

Abstract
In eukaryotes, heterochromatin regions are typically subjected to transcriptional silencing. DNA methylation has an important role in such silencing and has been studied extensively. However, little is known about how methylated heterochromatin regions are subjected to silencing. We conducted a genetic screen and identified an epcr (enhancer of polycomb‐related) mutant that releases heterochromatin silencing in Arabidopsis thaliana. We demonstrated that EPCR1 functions redundantly with its paralog EPCR2 and interacts with PWWP domain‐containing proteins (PWWPs), AT‐rich interaction domain‐containing proteins (ARIDs), and telomere repeat binding proteins (TRBs), thus forming multiple functionally redundant protein complexes named PEAT (PWWPs‐EPCRs‐ARIDs‐TRBs). The PEAT complexes mediate histone deacetylation and heterochromatin condensation and thereby facilitate heterochromatin silencing. In heterochromatin regions, the production of small interfering RNAs (siRNAs) and DNA methylation is repressed by the PEAT complexes. The study reveals how histone deacetylation, heterochromatin condensation, siRNA production, and DNA methylation interplay with each other and thereby maintain heterochromatin silencing.
Keywords: DNA methylation, EPL, heterochromatin silencing, histone deacetylation, PEAT complex, siRNA
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; RNA Biology
Introduction
Heterochromatin is highly condensed and typically subjected to transcriptional silencing in eukaryotes (Kim & Zilberman, 2014; Martienssen & Moazed, 2015). DNA methylation plays an important role in transcriptional silencing of heterochromatin (Law & Jacobsen, 2010; He et al, 2011; Matzke & Mosher, 2014). In Arabidopsis, the mechanisms of DNA methylation are well known. The DNA methyltransferases MET1, CMT3, and CMT2 are responsible for maintenance of DNA methylation at CG, CHG, and CHH (H is A, T, and C) sites, respectively (Ronemus et al, 1996; Bartee et al, 2001; Lindroth et al, 2001; Stroud et al, 2013; Zemach et al, 2013). The SNF2 type chromatin remodeling protein DDM1 facilitates maintenance of DNA methylation (Zemach et al, 2013). Decondensation of heterochromatin and release of silencing were observed in the mutants defective in maintenance of DNA methylation (Soppe et al, 2002; Lindroth et al, 2004), suggesting that maintenance of DNA methylation has a significant role in heterochromatin condensation and silencing.
DNA methylation is established by the de novo DNA methyltransferase DRM2 through RNA‐directed DNA methylation (RdDM) pathway in Arabidopsis (Law & Jacobsen, 2010; Matzke & Mosher, 2014). In the RdDM pathway, the DNA‐dependent RNA polymerases IV and V (Pol IV and Pol V) are responsible for the production of small interfering RNAs (siRNAs) and long non‐coding RNAs (lncRNAs), respectively (Haag & Pikaard, 2011). Both Pol IV‐dependent siRNAs and Pol V‐dependent lncRNAs are required for RdDM. Pol IV‐dependent siRNAs are not only produced from pericentromeric heterochromatin regions but also from transposable elements and DNA repeats that are dispersed throughout euchromatin regions (Zhang et al, 2007; Mosher et al, 2008). Pol V‐dependent lncRNAs are specifically enriched on edges of silenced transposable elements and are thought to determine boundaries of heterochromatin (Bohmdorfer et al, 2016). The RdDM pathway mediates de novo DNA methylation at CHH sites and to a lesser extent at CG and CHG sites (Stroud et al, 2013). In the RdDM pathway, Pol V but not Pol IV was reported to affect heterochromatin condensation (Pontes et al, 2009). Two conserved MORC family proteins, MORC1 and MORC6, were demonstrated to be required for heterochromatin condensation and silencing (Moissiard et al, 2012). We previously demonstrated that the Su(var)3‐9 homologs SUVH2 and SUVH9, which are RdDM components, interact with MORC1 and MORC6 and thereby link the RdDM pathway and heterochromatin condensation (Liu et al, 2016). However, it is thought that the RdDM pathway is unlikely to be involved in heterochromatin condensation at the whole‐genome level.
In addition to DNA methylation, the repressive histone marks H3K9me2 and H3K27me1 are enriched in heterochromatin regions (Zhou et al, 2010; Roudier et al, 2011). In the mutants defective in H3K9me2 and H3K27me1, decondensation of heterochromatin was observed (Soppe et al, 2002; Lindroth et al, 2004; Jacob et al, 2009), indicating that H3K9me2 and H3K27me1 are required for heterochromatin condensation. Histone acetylation, a histone mark related to transcriptional activation, is maintained at a relatively low level in heterochromatin regions (Zhou et al, 2010; Roudier et al, 2011). Acetylation of histone peptides can be added by histone acetyltransferases and removed by histone deacetylases. In Arabidopsis, there are 12 histone acetyltransferases, among which two MYST‐type histone acetyltransferases, HAM1 and HAM2, mediate histone acetylation specifically at H4K5 sites and are essential for gametophytic development (Pandey et al, 2002; Earley et al, 2007; Latrasse et al, 2008). In yeast and animals, the MYST‐type histone acetyltransferases were demonstrated to act as catalytic subunits of conserved NuA4/Tip60‐type histone acetyltransferase complexes (Doyon & Cote, 2004). There are 19 Arabidopsis histone deacetylases, in which only HDA6 is known to mediate heterochromatin condensation and transcriptional silencing (Aufsatz et al, 2002; Probst et al, 2004; Hollender & Liu, 2008; Liu et al, 2012). HDA6 is not only required for histone deacetylation but also for DNA methylation (Earley et al, 2010; To et al, 2011; Liu et al, 2012; Blevins et al, 2014). However, relatively little is known about how heterochromatin regions are specifically subjected to histone deacetylation, DNA methylation, and transcriptional silencing.
In this study, we carry out a reverse genetic screen to identify new chromatin regulators that are required for heterochromatin silencing. By combining genetic and biochemical methods, we identify multiple functionally redundant protein complexes and demonstrate that the complexes are required for heterochromatin condensation and silencing. The complexes, which we have termed PEAT, are composed of Pro‐Trp‐Trp‐Pro domain‐containing proteins (PWWPs), enhancer of polycomb‐related proteins (EPCRs), AT‐rich interaction domain‐containing proteins (ARIDs), and telomere repeat binding proteins (TRBs). We demonstrate that the complexes are involved in histone deacetylation and heterochromatin silencing, and show that they play a regulatory role in RdDM. The PEAT complexes' involvement in heterochromatin silencing occurs independently of their regulatory role in RdDM. Intriguingly, the complexes interact with both histone acetyltransferases (HAM1 and HAM2) and histone deacetylases (HDA6 and HDA9). The study provides a new insight for understanding how histone acetylation and heterochromatin condensation are regulated in eukaryotes.
Results
EPCR1 and EPCR2 are regulators of transcriptional silencing and development
With the goal of identifying unknown regulators that are required for transcriptional silencing of transposable elements and of other repetitive DNA sequences, we screened a collection of homozygous SALK T‐DNA insertion mutants and searched for mutants in which transcriptional silencing of the solo LTR (solo long terminal repeat) locus is released. The collection included mutants of the 550 chromatin‐related genes implicated in our previous study of chromatin regulation (Zhang et al, 2016). Although the screen did not identify any unknown mutants that strongly released transcriptional silencing of solo LTR, we found that this silencing was weakly released in a mutant (SALK_039205) harboring a T‐DNA insertion in AT4G32620, which encodes an enhancer of polycomb‐related protein that we here named EPCR1 (Fig 1A; Appendix Fig S1). EPCR1 has a close paralog, AT5G04670, which we named EPCR2 (Fig EV1A).
Figure 1. EPCR1 and EPCR2, two enhancer of polycomb‐related proteins, are required for transcriptional silencing and development.
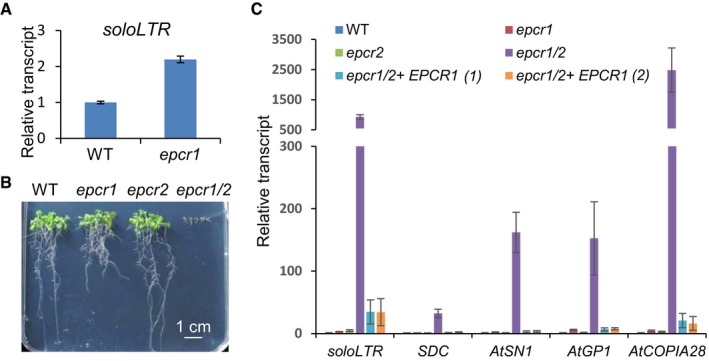
- The transcript level of solo LTR in the wild type and the epcr1 mutant. Error bars are standard deviations of three technical replicates. The experiment was independently performed three times, and similar results were obtained.
- The developmental phenotype of the epcr1 and epcr2 single mutants and the epcr1/2 double mutant relative to the wild type. Two‐week‐old seedlings grown on vertical MS medium plates are shown.
- The transcript levels of solo LTR, SDC, AtSN1, AtGP1, and AtCOPIA28 in the wild type, epcr1 and epcr2 single mutants, epcr1/2 double mutant, and two individual EPCR1 transgenic lines in the epcr1/2 double‐mutant background. Error bars are standard deviations of three biological replicates.
Figure EV1. Analysis of enhancer of polycomb‐like proteins in Arabidopsis .
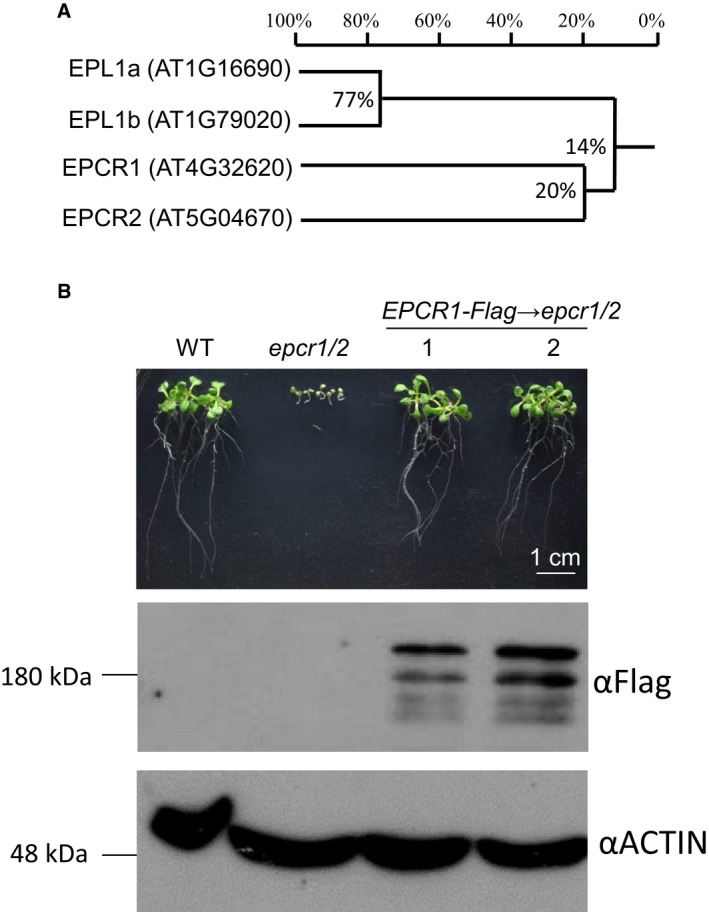
- Phylogenetic analysis of enhancer of polycomb‐like proteins in Arabidopsis.
- The EPCR1‐Flag transgene complements the developmental defects in the epcr1/2 mutant. We generated a construct harboring a native promoter‐driven EPCR1 genomic sequence and transformed the construct into the epcr1/2 mutant for complementation testing. Two independent transgenic lines were selected for analysis. Morphological phenotypes of 10‐day‐old seedlings are shown. Western blot shows the expression of EPCR1‐Flag protein in the epcr1/2 mutant, and ACTIN signals are shown as loading controls.
Source data are available online for this figure.
In view of the weak effect of epcr1 on transcriptional silencing of the solo LTR locus, we speculated that EPCR2 may function redundantly with EPCR1 and thereby mask the effect of the epcr1 mutant on transcriptional silencing. We obtained an epcr2 mutant (SALK_024125) and crossed it with epcr1 to obtain an epcr1 epcr2 (epcr1/2) double mutant (Appendix Fig S1). The epcr1 and epcr2 single mutants showed no obvious developmental defects, with the exception that the epcr1 mutant had shorter roots than the wild type (Fig 1B). In the epcr1/2 double mutant, however, plant development was halted at the cotyledon stage (Fig 1B), suggesting that EPCR1 and EPCR2 are functionally redundant and are required for early seedling development. Quantitative PCR (qPCR) analysis showed that the transcription of solo LTR was weakly induced in the epcr1 and epcr2 single mutants; this induction was much enhanced in the epcr1/2 double mutant (Fig 1C). Furthermore, we evaluated whether the epcr1 and epcr2 mutations affect the transcript levels of SDC, AtSN1, AtGP1, and AtCOPIA28, which are well‐known genomic loci that are silenced by DNA methylation. SDC is a protein‐coding gene that is redundantly silenced by CHG maintenance methylation and RdDM on tandem repeats of its promoter region (Henderson & Jacobsen, 2008). Our qPCR experiment indicated that the transcript levels of these loci were either not induced or were only weakly induced in the epcr1 and epcr2 single mutants, but the induction was markedly enhanced in the epcr1/2 double mutant (Fig 1C).
For complementation testing, we created a construct harboring a native promoter‐driven genomic EPCR1 sequence and transformed the construct into the EPCR1 −/− ;EPCR2 +/− plants. We successfully generated EPCR1 transgenic plants in the epcr1/2 double‐mutant background and found that the plants had no developmental defects (Fig EV1B), suggesting that EPCR1 complemented the developmental defect in the epcr1/2 double‐mutant background—the plants were able to grow beyond the cotyledon stage until flowering. Moreover, the EPCR1 transgene in the epcr1/2 double mutant significantly restored the transcriptional silencing phenotype even though EPCR2 is still defective in the transgenic plants (Fig 1C). These results demonstrate that EPCR1 and EPCR2 function redundantly in transcriptional silencing and early seedling development.
PWWPs, EPCRs, ARIDs, and TRBs form complexes in vivo
To understand how the EPCR1/2 proteins mediate transcriptional silencing, we created transgenic plants expressing EPCR1‐Flag fusion construct driven by the EPCR1 promoter (pEPCR1:EPCR1‐Flag). We then identified proteins that interact with EPCR1‐Flag using Flag‐tag‐based affinity purification in combination with mass spectrometry. We initially confirmed that the EPCR1‐Flag protein could be purified and identified via mass spectrometry, and then identified proteins that were co‐purified with it. These included an uncharacterized PWWP (Pro‐Trp‐Trp‐Pro)‐containing protein that we named PWWP1, three uncharacterized AT‐rich interaction domain‐containing proteins that we named ARID2, ARID3, and ARID4, and two previously characterized telomere repeat binding proteins, TRB1 and TRB2 (Schrumpfova et al, 2014) (Table 1; Dataset EV1). We next generated pARID2:ARID2‐Flag and pTRB1:TRB1‐Flag transgenic plants and conducted Flag‐tag‐based affinity purification experiments to identify proteins that interact with the ARID2‐Flag and TRB1‐Flag fusion proteins. ARID2‐Flag was co‐purified with AIRD2/3/4, EPCR1, PWWP1/2/3, and TRB1/2; TRB1‐Flag was co‐purified with TRB1, ARID2/3/4, EPCR1/2, and PWWP1 (Table 1; Dataset EV1). These results collectively indicate that EPCR1/2, ARID2/3/4, PWWP1/2/3, and TRB1/2 appear to interact with each other in Arabidopsis.
Table 1.
Identification of co‐purified proteins of EPCR1, ARID2, and TRB1 by mass spectrometry
| Gene | Protein | MW (Da) | EPCR1 pull‐down | ARID2 pull‐down | TRB1 pull‐down | |||
|---|---|---|---|---|---|---|---|---|
| Mascot | Spectra | Mascot | Spectra | Mascot | Spectra | |||
| Score | Score | Score | ||||||
| AT2G17410 | ARID2 | 86,517 | 2,103 | 46 | 5,735 | 122 | 1,132 | 45 |
| AT1G20910 | ARID3 | 44,238 | 997 | 25 | 1,716 | 44 | 320 | 8 |
| AT1G76510 | ARID4 | 48,010 | 904 | 22 | 2,341 | 53 | 483 | 16 |
| AT4G32620 | EPCR1 | 173,830 | 2,840 | 81 | 1,690 | 50 | 871 | 33 |
| AT5G04670 | EPCR2 | 87,699 | 0 | 0 | 0 | 0 | 417 | 13 |
| AT3G03140 | PWWP1 | 87,331 | 1,360 | 32 | 1,889 | 36 | 882 | 24 |
| AT1G51745 | PWWP2 | 65,004 | 0 | 0 | 1,419 | 27 | 0 | 0 |
| AT3G21295 | PWWP3 | 69,816 | 0 | 0 | 563 | 17 | 0 | 0 |
| AT1G49950 | TRB1 | 35,209 | 934 | 23 | 660 | 12 | 1,244 | 35 |
| AT5G67580 | TRB2 | 32,993 | 247 | 7 | 59 | 1 | 0 | 0 |
To confirm these protein–protein interactions, we crossed Flag‐ and Myc‐tagged EPCR1, ARID2, ARID3, and TRB1 transgenic plants with each other and conducted co‐immunoprecipitation (co‐IP) using anti‐Flag and anti‐Myc antibodies in their progeny. The co‐IP experiments confirmed that EPCR1 interacts with ARID2, ARID3, and TRB1, and confirmed that ARID2 interacts with TRB1 (Fig 2A–C). By yeast two‐hybrid combined with in vitro pull‐down assays, we verified these protein–protein interactions: PWWPs directly interact with ARIDs, EPCRs, and TRBs, while ARIDs, EPCRs, and TRB1 do not directly interact with each other (Fig 2D–G; Appendix Fig S2). Moreover, the yeast two‐hybrid results indicated that the self‐interaction and the interaction of paralogs occurred in the following sets of paralogous proteins: the ARIDs, the PWWPs, and the TRBs (Fig 2D). However, it remains to be determined whether these interactions occur in Arabidopsis.
Figure 2. EPCR1/2 interact with ARID2/3/4, PWWP1/2/3, and TRB1/2, and form complexes.
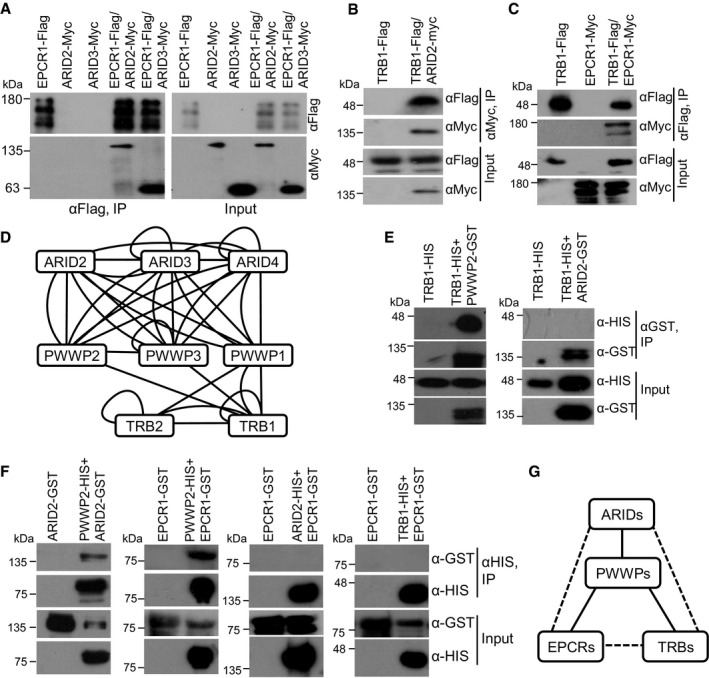
- The interaction between EPCR1 and ARID2 or ARID3. Arabidopsis plants carrying EPCR1‐Flag and ARID2‐Myc or ARID3‐Myc transgenes were used for co‐IP.
- The interaction between TRB1 and ARID2. Arabidopsis plants carrying TRB1‐Flag and/or ARID2‐Myc transgenes were used for co‐IP.
- The interaction between TRB1 and EPCR1. Arabidopsis plants carrying TRB1‐Flag and/or EPCR1‐Myc transgenes were used for co‐IP.
- The diagram of protein–protein interactions that were detected in yeast two‐hybrid assays. A line represents an interaction between two proteins. If a protein was indicated to interact with another protein when it was fused with both GAL4‐BD and GAL4‐AD, both straight and curved lines are shown between the two proteins. A lariat loop indicates that a protein can form a homodimer.
- The interaction between TRB1 and PWWP2 or ARID2 was detected by an GST pull‐down assay.
- The interaction between EPCR1, PWWP2, ARID2, and TRB1 was detected by an HIS pull‐down assay. The full‐length PWWP2, ARID2, and TRB1, and the EPCR1‐N terminal (1‐500 aa) were used for the interaction assay.
- The diagram indicates the interactions among EPCR1, PWWP2, ARID2, and TRB1 as determined by pull‐down assay, yeast two‐hybrid, and co‐IP. The solid line and the broken line indicate, respectively, direct and indirect protein–protein interactions.
Source data are available online for this figure.
To determine whether PWWP, EPCR, ARID, and TRB proteins form protein complexes in vivo, we performed gel filtration coupled with Western blotting (Fig EV2). The result indicated that PWWP2, EPCR1, ARID2, and TRB1 were all present in large‐size fractions (> 443 kDa) even though TRB1 was also shown in small‐size fractions (Fig EV2), supporting the notion that PWWP2, EPCR1, ARID2, and TRB1 form a multi‐subunit protein complex in Arabidopsis. Considering that there are paralogs of the PWWP, EPCR, ARID, and TRB proteins, and given the functional redundancy between the paralogs, we predict that these paralogs form multiple functionally redundant complexes, which we hereafter deem PEAT (PWWPs‐EPCRs‐ARIDs‐TRBs) complexes. It is worth noting that, as determined by affinity purification coupled with mass spectrometric analyses, ARID2 but not EPCR1 and TRB1 can co‐purify paralogs (Table 1). The results suggest that while the paralogs of the ARID paralogs can exist in one PEAT complex, the paralogs of the EPCR and TRB proteins are mutually exclusive in different PEAT complexes.
Figure EV2. PWWP, EPCR, ARID, and TRB proteins form complexes in vivo as determined by gel filtration.
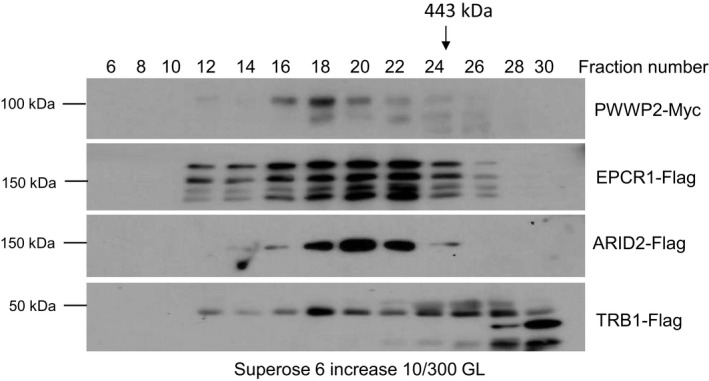
Proteins were extracted from PWWP2‐Myc, EPCR1‐Flag, ARID2‐Flag, and TRB1‐Flag transgenic plants in the wild‐type background. The proteins were eluted on a Superose 6 increase (10/300 GL) column. The epitope‐tagged proteins in different fractions were detected by antibodies against the Myc or Flag‐tag. Arrows indicate the fractions that correspond to the standard proteins 443 kDa.Source data are available online for this figure.
The PEAT complexes are required for transcriptional silencing and development
Considering the effects of EPCR1/2 on transcriptional silencing and development, and given the interactions of EPCR1/2 with ARID2/3/4, PWWP1/2/3, and TRB1/2, we next asked whether ARID2/3/4, PWWP1/2/3, and TRB1/2 also function in transcriptional silencing and development. We observed no obvious developmental defects in any of the following single mutants: arid2 (SALK_026835), arid3 (SALK_022359), arid4 (SALK_007400), pwwp1 (SAIL_342_C09), pwwp2 (SALK_136093), or pwwp3 (SALK_042581) (Appendix Fig S1). We predicted that, like EPCR1/2, the ARID2/3/4 proteins and/or the PWWP1/2/3 proteins may exhibit functional redundancy. To evaluate this, we obtained double and triple mutants of ARID2/3/4 by crossing. Although no obvious development defects were observed for any of the double mutants of ARID2/3/4 (Appendix Fig S3A), the development of the ARID2/3/4 triple mutant halted at the cotyledon stage in seedlings grown on MS medium plates (Fig 3A; Appendix Fig S3A). The halted development phenotype is highly similar for the arID2/3/4 mutant and the epcr1/2 mutant. Intriguingly, when we transferred plants from MS medium plates into soil at the cotyledon stage, the arid2/3/4 mutant, but not the epcr1/2 mutant, was able to continue development up until flowering and seed set. We also tried to generate double and triple mutants of PWWP1/2/3 by crossing. We successfully obtained pwwp1/2, pwwp1/3, and pwwp2/3 double mutants; these mutants showed no obvious developmental defects. We failed to obtain a pwwp1/2/3 triple mutant, suggesting that the PWWP1/2/3 proteins function redundantly and are required for normal gametophytic and/or sporophytic development. However, we identified a mutant with a genotype harboring homozygous T‐DNA insertions in PWWP1 and PWWP2 and a heterozygous T‐DNA insertion in PWWP3 (PWWP1 −/−;PWWP −/−;PWWP3 +/−) that showed pleiotropic developmental defects (Fig 3B). These results indicate that, like EPCR1/2, there is redundancy in the development‐related functions among the ARID2/3/4 proteins and among the PWWP1/2/3 proteins.
Figure 3. The PEAT complexes are required for transcriptional silencing and development.
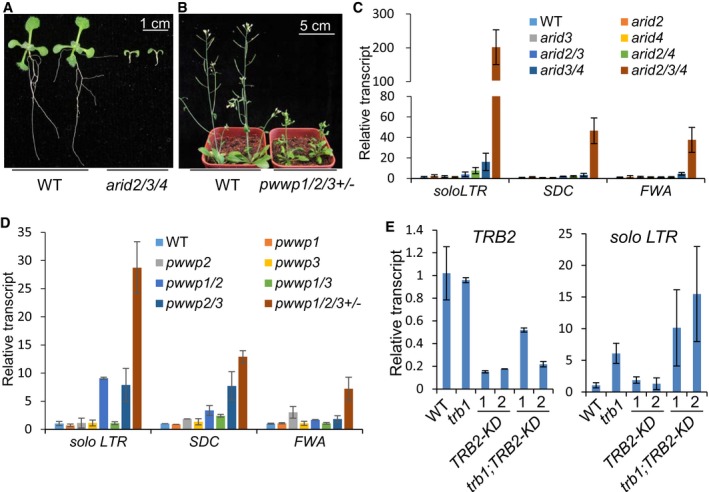
-
AThe developmental phenotype of the arid2/3/4 mutant as compared to the wild type. Two‐week‐old seedlings grown on MS medium are shown.
-
BThe developmental phenotype of the pwwp1/2/3 +/− mutant as compared to the wild type. One‐month‐old plants grown in soil are shown.
-
C, DThe effect of the arid2, arid3, and arid4 mutations, and the effect of the pwwp1, pwwp2, and pwwp3 mutations on the silencing of solo LTR, SDC, and FWA as determined by qPCR. Showing are results of three biological replicates with standard deviations.
-
EThe effect of the trb1 mutation and the TRB2 knockdown (TRB2‐KD) on the silencing of solo LTR, as determined by qPCR (right panel). The transcript level of TRB2 was evaluated to confirm the TRB2 knockdown (left panel). Error bars represent standard deviations of three biological replicates.
We evaluated the transcript levels of solo LTR, SDC, and FWA by qPCR to determine whether or not the arid2/3/4 and pwwp1/2/3 genes have functions relating to transcriptional silencing. Like SDC, FWA is a protein‐coding gene that is silenced by DNA methylation of tandem repeats in its promoter region (Soppe et al, 2000; Chan et al, 2006). In single and double mutants of ARid2/3/4, the transcript levels for the solo LTR, SDC, and FWA loci were either not induced or were only weakly induced; their levels were greatly enhanced in the arid2/3/4 triple mutant (Fig 3C). Similarly, although the transcript levels for the solo LTR, SDC, and FWA loci were either not induced or weakly induced in the single or double mutants of PWwp1/2/3, their levels were markedly induced in the PWWP1 −/−;PWWP −/−;PWWP3 +/− mutant (Fig 3D). These results demonstrate that, like EPCR1/2, the ARID2/3/4 proteins and the PWWP1/2/3 proteins function redundantly in transcriptional silencing.
The telomere repeat binding protein TRB1 was previously reported to facilitate maintenance of telomeres in Arabidopsis (Schrumpfova et al, 2014). Considering the function of EPCRs, ARIDs, and PWWPs in development and transcriptional silencing, we predicted that TRB1/2 may be required for development and transcriptional silencing in addition to their role in the maintenance of telomeres. We obtained a homozygous trb1 mutant (SALK_025147); it had no obvious developmental defects. We obtained a heterozygous trb2 mutant (GK‐103E02), but failed to identify a homozygous mutant in its segregation group, suggesting that TRB2 is required for gametophytic or sporophytic development. The requirement of TRB2 for the viability of plants is consistent with our finding that the PEAT complexes play an important role in development. To determine whether TRB2 is required for transcriptional silencing, we generated TRB2 knockdown lines (TRB2‐KD) to evaluate the function of TRB2 in transcriptional silencing. Fortunately, we obtained TRB2‐KD in both wild‐type and trb1 mutant backgrounds (Fig 3E). No obvious developmental defects were observed in the TRB2‐KD plants, suggesting that residual TRB2 expression levels in the TRB2‐KD lines are sufficient for maintaining a wild‐type development phenotype. Relative to wild‐type plants, the solo LTR transcript level was significantly increased in trb1 plants but not in TRB2‐KD plants (Fig 3E). Of note, the solo LTR transcript level was slightly higher in the trb1;TRB2‐KD plants than in the trb1 plants (Fig 3E). These results establish that TRB1 and TRB2 cooperate to mediate transcriptional silencing.
Considering that transcriptional silencing of the heterochromatic rDNA loci is reported to change in the early seedling development (Mathieu et al, 2003; Earley et al, 2010), it is possible that the release of transcriptional silencing in the PEAT mutants is indirectly caused by the cotyledon stage arrest of the PEAT mutants. To exclude the possibility, the wild type, arid2/3/4, and epcr1/2 mutant seedlings were grown for 2, 4, 7, and 10 days after germination and were harvested for RNA extraction followed by qPCR analysis (Appendix Fig S3B). The aforementioned qPCR analysis indicated that the silencing of the solo LTR, AtSN1, AtGP1, SDC, and AtCOPIA28 loci was released in the epcr1/2 mutant (Fig 1C). Our qPCR analysis indicated that although the expression levels of these loci were affected to a certain degree by developmental stages, the expression of all these loci was markedly increased in both the epcr1/2 and arid2/3/4 mutants compared to the wild type at different development stages (Fig EV3), suggesting that the release of silencing in the mutants of the PEAT complexes is not directly caused by the halted development of these mutants.
Figure EV3. Transcriptional silencing in the wild type and the arid2/3/4 and epcr1/2 mutant seedlings at different development stages.
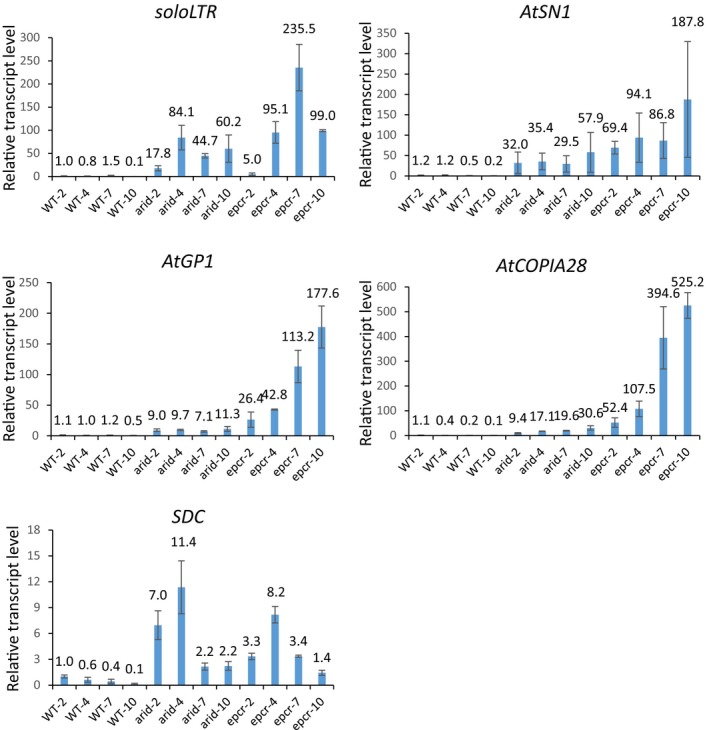
Transcript levels of solo LTR, AtSN1, AtGP1, AtCOPIA28, and SDC in the wild type and the ARID2/3/4, and epcr1/2 mutant seedlings grown for 2, 4, 7, and 10 days after germination. Shown are transcript levels of indicated loci, as determined by qPCR analysis. Error bars represent SD from three independent biological replicates.
The function of the PEAT complexes as determined by RNA‐seq
To assess the function of the PEAT complexes in transcription of TEs and genes at the whole‐genome level, we performed RNA deep sequencing (RNA‐seq) for epcr1/2, arID2/3/4 mutant, and wild‐type plants. We also included the RNA‐directed DNA methylation mutant nrpe1 (mutation in the largest subunit of Pol V), which is known to have increased TE transcription, in our RNA‐seq analysis. We identified 89 and 61 TEs that were significantly upregulated in the epcr1/2 and ARID2/3/4 mutants (P < 0.01; log2 (fold change) > 1; Cufflinks), respectively; only 24 and 22 TEs were significantly downregulated in these two mutants (P < 0.01; log2(fold change)< −1; Cufflinks) (Fig 4A; Dataset EV2). About two‐third of the upregulated TEs in arID2/3/4 (42/61) were also upregulated in epcr1/2 (Fig 4A and B), suggesting that EPCR1/2 and ARID2/3/4 have related functions in transcriptional silencing of TEs. It was notable that the number of upregulated TEs (64 upregulated TEs) in the nrpe1 mutant is comparable to that in the arid2/3/4 mutant and is fewer that in the epcr1/2 mutant (Fig 4A, Dataset EV2). Many upregulated TEs in the epcr1/2 and arid2/3/4 mutants were present in pericentromeric heterochromatin regions (Fig 4C), suggesting that the PEAT complexes are involved in heterochromatin silencing. Further analysis of the RNA‐seq data indicated that many protein‐coding genes are differentially expressed in the epcr1/2 (1,544 up and 2,006 down) and arid2/3/4 mutants (1,216 up and 1,426 down) (Fig 4A; Dataset EV2). There were 712 and 779 genes that were commonly up‐ and downregulated between the epcr1/2 and arid2/3/4 mutants (Fig 4A and B; Dataset EV2). Specifically, the RNA‐seq data indicated that several genes that are known to be required for shoot apical meristem formation and maintenance (STM, CUC1, CUC2, KNAT1, KNAT2, and KNAT6) are significantly upregulated in the arid2/3/4 and epcr1/2 mutants (Dataset EV2). To confirm the effect of arid2/3/4 and epcr1/2 on the expression of these genes, we carried out qPCR and demonstrated that the expression of these genes is significantly upregulated in the arid2/3/4 and epcr1/2 mutants at different developmental stages (Appendix Figs S3B and S4), suggesting that the developmental arrest of the epcr1/2 and arid2/3/4 mutants may be caused by the aberrant expression of these genes.
Figure 4. ARID2/3/4 and EPCR1/2 are required for transcriptional silencing.
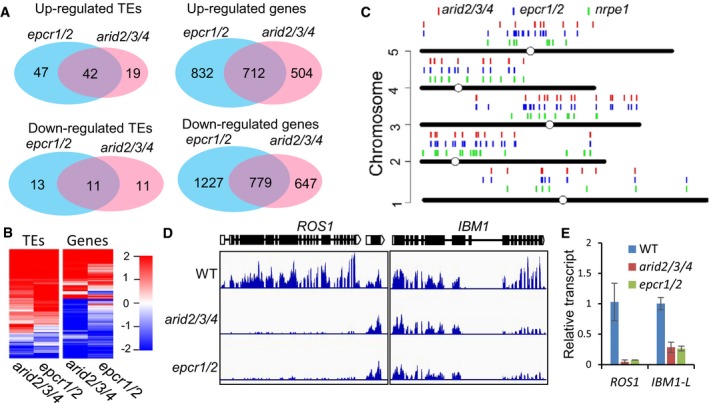
- Venn diagrams showing up‐ and downregulated genes and TEs in the epcr1/2 and arid2/3/4 mutants relative to the wild type, as determined by RNA‐seq. The data are from three independent biological replicates. All the overlaps are highly significant (P < 2.6 × 10−30) as determined by hypergeometric test.
- Heat maps showing differentially transcribed genes and TEs in the arid2/3/4 and epcr1/2 mutants relative to the wild type.
- Distribution of upregulated TEs in nrpe1, epcr1/2, and arid2/3/4 on the Arabidopsis chromosomes.
- Genome browser snapshots showing the transcript patterns of ROS1 and IBM1 in arid2/3/4 and epcr1/2 relative to the wild type.
- Effects of arid2/3/4 epcr1/2 on the transcript levels of ROS1 and IBM1‐L as determined by qPCR. The expression of ROS1 and the longer version of IBM1 (IBM1‐L) was evaluated by qPCR. ACT7 was amplified as an internal control. Bars represent SD from two independent experiments, each with three technical replications.
The DNA glycosylase gene ROS1 is responsible for active DNA demethylation and repression of transcriptional gene silencing (Gong et al, 2002; Zhu, 2009). The transcript level of ROS1 is known to be decreased in the DNA methylation mutants including met1 and RdDM mutants (Mathieu et al, 2007). DNA methylation in the ROS1 promoter region is required for ROS1 expression (Lei et al, 2015; Williams et al, 2015). Our transcriptome data indicated that the transcript level of ROS1 was markedly decreased in both the epcr1/2 and arid2/3/4 mutants (Fig 4D). The JmjC‐type histone H3K9 demethylase gene IBM1 has two different lengths of transcripts (IBM1‐L and IBM1‐S). DNA methylation in an IBM1 intronic region is required for accumulation of the long IBM1 transcript (IBM1‐L) but is not required for accumulation of the short one (IBM1‐S) (Rigal et al, 2012). In the mutants that are defective in DNA methylation and heterochromatin silencing such as met1, cmt3, and kyp/suvh4, the intronic DNA methylation is decreased, which represses the accumulation of the IBM1‐L transcript (Rigal et al, 2012). Our transcriptome data indicated that in epcr1/2 and arid2/3/4, the IBM1‐L transcript level was markedly decreased, whereas the IBM1‐S transcript level was not affected (Fig 4D). Further, we performed qPCR analysis and confirmed the effect of the PEAT mutations (epcr1/2 and arid2/3/4) on the expression of ROS1 and IBM1‐L (Fig 4E). These results demonstrate that the expression of ROS1 and IBM1‐L is affected in the PEAT mutants as well as in the mutants that are defective in DNA methylation and heterochromatin silencing, which is consistent with the observed role of the PEAT complexes in heterochromatin silencing.
The PEAT complexes interact with both histone acetyltransferases and histone deacetylases
To investigate the mechanism of how the PEAT complexes function in heterochromatin silencing, we searched for EPCR1‐Flag, ARID2‐Flag, and TRB1‐Flag interacting proteins that were identified by affinity purification in combination with mass spectrometry. We found two conserved MYST‐type histone acetyltransferases (HAM1 and HAM2) that were co‐purified with EPCR1‐Flag, ARID2‐Flag, and TRB1‐Flag (Table 2; Dataset EV1), suggesting that EPCR1, ARID2, and TRB1 interact with HAM1/2 in vivo. To confirm these interactions, we generated pHAM1:HAM1‐Myc and pHAM2:HAM2‐Myc transgenic plants and identified proteins that interact with HAM1/2 by affinity purification. PWWP1, EPCR1, ARID2/3/4, and TRB1/2 were co‐purified with HAM1‐Myc or HAM2‐Myc (Table 2; Dataset EV3), demonstrating that these PEAT components interact with HAM1/2. Furthermore, we generated transgenic plants expressing differentially tagged (Flag and Myc) versions of EPCR1, ARID2/3/4, TRB1, and HAM1/2, and then crossed these plants with each other. Our co‐IP analysis of the progeny from these crosses confirmed that the PEAT components EPCR1, ARID2/3/4, and TRB1 each interact with the HAM1 and HAM2 acetyltransferases in vivo (Fig 5A–C).
Table 2.
EPCR1, ARID2, and TRB1 interact with HAM1 and HAM2 but not with accessory subunits of the histone acetyltransferase
| Yeast orthologs | Affinity purification | ||||
|---|---|---|---|---|---|
| HAM1/2 | EPL1a/1b | EPCR1 | ARID2 | TRB1 | |
| Esa1 | HAM1/2 | + | + | + | + |
| Epl1 | EPL1a/1b | + | − | − | − |
| Eaf1 | ATEAF1a/1b | + | − | − | − |
| Eaf2 | ATSWC4 | + | − | − | − |
| Yaf9 | GAS41 | + | − | − | − |
| Yng2 | ING2 | + | − | − | − |
| Eaf3 | MRG1 | + | − | − | − |
| Arp4 | ARP4 | + | − | − | − |
| Eaf6 | AT4G14385 | + | − | − | − |
| Tra1 | AT4G36080 | + | − | − | − |
| Act1 | ACT1/2/11/12 | + | − | − | − |
| – | EPCR1/2 | − | + | + | + |
| – | ARID2/3/4 | − | + | + | + |
| – | PWWP1/2/3 | − | + | + | + |
| – | TRB1/2 | − | + | + | + |
Figure 5. The interaction of EPCR1, ARID2, ARID3, ARID4, and TRB1 with the histone acetyltransferases HAM1 and HAM2 and the histone deacetylases HDA9 and HDA6.
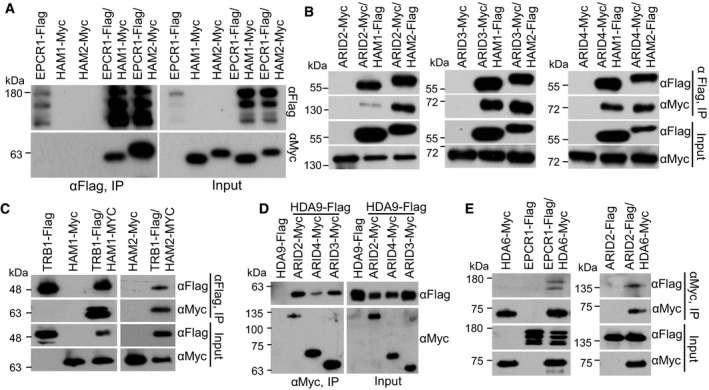
- The interaction of EPCR1 with HAM1 and HAM2. EPCR1‐Flag transgenic plants were crossed to HAM1‐Myc and HAM2‐Myc transgenic plants. The progeny were used to evaluate the interaction between EPCR1 with HAM1 and HAM2 by co‐IP.
- The interaction of ARID2, ARID3, and ARID4 with HAM1 and HAM2. ARID2‐Myc, ARID3‐Myc, and ARID4‐Myc transgenic plants were crossed to HAM1‐Flag and HAM2‐Flag transgenic plants, and their progeny were used for co‐IP.
- The interaction of TRB1 with HAM1 and HAM2. TRB1‐Flag transgenic plants were crossed to HAM1‐Myc and HAM2‐Myc transgenic plants, and their progeny were used for co‐IP.
- The interaction of HDA9 with ARID2, ARID3, and ARID4. ARID2‐Myc, ARID3‐Myc, and ARID4‐Myc transgenic plants were crossed to HDA9‐Flag transgenic plants, and their progeny were used for co‐IP.
- The interaction of HDA6 with EPCR1 and ARID2. EPCR1‐Flag and ARID2‐Flag transgenic plants were crossed to HDA6‐Myc transgenic plants, and their progeny were used for co‐IP.
Source data are available online for this figure.
In addition to their interaction with the PEAT components, our affinity purification experiments showed that HAM1‐Myc and HAM2‐Myc interact with 10 conserved accessory subunits of the NuA4 histone acetyltransferase complex (Table 2; Dataset EV3). Thus, as in yeast and animals, Arabidopsis HAM1 and HAM2 appear to function in the NuA4 histone acetyltransferase complex. Two enhancer of polycomb‐like proteins (EPL1a and EPL1b), which are distantly related to the PEAT subunits EPCR1/2, are conserved subunits of the NuA4 histone acetyltransferase complex (Table 2). We generated pEPL1a:EPL1a‐Flag and pEPL1b:EPL1b‐Flag transgenic plants and used affinity purification to identify proteins that interact with EPL1a and EPL1b. EPL1a and EPL1b were co‐purified with all the 10 other subunits of the NuA4 histone acetyltransferase complex but none of PEAT components (Table 2; Dataset EV3). Moreover, none of the 10 accessory subunits of the NuA4 histone acetyltransferase complex were identified via affinity purification of EPCR1‐Flag, ARID2‐Flag, or TRB1‐Flag (Table 2; Dataset EV1). These results show that the PEAT components interact with HAM1 and HAM2; further, they exclude the possibility that the PEAT components are part of the NuA4 histone acetyltransferase complex. HAM1/2 mediates histone H4K5 acetylation, which positively regulates transcriptional activation (Earley et al, 2007). Given that the PEAT complexes interact with HAM1/2, and considering that the PEAT complexes function in heterochromatin silencing, we predicted that the PEAT complexes somehow interfere with the histone acetylation activity of HAM1/2.
The RPD3‐like histone deacetylase HDA9 was among the proteins co‐purified with ARID2‐Flag (Dataset EV1). To validate the interaction of this deacetylase with ARID2/3/4, we generated HDA9‐Flag transgenic plants and crossed them with ARID2‐Myc, ARID3‐Myc, and ARID4‐Myc transgenic plants. Progeny harboring both transgenes were used for co‐IP analysis, which revealed that HDA9 interacts with ARID2, ARID3, and ARID4 (Fig 5D). However, HDA9 is known to regulate flowering time regulation, silique development, and leaf senescence but not heterochromatin silencing (Kim et al, 2013, 2016; Chen et al, 2016). Another RPD3‐like histone deacetylase, HDA6, was previously shown to be required for heterochromatin silencing (Hollender & Liu, 2008; Earley et al, 2010). We therefore tested whether or not the PEAT complexes interact with HDA6. Using transgenic plants harboring both HDA6‐Myc and EPCR1‐Flag or ARID2‐Flag, we performed co‐IP and determined that HDA6 interacts with EPCR1 and ARID2 (Fig 5E).
The PEAT complexes contribute to histone deacetylation and heterochromatin condensation
Since the PEAT complexes interact not only with the histone acetyltransferases HAM1/2 but also with the histone deacetylases HDA6/9, the involvement of the PEAT complexes in heterochromatin silencing may be related to regulation of histone acetylation. We performed chromatin immunoprecipitation sequencing (ChIP‐seq) to determine whether or not mutations in PEAT components affect histone H4K5 acetylation (H4K5Ac), a modification of a histone that can be added by the histone acetyltransferases HAM1/2 and can be removed by the histone deacetylase HAD6 (Earley et al, 2006, 2007). Our H4K5Ac ChIP‐seq analysis identified a number of histone hyperacetylated regions in the epcr1/2 and arid2/3/4 mutants relative to the wild type (Fig 6A). There was a significant overlap in the hyperacetylated chromatin regions of the epcr1/2 and arid2/3/4 mutants (P < 0.01, hypergeometric test) (Fig 6A). Since the PEAT mutants released silencing of TEs, we investigated whether or not histone H4K5Ac levels of TEs are affected in the PEAT mutants. Analyses of the H4K5Ac ChIP‐seq data indicated that H4K5Ac levels of TEs are significantly increased in the epcr1/2 and arid2/3/4 mutants relative to the wild type (Fig 6B and C). The hyperacetylation of TEs is consistent with the release of TE silencing in the arid2/3/4 and epcr1/2 mutants, suggesting that the PEAT components are required for histone deacetylation of TEs. Further, we carried out ChIP in combination with qPCR to determine whether the PEAT complexes directly bind to their target loci. In the ChIP–qPCR experiment, the anti‐Flag antibody was used to precipitate Flag‐tagged proteins in EPCR1‐Flag, ARID2‐Flag, and TRB1‐Flag transgenic plants. The result indicated that these PEAT components are enriched in their target loci including solo LTR, AtGP1, AtCOPIA28, and AT1TE42205 (Appendix Fig S5), suggesting that the PEAT complexes directly bind to their target loci and thereby mediate histone deacetylation and transcriptional silencing.
Figure 6. ARID2/3/4 and EPCR1/2 regulate histone acetylation, heterochromatin condensation, and siRNA accumulation.
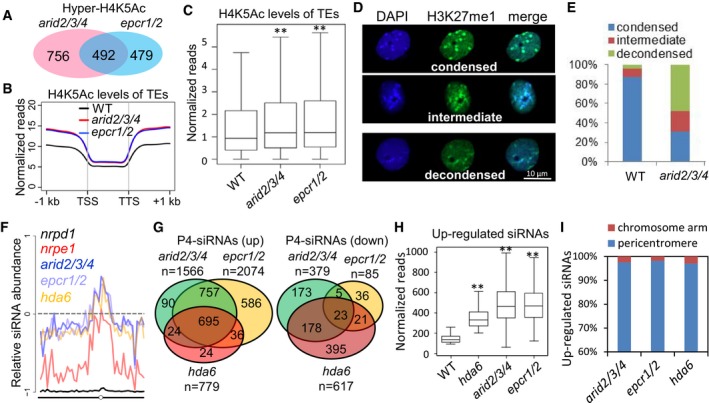
- Overlap among H4K5 hyperacetylated regions in the arid2/3/4 and epcr1/2 mutants relative to the wild type. The overlap is significant (P = 4.4 × 10−78) as determined by hypergeometric test.
- Metaplot of histone H4K5 acetylation levels of TEs in wild‐type, arid2/3/4, and epcr1/2 plants. Histone H4K5 acetylation levels are represented by normalized reads that obtained in the H4K5Ac ChIP‐seq analysis. TSS represents transcription start site, and TTS represents transcription termination site.
- Boxplot of histone H4K5 acetylation levels of TEs in wild‐type, arid2/3/4, and epcr1/2 plants. Asterisks indicate that H4K5Ac levels are significantly higher in the arid2/3/4 (P = 2.42 × 10−13) and epcr1/2 (P = 1.52 × 10−8) mutants than in the wild type as determined by Welch's two‐sample t‐test. Horizontal lines represent the median, and the bottom and top of the box represent the 25th and 75th percentile. The whiskers represent data range within 1.5× of the interquartile range.
- The nuclei with condensed, intermediate, or decondensed heterochromatin status, as marked by DAPI staining and H3K27me signals.
- Percentages of nuclei with condensed, intermediate, or decondensed heterochromatin signals in the wild type and the arid2/3/4 mutant. n = 113.
- Abundance of Pol IV‐dependent siRNAs in the epcr1/2, arid2/3/4, nrpd1, nrpe1, and hda6 mutants relative to the wild type [(Mut‐WT)/(Mut+WT)] from chromosome 3 of Arabidopsis.
- Venn diagrams showing the numbers of differentially expressed Pol IV‐dependent 24‐nt siRNA clusters (P4‐siRNAs) in arid2/3/4, epcr1/2, and hda6 relative to the wild type. All the overlaps are highly significant (P→0) as determined by hypergeometric test.
- Boxplot showing levels of Pol IV‐dependent siRNAs that are upregulated in the hda6 mutant. Asterisks indicate that siRNA levels in the mutants are significantly (P→0) upregulated compared to the wild type as determined by paired Student's t‐test. Horizontal lines represent the median, and the bottom and top of the box represent the 25th and 75th percentile. The whiskers represent data range within 1.5× of the interquartile range.
- Distribution of the Pol IV‐dependent siRNA regions in which siRNAs are upregulated in the mutants relative to the wild type. The pericentromeric region refers to 6 million base pairs of a pericentromeric region on each chromosome.
Considering the interaction of the PEAT components with the deacetylases HDA6 and HDA9, we predict that the PEAT complexes may mediate histone deacetylation of TEs by facilitating the function of the histone deacetylases. To explore how the PEAT complexes contribute to the function of the histone deacetylases, we carried out an in vitro histone deacetylation assay to test whether the PEAT complexes affect the activity of the histone deacetylase HDA6. However, although we detected the histone deacetylation activity of HDA6, its activity is not affected by addition of the PEAT complexes isolated by PWWP2, EPCR1, ARID2, and TRB1 from Arabidopsis seedlings (Appendix Fig S6), suggesting that the PEAT complexes do not affect the activity of the histone deacetylase HDA6 as determined by the in vitro assay. We predict that specific chromatin environments may be critical for the role of the PEAT complexes in the regulation of histone deacetylation and transcriptional silencing.
TRB2 was previously shown to interact with the histone deacetylases HDT4 and HDA6, thereby regulating the telomere length (Lee & Cho, 2016). By telomere length analysis, we demonstrated that the telomere length was significantly increased in epcr1/2 and to a lesser extent in arid2/3/4 (Appendix Fig S7), which is consistent with the previous study showing that the telomere length was increased in the trb2 mutant compared to the wild type (Lee & Cho, 2016). The result suggests that, like TRB2, ARID2/3/4 and EPCR1/2 act as negative regulators of telomere elongation, confirming the molecular and functional connection between TRB proteins and AIRD2/3/4 or EPCR1/2 as identified in this study.
In interphase Arabidopsis nuclei, heterochromatin regions form 8 to 10 condensed nuclear bodies that are densely stained by DAPI (4′,6‐diamidino‐2‐phenylindole). These dense bodies of condensed heterochromatin can also be identified using antibodies for repressive histone modifications including H3K9me2 and H3K27me1 (Soppe et al, 2002; Lindroth et al, 2004; Jacob et al, 2009). Heterochromatin condensation is disrupted in the histone deacetylase mutant hda6 and in mutants defective in DNA methylation such as met1 and ddm1 (Soppe et al, 2002; Probst et al, 2004; Tessadori et al, 2009; Earley et al, 2010). We used DAPI staining and immunostaining with an anti‐H3K27me1 antibody to determine whether or not the PEAT complexes are required for heterochromatin condensation, and found that in the wild type, heterochromatin regions were highly compact and form condensed foci in the nuclei (Fig 6D and E; Appendix Fig S8A and B). However, in the arid2/3/4 mutant, heterochromatin regions were clearly less compact and showed fewer condensed foci in the nuclei (Fig 6D and E; Appendix Fig S8A and B). These results imply that the function of the PEAT complexes in heterochromatin condensation is involved in heterochromatin silencing.
The PEAT complexes regulate the production of siRNAs
Previous studies suggest that the known mutants defective in DNA methylation and heterochromatin silencing such as met1, ddm1, and hda6, differently affect the production of Pol IV‐dependent siRNAs at particular chromatin loci (Lippman et al, 2004; Mathieu et al, 2007; Blevins et al, 2009, 2014; He et al, 2009; Pontes et al, 2009; Earley et al, 2010). Since the aforementioned results demonstrated that the PEAT complexes are required for heterochromatin silencing, we performed small RNA deep sequencing (sRNA‐seq) to determine whether or not the PEAT complexes affect the production of Pol IV‐dependent siRNAs. We identified genome loci from which fewer siRNAs were produced in the Pol IV mutant nrpd1 compared to the wild type. Consistent with previous studies (Zhang et al, 2007; Mosher et al, 2008), our sRNA‐seq analysis indicated that the Pol IV‐dependent siRNAs are enriched in pericentromeric heterochromatin regions and are also accumulated in dispersed loci in chromosome arms (Appendix Fig S9A; Dataset EV4). We next examined the production of the Pol IV‐dependent siRNAs in the PEAT mutants (epcr1/2 and arid2/3/4) and the hda6 mutant. Our sRNA‐seq analysis showed that, compared to the wild type, the production of Pol IV‐dependent siRNAs was either not affected or weakly reduced from Pol IV target loci dispersed in chromosome arms in each of these mutants (Fig 6F; Appendix Fig S9A and B). In pericentromeric heterochromatin regions, however, all of the mutants exhibited increased production of Pol IV‐dependent siRNAs (Fig 6F; Appendix Fig S9A and B). Analysis of these mutants suggests that, like HDA6, the PEAT complexes differentially regulate the production of Pol IV‐dependent siRNAs between chromosome arms and pericentromeric heterochromatin regions.
To further investigate how the PEAT complexes differentially affect the production of Pol IV‐dependent siRNAs, we used our sRNA‐seq data to identify Pol IV‐dependent siRNA regions (500 bp) that produced significantly (fold change ≥ 2; P < 0.01) different amounts of Pol IV‐dependent siRNAs in the PEAT and hda6 mutants compared to the wild type. We identified 1,566, 2,074, and 779 siRNA regions that produced significantly more Pol IV‐dependent siRNAs in the arid2/3/4, epcr1/2, and hda6 mutants, respectively, than in the wild type (Fig 6G). In the 1,566 siRNA regions identified in the arid2/3/4 mutant, 92.7% (1,452/1,566) are also identified in the epcr1/2 mutants (Fig 6G; Dataset EV4), supporting the notion that ARID2/3/4 and EPCR1/2 commonly repress the production of Pol IV‐dependent siRNAs. In the 779 siRNA regions identified in the hda6 mutant, 89.2% (695/779) are identified in both the arid2/3/4 and epcr1/2 mutants (Fig 6G; Dataset EV4). Our boxplot analysis indicated that, in the 779 siRNA regions that produced more Pol IV‐dependent siRNAs in the hda6 mutant, siRNAs are significantly upregulated not only in the hda6 mutant but also in the arid2/3/4 and epcr1/2 mutants (Fig 6H). Further, we analyzed the chromosome locations of the siRNA regions that produced more Pol IV‐dependent siRNAs in the arid2/3/4, epcr1/2, and hda6 mutants than in the wild type, indicating that most of these siRNA regions (> 95%) are present in pericentromeric heterochromatin regions (Figs 6I and EV4A). These results demonstrate that the PEAT complexes and HDA6 commonly repress the production of Pol IV‐dependent siRNAs in pericentromeric heterochromatin regions. We identified 379, 85, and 617 siRNA regions that produce less Pol IV‐dependent siRNAs in the arid2/3/4 and epcr1/2, and hda6 mutants than in the wild type (Fig 6G). These siRNA regions are present not only in pericentromeric heterochromatin regions but also in chromosome arms (Fig EV4A). These results suggest that chromosome arms predominantly contain the siRNA regions that produced less siRNAs in these mutants, which is consistent with the decreased siRNA accumulation on chromatin arms.
Figure EV4. Effect of the arid2/3/4, epcr1/2, and hda6 mutations on small RNA accumulation.

- Distribution of the Pol IV‐dependent siRNA regions that are differentially expressed in the arid2/3/4, epcr1/2, and hda6 mutants relative to the wild type. “Up” and “Down” refer to the Pol IV‐dependent siRNA regions in which the siRNAs are up‐ and downregulated, respectively, in the indicated mutants relative to the wild type.
- Relative levels of 21‐ and 24‐nt siRNAs from the 45S rDNA intergenic spacer locus IGS1 in the wild type, arid2/3/4, epcr1/2, and hda6 mutants. The RNA levels were indicated by normalized small RNA reads as determined by our small RNA‐seq analysis.
The production of siRNAs from the heterochromatic rDNA loci is overproduced in the hda6 mutant and the mutants (i.e., met1 and ddm1) that are defective in DNA methylation and heterochromatin condensation (Mathieu et al, 2007; Blevins et al, 2009; Pontes et al, 2009). Northern blotting analyses in a previous study show that 21‐nt and 24‐nt siRNAs from the intergenic spacers of the heterochromatic 45S rDNA loci are accumulated more in the hda6 mutant than in the wild type (Earley et al, 2010). While the production of 24‐nt siRNAs is dependent on Pol IV, the production of 21‐nt siRNAs is dependent on Pol II (Earley et al, 2010). We used our sRNA‐seq data to evaluate how the 24‐nt and 21‐nt siRNAs from the 45S rDNA intergenic spacers are affected in the PEAT and hda6 mutants. Our sRNA‐seq data indicated that both the 24‐nt and 21‐nt siRNAs from the 45S rDNA intergenic spacer sequence IGS1 were overproduced not only in the hda6 mutant but also in the arid2/3/4 and epcr1/2 mutants (Fig EV4B). The result suggests that, like the hda6 mutant, the PEAT mutants exhibit increased levels of both the 24‐nt and 21‐nt siRNAs, which supports the inference that the PEAT complexes affect the production of siRNAs in a similar manner with HDA6.
The PEAT complexes regulate DNA methylation
Considering the known role of DNA methylation in heterochromatin condensation and transcriptional silencing, we performed bisulfite sequencing to determine the effect of the epcr1/2 and arid2/3/4 mutations on DNA methylation at the whole‐genome level. At the whole‐genome level, DNA methylation is slightly decreased especially at CHH sites in the promoter regions of genes in both epcr1/2 and arid2/3/4 (Fig 7A). DNA methylation in the promoter regions of genes was known to be established by RdDM in Arabidopsis (Stroud et al, 2013). We therefore tested whether the epcr1/2 and arid2/3/4 mutations affect DNA methylation at RdDM target loci. The hypo‐DNA methylated regions (hypo‐DMRs) identified in the Pol V mutant nrpe1 were defined as RdDM target loci. Our boxplot analysis indicated that the DNA methylation levels were significantly decreased in both epcr1/2 and arid2/3/4 even though the decrease was much weaker in the epcr1/2 and arid2/3/4 than in nrpe1 (Fig 7D). Further, heat maps showed that DNA methylation was decreased at CHH sites and to a lesser extent at CG and CHG sites at a subset of RdDM target loci (Fig 7E), suggesting that the PEAT complexes are involved in DNA methylation at a subset of RdDM target loci. As previously reported (Blevins et al, 2014), the hda6 mutation also affects DNA methylation at a subset of RdDM target loci. Considering the interaction of the PEAT components and HDA6, we predict that the PEAT complexes may regulate RdDM through the same mechanism as HDA6.
Figure 7. Effect of the epcr1/2 and arid2/3/4 mutations on DNA methylation.
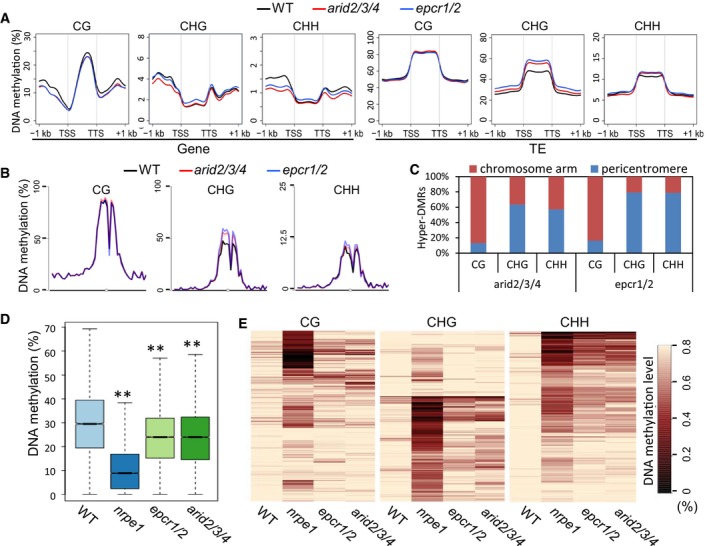
- Metaplot of CG, CHG, and CHH methylation of genes and TEs in the genomes of wild‐type, epcr1/2, and arid2/3/4 plants. H indicates C, A, or T.
- Metaplot of CG, CHG, and CHH methylation on chromosome 3 of Arabidopsis in wild‐type, epcr1/2, and arid2/3/4 plants.
- Distribution of arid2/3/4 and epcr1/2 hyper‐DMRs of CG, CHG, and CHH sites in pericentromeric regions and two chromosome arms. A pericentromeric region refers to 6 million base pairs of a centromere‐flanking region on each chromosome.
- Boxplot of DNA methylation in the wild type, nrpe1, epcr1/2, and arid2/3/4 mutants. The hypo‐DMRs identified in the nrpe1 mutant were defined as RdDM target loci. Asterisks indicate that DNA methylation is significantly (P < 0.001) decreased in the mutants as determined by paired Student's t‐test. Horizontal lines represent the median, and the bottom and top of the box represent the 25th and 75th percentile. The whiskers represent data range within 1.5× of the interquartile range.
- Heat maps of CG, CHG, and CHH methylation at RdDM target loci. The hypo‐DMRs identified in the nrpe1 mutant were defined as RdDM target loci and analyzed in the wild type, nrpe1, eprc1/2, and arid2/3/4 mutants. Black and light yellow indicate low methylation and high methylation, respectively.
Although DNA methylation is decreased at a subset of RdDM target loci in the PEAT mutants, DNA methylation in TE body is clearly increased especially at CHG and CHH sites (Fig 7A). Given that RdDM normally targets dispersed chromatin loci but not TEs and DNA repeats in pericentromeric heterochromatin regions (Stroud et al, 2013), the two seemingly contradictory results are actually reasonable. Consistent with the increased CHG and CHH methylation of TEs in the PEAT mutants, CHG and CHH methylation is specifically increased in pericentromeric heterochromatin regions but not in chromosome arms (Fig 7B; Appendix Fig S10). Further, CHG and CHH hyper‐DMRs (hyper‐DNA methylated regions) but not CG hyper‐DMRs identified in epcr1/2 and arid2/3/4 were found to be enriched in pericentromeric heterochromatin regions (Fig 7C; Appendix Fig S11; Dataset EV5), confirming that CHG and CHH methylation is increased in pericentromeric heterochromatin regions in arid2/3/4 and epcr1/2 relative to the wild type.
The increased CHG and CHH methylation in epcr1/2 and arid2/3/4 was accompanied by the increased production of Pol IV‐dependent siRNAs from pericentromeric heterochromatin regions (Fig 6F and I; Appendix Fig S9B), suggesting that the increased DNA methylation is likely to be mediated by the increased production of Pol IV‐dependent siRNAs through the RdDM pathway in the PEAT mutants. In the hda6 mutant, we found that DNA methylation was actually decreased in heterochromatin regions (Appendix Fig S12), while the production of Pol IV‐dependent siRNAs from heterochromatin regions was increased (Fig 6F and I; Appendix Fig S9B). By itself, this is not surprising—a previous study reported a similar decrease of DNA methylation in the hda6 mutant (Stroud et al, 2013). HDA6 was known to interact with the DNA methyltransferase MET1 and thereby facilitate maintenance of normal DNA methylation levels (To et al, 2011; Liu et al, 2012). We predict that, although the increased production of Pol IV‐dependent siRNAs in the hda6 mutant is also known to mediate DNA methylation through the RdDM pathway (Earley et al, 2010), it is not enough to compensate for the defect in the maintenance of DNA methylation.
Release of silencing in the PEAT mutants is independent of alteration in DNA methylation
To determine whether the release of transcriptional silencing that we observed in the epcr1/2 and arid2/3/4 mutants is correlated with any reduction in DNA methylation, we analyzed the DNA methylation of TEs and genes with increased expression in the PEAT mutants. Compared to the wild type, the mutants did not show significant reduction in DNA methylation at CG, CHG, and CHH sites among these TEs, whereas CHH methylation but not CG and CG methylation was significantly decreased among a subset of these genes (Fig 8A and B). Similarly, we examined the DNA methylation of TEs and genes with decreased expression in the PEAT mutants. The result showed that the DNA methylation of these TEs was not significantly affected and indicated that the DNA methylation of a subset of these genes was significantly reduced at CHH sites but not at CG and CHG sites (Appendix Fig S13A and B).
Figure 8. The PEAT complexes mediate heterochromatin silencing independently of affecting DNA methylation.
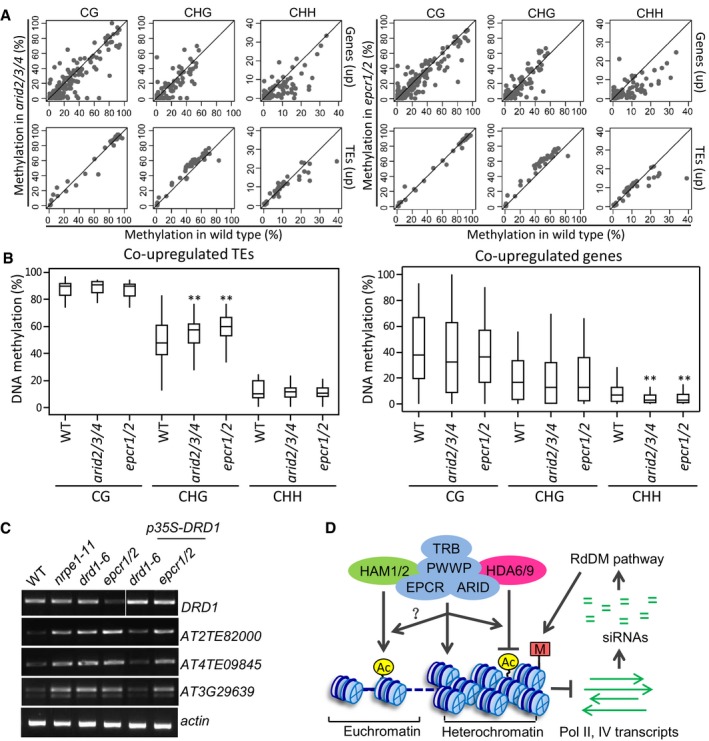
- Scatter plots showing the effect of the epcr1/2 and arid2/3/4 mutations on CG, CHG, and CHH methylation at the co‐upregulated TEs and genes in the epcr1/2 and arid2/3/4 mutants. TE methylation and gene DNA methylation refer, respectively, to methylation of the TE body and methylation of the 1‐kb‐gene‐promoter region.
- Box plots of CG, CHG, and CHH methylation in the wild type, arid2/3/4, and epcr1/2 mutants at the co‐upregulated TEs and genes in the arid2/3/4 and epcr1/2 mutants. Asterisks indicate statistical significance as determined by paired Student's t‐test. CHG methylation of co‐upregulated TEs is significantly increased in arid2/3/4 (P = 1.26 × 10−4) and epcr1/2 (P = 6.3 × 10−9); CHH methylation of co‐upregulated genes is significantly decreased in arid2/3/4 (P = 3.5 × 10−6) and epcr1/2 (P = 6.4 × 10−7). Horizontal lines represent the median, and the bottom and top of the box represent the 25th and 75th percentile. The whiskers represent data range within 1.5× of the interquartile range.
- Overexpression of DRD1 fails to restore the silencing of EPCR1/2 target loci in the epcr1/2 mutant background.
- Model for the role of the PEAT complexes in chromatin regulation. In heterochromatin regions, the PEAT complexes either directly mediate heterochromatin condensation or interact with the histone deacetylases HDA6 and HDA9 and thereby mediate heterochromatin condensation by histone deacetylation. Thus, the PEAT complexes repress transcription mediated by both Pol II and Pol IV. The repression of Pol II transcription is required for transcriptional silencing in heterochromatin regions. Moreover, the repression of Pol II and Pol IV transcription prevents overproduction of siRNAs that are responsible for DNA methylation. In euchromatin regions, we predict that the PEAT complexes may regulate histone acetylation by interaction with the histone acetyltransferases HAM1 and HAM2. However, further studies are required to elucidate how the molecular mechanisms of the regulation.
Source data are available online for this figure.
If the PEAT complexes facilitate DNA methylation and thereby mediate silencing of TEs, then one would expect to observe hypomethylation of the TEs with increased expression. Our results demonstrate that while silencing of these TEs is released in the PEAT mutants, DNA methylation of the corresponding TEs is not reduced. Although CHH methylation is reduced at a subset of the genes with increased expression in the PEAT mutants, release of silencing in many of these genes is not accompanied by reduced DNA methylation (Fig 8A; Dataset EV6). Specifically, SDC is known to be redundantly silenced by CMT3‐mediated CHG methylation and RdDM (Henderson & Jacobsen, 2008). Our whole‐genome data indicated that, while the silencing of SDC is significantly released in the PEAT mutants, its DNA methylation is not significantly affected at all cytosine sites (Appendix Fig S14). These results imply that the PEAT complexes may mediate heterochromatin silencing either at a downstream step of or in parallel to DNA methylation.
Previous studies indicated that the expression of ROS1 and IBM1‐L (the longer IBM1 transcript) is reduced in the mutants that are defective in DNA methylation and demonstrated that DNA methylation of the regulatory regions in the ROS1 promoter and the IBM1 intron is required for their expression (Mathieu et al, 2007; Rigal et al, 2012; Lei et al, 2015; Williams et al, 2015). Given the effect of arid2/3/4 and epcr1/2 on the expression of both ROS1 and IBM1‐L (Fig 4D and E), we evaluated whether the DNA methylation in ROS1 and IBM1 was affected by the arid2/3/4 and epcr1/2 mutations as determined by the whole‐genome DNA methylation data. We found that in the arid2/3/4 and epcr1/2 mutants, the DNA methylation in the ROS1 promoter was only weakly reduced, whereas the DNA methylation in the IBM1 intron was not affected (Fig EV5A). The results suggest that, in the PEAT mutants, the decrease in the expression of ROS1 and IBM1‐L is unlikely caused by alteration of DNA methylation in their regulatory regions. Considering that the PEAT components interact with the histone acetyltransferases HAM1/2, we examined whether the complex contributes to the expression of ROS1 and IBM1‐L through activating histone acetylation. Analysis of our whole‐genome histone H4K5 acetylation data indicated that the H4K5 acetylation levels of ROS1 and IBM1 are not affected in the epcr1/2 and arid2/3/4 mutants (Fig EV5B). Therefore, although the PEAT components interact with the histone acetyltransferases HAM1/2, its function in activation of ROS1 and IBM1 does not require histone acetylation. Given decondensation of heterochromatin and release of silencing in the PEAT mutants, we deduce that the PEAT complexes may regulate the expression of ROS1 and IBM1‐L through facilitating the heterochromatin formation in the ROS1 and IBM1 loci even though the complexes do not affect their DNA methylation. This finding is consistent with the results showing that the PEAT complexes are involved in heterochromatin condensation and silencing independently of alteration in DNA methylation at the whole‐genome level.
Figure EV5. Genome browser snapshots showing DNA methylation and histone H4K5 acetylation patterns of ROS1 and IBM1 in wild type, arid2/3/4, and epcr1/2 .
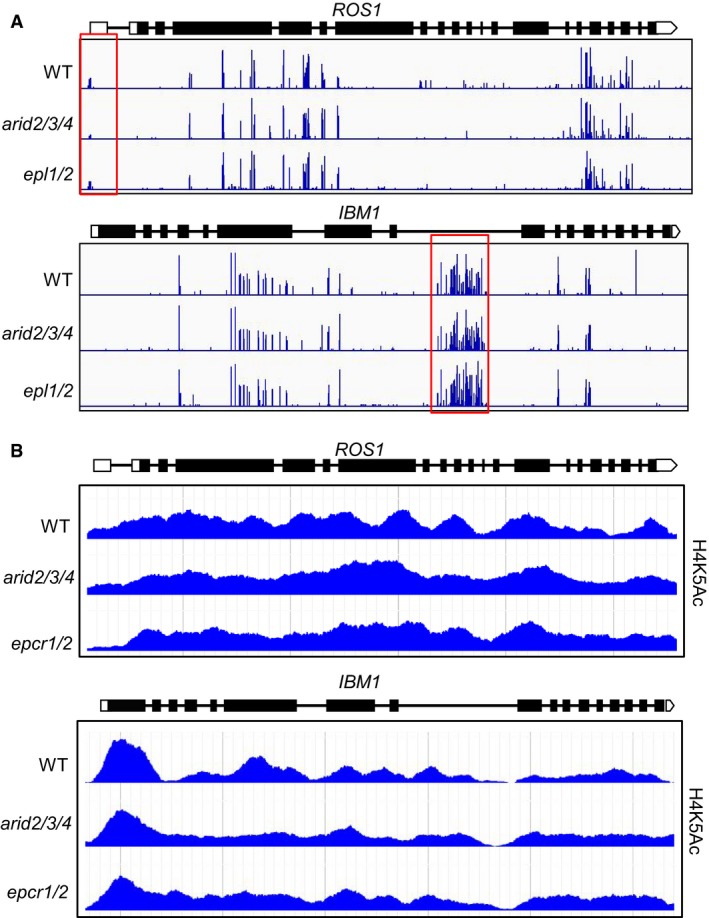
- DNA methylation of ROS1 and IBM1 as determined by the whole‐genome DNA methylation analysis. The length of blue lines represents DNA methylation levels of cytosine sites. Red boxes highlight genomic loci harboring DNA methylation that is required maintenance of the expression of ROS1 and IBM1‐L.
- Histone H4K5 acetylation of ROS1 and IBM1 as determined by the histone H4K5Ac ChIP‐seq experiment.
Among the differentially expressed genes of the epcr1/2 and arid2/3/4 mutants identified in our RNA‐seq analysis, we observed significantly reduced expression of DRD1 in both mutants; this gene encodes a component of the canonical RdDM pathway (Matzke & Mosher, 2014; Dataset EV2). To exclude the possibility that the release of silencing in the PEAT mutants could have resulted from reduced expression of DRD1, we generated a DRD1 overexpression construct, p35S‐DRD1, and introduced it into the epcr1/2 and drd1 mutant backgrounds. We performed RT–PCR to determine whether or not the drd1 mutation affects the silencing of loci that were identified by our RNA‐seq analysis and had upregulated expression in the epcr1/2 mutant. We identified three loci (1 gene and 2 TEs) that had co‐upregulated expression in both the epcr1/2 and drd1 mutants (Fig 8C; Dataset EV2). Further, we evaluated the expression of the three loci in the p35S‐DRD1‐transformed epcr1/2 and drd1 plants, and the results demonstrated that the overexpression of DRD1 rescued the silencing of the loci in the drd1 background but not in the epcr1/2 background (Fig 8C), establishing that the PEAT complexes are involved in silencing in a manner that is independent of DRD1. Given that DRD1 is a component of the RdDM pathway, we predicted that the involvement of the PEAT complexes in silencing is independent of the RdDM pathway. Further confirming this idea, our RNA‐seq analysis identified many loci in which silencing was released in the PEAT mutants but not in the RdDM mutant nrpe1 (Fig 4C; Dataset EV2). It is thus clear that the PEAT complexes mediate heterochromatin silencing via an RdDM‐independent mechanism.
Discussion
The enhancer of polycomb‐like protein (EPL) is a subunit of the conserved NuA4/Tip60 histone acetyltransferase complex that mediates histone acetylation and transcriptional activation in eukaryotes (Doyon & Cote, 2004). In yeast, loss‐of‐function mutations in Epl1 have reduced levels of both histone acetylation and transcription, suggesting that yeast Epl1 is a functional subunit of the NuA4 histone acetyltransferase complex (Boudreault et al, 2003). Moreover, yeast Epl1 was found to mediate silencing of telomere regions, although its molecular mechanism has remained elusive (Boudreault et al, 2003). In Arabidopsis, there are two EPL orthologs, EPL1a and EPL1b, and two distantly related proteins we have here named EPCR1 and EPCR2 (Fig EV1A). Affinity purification of EPL1a and EPL1b identified interactions between EPL1a/1b and all of the conserved subunits of the NuA4 histone acetyltransferase complex, demonstrating that EPL1a and EPL1b act as canonical subunits of the NuA4 histone acetyltransferase complex. However, affinity purification of EPCR1 identified interactions with the histone acetyltransferases HAM1 and HAM2 but not with accessory subunits of the NuA4 histone acetyltransferase complex (Table 2), suggesting that EPCR1 is not a subunit of the NuA4 histone acetyltransferase complex. Here, we demonstrate that EPCR1 and its close paralog EPCR2 interact with PWWPs, ARIDs, and TRBs, and form complexes that are required for heterochromatin silencing. Affinity purification showed that, like EPCR1, both ARID2 and TRB1 interact with the catalytic subunits HAM1/2, but not with the accessory subunits in the NuA4 histone acetyltransferase complex (Table 2). These results indicate that the PEAT components are not subunits of the NuA4 histone acetyltransferase complex in Arabidopsis.
Heterochromatin condensation blocks the transcriptional machinery binding to DNA, a process that is required for maintenance of transposon silencing. In Arabidopsis, DNA methylation and the repressive histone modifications H3K9me2 and H3K27me1 are enriched in heterochromatin regions, while activating histone modifications such as H3K4me3 and histone acetylation are absent or have low levels in heterochromatin regions (Cokus et al, 2008; Lister et al, 2008; Zhang et al, 2009; Zhou et al, 2010; Roudier et al, 2011). Decondensation of heterochromatin was observed in the DNA methylation mutants met1 and ddm1, in the histone H3K9 methyltransferase mutants kyp/suvh4, and in the histone H3K7 mono‐methyltransferase mutant atxr5/6 (Soppe et al, 2002; Lindroth et al, 2004; Jacob et al, 2009), suggesting that DNA methylation and the repressive histone modifications H3K9me2 and H3K27me1 are required for heterochromatin condensation. Decondensation of heterochromatin was also found in the hda6 mutant (Probst et al, 2004; Tessadori et al, 2009), suggesting a role of HDA6 in heterochromatin condensation. Considering the significant effect of hda6 on DNA methylation in pericentromeric heterochromatin regions (Earley et al, 2010; Stroud et al, 2013), decondensation of heterochromatin in hda6 may be at least partially caused by reduced DNA methylation. Here, we demonstrated that like HDA6, the PEAT complexes are involved in heterochromatin condensation (Fig 6D and E; Appendix Fig S8A and B). However, different from HDA6, the PEAT complexes are not required for the maintenance of DNA methylation in pericentromeric heterochromatin regions. The PEAT complexes may contribute to heterochromatin condensation via histone deacetylation that is catalyzed by HDA6. Further, the PEAT complexes may also influence heterochromatin condensation independently of HDA6. Additional experiments will be necessary to determine precisely how the PEAT complexes mediate heterochromatin condensation and transcriptional silencing.
Our affinity purification analysis demonstrates that the PEAT components are not subunits of the NuA4 histone acetyltransferase complex, and shows that ARID‐containing proteins are subunits of the PEAT complexes. In yeast and human, ARID‐containing proteins are present in the SWI/SNF chromatin remodeling protein complex (Wilsker et al, 2004), in which these proteins are responsible for binding to DNA and facilitating remodeling of nucleosomes. In Arabidopsis, there are 10 ARID‐containing proteins (Zhu et al, 2008). ARID1, which contains an ELM domain in addition to the ARID domain, was found to be required for sperm cell formation in Arabidopsis (Zheng et al, 2014). In rice, OsARID3, an hsp20 domain‐containing ARID protein was reported to be required for shoot meristem development (Xu et al, 2015). However, the molecular mechanism of these ARID domain‐containing proteins is largely unknown. Our affinity purification experiments in the present study demonstrate that the three Arabidopsis Hsp20 domain‐containing ARID proteins ARID2/3/4 function redundantly in the PEAT complexes and are required for development and heterochromatin silencing. Several genes required for shoot apical meristem formation and maintenance are aberrantly expressed in the PEAT mutants as determined by our RNA‐seq analysis. The aberrant expression of these genes may cause the developmental arrest of the PEAT mutants. Further studies are required to investigate how the PEAT complexes regulate the genes that are involved in shoot apical meristem formation and maintenance and thereby affect plant development.
In the PEAT complexes, we identified three previously uncharacterized PWWP domain‐containing proteins (Stec et al, 2000). In mammals, The PWWP domain binds to either DNA or methylated histones and is responsible for the chromatin‐related functions of these proteins (Alvarez‐Venegas & Avramova, 2012). In Arabidopsis, the PWWP domain is present in many proteins, including the histone H3K4 methyltransferases ATX1–ATX5. Our recent study report that a subfamily of PWWP domain‐containing proteins interacts with FVE and MSI5 and represses the expression of the flowering repressor gene FLC by facilitating histone H3K27 trimethylation (Zhou et al, 2018a). Here, we identify three PWWP domain‐containing paralogs and demonstrate that these proteins act as subunits of the PEAT complexes and function in heterochromatin silencing. As examined by a recent systematic profiling of histone readers in Arabidopsis (Zhao et al, 2018), the PWWP domains in these proteins are not shown to specifically associate with methylated histone peptides. Further studies will be required to characterize how the PWWP domain functions in the PEAT complexes to regulate heterochromatin silencing.
The telomeric DNA binding proteins TRB1 and TRB2 interact with double‐stranded telomeric repeats through the N‐terminal Myb domain and are involved in the formation and stability of telomeres (Schrumpfova et al, 2014). The trb1 mutant was identified as an enhancer of a polycomb mutant lhp1 (Zhou et al, 2016). TRB1 is a bivalent transcriptional regulator: It represses the transcription of polycomb group (PcG) target genes in the lhp1 mutant but activates the transcription of target genes that are regulated independently of PcG in wild‐type plants (Zhou et al, 2016). Although the trb1 mutation enhances the developmental defect phenotypes of the lhp1 mutant, the trb1 mutant does not show any significant developmental defects. Our study indicates that TRB1 and TRB2 function redundantly and act as subunits of the PEAT complexes. Further, the PEAT components are shown to interact with the histone deacetylases HDA6 and HDA9 and mediate histone deacetylation and heterochromatin silencing. This finding is consistent with a role for TRB1 in the repression of transcription as previously described (Zhou et al, 2016). Considering that the trb1 mutant was identified in a screen for mutants that enhance the polycomb phenotype of the lhp1 mutant (Zhou et al, 2016), the identification of trb1 is reminiscent of the Drosophila epl mutant that was originally identified by screening for mutants that enhance the polycomb mutant phenotype (Stankunas et al, 1998). Moreover, considering that both TRB1/2 and the EPL‐related proteins EPCR1/2 are subunits of the PEAT complexes, we predict that in addition to trb1, mutations in other subunits of the PEAT complexes may also enhance the lhp1 mutant phenotype in Arabidopsis.
A recent study reported the generation of a trb1/2/3 triple mutant and demonstrated that the mutant showed defects in early seedling development (Zhou et al, 2018b). In our study, TRB1 and TRB2 are identified as subunits of the PEAT complexes involved in both early seedling development and heterochromatin silencing. The developmental defects of the trb1/2/3 mutant reported by Zhou et al are highly similar with the developmental defects of the PWWP1/2/3, epcr1/2, and arid2/3/4 mutants observed in our study, supporting the notion that TRBs interact with PWWPs, EPCRs, and ARIDs and form redundant multi‐subunit complexes involved in early seedling development. In addition, we demonstrate that subunits of the PEAT complexes interact with the histone acetyltransferases HAM1/2 and the histone deacetylases HDA9 and HDA6 in vivo, suggesting that the PEAT complexes may regulate histone acetylation/deacetylation and thereby affect development and heterochromatin silencing. Different from our study, Zhou et al (2018b) reported that TRBs interact with the histone H3K27 trimethyltransferase CLF and thereby mediate H3K27 trimethylation. In future, it is necessary to investigate whether the interactions of TRBs with the histone acetyltransferases, the histone deacetylases, and the histone H3K27 trimethyltransferase are shared by the other subunits of the PEAT complexes. Moreover, it is interesting to study the possible role of the PEAT complexes in coordinating histone acetylation/deacetylation and H3K27 trimethylation during development.
The DNA glycosylase ROS1 is required for active DNA demethylation and repression of transcriptional gene silencing (Gong et al, 2002; Zhu, 2009). ROS1 expression is decreased in DNA methylation mutants including met1 and RdDM mutants (Mathieu et al, 2007). DNA methylation of a TE in the ROS1 promoter was shown to be required for ROS1 expression (Lei et al, 2015; Williams et al, 2015). IBM1 is a histone H3K9 demethylase that is responsible for protecting gene bodies from histone H3K9 dimethylation and CHG methylation (Saze et al, 2008; Miura et al, 2009). IBM1 has a hypermethylated intronic region, which is required for accumulation of the longer IBM1 (IBM1‐L) transcript but not of the shorter IBM1 (IBM1‐S) transcript (Rigal et al, 2012). DNA methylation is an important regulatory mechanism that is required for maintenance of the transcript levels of ROS1 and IBM1‐L (Rigal et al, 2012; Lei et al, 2015; Williams et al, 2015). However, it is unknown how DNA methylation confers transcriptional activation in ROS1 and IBM1‐L. Our results suggest that like components involved in DNA methylation and heterochromatin silencing, the PEAT complexes are required for maintenance of the transcript levels of ROS1 and IBM1‐L even though they do not affect DNA methylation. The PEAT complexes may act at a downstream step of or in parallel to DNA methylation to regulate the transcript levels of ROS1 and IBM1‐L. Previous studies suggest that the RNA‐binding protein IBM2/ASI1 promotes the IBM1‐L transcription by preventing the inhibitory effect of the IBM1 intronic heterochromatin (Saze et al, 2013; Wang et al, 2013). Given the interaction between the PEAT complexes and the histone acetyltransferases HAM1/2, it is possible that the PEAT complexes promote the IBM1‐L expression by facilitating histone acetylation. However, our study reveals that the suppressed expression of IBM1‐L in the PEAT mutants is not accompanied by reduction of histone acetylation, suggesting that the function of the PEAT complexes in the IBM1‐L expression is independent of histone acetylation. This finding supports the notion that, like the known components required for DNA methylation and heterochromatin silencing, the PEAT complexes may facilitate maintenance of the heterochromatin status in the IBM1 intron and thereby promote the expression of IBM1‐L.
In conclusion, this study indicates that the PEAT complexes interact with the histone deacetylase HDA6/9 and mediate histone deacetylation and heterochromatin condensation, resulting in repression of Pol II transcription and heterochromatin silencing (Fig 8D). Moreover, histone deacetylation and heterochromatin condensation mediated by the PEAT complexes suppress Pol IV transcription and thus prevent overproduction of Pol IV‐dependent siRNAs from pericentromeric heterochromatin regions. The prevention of the overproduction of Pol IV‐dependent siRNAs is required for maintenance of DNA methylation at a normal level in pericentromeric heterochromatin regions (Fig 8D). The PEAT complexes, as well as HDA6, are also required for the production of a subset of Pol IV‐dependent siRNAs. Unlike the PEAT complexes, HDA6 is required for the maintenance of DNA methylation. The role of HDA6 in DNA methylation was shown to facilitate Pol IV and Pol V recruitment to chromatin and thereby mediate the production of Pol IV‐dependent siRNAs (Blevins et al, 2014). It remains to be studied how the PEAT complexes facilitate the production of a subset of Pol IV‐dependent siRNAs independently alteration of DNA methylation. Moreover, the PEAT complexes are also shown to interact with the histone acetyltransferases HAM1/2. We predict that the interaction of the PEAT complexes with HAM1/2 may occur in euchromatin regions and thereby regulate the transcription of protein‐coding genes required for development (Fig 8D). Together, this study reveals a mechanism that mediates heterochromatin silencing via histone deacetylation and heterochromatin condensation in Arabidopsis. In future, it is necessary to investigate whether and how the PEAT complexes affect the function of the histone acetyltransferases and deacetylases and how the complexes regulate development.
Materials and Methods
Plant materials and mutant screening
With the exception of hda6/axe1‐5, which was described previously (Earley et al, 2010), all of the Arabidopsis T‐DNA insertion lines used in this study were obtained from the Arabidopsis Biological Resource Center (ABRC): arid2 (SALK_026835), arid3 (SALK_022359), arid4 (SALK_007400), epcr1 (SALK_039205), epcr2 (SALK_024125), pwwp1(SAIL_342_C09), pwwp2 (SALK_136093), pwwp3 (SALK_042581), trb1 (SALK_025147), trb2 (GK‐103E02). The double or triple mutants used in this study were generated by crossing single mutants. Due to the early seedling lethality of the epcr1/2 double‐mutant and arid2/3/4 triple‐mutant plants, these mutants were maintained in a heterozygous state. Arabidopsis seedlings were grown on MS medium plates with a 16 h light/8 h dark photoperiod at 22°C.
Full‐length ARID2, ARID3, ARID4, EPCR1, TRB1, HDA6, HDA9, HAM1, HAM2, EPL1a, and EPL1b as driven by their native promoters were introduced into the modified pCAMBIA1305 or pRI909 vectors, producing, respectively, 3xFlag‐ and C‐terminal 5xMyc‐tagged proteins. A DRD1 overexpression construct as driven by the 35S promoter was introduced into the pCAMBIA1300 vector. The TRB2 cDNA fragment (+1~+420 aa) was inserted into the pFGC5941 RNAi vector and transformed into the wild‐type and trb1 (SALK_025147) mutant plants for the knockdown of TRB2 in Arabidopsis. The constructs were introduced into the Agrobacterium strain GV3101 and transformed into Arabidopsis via the flower‐dipping method. All constructs were sequenced for verification, and the primers used for their construction are listed in Dataset EV7.
Affinity purification, mass spectrometric analysis, co‐immunoprecipitation, and gel filtration
For affinity purification, 3 g of seedling or flower tissue samples were ground in liquid nitrogen and homogenized in 15 ml of lysis buffer (50 mM Tris, pH 7.6, 150 mM NaCl, 5 mM MgCl2, 10% glycerol, 0.1% NP‐40, 0.5 mM DTT, 1 mM PMSF, with one tablet of Roche protease inhibitor cocktail per 50 ml). Following centrifugation, the supernatant was incubated with 100 μl of anti‐Flag M1 Agarose (Sigma, A4596) or anti‐c‐Myc Agarose (Sigma, A 7470) at 4°C for 2.5 h. The resins were washed five times with lysis buffer. The Flag bead‐bound proteins were then eluted with 3xFlag peptide (Sigma, F4799), whereas the Myc bead‐bound proteins were eluted with c‐Myc peptide (Sigma, M2435). The eluted proteins were run on a 10% SDS–PAGE gel and then subjected to silver staining with a ProteoSilver Silver Stain Kit (Sigma, PROT‐SIL1). The mass spectrometric analysis was performed as described previously (Zhang et al, 2012). Briefly, proteins on SDS–PAGE gels were de‐stained and digested in‐gel with trypsin at 37°C overnight. The digested peptides were eluted on a capillary column and introduced into an LTQ mass spectrometer equipped with a nano‐ESI ion source for analysis (Thermo Fisher).
For co‐immunoprecipitation analysis, 1 g of tissue from parental lines, as well as 1 g from F1 plants, was ground in liquid nitrogen and homogenized in 5 ml of lysis buffer. Following centrifugation, the supernatant was incubated with 50 μl of anti‐Flag M1 Agarose or anti‐c‐Myc Agarose at 4°C for 2.5 h. The resins were washed five times with lysis buffer, and the bead‐bound proteins were eluted with 3xFlag peptide (Sigma, F4799) or c‐Myc peptide (Sigma, M2435). Both eluted proteins and untreated supernatant control samples were separated on SDS–PAGE gel prior to immunoblotting.
For gel filtration, 0.4 g of seedlings was ground to a powder and suspended in 2.4 ml of lysis buffer. After centrifugation, the supernatant was passed through a 0.22‐μm filter, and 500 μl of the filtrate was loaded onto a Superose 6 increase (10/300 GL) column (GE Healthcare, 29‐0915‐96). The eluate was collected in a series of fractions (500 μl/fraction) and run on SDS–PAGE gel for immunoblotting.
Yeast two‐hybrid assay
The cDNA sequences of PWWP1, PWWP2, PWWP3, TRB1, and TRB2 were cloned in‐frame with the 3′‐terminal sequence of GAL4‐AD (pGADT7 vector) and the 3′‐terminal sequence of GAL4‐BD (pGBKT7) using a One‐step Cloning Kit (Vazyme Biotech, C112). The full‐length cDNA sequences of EPCR1 and ARID4 were separately cloned into both the pGADT7 and pGBKT7 vectors between the NdeI and XmaI sites. The full‐length cDNA sequences of ARID2 and ARID3 were separately cloned into both the pGADT7 and pGBKT7 vectors between the NcoI and BamHI sites. The primers used here are listed in Dataset EV7. The yeast strains AH109 and Y187 were transformed with the pGADT7 and pGBKT7 constructs and grown on synthetic dropout medium lacking, respectively, Leu and Trp. The positive clones from the synthetic dropout medium lacking Leu were mated for 16–20 h in YPD medium with the positive clones from the synthetic dropout medium lacking Trp. After mating, the mixture was spotted on synthetic dropout medium lacking both Leu and Trp. The positive yeast colonies were then spotted on synthetic dropout medium lacking Trp and Leu and spotted on synthetic dropout medium lacking Trp, Leu, and His. Growth of transformed, positive yeast strains on SD‐LWH indicates interaction between the GAL‐AD fusion protein and the GAL4‐BD fusion protein. A 5 mM solution of 3‐amino‐1,2,4‐triazole (3‐AT) was used to inhibit the background growth of transformed strains on the synthetic dropout medium lacking Trp, Leu, and His.
Protein expression and pull‐down assays in bacteria
The full‐length PWWP2, ARID2, TRB1, and the sequence for the EPCR1‐N terminal (1–500 aa) fragment were cloned into the pGEX‐6P‐1 and pET28a+ plasmids to generate, respectively, fusion constructs with 5′‐terminal GST or 6xHIS tags. The primers here are listed in Dataset EV7. The protein induction and pull‐down assays were performed as described previously (Han et al, 2016). Briefly, the constructs expressing both GST‐ and HIS‐tagged proteins were co‐transformed into the E. coli expression strain Transetta (DE3). 100 μM IPTG was used for protein induction. The bacteria in the culture were collected and suspended in 1.5 ml of protein extraction buffer (50 mM Tris–HCl [pH 8.0], 300 mM NaCl, 10% glycerol, 0.5% Tween‐20, 15 mM β‐mercaptoethanol, 1 mM PMSF, and 1 tablet of Roche protease inhibitor cocktail per 50 ml). The sample was sonicated for 1 min (4 s on and 6 s off) and then centrifuged at 13,000 g for 10 min at 4°C. Of the supernatant, 200 μl was used as input, and the remainder was incubated with 50 μl GST‐beads (GE, 17‐0756‐01) or HIS‐beads (Millipore, 70666‐4) for a pull‐down assay. The samples were boiled for 6 min at 100°C and separated on 7.5% SDS–PAGE gels prior to immunoblotting with GST antibody (Abmart, 12G8) or HIS antibody (Abmart, 10E2).
RNA deep sequencing and data analysis
Total RNA was extracted from 1 g samples of 10‐day‐old Arabidopsis seedlings with TRIzol reagent (Invitrogen) and sent to BGI (Shenzhen, China) for library preparation and deep sequencing. Three independent biological replicates were performed. For data analysis, after removing adapter and filtering low‐quality (q < 20) reads, clean reads were mapped to the TAIR10 Arabidopsis genome using TopHat v2.1.0 (Trapnell et al, 2009), allowing up to two mismatches. The differentially expressed genes and TEs were identified using cuffdiff (P < 0.01 and |log2(FC)|>=1) (Trapnell et al, 2010). The distribution of differentially expressed genes and TEs throughout the five Arabidopsis chromosomes was drawn using the quantsmooth R package in Bioconductor with minor modification. The raw RNA deep sequencing data have been deposited in the Gene Expression Omnibus (GEO) database (accession number GSE116066).
Small RNA deep sequencing and data analysis
Total RNA was extracted from 1 g samples of 10‐day‐old Arabidopsis seedlings using the TRIzol reagent (Invitrogen) and sent to BIONOVA (Beijing, China) for library preparation and small RNA deep sequencing. For data analysis, after adapter sequences were removed and low‐quality reads were filtered, 18–30 nt clean reads were mapped to the TAIR10 Arabidopsis genome using Bowtie (Langmead et al, 2009); only perfectly matched reads were retained for further analysis. The TAIR10 Arabidopsis genome was split into 500‐bp windows, and the read counts of 24‐nt reads in every window were normalized to reads per 10 million (RPTM) by the total number of clean reads that mapped to the nuclear genome. The regions whose combined expression level in wild‐type and mutant plants were < 100 RPTM were removed. The 500‐bp regions that showed fivefold lower expression in the nrpd1 mutant than wild type were defined as Pol IV‐dependent siRNA loci. The profile of Pol IV‐dependent siRNAs across chromosomes was plotted using 500‐kb windows. The raw small RNA deep sequencing data have been deposited in the GEO database (accession number GSE116067).
Whole‐genome bisulfite sequencing and data analysis
Genomic DNA was extracted from 10‐day‐old Arabidopsis seedlings with a DNeasy Plant Mini Kit (QIAGEN, 69104) and sent to BGI (Shenzhen, China) for bisulfite treatment, library preparation, and deep sequencing. For data analysis, reads were mapped to the TAIR10 Arabidopsis genome using Bismark, allowing two mismatches (Krueger & Andrews, 2011). Reads were retained only when they are uniquely mapped on the genome. Only cytosine sites that were covered by at least five reads were included for further analysis. The DNA methylation level of a cytosine site was represented by the percentage of the number of reads reporting a C relative to the total number of reads reporting a C or T. The methylation levels of genes and TEs were determined by pooling the reads. CG, CHG, and CHH methylation were each analyzed separately. The raw bisulfite sequencing data have been deposited in the GEO database (accession number GSE116064).
ChIP assays
The levels of histone H4K5Ac on chromatin were determined by ChIP‐seq analysis, using a previously described protocol with minor modifications (Rowley et al, 2013). Briefly, 2 g samples of 10‐day‐old seedlings were ground into powder in liquid nitrogen and homogenized in 25 ml Honda Buffer (20 mM HEPES‐KOH, pH 7.4, 0.44 M sucrose, 1.25% ficoll, 2.5% Dextran T40, 10 mM MgCl2, 0.5% Triton X‐100, 5 mM DTT, 1 mM PMSF, Roche protease inhibitor cocktail) and then cross‐linked with 0.5% formaldehyde (Sigma, F8775). After sonication, the chromatin was incubated with H4K5Ac (Abcam, ab51997) antibody and Dynabeads Protein A (Invitrogen, 100–01D) overnight with rotation at 4°C. The chromatin DNA was purified using a standard phenol–chloroform method, ChIP DNA was used for library generation and sequencing. For data analysis, raw reads were cleaned by removing adapter sequences and filtering low‐quality (q < 20) reads. Then, clean reads were mapped to the TAIR10 Arabidopsis genome using Bowtie (Langmead et al, 2009). Only perfectly and uniquely matched reads were retained. The SICER program was used to identify ChIP‐enriched peaks and determine differentially expressed peaks between wild‐type and mutant plants (Zang et al, 2009). The raw ChIP‐seq data have been deposited in the GEO database (accession number GSE116065).
For ARID2‐Flag, EPCR1‐Flag, and TRB1‐Flag ChIP assays, 1 g of 10‐day‐old seedlings were cross‐linked with 1% formaldehyde for 20 min under vacuum and ground into fine powder in liquid nitrogen. After sonication, chromatin was incubated with anti‐Flag (Sigma, F1804) antibody and Dynabeads Protein G (Invitrogen, 10003D) overnight with rotation at 4°C. The precipitated chromatin DNA was then recovered and purified for ChIP–qPCR. The ChIP signals on indicated loci were normalized by ACT2. Sequences of the primers used for ChIP–qPCR are indicated in Dataset EV7.
RNA expression analysis
Total RNA was extracted from 10‐day‐old Arabidopsis seedlings with TRIzol reagent (Invitrogen). After contaminating DNA was removed by DNase I (Takara), 1 μg of total RNA was used for reverse transcription using both oligo dT and random primers. The reverse transcription products (cDNA) were used for PCR. For qPCR, at least three biological replications were performed.
Immunofluorescence assay
Protoplasts were isolated from young Arabidopsis leaves, and nuclei were fixed in 4% formaldehyde and applied to slides. Histone H3K27me1 was immunolocalized as previously described (Jacob et al, 2009). In brief, after nuclei were blocked with 3% BSA in PBS, the primary antibody H3K27me1 (07‐448; Millipore) was diluted at 1:200 and incubated overnight at 4°C. Secondary anti‐rabbit Alexa Fluor 488 (Jackson ImmunoResearch, 711‐545‐152) was used at 1:500 dilutions and incubated for 2 h at room temperature. Chromatin was counterstained with DAPI in mounting medium. Images were acquired with a spinning‐disk confocal microscope and analyzed with Volocity software.
Telomere length analysis
The telomere length was determined by Terminal Restriction Fragmentation (TRF) analysis. Briefly, genomic DNA extracted from 10‐day‐old seedlings was digested overnight with the restriction enzymes MseI or Hinfl. The samples of digested DNA were subjected to Southern blotting. The telomeric repeat probe [5′‐(TTTAGGG)7] was directly synthesized, and the 5′ end was labeled by [γ‐32P]ATP using T4 polynucleotide kinase.
HDAC assay
For histone deacetylation (HDAC) assay, the histone H4 was acetylated by the histone acetyltransferase HAM1 purified from E. coli. The acetylated H4 was then used for the histone deacetylation assay. The proteins that were purified from transgenic plants were incubated with acetylated H4 at 30°C for 3 h with gentle shaking and terminated by addition of 2× SDS Laemmli sample buffer followed by heating at 100°C for 5 min. The reaction products were run on 12% SDS–PAGE gels and subjected to immunoblotting with anti‐H4K5ac (Abcam, ab51997) and anti‐H4 (Abcam, ab10158) antibodies.
Author contributions
L‐MT, C‐JZ, and X‐JH designed the research; L‐MT, C‐JZ, X‐MH, C‐RS, Y‐JL, J‐XZ, and LL performed research; L‐MT, C‐JZ, Y‐QL, SC, and X‐JH analyze data; L‐MT and X‐JH wrote the paper. All authors read and approved the final manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Dataset EV6
Dataset EV7
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 2
Source Data for Figure 5
Source Data for Figure 8
Acknowledgements
This work was supported by the National Key Research and Development Program of China (2016YFA0500801) and the 973 Program (2011CB812600) from the Chinese Ministry of Science and Technology.
The EMBO Journal (2018) 37: e98770
See also: https://doi.org/10.15252/embj.2018100573 (October 2018)
References
- Alvarez‐Venegas R, Avramova Z (2012) Evolution of the PWWP‐domain encoding genes in the plant and animal lineages. BMC Evol Biol 12: 101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aufsatz W, Mette MF, van der Winden J, Matzke M, Matzke AJ (2002) HDA6, a putative histone deacetylase needed to enhance DNA methylation induced by double‐stranded RNA. EMBO J 21: 6832–6841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartee L, Malagnac F, Bender J (2001) Arabidopsis cmt3 chromomethylase mutations block non‐CG methylation and silencing of an endogenous gene. Genes Dev 15: 1753–1758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blevins T, Pontes O, Pikaard CS, Meins F Jr (2009) Heterochromatic siRNAs and DDM1 independently silence aberrant 5S rDNA transcripts in Arabidopsis . PLoS ONE 4: e5932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blevins T, Pontvianne F, Cocklin R, Podicheti R, Chandrasekhara C, Yerneni S, Braun C, Lee B, Rusch D, Mockaitis K, Tang H, Pikaard CS (2014) A two‐step process for epigenetic inheritance in Arabidopsis . Mol Cell 54: 30–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohmdorfer G, Sethuraman S, Rowley MJ, Krzyszton M, Rothi MH, Bouzit L, Wierzbicki AT (2016) Long non‐coding RNA produced by RNA polymerase V determines boundaries of heterochromatin. Elife 5: e19092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreault AA, Cronier D, Selleck W, Lacoste N, Utley RT, Allard S, Savard J, Lane WS, Tan S, Cote J (2003) Yeast enhancer of polycomb defines global Esa1‐dependent acetylation of chromatin. Genes Dev 17: 1415–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SW, Zhang X, Bernatavichute YV, Jacobsen SE (2006) Two‐step recruitment of RNA‐directed DNA methylation to tandem repeats. PLoS Biol 4: e363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Lu L, Mayer KS, Scalf M, Qian S, Lomax A, Smith LM, Zhong X (2016) POWERDRESS interacts with HISTONE DEACETYLASE 9 to promote aging in Arabidopsis . Elife 5: e17214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cokus SJ, Feng S, Zhang X, Chen Z, Merriman B, Haudenschild CD, Pradhan S, Nelson SF, Pellegrini M, Jacobsen SE (2008) Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 452: 215–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyon Y, Cote J (2004) The highly conserved and multifunctional NuA4 HAT complex. Curr Opin Genet Dev 14: 147–154 [DOI] [PubMed] [Google Scholar]
- Earley K, Lawrence RJ, Pontes O, Reuther R, Enciso AJ, Silva M, Neves N, Gross M, Viegas W, Pikaard CS (2006) Erasure of histone acetylation by Arabidopsis HDA6 mediates large‐scale gene silencing in nucleolar dominance. Genes Dev 20: 1283–1293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earley KW, Shook MS, Brower‐Toland B, Hicks L, Pikaard CS (2007) In vitro specificities of Arabidopsis co‐activator histone acetyltransferases: implications for histone hyperacetylation in gene activation. Plant J 52: 615–626 [DOI] [PubMed] [Google Scholar]
- Earley KW, Pontvianne F, Wierzbicki AT, Blevins T, Tucker S, Costa‐Nunes P, Pontes O, Pikaard CS (2010) Mechanisms of HDA6‐mediated rRNA gene silencing: suppression of intergenic Pol II transcription and differential effects on maintenance versus siRNA‐directed cytosine methylation. Genes Dev 24: 1119–1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Z, Morales‐Ruiz T, Ariza RR, Roldán‐Arjona T, David L, Zhu JK (2002) ROS1, a repressor of transcriptional gene silencing in Arabidopsis, encodes a DNA glycosylase/lyase. Cell 111: 803–814 [DOI] [PubMed] [Google Scholar]
- Haag JR, Pikaard CS (2011) Multisubunit RNA polymerases IV and V: purveyors of non‐coding RNA for plant gene silencing. Nat Rev Mol Cell Biol 12: 483–492 [DOI] [PubMed] [Google Scholar]
- Han YF, Zhao QY, Dang LL, Luo YX, Chen SS, Shao CR, Huang HW, Li YQ, Li L, Cai T, Chen S, He XJ (2016) The SUMO E3 ligase‐like proteins PIAL1 and PIAL2 interact with MOM1 and form a novel complex required for transcriptional silencing. Plant Cell 28: 1215–1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He XJ, Hsu YF, Pontes O, Zhu J, Lu J, Bressan RA, Pikaard C, Wang CS, Zhu JK (2009) NRPD4, a protein related to the RPB4 subunit of RNA polymerase II, is a component of RNA polymerases IV and V and is required for RNA‐directed DNA methylation. Genes Dev 23: 318–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He XJ, Chen T, Zhu JK (2011) Regulation and function of DNA methylation in plants and animals. Cell Res 21: 442–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson IR, Jacobsen SE (2008) Tandem repeats upstream of the Arabidopsis endogene SDC recruit non‐CG DNA methylation and initiate siRNA spreading. Genes Dev 22: 1597–1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollender C, Liu Z (2008) Histone deacetylase genes in Arabidopsis development. J Integr Plant Biol 50: 875–885 [DOI] [PubMed] [Google Scholar]
- Jacob Y, Feng S, LeBlanc CA, Bernatavichute YV, Stroud H, Cokus S, Johnson LM, Pellegrini M, Jacobsen SE, Michaels SD (2009) ATXR5 and ATXR6 are H3K27 monomethyltransferases required for chromatin structure and gene silencing. Nat Struct Mol Biol 16: 763–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W, Latrasse D, Servet C, Zhou DX (2013) Arabidopsis histone deacetylase HDA9 regulates flowering time through repression of AGL19. Biochem Biophys Res Commun 432: 394–398 [DOI] [PubMed] [Google Scholar]
- Kim MY, Zilberman D (2014) DNA methylation as a system of plant genomic immunity. Trends Plant Sci 19: 320–326 [DOI] [PubMed] [Google Scholar]
- Kim YJ, Wang R, Gao L, Li D, Xu C, Mang H, Jeon J, Chen X, Zhong X, Kwak JM, Mo B, Xiao L, Chen X (2016) POWERDRESS and HDA9 interact and promote histone H3 deacetylation at specific genomic sites in Arabidopsis . Proc Natl Acad Sci USA 113: 14858–14863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger F, Andrews SR (2011) Bismark: a flexible aligner and methylation caller for Bisulfite‐Seq applications. Bioinformatics 27: 1571–1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory‐efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latrasse D, Benhamed M, Henry Y, Domenichini S, Kim W, Zhou DX, Delarue M (2008) The MYST histone acetyltransferases are essential for gametophyte development in Arabidopsis . BMC Plant Biol 8: 121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law JA, Jacobsen SE (2010) Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet 11: 204–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WK, Cho MH (2016) Telomere‐binding protein regulates the chromosome ends through the interaction with histone deacetylases in Arabidopsis thaliana . Nucleic Acids Res 44: 4610–4624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei M, Zhang H, Julian R, Tang K, Xie S, Zhu JK (2015) Regulatory link between DNA methylation and active demethylation in Arabidopsis . Proc Natl Acad Sci USA 112: 3553–3557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindroth AM, Cao X, Jackson JP, Zilberman D, McCallum CM, Henikoff S, Jacobsen SE (2001) Requirement of CHROMOMETHYLASE3 for maintenance of CpXpG methylation. Science 292: 2077–2080 [DOI] [PubMed] [Google Scholar]
- Lindroth AM, Shultis D, Jasencakova Z, Fuchs J, Johnson L, Schubert D, Patnaik D, Pradhan S, Goodrich J, Schubert I, Jenuwein T, Khorasanizadeh S, Jacobsen SE (2004) Dual histone H3 methylation marks at lysines 9 and 27 required for interaction with chromomethylase3. EMBO J 23: 4286–4296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippman Z, Gendrel AV, Black M, Vaughn MW, Dedhia N, McCombie WR, Lavine K, Mittal V, May B, Kasschau KD, Carrington JC, Doerge RW, Colot V, Martienssen R (2004) Role of transposable elements in heterochromatin and epigenetic control. Nature 430: 471–476 [DOI] [PubMed] [Google Scholar]
- Lister R, O'Malley RC, Tonti‐Filippini J, Gregory BD, Berry CC, Millar AH, Ecker JR (2008) Highly integrated single‐base resolution maps of the epigenome in Arabidopsis . Cell 133: 523–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Luo M, Wu K (2012) Epigenetic interplay of histone modifications and DNA methylation mediated by HDA6. Plant Signal Behav 7: 633–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ZW, Zhou JX, Huang HW, Li YQ, Shao CR, Li L, Cai T, Chen S, He XJ (2016) Two components of the RNA‐directed DNA methylation pathway associate with MORC6 and silence loci targeted by MORC6 in Arabidopsis . PLoS Genet 12: e1006026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martienssen R, Moazed D (2015) RNAi and heterochromatin assembly. Cold Spring Harb Perspect Biol 7: a019323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieu O, Jasencakova Z, Vaillant I, Gendrel AV, Colot V, Schubert I, Tourmente S (2003) Changes in 5S rDNA chromatin organization and transcription during heterochromatin establishment in Arabidopsis . Plant Cell 15: 2929–2939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieu O, Reinders J, Caikovski M, Smathajitt C, Paszkowski J (2007) Transgenerational stability of the Arabidopsis epigenome is coordinated by CG methylation. Cell 130: 851–862 [DOI] [PubMed] [Google Scholar]
- Matzke MA, Mosher RA (2014) RNA‐directed DNA methylation: an epigenetic pathway of increasing complexity. Nat Rev Genet 15: 394–408 [DOI] [PubMed] [Google Scholar]
- Miura A, Nakamura M, Inagaki S, Kobayashi A, Saze H, Kakutani T (2009) An Arabidopsis jmjC domain protein protects transcribed genes from DNA methylation at CHG sites. EMBO J 28: 1078–1086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moissiard G, Cokus SJ, Cary J, Feng S, Billi AC, Stroud H, Husmann D, Zhan Y, Lajoie BR, McCord RP, Hale CJ, Feng W, Michaels SD, Frand AR, Pellegrini M, Dekker J, Kim JK, Jacobsen SE (2012) MORC family ATPases required for heterochromatin condensation and gene silencing. Science 336: 1448–1451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosher RA, Schwach F, Studholme D, Baulcombe DC (2008) PolIVb influences RNA‐directed DNA methylation independently of its role in siRNA biogenesis. Proc Natl Acad Sci USA 105: 3145–3150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey R, Muller A, Napoli CA, Selinger DA, Pikaard CS, Richards EJ, Bender J, Mount DW, Jorgensen RA (2002) Analysis of histone acetyltransferase and histone deacetylase families of Arabidopsis thaliana suggests functional diversification of chromatin modification among multicellular eukaryotes. Nucleic Acids Res 30: 5036–5055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontes O, Costa‐Nunes P, Vithayathil P, Pikaard CS (2009) RNA polymerase V functions in Arabidopsis interphase heterochromatin organization independently of the 24‐nt siRNA‐directed DNA methylation pathway. Mol Plant 2: 700–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Probst AV, Fagard M, Proux F, Mourrain P, Boutet S, Earley K, Lawrence RJ, Pikaard CS, Murfett J, Furner I, Vaucheret H, Mittelsten Scheid O (2004) Arabidopsis histone deacetylase HDA6 is required for maintenance of transcriptional gene silencing and determines nuclear organization of rDNA repeats. Plant Cell 16: 1021–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigal M, Kevei Z, Pélissier T, Mathieu O (2012) DNA methylation in an intron of the IBM1 histone demethylase gene stabilizes chromatin modification patterns. EMBO J 31: 2981–2993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronemus MJ, Galbiati M, Ticknor C, Chen J, Dellaporta SL (1996) Demethylation‐induced developmental pleiotropy in Arabidopsis . Science 273: 654–657 [DOI] [PubMed] [Google Scholar]
- Roudier F, Ahmed I, Bérard C, Sarazin A, Mary‐Huard T, Cortijo S, Bouyer D, Caillieux E, Duvernois‐Berthet E, Al‐Shikhley L, Giraut L, Després B, Drevensek S, Barneche F, Dèrozier S, Brunaud V, Aubourg S, Schnittger A, Bowler C, Martin‐Magniette ML et al (2011) Integrative epigenomic mapping defines four main chromatin states in Arabidopsis . EMBO J 30: 1928–1938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowley MJ, Bohmdorfer G, Wierzbicki AT (2013) Analysis of long non‐coding RNAs produced by a specialized RNA polymerase in Arabidopsis thaliana . Methods 63: 160–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saze H, Shiraishi A, Miura A, Kakutani T (2008) Control of genic DNA methylation by a jmjC domain‐containing protein in Arabidopsis thaliana . Science 319: 462–465 [DOI] [PubMed] [Google Scholar]
- Saze H, Kitayama J, Takashima K, Miura S, Harukawa Y, Ito T, Kakutani T (2013) Mechanism for full‐length RNA processing of Arabidopsis genes containing intragenic heterochromatin. Nat Commun 4: 2301 [DOI] [PubMed] [Google Scholar]
- Schrumpfova PP, Vychodilova I, Dvorackova M, Majerska J, Dokladal L, Schorova S, Fajkus J (2014) Telomere repeat binding proteins are functional components of Arabidopsis telomeres and interact with telomerase. Plant J 77: 770–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soppe WJ, Jacobsen SE, Alonso‐Blanco C, Jackson JP, Kakutani T, Koornneef M, Peeters AJ (2000) The late flowering phenotype of fwa mutants is caused by gain‐of‐function epigenetic alleles of a homeodomain gene. Mol Cell 6: 791–802 [DOI] [PubMed] [Google Scholar]
- Soppe WJ, Jasencakova Z, Houben A, Kakutani T, Meister A, Huang MS, Jacobsen SE, Schubert I, Fransz PF (2002) DNA methylation controls histone H3 lysine 9 methylation and heterochromatin assembly in Arabidopsis . EMBO J 21: 6549–6559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankunas K, Berger J, Ruse C, Sinclair DA, Randazzo F, Brock HW (1998) The enhancer of polycomb gene of Drosophila encodes a chromatin protein conserved in yeast and mammals. Development 125: 4055–4066 [DOI] [PubMed] [Google Scholar]
- Stec I, Nagl SB, van Ommen GJ, den Dunnen JT (2000) The PWWP domain: a potential protein–protein interaction domain in nuclear proteins influencing differentiation? FEBS Lett 473: 1–5 [DOI] [PubMed] [Google Scholar]
- Stroud H, Greenberg MV, Feng S, Bernatavichute YV, Jacobsen SE (2013) Comprehensive analysis of silencing mutants reveals complex regulation of the Arabidopsis methylome. Cell 152: 352–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessadori F, van Zanten M, Pavlova P, Clifton R, Pontvianne F, Snoek LB, Millenaar FF, Schulkes RK, van Driel R, Voesenek LA, Spillane C, Pikaard CS, Fransz P, Peeters AJ (2009) Phytochrome B and histone deacetylase 6 control light‐induced chromatin compaction in Arabidopsis thaliana . PLoS Genet 5: e1000638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- To TK, Kim JM, Matsui A, Kurihara Y, Morosawa T, Ishida J, Tanaka M, Endo T, Kakutani T, Toyoda T, Kimura H, Yokoyama S, Shinozaki K, Seki M (2011) Arabidopsis HDA6 regulates locus‐directed heterochromatin silencing in cooperation with MET1. PLoS Genet 7: e1002055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL (2009) TopHat: discovering splice junctions with RNA‐Seq. Bioinformatics 25: 1105–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L (2010) Transcript assembly and quantification by RNA‐Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28: 511–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Duan CG, Tang K, Wang B, Zhang H, Lei M, Lu K, Mangrauthia SK, Wang P, Zhu G, Zhao Y, Zhu JK (2013) RNA‐binding protein regulates plant DNA methylation by controlling mRNA processing at the intronic heterochromatin‐containing gene IBM1. Proc Natl Acad Sci USA 110: 15467–15472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams BP, Pignatta D, Henikoff S, Gehring M (2015) Methylation‐sensitive expression of a DNA demethylase gene serves as an epigenetic rheostat. PLoS Genet 11: e1005142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilsker D, Patsialou A, Zumbrun SD, Kim S, Chen Y, Dallas PB, Moran E (2004) The DNA‐binding properties of the ARID‐containing subunits of yeast and mammalian SWI/SNF complexes. Nucleic Acids Res 32: 1345–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Zong W, Hou X, Yao J, Liu H, Li X, Zhao Y, Xiong L (2015) OsARID3, an AT‐rich interaction domain‐containing protein, is required for shoot meristem development in rice. Plant J 83: 806–817 [DOI] [PubMed] [Google Scholar]
- Zang C, Schones DE, Zeng C, Cui K, Zhao K, Peng W (2009) A clustering approach for identification of enriched domains from histone modification ChIP‐Seq data. Bioinformatics 25: 1952–1958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemach A, Kim MY, Hsieh PH, Coleman‐Derr D, Eshed‐Williams L, Thao K, Harmer SL, Zilberman D (2013) The Arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1‐containing heterochromatin. Cell 153: 193–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Henderson IR, Lu C, Green PJ, Jacobsen SE (2007) Role of RNA polymerase IV in plant small RNA metabolism. Proc Natl Acad Sci USA 104: 4536–4541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Bernatavichute YV, Cokus S, Pellegrini M, Jacobsen SE (2009) Genome‐wide analysis of mono‐, di‐ and trimethylation of histone H3 lysine 4 in Arabidopsis thaliana . Genome Biol 10: R62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CJ, Ning YQ, Zhang SW, Chen Q, Shao CR, Guo YW, Zhou JX, Li L, Chen S, He XJ (2012) IDN2 and its paralogs form a complex required for RNA‐directed DNA methylation. PLoS Genet 8: e1002693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CJ, Hou XM, Tan LM, Shao CR, Huang HW, Li YQ, Li L, Cai T, Chen S, He XJ (2016) The Arabidopsis acetylated histone‐binding protein BRAT1 forms a complex with BRP1 and prevents transcriptional silencing. Nat Commun 7: 11715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Zhang B, Yang M, Zhu J, Li H (2018) Systematic profiling of histone readers in Arabidopsis thaliana . Cell Rep 22: 1090–1102 [DOI] [PubMed] [Google Scholar]
- Zheng B, He H, Zheng Y, Wu W, McCormick S (2014) An ARID domain‐containing protein within nuclear bodies is required for sperm cell formation in Arabidopsis thaliana . PLoS Genet 10: e1004421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Wang X, He K, Charron JB, Elling AA, Deng XW (2010) Genome‐wide profiling of histone H3 lysine 9 acetylation and dimethylation in Arabidopsis reveals correlation between multiple histone marks and gene expression. Plant Mol Biol 72: 585–595 [DOI] [PubMed] [Google Scholar]
- Zhou Y, Hartwig B, James GV, Schneeberger K, Turck F (2016) Complementary activities of TELOMERE REPEAT BINDING proteins and polycomb group complexes in transcriptional regulation of target genes. Plant Cell 28: 87–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou JX, Liu ZW, Li YQ, Li L, Wang B, Chen S, He XJ (2018a) Arabidopsis PWWP domain proteins mediate H3K27 trimethylation on FLC and regulate flowering time. J Integr Plant Biol 60: 362–368 [DOI] [PubMed] [Google Scholar]
- Zhou Y, Wang Y, Krause K, Yang T, Dongus JA, Zhang Y, Turck F (2018b) Telobox motifs recruit CLF/SWN‐PRC2 for H3K27me3 deposition via TRB factors in Arabidopsis . Nat Genet 50: 638–644 [DOI] [PubMed] [Google Scholar]
- Zhu H, Chen T, Zhu M, Fang Q, Kang H, Hong Z, Zhang Z (2008) A novel ARID DNA‐binding protein interacts with SymRK and is expressed during early nodule development in Lotus japonicas . Plant Physiol 148: 337–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu JK (2009) Active DNA demethylation mediated by DNA glycosylases. Annu Rev Genet 43: 143–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Dataset EV6
Dataset EV7
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 2
Source Data for Figure 5
Source Data for Figure 8