

Abstract
Although covalent protein binding is established as the pivotal event underpinning acetaminophen (APAP) toxicity, its mechanistic details remain unclear. In this study, we demonstrated that APAP induces widespread protein glutathionylation in a time-, dose- and bioactivation-dependent manner in HepaRG cells. Proteo-metabonomic mapping provided evidence that APAP-induced glutathionylation resulted in functional deficits in energy metabolism, elevations in oxidative stress and cytosolic calcium, as well as mitochondrial dysfunction that correlate strongly with the well-established toxicity features of APAP. We also provide novel evidence that APAP-induced glutathionylation of carnitine O-palmitoyltransferase 1 (CPT1) and voltage-dependent anion-selective channel protein 1 are respectively involved in inhibition of fatty acid β-oxidation and opening of the mitochondrial permeability transition pore. Importantly, we show that the inhibitory effect of CPT1 glutathionylation can be mitigated by PPARα induction, which provides a mechanistic explanation for the prophylactic effect of fibrates, which are PPARα ligands, against APAP toxicity. Finally, we propose that APAP-induced protein glutathionylation likely occurs secondary to covalent binding, which is a previously unknown mechanism of glutathionylation, suggesting that this post-translational modification could be functionally implicated in drug-induced toxicity.
Keywords: Post-translational modifications, Metabolomics, Chemical biology, Protein Modification, Protein adducts, Acetaminophen, Drug-induced toxicity, Hepatotoxicity, Protein glutathionylation
Protein glutathionylation is a relatively understudied post-translational modification of cysteine residues. A highly dynamic process, it involves both chemical (via reaction with reactive oxygen/nitrogen species) and enzymatic (glutathione-S-transferase π or glutaredoxin 1 (Grx1))1 mediators. Conversely, deglutathionylation is exclusively an enzymatic process mediated by Grx1, glutaredoxin 2, sulfiredoxin and protein disulfide isomerase (1). As glutathionylation induces a change in the size and charge of the cysteine residue, it confers various functional changes including selective modulation of enzymatic activity and alteration in DNA binding by transcription factors (1). In other instances, glutathionylation does not impact protein function discernibly but protects cysteine thiols from permanent damage due to irreversible oxidation. To date, research in protein glutathionylation is centered on pathological conditions where oxidative stress is featured prominently, such as cardiovascular and neurological diseases (1).
Acetaminophen (APAP), an over-the-counter antipyretic and analgesic is the leading cause of acute liver failure in the United States (2). APAP undergoes oxidation by CYP2E1 in the liver to form N-acetyl-p-benzoquinone imine (NAPQI), a reactive metabolite that binds covalently to protein thiols (Fig. 1A). The crucial role of covalent binding in APAP-induced hepatotoxicity is well-established with three key evidences: (1) toxicity is attenuated and amplified when covalent binding is diminished and magnified by CYP450 inhibitors and inducers respectively (3), (2) peak covalent binding preceded hepatic necrosis by 1–2 h consistently (3), and (3) patients experiencing the most severe toxicity exhibit the highest levels of APAP adducts in plasma, correlating with hepatic transaminase levels (4). Despite clear causality, there remains a significant disconnect between the initial covalent adduction and the toxic sequelae. For example, over 30 hepatic proteins including glutathione peroxidase (GPx) were reported to be adducted, but the activities of these proteins were only moderately inhibited (4), whereas GPx−/− knockout mice exposed to APAP did not exhibit attenuation of liver injury (5). Conversely, activities of calcium-dependent ATPases, plasma membrane Na+-K+-ATPase and protein phosphatases decrease after exposure to hepatotoxic doses of APAP, but with no evidence of adduction by APAP (6). Therefore, covalent protein adduction is critical yet insufficient to account for APAP-induced hepatotoxicity.
Fig. 1.

Profiling of APAP-induced glutathionylation. A, Key events in APAP-induced hepatotoxicity. The molecular initiating event begins with generation of NAPQI by CYP2E1, resulting in covalent adduction to protein thiols. The magnitude of covalent binding correlates with the degree of hepatotoxicity, and can be modulated by CYP450 inhibition or induction. Widespread covalent binding is thought to result in the key events listed under cellular responses, culminating in necrosis and liver injury. B, Mechanisms of APAP-induced glutathionylation: (1) via formation of a covalent ipso adduct between a protein or non-protein thiol with NAPQI, followed by glutathionylation, or (2) via thiol S-oxidation or S-nitrosylation by oxidative stress. It should be noted that the ipso adduct can also undergo an alternative rearrangement to the well-known thioether conjugates of APAP (9) which is not illustrated here for simplicity.
Seminal work by Nelson and colleagues indicated that APAP treatment depleted free protein thiols, which was greater than that expected due to covalent binding of thiols alone; whereas treatment of liver homogenates with dithiothreitol restored free thiol levels (7). Although APAP causes mitochondrial oxidative stress, which could account for the loss of protein thiols due to oxidation, subsequent investigations revealed the formation of an ipso conjugate of NAPQI with GSH. Notably, the ipso GSH adduct reacts with a protein thiol, resulting in glutathionylation independent of a chemical reaction with oxidants (8). Similarly, an ipso protein adduct reacts with free GSH, also culminating in protein glutathionylation (Fig. 1B). Glutathionylated regions were also observed in liver sections of mice exposed to APAP, which overlapped with regions of APAP protein adduction (9). These reports provide the first evidence that besides oxidative stress, protein glutathionylation can also occur due to covalent binding of a reactive metabolite to a protein thiol. Given that glutathionylation can modulate protein function, we hypothesized that this post-translational modification could account for aspects of APAP-induced hepatotoxicity inadequately explained by covalent adduction. Using metabolically competent HepaRG hepatocytes as a model for the APAP-liver injury axis, we demonstrate that aberrant glutathionylation patterns induced by APAP can be comprehensively mapped to its known toxicity endpoints in a time-, dose- and bioactivation-dependent manner, thus illustrating that aberrant protein glutathionylation is potentially a novel and previously unrecognized mechanism of drug-induced hepatotoxicity.
EXPERIMENTAL PROCEDURES
Reagents
Acetaminophen (APAP), bovine serum albumin, carnitine, dithiothreitol (DTT), fenofibrate, glyceraldehyde-3-phosphate, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), reduced l-glutathione (GSH), hydrogen peroxide, ketoconazole, palmitoyl carnitine, palmitoyl-CoA, sodium diethyldithiocarbamate trihydrate (DEDC), sodium dodecyl sulfate (SDS), trichloroacetic acid (TCA) and tris(2-carboyxethyl)phosphine (TCEP) hydrochloride were purchased from Sigma-Aldrich (St Louis, MO). NADPH tetrasodium salt was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Etomoxir and glutaredoxin-1 C14S mutant (Grx1) was purchased from Cayman Chemicals (Ann Arbor, MA). Diamide was obtained from Tokyo Chemical Industries (Tokyo, Japan). N-acetyl-p-benzoquinone imine (NAPQI) was obtained from Toronto Research Chemicals (Ontario, Canada). Glutathione reductase (GR) was obtained from Roche Diagnostics (Basel, Switzerland). Sequencing Grade Modified Trypsin was purchased from Promega (Madison, WI). Heat-inactivated fetal bovine serum (FBS), l-glutamine, minimum essential medium/Earle's balanced salt solution (MEM/EBSS) and MEM non-essential amino acids, were obtained from Hyclone (Logan, UT). Penicillin G and streptomycin were obtained from Pan Biotech (Dorset, UK). Pierce BCA protein assay kit and mitochondrial isolation kit for cultured cells was obtained from Thermo Scientific (Waltham, MA). Phosphate-buffered saline (PBS) was obtained from Vivantis (Selangor, Malaysia). Cleavable ICAT (isotope-coded affinity tag) reagents were purchased from ABSciex (Framingham, MA). Filter-aided sample preparation (FASP) kits were obtained from Expedeon (Cambridgeshire, UK). Water was purified using a Milli-Q water purification system (Millipore, Bedford, MA). All other solvents and reagents used were of analytical grades.
Experimental Design and Statistical Rationale
This study aimed to define protein glutathionylation patterns induced by APAP in HepaRG cells. This was examined temporally, across different doses and in the presence of a CYP2E1 inhibitor (Fig. 2) For the global relative quantitation of protein glutathionylation experiments, biological triplicates of each treatment condition were collected across 3 cell passages. Each sample was processed and measured once using mass spectrometry.
Fig. 2.

Overall experimental design for profiling APAP-induced glutathionylation. A, HepaRG cells were treated either with APAP or a vehicle control in biological triplicates, followed by harvesting of lysate in ice-cold 10% TCA and GluICAT profiling to identify glutathionylated proteins. B, APAP-induced glutathionylation was profiled longitudinally (3–24 h) using 30 mm APAP. The effect of dose (sub-toxic 0.5 mm versus 30 mm APAP at 3 h) was also examined. Finally the influence of APAP bioactivation by CYP2E1 on glutathionylation was investigated using the CYP2E1 inhibitor DEDC. C, Proteo-metabonomic mapping was applied to infer the functional consequences of protein glutathionylation and obtain insight into the perturbed pathways related to APAP toxicity.
HepaRG Cell Culture
HepaRG cells at passage 13 were obtained from Biopredic International (Rennes, France). The cells were seeded in T-75 flasks at 2 × 106 undifferentiated cells/flask and grown to confluence in 710 growth medium (Biopredic, Rennes, France). The cells were cultured under a humidified atmosphere of 5% CO2 at 37 °C for 14 days before passaging. Medium was renewed every 3 days. Passages 13 and 14 were neither differentiated nor used as per the vendor's instructions. Differentiation was induced on day 14 by replacing 710 growth medium with 720 differentiation medium (Biopredic) for a further 2 weeks. The cells were maintained up to 4 weeks after differentiation and were used at passage 15–18 for drug treatment and biochemical analyses.
HepG2 Cell Culture
HepG2 human hepatocellular carcinoma cells (American Type Culture Collection, Manassas, VA) were cultured in T-175 culture flasks in culture media consisting of MEM/EBSS supplemented with 10% heat-inactivated FBS, 2 mm l-glutatmine, 100 U/ml Penicillin G, 100 μg/ml streptomycin and MEM non-essential amino acids. The cells were cultured under a humidified atmosphere of 5% CO2 at 37 °C. Culture medium was changed every 3 days and cells were subcultured weekly.
Biochemical Analyses
All biochemical analyses were performed in a 96-well plate format. Undifferentiated HepaRG cells were seeded in 96-well plates at a density of 9000 cells/well and then differentiated as described above. After differentiation, 720 differentiation medium, which contains 1.7% DMSO, was replaced with HepaRG Tox Working Medium (Life Technologies, Carlsbad, CA) which contains 0.5% DMSO and maintained for an additional week. Cells were treated with 30 mm APAP for 3, 6, 12 and 24 h for all assays. Cell viability was assessed by the CellTiter-Glo Luminescent Cell Viability Assay (Promega, Madison, WI). Mitochondrial ROS generation was measured using MitoSOX Red Mitochondrial Superoxide Indicator (Life Technologies). Mitochondrial superoxide was measured at λex = 396 nm and nonspecific oxidation at λex = 510 nm, whereas λem = 580 nm was used for both (10). Cellular percentage of GSH relative to untreated control as an indicator of cell health and oxidative stress was quantified using the luminescence-based system of GSH/GSSG-Glo Assay (Promega). All biochemical assays were performed according to manufacturer's instructions.
APAP Treatment of HepaRG Cells
Undifferentiated HepaRG cells were seeded in 6-well plates at a density of 2 × 105 cells/well, differentiated, and then maintained in HepaRG Tox Working Medium for 1 week in a total volume of 2 ml per well as mentioned above. To investigate the time-dependence of APAP glutathionylation, cells were treated with 30 mm APAP for 3, 6, 12 and 24 h. To determine dose-dependence of APAP glutathionylation, cells were treated with 0.5 mm or 30 mm APAP for 3 h. To assess the influence of CYP2E1 bioactivation on APAP glutathionylation, cells were pretreated with DEDC, a CYP2E1 inhibitor for 30 min. The culture media was then replaced with DEDC-free media containing 0.5 mm APAP for 3 h. For vehicle-treated controls, cells were treated with 1.9% ethanol for 6 h. Every vehicle or drug treatment was performed in triplicates across 3 cell passages.
Harvesting of APAP-treated Cell Lysate
Drug-containing culture media was removed from each well at the appropriate time-points and cells were rinsed twice with ice-cold PBS. 500 μl of ice-cold 10% (w/v) TCA was added to each well and kept on ice for 10 min. Cells were then scraped and the suspension was transferred into a 1.5 ml microtube. Each well was rinsed with 500 μl of ice-cold 10% (w/v) TCA and added to the same microtube. All tubes were kept on ice for 30 min and vortexed every 10 min. Samples were stored at −80 °C until further processing.
GluICAT Processing of Glutathionylated Proteins
Cell lysates were centrifuged at 14,000 × g for 30 min at 4 °C. Denaturing alkylation buffer (DAB) containing 6 m urea, 200 mm Tris-HCI, pH 8.5, 10 mm EDTA, and 4% (w/v) SDS was freshly prepared before use. The protein pellet was rinsed once with 500 μl ice-cold 10% (w/v) TCA, followed by a second rinse with 200 μl ice-cold 5% (w/v) TCA. The pellet was then redissolved in 80 μl DAB and quantified using the BCA assay. An aliquot containing 100 μg of protein was removed and additional DAB added to a final volume of 80 μl, followed by alkylation with light ICAT reagent for 2 h. Proteins were precipitated using 500 μl of −20 °C cold acetone and maintained at −20 °C overnight. The acetone precipitate was centrifuged at 14,000 × g for 30 min at 4 °C, and the protein pellet was rinsed twice with 500 μl−20 °C cold acetone to remove excess light ICAT. The protein pellet was resolubilized in DAB and prepared for deglutathionylation as follows: the protein solution was loaded onto a spin column and SDS was removed by washing 3 times with 6 m urea, followed by 3 washes with 50 mm ammonium bicarbonate to condition the membrane for the subsequent deglutathionylation step. To effect deglutathionylation, the following mixture was added: 10 mm GSH, 10 mm NADPH, 4 U/ml GR and 0.5 U/ml Grx1 in 100 μl 50 mm ammonium bicarbonate and incubated for 1 h at 37 °C on the spin column. Excess GSH was removed by washing 4 times with 50 mm ammonium bicarbonate. Newly reduced cysteines were labeled with heavy ICAT reagent for 2 h, followed by removal of excess heavy ICAT by washing 3 times with 50 mm ammonium bicarbonate. Proteins were then digested with trypsin (1:30 trypsin/protein ratio) for 12–16 h at 37 °C. Peptides were eluted with 50 mm ammonium bicarbonate and 0.5 m sodium chloride. Peptides were purified using avidin affinity chromatography. Finally, the biotin tag was cleaved, and samples were desalted using UltraMicro Spin Columns (Nest Group, Southborough, MA) before LC/MS/MS analysis.
LC/MS/MS Detection of GluICAT-treated Glutathionylated Proteins
LC/MS/MS was performed using an Ultimate 3000 nanoLC system (Dionex, Thermo Fisher Scientific, MA) coupled to an ABSciex 5600 TripleTOF (ABSciex). A 50 cm × 75 μm i.d. Acclaim PepMap RSLC C18 column was employed (Dionex). This column was connected to a spray tip (New Objectives, Woburn, MA), which was directly coupled to the nano-spray interface of the mass spectrometer. Samples were loaded onto a trap column (Acclaim PepMap 100 C18, 2 cm × 75 μm i.d., Dionex) at a flow rate of 5 μl/min. After a 3 min wash with loading buffer (2/98 v/v of ACN/water with 0.1% formic acid), the system was switched into line with the C18 analytical capillary column. A step linear gradient of mobile phase B (2/98 v/v of water/ACN with 0.1% formic acid) from 5% to 25% over 10 min, 25–60% for 9 min, and lastly, 60–95% over 1 min at flow rate of 300 nL/min was utilized for this analysis. Third generation Nanospray Source was installed, and other instrumentation settings were as follows: ionspray voltage floating, 2200 V; curtain gas, 30 psi; ion source gas 1, 12 psi; interface heater temperature, 125 °C; declustering potential, 100 V. Data was acquired using TOF MS + Hi Sensitivity product ion with Analyst TF 1.6 software (ABSciex). TOF-MS scan (experiment 1) parameters were set as follows: 0.25 s TOF MS accumulation time in the mass range of 350 to 1250 Da was followed by MS/MS scan based on the following parameters: mass range was set at 100 to 1500 Da; switching criteria were set to ions greater than m/z = 350 and smaller than m/z = 1250 with charge state of 2 to 5 and an abundance threshold of greater than 120 cps. Former target ions were excluded for 4 s and excluded after 1 repeat. The maximum number of candidate ions to monitor per cycle was 20 spectra with an accumulation time of 100 ms. All injections were performed in triplicates.
Database Search Parameters, Acceptance Criteria for Protein Identification and ICAT Relative Quantitation
The MS data was processed using ProteinPilot software 4.5 (ABSciex) with database search using Uniprot (Sept 2010 release, 40516 proteins searched). Searching parameters were as follows: (1) sample type: cleavable ICAT; (2) Cys Alkylation: none; (3) digestion: trypsin; (4) instrument: TripleTOF 5600; (5) species: Homo sapiens; (6) ID focus: biological modifications. All peptides identified from the database were filtered according to the following criteria: at least 95% confidence, and possessing a heavy-to-light (H/L) ratio of at least 0.01. Peptides that did not meet the criteria were removed from further analysis. The average H:L value across triplicate sets of peptide data from the vehicle-treated control arm were computed. Peptides were cross-matched between those in the control list and each drug-treated replicate, and the quotient of [H/L treated/H/L control] for each peptide in each treatment replicate was obtained. A particular peptide was considered to be glutathionylated only if the [H/L treated/H/L control] ratio exceeded 1.5-fold in at least 2 of 3 drug-treated replicates and subjected to further pathway analysis described below.
Bioinformatics Analysis
Glutathionylated proteins were submitted for pathway analysis and searched by UniProt accession number using the Reactome Pathway Knowledgebase pathway analysis tool (version 53) (14). A false discovery rate (FDR) less than 5% was applied to returned pathways. Manual inspection of the glutathionylated proteins to identify candidates relevant to the known manifestations of APAP toxicity was also performed. Enrichment analysis was also performed using the PANTHER and GO Ontology database to determine the molecular function and subcellular localization of glutathionylated proteins respectively. Proteo-metabonomic mapping was applied to pathways meeting the FDR criteria to ascertain the relevance of these pathways to APAP toxicology.
To obtain an overview of APAP-induced metabolite perturbations, we sought to capitalize on the large number of published metabonomics studies on this subject. A comprehensive literature search to retrieve references related to metabonomics studies of APAP toxicity was performed using the Web of Science portal (Thomson Reuters, Philadelphia, PA) with a combination of the keywords “acetaminophen,” “paracetamol,” “metabolomics,” “metabonomics,” “metabolic,” “phenotyping,” “profiling,” “biomarkers,” “toxicity,” “hepatotoxicity,” or “dysfunction.” A list of metabolite biomarkers was compiled from reports of APAP-induced metabolic perturbations and used to manually annotate glutathionylated protein pathways obtained from the Reactome search. Mapped proteomic-metabonomic pathways were then examined for their biological relevance and relationship to the known manifestations of APAP toxicity.
Measurement of Mitochondrial CPT1 Activity in Untreated and Fenofibrate-treated HepG2 Cells
When HepG2 cells reached ∼80% confluency, cells were cultured for 48 h in serum-free culture media supplemented with 150 μm fenofibrate. HepG2 cells in the untreated arm underwent the same procedure in serum-free culture media supplemented with 0.2% DMSO. Mitochondria were isolated from untreated and fenofibrate-treated HepG2 cells using the mitochondrial isolation kit for cultured cells. The mitochondrial pellet was resuspended in assay buffer consisting of 117 mm tris-buffered saline (pH 7.4), 0.28 mm GSH, 4.4 mm magnesium chloride, 16.7 mm potassium chloride, 5 mm potassium cyanide and 0.1% bovine serum albumin and maintained on ice. Mitochondrial protein content was quantified using the BCA assay and diluted to a concentration of 310 μg/ml. For the measurement of carnitine O-palmitoyltransferase 1 (CPT1) activity, 6.25 μg of mitochondrial suspension was mixed with 25 μm palmitoyl-CoA and pre-incubated for 10 min at 37 °C with either 2 mm diamide, 2 mm NAPQI, 20 nm etomoxir or DMSO as a vehicle control. Reactions were initiated with 25 μm carnitine and allowed to proceed at 37 °C for 10 min. Reactions were quenched using 50 μl of ice-cold ACN containing 2 μm ketoconazole as an internal standard followed by LC/MS/MS analysis.
LC/MS/MS Detection of Palmitoyl Carnitine as a Marker of CPT1 Activity
Samples were analyzed using an Agilent 1290 Infinity LC system interfaced with an ABSciex Triple Quad 3500 triple quadrupole mass spectrometer equipped with a TurboIonSpray source (ABSciex). Mobile phase A was 50 mm ammonium formate in Milli-Q water and mobile phase B was 50 mm ammonium formate in 20/80 v/v Milli-Q water/ACN. Mobile phases were delivered at flow rate of 0.6 ml/min. Both mobile phases were pumped through a Waters Acquity UPLC BEH C18 130Å column (1.7 μm, 50 mm × 2.1 mm i.d.; Waters, Milford, MA) at a flow rate of 0.6 ml/min. Elution conditions were as follows: linear gradient 70% to 100% B (0–1 min), held at 100% B (1–2 min) and 70% B (2–2.5 min). Using an autosampler thermostatted at 4 °C, 2 μl of each sample was injected into the UPLC column maintained at 45 °C. The needle was flushed with ACN for 10 s post-injection to minimize carry-over effect. Tandem mass spectrometry was operated in the positive ion electrospray ionization (ESI +ve) mode. MS source conditions were: curtain gas 25 psi; ionspray voltage, 3000 V; temperature, 600 °C; ion source gas 1, 30 psi; and ion source gas 2, 60 psi. Compound-dependent MS parameters were optimized using pure standards and are summarized in supplemental Table S1. Multiple reaction monitoring (MRM) transition was applied for semi-quantitation based on analyte-to-internal standard peak area ratios. Data were acquired using ABSciex Analyst software version 1.6, whereas integration of chromatographic peaks was performed using ABSciex MultiQuant software version 3.0. Data from the biochemical analyses and CPT1 activity assay were expressed as mean ± standard deviation (S.D.). Comparisons between multiple groups were performed with one-way analysis of variance (ANOVA) followed by a post-hoc Bonferroni test, where p < 0.05 was considered significant.
Molecular Dynamics Simulations of Native and Glutathionylated VDAC1
Native VDAC1 structure was taken from the RSCB Protein Data Bank (PDB ID: 2jk4), whereas glutathionylated VDAC1 was constructed using Discovery Studio. Both proteins were embedded in a membrane patch of 200 POPC lipid molecules. The protein molecules were modeled using AMBER99sb force field (11), and the lipid molecules were described by AMBER14 lipid parameters (12). TIP3P water model was used to solvate the protein-membrane system and 0.1 m counter ions were added to neutralize the system. Each system was first subjected to 500 steps of energy minimization, followed by 100 ps of position restrained molecular dynamics simulations. Then 400 ns of molecular dynamics simulation for each protein was performed. During the molecular dynamics simulations, both short-range electrostatic interactions and Van der Waals interactions were calculated using a cut-off value of 0.8 nm, whereas the long-range electrostatic interactions were calculated using the particle mesh Ewald methods (13). The simulations were carried out in NPT ensemble with temperature and pressure maintained at 300 K and 1 bar, respectively. The simulations were performed using GROMACS package (14).
RESULTS
Development and Validation of the GluICAT Methodology
We modified the OxICAT methodology by Leichert et al. (15) to differentially label: (1) native cysteines using the 12C-labeled (light) isotope-coded affinity tag (ICAT), and (2) glutathionylated cysteines using 13C-labeled (heavy) ICAT. Unlike native cysteines, glutathionylated cysteines are treated with Grx1 to reverse glutathionylation prior to labeling. In this approach, which we termed as GluICAT, the extent of glutathionylation of a particular peptide in a single sample is determined by the heavy-to-light ICAT (H:L) ratio, where a high H:L ratio indicates a greater degree of glutathionylation (Fig. 3A). To validate GluICAT in distinguishing native and glutathionylated proteins, we artificially glutathionylated glyceraldehyde-3-phosphate dehydrogenase (GAPDH-SG) via treatment with hydrogen peroxide in the presence of GSH. Based on GluICAT analysis, native GAPDH and GAPDH-SG (+Grx1) yielded H:L ratios of 0.51 and 8.79 respectively (Fig. 3B). A protein is typically considered redox modified (oxidized, nitrosylated or glutathionylated) if it exhibits a signal 1.5-fold greater than that of the untreated control (15–17). The 17-fold greater H:L ratio of GAPDH-SG (+Grx1) corroborated its greater degree of glutathionylation and validated GluICAT in identifying glutathionylated proteins. GAPDH-SG that did not undergo Grx1-mediated deglutathionylation (—Grx1) yielded lower H:L ratio (3.61) as compared with +Grx1 (8.79), further validating the GluICAT method. The seemingly higher than expected H:L ratio of GAPDH-SG (—Grx1) compared with untreated GAPDH is attributed to a lower fraction of reduced cysteine thiols tagged with light ICAT, resulting in a smaller denominator which amplified the H:L ratio.
Fig. 3.
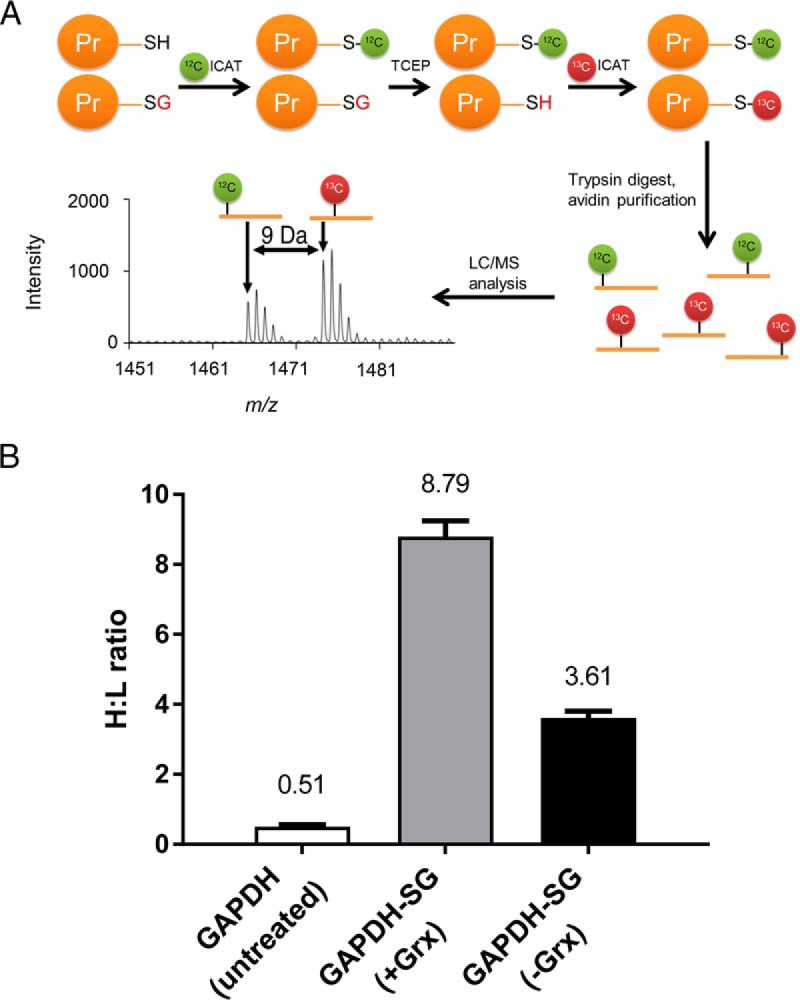
Development and validation of GluICAT methodology. A, Schematic illustrating the GluICAT strategy of differential tagging of reduced and glutathionylated thiols. Reduced thiols are tagged with light (12C) ICAT, whereas glutathionylated thiols are reduced by Grx1 then tagged with heavy (13C) ICAT, and subsequently detected by LC/MS/MS. B, H:L ratios for GAPDH (untreated) and GAPDH-SG (with (+) or without (−) Grx1) from the tryptic GAPDH peptide VPTPNVSVVDLTCR (mean of 3 replicates).
Identification of Proteins Glutathionylated by APAP
We established in a preliminary experiment that APAP induces characteristic decreases in cell viability and GSH levels, as well as an increase in superoxide formation in a time-dependent manner across 3, 6, 12 and 24 h in HepaRG cells (supplemental Fig. S1). To characterize reactive metabolite-induced protein glutathionylation, we next performed a proteome-wide screen of HepaRG cells treated with 30 mm APAP across the same 4 time points using the validated GluICAT method. A total of 898 peptides corresponding to 588 proteins were found to be glutathionylated at any one of the 4 time points monitored across 3 biological replicates. 49% of these proteins exhibit catalytic activity (Fig. 4), whereas 29% are mitochondrial proteins and 13% reside in the endoplasmic reticulum. Following pathway analysis of glutathionylated proteins, 3 major pathways emerged: (1) energy metabolism, (2) protein turnover, and (3) defense against cellular stress. Further manual analysis uncovered proteins relevant to calcium dynamics and mitochondrial permeability transition pore (MPTP) formation. Proteins and peptides found to be glutathionylated by APAP temporally in these pathways are summarized in Fig. 5 (fold-change values and peptide sequences provided in supplemental Tables S2–S5). Collectively, these groups comprise 245 peptides and 186 proteins, ∼27 and 31% of total glutathionylated peptides and proteins respectively.
Fig. 4.
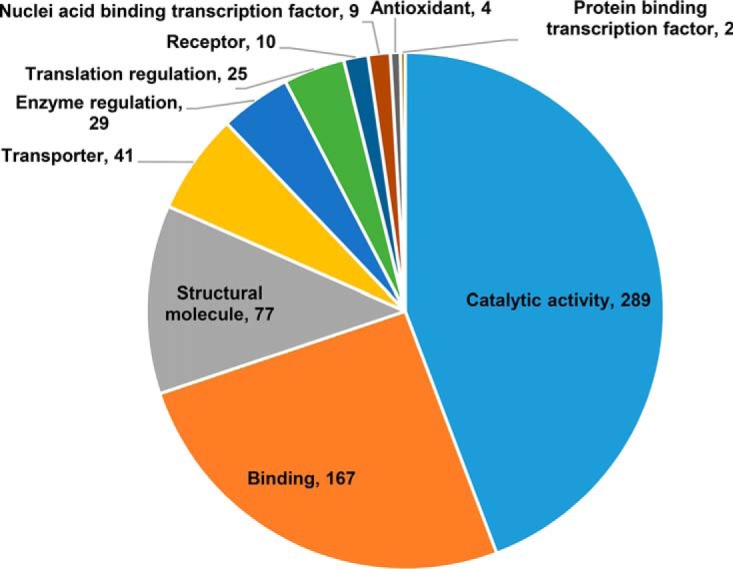
Gene ontology enrichment analysis. Distribution of protein functions for proteins glutathionylated by APAP at any time point. Numerals indicate number of proteins in each category. Some proteins may be listed in more than one category.
Fig. 5.

Identification of pathways glutathionylated by APAP. Heat map representing the H:L fold-change ratios for glutathionylated proteins in (A) energy metabolism, (B) protein turnover, (C) defense against cellular stress, and (D) calcium dynamics and mitochondrial permeability transition pore (MPTP). Duplicated gene abbreviations indicate multiple glutathionylated peptides for the same protein (see supplemental Tables S2–S5 for further details).
Proteo-metabonomic Mapping of APAP-induced Glutathionylation
Although omics-level studies often yield an abundance of information, unraveling the biological significance of these findings remains an analytical bottleneck. Given that 49% of glutathionylated proteins in our dataset comprised catalytic proteins, we reasoned that a juxtaposition of these protein pathways against APAP-induced metabolic perturbations would yield broad inferences regarding the functional consequences of glutathionylation. A total of 19 primary research articles investigating APAP-induced metabolic alterations were retrieved from literature, which collectively reported 81 metabolites that were perturbed by APAP (supplemental Table S6). Despite the diversity in models (mouse, rat, pig, and human) and biological matrices (serum, plasma, whole blood, urine, and liver), the perturbed metabolic pathways are remarkably coherent, confirming the robustness of APAP toxicity metabotypes.
Proteo-metabonomic mapping yielded insights primarily related to the inhibition of energy metabolism pathways. For example, fatty acids and acylcarnitine levels were consistently elevated, whereas the proteins responsible for their activation, transport, and downstream oxidation were glutathionylated, suggesting that these proteins are inhibited via glutathionylation (Fig. 6A). Similarly, the Krebs cycle and oxidative phosphorylation proteins were extensively glutathionylated, whereas multiple studies reported a decrease in Krebs cycle substrates as well as a profound decrease in ATP, suggesting glutathionylation inhibits these enzymes (Fig. 6B and 6C). For other pathways, proteo-metabonomic mapping did not reveal insights into the functional consequences of glutathionylation for several reasons: (1) low numbers of relevant metabolic derangements (e.g. glycolysis, Fig. 6D), (2) metabolites are involved in multiple pathways, where the metabolic perturbations could not be assigned to one particular glutathionylated pathway (e.g. amino acids), (3) proteins interact with a broad range of endogeneous and exogeneous substrates (e.g. Phase I and II detoxification proteins), or (4) pathways do not intersect with small molecule metabolite fluxes (e.g. MPTP). In such cases an extensive literature search was performed to extract further insights into the functional effect of glutathionylation.
Fig. 6.

Proteo-metabonomic mapping of glutathionylated proteins. Glutathionylated proteins in each map are marked with an asterisk, and up/down arrows indicate the direction of change (increase/decrease) for relevant metabolites. Where divergent changes were reported in literature, the corresponding biological matrices used to perform the measurements are indicated in italics. A, Fatty acid β-oxidation. B, Krebs cycle. C, Oxidative phosphorylation. D, Glycolysis.
Glutathionylation of CPT1 Inhibits Its Activity Whereas Treatment with Fenofibrate Attenuates the Inhibition
Although our analysis suggested that proteins involved in fatty acid activation, transport and β-oxidation are amenable to inhibition by glutathionylation, there is currently no direct evidence to confirm our inferences apart from the inhibition of mitochondrial carnitine/acylcarnitine carrier (CAC) by glutathionylation (18). We measured the functional activity of CPT1 in mitochondria isolated from HepG2 hepatocytes, which was subsequently exposed to the glutathionylating agent diamide, NAPQI and etomoxir (CPT1 inhibitor; positive control). Exposure of the mitochondria to etomoxir resulted in a 35% decrease in CPT1 activity, validating the experimental design. Diamide and NAPQI resulted in a 40 and 74% decrease in CPT1 activity respectively (Fig. 7A). These observations confirmed that CPT1 is functionally inhibited by glutathionylation, suggesting that APAP-induced glutathionylation impairs the fatty acid activation pathway.
Fig. 7.

CPT1 activity of isolated mitochondria from HepG2 cells. A, CPT1 activity following exposure to 2 mm diamide, 2 mm NAPQI and 20 nm etomoxir normalized to vehicle control (DMSO). B, CPT1 activity following pre-treatment with 100 μm fenofibrate. Activity in each treatment arm is normalized to the corresponding arm in the absence of fenofibrate pre-treatment. *p < 0.05, **p < 0.01, ****p < 0.0001. Error bars indicate standard deviations.
PPARα induction was reported to protect mice against APAP toxicity (19), however the mechanism remains unclear. We isolated mitochondria from HepG2 cells pre-treated with fenofibrate (PPARα inducer) and measured CPT1 activity following exposure to diamide, NAPQI and etomoxir. Remarkably, CPT1 activities in each treatment arm were at least 2-fold higher than in the absence of fenofibrate pre-treatment (Fig. 7B). This observation is attributed to the dampening of the inhibitory effect of glutathionylation on these enzymes via the up-regulation of fatty acid β-oxidation genes by fenofibrate. These findings further established glutathionylation as the mechanistic link between the impairment of fatty acid β-oxidation and the prophylactic effects of PPARα inducers.
Glutathionylation Modulates MPTP Permeability by Shifting the Conformation of VDAC to an Open State
Given that MPTP opening is a major event in APAP toxicity, we sought to understand the effect of glutathionylation on the voltage-dependent anion-selective channel protein (VDAC) family (Fig. 5D; supplemental Table S5), which is regarded to be involved in MPTP formation. As human VDAC1–3 share high structural homology (20), VDAC1 which possesses a crystal structure was selected as a representative template for computational evaluation. VDAC1 is known to adopt open and closed conformations (Fig. 8A), which in turn modulates pore permeability. Molecular dynamics simulations revealed that glutathionylation of Cys232 of VDAC1 stabilizes the open conformation, suggesting enhanced permeability to metabolites and reactive oxygen species such as superoxides (Fig. 8B). The preference for an open state arises from the increased hydrogen bonding between the carboxylic group of the glutathionyl segment and the basic residues of the VDAC1 protein. The interactions of the glutathionyl segment with the N-terminal segment (residues 1–29) stabilize its conformation, as shown in the reduced pairwise root mean square deviation (RMSD) and the secondary structure evolution (supplemental Fig. S2A and S2B). However, the pairwise RMSD for the whole protein increases marginally, suggesting that the overall structure becomes more flexible.
Fig. 8.

Structural characterization of native and glutathionylated VDAC1 from molecular dynamics simulations. A, Native VDAC1 in open (left) and closed (right) conformations. B, Glutathionylated VDAC1 in open conformations and the hydrogen bonds between the glutathionyl group with the VDAC1 protein. The glutathionyl group is displayed in ball-and-stick mode, and the residues involved in hydrogen bonding are displayed in sticks. Hydrogen bonds are represented using dashed red lines.
Glutathionylation May Increase Cationic Binding to and Translocation through VDAC
To understand the interactions of the VDAC1 with charged molecules, the electrostatic potential map of the VDAC1 surface was calculated (supplemental Fig. S2C). Because the glutathionyl group carries a net negative charge, glutathionylated VDAC1 displays a more negative surface potential, which reduces the free energy barrier of binding and translocation of cations such as calcium across the pore. Molecular dynamics simulations also revealed that the location of the negative glutathionyl group was found to be located close to the calcium binding site Glu206, which may further enhance calcium binding.
Longitudinal Progression of APAP-induced Protein Glutathionylation
The highest number of glutathionylated peptides was observed at 3 h, followed by a 50% decrease at 6 h, and was stable thereafter until 24 h (Fig. 9A). Although the total number of glutathionylated peptides remained steady from 6–24 h, the number of glutathionylated peptides in the 3 key pathways of energy metabolism, protein turnover and defense against cellular stress continued to decrease over time. The temporal decrease in the number of glutathionylated peptides that are relevant to APAP toxicity appeared to be balanced by an increase in glutathionylation of other peptides. The percentages of peptides that did not fall into these 3 pathways were 67% at 3 h, 65% at 6 h, and increased to 78% at 12 h and 84% at 24 h.
Fig. 9.

Glutathionylation of peptides classified under energy metabolism, protein turnover and defense against cellular stress pathways. A, Number of glutathionylated peptides as a function of time post-dose. B, Number of glutathionylated peptides among different APAP treatment conditions.
APAP-induced Protein Glutathionylation Is Dose- and Bioactivation-dependent
The glutathionylation patterns associated with treatments using 0.5 mm APAP and 0.5 mm APAP + diethyldithiocarbamate (DEDC; CYP2E1 inhibitor) were compared with that established for 30 mm APAP at the 3 h time point. For all three key pathways, 0.5 mm APAP and 0.5 mm APAP + DEDC treatments resulted in a decrease in H:L fold-change values compared with that of 30 mm APAP (Fig. 9B). Comparing 30 mm APAP against 0.5 mm APAP, there was a small percentage decrease (7%) in total number of glutathionylated peptides, which became more prominent (26%) when the data was compared against 0.5 mm APAP + DEDC treatment. When focusing on the peptides glutathionylated within the 3 key pathways, the differences among the 3 treatments became apparent. In all 3 pathways, the number of glutathionylated peptides decreased by ∼50% in the 0.5 mm APAP and 0.5 mm APAP + DEDC treatment groups when compared against 30 mm APAP. When comparing the effect of the CYP2E1 inhibitor on APAP-induced glutathionylation (0.5 mm APAP versus 0.5 mm APAP + DEDC), there was no change in the number of glutathionylated peptides involved in energy metabolism; however, the number of glutathionylated peptides involved in protein turnover, as well as defense against cellular stress decreased by a further 30%.
DISCUSSION
APAP-induced hepatotoxicity is characterized by the following classical features: (1) impaired energy metabolism, (2) elevated oxidative stress, (3) sustained increase in cytosolic calcium, and (4) MPTP opening and DNA fragmentation, which culminate in the classical necrosis and liver injury (Fig. 1A). Although research in the past three decades has uncovered much knowledge regarding the toxicity pathways leading to these endpoints, the precise mechanism by which covalent binding via NAPQI initiates these cellular responses remains an unresolved enigma. Although there have been numerous proteomic studies examining changes in protein expression upon exposure to APAP (21–25), thus far the identified proteins are thought to be involved in a compensatory response to toxicity, rather than directly mediating APAP toxicity. Given the evidence that formation of the ipso covalent adduct could induce protein glutathionylation, we developed the GluICAT method and applied it to the HepaRG model of APAP toxicity by McGill et al. (26) to explore the relationship between this post-translational modification and APAP toxicity. Here our findings suggest that aberrant protein glutathionylation induced by NAPQI, secondary to covalent binding could be responsible for initiating and driving these toxicity phenotypes.
Impaired Energy Metabolism
Remarkably, nearly one-third of glutathionylated proteins in our study were localized in the mitochondria, whereas mitochondrial proteins typically account for only 7% of the total liver proteome (27). The enrichment of mitochondrial proteins in our dataset agrees with the central role that mitochondrial dysfunction plays in APAP-induced hepatotoxicity (28). Indeed, the impairment of mitochondrial cellular energetics is the key hallmark of APAP toxicity (29), but the underlying mechanism remains undefined. Here we provide novel evidence that APAP-induced glutathionylation results in an extensive inhibition of the major energy metabolism pathways. We report for the first time that CPT1, the rate-limiting and key regulator of mitochondrial fatty acid β-oxidation is inhibited by glutathionylation. Besides CPT1, CAC which transports acylcarnitines into the mitochondria is also inhibited by glutathionylation (18). The combined inhibition of fatty acid activation and uptake suggests a significant impairment of fatty acid β-oxidation, which corroborates reports of elevated fatty acids and acylcarnitines in APAP toxicity. Separately, pretreatment of mice with the PPARα inducer clofibrate, whether acute (24 h) or chronic (10 days) prior to APAP exposure conferred protection against APAP hepatotoxicity, which was unrelated to the degree of covalent binding or GSH depletion (19, 30–32). Interestingly, genes involved in fatty acid β-oxidation including CPT1, fatty acid ligases and acyl-CoA dehydrogenases are induced by PPARα, and the PPARα agonist Wy-14,643 resulted in protection from APAP toxicity, accompanied by a parallel attenuation of the increase in palmitoylcarnitines (33). Using CPT1 activity as a marker of fatty acid β-oxidation fitness, our findings demonstrate that the inhibitory effect of glutathionylation is attenuated by the up-regulation of proteins in the affected pathway.
Our inference of a similar inhibitory effect on the Krebs cycle agrees with literature reports that glutathionylation of aconitate hydratase (34), 2-oxoglutarate dehydrogenase E2 subunit (35), isocitrate dehydrogenase (36) and succinate dehydrogenase (37) resulted in enzymatic inhibition. APAP is known to deplete Krebs cycle substrates, as supplementation with GSH and N-acetylcysteine post-APAP treatment is thought to provide energy substrates in the Krebs cycle (38). Importantly, succinate dehydrogenase is highly sensitive to inhibition by NAPQI (29), which we demonstrate is mediated by glutathionylation.
Last, mitochondrial complexes I, II, and III were found to be inhibited by NAPQI (29, 39–41), whereas covalent binding of APAP to ATP synthase F1 α subunit has been reported (42) which was accompanied by its inhibition (43). In parallel with these observations, complexes I-III and the ATP synthase F0 complex subunit B1 was found to be glutathionylated by 30 mm APAP in our study, and complexes I, II and ATP synthase F1 subunit α are known to be inhibited by glutathionylation (37, 44, 45). Collectively, glutathionylation of the electron transport chain and ATP synthase accounts for the profound decrease in ATP levels observed with APAP. Importantly, after APAP treatment in vivo (7, 46), hepatic ATP levels were reported to decline rapidly (maximally within 1–1.5 h) followed by a partial recovery, which is consistent with our findings where glutathionylation of energy metabolism proteins is greatest at 3 h and declines thereafter (Fig. 9A), possibly indicating restoration of ATP synthesis.
In the case of glycolysis, the numbers of mapped metabolites alone are insufficient to make a judgement with regards to the functional consequence of glutathionylation of this pathway; however the glycolytic enzymes identified as glutathionylated in our study were all reported to be inhibited by glutathionylation (47, 48).
Elevated Oxidative Stress
Glutathionylation of complex I of the electron transport chain and 2-oxoglutarate dehydrogenase E1 subunit is known to aberrantly amplify mitochondrial superoxide formation (49, 50), which in turn generates peroxynitrite. Separately, APAP-induced oxidants (including H2O2, superoxide and peroxynitrite) are believed to trigger c-jun-N-terminal kinase activation (28), which translocates to the mitochondria and further amplifies oxidative stress, triggering the opening of the MPTP, causing the collapse of mitochondrial membrane potential, loss of ATP synthesis capacity, rupture of the outer mitochondrial membrane, release of endonucleases and DNA fragmentation (28). A recent review by Jaeschke and colleagues (51) attributed the origin of APAP-induced oxidative stress to oxidant leakage from complex I of the electron transport chain, but the authors acknowledged that the molecular events triggering mitochondrial oxidative stress remain unclear. Here, we advance the explanation that aberrant glutathionylation of complex I and 2-oxoglutarate dehydrogenase is the source of increased superoxide formation. Importantly, the timeline of APAP glutathionylation of these proteins, which occurs within 3 h, agrees with reports of an initial increase in mitochondrial oxidative stress in the first 2 h of exposure to APAP (6). Finally, the protein deglycase DJ-1 is known to play a critical role in maintaining mitochondrial homeostasis and quenching oxidative stress, thereby exerting a cytoprotective activity toward oxidative insults (52). In Parkinson's disease models, loss of DJ-1 results in reduced mitochondrial membrane potential, increased mitochondrial fragmentation, and reduced mitochondrial connectivity (53). Glutathionylation of DJ-1 is known to increase its degradation (54), and here we propose that besides an increase in mitochondrial oxidative stress, glutathionylation also diminishes the ability of the mitochondria to resist the oxidative insult, ultimately culminating in mitochondrial injury and dysfunction.
Sustained Increase in Cytosolic Calcium
Numerous studies have demonstrated APAP-induced calcium dysregulation, characterized by a 10-fold increase in cytosolic and mitochondrial calcium to micromolar levels (55–57), which occurred as early as 45 min after APAP exposure (56). This increase strongly paralleled the time-course of covalent binding (58), preceded cytotoxicity (57) and was attributed to inhibition of PMCA via thiol oxidation, resulting in diminished extrusion of intracellular calcium (56, 58, 59). Addition of the reducing agent dithiothreitol was shown to reverse PMCA inhibition, restore cytosolic calcium to baseline levels and prevent APAP-induced cytotoxicity (60). Here we demonstrate that PMCA is glutathionylated by APAP, which functionally results in its inhibition (61). Separately, Kheradpezhouh et al. reported that the transient receptor potential melastatin 2 (TRPM2) mediated the hepatocellular uptake of calcium after in vitro exposure to APAP or H2O2 for 1 h, whereas calcium influx was almost completely blocked by TRPM2 inhibitors (62). NAPQI is known to trigger NAD+ hydrolysis to ADP-ribose (63), which is the ligand that activates TRPM2. We propose that it is the combination of both increased calcium influx via TRPM2 activation, as well as concurrent inhibition of calcium efflux via PMCA glutathionylation that explains the increase in cytosolic calcium observed in APAP toxicity.
MPTP Opening and DNA Fragmentation
MPTP opening is a key feature in APAP toxicity and represents the “point of no return” in the toxic sequelae as it results in the collapse of mitochondrial membrane potential and translocation of endonucleases to the nucleus which results in DNA fragmentation. Opening of the pore is known to be triggered by several events, including high mitochondrial oxidative stress and elevated mitochondrial calcium (secondary to increased cytosolic calcium). Although we observed glutathionylation of VDAC1–3 and adenine nucleotide translocase (ANT) 1/2 (thought to either be the main pore-forming components or to regulate pore opening), their precise contribution to MPTP opening remains an open question. Our molecular dynamics simulations reveal for the first time that glutathionylation of Cys232 of VDAC1 favors an open conformation, which is suggestive of pore-forming activity. Additionally, we found that ANT was glutathionylated at 3–6 h, whereas VDAC was generally glutathionylated at 12–24 h. McGill et al. reported that MPTP opening occurred at 12–24 h after exposure to APAP in HepaRG cells (26). The simulations and time-course data suggest that VDAC glutathionylation could be implicated in MPTP opening. Interestingly VDAC2 was found to be adducted by NAPQI (22), which agrees with the proposed relationship between protein adduction and glutathionylation. Overall, these evidences collectively indicate that protein glutathionylation triggers a series of direct and indirect events that culminate in MPTP opening and mitochondrial dysfunction.
Although not directly related to MPTP opening or APAP toxicity, the modification of the electrostatic potential of VDAC to favor binding and translocation of cationic molecules is an interesting observation. VDAC is known to regulate calcium permeability through the outer mitochondrial membrane and possesses two calcium-binding sites (Glu206 and Glu76) (64). The proximity of the negative glutathionyl group to Glu206 is suggestive of a regulatory role in mitochondrial calcium flux, particularly because VDAC1 (but not VDAC2 or 3) (65) provides the route for calcium entry into mitochondria upon apoptotic stimuli. Such findings create new research opportunities to further investigate the relationship between the closely related fields of oxidative stress, mitochondrial dysfunction, and apoptosis.
In contrast to energy metabolism, there are limited reports regarding the effect of glutathionylation on protein turnover and defense against cellular stress. APAP can inhibit protein synthesis and degradation (66), whereas glutathionylation of both the E2 ubiquitin-conjugating enzyme and the 26S proteasome results in inhibition of ubiquitination and proteasomal degradation (67, 68). Separately, glutathionylation inhibited cytosolic superoxide dismutase 1 (69), whereas that of heat shock protein 70 (70) resulted in a gain in activity. Glutathionylation of proteins in these pathways may contribute to APAP toxicity and requires further investigation.
The ability of APAP to directly induce protein glutathionylation via covalent binding, as well as indirectly via oxidative stress is a unique feature among known inducers of glutathionylation. Here we provide evidence which indicates that glutathionylation that is relevant to APAP toxicity is driven by the former rather than the latter. First, there is a 35% overlap between proteins that are known to be covalently bound to APAP and proteins that were glutathionylated in our study (supplemental Table S7). Additionally, Yang et al. provided evidence that regions of covalent binding overlapped with regions of protein glutathionylation in mice treated with APAP (9), whereas Nelson and colleagues mechanistically elucidated the formation of the ipso adduct that provokes glutathionylation (8). Second, the time-course of glutathionylation more closely matches that of covalent binding than oxidative stress. APAP-induced protein glutathionylation measured in our study peaked at 3 h (Fig. 9A), whereas covalent binding as measured by APAP-cysteine conjugates peaked at 6 h (26) and oxidative stress was maximal at 12 h (supplemental Fig. S1). Furthermore, Yang et al. reported that glutathionylation was highest in mice between 3–6 h, followed by a decline (9). Finally, glutathionylation exhibited dose- and bioactivation-dependence. Our results suggest that glutathionylation of proteins relevant to APAP toxicity was decreased as a result of exposure to a sub-toxic dose of 0.5 mm APAP. Although there was only a small decrease in total number of glutathionylated peptides with 0.5 mm APAP, there was a far larger decrease for the subset of toxicologically important peptides. When treated with CYP2E1 inhibitor (DEDC), which was reported to ameliorate APAP toxicity by suppressing NAPQI generation, the extent of protein glutathionylation further decreased, although it was not completely abolished for several reasons: (1) the use of DEDC itself may introduce another source of oxidative stress (71, 72), although we attempted to limit this by using a low DEDC concentration, and (2) NAPQI formation can also be mediated by other CYP450 enzymes, including CYP3A4, although the contributions from these enzymes are reported to be low (73). Taken together, our work provides further evidence that the reactive metabolite NAPQI can directly induce protein glutathionylation, a previously unrecognized mechanism of this modification (Fig. 1B).
Although McGarry and colleagues suggested that protein glutathionylation confers a protective effect to Gstp1/2−/− mice against APAP toxicity (74), it is difficult to compare with our findings for the following reasons. Our findings are drawn from a straightforward comparison between APAP-exposed and control HepaRG cells (rather than against a more complicated background of GSTP knockout). Although the authors chose the Gstp1/2−/− mice model to investigate if glutathionylation might account for the known resistance of Gstp1/2−/− mice to APAP toxicity, the ability of Gstp to modulate glutathionylation is itself a potential confounding factor in interpreting the results. Importantly, their study design is based on a comparison between wildtype and Gstp knockout mice, thus their findings need to be interpreted in the context of Gstp influence on glutathionylation patterns, rather than the role of glutathionylation in APAP toxicity which is the research question we have investigated. Finally, we cannot discount the possibility of species differences that led to their inference of the protective effect of glutathionylation under Gstp-null conditions.
Although the evidences we provide here collectively point toward a potentially causative role for glutathionylation in APAP toxicity, we recognize that more work is needed to definitively establish the precise role of glutathionylation in the etiology of APAP toxicity. For example, the relative contributions of direct covalent binding, protein glutathionylation and oxidative stress (which we suggest are interlinked) to the molecular perturbations and cellular dysregulations induced by APAP are still unknown but need to be worked out in future investigations. Additionally, although we have worked with an established in vitro cell model which is easier to handle and manipulate, our findings need to be further investigated in an appropriate animal model (e.g. mouse) which can respond dynamically with corresponding compensatory mechanisms to the toxicological insult. Finally, due to limited knowledge of the functional consequences of glutathionylation, we have not thoroughly explored the rest of the glutathionylated proteome found in our study. These are exciting avenues for future research, and we recognize that the functional “map” we propose here can be further expanded to provide a more holistic picture of the consequences of APAP-induced glutathionylation. For example, an important line of investigation would be to examine whether the deglutathionylation enzymes (e.g. protein disulfide isomerase) themselves are affected by glutathionylation and to assess their contribution to resisting APAP toxicity.
In conclusion, we provide evidence that NAPQI, the reactive metabolite of APAP directly induces widespread glutathionylation through covalent binding, independent of a thiol reaction with oxidants. Notably, our study provides the first example that this post-translational modification could be functionally implicated as an unrecognized mechanism of drug-induced hepatotoxicity. This raises further questions whether this feature is unique to NAPQI, or generalizable to the quinone imine electrophile. Additional work using other quinone imines is needed to evaluate this relationship, and potentially establish aberrant protein glutathionylation as a predictive marker of drug-induced hepatotoxicity.
DATA AVAILABILITY
Proteomics data have been deposited at jPOST (https://repository.jpostdb.org project ID: JPST000435).
Supplementary Material
Acknowledgments
We thank the late Professor Sidney Nelson for discussions that sparked the idea of profiling protein glutathionylation related to acetaminophen toxicity. E.C.Y.C. acknowledges the Singapore Ministry of Health, National Medical Research Training Fellowship that sponsored his sabbatical training at the School of Pharmacy, University of Washington in 2012 where this project was initiated. We also express our appreciation to Mr. Deng Lu for his kind assistance in preparing the heat map figures.
Footnotes
* This work was supported by the Singapore Ministry of Education Tier 1 Grant to E.C.Y.C [grant number R-148-000-204-112], the Singapore National Medical Research Council (NMRC) Centre Grants CG 2013 and CG 2017 to the Singapore Eye Research Institute, and the SingHealth Foundation to the proteomics core facility at the Singapore Eye Research Institute. J.C.Y.C was supported by the National University of Singapore President's Graduate Fellowship.
 This article contains supplemental Figures and Tables. The authors declare that there is no conflict of interest.
This article contains supplemental Figures and Tables. The authors declare that there is no conflict of interest.
1 The abbreviations used are:
- Grx1
- glutaredoxin 1
- ANT
- adenine nucleotide translocase
- APAP
- acetaminophen
- ATP
- adenosine triphosphate
- CAC
- carnitine/acylcarnitine carrier
- CoA
- coenzyme A
- CPT1
- carnitine O-palmitoyltransferase 1
- CYP450
- cytochrome P450
- DEDC
- diethyldithiocarbamate
- DNA
- deoxyribonucleic acid
- GPx
- glutathione peroxidase
- GSH
- glutathione
- MPTP
- mitochondrial permeability transition pore
- NAPQI
- N-acetyl-p-benzoquinone imine
- PMCA
- plasma membrane calcium ATPase
- PPARα
- peroxisome proliferator-activated receptor α
- RMSD
- root mean square deviation
- TRPM2
- transient receptor potential melastatin 2
- VDAC1
- voltage-dependent anion-selective channel protein 1.
REFERENCES
- 1. Mieyal J. J., Gallogly M. M., Qanungo S., Sabens E. A., and Shelton M. D. (2008) Molecular mechanisms and clinical implications of reversible protein S-glutathionylation. Antioxidants Redox Signaling 10, 1941–1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Larson A. M., Polson J., Fontana R. J., Davern T. J., Lalani E., Hynan L. S., Reisch J. S., Schiodt F. V., Ostapowicz G., Shakil A. O., Lee W. M., and Acute Liver Failure Study, G. (2005) Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 42, 1364–1372 [DOI] [PubMed] [Google Scholar]
- 3. Jollow D. J., Mitchell J. R., Potter W. Z., Davis D. C., Gillette J. R., and Brodie B. B. (1973) Acetaminophen-induced hepatic necrosis. II. Role of covalent binding in vivo. The Journal Of Pharmacology And Experimental Therapeutics 187, 195–202 [PubMed] [Google Scholar]
- 4. Hinson J. A., Roberts D. W., and James L. P. (2010) Mechanisms of acetaminophen-induced liver necrosis. Handbook Exp. Pharmacol. 196, 369–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Knight T. R., Ho Y. S., Farhood A., and Jaeschke H. (2002) Peroxynitrite is a critical mediator of acetaminophen hepatotoxicity in murine livers: protection by glutathione. J. Pharmacol. Exp. Therapeutics 303, 468–475 [DOI] [PubMed] [Google Scholar]
- 6. Nelson S. D., and Bruschi S. A. (2002) Mechanisms of Acetaminophen-induced Liver Disease. In Drug-induced Liver Disease (Kaplowitz N., ed), Chapter 18, pages 287–325, Taylor & Francis Group, Abingdon, UK. [Google Scholar]
- 7. Tirmenstein M. A., and Nelson S. D. (1990) Acetaminophen-induced oxidation of protein thiols. Contribution of impaired thiol-metabolizing enzymes and the breakdown of adenine nucleotides. J. Biol. Chem. 265, 3059–3065 [PubMed] [Google Scholar]
- 8. Chen W., Shockcor J. P., Tonge R., Hunter A., Gartner C., and Nelson S. D. (1999) Protein and nonprotein cysteinyl thiol modification by N-acetyl-p-benzoquinone imine via a novel ipso adduct. Biochemistry 38, 8159–8166 [DOI] [PubMed] [Google Scholar]
- 9. Yang X., Greenhaw J., Ali A., Shi Q., Roberts D. W., Hinson J. A., Muskhelishvili L., Beger R., Pence L. M., Ando Y., Sun J., Davis K., and Salminen W. F. (2012) Changes in mouse liver protein glutathionylation after acetaminophen exposure. J. Pharmacol. Exp. Therapeutics 340, 360–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Robinson K. M., Janes M. S., Pehar M., Monette J. S., Ross M. F., Hagen T. M., Murphy M. P., and Beckman J. S. (2006) Selective fluorescent imaging of superoxide in vivo using ethidium-based probes. Proc. Natl. Acad. Sci. U.S.A. 103, 15038–15043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hornak V., Abel R., Okur A., Strockbine B., Roitberg A., and Simmerling C. (2006) Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 65, 712–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dickson C. J., Madej B. D., Skjevik A. A., Betz R. M., Teigen K., Gould I. R., and Walker R. C. (2014) Lipid14: the amber lipid force field. J. Chem. Theory Comput. 10, 865–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Essmann U., Perera L., Berkowitz M. L., Darden T., Lee H., and Pedersen L. G. (1995) A smooth particle mesh Ewald method. J. Chem. Physics 103, 8577–8593 [Google Scholar]
- 14. Van Der Spoel D., Lindahl E., Hess B., Groenhof G., Mark A. E., and Berendsen H. J. (2005) GROMACS: fast, flexible, and free. J. Comput. Chem. 26, 1701–1718 [DOI] [PubMed] [Google Scholar]
- 15. Leichert L. I., Gehrke F., Gudiseva H. V., Blackwell T., Ilbert M., Walker A. K., Strahler J. R., Andrews P. C., and Jakob U. (2008) Quantifying changes in the thiol redox proteome upon oxidative stress in vivo. Proc. Natl. Acad. Sci. U.S.A. 105, 8197–8202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Su D., Gaffrey M. J., Guo J., Hatchell K. E., Chu R. K., Clauss T. R., Aldrich J. T., Wu S., Purvine S., Camp D. G., Smith R. D., Thrall B. D., and Qian W. J. (2014) Proteomic identification and quantification of S-glutathionylation in mouse macrophages using resin-assisted enrichment and isobaric labeling. Free Radical Biol. Med. 67, 460–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Go Y. M., Roede J. R., Orr M., Liang Y., and Jones D. P. (2014) Integrated redox proteomics and metabolomics of mitochondria to identify mechanisms of cd toxicity. Toxicol. Sci. 139, 59–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Giangregorio N., Palmieri F., and Indiveri C. (2013) Glutathione controls the redox state of the mitochondrial carnitine/acylcarnitine carrier Cys residues by glutathionylation. Biochim. Biophys. Acta 1830, 5299–5304 [DOI] [PubMed] [Google Scholar]
- 19. Chen C., Hennig G. E., Whiteley H. E., Corton J. C., and Manautou J. E. (2000) Peroxisome proliferator-activated receptor alpha-null mice lack resistance to acetaminophen hepatotoxicity following clofibrate exposure. Toxicol. Sci. 57, 338–344 [DOI] [PubMed] [Google Scholar]
- 20. Messina A., Reina S., Guarino F., and De Pinto V. (2012) VDAC isoforms in mammals. Biochim. Biophys. Acta 1818, 1466–1476 [DOI] [PubMed] [Google Scholar]
- 21. Eakins R., Walsh J., Randle L., Jenkins R. E., Schuppe-Koistinen I., Rowe C., Starkey Lewis P., Vasieva O., Prats N., Brillant N., Auli M., Bayliss M., Webb S., Rees J. A., Kitteringham N. R., Goldring C. E., and Park B. K. (2015) Adaptation to acetaminophen exposure elicits major changes in expression and distribution of the hepatic proteome. Sci. Rep. 5, 16423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bruderer R., Bernhardt O. M., Gandhi T., Miladinovic S. M., Cheng L. Y., Messner S., Ehrenberger T., Zanotelli V., Butscheid Y., Escher C., Vitek O., Rinner O., and Reiter L. (2015) Extending the limits of quantitative proteome profiling with data-independent acquisition and application to acetaminophen-treated three-dimensional liver microtissues. Mol. Cell. Proteomics 14, 1400–1410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ruepp S. U., Tonge R. P., Shaw J., Wallis N., and Pognan F. (2002) Genomics and proteomics analysis of acetaminophen toxicity in mouse liver. Toxicol. Sci. 65, 135–150 [DOI] [PubMed] [Google Scholar]
- 24. Stamper B. D., Mohar I., Kavanagh T. J., and Nelson S. D. (2011) Proteomic analysis of acetaminophen-induced changes in mitochondrial protein expression using spectral counting. Chem. Res. Toxicol. 24, 549–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fountoulakis M., Berndt P., Boelsterli U. A., Crameri F., Winter M., Albertini S., and Suter L. (2000) Two-dimensional database of mouse liver proteins: changes in hepatic protein levels following treatment with acetaminophen or its nontoxic regioisomer 3-acetamidophenol. Electrophoresis 21, 2148–2161 [DOI] [PubMed] [Google Scholar]
- 26. McGill M. R., Yan H. M., Ramachandran A., Murray G. J., Rollins D. E., and Jaeschke H. (2011) HepaRG cells: a human model to study mechanisms of acetaminophen hepatotoxicity. Hepatology 53, 974–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kampf C., Mardinoglu A., Fagerberg L., Hallstrom B. M., Edlund K., Lundberg E., Ponten F., Nielsen J., and Uhlen M. (2014) The human liver-specific proteome defined by transcriptomics and antibody-based profiling. FASEB J. 28, 2901–2914 [DOI] [PubMed] [Google Scholar]
- 28. Jaeschke H., McGill M. R., and Ramachandran A. (2012) Oxidant stress, mitochondria, and cell death mechanisms in drug-induced liver injury: lessons learned from acetaminophen hepatotoxicity. Drug Metabolism Rev. 44, 88–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Burcham P. C., and Harman A. W. (1991) Acetaminophen toxicity results in site-specific mitochondrial damage in isolated mouse hepatocytes. J. Biol. Chem. 266, 5049–5054 [PubMed] [Google Scholar]
- 30. Manautou J. E., Hoivik D. J., Tveit A., Hart S. G., Khairallah E. A., and Cohen S. D. (1994) Clofibrate pretreatment diminishes acetaminophen's selective covalent binding and hepatotoxicity. Toxicol. Appl. Pharmacol. 129, 252–263 [DOI] [PubMed] [Google Scholar]
- 31. Manautou J. E., Emeigh Hart S. G., Khairallah E. A., and Cohen S. D. (1996) Protection against acetaminophen hepatotoxicity by a single dose of clofibrate: effects on selective protein arylation and glutathione depletion. Fundamental Appl. Toxicol. 29, 229–237 [DOI] [PubMed] [Google Scholar]
- 32. Nicholls-Grzemski F. A., Calder I. C., Priestly B. G., and Burcham P. C. (2000) Clofibrate-induced in vitro hepatoprotection against acetaminophen is not due to altered glutathione homeostasis. Toxicol. Sci. 56, 220–228 [DOI] [PubMed] [Google Scholar]
- 33. Patterson A. D., Shah Y. M., Matsubara T., Krausz K. W., and Gonzalez F. J. (2012) Peroxisome proliferator-activated receptor alpha induction of uncoupling protein 2 protects against acetaminophen-induced liver toxicity. Hepatology 56, 281–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Han D., Canali R., Garcia J., Aguilera R., Gallaher T. K., and Cadenas E. (2005) Sites and mechanisms of aconitase inactivation by peroxynitrite: modulation by citrate and glutathione. Biochemistry 44, 11986–11996 [DOI] [PubMed] [Google Scholar]
- 35. McLain A. L., Cormier P. J., Kinter M., and Szweda L. I. (2013) Glutathionylation of alpha-ketoglutarate dehydrogenase: the chemical nature and relative susceptibility of the cofactor lipoic acid to modification. Free Radical Biol. Med. 61, 161–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kil I. S., and Park J. W. (2005) Regulation of mitochondrial NADP+-dependent isocitrate dehydrogenase activity by glutathionylation. J. Biol. Chem. 280, 10846–10854 [DOI] [PubMed] [Google Scholar]
- 37. Chen Y. R., Chen C. L., Pfeiffer D. R., and Zweier J. L. (2007) Mitochondrial complex II in the post-ischemic heart: oxidative injury and the role of protein S-glutathionylation. J. Biol. Chem. 282, 32640–32654 [DOI] [PubMed] [Google Scholar]
- 38. Saito C., Zwingmann C., and Jaeschke H. (2010) Novel mechanisms of protection against acetaminophen hepatotoxicity in mice by glutathione and N-acetylcysteine. Hepatology 51, 246–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Meyers L. L., Beierschmitt W. P., Khairallah E. A., and Cohen S. D. (1988) Acetaminophen-induced inhibition of hepatic mitochondrial respiration in mice. Toxicol. Appl. Pharmacol. 93, 378–387 [DOI] [PubMed] [Google Scholar]
- 40. Donnelly P. J., Walker R. M., and Racz W. J. (1994) Inhibition of mitochondrial respiration in vivo is an early event in acetaminophen-induced hepatotoxicity. Arch. Toxicol. 68, 110–118 [DOI] [PubMed] [Google Scholar]
- 41. Ramsay R. R., Rashed M. S., and Nelson S. D. (1989) In vitro effects of acetaminophen metabolites and analogs on the respiration of mouse liver mitochondria. Arch. Biochem. Biophys. 273, 449–457 [DOI] [PubMed] [Google Scholar]
- 42. Qiu Y., Benet L. Z., and Burlingame A. L. (1998) Identification of the hepatic protein targets of reactive metabolites of acetaminophen in vivo in mice using two-dimensional gel electrophoresis and mass spectrometry. J. Biol. Chem. 273, 17940–17953 [DOI] [PubMed] [Google Scholar]
- 43. Parmar D. V., Ahmed G., Khandkar M. A., and Katyare S. S. (1995) Mitochondrial ATPase: a target for paracetamol-induced hepatotoxicity. Eur. J. Pharmacol. 293, 225–229 [DOI] [PubMed] [Google Scholar]
- 44. Mailloux R. J., Xuan J. Y., McBride S., Maharsy W., Thorn S., Holterman C. E., Kennedy C. R., Rippstein P., deKemp R., da Silva J., Nemer M., Lou M., and Harper M. E. (2014) Glutaredoxin-2 is required to control oxidative phosphorylation in cardiac muscle by mediating deglutathionylation reactions. J. Biol. Chem. 289, 14812–14828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang S. B., Foster D. B., Rucker J., O'Rourke B., Kass D. A., and Van Eyk J. E. (2011) Redox regulation of mitochondrial ATP synthase: implications for cardiac resynchronization therapy. Circulation Res. 109, 750–757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jaeschke H. (1990) Glutathione disulfide formation and oxidant stress during acetaminophen-induced hepatotoxicity in mice in vivo: the protective effect of allopurinol. J. Pharmacol. Exp. Therapeutics 255, 935–941 [PubMed] [Google Scholar]
- 47. Tew K. D., Manevich Y., Grek C., Xiong Y., Uys J., and Townsend D. M. (2011) The role of glutathione S-transferase P in signaling pathways and S-glutathionylation in cancer. Free Radical Biol. Med. 51, 299–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kehr S., Jortzik E., Delahunty C., Yates J. R. 3rd, Rahlfs S., and Becker K. (2011) Protein S-glutathionylation in malaria parasites. Antioxidants Redox Signaling 15, 2855–2865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Taylor E. R., Hurrell F., Shannon R. J., Lin T. K., Hirst J., and Murphy M. P. (2003) Reversible glutathionylation of complex I increases mitochondrial superoxide formation. J. Biol. Chem. 278, 19603–19610 [DOI] [PubMed] [Google Scholar]
- 50. Mailloux R. J., Craig Ayre D., and Christian S. L. (2016) Induction of mitochondrial reactive oxygen species production by GSH mediated S-glutathionylation of 2-oxoglutarate dehydrogenase. Redox Biol. 8, 285–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Du K., Ramachandran A., and Jaeschke H. (2016) Oxidative stress during acetaminophen hepatotoxicity: Sources, pathophysiological role and therapeutic potential. Redox Biol. 10, 148–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Junn E., Jang W. H., Zhao X., Jeong B. S., and Mouradian M. M. (2009) Mitochondrial localization of DJ-1 leads to enhanced neuroprotection. J. Neurosci. Res. 87, 123–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Krebiehl G., Ruckerbauer S., Burbulla L. F., Kieper N., Maurer B., Waak J., Wolburg H., Gizatullina Z., Gellerich F. N., Woitalla D., Riess O., Kahle P. J., Proikas-Cezanne T., and Kruger R. (2010) Reduced basal autophagy and impaired mitochondrial dynamics due to loss of Parkinson's disease-associated protein DJ-1. PloS ONE 5, e9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Johnson W. M., Golczak M., Choe K., Curran P. L., Miller O. G., Yao C., Wang W., Lin J., Milkovic N. M., Ray A., Ravindranath V., Zhu X., Wilson M. A., Wilson-Delfosse A. L., Chen S. G., and Mieyal J. J. (2016) Regulation of DJ-1 by glutaredoxin 1 in vivo: implications for Parkinson's disease. Biochemistry 55, 4519–4532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lauterburg B. H. (1987) Early disturbance of calcium translocation across the plasma membrane in toxic liver injury. Hepatology 7, 1179–1183 [DOI] [PubMed] [Google Scholar]
- 56. Tsokos-Kuhn J. O., Todd E. L., McMillin-Wood J. B., and Mitchell J. R. (1985) ATP-dependent calcium uptake by rat liver plasma membrane vesicles. Effect of alkylating hepatotoxins in vivo. Mol. Pharmacol. 28, 56–61 [PubMed] [Google Scholar]
- 57. Bruschi S. A., and Priestly B. G. (1990) Implication of alterations in intracellular calcium ion homoeostasis in the advent of paracetamol-induced cytotoxicity in primary mouse hepatocyte monolayer cultures. Toxicol. In Vitro 4, 743–749 [DOI] [PubMed] [Google Scholar]
- 58. Tirmenstein M. A., and Nelson S. D. (1989) Subcellular binding and effects on calcium homeostasis produced by acetaminophen and a nonhepatotoxic regioisomer, 3′-hydroxyacetanilide, in mouse liver. J. Biol. Chem. 264, 9814–9819 [PubMed] [Google Scholar]
- 59. Moore M., Thor H., Moore G., Nelson S., Moldeus P., and Orrenius S. (1985) The toxicity of acetaminophen and N-acetyl-p-benzoquinone imine in isolated hepatocytes is associated with thiol depletion and increased cytosolic Ca2+. J. Biol. Chem. 260, 13035–13040 [PubMed] [Google Scholar]
- 60. Nicotera P., Rundgren M., Porubek D. J., Cotgreave I., Moldeus P., Orrenius S., and Nelson S. D. (1989) On the role of Ca2+ in the toxicity of alkylating and oxidizing quinone imines in isolated hepatocytes. Chem. Res. Toxicol. 2, 46–50 [DOI] [PubMed] [Google Scholar]
- 61. Lock J. T., Sinkins W. G., and Schilling W. P. (2011) Effect of protein S-glutathionylation on Ca2+ homeostasis in cultured aortic endothelial cells. Am. J. Physiol. Heart Circulatory Physiol. 300, H493–H506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kheradpezhouh E., Ma L., Morphett A., Barritt G. J., and Rychkov G. Y. (2014) TRPM2 channels mediate acetaminophen-induced liver damage. Proc. Natl. Acad. Sci. U.S.A. 111, 3176–3181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Weis M., Kass G. E., Orrenius S., and Moldeus P. (1992) N-acetyl-p-benzoquinone imine induces Ca2+ release from mitochondria by stimulating pyridine nucleotide hydrolysis. J. Biol. Chem. 267, 804–809 [PubMed] [Google Scholar]
- 64. Israelson A., Abu-Hamad S., Zaid H., Nahon E., and Shoshan-Barmatz V. (2007) Localization of the voltage-dependent anion channel-1 Ca2+-binding sites. Cell Calcium 41, 235–244 [DOI] [PubMed] [Google Scholar]
- 65. De Stefani D., Bononi A., Romagnoli A., Messina A., De Pinto V., Pinton P., and Rizzuto R. (2012) VDAC1 selectively transfers apoptotic Ca2+ signals to mitochondria. Cell Death Differentiation 19, 267–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gwynn J., Fry J. R., and Bridges J. W. (1979) The effects of paracetamol and other foreign compounds on protein synthesis in isolated adult rat hepatocytes Proc. Biochem. Soc. Trans. 7, 117–119 [DOI] [PubMed] [Google Scholar]
- 67. Xiong Y., Uys J. D., Tew K. D., and Townsend D. M. (2011) S-glutathionylation: from molecular mechanisms to health outcomes. Antioxidants Redox Signaling 15, 233–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hohn T. J., and Grune T. (2014) The proteasome and the degradation of oxidized proteins: part III-Redox regulation of the proteasomal system. Redox Biol. 2, 388–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wilcox K. C., Zhou L., Jordon J. K., Huang Y., Yu Y., Redler R. L., Chen X., Caplow M., and Dokholyan N. V. (2009) Modifications of superoxide dismutase (SOD1) in human erythrocytes: a possible role in amyotrophic lateral sclerosis. J. Biol. Chem. 284, 13940–13947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Fedoroff N. (2006) Redox regulatory mechanisms in cellular stress responses. Ann. Botany 98, 289–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ishiyama H., Ogino K., Shimomura Y., Kanbe T., and Hobara T. (1990) Hepatotoxicity of diethyldithiocarbamate in rats. Pharmacol. Toxicol. 67, 426–430 [DOI] [PubMed] [Google Scholar]
- 72. Ishiyama H., Ogino K., and Hobara T. (1995) Role of Kupffer cells in rat liver injury induced by diethyldithiocarbamate. Eur. J. Pharmacol. 292, 135–141 [DOI] [PubMed] [Google Scholar]
- 73. Patten C. J., Thomas P. E., Guy R. L., Lee M., Gonzalez F. J., Guengerich F. P., and Yang C. S. (1993) Cytochrome P450 enzymes involved in acetaminophen activation by rat and human liver microsomes and their kinetics. Chem. Res. Toxicol. 6, 511–518 [DOI] [PubMed] [Google Scholar]
- 74. McGarry D. J., Chakravarty P., Wolf C. R., and Henderson C. J. (2015) Altered protein S-glutathionylation identifies a potential mechanism of resistance to acetaminophen-induced hepatotoxicity. J. Pharmacol. Exp. Therapeutics 355, 137–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Proteomics data have been deposited at jPOST (https://repository.jpostdb.org project ID: JPST000435).