

Abstract
The eukaryotic replisome disassembles parental chromatin at DNA replication forks, but then plays a poorly understood role in the re‐deposition of the displaced histone complexes onto nascent DNA. Here, we show that yeast DNA polymerase α contains a histone‐binding motif that is conserved in human Pol α and is specific for histones H2A and H2B. Mutation of this motif in budding yeast cells does not affect DNA synthesis, but instead abrogates gene silencing at telomeres and mating‐type loci. Similar phenotypes are produced not only by mutations that displace Pol α from the replisome, but also by mutation of the previously identified histone‐binding motif in the CMG helicase subunit Mcm2, the human orthologue of which was shown to bind to histones H3 and H4. We show that chromatin‐derived histone complexes can be bound simultaneously by Mcm2, Pol α and the histone chaperone FACT that is also a replisome component. These findings indicate that replisome assembly unites multiple histone‐binding activities, which jointly process parental histones to help preserve silent chromatin during the process of chromosome duplication.
Keywords: DNA polymerase alpha, DNA replication, histone chaperone, histones, replisome
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; DNA Replication, Repair & Recombination
Introduction
A subset of the many factors required to duplicate chromosomes assembles into a large and highly dynamic molecular machine called the replisome (Bell & Labib, 2016; Burgers & Kunkel, 2017; Kunkel & Burgers, 2017; Riera et al, 2017). Bacterial, viral and eukaryotic replisomes vary widely in their composition, but a common feature in all cases is that replisome assembly connects the DNA helicase that unwinds the parental DNA duplex to one or more of the DNA polymerases that synthesize the two daughter strands. Such physical connections between helicase and polymerases can be direct or else be mediated by other factors, and can serve to couple the rates of DNA unwinding and DNA synthesis, thus minimizing the exposure of single‐strand DNA at replication forks. In addition, work with Escherichia coli showed that replisome assembly allows DNA polymerase action to propel the DNA helicase forwards and thus stimulate the rate of fork progression (Kim et al, 1996).
Whereas the E. coli replisome is well characterized and the role of each component is defined, the eukaryotic replisome remains an enigma and contains around twice as many subunits, many of which are still of unknown biochemical function. The greater complexity of the eukaryotic replisome reflects the additional challenges of duplicating chromosomes that contain vast amounts of DNA packaged with histones into chromatin, which must be disrupted and then re‐established during replication, whilst preserving epigenetic information that controls patterns of gene expression (Hammond et al, 2017; Miller & Costa, 2017). Very little is known about the activities within the replisome that contributes to the processing of parental chromatin during chromosome duplication. Moreover, the links between parental histone processing at replication forks and the preservation of gene silencing are largely unexplored.
The eukaryotic replisome connects the CMG helicase (Cdc45‐MCM‐GINS) to two of the three DNA polymerases that are essential for DNA synthesis at eukaryotic replication forks, namely DNA polymerase epsilon (Pol ε) that synthesizes the leading strand and DNA polymerase alpha (Pol α) that initiates each new Okazaki fragment during lagging strand synthesis. Pol ε binds directly to the CMG helicase and stimulates the rate of fork progression (Kang et al, 2012; Sengupta et al, 2013; Georgescu et al, 2014; Langston et al, 2014), analogous to the stimulation of helicase by polymerase in the E. coli replisome (Kim et al, 1996). In contrast, Pol α is connected indirectly to the CMG helicase by the trimeric adaptor known as Ctf4 (Zhu et al, 2007; Gambus et al, 2009; Tanaka et al, 2009a), each protomer of which can bind to short “Ctf4‐Interacting Peptides” (or CIP‐boxes) in client proteins such as the Sld5 subunit of yeast CMG, or the Pol1 catalytic subunit of Pol α (Simon et al, 2014; Villa et al, 2016). The tethering of Pol α to the eukaryotic replisome by Ctf4 was originally thought to promote efficient priming of Okazaki fragments during lagging strand DNA synthesis. However, Ctf4 has no apparent impact on DNA synthesis in vitro, using a reconstituted replisome system based on purified budding yeast proteins (Yeeles et al, 2015, 2017). Moreover, recent work indicates that trimeric Ctf4 represents a hub in the eukaryotic replisome with roles that go beyond DNA synthesis (Fumasoni et al, 2015; Samora et al, 2016; Villa et al, 2016). In addition to Pol α, budding yeast Ctf4 recruits other CIP‐box proteins such as the Chl1 helicase that helps to establish sister chromatid cohesion (Samora et al, 2016) and the Dna2 and Tof2 proteins that help to maintain the integrity of the rDNA repeats on chromosome 12 (Villa et al, 2016). Here, we show that replisome tethering of Pol α via Ctf4 is dispensable for efficient DNA synthesis in budding yeast cells, and instead is required to preserve epigenetic gene silencing at telomeres and the silent mating‐type genes, in a manner that is dependent upon a novel histone‐binding motif in the amino‐terminal region of the Pol1 DNA polymerase subunit. These findings expand our view of the eukaryotic replication machinery and show how the coupling of a DNA polymerase to the helicase within the replisome can contribute to functions beyond DNA synthesis.
Results
Displacement of Pol α from Ctf4 does not perturb DNA replication in vivo
We previously showed that Pol α was no longer able to associate with Ctf4 in pol1‐4A cells with mutations in the CIP‐box motif of the catalytic subunit (Simon et al, 2014). However, pol1‐4A cells lack multiple phenotypes of ctf4∆ strains, which instead reflect the recruitment by Ctf4 of other CIP‐box proteins to the replisome (Samora et al, 2016; Villa et al, 2016). To examine in more detail the consequences of displacing Ctf4‐tethered Pol α from the replisome, we synchronized control cells and pol1‐4A in G1‐phase and monitored DNA synthesis and replisome assembly when cells entered S‐phase. As shown in Fig 1, the kinetics of DNA synthesis were very similar in pol1‐4A and control cells (Fig 1A), and replisome assembly was normal except that Pol α was largely displaced (Fig 1B; Appendix Fig S1 shows that the weakened association of Pol1‐4A with the replisome is equivalent to the situation previously reported for ctf4∆ cells (Sengupta et al, 2013), reflecting a residual Ctf4‐independent link between Pol α and the CMG helicase).
Figure 1. Ctf4‐dependent tethering of Pol alpha to the replisome is dispensable for efficacious DNA synthesis in budding yeast cells.
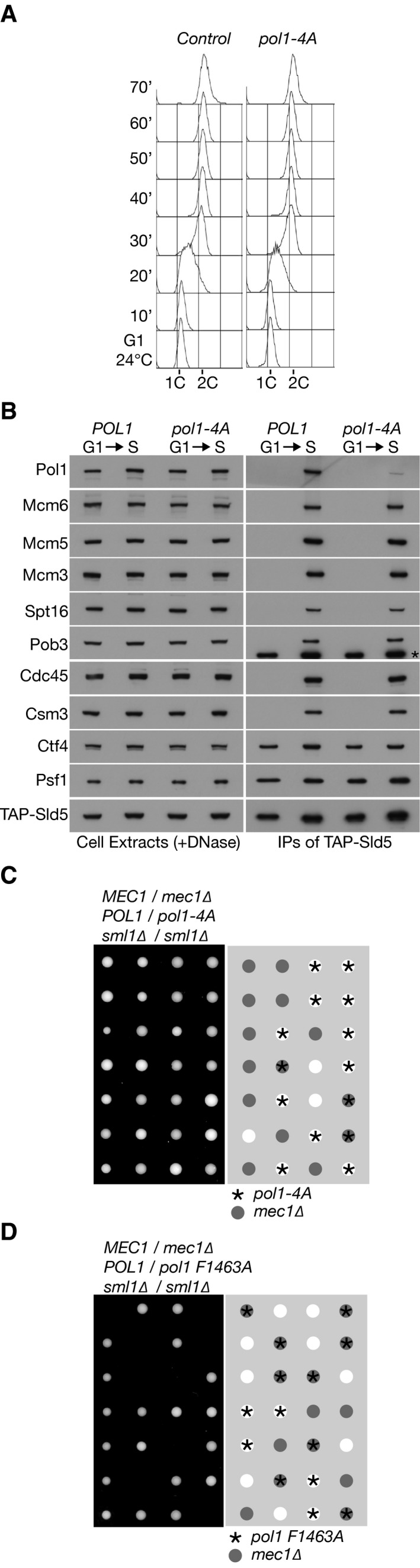
- Control (YCE542) and pol1‐4A (YCE544) cells were synchronized in G1‐phase at 24°C then released into S‐phase for the indicated times. DNA content was measured by flow cytometry throughout the experiment.
- In a similar experiment to that described above, samples from the G1‐phase and 30′ S‐phase timepoints were used to prepare cell extracts, from which the CMG helicase component Sld5 was isolated by immunoprecipitation. The associated replisome components were monitored by immunoblotting. The asterisk indicates a non‐specific band in the anti‐Pob3 immunoblot (corresponding to TAP‐Sld5).
- Diploid cells of the indicated genotype were sporulated, and then, asci were dissected on rich medium. The image of the resultant tetrads was taken after 2 days growth at 30°C, and the genotype of each colony was determined by replica plating to selective media, indicating that the growth of pol1‐4A is not affected by combination with mec1∆.
- Equivalent analysis showing that pol1‐F1463A is synthetic lethal with mec1∆.
As a more sensitive assay for impaired DNA synthesis, we combined pol1‐4A with deletion of the MEC1 gene, the yeast orthologue of the ATR checkpoint kinase, which becomes essential in cells that have the slightest defect in DNA replication. Colony growth of mec1∆ cells was unaffected by the pol1‐4A mutations (Fig 1C), indicating that Pol α is still very efficient at priming nascent DNA during leading and lagging strand synthesis in pol1‐4A cells, even though it is no longer tethered to the replisome by Ctf4. As a control, we confirmed that mec1∆ is synthetic lethal with the pol1‐F1463A point mutation (Fig 1D), which displaces primase from the carboxyl terminus of Pol1 (Kilkenny et al, 2012). Previous work indicated that the progression of chromosome replication in pol1‐F1463A is very similar to control cells, but cell viability is absolutely dependent upon Mec1, pointing to a subtle defect in DNA synthesis at replication forks (Kilkenny et al, 2012).
Overall, these findings indicated that replisome tethering of Pol α by Ctf4 is dispensable in vivo for efficient DNA synthesis and instead fulfils a different role during chromosome replication.
Replisome tethering of Pol α is required to preserve gene silencing at sub‐telomeric and mating‐type loci
One possible function for Ctf4‐dependent replisome tethering of Pol α was suggested by analogy with the rDNA‐associated protein Tof2 and the Dna2 nuclease, which are recruited to the replisome by Ctf4 as part of a mechanism that preserves the integrity of the large array of rDNA repeats (Villa et al, 2016). As seen for Pol α, mutation of the CIP‐boxes of Tof2 and Dna2 disrupted their interaction with Ctf4 but did not perturb DNA replication in yeast cells. Instead, the tof2‐4A and dna2‐4A alleles both led to a reduction in the size of chromosome 12 (Villa et al, 2016). However, this phenotype was not observed with pol1‐4A cells (CE and KL, unpublished data), indicating that Ctf4 tethers Pol α to the replisome in order to support some other function.
A second possibility was indicated by the observation that Pol α co‐purifies from yeast cell extracts with histone complexes that have been released from chromatin (Foltman et al, 2013). This suggested that Pol α might have an associated histone‐binding activity that contributes to chromatin replication when Pol α is tethered to the replisome by Ctf4, analogous to the role of the histone‐binding motif of the Mcm2 subunit of the CMG helicase (Ishimi et al, 1998; Foltman et al, 2013; Huang et al, 2015; Richet et al, 2015), which is required to preserve gene silencing near budding yeast telomeres (Foltman et al, 2013). Similar to the phenotype of pol1‐4A cells, mutation of the conserved histone‐binding motif of Mcm2 is not synthetic lethal with mec1∆ (Foltman et al, 2013) and thus does not perturb DNA replication in yeast cells.
If the ADE2 marker gene is placed near a telomere in a wild‐type yeast strain, it is expressed in some cells and repressed in others for many generations (Fig 2A), leading to colonies with white sectors (expressing ADE2) and red sectors (repression of ADE2). However, when a haploid strain with telomeric ADE2 was crossed to the mcm2‐3A histone‐binding mutant and the resulting diploid strain was sporulated, the mcm2‐3A progeny that inherited the marker gene produced pure white colonies (Foltman et al, 2013), in which all cells express sub‐telomeric ADE2 (Fig 2B). Strikingly, we found in similar experiments that pol1‐4A is also defective in the maintenance of telomeric silencing (Fig 2C), and the same is true for ctf4∆ cells (Suter et al, 2004). Moreover, this phenotype was not produced by mutation of the CIP‐box in other Ctf4 partners such as Tof2 or Dna2 (Fig EV1). These findings indicated that Ctf4‐dependent tethering of Pol α to the replisome is required to preserve gene silencing near a telomere, but is not required for Pol α to fulfil its role in DNA synthesis. Consistent with this view, telomere length is normal in pol1‐4A cells (Appendix Fig S2; compare pol1‐4A and control), whereas telomere length increases as a consequence of defects in Pol α catalytic function (Adams & Holm, 1996), or following the displacement of primase from Pol α (Appendix Fig S2, pol1‐F1463A).
Figure 2. Displacement of Pol alpha from the replisome leads to a loss of gene silencing at sub‐telomeric or mating‐type loci.

-
A–CTetrad analysis of meiotic progeny of the indicated diploids (heterozygous for sub‐telomeric ADE2), processed as described in Materials and Methods. Sectored colonies represent epigenetic variation in the expression of the ADE2 marker at the sub‐telomeric location. White colonies indicate expression of ADE2 in all cells, and red colonies lack the ADE2 marker (as denoted in the right‐hand panels). We examined a total of 19 mcm2‐3A VR::ADE2‐TEL colonies and 11 pol1‐4A VR::ADE2‐TEL colonies.
-
D–FEquivalent analysis of the meiotic progeny of diploid cells that were heterozygous for an insertion of the ADE2 marker at HMR locus on chromosome 3 (denoted HMR::2EDA since the orientation of the ADE2 marker was such that the promoter was distal to the HMR‐E silencer element). We examined a total of 19 mcm2‐3A HMR::2EDA colonies and 12 pol1‐4A HMR::2EDA colonies.
Figure EV1. Sub‐telomeric silencing is not affected by CIP‐box mutations in the Ctf4 partners Dna2 or Tof2, or by deletion of the TEL1 gene.

The indicated diploids were processed as described above for Fig 2.
Gene silencing in budding yeast cells also occurs at the silent mating‐type genes on chromosome 3 (Haber, 2012). The ADE2 marker gene is very strongly repressed when inserted at the HMR mating‐type locus in control cells and therefore produces red colonies that are only slightly paler than colonies of control cells (Fig 2D; ADE2 replaces the silenced MAT a 2 gene and has the same orientation, designated HMR::2EDA). However, we found that HMR‐2EDA is largely de‐repressed in mcm2‐3A and pol1‐4A cells, which thus grew as very pale pink colonies (Fig 2E and F; note that the control HMR‐2EDA cells sometimes produced dark pink rather than red colonies in this experiment, probably due to partial de‐repression of HMR‐2EDA in the parental diploid cells that were heterozygous for mcm2‐3A or pol1‐4A). These data indicate that the histone‐binding activities of the replisome are important to preserve gene silencing at the mating‐type loci, in addition to being important for telomeric silencing (Fig EV2B shows that the same is true in HMR::ADE2 cells in which ADE2 is inserted in the opposite orientation to HMR::2EDA, leading to weaker gene silencing).
Figure EV2. Replisome‐tethered histone‐binding activity of Mcm2 and Pol alpha is required for gene silencing at the HMR mating‐type locus.

-
A–ETetrad analysis of meiotic progeny of the indicated diploids (heterozygous for insertion of the ADE2 marker gene at the HMR locus on chromosome 3), processed as described in Materials and Methods. The promoter of the ADE2 marker was positioned proximal to the HMR‐E silencer element (denoted HMR::ADE2), and this corresponded to weaker silencing of ADE2 compared to the HMR::2EDA strains in Fig 2D–F. We examined a total of 16 mcm2‐3A HMR::ADE2 colonies, 16 pol1‐4A HMR::ADE2 colonies, 15 pol1‐2A2 HMR::ADE2 colonies and 16 pol1‐6A HMR::ADE2 colonies.
Finally, we tested whether the mcm2‐3A and pol1‐4A mutations affected silencing of a URA3 marker gene that had been inserted into one copy of the rDNA repeats on chromosome 12. Interestingly, gene silencing in the rDNA is mechanistically distinct from silencing at telomeres and the silent mating‐type loci (Srivastava et al, 2016). The URA3 gene was silenced when inserted into either the NTS1 or NTS2 sequences of an rDNA repeat, and silencing at NTS1 was relieved in cells that lack the type I topoisomerase Top1 (Fig EV3), as reported previously (Huang et al, 2006). However, gene silencing at NTS1 and NTS2 persisted in mcm2‐3A and pol1‐4A cells. Therefore, although Mcm2 and replisome‐tethered Pol α are both required to preserve gene silencing at telomeric and mating‐type loci in budding yeast cells, they are dispensable for the mechanistically distinct phenomenon of rDNA silencing.
Figure EV3. Histone‐binding activities of replisome proteins are largely dispensable for gene silencing within the rDNA repeats.

Serial dilutions of the indicated strains, in which the “modified URA3 marker” (mURA3) was inserted at the leu2 locus or else within the “Non‐transcribed spacers” of an rDNA repeat (NTS1 and NTS2), were grown on non‐selective or selective medium, as described in Materials and Methods. In control cells, mURA3 can be expressed from the leu2 locus but is repressed when present at the NTS1 or NTS2 element of an rDNA repeat. Repression at NTS1 was lost in top1∆ cells, as seen previously (Huang et al, 2006), but an equivalent defect was not observed in mcm2‐3A, pol1‐2A2, pol1‐4A, pol1‐6A, mcm2‐3A pol1‐6A, dpb3∆ or dpb4∆.
The amino terminus of the DNA polymerase subunit of Pol α contains a conserved histone‐binding motif
The ability of Pol α to interact in yeast cell extracts with chromatin‐derived histone complexes might reflect a novel histone‐binding activity in Pol α, but could also be due to the previously described association of Pol α with the histone chaperone FACT (Miles & Formosa, 1992). Consistent with the former possibility, we noticed that the extended amino‐terminal tail of the Pol α catalytic subunit in eukaryotes contains a motif that resembles the histone‐binding element in Mcm2 (Fig 3A), in that it contains two conserved aromatic residues that are separated by eight amino acids and are flanked by conserved acidic residues, adjacent to a predicted alpha helix (Foltman et al, 2013). However, the two motifs also have important differences, since the orientation of the alpha helix and the adjacent pair of aromatic residues is reversed between Mcm2 and Pol α, and the Pol1 tail has an additional pair of conserved tyrosines that overlap with the predicted alpha helix.
Figure 3. The amino terminus of Pol1 binds chromatin‐derived histone complexes, together with Mcm2 and FACT.
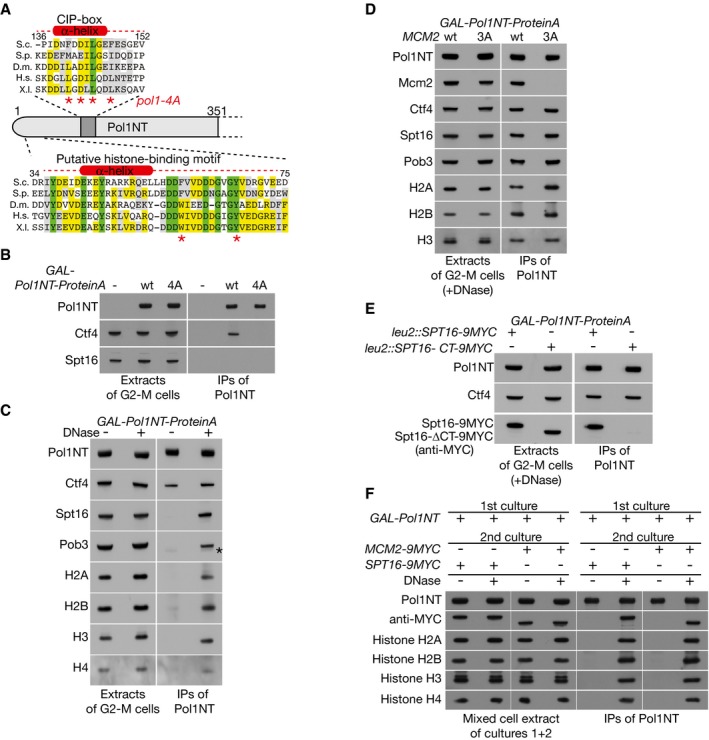
- Sequence alignment indicating the location of the CIP‐box and a newly identified putative histone‐binding motif in the amino‐terminal tail of budding yeast Pol1. Asterisks under the CIP‐box indicate residues mutated in the pol1‐4A allele, whereas those under the novel histone‐binding motif denote two conserved aromatic residues separated by eight amino acids, as in the histone‐binding motif of Mcm2.
- Control cells (YSS3), cells expressing ProteinA‐tagged Pol1NT (YCE39) and cells expressing ProteinA‐tagged Pol1NT‐4A (YCE217) were synchronized in G2‐M phase at 30°C, before the isolation of the ProteinA‐tagged Pol1 tails on IgG beads. Pol1NT interacts with Ctf4, dependent upon the CIP‐box, but does not interact with FACT (for which the Spt16 subunit is shown).
- Cells expressing ProteinA‐tagged Pol1NT (YCE39) were grown as above, but the resultant extracts were then incubated +/−DNase, before isolation of Pol1NT. The associated proteins were monitored by immunoblotting—the asterisk in the Pob3 blot indicates faint cross‐reactivity with ProteinA‐Pol1NT.
- Analogous experiment comparing expression of GAL‐Pol1NT in control cells (YCE39) or mcm2‐3A cells (YCE238). After release of histones from chromatin by DNase treatment, the co‐purification of Mcm2 with Pol1NT depends on the H3‐H4 binding motif of Mcm2, which is mutated in mcm2‐3A.
- Similar experiment comparing expression of GAL‐Pol1NT in cells with a second copy of SPT16 at the leu2 locus, expressing either wild‐type Spt16 (YCE248) or Spt16 with a small deletion at the carboxyl terminus (YCE250) that abolishes interaction of FACT with H3‐H4 tetramers.
- Cultures of GAL‐Pol1NT (YCE39), MCM2‐9MYC (YMP154‐1) and SPT16‐9MYC (YMP177‐1) were arrested in G2‐M phase at 30°C, and then, equal volumes were mixed as indicated. Cell extracts were prepared, and Pol1NT was isolated on IgG‐coated magnetic beads. Release of histones from chromatin by DNase treatment allowed Pol1NT from the first culture to form ternary complexes containing not only histones but also Mcm2‐9MYC or Spt16‐9MYC from the second cultures.
To test the ability of the amino‐terminal tail of budding yeast Pol1 to associate with histone complexes and FACT, we expressed the first 351 amino acids of Pol1 in yeast cells as a fusion to Protein A, and then isolated the fusion protein from cell extracts, with or without prior DNase treatment to release histone complexes from chromatin. As shown in Fig 3B, the amino‐terminal fragment of Pol1 (hereafter termed Pol1NT) associated specifically with Ctf4, dependent upon the integrity of the CIP‐box (Fig 3B, compare Pol1NT‐4A with wt Pol1NT), but did not associate with FACT, which must instead bind to some other region of Pol α. However, upon digestion of genomic DNA to release histone complexes from chromatin, Pol1NT co‐purified not only with histones but also with FACT in yeast cell extracts (Fig 3C, +DNase; Appendix Fig S3 shows the salt sensitivity of the observed complexes). These findings suggested that Pol1NT shares the ability of Mcm2 to associate with histone complexes that have been released from chromatin and also indicated that the same histone‐containing complexes can be bound by FACT.
To explore this further, we expressed Pol1NT (Pol1 1–351) and Mcm2NT (Mcm2 1–200) in yeast cells as fusions to Protein A and also included Protein A‐fused Mcm4NT (Mcm4 1–186) as a negative control. We isolated the fusion proteins by immunoprecipitation from yeast extracts with or without DNase treatment, before analysis of the associated factors by mass spectrometry. As summarized in Table 1, the treatment of yeast extracts with DNase greatly stimulated the detectable association of both Pol1NT and Mcm2NT, but not Mcm4NT, with histones and FACT. In addition, Mcm2 (but not Mcm4) was greatly enriched in the Pol1NT immunoprecipitates after DNase treatment of extracts. Incidentally, we found that the Tra1 subunit of the SAGA and NuA4 histone acetyltransferases was also enriched in the immunoprecipitates of Pol1NT and Mcm2NT after DNase treatment, suggesting that Tra1 might also bind to histone complexes released from chromatin, presumably on distinct sites to Mcm2NT and Pol1NT.
Table 1.
Mass spectrometry analysis of proteins co‐purifying with Pol1NT, Mcm2NT or Mcm4NT, +/−DNase treatment to release histone complexes from chromatin
| Identified protein | MS analysis of IPs from G2‐M cell extracts (spectral counts) | ||||
|---|---|---|---|---|---|
| Pol1 (1–351) +DNase | Pol1 (1–351) −DNase | Mcm2 (1–200) +DNase | Mcm2 (1–200) −DNase | Mcm4 (1–186) +DNase | |
| Pol1NT | 1,570 | 854 | 7 | 0 | 0 |
| Mcm2 | 436 | 0 | 258 | 218 | 6 |
| Mcm4 | 38 | 11 | 14 | 14 | 681 |
| Ctf4 | 938 | 564 | 0 | 0 | 0 |
| Tel1 | 407 | 229 | 18 | 0 | 0 |
| Histone H2A | 95 | 0 | 46 | 0 | 16 |
| Histone H2B | 441 | 3 | 140 | 3 | 33 |
| Histone H3 | 275 | 2 | 178 | 5 | 13 |
| Histone H4 | 287 | 4 | 275 | 4 | 28 |
| Spt16 | 2,255 | 53 | 1,558 | 5 | 201 |
| Pob3 | 812 | 8 | 486 | 0 | 46 |
| Tra1 | 600 | 32 | 1,105 | 44 | 86 |
| Spt5 | 396 | 22 | 38 | 3 | 26 |
The indicated protein fragments were expressed in G2‐M‐arrested cells and then isolated by immunoprecipitation on IgG‐coated magnetic beads. The resultant material was resolved in a 4–12% gradient gel, which was then stained with colloidal Coomassie blue, before each lane was cut into 40 bands for mass spectrometry analysis. The table summarizes the spectral counts that were detected for each factor that specifically co‐purified with the indicated fragment.
As expected, we found that Pol1NT interacted specifically with Ctf4 regardless of DNase treatment. Moreover, Pol1NT interacted with the Tel1 checkpoint kinase in a similar manner to Ctf4 (Table 1), indicating that Tel1 is a novel partner of Pol1NT. However, subsequent experiments indicated that neither Ctf4 (Fig EV4A) nor Tel1 (Fig EV4B) was required for Pol1NT to associate with histone complexes released from chromatin. Consistent with this, tel1∆ was not associated with a loss of telomeric silencing (Fig EV1), though it did reduce telomere length (Appendix Fig S2), as previously reported (Lustig & Petes, 1986; Greenwell et al, 1995).
Figure EV4. Neither Ctf4 nor Tel1 is required for the histone‐binding activity of Pol1NT .
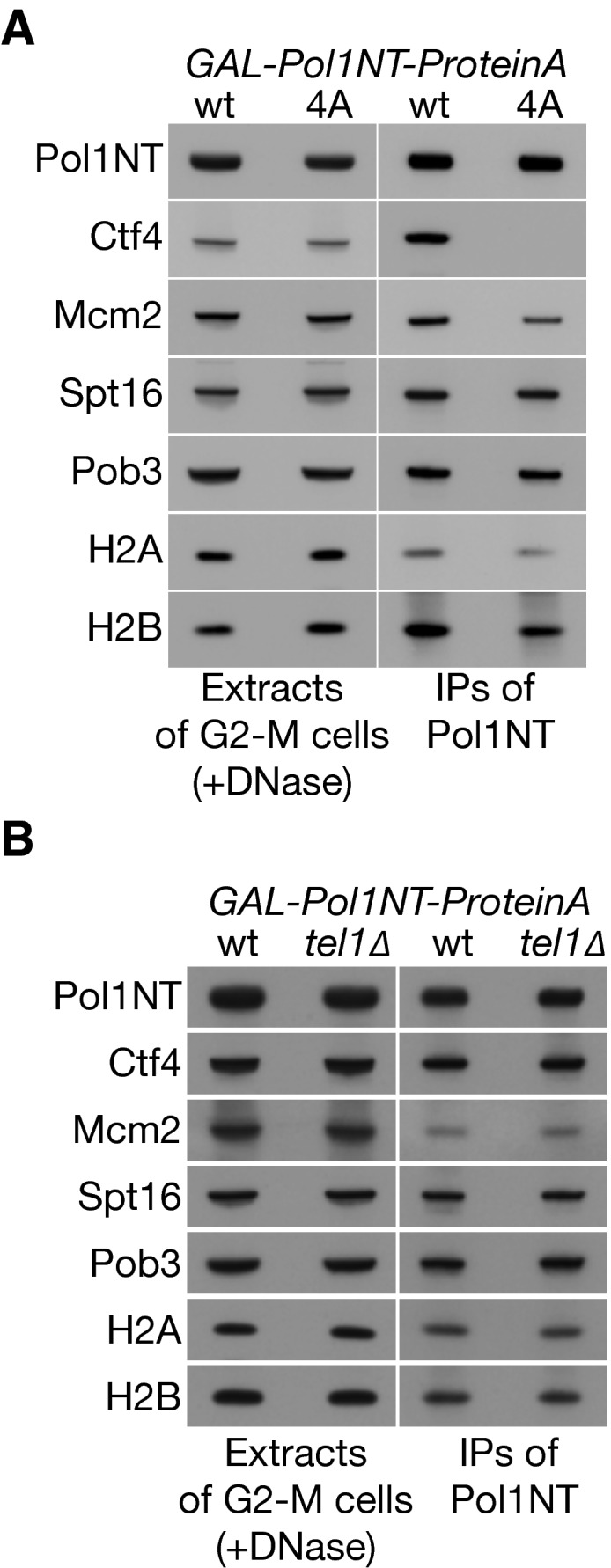
- Mutation of the CIP‐box of Pol1 displaces Ctf4 without affecting the interaction with histones released from chromatin.
- Although Tel1 is a novel partner of Pol1NT, deletion of the TEL1 gene does not affect histone‐binding by Pol1NT.
The presence of Mcm2 in the Pol1NT immunoprecipitates was dependent not only upon the release of histones from chromatin by DNase treatment, but also required the integrity of the conserved histone‐binding motif in the Mcm2 tail (Fig 3D, mcm2‐3A). Similarly, the co‐purification of FACT with Pol1NT, upon release of histones from chromatin, was abolished by mutation of the carboxyl terminus of the Spt16 subunit (Fig 3E and Appendix Fig S4), corresponding to a small truncation that was previously shown to abolish the association of FACT with H3‐H4 tetramers (Tsunaka et al, 2016). These findings indicated that Pol1NT, Mcm2NT and FACT are able to bind to different regions of the same histone complexes that are released from chromatin. To confirm that these interactions can indeed occur in vitro upon release of histone complexes from DNA, we mixed a yeast culture expressing Pol1NT with cells expressing either Mcm2‐9MYC or Spt16‐9MYC, before making mixed cell extracts. In the absence of DNase treatment, Pol1NT from the first cell culture did not interact with either Mcm2‐9MYC or SPT16‐9MYC from the second cultures (Fig 3F, −DNase). In contrast, DNase treatment of the cell extracts allowed Pol1NT from the first culture not only to associate in vitro with histones, but also to co‐purify with tagged Mcm2 or Spt16 from the second culture (Fig 3F, +DNase). Overall, these findings indicated that Pol1NT is able to interact in vitro with chromatin‐derived histone complexes, which can also be co‐chaperoned by Mcm2 and FACT.
Mutation to alanine of six conserved residues in Pol1NT (Y37, Y45, F58, D62, G66 and Y67), within the region of similarity with the histone‐binding motif of Mcm2, greatly reduced the ability of Pol1NT to co‐purify with histones, FACT and Mcm2, without affecting the interaction of Pol1NT with Ctf4 (Fig 4A and B, and Appendix Fig S5A and B). Further analysis of subsets of these mutations (Appendix Fig S5C) highlighted the importance of the two conserved aromatic residues F58 and Y67 that are located downstream of the predicted alpha helix (Fig 4A and B, Pol1‐2A), along with neighbouring acidic amino acids (Fig 4A and B, Pol1‐2A2 containing the F58A and D62A mutations). In addition, the pair of conserved tyrosines that overlap with the alpha helix also contributed to the observed histone‐binding activity (Appendix Fig S5, Y37A Y45A).
Figure 4. A conserved motif at the amino terminus of Pol1 is required to bind chromatin‐derived histone complexes, together with Mcm2 and FACT.
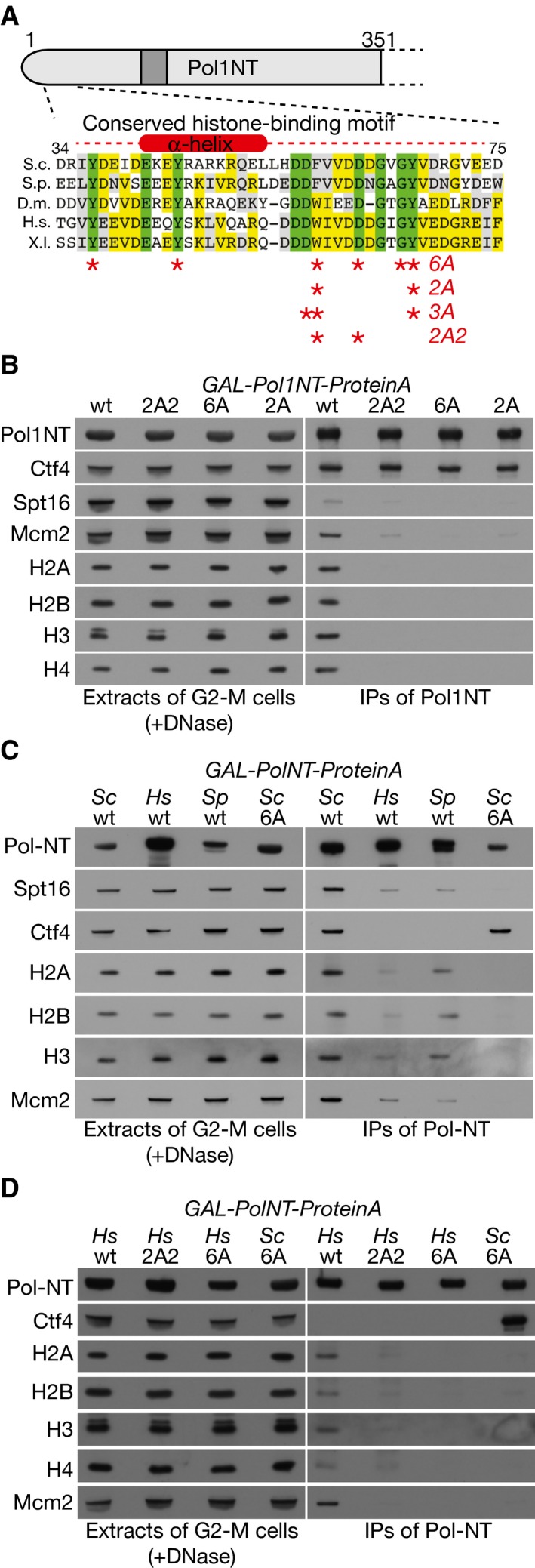
- Asterisks correspond to the residues in the newly identified histone‐binding motif that were mutated to create the indicated alleles.
- The amino‐terminal tails of Pol1‐2A (YCE1084), Pol1‐2A2 (YCE232) and Pol1‐6A (YCE226) still interact with Ctf4, but are unable to associate with histones released from chromatin, and do not co‐purify with FACT.
- The amino‐terminal tail of the DNA polymerase alpha catalytic subunit from fission yeast (YCE378; Sp = Schizosaccharomyces pombe) and human (YCE407; Hs = Homo sapiens) bind specifically to histone complexes derived from budding yeast chromatin, by comparison with the Pol1‐6A budding yeast tail with a mutated histone‐binding site.
- Mutation of conserved residues in the histone‐binding motif (YCE425 expresses the 2A2 allele and YCE435 corresponds to the 6A allele) kills the ability of Hs‐Pol1NT to associate with histone complexes derived from budding yeast chromatin.
To test whether the corresponding region of Pol α from other species is able to bind to chromatin‐derived histone complexes, we expressed the fission yeast and human equivalents of Pol1NT in budding yeast cells, as fusions to Protein A. Neither of the fusion proteins bound to budding yeast Ctf4, probably due to the evolutionary divergence of the human and fission yeast CIP‐box sequences. However, both proteins co‐purified with yeast histones, FACT and Mcm2, reflecting the high conservation of histone sequences amongst diverse eukaryotic species (Fig 4C; note that the binding of fission yeast and human Pol α tails to budding yeast histone complexes was weaker than seen with wild‐type ScPol1NT, but was still clearly specific in comparison with ScPol1NT‐6A). Moreover, the ability of HsPolα‐NT to bind to yeast‐chromatin‐derived histone complexes, together with Mcm2 and FACT, was abrogated by mutations in conserved residues within the novel histone‐binding motif (Fig 4D). Overall, these findings indicated that Pol1NT contains an evolutionarily conserved histone‐binding motif that is capable of binding to chromatin‐derived histone complexes, on distinct sites to those bound by Mcm2 and FACT.
To explore which histones are bound by the novel motif in Pol α, we took advantage of the fact that HeLa nuclei contain high concentrations of free histones that are available to bind to histone chaperones, whereas in budding yeast cells, the non‐nucleosomal pool of free histones is extremely low. We incubated extracts of HeLa nuclei with bead‐bound versions of recombinant HsMCM2‐NT (amino acids 43–160 of human MCM2), wild‐type HsPOLA1‐NT (amino acids 1–110 of human POLA1), or HsPOLA1‐3A‐NT (Fig 4A shows the location of the 3A mutations within the conserved histone‐binding motif of HsPOLA1‐NT). After recovery of the bead‐bound versions of the HsMCM2‐NT and HsPOLA1‐NT tails, the major associated proteins were visualized by Coomassie staining (Fig 5A) and then identified by mass spectrometry and immunoblotting (AD, JM and LP, unpublished data). HsMCM2‐NT bound specifically to histones H3‐H4, as observed previously (Huang et al, 2015; Richet et al, 2015). In contrast, HsPOLA1‐NT bound preferentially to histones H2A‐H2B (Fig 5A), and the observed association was diminished by the 3A mutations of conserved residues, including those that we showed to be important for the association of yeast Pol1NT with chromatin‐derived histone complexes. To confirm and quantify the interaction of HsPOLA1‐NT with histones H2A‐H2B, we monitored the interaction of recombinant HsPOLA1‐NT and H2A‐H2B by isothermal titration calorimetry (ITC), as described in Materials and Methods. These experiments indicated that HsPOLA1‐NT bound with 1:1 stoichiometry to an H2A‐H2B heterodimer, with a dissociation constant of 19 nM (Fig 5B). Moreover, binding was abolished by mutation of six conserved residues in the histone‐binding motif (equivalent to the budding yeast Pol1‐6A mutant), confirming the specificity of the observed interaction (Fig 5C). Overall, therefore, these data demonstrate that the amino‐terminal tail of the DNA polymerase subunit of human Pol α contains a conserved motif that binds specifically to histones H2A‐H2B. Moreover, our data indicate that Pol α, Mcm2 and FACT are able to bind simultaneously to distinct sites on the surface of chromatin‐derived histone complexes that contain all four yeast histones, after release of the latter from DNA.
Figure 5. The histone‐binding motif in the amino‐terminal tail of the DNA polymerase subunit of Pol α is specific for histones H2A‐H2B.
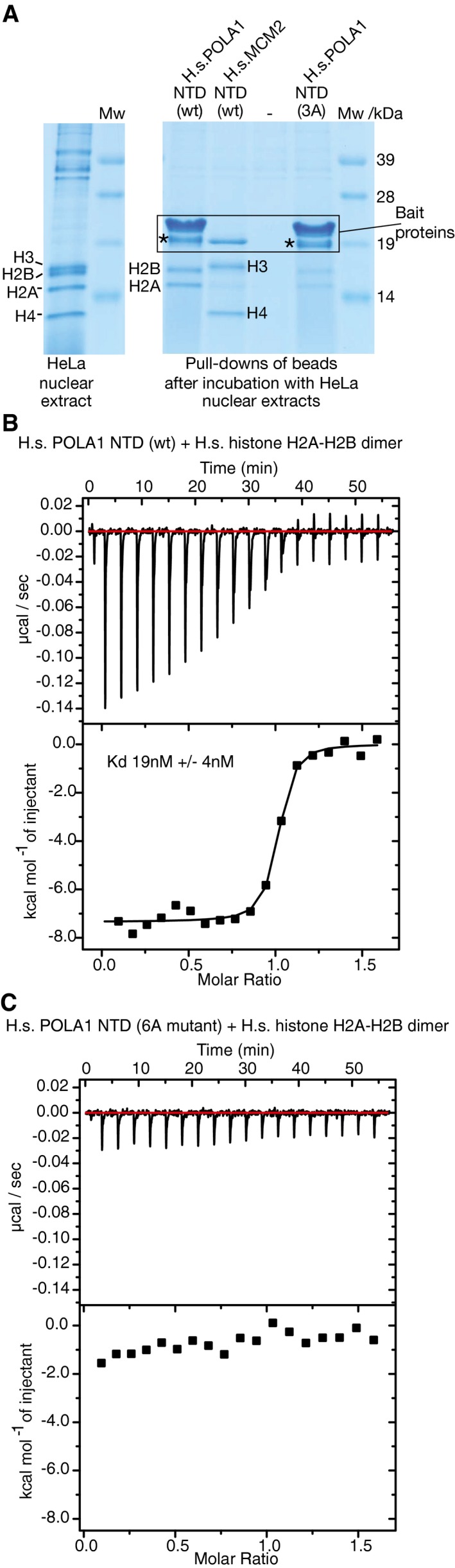
- Recombinant versions of the amino‐terminal tail of human POLA1 (amino acids 1–110 of the polymerase subunit of Pol α) or human MCM2 (residues 43–160) were purified from Escherichia coli as fusions to a Twin‐Strep‐tag. The Strep‐Tactin beads carrying the purified tails were then incubated with a high‐salt extract of HeLa nuclei, before the beads were washed and the bound proteins eluted in d‐Desthiobiotin buffer. The eluted proteins were revealed by SDS–PAGE and Coomassie staining, and relevant bands were identified by mass spectrometry and confirmed by immunoblotting. The asterisk denotes a degradation product of the POLA1‐NTD.
- The interaction of recombinant H.s. POLA1‐NTD with a complex of recombinant histones H2A‐H2B was monitored by isothermal titration calorimetry. The experiment was performed three times, and the figure shows a representative example.
- Similar analysis of the interaction of H.s. POLA1‐NTD (6A mutant) with a complex of recombinant human histones H2A‐H2B.
Mutation of the histone‐binding motif in budding yeast Pol1 disrupts gene silencing without affecting DNA synthesis or the tethering of Pol α to the replisome
To investigate the in vivo role of the conserved histone‐binding motif in budding yeast Pol1NT, we introduced the pol1‐6A or pol1‐2A2 mutations into the endogenous POL1 locus in yeast cells, in order to compare the phenotypes of mutating the histone‐binding motif of Pol1 with the pol1‐4A allele that displaces Pol α from Ctf4 in the replisome. Replisome assembly was normal in pol1‐6A (Fig 6A and Appendix Fig S6A) and pol1‐2A2 cells (Appendix Fig S6C), including the tethering of Pol α to the CMG helicase, and the kinetics of DNA replication were indistinguishable from control cells (Fig 6B and Appendix Fig S6D). Moreover, neither pol1‐2A2 nor pol1‐6A caused sensitivity to the replication inhibitor hydroxyurea (Fig EV5), and the viability of pol1‐2A2 and pol1‐6A was independent of the Mec1 checkpoint kinase (Fig 6C and D). These data indicate that the ability of Pol1NT to bind to chromatin‐derived histone complexes is dispensable for efficient DNA synthesis during chromosome replication in budding yeast cells. Importantly, however, gene silencing near a telomere and at the HMR mating‐type locus was defective in pol1‐2A2 and pol1‐6A cells (Figs 7A–D and EV2D and E), whereas rDNA silencing was not abolished (Fig EV3). Therefore, the phenotype of a replisome in which Pol α has a defective histone‐binding motif resembles the phenotype of displacing Pol α from the replisome (pol1‐4A, described above). These findings indicate that Ctf4‐dependent replisome tethering of Pol α in budding yeast is dispensable for DNA synthesis at replication forks and instead contributes to histone processing and gene silencing.
Figure 6. The histone‐binding motif in the amino‐terminal tail of budding yeast Pol1 is dispensable for replisome tethering of Pol alpha and for efficacious DNA synthesis.
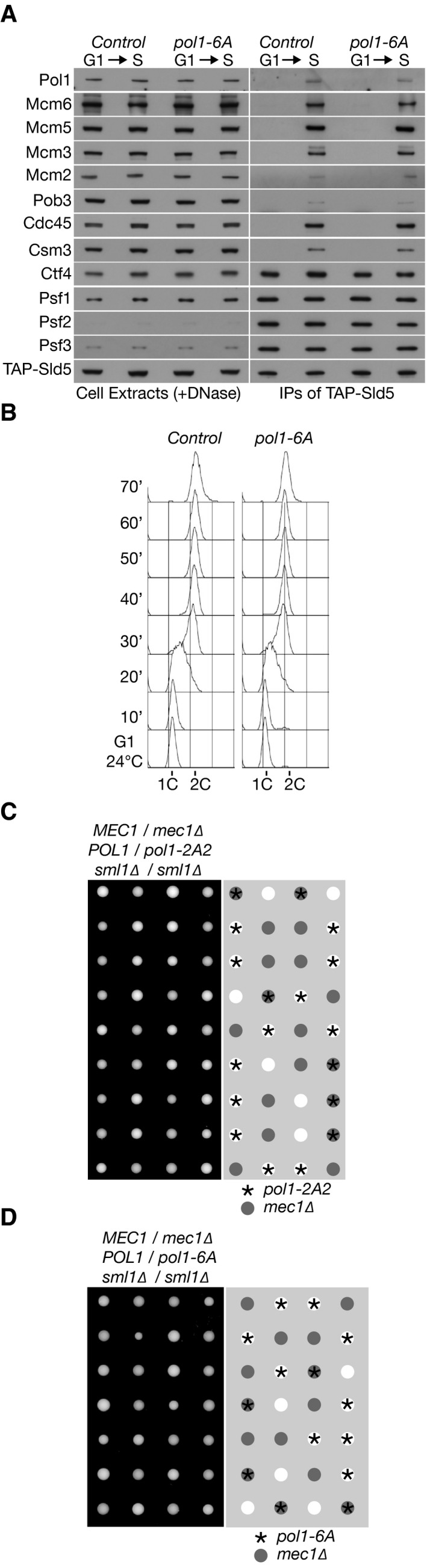
-
AControl (YCE542) and pol1‐6A (YCE546) cells were synchronized in G1‐phase at 24°C and then released into S‐phase for 30′, before replisome assembly was monitored as above, via immunoprecipitation of the Sld5 subunit of the CMG helicase.
- B
-
C, DThe growth of pol1‐2A2 and pol1‐6A was not affected by combination with mec1∆.
Figure EV5. Mutations in the histone‐binding motifs of Mcm2 or Pol1 do not cause sensitivity to hydroxyurea treatment.
Serial dilutions of cells were grown on the indicated media for 2–3 days at 30°C before imaging.
Figure 7. The histone‐binding activity of Pol1 is required for gene silencing at sub‐telomeric and mating‐type loci.

-
A, BThe indicated strains were analysed as in Fig 2A and B. The pol1‐2A2 and pol1‐6A alleles are defective in sub‐telomeric gene silencing. We examined a total of 15 pol1‐2A2 VR::ADE2‐TEL colonies and 19 pol1‐6A VR::ADE2‐TEL colonies.
-
C, DMating‐type silencing was analysed as in Fig 2D–F. Silencing was found to be defective in the pol1‐2A2 and pol1‐6A strains. We examined a total of 15 pol1‐2A2 HMR::2EDA colonies and 13 pol1‐6A HMR::2EDA colonies.
Discussion
Long before the identification of the CMG helicase first made possible the characterization of the eukaryotic replisome, work with eukaryotic DNA viruses suggested that the tethering of Pol α to the replicative helicase might be an important feature of replisome assembly in eukaryotic cells. For example, Pol α was shown to interact directly with the replicative helicases of SV40 (Dornreiter et al, 1990) and bovine papillomaviruses (Park et al, 1994; Bonne‐Andrea et al, 1995), as part of a minimal replisome in which the viral helicase associates with eukaryotic Pol α and topoisomerase 1, but not with the leading strand polymerase or with other eukaryotic replisome components that interact with the CMG helicase. However, tethering of Pol α to the eukaryotic replisome is more complex than for eukaryotic viruses, since it is largely dependent upon the Ctf4 protein (Zhu et al, 2007; Gambus et al, 2009; Tanaka et al, 2009a), in addition to a residual interaction of Pol α with the CMG helicase (Appendix Fig S1, Sengupta et al, 2013). This role for Ctf4 was first described for budding yeast but is likely to be conserved broadly in other species, since Ctf4 from fission yeast, human and frog has also been reported to interact with Pol α (Zhu et al, 2007; Tanaka et al, 2009b; Kilkenny et al, 2017).
Recent in vitro studies of a reconstituted budding yeast replisome system suggested that Pol α synthesizes RNA‐DNA hybrids in a distributive fashion at eukaryotic DNA replication forks, at least on a naked DNA template, implying that replisome tethering of Pol α by Ctf4 does not on its own ensure efficient priming (Yeeles et al, 2017). Consistent with this idea, Ctf4 is dispensable for synthesis of the lagging strand by the reconstituted yeast replisome (Yeeles et al, 2015). Efficient priming by Pol α might instead depend on functional interactions with the CMG helicase (Georgescu et al, 2015; Taylor & Yeeles, 2018), the single‐strand DNA‐binding protein RPA (Dornreiter et al, 1992; Nasheuer et al, 1992; Braun et al, 1997), the DNA template or even chromatin (Kurat et al, 2017).
Our data indicate that Ctf4‐dependent tethering of Pol α and other factors to the eukaryotic replisome has evolved to serve functions that go beyond DNA synthesis. Strikingly, CIP‐box mutations in the known partners of budding yeast Ctf4 have no impact on DNA synthesis, but instead lead to phenotypes that reflect the roles of Ctf4 in other aspects of chromosome replication, such as sister chromatid cohesion (Samora et al, 2016), preserving the length of chromosome 12 that contains the rDNA (Villa et al, 2016), or the maintenance of gene silencing (this study). These findings highlight the role of Ctf4 as a hub in the eukaryotic replisome that coordinates diverse processes with DNA synthesis. Moreover, these data emphasize the notion that replisome assembly in eukaryotes evolved not simply to facilitate timely and efficient DNA synthesis, but also to promote chromosome replication in all its complexity, by coupling the CMG helicase to other processes that can only be realized as part of a dynamic multi‐protein assembly.
Our data indicate that the amino‐terminal tail of the Pol α DNA polymerase subunit contains a novel histone‐binding activity. Unexpectedly, given that the identification of the motif was based on its partial resemblance to the H3‐H4 binding motif of human Mcm2, we show that the histone‐binding motif of human Pol α binds to histones H2A‐H2B. The Pol α histone‐binding motif appears to be more complex than the equivalent element in Mcm2‐NT, and it will be fascinating in future studies to investigate the structural basis for histone binding by Pol α‐NT, for both the human and yeast proteins.
Despite the differences in histone binding by Mcm2 and Pol α, mutation of the histone‐binding motif in either protein produces very similar phenotypes in budding yeast cells. Firstly, DNA synthesis is not impaired (Fig 6 and Appendix Fig S6D), even when both motifs are mutated simultaneously (Fig EV5). Instead, the histone‐binding activities of both Mcm2 and Pol1 are required to preserve gene silencing in telomeric and mating‐type loci (Figs 7A–D and EV2), and yet are largely dispensable for gene silencing in the rDNA repeats (Fig EV3).
Both at telomeres and at the silent mating‐type genes, a complex of the silent information repressor (SIR) proteins Sir2‐Sir3‐Sir4 is first recruited to specific sites and then spreads to mediate gene silencing over a distance of several kilobases (Haber, 2012; Wellinger & Zakian, 2012). The initial recruitment of Sir2‐Sir3‐Sir4 at telomeres is mediated by the Rap1 protein bound to the telomeric DNA repeats (Wellinger & Zakian, 2012), whereas at the silent mating‐type loci, the origin recognition complex promotes the recruitment of the Sir1 protein (Haber, 2012), which in turn loads the first Sir2‐Sir3‐Sir4 complex. Once recruited to telomeres and to mating‐type silencers, Sir2 deacetylates lysine residues in the amino‐terminal tails of histones H3 and H4, within the adjacent nucleosomes. Deacetylation of H4K16 is particularly important and promotes the stable binding of Sir2‐Sir3‐Sir4, initiating an iterative process by which Sir2‐Sir3‐Sir4 are able to spread away from the initial site of recruitment, driven by successive rounds of histone deacetylation by Sir2 and the consequent and stable binding of Sir2‐3‐4. Across the region to which the Sir proteins are bound, they mediate gene silencing by inhibiting the recruitment of transcription factors and tethering the loci to the nuclear periphery.
Our data indicate that Mcm2 and Ctf4‐tethered Pol α are required to preserve the functional integrity of chromatin regions that are silenced by Sir2‐3‐4. We suggest that the replisome's histone‐binding activities help to retain parental histone complexes after their initial displacement from DNA at replication forks, so that the histones can be re‐deposited close to their original location. If the histone‐binding activity of the replisome is abrogated by mutation, a higher proportion of parental histones from other loci, with acetylated H4 and H3 tails, might then be deposited onto nascent DNA across the previously silenced regions, thus leading to displacement of Sir2‐3‐4 and the disruption of gene silencing. In such circumstances, it is likely that there is insufficient time before the next S‐phase for Sir2‐Sir3‐Sir4 to spread once again across the formerly silenced region, thus explaining the gene silencing defect in the histone‐binding mutants.
Gene silencing in the rDNA is considerably more complex than at the telomeric and silent mating‐type loci (Huang & Moazed, 2003; Huang et al, 2006; Srivastava et al, 2016). Although the Sir2 histone deacetylase still plays an important role, both Sir3 and Sir4 are dispensable for rDNA silencing. This is because Sir2 is recruited to the rDNA by the RENT complex, which in turn associates with Fob1 that is bound to the replication fork barrier (Srivastava et al, 2016). Moreover, the Tof2 protein is also recruited by Fob1 and supports a Sir2‐independent mechanism of silencing. The latter pathway is understood poorly, but our data suggest that rDNA silencing does not require the faithful retention of parental histone complexes close to their original location during the passage of a DNA replication fork, implying that rDNA silencing can be re‐established de novo after the passage of a replication fork, before the S‐phase of the subsequent cell cycle.
When the parental DNA duplex is unwound at eukaryotic DNA replication forks, parental histones are released transiently into the nucleoplasm before re‐associating with nascent DNA (Radman‐Livaja et al, 2011; Kurat et al, 2017). Very little is known about the mechanisms of parental histone transfer during chromosome replication, but past work showed that an intact tetramer of parental histones H3‐H4 is preserved on nascent DNA after replication (Prior et al, 1980; Yamasu & Senshu, 1990; Vestner et al, 2000; Katan‐Khaykovich & Struhl, 2011), and a recent study highlighted a role for the eukaryotic replisome in ensuring that histones H3‐H4 are retained close to their original genomic location after the passage of a DNA replication fork (Madamba et al, 2017).
We favour a model in which the unwinding of the initial segment of nucleosomal DNA by the CMG helicase allows FACT to use its multiple histone‐binding activities to destabilize histone‐DNA interactions within the remainder of the nucleosome. Firstly, FACT detaches a dimer of H2A‐H2B from the octamer (Belotserkovskaya et al, 2003; Tsunaka et al, 2016), and binds to the H3‐H4 tetramer of the resulting histone hexamer (Tsunaka et al, 2016), destabilizing histone‐DNA interactions and promoting the continued unwinding of nucleosomal DNA by CMG (Kurat et al, 2017). After the DNA has been stripped away, the histone hexamer is then chaperoned and retained in the vicinity of the replication fork (potentially together with the detached H2A‐H2B dimer), by the binding of multiple replisome components including FACT, Mcm2 and Pol α, before the local re‐deposition of the parental histones onto nascent DNA behind the replisome (Kurat et al, 2017). The latter process is coupled to the assembly of additional nucleosomes from newly synthesized histones, and the rapid exchange of H2A‐H2B in nascent chromatin then leads to the observed situation, whereby parental H3‐H4 tetramers are inherited stably during chromosome replication, whereas H2A‐H2B are considerably more dynamic.
Whereas human FACT and Mcm2 are known to bind to adjacent non‐overlapping sites on the H3‐H4 tetramer (Huang et al, 2015; Richet et al, 2015; Tsunaka et al, 2016), our data indicate that human Pol α binds to H2A‐H2B, and all three replisome components from budding yeast are able to bind simultaneously to the same chromatin‐derived histone complexes (perhaps to a histone hexamer, after FACT detaches an H2A‐H2B dimer from the octamer). The ability of multiple replisome components to bind to different surfaces on the same nucleosome‐derived histone complexes creates the possibility of a relay mechanism, whereby parental histone complexes are passed from one replisome component to another before deposition on nascent DNA immediately behind the replisome. Alternatively, simultaneous binding by multiple replisome components might be required to chaperone the parental histone complexes that have been released from DNA.
Consistent with our proposed role for multiple replisome components in the processing of parental histones, recent structural work indicates that both Ctf4‐Pol α and the amino‐terminal tail of Mcm2 are located at the front face of the yeast replisome (Georgescu et al, 2017; Douglas et al, 2018) and thus are orientated towards the site from which parental histones are released during DNA unwinding at replication forks. The precise location of FACT in the replisome has yet to be determined, but FACT is part of the replisome progression complex that assembles around the CMG helicase (Gambus et al, 2006). It is likely that the replisome can accommodate at least one additional copy of FACT, tethered to Pol α (Miles & Formosa, 1992), plus a further chaperone for histone complexes released from chromatin, namely the Dpb3‐Dpb4 subunits of Pol ε (Tackett et al, 2005). As seen upon mutation of the histone‐binding activity of Mcm2 or Pol α, cells lacking Dpb3–Dpb4 are viable in the absence of the Mec1 checkpoint kinase (Appendix Fig S7) and are defective in sub‐telomeric silencing (Iida & Araki, 2004) but not rDNA silencing (Fig EV3).
In the future, it will be of particular interest to study organisms with more complex epigenetic regulation of gene expression than Saccharomyces cerevisiae, since such studies might reveal more complex phenotypes upon mutation of the replisome's histone‐binding activities. For example, we note that a recent study of the plant orthologue of Ctf4 (Zhou et al, 2017) found that it was required to preserve gene silencing that is dependent upon trimethylation of histone H3K27—an epigenetic mark not found in budding yeast. Moreover, previous work showed that mutations in fission yeast Pol alpha also lead to defects in gene silencing, reflecting displacement from chromatin of the Swi6 chromodomain protein that binds to H3K9Me—another mark that is absent in S. cerevisiae (Nakayama et al, 2001). Nevertheless, the budding yeast replisome provides an important model system with which to identify histone‐binding activities that are highly conserved in other species, thus defining motifs and residues that can be studied subsequently in metazoa or plants.
Materials and Methods
Yeast strains and growth
All S. cerevisiae strains used in this study are listed in Appendix Table S1. Cells were grown in rich medium (1% yeast extract, 2% peptone) supplemented with 2% glucose (YPD), 2% raffinose (YPRaff) or 2% galactose (YPGal). G1 phase samples were obtained by arresting exponentially growing cells (7 × 106/ml) with 7.5 μg/ml α‐factor (Pepceuticals) until 90% were unbudded and S samples by releasing G1‐arrested cells for 30′ (at 24°C) into fresh medium lacking mating pheromone. To arrest cells in G2‐M phase, a mid‐exponential culture was treated with 5 μg/ml nocodazole (Sigma‐Aldrich, M1404) for 2 h.
Dilution spotting
For the experiments in Figs EV4 and EV5, cells were diluted to 3 × 106, 3 × 105, 3 × 104 and 3 × 103 cells/ml in PBS. Subsequently, spots containing 50 × 104, 50 × 103, 50 × 102 or 50 cells were then placed on the indicated media (YPD or YPD + 100 mM hydroxyurea), before growth for 2 days at 30°C.
Measurement of DNA content
Cells were fixed and prepared for flow cytometry as described previously (Labib et al, 1999) and then analysed with a FACSCanto II flow cytometer (Becton Dickinson) and FlowJo software (Tree Star).
Immunoprecipitation of proteins from yeast cell extracts
Frozen yeast pellets from 1 l cultures (except for Figs 1B and 6A, and Appendix Figs S1 and S6C, for which 250 ml culture was used) were prepared as described previously (Maric et al, 2014) in 100 mM Hepes‐KOH pH 7.9, 100 mM KOAc, 10 mM Mg(OAc)2, 2 mM EDTA, 2 mM β‐glycerophosphate, 2 mM NaF, 1 mM DTT, 1% protease inhibitor cocktail (P8215, Sigma‐Aldrich), 1× “Complete protease inhibitor Cocktail” (05056489001, Roche; a 25× stock was made by dissolving one tablet in 1 ml of water). For each sample, 2–3 g of frozen cells was ground in a SPEX SamplePrep 6780 Freezer/Mill. Chromosomal DNA was digested with Universal nuclease (123991963, Fisher; 1,600 U/ml, except for Figs 1 and 6, Appendix Figs S1 and S6 where 400 U/ml was used) for 30 min at 4°C, before two consecutive centrifugations at 25,000 × g for 30 min and 100,000 × g for 1 h. The resulting cell extract was added to magnetic beads (Dynabeads M‐270 Epoxy, 14302D, Life Technologies) coupled to rabbit IgG (S1265, Sigma‐Aldrich) and incubated for 2 h at 4°C. Proteins were separated on a 4–12% Bis‐Tris NuPAGE gel (WG1402BOX, Life Technologies) in 1× MES and detected by immunoblotting, using previously characterized antibodies (Gambus et al, 2006, 2009; Foltman et al, 2013; Maric et al, 2014).
For mass spectrometry analysis, gels were stained with colloidal Coomassie (Simply Blue, LC6060, Thermo Fisher) and each sample lane was then cut into 40 bands before digestion with trypsin. Peptides were analysed by nanoliquid chromatography–tandem mass spectrometry with an Orbitrap Velos (MS Bioworks).
Gene silencing assays at sub‐telomeric, mating type or rDNA loci
The insertion of the ADE2 marker near the right telomere of chromosome 5 was described previously (Iida & Araki, 2004; Foltman et al, 2013). We also generated an insertion of ADE2 at the HMR locus on chromosome 3 (Foltman et al, 2013), but further analysis showed that the integration of ADE2 was aberrant, as the marker did not segregate correctly in subsequent crosses (CE, unpublished data). This explains why we did not previously detect a defect in mating‐type silencing in mcm2‐3A cells (Foltman et al, 2013). In contrast, the HMR::ADE2 and HMR::2EDA strains in this study were derived by crossing the previously described strains YLS409 (HMR::ADE2 with the ADE2 promoter proximal to the HMR‐E silencer element) and YLS410 (HMR::2EDA with the ADE2 promoter distal to the HMR‐E silencer element). The YLS409 and YLS410 strains (Sussel et al, 1993) were kindly provided by Professor David Shore (University of Geneva).
Diploid cells were generated that were heterozygous for the insertions of ADE2 at the telomeric or mating‐type loci. The cells were then sporulated and micro‐dissected on YPD plates, using the Singer MSM system (Singer Instruments). The colonies were grown for 3 days at 30°C, followed by a further 3 days of incubation at 4°C to intensify the red colour associated with ADE2 expression.
The rDNA silencing assays in Fig EV3 were based on strains generously provided by Danesh Moazed (Huang et al, 2006). Serial dilutions were made as described above, and then plated on “Synthetic Complete” medium (SC medium, non‐selective for the URA3 marker), or the equivalent mediums lacking Uracil (SC‐Ura, selective for the URA3 marker) for 2 days at 30°C, to detect expression of the “modified URA3” (mURA3) marker gene (Smith & Boeke, 1997) at the leu2 locus (leu2::mURA3) or within the non‐transcribed spacers 1 and 2 of an rDNA repeat (NTS1::mURA3 and NTS2::mURA3).
Telomere length assay
Yeast strains were grown in YPD medium until they reached 1.5–2 × 107 cells/ml. Genomic DNA from each strain was isolated using the MasterPure Yeast DNA Purification Kit (MPY80200, Epicentre), and then, 6 μg was digested with XhoI for 12 h at 37°C. The digested DNA was resolved in a 0.8% agarose gel in 0.5× TBE for 24 h at 25 V, then transferred to a nitrocellulose membrane and hybridized with a probe specific to the Y' element of the telomere. The probe was labelled with 32P‐dCTP using a Random Primed DNA labelling Kit (Roche 11 004 760 001). The membrane was exposed to BAS Imaging Plates (Fujifilm), which were then analysed using a Fujifilm FLA‐5100 scanner and AIDA Image Analysis software (Raytest).
Cloning and expression of human POLA1‐NTD and MCM2‐NTD
Synthetic gene fragments encoding the wild‐type and mutated human POLA1 N‐terminal domain (HsPOLA1‐NTD) residues M1 to G110 and human MCM2 N‐terminal domain (HsMCM2‐NTD) residues G43 to D160, followed by a Twin‐Strep tag, were purchased from Integrated DNA Technologies (IDT; Iowa, USA). The synthetic DNA fragments were designed with appropriate restriction sites at their 5′ and 3′ termini to allow insertion of HsPOLA1NTD‐Twin‐Strep into the expression vectors pOP5MP (for HsPOLA1NTD‐Twin‐Strep) and pOP3MP (for HsMCM2NTD‐Twin‐Strep), both of which introduce an N‐terminal 8His‐MBP tag that is cleavable by PreScission Protease (http://hyvonen.bioc.cam.ac.uk/pOP-vectors/); note that the C‐terminal Avi‐tag in pOPMP3 was removed during the cloning procedure. The amino acid sequences encoded by the resulting constructs are provided in Appendix Fig S8.
The expression plasmids were transformed into E. coli Rosetta2 (DE3) cells and grown in 1 l of 2× YT medium (Formedium; Hunstanton, UK) + 50 μg/ml Ampicillin (Melford; Chelsworth, UK) at 37°C to an OD600 of about 0.4. The temperature was lowered to 20°C and after continued growth to an OD600 of about 0.7, and protein expression was induced overnight by addition of 1 mM isopropyl‐β‐D‐thiogalactopyranoside (Generon; Slough, UK). Cells were harvested by centrifugation at 4,000 × g for 10 min and the pellet was resuspended in 25 ml buffer A [50 mM Tris–HCl pH 8.0, 500 mM NaCl, 2 mM imidazole + 1 tablet EDTA‐free SIGMAFAST protease inhibitors per 100 ml (Sigma; St. Louis, USA)] and then snap‐frozen in liquid nitrogen before storage at −80°C.
Purification of HsPOLA1‐NTD and HsMCM2‐NTD
Cell pellets were thawed and 200 μl benzonase (25 U/μl, Merck; USA) were added. The cells were lysed by sonicating for 12 pulses of 10 s on and 20 s off, in iced water at 50% amplitude (Sonics VCX130; CT, USA). The lysed cells were centrifuged at 35,000 × g for 45 min at 4°C, and the supernatant was then filtered (0.45 μm). The His‐tagged proteins were purified by nickel affinity chromatography on a 1 ml gravity‐flow Ni‐NTA column (Qiagen; MD, USA). The column was equilibrated with 10 column volumes of equilibration buffer (20 mM Tris–HCl pH 8.0, 300 mM NaCl, 2 mM imidazole), and the filtered supernatant was loaded onto the column. The flow‐through was discarded, and the column was then washed with 20 column volumes of equilibration buffer and 20 column volumes of wash buffer (20 mM Tris–HCl pH 8.0, 300 mM NaCl, 10 mM imidazole). Proteins were then eluted with 10 column volumes of elution buffer (20 mM Tris–HCl pH 8.0, 300 mM NaCl, 300 mM imidazole), and 1 ml fractions were collected. Fractions containing the His‐tagged protein were pooled, concentrated and loaded on a size‐exclusion chromatography column (HiLoad 16/60 Superdex 200; GE Life‐Science, USA) pre‐equilibrated in SEC buffer (20 mM Tris–HCl pH 8, 300 mM NaCl). Peak fractions containing the purified proteins were pooled and incubated overnight at 4°C in the presence of GST‐tagged PreScission Protease to remove His‐MBP tandem tags. The digested proteins were then loaded onto a 1 ml GSTrap HP column (GE Life‐Science; USA) to remove the GST‐tagged PreScission Protease, followed by a 1 ml gravity‐flow Ni‐NTA column to remove the His‐tagged MBP. The purity of proteins was determined by SDS polyacrylamide gel electrophoresis and protein concentration was determined by UV spectroscopy, using theoretical ε280 (http://web.expasy.org/protparam/), and amino acid analysis (http://www3.bioc.cam.ac.uk/pnac/aaa.html).
Preparation of high‐salt HeLa nuclear extracts
The procedure for preparing HeLa cell extracts was adapted from a previously described protocol (Abmayr et al, 2006). All operations were performed at 0–4°C. Initially, 5 × 109 HeLa cells (IpraCell; Mons, Belgium) were washed in 50 ml PBS buffer, and the cell pellet was then resuspended in five packed cell volumes of lysis buffer (10 mM Tris–HCl pH 8.0, 100 mM NaCl, 0.15 mM spermine, 0.5 mM spermidine, 0.5 M sucrose, 2 mM EDTA, 0.1% Igepal, 2 mM DTT and 1 tablet SIGMAFAST protease inhibitors per 100 ml). Cells were homogenized with a glass Dounce homogenizer with 15 up‐and‐down strokes using a type B pestle. Nuclei were then collected by centrifuging at 3,000 × g for 10 min and were resuspended in one packed nuclear volume of low‐salt buffer (20 mM HEPES‐NaOH pH 7.8, 25% glycerol, 1.5 mM MgCl2, 20 mM KCl, 1 mM EDTA, 2 mM DTT, one tablet SIGMAFAST protease inhibitors per 50 ml). Following incubation for 10 min on rollers, the sample was spun at 5,000 × g for 10 min and the nuclear pellet was slowly resuspended in two packed‐nuclear‐volumes of high‐salt buffer 1 (20 mM HEPES‐NaOH pH 7.8, 25% glycerol, 1.5 mM MgCl2, 1 M KCl, 1 mM EDTA, 2 mM DTT and one tablet SIGMAFAST protease inhibitors per 50 ml) and then homogenized with 10 up‐and‐down strokes. Nuclei were incubated for 30 min with continuous rotation and 200 mg of polyethyleneimine (Polysciences; IL, USA) was added. The nuclear debris and DNA were pelleted at 50,000 × g for 30 min, and the supernatant was collected. 50 mg of polyethyleneimine was added to the supernatant that, after a 2‐h incubation on rollers, was then dialysed against high‐salt buffer 2 (20 mM HEPES‐NaOH pH 7.8, 25% glycerol, 1.5 mM MgCl2, 1 M NaCl, 1 mM EDTA, 2 mM DTT) for 2 h. Following centrifugation at 50,000 × g for 30 min, the supernatant, corresponding to the high‐salt nuclear extract, was collected, aliquoted and stored at −80°C.
Pull‐down assays for HsPOLA1‐NTD and HsMCM2‐NTD
The purified HsPOLA1‐NTD and HsMCM2‐NTD fragments were bound to beads via the Twin‐Strep tag at the C‐terminal end of each protein. All operations were performed at 0–4°C, using the “pull‐down buffer” (PD buffer) containing 20 mM Tris–HCl pH 8.0, 150 mM NaCl, 1 mM DTT, 5% glycerol and 0.005% Tween‐20 plus one tablet SIGMAFAST protease inhibitors per 50 ml buffer. For each pull‐down assay, 300 μl Strep‐Tactin Superflow 50% suspension resin (IBA; Goettingen, Germany) was washed twice in 1 ml PD buffer, before addition of 45 μl of Twin‐Strep‐tagged bait at 40 μM concentration, together with 150 μl PD buffer. Following a 1‐h incubation on rollers, the samples were washed twice with 1 ml PD buffer. 150 μl High‐salt HeLa nuclear extract was then added to the resin, together with 150 μl PD buffer, and after a 1‐h incubation on rollers, the samples were washed twice with 1 ml PD buffer. Elution was performed by adding 150 μl elution buffer (7.5 mM d‐Desthiobiotin (Sigma) dissolved in PD buffer) to the pelleted resin. Following a 15‐min incubation, the resin was spun down at 5,000 g for 3 min and 90 μl of the supernatant was acid precipitated by the addition of 30 μl, ice‐cold, trichloroacetic acid and incubation for 30 min on ice. The precipitated proteins were pelleted at 20,000 g for 10 min, the supernatant was aspirated off and the pellets washed once with 1 ml acidified acetone (10 mM HCl in acetone) and twice in pure acetone. The pellets were air‐dried overnight, resuspended in denaturing gel loading buffer containing fresh 8 M urea and analysed on Bis‐Tris 20% acrylamide gels (29:1 Acrylamide : Bis‐Acrylamide). Gel bands for the pulled‐down proteins were identified by MALDI fingerprinting (http://www3.bioc.cam.ac.uk/pnac/proteomics.html).
Cloning and expression of human histones H2A and H2B
Synthetic genes coding for human histones H2A type 1 (UniProt P0C0S8) and human H2B type 1‐A (UniProt Q96A08) were purchased from Integrated DNA Technologies (IDT; Iowa, USA). The synthetic genes were codon‐optimized for expression in E. coli, with appropriate restriction sites at their 5′ and 3′ termini to allow insertion into the dual expression vector pRSFDuet (Novagen, Merck; Darmstadt, Germany). The synthetic H2A gene was inserted into the Multiple Cloning Site 1 (MCS1) downstream from the vector hexa‐histidine site, followed by a PreScission protease site. The synthetic H2B gene was cloned into MCS2.
The expression plasmid was transformed into E. coli Rosetta2 (DE3) cells, and H2A‐H2B expression was induced as described above for HsPOLA1‐NTD and HsMCM2‐NTD. The harvested cell pellet was resuspended in 25 ml (per 1 l cell culture) of lysis buffer (50 mM sodium phosphate pH 7.8, 2 M NaCl, 20 mM imidazole), then snap‐frozen in liquid nitrogen and stored at −80°C.
Purification of human H2A‐H2B dimer
The thawed cell pellet was lysed by sonicating at 50% amplitude (Sonics VCX130; CT, USA) for 12 pulses of 20 s on and 40 s off, in iced water. The lysed cell extract was centrifuged at 35,000 × g for 45 min at 4°C, and the supernatant was then filtered (0.45 μm). The His‐tagged proteins were purified by nickel affinity chromatography on a 1 ml HisTrap column (GE Life‐Science, USA). The column was equilibrated with five column volumes of lysis buffer, and the filtered supernatant was loaded onto the column. The flow‐through was discarded, and the column was washed with 20 column volumes of lysis buffer. The H2A‐H2B dimer was eluted with 50 mM sodium phosphate pH 7.8, 2 M NaCl, 300 mM imidazole, and 1 ml fractions were collected. Fractions containing the His‐tagged H2A‐H2B dimer were pooled, concentrated and loaded on a size‐exclusion chromatography column (HiLoad 16/60 Superdex 75; GE Life‐Science, USA) pre‐equilibrated in 20 mM Tris–HCl pH 8.0, 2 M NaCl, 0.1 mM TCEP). The peak fractions containing the purified dimer were pooled and concentrated in an Amicon Ultra‐15 spin column (3000 MWCO; Merck; Darmstadt, Germany) at 4°C to a volume of about 2 ml (≅ 100 μM H2A‐H2B dimer). The NaCl concentration was reduced from 2 M to 0.25 M by twofold dilution steps with 20 mM Tris–HCl pH 8.0, 1 mM DTT and the sample was then incubated overnight at 4°C in the presence of GST‐tagged PreScission Protease, in order to remove the hexa‐histidine tag. The digested dimer was loaded onto a 1 ml GSTrap HP column (GE Life‐Science; USA) to remove the GST‐tagged PreScission protease, and the flow‐through was then loaded onto a 1 ml HiTrap‐Heparin column (GE Life‐Science, USA). The column was equilibrated with 20 mM Tris–HCl pH 8.0, 100 mM NaCl, 0.1 mM TCEP, and H2A‐H2B dimer was eluted with a 20‐column volume gradient that ranged from the above “low‐salt” buffer to 20 mM Tris–HCl pH 8.0, 2 M NaCl, 0.1 mM TCEP “high‐salt” buffer. Fractions containing pure H2A‐H2B dimer were pooled, concentrated, snap‐frozen in liquid nitrogen and stored at −80°C. The purity of the proteins was determined by SDS polyacrylamide gel electrophoresis and the protein concentration was determined by UV spectroscopy, using the theoretical ε280 value (http://web.expasy.org/protparam/). The integrity of the human H2A‐H2B dimer was determined by multi‐angle light scattering (MALS) in 20 mM Tris–HCl pH 8.0, 150 mM NaCl, 5% glycerol, 0.005% Tween 20 (not shown).
Isothermal titration calorimetry
Isothermal titration calorimetry experiments were conducted using a MicroCal iTC200 instrument (Malvern Panalytical; Malvern, UK). Prior to the experiments, human H2A‐H2B dimer, wild‐type HsPOLA1‐NTD and HsPOLA1‐NTD‐6A were re‐purified on a Superdex 75 10/300 GL column (GE Life‐Science, USA) equilibrated in ITC buffer (20 mM Tris–HCl pH 8.0, 150 mM NaCl, 5% glycerol, 0.005% Tween 20). Titration experiments were carried out at 25°C using 19 injections and 750 r.p.m. The first injection was 0.4 μl at 0.5 μl/s and was discarded from the data analysis. All the following injections were 2 μl at 0.5 μl/s with 180‐s spacing between each injection. 80 μM Pol1‐NTD fragments were injected into 10 μM H2A‐H2B dimer in the cell. For controls, 80 μM Pol1‐NTD constructs were injected into ITC buffer and the heat evolved was subtracted from the heat produced by the interaction of HsPol1NTD with the H2A‐H2B dimer. The data were analysed using a single‐site binding model and a non‐linear regression analysis using ORIGIN software (OriginLab Corporation, USA). The variables for ΔH (enthalpy change), N (stoichiometry) and Ka (association equilibrium constant) were allowed to “float” during the analysis.
Author contributions
LP noticed the similarity between the amino terminus of Pol1 and the histone‐binding motif of Mcm2. KL and CE conceived and designed the project and CE performed all experiments, except for those in Fig 5 and Appendix Fig S8 that were carried out by JDM and AD. KL wrote the manuscript with contributions and critical comments from CE, JM and LP.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
We gratefully acknowledge the support of the Wellcome Trust (references 102943/Z/13/Z and 104641/Z/14/Z for investigator awards to KL and LP respectively, plus reference 097945/B/11/Z for flow cytometry), and the Medical Research Council (core grant MC_UU_12016/13). We thank Magda Foltman for assistance in the early stages of this project; Arturo Calzada for the telomeric DNA probe; Hiroyuki Araki, Danesh Moazed and David Shore for strains; Fabrizio Villa for help with running CHEF gels; and MRC PPU Reagents and Services (https://mrcppureagents.dundee.ac.uk) for plasmids and antibodies.
The EMBO Journal (2018) 37: e99021
References
- Abmayr SM, Yao T, Parmely T, Workman JL (2006) Preparation of nuclear and cytoplasmic extracts from mammalian cells. Curr Protoc Mol Biol Chapter 12: Unit 12.1 [DOI] [PubMed] [Google Scholar]
- Adams AK, Holm C (1996) Specific DNA replication mutations affect telomere length in Saccharomyces cerevisiae . Mol Cell Biol 16: 4614–4620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell SP, Labib K (2016) Chromosome duplication in Saccharomyces cerevisiae . Genetics 203: 1027–1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belotserkovskaya R, Oh S, Bondarenko VA, Orphanides G, Studitsky VM, Reinberg D (2003) FACT facilitates transcription‐dependent nucleosome alteration. Science 301: 1090–1093 [DOI] [PubMed] [Google Scholar]
- Bonne‐Andrea C, Santucci S, Clertant P, Tillier F (1995) Bovine papillomavirus E1 protein binds specifically DNA polymerase alpha but not replication protein A. J Virol 69: 2341–2350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun KA, Lao Y, He Z, Ingles CJ, Wold MS (1997) Role of protein‐protein interactions in the function of replication protein A (RPA): RPA modulates the activity of DNA polymerase alpha by multiple mechanisms. Biochemistry 36: 8443–8454 [DOI] [PubMed] [Google Scholar]
- Burgers PMJ, Kunkel TA (2017) Eukaryotic DNA replication fork. Annu Rev Biochem 86: 417–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dornreiter I, Hoss A, Arthur AK, Fanning E (1990) SV40 T antigen binds directly to the large subunit of purified DNA polymerase alpha. EMBO J 9: 3329–3336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dornreiter I, Erdile LF, Gilbert IU, von Winkler D, Kelly TJ, Fanning E (1992) Interaction of DNA polymerase alpha‐primase with cellular replication protein A and SV40 T antigen. EMBO J 11: 769–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas ME, Ali FA, Costa A, Diffley JFX (2018) The mechanism of eukaryotic CMG helicase activation. Nature 555: 265–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foltman M, Evrin C, De Piccoli G, Jones RC, Edmondson RD, Katou Y, Nakato R, Shirahige K, Labib K (2013) Eukaryotic replisome components cooperate to process histones during chromosome replication. Cell Rep 3: 892–904 [DOI] [PubMed] [Google Scholar]
- Fumasoni M, Zwicky K, Vanoli F, Lopes M, Branzei D (2015) Error‐free DNA damage tolerance and sister chromatid proximity during DNA replication rely on the Polalpha/Primase/Ctf4 Complex. Mol Cell 57: 812–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambus A, Jones RC, Sanchez‐Diaz A, Kanemaki M, van Deursen F, Edmondson RD, Labib K (2006) GINS maintains association of Cdc45 with MCM in replisome progression complexes at eukaryotic DNA replication forks. Nat Cell Biol 8: 358–366 [DOI] [PubMed] [Google Scholar]
- Gambus A, van Deursen F, Polychronopoulos D, Foltman M, Jones RC, Edmondson RD, Calzada A, Labib K (2009) A key role for Ctf4 in coupling the MCM2‐7 helicase to DNA polymerase alpha within the eukaryotic replisome. EMBO J 28: 2992–3004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgescu RE, Langston L, Yao NY, Yurieva O, Zhang D, Finkelstein J, Agarwal T, O'Donnell ME (2014) Mechanism of asymmetric polymerase assembly at the eukaryotic replication fork. Nat Struct Mol Biol 21: 664–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgescu RE, Schauer GD, Yao NY, Langston LD, Yurieva O, Zhang D, Finkelstein J, O'Donnell ME (2015) Reconstitution of a eukaryotic replisome reveals suppression mechanisms that define leading/lagging strand operation. Elife 4: e04988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgescu R, Yuan Z, Bai L, de Luna Almeida Santos R, Sun J, Zhang D, Yurieva O, Li H, O'Donnell ME (2017) Structure of eukaryotic CMG helicase at a replication fork and implications to replisome architecture and origin initiation. Proc Natl Acad Sci USA 114: E697–E706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwell PW, Kronmal SL, Porter SE, Gassenhuber J, Obermaier B, Petes TD (1995) TEL1, a gene involved in controlling telomere length in S. cerevisiae, is homologous to the human ataxia telangiectasia gene. Cell 82: 823–829 [DOI] [PubMed] [Google Scholar]
- Haber JE (2012) Mating‐type genes and MAT switching in Saccharomyces cerevisiae . Genetics 191: 33–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond CM, Stromme CB, Huang H, Patel DJ, Groth A (2017) Histone chaperone networks shaping chromatin function. Nat Rev Mol Cell Biol 18: 141–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Moazed D (2003) Association of the RENT complex with nontranscribed and coding regions of rDNA and a regional requirement for the replication fork block protein Fob1 in rDNA silencing. Genes Dev 17: 2162–2176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Brito IL, Villen J, Gygi SP, Amon A, Moazed D (2006) Inhibition of homologous recombination by a cohesin‐associated clamp complex recruited to the rDNA recombination enhancer. Genes Dev 20: 2887–2901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Stromme CB, Saredi G, Hodl M, Strandsby A, Gonzalez‐Aguilera C, Chen S, Groth A, Patel DJ (2015) A unique binding mode enables MCM2 to chaperone histones H3‐H4 at replication forks. Nat Struct Mol Biol 22: 618–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iida T, Araki H (2004) Noncompetitive counteractions of DNA polymerase epsilon and ISW2/yCHRAC for epigenetic inheritance of telomere position effect in Saccharomyces cerevisiae . Mol Cell Biol 24: 217–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimi Y, Komamura Y, You Z, Kimura H (1998) Biochemical function of mouse minichromosome maintenance 2 protein. J Biol Chem 273: 8369–8375 [DOI] [PubMed] [Google Scholar]
- Kang YH, Galal WC, Farina A, Tappin I, Hurwitz J (2012) Properties of the human Cdc45/Mcm2‐7/GINS helicase complex and its action with DNA polymerase epsilon in rolling circle DNA synthesis. Proc Natl Acad Sci USA 109: 6042–6047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katan‐Khaykovich Y, Struhl K (2011) Splitting of H3‐H4 tetramers at transcriptionally active genes undergoing dynamic histone exchange. Proc Natl Acad Sci USA 108: 1296–1301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny ML, De Piccoli G, Perera RL, Labib K, Pellegrini L (2012) A conserved motif in the C‐terminal tail of DNA polymerase alpha tethers primase to the eukaryotic replisome. J Biol Chem 287: 23740–23747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny ML, Simon AC, Mainwaring J, Wirthensohn D, Holzer S, Pellegrini L (2017) The human CTF4‐orthologue AND‐1 interacts with DNA polymerase alpha/primase via its unique C‐terminal HMG box. Open Biol 7: 170217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Dallmann HG, McHenry CS, Marians KJ (1996) Coupling of a replicative polymerase and helicase: a tau‐DnaB interaction mediates rapid replication fork movement. Cell 84: 643–650 [DOI] [PubMed] [Google Scholar]
- Kunkel TA, Burgers PMJ (2017) Arranging eukaryotic nuclear DNA polymerases for replication: specific interactions with accessory proteins arrange Pols alpha, delta, and in the replisome for leading‐strand and lagging‐strand DNA replication. BioEssays 39: 1700070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurat CF, Yeeles JT, Patel H, Early A, Diffley JF (2017) Chromatin controls DNA replication origin selection, lagging‐strand synthesis, and replication fork rates. Mol Cell 65: 117–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labib K, Diffley JFX, Kearsey SE (1999) G1‐phase and B‐type cyclins exclude the DNA‐replication factor Mcm4 from the nucleus. Nat Cell Biol 1: 415–422 [DOI] [PubMed] [Google Scholar]
- Langston LD, Zhang D, Yurieva O, Georgescu RE, Finkelstein J, Yao NY, Indiani C, O'Donnell ME (2014) CMG helicase and DNA polymerase epsilon form a functional 15‐subunit holoenzyme for eukaryotic leading‐strand DNA replication. Proc Natl Acad Sci USA 111: 15390–15395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lustig AJ, Petes TD (1986) Identification of yeast mutants with altered telomere structure. Proc Natl Acad Sci USA 83: 1398–1402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madamba EV, Berthet EB, Francis NJ (2017) Inheritance of histones H3 and H4 during DNA replication in vitro . Cell Rep 21: 1361–1374 [DOI] [PubMed] [Google Scholar]
- Maric M, Maculins T, De Piccoli G, Labib K (2014) Cdc48 and a ubiquitin ligase drive disassembly of the CMG helicase at the end of DNA replication. Science 346: 1253596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles J, Formosa T (1992) Evidence that POB1, a Saccharomyces cerevisiae protein that binds to DNA polymerase alpha, acts in DNA metabolism in vivo . Mol Cell Biol 12: 5724–5735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller TC, Costa A (2017) The architecture and function of the chromatin replication machinery. Curr Opin Struct Biol 47: 9–16 [DOI] [PubMed] [Google Scholar]
- Nakayama J, Allshire RC, Klar AJ, Grewal SI (2001) A role for DNA polymerase alpha in epigenetic control of transcriptional silencing in fission yeast. EMBO J 20: 2857–2866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasheuer HP, Vonwinkler D, Schneider C, Dornreiter I, Gilbert I, Fanning E (1992) Purification and functional‐characterization of bovine rp‐a in an invitro sv40‐dna replication system. Chromosoma 102: S52–S59 [DOI] [PubMed] [Google Scholar]
- Park P, Copeland W, Yang L, Wang T, Botchan MR, Mohr IJ (1994) The cellular DNA polymerase alpha‐primase is required for papillomavirus DNA replication and associates with the viral E1 helicase. Proc Natl Acad Sci USA 91: 8700–8704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prior CP, Cantor CR, Johnson EM, Allfrey VG (1980) Incorporation of exogenous pyrene‐labeled histone into Physarum chromatin: a system for studying changes in nucleosomes assembled in vivo . Cell 20: 597–608 [DOI] [PubMed] [Google Scholar]
- Radman‐Livaja M, Verzijlbergen KF, Weiner A, van Welsem T, Friedman N, Rando OJ, van Leeuwen F (2011) Patterns and mechanisms of ancestral histone protein inheritance in budding yeast. PLoS Biol 9: e1001075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richet N, Liu D, Legrand P, Velours C, Corpet A, Gaubert A, Bakail M, Moal‐Raisin G, Guerois R, Compper C, Besle A, Guichard B, Almouzni G, Ochsenbein F (2015) Structural insight into how the human helicase subunit MCM2 may act as a histone chaperone together with ASF1 at the replication fork. Nucleic Acids Res 43: 1905–1917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riera A, Barbon M, Noguchi Y, Reuter LM, Schneider S, Speck C (2017) From structure to mechanism‐understanding initiation of DNA replication. Genes Dev 31: 1073–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samora CP, Saksouk J, Goswami P, Wade BO, Singleton MR, Bates PA, Lengronne A, Costa A, Uhlmann F (2016) Ctf4 links DNA replication with sister chromatid cohesion establishment by recruiting the Chl1 helicase to the replisome. Mol Cell 63: 371–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta S, van Deursen F, de Piccoli G, Labib K (2013) Dpb2 integrates the leading‐strand DNA polymerase into the eukaryotic replisome. Curr Biol 23: 543–552 [DOI] [PubMed] [Google Scholar]
- Simon AC, Zhou JC, Perera RL, van Deursen F, Evrin C, Ivanova ME, Kilkenny ML, Renault L, Kjaer S, Matak‐Vinkovic D, Labib K, Costa A, Pellegrini L (2014) A Ctf4 trimer couples the CMG helicase to DNA polymerase alpha in the eukaryotic replisome. Nature 510: 293–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JS, Boeke JD (1997) An unusual form of transcriptional silencing in yeast ribosomal DNA. Genes Dev 11: 241–254 [DOI] [PubMed] [Google Scholar]
- Srivastava R, Srivastava R, Ahn SH (2016) The epigenetic pathways to ribosomal DNA silencing. Microbiol Mol Biol Rev 80: 545–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sussel L, Vannier D, Shore D (1993) Epigenetic switching of transcriptional states: cis‐ and trans‐acting factors affecting establishment of silencing at the HMR locus in Saccharomyces cerevisiae . Mol Cell Biol 13: 3919–3928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter B, Tong A, Chang M, Yu L, Brown GW, Boone C, Rine J (2004) The origin recognition complex links replication, sister chromatid cohesion and transcriptional silencing in Saccharomyces cerevisiae . Genetics 167: 579–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tackett AJ, Dilworth DJ, Davey MJ, O'Donnell M, Aitchison JD, Rout MP, Chait BT (2005) Proteomic and genomic characterization of chromatin complexes at a boundary. J Cell Biol 169: 35–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka H, Katou Y, Yagura M, Saitoh K, Itoh T, Araki H, Bando M, Shirahige K (2009a) Ctf4 coordinates the progression of helicase and DNA polymerase alpha. Genes Cells 14: 807–820 [DOI] [PubMed] [Google Scholar]
- Tanaka H, Kubota Y, Tsujimura T, Kumano M, Masai H, Takisawa H (2009b) Replisome progression complex links DNA replication to sister chromatid cohesion in Xenopus egg extracts. Genes Cells 14: 949–963 [DOI] [PubMed] [Google Scholar]
- Taylor RG, Yeeles JT (2018) The initial response of a eukaryotic replisome to DNA damage. Mol Cell 70: 1067–1080.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunaka Y, Fujiwara Y, Oyama T, Hirose S, Morikawa K (2016) Integrated molecular mechanism directing nucleosome reorganization by human FACT. Genes Dev 30: 673–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vestner B, Waldmann T, Gruss C (2000) Histone octamer dissociation is not required for in vitro replication of simian virus 40 minichromosomes. J Biol Chem 275: 8190–8195 [DOI] [PubMed] [Google Scholar]
- Villa F, Simon AC, Ortiz Bazan MA, Kilkenny ML, Wirthensohn D, Wightman M, Matak‐Vinkovic D, Pellegrini L, Labib K (2016) Ctf4 is a hub in the eukaryotic replisome that links multiple CIP‐box proteins to the CMG helicase. Mol Cell 63: 385–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellinger RJ, Zakian VA (2012) Everything you ever wanted to know about Saccharomyces cerevisiae telomeres: beginning to end. Genetics 191: 1073–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasu K, Senshu T (1990) Conservative segregation of tetrameric units of H3 and H4 histones during nucleosome replication. J Biochem 107: 15–20 [DOI] [PubMed] [Google Scholar]
- Yeeles JT, Deegan TD, Janska A, Early A, Diffley JF (2015) Regulated eukaryotic DNA replication origin firing with purified proteins. Nature 519: 431–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeeles JT, Janska A, Early A, Diffley JF (2017) How the eukaryotic replisome achieves rapid and efficient DNA replication. Mol Cell 65: 105–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Tergemina E, Cui H, Forderer A, Hartwig B, Velikkakam James G, Schneeberger K, Turck F (2017) Ctf4‐related protein recruits LHP1‐PRC2 to maintain H3K27me3 levels in dividing cells in Arabidopsis thaliana . Proc Natl Acad Sci USA 114: 4833–4838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Ukomadu C, Jha S, Senga T, Dhar SK, Wohlschlegel JA, Nutt LK, Kornbluth S, Dutta A (2007) Mcm10 and And‐1/CTF4 recruit DNA polymerase alpha to chromatin for initiation of DNA replication. Genes Dev 21: 2288–2299 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File