

Abstract
Key points
ERG3 channels have a high expression level in the central nervous system.
Knockdown of ERG3 channels enhances neuronal intrinsic excitability (caused by decreased fast afterhyperpolarization, shortened delay time to the generation of an action potential and enhanced summation of somatic excitatory postsynaptic potentials) in hippocampal CA1 pyramidal neurons and dentate gyrus granule cells.
The expression of ERG3 protein is reduced in human and mouse hippocampal epileptogenic foci.
Knockdown of ERG3 channels in hippocampus enhanced seizure susceptibility, while mice treated with the ERG channel activator NS‐1643 were less prone to epileptogenesis.
The results provide strong evidence that ERG3 channels have a crucial role in the regulation of neuronal intrinsic excitability in hippocampal CA1 pyramidal neurons and dentate gyrus granule cells and are critically involved in the onset and development of epilepsy.
Abstract
The input–output relationship of neuronal networks depends heavily on the intrinsic properties of their neuronal elements. Profound changes in intrinsic properties have been observed in various physiological and pathological processes, such as learning, memory and epilepsy. However, the cellular and molecular mechanisms underlying acquired changes in intrinsic excitability are still not fully understood. Here, we demonstrate that ERG3 channels are critically involved in the regulation of intrinsic excitability in hippocampal CA1 pyramidal neurons and dentate gyrus granule cells. Knock‐down of ERG3 channels significantly increases neuronal intrinsic excitability, which is mainly caused by decreased fast afterhyperpolarization, shortened delay time to the generation of an action potential and enhanced summation of somatic excitatory postsynaptic potentials. Interestingly, the expression level of ERG3 protein is significantly reduced in human and mouse brain tissues with temporal lobe epilepsy. Moreover, ERG3 channel knockdown in hippocampus significantly enhanced seizure susceptibility, while mice treated with the ERG channel activator NS‐1643 were less prone to epileptogenesis. Taken together, our results suggest ERG3 channels play an important role in determining the excitability of hippocampal neurons and dysregulation of these channels may be involved in the generation of epilepsy. ERG3 channels may thus be a novel therapeutic target for the prevention of epilepsy.
Keywords: ERG channels, Intrinsic excitability, Epilepsy
Key points
ERG3 channels have a high expression level in the central nervous system.
Knockdown of ERG3 channels enhances neuronal intrinsic excitability (caused by decreased fast afterhyperpolarization, shortened delay time to the generation of an action potential and enhanced summation of somatic excitatory postsynaptic potentials) in hippocampal CA1 pyramidal neurons and dentate gyrus granule cells.
The expression of ERG3 protein is reduced in human and mouse hippocampal epileptogenic foci.
Knockdown of ERG3 channels in hippocampus enhanced seizure susceptibility, while mice treated with the ERG channel activator NS‐1643 were less prone to epileptogenesis.
The results provide strong evidence that ERG3 channels have a crucial role in the regulation of neuronal intrinsic excitability in hippocampal CA1 pyramidal neurons and dentate gyrus granule cells and are critically involved in the onset and development of epilepsy.
Introduction
Neuroplasticity plays an important role in regulating several brain functions such as learning and memory (Kandel, 2001; Zhang & Linden, 2003; Losonczy et al. 2008; Mozzachiodi et al. 2008; Caroni et al. 2012). Intrinsic plasticity, which is one type of neuroplasticity, is a phenomenon where the firing efficiency of postsynaptic neurons in response to synaptic inputs can be altered in an activity‐dependent manner. One important feature of intrinsic plasticity is to modify the input–output information flow from dendrites to axon terminals, through regulating expression patterns or biophysical properties of multiple ion channels located in different neuronal compartments (Hausser et al. 2000; Daoudal & Debanne, 2003; Lai & Jan, 2006; Nelson & Turrigiano, 2008; Sweatt, 2016). Impairment of intrinsic plasticity is involved in a range of neurological and psychiatric disorders such as neuropathic pain, addiction and epilepsy (Chen et al. 2002; Beck & Yaari, 2008; Wu et al. 2008; Jung et al. 2011; Surges et al. 2012; Shah et al. 2013; Kourrich et al. 2015). Despite the important role of intrinsic neuroplasticity in physiological and pathological processes in the brain, the underlying mechanisms are still poorly understood.
Ether‐à‐go‐go‐related gene‐encoded channels (ERG, Kv11) are voltage‐gated potassium channels with unusual kinetics in that their inactivation rates vastly exceed activation rates, so that the greatest conductance is generated on repolarization rather than depolarization, which makes the channel current inwardly rectifying (Shibasaki, 1987; Sanguinetti et al. 1995; Trudeau et al. 1995; Smith et al. 1996). The ERG channel family consists of three cloned members, ERG1 (Kv11.1), ERG2 (Kv11.2) and ERG3 (Kv11.3) (Shi et al. 1997). ERG1 protein is extensively studied for its role in the repolarization of the ventricular action potential, and these channels also contribute to the pacemaker potential in the sinoatrial node (Sanguinetti & Tristani‐Firouzi, 2006). ERG2 protein has a relatively low expression in the whole body, and ERG3 protein is mainly expressed in the central nervous system (CNS; Guasti et al. 2005). ERG channels were found to regulate firing frequency in cerebellar Purkinje cells (Sacco et al. 2003) and action potential threshold in mouse auditory brainstem neurons (Hardman & Forsythe, 2009), while the neuronal excitability of neocortical pyramidal cells was also regulated by ERG current (Cui & Strowbridge, 2017). In addition, the ERG channel blocker E‐4031 has significant effects on neuronal excitability of hippocampal CA1 pyramidal neurons (Fano et al. 2012). Although ERG3 channels are highly expressed in the CNS (Shi et al. 1997; Polvani et al. 2003; Bauer & Schwarz, 2018), previous investigations have mainly focused on the mixture of currents mediated by the three ERG channel subtypes and the function of ERG3 channels in the CNS is still not clear.
Here, we characterize the expression and functional properties of ERG3 channels in hippocampal neurons. Western blots and RT‐PCR experiments showed that the ERG3 protein and mRNA are predominantly expressed in hippocampus and that the ERG3 mRNA level is much higher than that of ERG1 and ERG2. Immunofluorescence data showed that the ERG3 protein is predominantly localized to the proximal apical dendrites and soma of excitatory neurons. Knockdown of ERG3 channels with ERG3 short hairpin RNA (shRNA) significantly increased neuronal intrinsic excitability in dentate gyrus (DG) granule cells and CA1 pyramidal neurons, which was mainly caused by decreased fast afterhyperpolarization (AHP), shortened delay time to the generation of an action potential and enhanced somatic excitatory postsynaptic potential (EPSP) summation. Interestingly, functional ERG‐mediated currents and ERG3 protein were significantly reduced after status epilepticus in our mouse epilepsy model. Moreover, knocking down ERG3 channels enhanced seizure susceptibility, while mice treated with the ERG channel activator NS‐1643 were less prone to epileptogenesis. Finally, examination of brain tissues obtained from human patients with temporal lobe epilepsy revealed decreased expression of ERG3 channels, supporting the hypothesis of an important role of ERG3 channels in suppressing epileptogenesis. Together, our results show a novel finding of ERG3 channels regulating neuronal intrinsic excitability in the hippocampus, and suggest ERG3 channels might be a potential therapeutic target for the prevention and treatment of temporal lobe epilepsy (TLE).
Methods
Ethical approval
This study was approved by the Institutional Animal Care and Use Committee at Peking University (LA20141200). All animals were handled in strict accordance with two guidelines: Guide for the Care and Use of Laboratory Animals and Principles for the Utilization and Care of Vertebrate Animals. Every effort was made to minimize animal suffering and the number of animals used. The study complied with the ethical principles of The Journal of Physiology, and the experiments complied with The Journal’s animal ethics principles and regulation checklist (Grundy, 2015).
Animals
C57BL/6 male mice (6–8 weeks, 18–23 g) were purchased from Charles River Laboratories (Beijing, China). Animals were housed under a standard artificial 12 h light/dark cycle with water and food provided ad libitum. For the shRNA injection surgical procedures, mice were anaesthetized with sodium pentobarbital (50 mg kg−1) administered i.p. The adequacy of the anaesthesia was judged by the lack of reflex in response to a firm tail pinch. Additional anaesthetic was administered during the surgery if necessary (25% of the original dose, i.p.). For the electrophysiology studies, mice were anaesthetized with chloral hydrate (500 mg kg−1) and perfused intracardially with ice‐cold ‘cutting solution’, then the brain was removed and submerged in ‘cutting solution’ for the preparation of slices. The experimenters were blind to viral treatment or drug treatment condition during behavioural testing. Electrophysiology recordings, anatomical investigation and the adeno‐associated virus (AAV)‐infection experiments were all performed in the dorsal part of the hippocampus.
Acute slice preparation and electrophysiological recordings
Horizontal slices containing hippocampus were obtained from 6‐ to 8‐week‐old male C57/BL6 mice. In accordance with previous studies (Huang et al. 2009, 2011), the brain was removed and submerged in ice‐cold ‘cutting solution’ containing (mm): 110 choline chloride, 2.5 KCl, 0.5 CaCl2, 7 MgCl2, 25 NaHCO3, 1.25 NaH2PO4 and 10 glucose. Next, the brain was cut into 350 μm slices with a vibrating blade microtome (WPI, Sarasota, FL, USA). Slices were incubated in oxygenated (95% O2 and 5% CO2) ‘recording solution’ containing (mm): 125 NaCl, 2.5 KCl, 2 CaCl2, 2 MgCl2, 25 NaHCO3, 1.25 NaH2PO4 and 10 glucose (315 mOsm, pH 7.4, 37 °C) for 15 min, and then stored at room temperature in recording solution. Slices were subsequently transferred to a submerged chamber containing ‘recording solution’ maintained at 34–36°C. Whole‐cell recordings were obtained from visually identified hippocampal DG and CA1 neurons under a water‐immersed ×40 objective of an Olympus BX51WI microscope. Pipettes had resistances of 5–8 MΩ. Series resistance was in the order of 10–30 MΩ, which was compensated by 60–80% during the experiments. Recordings were discarded if the series resistance increased by more than 20% during the course of the recordings. For voltage‐clamp and current‐clamp recordings, electrophysiological recordings were made using a Multiclamp 700B amplifier (Molecular Devices, Sunnyvale, CA, USA). Recordings were filtered at 10 kHz and sampled at 50 kHz. Data were acquired and analysed using pCLAMP 10.0 (Molecular Devices). In our whole‐cell current‐clamp recordings (with an Axon 700B amplifier, Molecular Devices, San Jose, CA, USA), we initially applied a 100 ms, 20 pA test pulse to the recording neurons right after breaking into the whole‐cell configuration. A fast‐rising component and a slow‐rising component of voltage response were clearly visible. Then we zoomed into the fast‐rising component of voltage responses and turned up the pipette capacitance neutralization slowly to shorten the rise time of the fast‐rising component until the oscillations of voltage responses appeared. Subsequently, we decreased the capacitance compensation just until the oscillations disappeared. For bridge balance of current‐clamp recordings, we increased the value of bridge balance slowly until the fast component of the voltage response disappeared, and the slow‐rising component appeared to rise directly from the baseline. Series resistance and pipette capacitance were compensated using the bridge balance and pipette capacitance neutralization options in the Multiclamp 700B command software. The bridge balance value was between 20 and 30 MΩ and the pipette capacitance neutralization value was between 3 and 5 pF.
ERG currents were recorded under whole‐cell voltage‐clamp condition using a high [K+] (40 mm, substituted for Na+) and Ca2+‐free (replaced with MgCl2) solution to enhance the ERG channel's conductance, which is consistent with a previous study (Hirdes et al. 2009). The internal pipette solution contained (mm): 118 KMeSO4, 15 KCl, 10 Hepes, 2 MgCl2, 0.2 EGTA, 4 Na2ATP, 0.3 Tris‐GTP and 14 Tris‐phosphocreatine (pH 7.3 with CsOH); 500 nm tetrodotoxin (TTX, Abcam, Cambridge, UK), 15 μm ZD7288 (Abcam) and 100 μm CdCl2 (Abcam) were added to block voltage‐activated sodium channels, HCN channels and voltage‐gated calcium channels, respectively. E‐4031 (N‐[4‐[[1‐[2‐(6‐methyl‐2‐pyridinyl)ethyl]‐4‐piperidinyl]carbonyl]phenyl]methanesulfonamide dihydrochloride; 10 μm) was extracellularly perfused (10 μm) in the high potassium solution.
Control tissues or tissues with TLE
Patients (n = 355) with medically intractable TLE underwent phased presurgical assessment at Shengjing Hospital affiliated to China Medical University. Epilepsy diagnosis (including types and localization) was determined by clinical history, imaging examination (including magnetic resonance imaging and/or positron emission tomography), electroencephalography (EEG; including scalp and/or intracranial EEG), and psychological assessment. Patients with TLE caused by stroke, tumour, injury and malformations were excluded in this study. In those selected for surgery, the hippocampus was resected according to standard procedures. Between July 2010 and February 2016, 246 hippocampi were resected. The study, using randomly chosen clinical samples, which include eight paired epileptogenic tissues and matched adjacent normal tissues from the same patient, was approved by the Ethics Committee of Shengjing Hospital affiliated to China Medical University. Informed consent was obtained from all subjects or their relatives. Before surgical resection of hippocampi, the multi‐channel intracranial EEG electrodes were implanted to localize the origin of epilepsy. The ‘epileptic tissues’ in this study were from originating sites of spontaneous seizure which were determined by their electrical seizure‐like activities recorded with intracranial EEG electrodes. Tissues adjacent to ‘epileptic tissues’ serve as the ‘controls’, which were normally the adjacent entorhinal cortical area. The control tissues could also show abnormal electrical activities during a seizure, but the occurrence of such activities is significantly later than that in epileptic tissues and we thus consider it as a secondary effect. Tissues were frozen in liquid nitrogen immediately after surgical removal and maintained at −80°C until mRNA and protein extraction. These eight pairs of samples come from patients of both sexes and age from 26 to 57; the detailed information of these patients is provided in Table 4.
Table 4.
Demographic and clinical characteristics of selected patients with mesial TLE
| Patient* (number) | Age (years) | Sex | Time since first unprovoked seizure (years) | Medication history | MRI examination |
|---|---|---|---|---|---|
| 1 (SE1/NC1) | 57 | Male | 4 | Carbamazepine, topiramate | Normal |
| 2 (SE2/NC2) | 26 | Male | 18 | Carbamazepine | L temporal‐lobe atrophy, no signal changes |
| 3 (SE3/NC3) | 29 | Male | 5 | Lamotrigine, gabapentin | Normal |
| 4 (SE4/NC4) | 42 | Female | 31 | Carbamazepine, phenobarbital | L temporal‐lobe and hippocampal atrophy, no signal changes |
| 5 (SE5/NC5) | 29 | Male | 2 | Valporate | Normal |
| 6 (SE6/NC6) | 31 | Female | 11 | Carbamazepine, phenobarbita | L temporal‐lobe and hippocampal atrophy, signal changes |
| 7 (SE7/NC7) | 39 | Female | 15 | Carbamazepine, valporate | L hippocampal atrophy, signal changes |
| 8 (SE8/NC8) | 34 | Female | 15 | Valporate | Normal |
*All selected patients were seizure‐free for periods of 2 or 3 years. Epileptic foci (named SE1–SE8): seizure onset tissues from hippocampus confirmed by scalp and intracranial EEG. Type of surgery: anterior temporal lobe and hippocampus–amygdala resection.
Adeno‐associated virus design
We used AAV carrying shRNA targeting mouse ERG3. AAV vectors (WZ004) were obtained from Shanghai Genechem. AAV serotype 9 was used in the experiment, since previous experiments showed that AAV9 has the highest transduction rate in hippocampus (Aschauer et al. 2013). The sequence of the components in the AAV vector was U6 promoter–shRNA–CMV bGlobin–eGFP–3*Flag. Adult mouse hippocampal DG granule neurons were transduced with proper AAV containing non‐silencing shRNA or ERG3 shRNA. The three ERG3‐shRNA sequences are: ERG3‐shRNA no. 1: 5′‐GCAGTCAAGTTCCCAACTA‐3′; ERG3‐shRNA no. 2: 5′‐GCACCCAAGGTTAAAGAAA‐3′; and ERG3‐shRNA no. 3: 5′‐GCAAGTAAAGGCTGTCTTA‐3′. The non‐silencing shRNA sequence is 5′‐CGCTGAGTACTTCGAAATGTC‐3′. The effect of shRNA was tested by ERG3‐overexpressed HEK‐293 cells. Since western blot experiment showed three shRNAs respectively caused 69.1 ± 4.6%, 57.7 ± 0.17% and 85.9 ± 2.0% decrease of ERG3 protein expression in HEK‐293 cells, ERG3‐shRNA no. 3 was used in further experiments. The infection efficiency was also confirmed by the expression of a green fluorescent protein under microscopy and the electrophysiology recordings from shRNA‐transduced DG neurons.
Stereotaxically guided AAV injection and EEG recordings
Animals (mice of 6–8 weeks) were deeply anaesthetized by intraperitoneal injection of sodium pentobarbital (50 mg kg−1 body weight) and secured in the stereotaxic apparatus (RWD Ltd, Shenzheng, China). AAV was bilaterally injected into dorsal hippocampus DG area (coordinates, bregma: anterior–posterior, −1.60 mm; dorsal–ventral, −2.00 mm; lateral, ±1.30 mm, 600 nl per side) or CA1 area (coordinates, bregma: anterior–posterior, −2.00 mm; dorsal–ventral, −2.50 mm; lateral, ±2.40 mm, 600 nl per side). Ten minutes after microinjection, the needle was retracted and after another 10 min, the wound was sutured. For those mice prepared for the seizure susceptibility measurement, depth electrodes (Plastics One, Roanoke, VA, USA) were implanted into the DG region (with the same coordinates stated above). All animals were monitored for at least an hour post‐surgery and at 12 h intervals for the next 5 days. Since ERG3‐shRNA was strongly expressed from 7 days post‐injection (DPI) and lasted at least 2 months, the tested mice were used at 14 DPI. EEG was recorded during the measurement of seizure susceptibility with Omniplex‐D Neuronal data acquisition system (Neurolog, Houston, TX, USA).
Western blot
Mouse hippocampal tissues were collected from the kainic acid model mice and control mice. There were three mice in each group used in this experiment, and the mice were anaesthetized with chloral hydrate (500 mg kg−1) then decapitated for the acquisition of hippocampal tissues. For human tissues, five pairs of hippocampal and adjacent normal tissues were used in this experiment (patients 1–5). Tissues were homogenized in RIPA buffer (containing 50 mm Tris–HCl, pH 8.0, 150 mm NaCl, 1% NP‐40, 0.1% SDS, 0.5% sodium deoxycholate) on ice and then centrifuged at 13680 g for 3 min at 4°C. The supernatant was stored at −20°C until use. Protein concentration was measured by the BCA method. The lysate was mixed with 5× SDS sample buffer (200 mm Tris–HCl pH 6.8, 10% SDS, 25% glycerol, 5% 2‐mercaptoethanol, 0.05% bromophenol blue) and boiled at 70°C for 8 min. Approximately 35 μg protein of each sample was loaded on 8% SDS–PAGE and run at 120 V constant voltage. A constant current of 280 mA was used for transblotting to a poly(vinylidene difluoride) membrane (Millipore, Billerica, MA, USA). Blots were probed with primary antibodies (1:1500) overnight at 4°C. After washing three times, blots were then incubated with goat anti‐rabbit secondary antibody (1:20,000) at room temperature for 2 h. Chemiluminescence was used to visualize protein bands. Antibodies used were anti‐ERG3 (Abcam, ab80455 lot: R199256‐2; for mouse sample assessments), anti‐ERG3 (Alomone Laboratories, Jerusalem, Israel, APC‐112 lot: AN‐02; for human sample assessments), anti‐actin and anti‐glyceraldehyde 3‐phosphate dehydrogenase (GAPDH; Beijing Biodragon Immunotechnologies Co., Ltd, Beijng, China). Blocked antibody was prepared by adding the same concentration of control peptide into the diluted antibody and incubated in room temperature for 30 min. The images were captured and quantified using Tanon GIS digital gel processing system (Tanon Science & Technology, Shanghai, China).
RNA extraction and RT‐PCR
Total RNA was prepared from the hippocampi of epilepsy and control mice. Mice were anaesthetized with chloral hydrate (500 mg kg−1) then decapitated for the acquisition of hippocampal tissues. RNA was extracted using the RNAprep pure micro kit (Tiangen DP420, Tiangen, Beijing, China). After RNA extraction, RNA was reverse‐transcribed with AT301‐03 (Transgene, Beijing, China), in accordance with the manufacturer's protocol. And the ERG3 cDNA was analysed by qPCR with the following sets of primers: ERG3‐FW (5′‐CCAGGAAACTGGACCGATACT‐3′), ERG3‐RV (5′‐CCAATCGCATACCAGATGCAA‐3′), mActin‐FW (5′‐GGCTGTATTCCCCTCCATCG‐3′), mActin‐RV (5′‐CCAGTTGGTAACAATGCCATGT‐3′). qPCR reactions were performed using qPCR mix (Transgene AQ141) in the MX3005p machine and SYBR Green was used during the experiment. Expression levels were calculated using MxPro software. Actin and GAPDH genes were used as controls to normalize expression levels.
ERG3 fluorescence staining
Mice were killed by perfusion with 1% paraformaldehyde and 1% sucrose (w/v) in 0.1 m phosphate buffer (pH 7.4) after deep anaesthesia with sodium pentobarbital (50 mg kg−1). The brain was removed and post‐fixed in the same fixative for 2 h, and subsequently immersed in 30% sucrose in 0.1 m phosphate buffer for 48 h. Cryostat horizontal sections (50 μm) were obtained using a freezing microtome. The sections were rinsed in 0.01 m phosphate‐buffered saline (PBS, pH 7.4) and incubated in a blocking solution (5% normal goat serum, 0.3% Triton X‐100 in PBS, v/v) at 20–25°C for 2 h, followed by overnight incubation at 4°C with primary antibody to NeuN (1:300, Millipore, Mab377 lot 2829834), ERG3 (1:100, Alomone, APC‐112 lot: AN‐02), glutamic acid decarboxylase of molecular mass 67 kDa (GAD67; 1:300, Millipore, Mab5406 lot 2844575), glial fibrillary acidic protein (GFAP; 1:300, Abcam ab10062 lot GR270398‐8) separately in 0.1% Triton (v/v). After a complete wash in PBS, the sections were incubated in Alexa 488‐conjugated goat anti‐rabbit IgG and Alexa 594 goat anti‐mouse IgG in 0.1% Triton (1:1000; Molecular Probes, Thermo Fisher Scientific, Waltham, MA, USA) at 20–25°C for 2 h. Images were taken in the linear range of the photomultiplier with a laser scanning confocal microscope (Nikon A1r, Nikon, Melville, NY, USA) and the projection of z stack images (0.4 μm per image) was used in the figures. Blocked antibody was prepared by adding the same concentration of control peptide into the diluted antibody and incubating at room temperature for 30 min.
ERG3 immunohistochemistry
Three pairs of human epilepsy tissues and normal tissues (patients 6–8) and three mice were used for the immunohistochemistry experiment. Mice were deeply anaesthetized by sodium pentobarbital (50 mg kg−1) and transcardially perfused using PBS followed by 4% paraformaldehyde in PBS. Brains were post‐fixed in fixative and stored in 30% sucrose in PBS overnight for cryoprotection. Brains were embedded and mounted in paraffin and 20 μm sections were cut using a cryostat (Leica RM2016, Leica, Buffalo Grove, IL, USA). Sections cut from formalin‐fixed paraffin‐embedded blocks were deparaffinized and rehydrated with serial passage through changes of xylene and graded ethanols. All slides were subjected to heat‐induced epitope retrieval in a citrate buffer kit. Endogenous peroxidase in tissues was blocked by incubation of slides in 3% hydrogen peroxide solution for 25 min prior to incubation with primary antibody. Then slides were incubated with blocking solution (3% bovine serum albumin in phosphate‐buffered saline with Tween 20) for 30 min followed by primary antibody incubation overnight at 4°C with ERG3 antibody (Alomone, APC‐112, lot: AN‐02). The next day, Antigen‐antibody binding was visualized via application of the DAKO EnVision Detection Kit (DAKO K5007, Agilent, Santa Clara, CA, USA). After that, the slices were washed with PBS followed by counterstaining with haematoxylin and coverslipped for review. Blocked antibody was prepared by adding the same concentration of control peptide into the diluted antibody and incubating at room temperature for 30 min. The results of immunohistochemistry experiments were analysed by Image‐pro plus 6.0 (Media Cybernetics, Inc., Rockville, MD, USA).
Kainic acid‐induced status epilepticus
Kainic acid (Sigma‐Aldrich, St Louis, MO, USA) was intraperitoneally administered to produce class V seizures. The dose of kainite acid used was 20 mg kg−1 for mice (6–8 weeks). To assess epilepsy susceptibility, seizures were rated using a modified Racine scale: (1) immobility followed by facial clonus; (2) masticatory movements and head nodding; (3) continuous body tremor or wet‐dog shakes; (4) unilateral or bilateral forelimb clonus; (5) rearing and falling. Status epilepticus was terminated 1 h after onset with the use of sodium pentobarbital (30 mg kg−1; Sigma‐Aldrich). Control groups were treated with sodium pentobarbital only (30 mg kg−1).
Protocols and statistical methods
In the current‐clamp recording, αEPSPs were generated by current injection of the order:
where A is the amplitude of the current injected and τ is the rise time constant. The decay time constant of αEPSPs was calculated by fitting the trace with double‐exponential function:
where τ1 and τ2 represent time constants of the initial and falling phase of the αEPSPs. The summation ratio of EPSPs was calculated as the ratio of the peak of the fifth EPSP to that of the first EPSP.
Presynaptic axons in stratum radiatum of CA1 and stratum moleculare of DG were stimulated using a tungsten electrode (A‐M Systems, Sequim, WA, USA), with a tip diameter of ∼5 μm, connected to a stimulus unit (Digitimer, Welwyn Garden City, UK). Stimulation electrodes were placed at ∼100 μm from the soma. Subthreshold EPSPs were elicited by 100 μs current injections that were able to cause approximately 5 mV depolarization for the recording neuron. The decay time constant of evoked EPSPs was calculated by fitting the trace with single‐exponential function:
where τ represent time constants of the decay phase of the evoked EPSPs.
The firing rate of neurons was determined as the number of action potentials that could be elicited by a 400 ms depolarizing current injection (50–250 pA for granule cells; 100–400 pA for pyramidal neurons). Fast AHPs were measured as the most negative membrane potential (relative to the threshold of action potential) from the first, second, third and fourth action potentials with a constant current injection (200 pA for granule cells; 300 pA for pyramidal neurons). Action potential amplitude was also measured relative to the action potential threshold. The input resistance was calculated from voltage responses to current injections (400 ms; −100 pA). Delay time in the initiation of an action potential was measured from the time of current injection to the time of action potential threshold. Spike half‐width was calculated as spike duration at 50% of the spike amplitude. The membrane time constant was measured by fitting a single exponential function to the slow phase of the charging curve produced by application of the negative current pulse (−100 pA). The threshold of action potential was determined by the phase plane plots. For phase plot analysis, changes of the membrane potential with time (dV/dt measured as mV ms−1; y‐axis) are plotted against the instantaneous membrane potential (measured as mV; x‐axis). A single action potential (AP) is represented as a loop in which the starting point of the loop, where the point before the first derivative of the trace was no longer equal to zero, represents the threshold membrane potential (AP threshold) (Trombin et al. 2011).
Analysis of the voltage dependence of activation and inactivation was performed using Prism 6 software (GraphPad Software, Inc., La Jolla, CA, USA). Activation parameters were determined by fitting the total charge of tail current and test potential with the
Boltzmann function:
Where X is the test potential, V 0.5 is the potential of half‐maximal channel activation, b is the least current, a is the max current and k is the slope factor for activation. For the measurement of the steady‐state inactivation of ERG channels, the relative conductance values for the single experiments were calculated from the fully activated current–voltage relationship using values of the reversal potential estimated by interpolation. The inactivation curve is also drawn by fitting the EC50 equation, yielding inactivation curves with V 0.5 as the potential of half‐maximal channel inactivation and k as the slope factor.
Group data are represented as the mean ± SEM. Comparisons between two groups were made using Student's paired or unpaired two‐tailed t test as appropriate. Statistical significance of differences at P < 0.05 is indicated as an asterisk (*), P < 0.01 is indicated as two asterisks (**) and P < 0.001 is shown with three asterisks (***) in all figures.
Results
ERG3 channels are highly expressed in brain
In order to characterize the expression pattern of the ERG3 protein in adult mouse brain, immunohistochemistry (IHC) and immunofluorescence (IF) were used for the investigation. As shown in Fig. 1 A, IHC experiments showed that ERG3 protein was expressed in the hippocampus of adult mice. In CA1, the stratum pyramidale layer (Fig. 1 A) was most intensely stained by the ERG3 antibody. The DG stratum granulosum was also stained with the ERG3 antibody, but the labelling intensity was weaker than CA1 stratum pyramidale layer (Fig. 1 A). In the polymorphic layer and stratum radiatum, stratum oriens and stratum moleculare, a few sparse neurons were also stained with the antibody to ERG3 protein. In addition, IHC images showed that CHO cells expressing ERG3 channels have a strong staining with ERG3 antibody. We further stained the CHO cells expressing ERG3 channels with ERG3 antibody that was incubated with control antigen. The results showed that the staining of ERG3 channels was almost absent (Fig. 1 B). Moreover, the results of a western blot assay suggested ERG3‐expressing HEK‐293 cells stained strongly with ERG3 antibody, while ERG1‐overexpressing cells and control cells did not show this (Fig. 1 C). Both of these experiments suggest this ERG3 antibody was specific to ERG3 protein.
Figure 1. ERG3 protein is expressed in hippocampus.
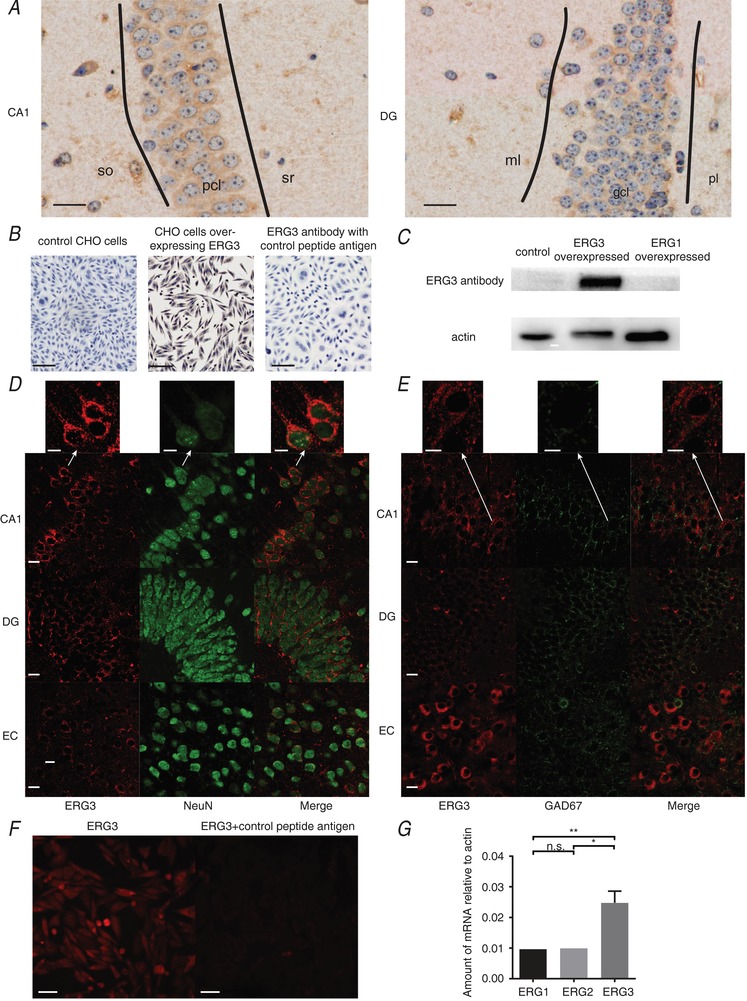
A, IHC images showing the expression of the ERG3 protein in DG and CA1 areas of the mouse hippocampus. Scale bar, 20 μm. gcl, stratum granulosum; ml, stratum moleculare; pcl, stratum pyramidale layer; pl, polymorphic layer; so, stratum oriens; sr, stratum radiatum. B, IHC images showing CHO cells overexpressing ERG3 are heavily stained with ERG3 antibody, while control CHO cell and the negative control groups show almost no stain at all. IHC and image capturing conditions are identical in these groups. Scale bar, 50 μm. C, western blot analysis of HEK‐293 cells with ERG3 overexpression, ERG1 overexpression and control group. D, double IF staining for ERG3 (red) and NeuN (green) in entorhinal cortex and hippocampal DG and CA1 regions of mice. Scale bar, 10 μm. Arrows show a magnified image, indicating ERG3 protein is expressed in the cytoplasm and proximal part of apical dendrites of neurons. E, double staining of ERG3 (red) and GAD67 (green) in entorhinal cortex and hippocampal DG and CA1 regions of mice. Scale bar, 10 μm. Arrows show magnified images, indicating most ERG3 positive cells lack staining for GAD67. F, IF staining of ERG3‐overexpressing CHO cells with the ERG3 antibody and the antibody incubated with control antigen. IF and image capturing conditions are identical in these two groups. G, quantification of real‐time RT qPCR experiment of hippocampal tissues obtained from control mice. n = 9 from 9 mice; n.s., P > 0.05; * P < 0.05; ** P < 0.01; ordinary one‐way ANOVA with Tukey's multiple comparison test. Data are presented as means ± SEM.
Furthermore, double‐labelling IF showed ERG3‐positive cells typically expressed NeuN (a neuron marker) in hippocampal CA1 and DG areas (Fig. 1 D), suggesting that ERG3 protein was present in neurons. Occasionally, we found a few ERG3 positive cells were also positive for GAD67 (a GABAergic neuron marker) in DG and entorhinal cortex areas (Fig. 1 E). However, most ERG3‐positive neurons did not show staining for GAD67 protein, suggesting ERG3 protein was predominately expressed in excitatory neurons. Moreover, in agreement with a previous report (Guasti et al. 2005), ERG3 staining was most intense in the proximal apical dendrites and soma, which is clearly seen in the CA1 region (Fig. 1 D). Consistent with the IHC experiment, CHO cells overexpressing ERG3 showed strong staining with ERG3 antibody in the IF experiment, but the staining was absent using the antibody incubated with control antigen (Fig. 1 F). This result further confirmed that this antibody is specific to ERG3 protein. In addition, RT‐qPCR experiments showed that the ERG3 mRNA level was significantly higher than those of ERG1 and ERG2 in hippocampus (Fig. 1 G), which is in accordance with a prior study (Polvani et al. 2003). In conclusion, ERG3 protein has a high expression level in hippocampal areas and mainly localizes to soma and proximal apical dendrites.
Knockdown of ERG3 channels enhances neuronal intrinsic excitability
To examine whether ERG3 channels are involved in regulating neuronal intrinsic excitability, three different ERG3 short hairpin RNAs (ERG3‐shRNA) were designed to suppress the expression level of ERG3 protein, and the most effective one was used in our experiments (Fig. 2 A). Using stereotactic micro‐injection of AAV into the hippocampal DG or CA1 region of 6‐ to 8‐week‐old mice, ERG3‐shRNA was expressed in vivo in the hippocampal CA1 or DG region from 7 DPI and lasted at least 2 months, during which period the viruses spread mediolaterally 0.71–0.92 mm and anteroposteriorly 1.05–1.40 mm; about 60% of neurons were successfully transduced (Fig. 2 B and C). The efficiency of ERG3 silencing was confirmed by western blots and whole‐cell voltage‐clamp recordings. Western blot assay suggested that of all three shRNAs, the most effective ERG3‐shRNA caused an 85.9 ± 2.0% decrease of ERG3 protein in the ERG3 overexpressing HEK‐293 cells, and thus this shRNA was used in other experiments. A 56 ± 9% decrease of functional ERG‐mediated currents was observed in ERG3‐shRNA‐transduced mouse hippocampal neurons (Fig. 2 E–G), suggesting that a (unknown) proportion of the ERG3 channels were down‐regulated by AAV‐carried ERG3‐shRNA. We next investigated whether knockdown of ERG3 protein had any effect on the neuronal excitability of hippocampal neurons. For this purpose, whole‐cell current‐clamp recordings were used in mouse brain slices transduced with non‐silencing shRNA or ERG3‐shRNA expressing virus on 14–21 DPI. Hippocampal DG granule cells and CA1 pyramidal neurons were identified by their localization, morphological properties and input resistances (R N) of 244.9 ± 14 MΩ (n = 8) for DG granule cells and 103.9 ± 8.7 MΩ for CA1 pyramidal neurons (n = 10) (Fig. 3 A), which was consistent with previous reports (Lubke et al. 1998; Dyhrfjeld‐Johnsen et al. 2007). Interestingly, neurons transduced with ERG3‐shRNA exhibited a significant increase in the number of action potentials evoked by depolarizing current injections (Fig. 3 B), suggesting ERG3 knockdown clearly increased neuronal excitability. We further performed current‐clamp recordings to measure the minimum positive current injection required to induce a single AP in hippocampal DG granule cells and CA1 pyramidal neurons. The results showed that infection with virus expressing ERG3‐shRNA significantly reduced the delay time to the initiation of an action potential (Fig. 4 D). The longer delay time in control neurons could be caused by the sub‐threshold tonic opening of ERG3 channels. In addition, the fast AHP was also investigated by applying 400 ms depolarizing current injections of 200 pA for DG granule cells and 300 pA for CA1 pyramidal neurons. Current‐clamp recordings showed that the absolute values of fast AHP for the second, third and fourth action potentials in a train were smaller in ERG3‐shRNA‐transduced neurons, while the fast AHP of the first action potential was not changed (Fig. 4 E). Other intrinsic membrane properties of neurons were not affected (Tables 1 and 2). This phenomenon could be caused by the unique gating kinetics of ERG3 channels (Sanguinetti et al. 1995; Smith et al. 1996), in that their inactivation rate is faster than the activation rate so that the channel mainly opens in the repolarization phase, which contributes to the fast AHP of neurons. The duration of the first action potential only activates a small fraction of ERG3‐mediated currents due to the slow activation and fast inactivation kinetics of ERG channels (Mauerhofer & Bauer, 2016), and thus the fast AHP of the first action potential was not altered.
Figure 2. Injection of AAV carrying ERG3 shRNA significantly decreased the expression of ERG3 channels.
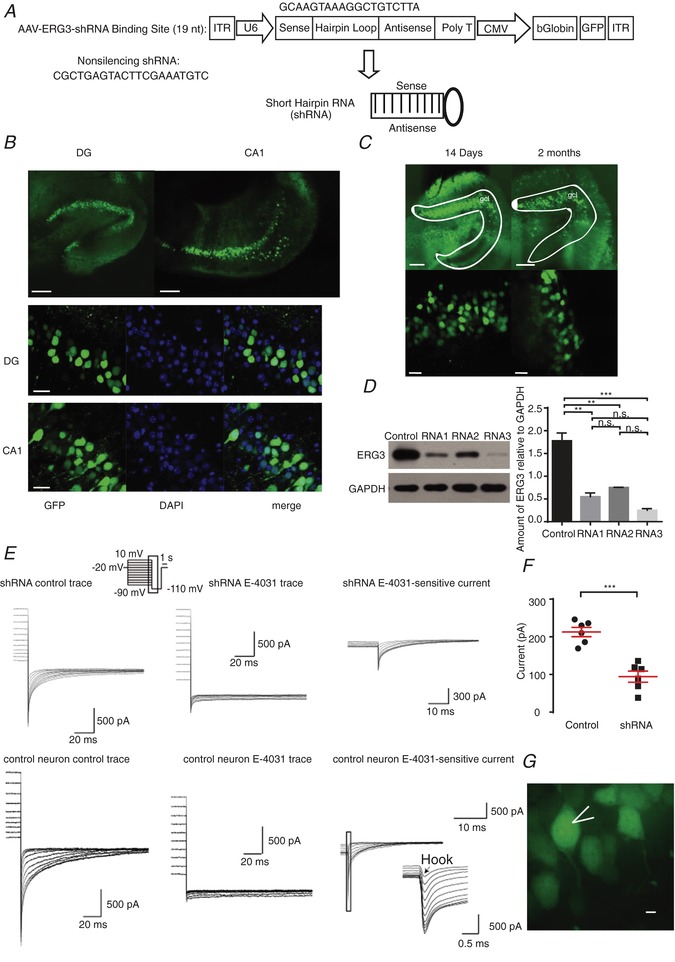
A, schematic diagram showing the lentiviral shRNA expression vector system. The 19‐nucleotide shRNA is specific to the mouse ERG3 mRNA. B, GFP fluorescence showing the expression of AAV in DG and CA1 areas. The rate of successful transduction was measured by the ratio of GFP positive cells to the DAPI positive cells. Scale bar, 100 μm (upper panels), 25 μm (lower panels). C, expression of AAV over time in the dorsal hippocampal DG area. Scale bar, 100 μm (upper panels), 20 μm (lower panels). gcl, DG stratum granulosum. D, western blotting and quantification showing ERG3 expression levels were reduced after the injection of ERG3‐shRNA. n.s., P > 0.05; ** P < 0.01; *** P < 0.001; ordinary one‐way ANOVA. The protein level of shRNA no. 3 group showed a significant decrease compared with control group, and the significance level was P < 0.001 (Dunnett's and Tukey's multiple comparison test). E, representative ERG current traces recorded in different cells from control mice and mice transduced with ERG3‐shRNA. The ERG‐mediated current was obtained by subtracting the E‐4031 (ERG channel blocker) trace from control trace. Note that the ERG current is significantly smaller in mice transduced with ERG3‐shRNA than control mouse. F, quantification of the tail current amplitudes recorded in different cells from control mice and mice transduced with ERG3‐shRNA. The amplitude is calculated by subtracting the tail current from the steady state current and the tail current is measured at 5 ms after the voltage was changed to −110 mV. n = 6 neurons from 5 control mice; n = 6 neurons from 3 ERG3‐shRNA injected mice. *** P < 0.001, unpaired two‐tailed Student's t test. G, representative morphological images of ERG3‐shRNA‐transduced rat DG neurons. Scale bar, 5 μm. Data are presented as means ± SEM.
Figure 3. Knocking down ERG3 protein significantly enhanced neuronal intrinsic excitability.
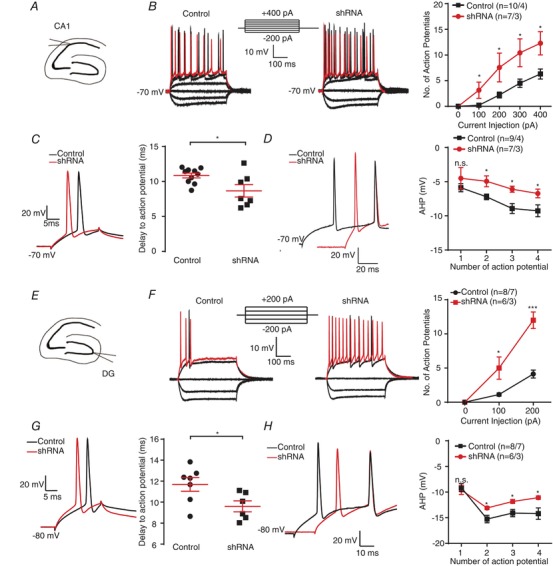
A, schematic diagram of CA1 region. B, representative current‐clamp recordings obtained from normal control (left) and ERG3‐shRNA tranduced mouse CA1 neurons (right). A series of 400 ms hyperpolarizing and depolarizing steps in 100 pA increments were applied to produce the traces. For comparison, the cells were held at −70 mV membrane potential. The mean numbers of action potentials generated in the response of depolarizing current pulses are shown in the right panel. The 400 pA current trace is shown in red. These data were recorded from 10 neurons from 4 control mice, and 7 neurons from 3 ERG3‐shRNA injected mice. * P < 0.05, unpaired two‐tailed Student's t test. C, typical action potential traces (left) and the delay to action potential (right) obtained from control and ERG3‐shRNA‐transduced mouse CA1 neurons. n = 10 neurons from 4 control mice; n = 7 neurons from 3 ERG3‐shRNA injected mice. * P < 0.05, unpaired two‐tailed Student's t test. D, representative traces of CA1 neurons from normal control and ERG3‐shRNA‐transduced mice in response to 300 pA positive current injection (left). The averaged fast afterhyperpolarization (AHP) of first to fourth action potential recorded in CA1 neurons from normal control and ERG3‐shRNA‐transduced mice (right). n = 9 neurons from 4 control mice; n = 7 neurons from 3 ERG3‐shRNA injected mice. * P < 0.05, unpaired two‐tailed Student's t test. E, schematic diagram of DG region. F, representative current‐clamp recordings obtained from normal control (left) and ERG3‐shRNA‐transduced mouse DG neurons (right). A series of 400 ms hyperpolarizing and depolarizing steps in 100 pA increments were applied to produce the traces. For comparison, the cells were held at −80 mV membrane potential. The mean number of action potentials generated in the response of depolarizing current pulses is shown in the right panel. The 200 pA current trace is shown in red. n = 8 neurons from 7 control mice; n = 6 neurons from 3 ERG3‐shRNA injected mice. * P < 0.05, *** P < 0.001, unpaired two‐tailed Student's t test. G, typical spikes (left) and the delay to action potential (right) obtained from control and ERG3‐shRNA‐transduced mouse DG neurons. n = 7 neurons from 6 control mice; n = 6 neurons from 3 ERG3‐shRNA injected mice. * P < 0.05, unpaired two‐tailed Student's t test. H, left, representative traces of DG neurons from normal control and ERG3‐shRNA‐transduced mice in response to 200 pA positive current injection. Right, the averaged fast afterhyperpolarization (AHP) of first to fourth action potential recorded in CA1 neurons from normal control and ERG3‐shRNA infected mice. n = 8 neurons from 7 control mice; n = 6 neurons from 3 ERG3‐shRNA injected mice. * P < 0.05, unpaired two‐tailed Student's t test. Data are presented as means ± SEM. [Color figure can be viewed at http://wileyonlinelibrary.com]
Figure 4. Pharmacological blockade of ERG channel enhances neuronal excitability in DG granule cells.
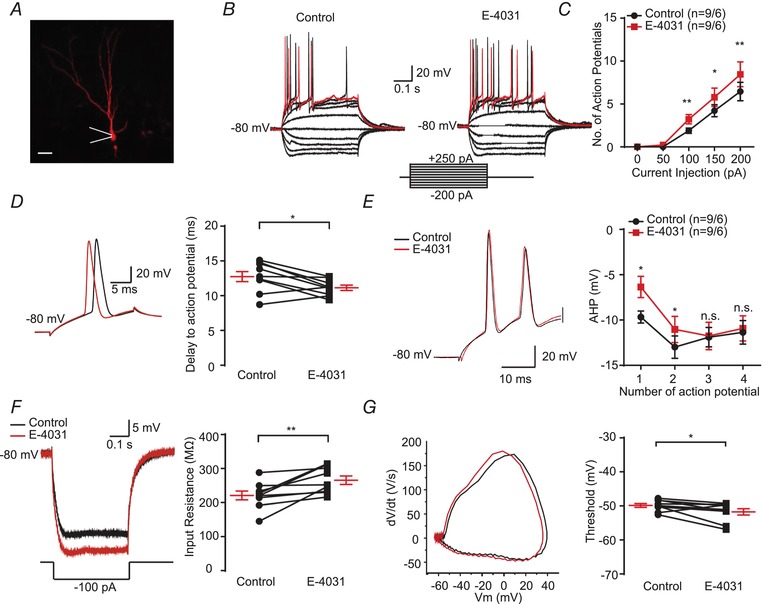
A, example of DG granule cell that was labelled with Neurobiotin. Scale bar, 25 μm. B and C, representative current‐clamp recordings obtained from mouse DG neurons with (right) or without (left) the presence of E‐4031. A series of 400 ms hyperpolarizing and depolarizing steps in 50 pA increments were applied to produce the traces. For comparison, the cells were held at −80 mV membrane potential. The mean number of action potentials generated in the response of depolarizing current pulses is shown in the right panel. The 250 pA current trace is shown in red. n = 9 neurons from 6 mice were recorded. * P < 0.05, ** P < 0.01, paired two‐tailed Student's t test. D, typical spikes (left), and quantification of the delay to action potential recorded in mouse DG neurons with or without the presence of E‐4031 (right). n = 9 neurons from 6 mice were recorded. * P < 0.05, paired two‐tailed Student's t test. E, left, representative traces of DG neurons with or without the presence of E‐4031 in response to 200 pA positive current injection. Right, the averaged afterhyperpolarization (AHP) of first to fourth action potential recorded in DG neurons with or without the presence of E‐4031 (right). n = 9 neurons from 6 mice were recorded. * P < 0.05, paired two‐tailed Student's t test. F, typical trace (left) and the input resistance (right) obtained in mouse DG neurons with or without the presence of E‐4031. n = 9 neurons from 6 mice were recorded. ** P < 0.01, paired two‐tailed Student's t test. G, associated phase plane plots of the single action potential (left) and the average spike threshold (right) obtained in mouse DG neurons with or without the presence of E‐4031. n = 9 neurons from 6 mice were recorded. * P < 0.05, paired two‐tailed Student's t test. Data are presented as means ± SEM. [Color figure can be viewed at http://wileyonlinelibrary.com]
Table 1.
Electrophysiological properties of CA1 pyramidal neurons of mice
| Parameter | Wild‐type | ERG3‐shRNA |
|---|---|---|
| Action potential threshold (mV) | 50.2 ± 0.8 (n = 10) | −49.3 ± 1.2 (n = 7) |
| Amplitude (mV) | 108.0 ± 2.0 (n = 10) | 102.0 ± 3.0 (n = 7) |
| Input resistance (MΩ) | 108.7 ± 10 (n = 10) | 103.9 ± 8.7 (n = 7) |
| Delay to action potential (ms) | 10.9 ± 0.4 (n = 10) | 8.7 ± 0.9 (n = 7)* |
| First action potential fast AHP (mV) | −5.9 ± 0.6 (n = 9) | −4.5 ± 1.6 (n = 7) |
| Second action potential fast AHP (mV) | −7.2 ± 0.4 (n = 9) | −5.0 ± 0.8 (n = 7)* |
| Third action potential fast AHP (mV) | −9.3 ± 0.9 (n = 7) | −6.7 ± 0.6 (n = 6)** |
| Forth action potential fast AHP (mV) | −4.5 ± 1.6 (n = 9) | −5.9 ± 0.6 (n = 7)* |
| Resting membrane potential (mV) | −70.1 ± 1.2 (n = 7) | −68.3 ± 1.3 (n = 11) |
| Half‐width of action potential (ms) | 1.96 ± 0.12 (n = 10) | 1.84 ± 0.11 (n = 11) |
| Membrane time constant (ms) | 31.6 ± 3.2 (n = 6) | 27.5 ± 2.5 (n = 10) |
Data are reported as means ± SEM for cells of CA1 pyramidal neurons of mice. * P < 0.05, ** P < 0.01, Student's t test.
Table 2.
Electrophysiological properties of DG granule cells of mice
| Parameter | Wild‐type | ERG3‐shRNA |
|---|---|---|
| Action potential threshold (mV) | −51.4 ± 0.7 (n = 8) | −49.6 ± 1.3 (n = 6) |
| Amplitude (mV) | 117.5 ± 1.6 (n = 8) | 115.6 ± 2.3 (n = 6) |
| Input resistance (MΩ) | 244.9 ± 14 (n = 8) | 215.3 ± 12 (n = 6) |
| Delay to action potential (ms) | 11.7 ± 0.7 (n = 7) | 9.6 ± 0.5 (n = 6)* |
| First action potential fast AHP (mV) | −9.1 ± 0.8 (n = 8) | −9.6 ± 0.9 (n = 6) |
| Second action potential fast AHP (mV) | −12.3 ± 0.7 (n = 8) | −13.1 ± 0.3 (n = 6)* |
| Third action potential fast AHP (mV) | −14.1 ± 0.7 (n = 8) | −11.9 ± 0.4 (n = 6)* |
| Forth action potential fast AHP (mV) | −14.2 ± 1.1 (n = 7) | −11.1 ± 0.4 (n = 6)* |
| Resting membrane potential (mV) | −76.9 ± 1.2 (n = 9) | −78.0 ± 1.7 (n = 7) |
| Half‐width of action potential (ms) | 1.73 ± 0.09 (n = 8) | 1.71 ± 0.15 (n = 6) |
| Membrane time constant (ms) | 14.0 ± 1.3 (n = 8) | 13.7 ± 1.1 (n = 5) |
Data are reported as means ± SEM for cells of DG granule cells of mice. * P < 0.05, Student's t test.
To further confirm the function of ERG3 channels in neuronal excitability, a pharmacological blocker of ERG channels, E‐4031, was applied. Comparably, increased neuronal excitability and decreased delay time to the initiation of an action potential were also observed in the presence of the ERG channel blocker, E‐4031 (Fig. 4 A–E). Moreover, bath‐application of 10 μm E‐4031 also increased neuronal input resistance and decreased threshold of the first action potential in response to current injection (Fig. 4 F and G; Table 3). The differences between the effects caused by pharmacological blockade of ERG channels and ERG3‐shRNA knockdown could be due to several reasons. Firstly, the ERG3‐shRNA‐transduced neurons could have compensatory changes in the expression level of other ion channels, and thus the compensation could reduce the effect of ERG3‐shRNA on neuronal intrinsic excitability. Besides, the E‐4031 is not specific to ERG3 channels and also blocks ERG1‐ and ERG2‐mediated potassium channels and other channels (Charpentier et al. 1998; Li et al. 2007). Therefore, E‐4031 may exert broader effects on intrinsic excitability than that of shRNA knockdown. In conclusion, the above data indicated that ERG3 channels inhibit neuronal intrinsic excitability in hippocampal DG granule cells and CA1 pyramidal neurons.
Table 3.
Electrophysiological properties of DG granule cells of mice with the presence of E‐4031
| Parameter | Control | E‐4031 |
|---|---|---|
| Action potential threshold (mV) | −49.9 ± 0.5 (n = 9) | −51.8 ± 1.0 (n = 9)* |
| Amplitude (mV) | 115.9 ± 2.3 (n = 9) | 111.2 ± 2.3 (n = 9) |
| Input resistance (MΩ) | 221.0 ± 13 (n = 9) | 265.7 ± 13 (n = 9)** |
| Delay to action potential (ms) | 12.7 ± 0.7 (n = 9) | 11.2 ± 0.4 (n = 9)* |
| First action potential fast AHP (mV) | −9.7 ± 0.7 (n = 9) | −6.4 ± 1.2 (n = 9)** |
| Second action potential fast AHP (mV) | −13.0 ± 1.2 (n = 9) | −11.0 ± 1.4 (n = 9)* |
| Third action potential fast AHP (mV) | −11.9 ± 1.1 (n = 9) | −11.8 ± 1.5 (n = 9) |
| Forth action potential fast AHP (mV) | −11.4 ± 1.3 (n = 9) | −10.9 ± 1.4 (n = 9) |
| Resting membrane potential (mV) | −80.2 ± 0.15 (n = 7) | −80.0 ± 0.04 (n = 7) |
| Half‐width of action potential (ms) | 1.67 ± 0.07 (n = 9) | 1.73 ± 0.08 (n = 9) |
| Membrane time constant (ms) | 17.3 ± 0.7 (n = 5) | 17.1 ± 1.1 (n = 5) |
Data are reported as means ± SEM for DG granule cells of mice. * P < 0.05, ** P < 0.01, Student's t test.
Knockdown of ERG3 channels enhances temporal summation of EPSPs
The temporal summation of EPSPs is crucial in regulating neuronal intrinsic excitability (Magee, 1999; Shah et al. 2004; Williams & Mitchell, 2008; Huang et al. 2009; Stuart & Spruston, 2015). Since ERG3 protein is mainly localized to the proximal apical dendrites and soma, we sought to investigate whether ERG3 channels were involved in regulating the temporal summation of somatic EPSPs. Artificial EPSPs were simulated by current injection of a defined α function (referred to as αEPSP). Compared with control neurons, ERG3‐shRNA‐transduced neurons in both CA1 pyramidal neurons and DG granule cells showed a significantly slower decay time constant (τ), suggesting that temporal summation of EPSPs was enhanced in ERG3‐knockdown neurons (Fig. 5 A and E). In addition, current injection of a 20 Hz train of αEPSPs showed the summation ratio was significantly higher in ERG3‐shRNA‐transduced neurons than control neurons (Fig. 5 B and F). Consistently, the pharmacological blockade of ERG channels with E‐4031 also showed elevated decay time constant and summation ratio in hippocampal DG granule cells (Fig. 5 C and D). Besides, in order to see if ERG3 channels are involved in the regulation of evoked EPSPs, we used a tungsten electrode to stimulate the dendrites of DG granule cells and CA1 pyramidal neurons to record the evoked EPSPs in the soma of these neurons. Results showed that ERG3‐shRNA‐transduced neurons showed an elevated decay time constant of evoked EPSCs compared to the control neurons, suggesting ERG3 channels regulate EPSP propagation from dendrites to soma in these neurons (Fig. 5 G and H). Together, these results suggested that genetic knockdown of ERG3 channels or pharmacological blockade of ERG channels enhances EPSP temporal summation, and thus contributes to the enhancement of neuronal intrinsic excitability.
Figure 5. ERG3 deficiency leads to increased EPSPs summation.
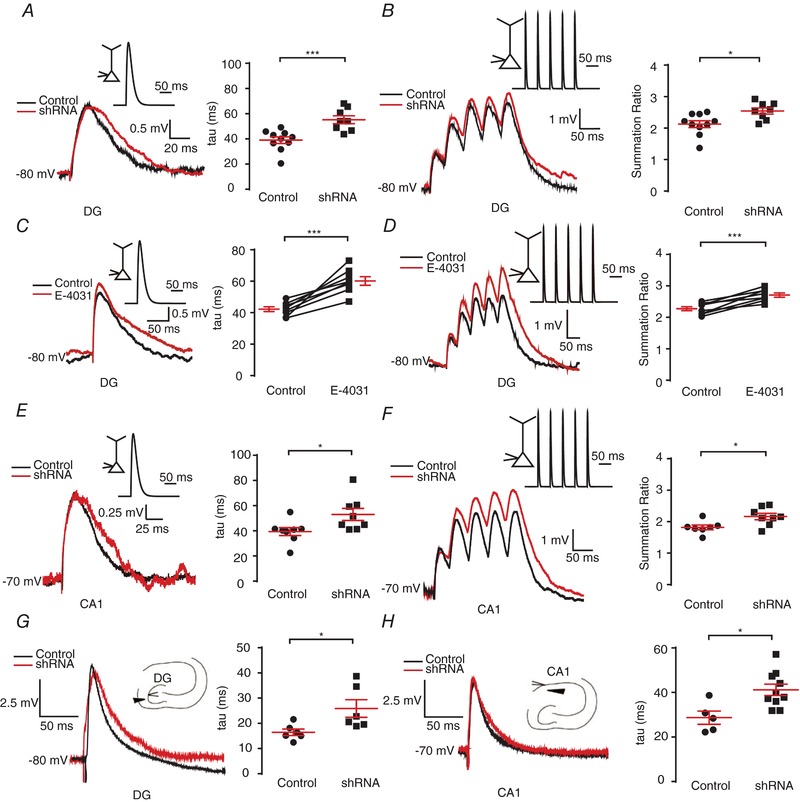
A and B, single (A) and 20 Hz trains (B) of αEPSPs were generated by current injection (see Methods) at the soma of DG neurons from normal control and ERG3‐shRNA‐transduced mice. All recordings were obtained at a fixed potential of −80 mV. The average decay time constant (τ, obtained by fitting a two‐exponential equation to the decay phase; see Methods) of single αEPSPs and the mean summation ratio (ratio of amplitude of 5th αEPSP to 1st αEPSP) of 20 Hz trains of αEPSPs obtained in normal control and ERG3‐shRNA‐transduced mice are shown in the panels on the right. Each trace is an average of 3 traces. n = 10 neurons from 6 control mice; n = 8 neurons from 3 ERG3‐shRNA‐injected mice. * P < 0.05, *** P < 0.001, unpaired two‐tailed Student's t test. C and D, single (C) and 20 Hz trains (D) of αEPSPs were generated by current injection (see Methods) at the soma of DG neurons with or without the presence of E‐4031. All recordings were obtained at a fixed potential of −80 mV. The average decay time constant (τ, obtained by fitting a two‐exponential equation to the decay phase) and amplitude (obtained by fitting a two‐exponential equation to the decay phase) of single αEPSPs and the mean summation ratio (ratio of amplitude of 5th αEPSP to 1st αEPSP) of 20 Hz trains of αEPSPs obtained with or without the presence of E‐4031 are shown on the panels on the right. Each trace is an average of three traces. n = 7 neurons from 5 mice were recorded. *** P < 0.001, paired two‐tailed Student's t test. E‐F, Single (E) and 20 Hz (F) trains of αEPSPs were generated by current injection (see Methods) at the soma of CA1 neurons from normal control and ERG3‐shRNA‐transduced mice. All recordings were obtained at a fixed potential of −70 mV. The average decay time constant (τ, obtained by fitting a two‐exponential equation to the decay phase; see Methods) of single αEPSPs and the mean summation ratio (ratio of amplitude of 5th αEPSP to 1st αEPSP) of 20 Hz trains of αEPSPs obtained in normal control and ERG3‐shRNA‐transduced mice are shown in the panels on the right. Each trace is an average of 3 traces. n = 8 neurons from 3 control mice; n = 7 neurons from 3 ERG3‐shRNA‐injected mice. * P < 0.05, unpaired two‐tailed Student's t test. G, fibre stimulation‐evoked EPSP in DG granule cells (left), and the quantification of decay time constant (right). The relative position of stimulation electrode and recorded neuron is shown in the schematic diagram. Each trace is an average of 10 traces. The decay time constants were obtained by fitting single‐exponential equation to the decay phase. * P < 0.05, unpaired two‐tailed Student's t test. H, fibre stimulation‐evoked EPSP in CA1 pyramidal neurons (left), and the quantification of decay time constant (right). The relative position of stimulation electrode and recorded neuron is shown in the schematic diagram. Each trace is an average of 10 traces. The decay time constants were obtained by fitting single‐exponential equation to the decay phase. * P < 0.05, unpaired two‐tailed Student's t test. Data are presented as means ± SEM. [Color figure can be viewed at http://wileyonlinelibrary.com]
The expression of ERG3 protein is reduced in human and mouse hippocampal epileptogenic foci
Having established that knockdown of ERG3 channels or pharmacological block of ERG channels effectively increased neuronal intrinsic excitability, we next examined whether ERG3 channels play a role in epilepsy because previous reports have indicated that the ERG channel corresponds with the previously identified ‘seizure gene’, mutations of which lead to hyperactivity in the motor system and thus cause a temperature‐sensitive paralytic phenotype (Jackson et al. 1984; Kasbekar et al. 1987; Titus et al. 1997; Wang et al. 1997). Firstly, we examined the expression level of ERG3 protein in human hippocampal epileptogenic foci. Western blots of paired epileptogenic hippocampal tissues and matched adjacent normal tissues showed expression of ERG3 protein was significantly reduced in epileptogenic tissues (Fig. 6 A and Table 4). Besides, ERG3 antibody incubated with control antigen could hardly detect ERG3 protein any more in a western blot experiment, suggesting that the ERG3 antibody is specific to ERG3 protein (Fig. 6 B). Furthermore, immunostaining experiments confirmed that the reduced expression of ERG3 protein occurred at the single neuronal level in the epileptic hippocampus (Fig. 6 C).
Figure 6. The expression of the ERG3 protein is down‐regulated in temporal lobe epilepsy.
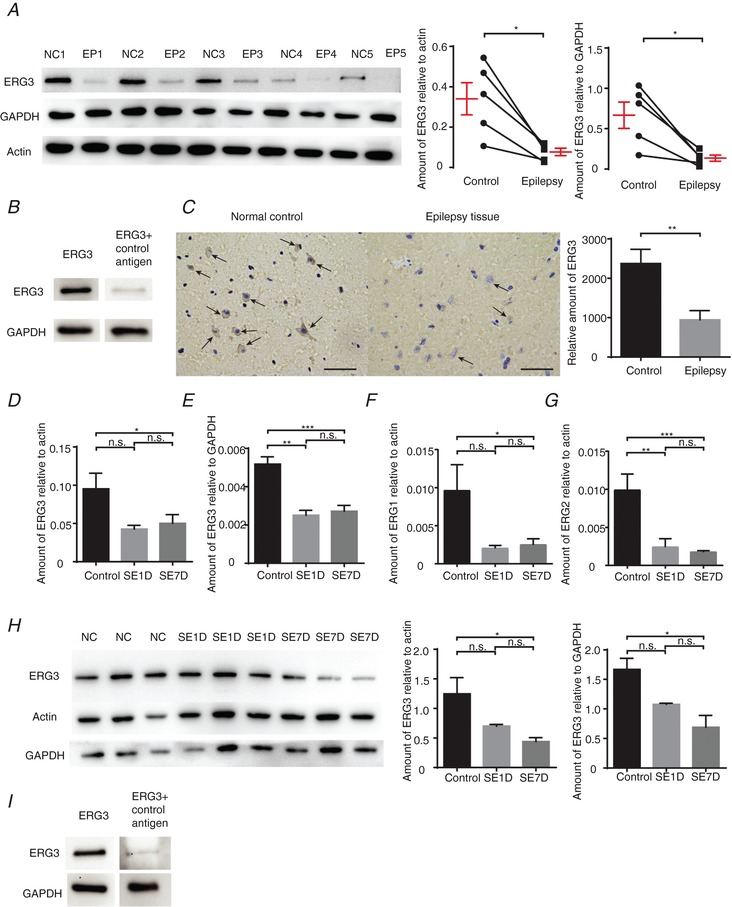
A, left, western blot analysis of ERG3 protein in the hippocampus from human epilepsy patient (EP) hippocampal tissues and adjacent normal control (NC) tissues. Right, quantification of the results by normalizing the protein levels of ERG3 to that of GAPDH and actin in EP and NC groups (n = 5). * P < 0.05, paired two‐tailed Student's t test. B, western blot analysis of ERG3 protein in the hippocampus from human patients with ERG3 antibody and control antigen. C, IHC results showing ERG3 protein was decreased in the human epilepsy tissues and adjacent normal control tissues (left), and quantification (right). Scale bar, 50 μm. n = 3, ** P < 0.01, paired two‐tailed Student's t test. Arrows suggest ERG3 protein is expressed in the cytoplasm of neurons. D and E, hippocampal tissues were obtained from control mice or kainic model mice 1 day after status epilepticus (SE1D) or 7 days after status epilepticus (SE7D). For these tissues, the mRNA and protein levels of ERG3 were examined by real‐time RT‐PCR, and the ERG3 mRNA level had decreased compared with GAPDH (right) and actin (left). n = 3 from 3 mice in control group; n = 5 from 5 mice in SE1D group; n = 4 from 4 mice in SE7D group. n.s., P > 0.05; * P < 0.05; ** P < 0.01; *** P < 0.001, ordinary one‐way ANOVA with Tukey's multiple comparison test. F and G, mouse tissue was also examined by real‐time RT‐PCR for the mRNA level of ERG1 (F) and ERG2 (G). n = 9 from 9 mice were used in each group. n.s., P > 0.05; * P < 0.05; ** P < 0.01; *** P < 0.001; ordinary one‐way ANOVA with Tukey's multiple comparison test. H, mouse tissue was also examined by western blot analysis (left) and quantified (right). n = 3 from 3 mice were used in each group. n.s., P > 0.05; * P < 0.05; ordinary one‐way ANOVA with Tukey's multiple comparison test. I, western blot analysis of ERG3 protein in the mouse tissue with ERG3 antibody and control antigen. Data are presented as means ± SEM. [Color figure can be viewed at http://wileyonlinelibrary.com]
In order to further confirm the phenomenon that ERG3 protein expression is decreased in epilepsy, we also measured the expression level of ERG3 protein in the mouse kainic acid (KA) epilepsy model, which is a commonly used animal preparation for studying temporal lobe epilepsy (Bernard et al. 2001; Smith & Dudek, 2002; Powell et al. 2008). In this model, a single episode of continuous seizures was induced by the administration of KA and terminated using sodium pentobarbital. Following a 2–4‐week seizure‐free period after the termination of the KA‐induced seizures, the animals developed a type of chronic temporal lobe epilepsy that mimics the prominent clinical and pathological features of the human disorder (Smith & Dudek, 2002). All experiments were done at two time points: 1 day (SE1D) and 7 days (SE7D) after the termination of seizures. These two time points represented the early and middle epochs during the latent period. Western blot and RT‐qPCR results showed that the triggering of seizures in mice led to a decrease of ERG3 mRNA at 1 day and 7 days after KA treatment (Fig. 6 D and E). Interestingly, the reduced mRNA level of ERG1 and ERG2 were also observed in SE1D and SE7D mice (Fig. 6 F and G). A western blot assay also showed that the ERG3 protein level was reduced in SE1D and SE7D mice compared with control mice (Fig. 6 H) and the antigen incubated antibody could barely detect the mouse ERG3 protein any more (Fig. 6 I). Together, these results suggested that the expression level of ERG3 protein is significantly reduced in human and mouse brain tissues with temporal lobe epilepsy, indicating ERG3 channels may be involved in epileptogenesis.
Functional ERG‐mediated currents were reduced in the hippocampus during epileptogenesis
To further confirm the phenomenon that ERG3 channel expression is reduced during epileptogenesis, we recorded ERG‐mediated currents in control and KA‐treated epileptic neurons. Whole‐cell voltage‐clamp recordings were made from hippocampal DG granule cells and CA1 pyramidal neurons in acute horizontal brain slices. The ERG‐mediated currents were obtained by subtraction of the currents resistant to E‐4031 from control currents (Fig. 7 A). Interestingly, the peak ERG currents in hippocampal DG neurons and CA1 neurons were persistently decreased at SE1D and SE7D neurons in comparison with control neurons (Fig. 7 B and D). The voltage dependence of ERG channel activation was determined using a series of 5 s depolarizing pulses between −90 and +10 mV, applied from a holding potential of −20 mV and the tail currents were measured at −110 mV. There were no significant differences in activation properties of ERG‐mediated currents between SE1D neurons, SE7D neurons and control neurons (Fig. 7 C and E), suggesting a persistent reduction in amplitude of ERG‐mediated currents in hippocampal DG neurons during epileptogenesis which was independent of the channel activation properties. In addition, we assessed the steady‐state inactivation of ERG channelsby hyperpolarizing neurons to a series of voltages from a holding potential of +40 mV (Fig. 7 F). Intriguingly, the inactivation kinetics recorded in control group, SE1D and SE7D did not show any significant differences (Fig. 7 G). Together, these results showed the functional ERG‐mediated currents were reduced during the latent period of KA model mice, which is mainly due to the reduced expression of ERG protein.
Figure 7. Functional ERG‐mediated current is significantly reduced in kainic acid‐induced epilepsy model mouse neurons.
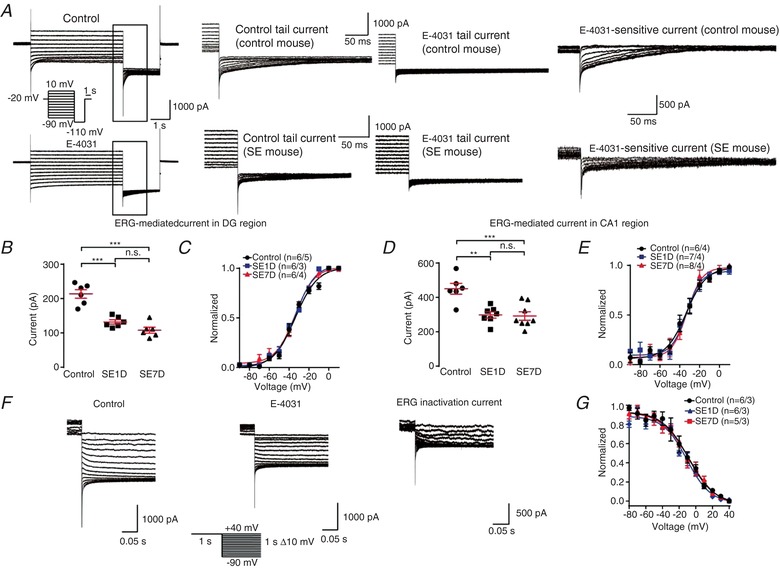
A, representative ERG current traces recorded in different cells from control mice and kainic acid model (SE) mice. From a holding potential of −20 mV, channel activation was assessed by 4 s test pulses between −90 and +10 mV with 10 mV increments, followed by a constant pulse to −110 mV (see pulse diagram). The ERG‐mediated current was obtained by subtracting the E‐4031(ERG channel blocker) trace from control trace. Note that the ERG current is significantly smaller in SE mouse than control mouse. B, quantification of the tail current amplitudes recorded in DG region. The amplitude was calculated by substracting the tail current from the steady state current and the tail current was measured at 5 ms after the voltage was changed to −110 mV. The ERG currents were calculated by subtracting the current 5 ms after the time of voltage change from the steady state current. n = 5 neurons from 5 control mice; n = 5 neurons from 3 SE1D mice; n = 6 neurons from 4 SE7D mice. n.s., P > 0.05; *** P < 0.001; ordinary one‐way ANOVA with Tukey's multiple comparison test. C, tail current amplitudes recorded in DG region plotted against the potential of the preceding test pulse. Data were normalized to the maximum tail current amplitudes, averaged and fitted with a Boltzmann function to yield activation curves. The V 0.5 for the control, SE1D and SE7D groups are −33.65 ± 1.37, −33.74 ± 0.89 and −33.97 ± 1.36 mV, respectively. There is no significant difference between three groups. n = 5 neurons from 5 control mice; n = 5 neurons from 3 SE1D mice; n = 6 neurons from 4 SE7D mice. D, quantification of the tail current amplitudes recorded in CA1 region. The amplitude was calculated by substracting the tail current from the steady state current and the tail current was measured at 5 ms after the voltage was changed to −110 mV. n = 6 neurons from 4 control mice; n = 7 neurons from 4 SE1D mice; n = 8 neurons from 4 SE7D mice. n.s., P > 0.05; ** P < 0.01; *** P < 0.001; ordinary one‐way ANOVA with Tukey's multiple comparison test. E, peak tail current amplitudes recorded in CA1 region plotted against the potential of the preceding test pulse. Data were normalized to the maximum tail current amplitudes, averaged and fitted with a Boltzmann function to yield activation curves. The V 0.5 for the control, SE1D and SE7D groups are −33.47 ± 1.97, −33.25 ± 1.72 and −32.67 ± 1.79 mV, respectively. There is no significant difference between the three groups. n = 6 neurons from 4 control mice; n = 7 neurons from 4 SE1D mice; n = 8 neurons from 4 SE7D mice. F, a representative of fully activated ERG currents recorded in different cells from control mice and kainic acid model (SE) mice. ERG currents were elicited by 1 s depolarizations to +40 mV, followed by variable 1 s test pulses from +40 to −90 mV with 10 mV decrements (see pulse diagram). G, normalized current amplitudes of the experiments were used to calculate the relative conductance as a measure of the voltage dependence of ERG steady‐state inactivation. Data were normalized to the conductance value at −90 mV before averaging and fitted with a Boltzmann function to yield inactivation curves. The V 0.5 for the control, SE1D and SE7D groups are −8.86 ± 2.73, −7.74 ± 3.40 and −10.63 ± 2.12 mV, respectively. There is no significant difference in fitting results between the three groups. n = 6 neurons from 3 control mice; n = 6 neurons from 3 SE1D mice; n = 5 neurons from 4 SE7D mice. Data are presented as means ± SEM. [Color figure can be viewed at http://wileyonlinelibrary.com]
In order to further confirm the consequences of down‐regulation of ERG3 protein in the latent period of epilepsy, we performed current‐clamp experiments from neurons obtained from SE7D DG granule cells. The ERG channel blocker E‐4031 had no effect on neuronal firing, fast AHP and temporal EPSP summation of SE7D neurons, indicating the ERG protein was already down‐regulated in SE7D neurons (Fig. 8 A–D). In conclusion, these results further confirmed that the expression of ERG channels was reduced in the latent period of temporal lobe epilepsy, and the reduced expression of ERG channels may facilitate the development of temporal lobe epilepsy.
Figure 8. The function of ERG channels was impaired in kainic model mice.
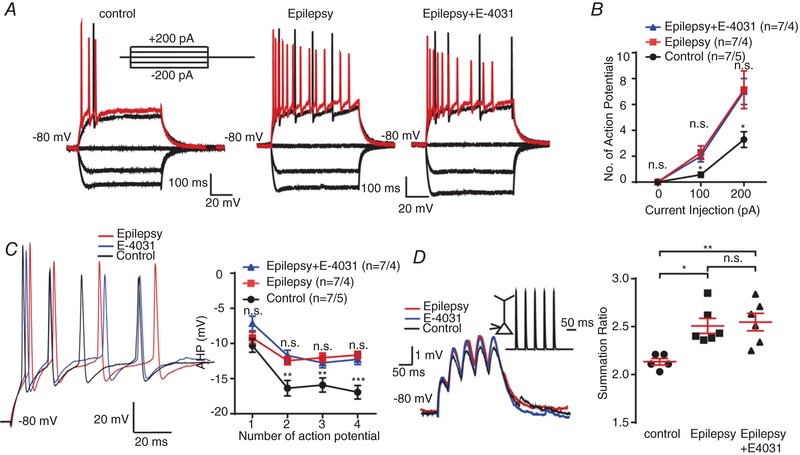
A and B, representative current‐clamp recordings obtained from control mouse (right) and epilepsy mouse DG granule cells with (left) or without (middle) the presence of E‐4031. A series of 400 ms hyperpolarizing and depolarizing steps in 100 pA increments were applied to produce the traces. For comparison, the cells were held at −80 mV membrane potential. The mean number of action potentials generated in the response of depolarizing current pulses is shown in B. n = 7 neurons from 4 epilepsy model mice; n = 7 neurons from 5 control mice. n.s., P > 0.05; * P < 0.05; one‐way ANOVA with Tukey's multiple‐comparison test. According to the multiple‐comparison test, the neuronal excitability did not alter after the application of E‐4031, while the ANOVA of control, epilepsy and epilepsy+E‐4031 groups showed significant change of neuronal excitability. C, left, representative traces of DG neurons from control mice (black) and epilepsy model mice with (blue) or without (red) the presence of E‐4031 in response to 200 pA positive current injection. Right, the quantification of averaged fast afterhyperpolarization (AHP) of first to fourth action potential recorded in control and epilepsy mouse DG granule cells (right panel). n = 7 neurons from 4 epilepsy model mice; n = 7 neurons from 5 control mice. n.s., P > 0.05; ** P < 0.01; *** P < 0.001; one‐way ANOVA with Tukey's multiple‐comparison test. According to multiple‐comparison test, the fAHP did not alter after the application of E‐4031, but in the second, third and fourth action potentials, the ANOVA of control, epilepsy and epilepsy+E‐4031 groups showed significant change of fAHP. D, 20 Hz trains of αEPSPs were generated by current injection at the soma of DG neurons in control (black) and epilepsy mouse DG granule cells with (blue) or without (red) the presence of E‐4031. All recordings were obtained at a fixed potential of −80 mV. The mean summation ratio (ratio of the amplitude of 5th αEPSP to 1st αEPSP) of 20 Hz trains of αEPSPs are shown in the panel on the right. Each trace is an average of three traces. n = 5 from 3 control mice; n = 6 neurons from 4 epilepsy model mice. n.s., P > 0.05; * P < 0.05; ** P < 0.01; ordinary one‐way ANOVA with Tukey's multiple‐comparison test. Data are presented as means ± SEM. [Color figure can be viewed at http://wileyonlinelibrary.com]
ERG3 channels suppress seizure susceptibility
In order to test if ERG3 channels are involved in epileptic encephalopathy, and further assess the pathophysiological consequences of reduced expression of ERG3 channels in vivo, we next examined whether knockdown of ERG3 channels would affect seizure susceptibility in animal models.
To investigate whether there were differences in seizure threshold between ERG3 knockdown and wild‐type mice, we used the KA epilepsy model to evaluate the seizure susceptibility of testing mice (Racine, 1972; Ben‐Ari & Cossart, 2000; Smith & Dudek, 2002). EEG was also employed to confirm the onset of status epilepticus (Fig. 9 A). In accordance with a previous report (Huang et al. 2009), 20 mg kg−1 KA was intraperitoneally administrated into the control and shRNA‐transduced mice. Results showed KA induced class 4 seizures (defined by the Racine scale) in wild‐type mice in 68 ± 9.2 min (n = 10), while the same concentration of KA caused class 4 seizures in 31 ± 9.2 min (n = 7) in ERG3 knockdown mice (Fig. 9 B). In addition, seizure progression was considerably faster in ERG3‐shRNA‐transduced mice, with more of them developing into tonic hindlimb extension compared to controls (Fig. 9 C and D). In order to assess the effect of ERG3 channel activation, we used 30 mg kg−1 KA to elicit a faster onset of seizure. Administration of the ERG channel enhancer, NS‐1643 (Bilet & Bauer, 2012) (30 min before the injection of KA), resulted in significantly slower seizure progression and reduced maximum seizure severity (Fig. 9 E–G). Clearly, these results suggested that altered expression or function of ERG3 channels in hippocampus had a substantial impact on the induction and development of acute epileptic seizures, indicating ERG3 channels could be a potential drug target to treat epilepsy.
Figure 9. ERG3 suppresses seizure susceptibility in temporal lobe epilepsy.
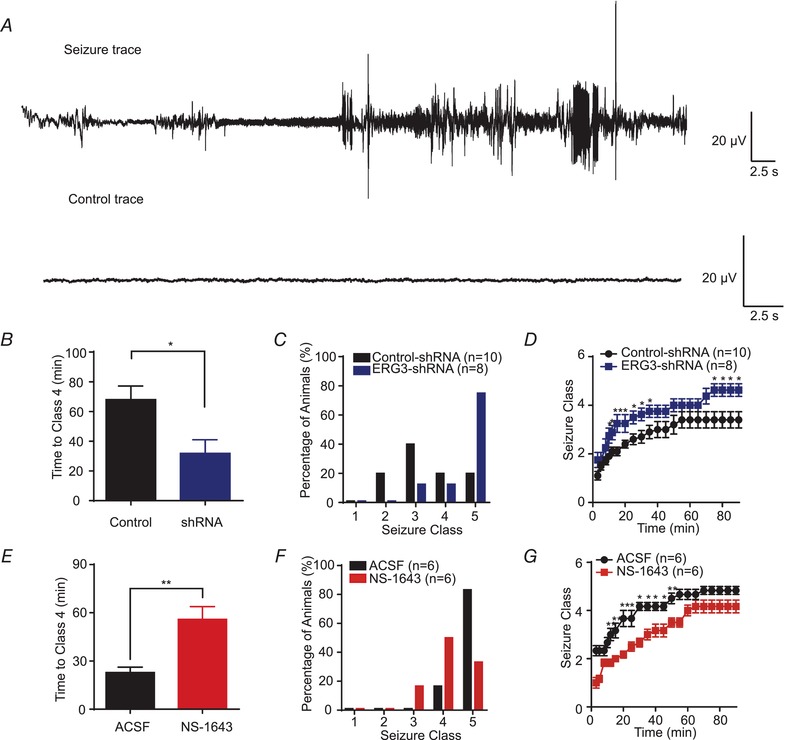
A, examples of in vivo EEG recordings obtained from ERG3‐shRNA‐transduced and wild‐type mice. B, graph depicting the time taken to reach class 4 seizure after kainic acid (KA) administration from ERG3‐shRNA‐transduced and wild‐type mice. * P < 0.05, unpaired two‐tailed Student's t test. C, the incidence of maximum seizure class reached during the course of the experiments in D. D, graph depicting the seizure progression in ERG3‐shRNA‐transduced and wild‐type mice, illustrated as mean maximum seizure class (defined by Racine scale, see Methods) reached in different time points after KA administration. * P < 0.05, unpaired two‐tailed Student's t test. E, graph depicting the time taken to reach class 4 seizure after KA administration from NS‐1643‐injected and wild‐type mice. ** P < 0.01, unpaired two‐tailed Student's t test. F, the incidence of maximum seizure class reached during the course of the experiments in G. G, graph depicting the seizure progression in NS‐1643 (ERG channel opener) injected and wild‐type mice, illustrated as mean maximum seizure class reached in different time points after KA administration. * P < 0.05; ** P < 0.01; unpaired two‐tailed Student's t test. Data are presented as means ± SEM. [Color figure can be viewed at http://wileyonlinelibrary.com]
Discussion
Despite the relatively high expression of ERG3 channels that has been found in the brain, their function in neuronal intrinsic excitability remains unclear (Guasti et al. 2005). Here, we demonstrate that ERG3 channels play an important role in regulating intrinsic excitability in hippocampal neurons (Fig. 3 B and H). ERG3‐shRNA‐transduced neurons show elevated neuronal excitability, which is associated with decreased action potential fast AHP, reduced time to initiate an action potential and enhanced somatic summation of EPSPs (Figs 3 C, D, G and H and 5 A–H). Consistently, pharmacological blockade of ERG channels also showed increased neuronal excitability in hippocampal neurons (Fig. 4 C). In addition, a reduced expression of ERG3 channels has been observed in both patient epileptic tissues and mouse epileptic tissues (Fig. 6 A and H) and functional ERG3‐mediated currents were decreased in hippocampal neurons of the mouse TLE model when compared to control neurons (Fig. 7 B–E). Finally, we have shown that ERG3‐shRNA‐transduced mice showed increased seizure susceptibility, and the ERG channel opener NS‐1643 decreased seizure susceptibility (Fig. 9 B–G). In conclusion, our results demonstrate that ERG3 channels have a key role in regulating neuronal intrinsic excitability in hippocampal neurons, indicating ERG3 channels could be a potential drug target to treat some forms of epilepsy.
The distribution of ERG3 channels in hippocampus
Our results showed the mRNA level of ERG3 is significantly higher than that of ERG1 and ERG2, which is collectively in accordance with in situ hybridization data gathered in the mouse CNS by Polvani et al. (2003). Therefore, we mainly focused on investigating the function of ERG3 channels in this study. Firstly, IF and IHC was used in order to examine the expression pattern of ERG3 protein in hippocampal neurons. The subcellular localization of ERG3 protein is closely associated with its function of regulating neuronal excitability. Although the action potential is generated in the axon initial segment (AIS), the neuronal soma also plays an important role in the initiation of an action potential, since the soma is electrotonically coupled with the AIS. When the expression of ERG3 protein was altered, the delay time to the initiation of an action potential could be influenced by a modified electrotonic spread pattern of the soma depolarization to the AIS (Kole & Stuart, 2012). More importantly, the fast speed of inactivation and slow speed of activation make ERG3 channels mainly function in the repolarization rather than depolarization phase of an action potential. This unique gating kinetics of ERG3 channels suggests that this channel is involved in fast AHP regulation, which can profoundly alter neuronal excitability. Furthermore, somatic EPSP summation also contributes to the change in neuronal excitability (Magee, 1999; Frick et al. 2004). The increased somatic EPSP summation indicates, at a given membrane potential, incoming synaptic potentials in dendrites are more likely to give rise to action potentials. Thus, by altering EPSP summation, ERG3 channels can also influence neuronal excitability. However, since ERG3 protein was also observed to localize in proximal dendrites, it is possible that ERG3 channels can also influence dendritic summation of EPSPs. Further experiments will be needed to investigate the changes caused by ERG3 channels in dendritic EPSP summation in order to test this hypothesis.
The ERG3 channels are involved in intrinsic plasticity of neurons
All CNS neurons have the ability to transform input information to an output signal. More importantly, neurons are capable of modifying their input–output properties in an activity‐dependent manner, and the modification of postsynaptic neuron firing in response to depolarization by EPSPs has been termed intrinsic plasticity (Zhang & Linden, 2003; Losonczy et al. 2008). Persistent changes in intrinsic excitability have been reported in several cases in which activity is regulated through ion channels. For instance, Kv4.2 channels, HCN1 channels, and Kv7 channels are all involved in regulating intrinsic properties of hippocampal neurons and are thereby connected with multiple physiological and pathological processes (Frick et al. 2004; Shah et al. 2004; Yue & Yaari, 2004). By regulating dendritic and somatic EPSP summation, action potential threshold and neuronal firing mode, these channels have a crucial part to play in regulating neuronal intrinsic excitability and are thereby related to epilepsy, neuropathic pain, and learning and memory (Bernard et al. 2001; Fan et al. 2005). In the case of ERG3 channels, our results have shown that this channel can regulate neuronal intrinsic excitability and somatic EPSP summation in hippocampal neurons. From this point of view, it is likely that ERG3 channels could also be involved in many other processes, such as neuropathic pain or learning and memory. We have shown that ERG3 channels have a crucial role in epilepsy, but whether this channel is linked with other brain functions remains unknown.
The ERG3 channels can be a potential drug target to treat epilepsy
Epilepsy is one of the most common and also heterogeneous neurological disorders. Despite the large number of patients, our knowledge of epilepsy is still very limited. Existing drugs can only control epilepsy in about 70% of patients, with 30% of patients developing drug‐resistant epilepsy (Tellez‐Zenteno & Hernandez‐Ronquillo, 2012). Therefore, it is imperative to find new drug targets to treat epilepsy. Since ion channels can profoundly alter neuronal excitability, it is possible to use ion channel modulators to reverse the hyperexcited state of neurons in epilepsy. For example, the Kv7 channel opener retigabine has been used as an anticonvulsant drug for the treatment of epilepsy (Cooper et al. 2000), indicating potassium channels can be promising drug targets to treat epilepsy.
Our results have shown that ERG3 channels play an important role in epileptogenesis, and ERG3‐shRNA and the ERG channel opener NS‐1643 can influence susceptibility to epilepsy. Still, we cannot exclude the possibility that the anti‐seizure effect of NS‐1643 is through the activation of ERG1 or ERG2, and ERG1 and ERG2 channels may also be involved in the epileptogenesis. But since ERG3 channels are the main subtype of ERG channels in hippocampus, the anti‐seizure effect of NS‐1643 may be mainly due to the activation of ERG3‐channels. In addition, in patient epileptic samples, reduced ERG3 channel expression has also been observed, indicating the effect of ERG3 channels is also applicable to humans rather than just animals. Therefore, ERG3 channels could be a potential drug target for epilepsy. However, if a drug is to be developed to treat epilepsy by targeting ERG3 channels, there are certain problems need to be conquered. Firstly, the drug needs to be selective to ERG3 and have no effect or little effect on ERG1, since it is well recognized that ERG1 channel modulators can change the heart rhythm and can be lethal (Sanguinetti & Tristani‐Firouzi, 2006). Secondly, we have only investigated the role of ERG3 in the hippocampus, and the change of ERG3 channels in other brain regions may be different; thus, an ERG3 channel modulator may also have other neurological effects. Nevertheless, our study suggests that ERG3 channels have a critical role in temporal lobe epilepsy, and targeting ERG3 channels could be a promising strategy for prevention and treatment of some types of epilepsy.
Additional information
Competing interests
The authors declare no competing financial interests.
Author contributions
K.X. performed and analysed immunostaining of hippocampus, morphological experiments, voltage clamp recordings, part of current‐clamp recordings, part of virus microinjections, RT‐qPCR and western blotting. Z.S. performed and analysed part of virus microinjections, kainic acid‐induced status epilepticus model and part of current clamp recordings. W.M. provided human epileptic tissues and helped with western blot experiments. Y.S. helped with the RT‐qPCR and western blot experiments and data analysis. S.L. and M.F. performed and analysed EEG recordings. X.J. and J.Z. helped with the recording of current clamp experiments. Z.H. designed the experiments. Z.H., K.X. and W.Y. wrote the paper. Q.C. performed part of RT‐qPCR assay. All authors have read and approved the final version of this manuscript and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by grants (973 Program: 2015CB559200 to Z.H.) from the Ministry of Science and Technology of China, grants (81371432 to Z.H.) from the National Natural Science Foundation of China and Beijing Natural Science Foundation (7182087).
Acknowledgements
We thank Dr Alasdair Gibb for valuable comments on this work. We also thank the support of Memprotein Ltd for the gift of ERG3 plasmid.
Biography
Kuo Xiao and Zhiming Sun received their bachelor's degrees in Peking University. Currently, they are working for their master's degree in Peking University. Their work is mainly concerned with ion channels and central nervous system diseases.

Edited by: Ole Paulsen & Katalin Toth
K. Xiao and Z. Sun contributed equally to this work.
References
- Aschauer DF, Kreuz S & Rumpel S (2013). Analysis of transduction efficiency, tropism and axonal transport of AAV serotypes 1, 2, 5, 6, 8 and 9 in the mouse brain. PLoS One 8, e76310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer CK & Schwarz JR (2018). Ether‐à‐go‐go K+ channels: effective modulators of neuronal excitability. J Physiol 596, 769–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck H & Yaari Y (2008). Plasticity of intrinsic neuronal properties in CNS disorders. Nat Rev Neurosci 9, 357–369. [DOI] [PubMed] [Google Scholar]
- Ben‐Ari Y & Cossart R (2000). Kainate, a double agent that generates seizures: two decades of progress. Trends Neurosci 23, 580–587. [DOI] [PubMed] [Google Scholar]
- Bernard C, Marsden DP & Wheal HV (2001). Changes in neuronal excitability and synaptic function in a chronic model of temporal lobe epilepsy. Neuroscience 103, 17–26. [DOI] [PubMed] [Google Scholar]
- Bilet A & Bauer CK (2012). Effects of the small molecule HERG activator NS1643 on Kv11.3 channels. PLoS One 7, e50886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caroni P, Donato F & Muller D (2012). Structural plasticity upon learning: regulation and functions. Nat Rev Neurosci 13, 478–490. [DOI] [PubMed] [Google Scholar]
- Charpentier F, Merot J, Riochet D, Le Marec H & Escande D (1998). Adult KCNE1‐knockout mice exhibit a mild cardiac cellular phenotype. Biochem Biophys Res Commun 251, 806–810. [DOI] [PubMed] [Google Scholar]
- Chen K, Aradi I, Santhakumar V & Soltesz I (2002). H‐channels in epilepsy: new targets for seizure control? Trends Pharmacol Sci 23, 552–557. [DOI] [PubMed] [Google Scholar]
- Cooper EC, Aldape KD, Abosch A, Barbaro NM, Berger MS, Peacock WS, Jan YN & Jan LY (2000). Colocalization and coassembly of two human brain M‐type potassium channel subunits that are mutated in epilepsy. Proc Natl Acad Sci U S A 97, 4914–4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui ED & Strowbridge BW (2017). Modulation of Ether‐à‐go‐go Related Gene (ERG) current governs intrinsic persistent activity in rodent neocortical pyramidal cells. J Neurosci 38, 423–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daoudal G & Debanne D (2003). Long‐term plasticity of intrinsic excitability: learning rules and mechanisms. Learning Mem 10, 456–465. [DOI] [PubMed] [Google Scholar]
- Dyhrfjeld‐Johnsen J, Santhakumar V, Morgan RJ, Huerta R, Tsimring L & Soltesz I (2007). Topological determinants of epileptogenesis in large‐scale structural and functional models of the dentate gyrus derived from experimental data. J Neurophysiol 97, 1566–1587. [DOI] [PubMed] [Google Scholar]
- Fan Y, Fricker D, Brager DH, Chen X, Lu HC, Chitwood RA & Johnston D (2005). Activity‐dependent decrease of excitability in rat hippocampal neurons through increases in I h . Nat Neurosci 8, 1542–1551. [DOI] [PubMed] [Google Scholar]
- Fano S, Caliskan G & Heinemann U (2012). Differential effects of blockade of ERG channels on gamma oscillations and excitability in rat hippocampal slices. Eur J Neurosci 36, 3628–3635. [DOI] [PubMed] [Google Scholar]
- Frick A, Magee J & Johnston D (2004). LTP is accompanied by an enhanced local excitability of pyramidal neuron dendrites. Nat Neurosci 7, 126–135. [DOI] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology . J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guasti L, Cilia E, Crociani O, Hofmann G, Polvani S, Becchetti A, Wanke E, Tempia F & Arcangeli A (2005). Expression pattern of the ether‐a‐go‐go‐related (ERG) family proteins in the adult mouse central nervous system: evidence for coassembly of different subunits. J Comp Neurol 491, 157–174. [DOI] [PubMed] [Google Scholar]
- Hardman RM & Forsythe ID (2009). Ether‐à‐go‐go‐related gene K+ channels contribute to threshold excitability of mouse auditory brainstem neurons. J Physiol 587, 2487–2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausser M, Spruston N & Stuart GJ (2000). Diversity and dynamics of dendritic signaling. Science 290, 739–744. [DOI] [PubMed] [Google Scholar]
- Hirdes W, Napp N, Wulfsen I, Schweizer M, Schwarz JR & Bauer CK (2009). Erg K+ currents modulate excitability in mouse mitral/tufted neurons. Pflugers Arch 459, 55–70. [DOI] [PubMed] [Google Scholar]
- Huang Z, Lujan R, Kadurin I, Uebele VN, Renger JJ, Dolphin AC & Shah MM (2011). Presynaptic HCN1 channels regulate Cav3.2 activity and neurotransmission at select cortical synapses. Nat Neurosci 14, 478–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Walker MC & Shah MM (2009). Loss of dendritic HCN1 subunits enhances cortical excitability and epileptogenesis. J Neurosci 29, 10979–10988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson FR, Wilson SD, Strichartz GR & Hall LM (1984). Two types of mutants affecting voltage‐sensitive sodium channels in Drosophila melanogaster . Nature 308, 189–191. [DOI] [PubMed] [Google Scholar]
- Jung S, Warner LN, Pitsch J, Becker AJ & Poolos NP (2011). Rapid loss of dendritic HCN channel expression in hippocampal pyramidal neurons following status epilepticus. J Neurosci 31, 14291–14295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandel ER (2001). The molecular biology of memory storage: a dialogue between genes and synapses. Science 294, 1030–1038. [DOI] [PubMed] [Google Scholar]
- Kasbekar DP, Nelson JC & Hall LM (1987). Enhancer of seizure: a new genetic locus in Drosophila melanogaster defined by interactions with temperature‐sensitive paralytic mutations. Genetics 116, 423–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kole MH & Stuart GJ (2012). Signal processing in the axon initial segment. Neuron 73, 235–247. [DOI] [PubMed] [Google Scholar]
- Kourrich S, Calu DJ & Bonci A (2015). Intrinsic plasticity: an emerging player in addiction. Nat Rev Neurosci 16, 173–184. [DOI] [PubMed] [Google Scholar]
- Lai HC & Jan LY (2006). The distribution and targeting of neuronal voltage‐gated ion channels. Nat Rev Neurosci 7, 548–562. [DOI] [PubMed] [Google Scholar]
- Li SZ, Wu F, Wang B, Wei GZ, Jin ZX, Zang YM, Zhou JJ & Wong TM (2007). Role of reverse mode Na+/Ca2+ exchanger in the cardioprotection of metabolic inhibition preconditioning in rat ventricular myocytes. Eur J Pharmacol 561, 14–22. [DOI] [PubMed] [Google Scholar]
- Losonczy A, Makara JK & Magee JC (2008). Compartmentalized dendritic plasticity and input feature storage in neurons. Nature 452, 436–441. [DOI] [PubMed] [Google Scholar]
- Lubke J, Frotscher M & Spruston N (1998). Specialized electrophysiological properties of anatomically identified neurons in the hilar region of the rat fascia dentata. J Neurophysiol 79, 1518–1534. [DOI] [PubMed] [Google Scholar]
- Magee JC (1999). Dendritic I h normalizes temporal summation in hippocampal CA1 neurons. Nat Neurosci 2, 508–514. [DOI] [PubMed] [Google Scholar]
- Mauerhofer M & Bauer CK (2016). Effects of temperature on heteromeric Kv11.1a/1b and Kv11.3 channels. Biophys J 111, 504–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozzachiodi R, Lorenzetti FD, Baxter DA & Byrne JH (2008). Changes in neuronal excitability serve as a mechanism of long‐term memory for operant conditioning. Nat Neurosci 11, 1146–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson SB & Turrigiano GG (2008). Strength through diversity. Neuron 60, 477–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polvani S, Masi A, Pillozzi S, Gragnani L, Crociani O, Olivotto M, Becchetti A, Wanke E & Arcangeli A (2003). Developmentally regulated expression of the mouse homologues of the potassium channel encoding genes m‐erg1, m‐erg2 and m‐erg3 . Gene Express Patt 3, 767–776. [DOI] [PubMed] [Google Scholar]
- Powell KL, Ng C, O'Brien TJ, Xu SH, Williams DA, Foote SJ & Reid CA (2008). Decreases in HCN mRNA expression in the hippocampus after kindling and status epilepticus in adult rats. Epilepsia 49, 1686–1695. [DOI] [PubMed] [Google Scholar]
- Racine RJ (1972). Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol 32, 281–294. [DOI] [PubMed] [Google Scholar]
- Sacco T, Bruno A, Wanke E & Tempia F (2003). Functional roles of an ERG current isolated in cerebellar Purkinje neurons. J Neurophysiol 90, 1817–1828. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Jiang C, Curran ME & Keating MT (1995). A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell 81, 299–307. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC & Tristani‐Firouzi M (2006). hERG potassium channels and cardiac arrhythmia. Nature 440, 463–469. [DOI] [PubMed] [Google Scholar]
- Shah MM, Anderson AE, Leung V, Lin X & Johnston D (2004). Seizure‐induced plasticity of h channels in entorhinal cortical layer III pyramidal neurons. Neuron 44, 495–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah MM, Huang Z & Martinello K (2013). HCN and KV7 (M‐) channels as targets for epilepsy treatment. Neuropharmacology 69, 75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi W, Wymore RS, Wang HS, Pan Z, Cohen IS, McKinnon D & Dixon JE (1997). Identification of two nervous system‐specific members of the erg potassium channel gene family. J Neurosci 17, 9423–9432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibasaki T (1987). Conductance and kinetics of delayed rectifier potassium channels in nodal cells of the rabbit heart. J Physiol 387, 227–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith BN & Dudek FE (2002). Network interactions mediated by new excitatory connections between CA1 pyramidal cells in rats with kainate‐induced epilepsy. J Neurophysiol 87, 1655–1658. [DOI] [PubMed] [Google Scholar]
- Smith PL, Baukrowitz T & Yellen G (1996). The inward rectification mechanism of the HERG cardiac potassium channel. Nature 379, 833–836. [DOI] [PubMed] [Google Scholar]
- Stuart GJ & Spruston N (2015). Dendritic integration: 60 years of progress. Nat Neurosci 18, 1713–1721. [DOI] [PubMed] [Google Scholar]
- Surges R, Kukley M, Brewster A, Ruschenschmidt C, Schramm J, Baram TZ, Beck H & Dietrich D (2012). Hyperpolarization‐activated cation current I h of dentate gyrus granule cells is upregulated in human and rat temporal lobe epilepsy. Biochem Biophys Res Commun 420, 156–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweatt JD (2016). Neural plasticity and behavior – sixty years of conceptual advances. J Neurochem 139(Suppl 2), 179–199. [DOI] [PubMed] [Google Scholar]
- Tellez‐Zenteno JF & Hernandez‐Ronquillo L (2012). A review of the epidemiology of temporal lobe epilepsy. Epilepsy Res Treat 2012, 630853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titus SA, Warmke JW & Ganetzky B (1997). The Drosophila erg K+ channel polypeptide is encoded by the seizure locus. J Neurosci 17, 875–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trombin F, Gnatkovsky V & de Curtis M (2011). Changes in action potential features during focal seizure discharges in the entorhinal cortex of the in vitro isolated guinea pig brain. J Neurophysiol 106, 1411–1423. [DOI] [PubMed] [Google Scholar]
- Trudeau MC, Warmke JW, Ganetzky B & Robertson GA (1995). HERG, a human inward rectifier in the voltage‐gated potassium channel family. Science 269, 92–95. [DOI] [PubMed] [Google Scholar]
- Wang XJ, Reynolds ER, Deak P & Hall LM (1997). The seizure locus encodes the Drosophila homolog of the HERG potassium channel. J Neurosci 17, 882–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SR & Mitchell SJ (2008). Direct measurement of somatic voltage clamp errors in central neurons. Nat Neurosci 11, 790–798. [DOI] [PubMed] [Google Scholar]
- Wu WW, Chan CS, Surmeier DJ & Disterhoft JF (2008). Coupling of L‐type Ca2+ channels to KV7/KCNQ channels creates a novel, activity‐dependent, homeostatic intrinsic plasticity. J Neurophysiol 100, 1897–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue C & Yaari Y (2004). KCNQ/M channels control spike afterdepolarization and burst generation in hippocampal neurons. J Neurosci 24, 4614–4624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W & Linden DJ (2003). The other side of the engram: experience‐driven changes in neuronal intrinsic excitability. Nat Rev Neurosci 4, 885–900. [DOI] [PubMed] [Google Scholar]