory cardiomyopathy (n = 48).
Table S1. Classification of cardiomyopathies according to clinicopathological and histopathological criteria.
{kind=link}
Table S2. Clinical outcome during follow‐up.
{kind=link}
Acknowledgements
We thank Diana Lombardi and Doris Rilling for their excellent organization and support.
Mueller, K. A. L. , Patzelt, J. , Sauter, M. , Maier, P. , Gekeler, S. , Klingel, K. , Kandolf, R. , Seizer, P. , Gawaz, M. , Geisler, T. , and Langer, H. F. (2018) Myocardial expression of the anaphylatoxin receptor C3aR is associated with cardiac inflammation and prognosis in patients with non‐ischaemic heart failure. ESC Heart Failure, 5: 846–857. 10.1002/ehf2.12298.
Contributor Information
Tobias Geisler, Email: tobias.geisler@med.uni-tuebingen.de.
Harald F. Langer, Email: harald.langer@med.uni-tuebingen.de
References
- 1. Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, Moss AJ, Seidman CE, Young JB, American Heart Association , Council on Clinical Cardiology, Heart Failure and Transplantation Committee , Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups , Council on Epidemiology and Prevention . Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 2006; 113: 1807–1816. [DOI] [PubMed] [Google Scholar]
- 2. Fildes JE, Shaw SM, Yonan N, Williams SG. The immune system and chronic heart failure: is the heart in control? J Am Coll Cardiol 2009; 53: 1013–1020. [DOI] [PubMed] [Google Scholar]
- 3. Zhang Y, Bauersachs J, Langer HF. Immune mechanisms in heart failure. Eur J Heart Fail 2017; 19: 1379–1389. [DOI] [PubMed] [Google Scholar]
- 4. Fairweather D, Frisancho‐Kiss S, Njoku DB, Nyland JF, Kaya Z, Yusung SA, Davis SE, Frisancho JA, Barrett MA, Rose NR. Complement receptor 1 and 2 deficiency increases coxsackievirus B3‐induced myocarditis, dilated cardiomyopathy, and heart failure by increasing macrophages, IL‐1β, and immune complex deposition in the heart. J Immunol 2006; 176: 3516–3524. [DOI] [PubMed] [Google Scholar]
- 5. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol 2010; 11: 785–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mollnes TE, Song WC, Lambris JD. Complement in inflammatory tissue damage and disease. Trends Immunol 2002; 23: 61–64. [DOI] [PubMed] [Google Scholar]
- 7. Frustaci A, Chimenti C, Calabrese F, Pieroni M, Thiene G, Maseri A. Immunosuppressive therapy for active lymphocytic myocarditis: virological and immunologic profile of responders versus nonresponders. Circulation 2003; 107: 857–863. [DOI] [PubMed] [Google Scholar]
- 8. Kaya Z, Afanasyeva M, Wang Y, Dohmen KM, Schlichting J, Tretter T, Fairweather DL, Holers VM, Rose NR. Contribution of the innate immune system to autoimmune myocarditis: a role for complement. Nat Immunol 2001; 2: 739–745. [DOI] [PubMed] [Google Scholar]
- 9. Ward PA. Functions of C5a receptors. J Mol Med 2009; 87: 375–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Langer HF, Chung KJ, Orlova VV, Choi EY, Kaul S, Kruhlak MJ, Alatsatianos M, DeAngelis RA, Roche PA, Magotti P, Li X, Economopoulou M, Rafail S, Lambris JD, Chavakis T. Complement‐mediated inhibition of neovascularization reveals a point of convergence between innate immunity and angiogenesis. Blood 2010; 116: 4395–4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Frey A, Ertl G, Angermann CE, Hofmann U, Stork S, Frantz S. Complement C3c as a biomarker in heart failure. Mediators Inflamm 2013; 2013: 716902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gao CQ, Sawicki G, Suarez‐Pinzon WL, Csont T, Wozniak M, Ferdinandy P, Schulz R. Matrix metalloproteinase‐2 mediates cytokine‐induced myocardial contractile dysfunction. Cardiovasc Res 2003; 57: 426–433. [DOI] [PubMed] [Google Scholar]
- 13. Riedemann NC, Guo RF, Neff TA, Laudes IJ, Keller KA, Sarma VJ, Markiewski MM, Mastellos D, Strey CW, Pierson CL, Lambris JD, Zetoune FS, Ward PA. Increased C5a receptor expression in sepsis. J Clin Invest 2002; 110: 101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Manthey HD, Thomas AC, Shiels IA, Zernecke A, Woodruff TM, Rolfe B, Taylor SM. Complement C5a inhibition reduces atherosclerosis in ApoE−/− mice. FASEB journal: official publication of the Federation of American Societies for Experimental Biology 2011; 25: 2447–2455. [DOI] [PubMed] [Google Scholar]
- 15. Leone O, Veinot JP, Angelini A, Baandrup UT, Basso C, Berry G, Bruneval P, Burke M, Butany J, Calabrese F, d'Amati G, Edwards WD, Fallon JT, Fishbein MC, Gallagher PJ, Halushka MK, McManus B, Pucci A, Rodriguez ER, Saffitz JE, Sheppard MN, Steenbergen C, Stone JR, Tan C, Thiene G, van der Wal AC, Winters GL. 2011 consensus statement on endomyocardial biopsy from the Association for European Cardiovascular Pathology and the Society for Cardiovascular Pathology. Cardiovasc Pathol 2012; 21: 245–274. [DOI] [PubMed] [Google Scholar]
- 16. Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JGF, Coats AJS, Falk V, González‐Juanatey JR, Harjola VP, Jankowska EA, Jessup M, Linde C, Nihoyannopoulos P, Parissis JT, Pieske B, Riley JP, Rosano GMC, Ruilope LM, Ruschitzka F, Rutten FH, van der Meer P, ESC Scientific Document Group . ESC guidelines for the diagnosis and treatment of acute and chronic heart failure: the Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail 2016; 37: 2129–2200. [DOI] [PubMed] [Google Scholar]
- 17. Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, Dubourg O, Kühl U, Maisch B, McKenna W, Monserrat L, Pankuweit S, Rapezzi C, Seferovic P, Tavazzi L, Keren A. Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2008; 29: 270–276. [DOI] [PubMed] [Google Scholar]
- 18. Muller KA, Muller I, Kramer U, Kandolf R, Gawaz M, Bauer A, Zuern CS. Prognostic value of contrast‐enhanced cardiac magnetic resonance imaging in patients with newly diagnosed non‐ischemic cardiomyopathy: cohort study. PloS one 2013; 8: e57077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gulati A, Jabbour A, Ismail TF, Guha K, Khwaja J, Raza S, Morarji K, Brown TDH, Ismail NA, Dweck MR, di Pietro E, Roughton M, Wage R, Daryani Y, O'Hanlon R, Sheppard MN, Alpendurada F, Lyon AR, Cook SA, Cowie MR, Assomull RG, Pennell DJ, Prasad SK. Association of fibrosis with mortality and sudden cardiac death in patients with nonischemic dilated cardiomyopathy. JAMA 2013; 309: 896–908. [DOI] [PubMed] [Google Scholar]
- 20. Kallianos K, Moraes GL, Ordovas KG. Prognostic role of MR imaging in nonischemic myocardial disease. Magn Reson Imaging Clin N Am 2015; 23: 89–94. [DOI] [PubMed] [Google Scholar]
- 21. Zuern CS, Muller KA, Seizer P, Geisler T, Banya W, Klingel K, Kandolf R, Bauer A, Gawaz M, May AE. Cyclophilin A predicts clinical outcome in patients with congestive heart failure undergoing endomyocardial biopsy. Eur J Heart Fail 2013; 15: 176–184. [DOI] [PubMed] [Google Scholar]
- 22. Cooper LT, Baughman KL, Feldman AM, Frustaci A, Jessup M, Kuhl U, Levine GN, Narula J, Starling RC, Towbin J, Virmani R, American Heart Association , American College of Cardiology , European Society of Cardiology . The role of endomyocardial biopsy in the management of cardiovascular disease: a scientific statement from the American Heart Association, the American College of Cardiology, and the European Society of Cardiology. Circulation 2007; 116: 2216–2233. [DOI] [PubMed] [Google Scholar]
- 23. Basso C, Calabrese F, Angelini A, Carturan E, Thiene G. Classification and histological, immunohistochemical, and molecular diagnosis of inflammatory myocardial disease. Heart Fail Rev 2013; 18: 673–681. [DOI] [PubMed] [Google Scholar]
- 24. Klingel K, Stephan S, Sauter M, Zell R, McManus B, Bültmann B, Kandolf R. Pathogenesis of murine enterovirus myocarditis: virus dissemination and immune cell targets. J Virol 1996; 70: 8888–8895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mahrholdt H, Wagner A, Deluigi CC, Kispert E, Hager S, Meinhardt G, Vogelsberg H, Fritz P, Dippon J, Bock CT, Klingel K, Kandolf R, Sechtem U. Presentation, patterns of myocardial damage, and clinical course of viral myocarditis. Circulation 2006; 114: 1581–1590. [DOI] [PubMed] [Google Scholar]
- 26. Caforio AL, Pankuweit S, Arbustini E, Basso C, Gimeno‐Blanes J, Felix SB, Fu M, Heliö T, Heymans S, Jahns R, Klingel K, Linhart A, Maisch B, McKenna W, Mogensen J, Pinto YM, Ristic A, Schultheiss HP, Seggewiss H, Tavazzi L, Thiene G, Yilmaz A, Charron P, Elliott PM, European Society of Cardiology Working Group on Myocardial and Pericardial Diseases . Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2013; 34: 2636‐2648, 2648a–2648d. [DOI] [PubMed] [Google Scholar]
- 27. Mueller KA, Mueller II, Eppler D, Zuern CS, Seizer P, Kramer U, Koetter I, Roecken M, Kandolf R, Gawaz M, Geisler T, Henes JC, Klingel K. Clinical and histopathological features of patients with systemic sclerosis undergoing endomyocardial biopsy. PloS One 2015; 10: e0126707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lang C, Sauter M, Szalay G, Racchi G, Grassi G, Rainaldi G, Mercatanti A, Lang F, Kandolf R, Klingel K. Connective tissue growth factor: a crucial cytokine‐mediating cardiac fibrosis in ongoing enterovirus myocarditis. J Mol Med 2008; 86: 49–60. [DOI] [PubMed] [Google Scholar]
- 29. Kindermann I, Kindermann M, Kandolf R, Klingel K, Bultmann B, Muller T, Lindinger A, Bohm M. Predictors of outcome in patients with suspected myocarditis. Circulation 2008; 118: 639–648. [DOI] [PubMed] [Google Scholar]
- 30. Frangogiannis NG. Regulation of the inflammatory response in cardiac repair. Circ Res 2012; 110: 159–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kitamura H, Cambier S, Somanath S, Barker T, Minagawa S, Markovics J, Goodsell A, Publicover J, Reichardt L, Jablons D, Wolters P, Hill A, Marks JD, Lou J, Pittet JF, Gauldie J, Baron JL, Nishimura SL. Mouse and human lung fibroblasts regulate dendritic cell trafficking, airway inflammation, and fibrosis through integrin αvβ8‐mediated activation of TGF‐β. J Clin Invest 2011; 121: 2863–2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gombos T, Forhecz Z, Pozsonyi Z, Széplaki G, Kunde J, Füst G, Jánoskuti L, Karádi I, Prohászka Z. Complement anaphylatoxin C3a as a novel independent prognostic marker in heart failure. Clin Res Cardiol 2012; 101: 607–615. [DOI] [PubMed] [Google Scholar]
- 33. Aukrust P, Yndestad A, Ueland T, Damas JK, Froland SS, Gullestad L. The role of intravenous immunoglobulin in the treatment of chronic heart failure. Int J Cardiol 2006; 112: 40–45. [DOI] [PubMed] [Google Scholar]
- 34. Westermann D, Lindner D, Kasner M, Zietsch C, Savvatis K, Escher F, von Schlippenbach J, Skurk C, Steendijk P, Riad A, Poller W, Schultheiss HP, Tschope C. Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circ Heart Fail 2011; 4: 44–52. [DOI] [PubMed] [Google Scholar]
- 35. Yeh ET, Willerson JT. Coming of age of C‐reactive protein: using inflammation markers in cardiology. Circulation 2003; 107: 370–371. [DOI] [PubMed] [Google Scholar]
- 36. Sadahiro T, Kohsaka S, Okuda S, Inohara T, Shiraishi Y, Kohno T, Yoshikawa T, Fukuda K. MRI and serum high‐sensitivity C reactive protein predict long‐term mortality in non‐ischaemic cardiomyopathy. Open Heart 2015; 2: e000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ronnow BS, Reyna SP, Muhlestein JB, Horne BD, Allen Maycock CA, Bair TL, Carlquist JF, Kfoury AG, Anderson JL, Renlund DG, International Heart Collaborative Study Group . C‐reactive protein predicts death in patients with non‐ischemic cardiomyopathy. Cardiology 2005; 104: 196–201. [DOI] [PubMed] [Google Scholar]
- 38. Zimmermann O, Bienek‐Ziolkowski M, Wolf B, Vetter M, Baur R, Mailänder V, Hombach V, Torzewski J. Myocardial inflammation and non‐ischaemic heart failure: is there a role for C‐reactive protein? Basic Res Cardiol 2009; 104: 591–599. [DOI] [PubMed] [Google Scholar]
- 39. Frustaci A, Pieroni M, Chimenti C. The role of endomyocardial biopsy in the diagnosis of cardiomyopathies. Ital Heart J 2002; 3: 348–353. [PubMed] [Google Scholar]
- 40. Kawai C. From myocarditis to cardiomyopathy: mechanisms of inflammation and cell death: learning from the past for the future. Circulation 1999; 99: 1091–1100. [DOI] [PubMed] [Google Scholar]
- 41. Rutschow S, Li J, Schultheiss HP, Pauschinger M. Myocardial proteases and matrix remodeling in inflammatory heart disease. Cardiovasc Res 2006; 69: 646–656. [DOI] [PubMed] [Google Scholar]
- 42. Mueller KA, Tavlaki E, Schneider M, Jorbenadze R, Geisler T, Kandolf R, Gawaz M, Mueller II, Zuern CS. Gremlin‐1 identifies fibrosis and predicts adverse outcome in patients with heart failure undergoing endomyocardial biopsy. J Card Fail 2013; 19: 678–684. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Flow chart depicting patient selection criteria and decision for endomyocardial biopsy.
Figure S2. The degree of cardiac fibrosis detected by endomyocardial biopsy and positive late gadolinium enhancement in cardiac MRI was not associated with the expression of anaphylatoxin receptor C3aR within the myocardium. Values are presented as percentages of patients (%). (A) The degree of myocardial fibrosis detected by EMB was not different, if staining was positive or negative for C3aR (P = 0.691). (B) Similarly, presence of late gadolinium enhancement in cardiac MRI was not associated with the expression of C3aR within the myocardium (P = 0.84).
Figure S3. No correlation was observed between the number of C3aR positive cells and left ventricular ejection fraction. Values are presented as Pearson's correlation coefficient r. (A) Correlation between the number of C3aR positive cells and LVEF for the complete collective (n = 102, inflammatory and non‐inflammatory cardiomyopathy). (B) Correlation between the number of C3aR positive cells and LVEF for the subgroup with inflammatory cardiomyopathy (n = 54). (C) Correlation between the number of C3aR positive cells and LVEF for the subgroup with non‐inflammatory cardiomyopathy (n = 48).
Figure S4. There is no correlation between the number of C3aR positive cells with left ventricular end‐diastolic diameter (LVEDD). Values are presented as Pearson's correlation coefficient r. (A) Correlation between the number of C3aR positive cells and LVEDD for the complete collective (n = 102, inflammatory and non‐inflammatory cardiomyopathy). (B) Correlation between the number of C3aR positive cells and LVEDD for the subgroup with inflammatory cardiomyopathy (n = 54). (C) Correlation between the number of C3aR positive cells and LVEDD for the subgroup with non‐inflammatory cardiomyopathy (n = 48).
Table S1. Classification of cardiomyopathies according to clinicopathological and histopathological criteria.
Table S2. Clinical outcome during follow‐up.