

Abstract
Antibody-mediated blockade of cluster of differentiation 47 (CD47)–thrombospondin-1 (TSP-1) interactions blocks osteoclast formation in vitro and attenuates parathyroid hormone (PTH)-induced hypercalcemia in vivo in mice. Hypercalcemia in this model reflects increased bone resorption. TSP-1 has two cell-associated binding partners, CD47 and CD36. The roles of these two molecules in mediating the effects of TSP1 in osteoclasts are unclear. Osteoclast formation was attenuated but not absent when preosteoclasts isolated from CD47−/− mice were cocultured with WT osteoblasts. Suppressing CD36 in osteoclast progenitors also attenuated osteoclast formation. The hypercalcemic response to a PTH infusion was blunted in CD47−/−/CD36−/− (double knockout (DKO)) female mice but not CD47−/− mice or CD36−/− animals, supporting a role for both CD47 and CD36 in mediating this effect. Consistent with this, DKO osteoclasts had impaired resorptive activity when analyzed in vitro. Inhibition of nitric oxide (NO) signaling is known to promote osteoclastogenesis, and TSP-1 suppresses NO production and signaling. An anti-TSP-1 antibody increased NO production in osteoclasts, and the inhibitory effect of anti-TSP-1 on osteoclastogenesis was completely rescued by l-nitroarginine methyl ester (l-NAME), a competitive NO synthase inhibitor. Supportive of an important role for CD36 in mediating the pro-osteoclastogenic effects of TSP-1, engaging CD36 with a synthetic agonist, p907, suppressed NO production in anti-TSP-1–treated cultures, allowing osteoclast maturation to occur. These results establish that CD36 and CD47 both participate in mediating the actions of TSP-1 in osteoclasts and establish a physiologically relevant cross-talk in bone tissue between these two molecules.
Keywords: osteoblast, osteoclast, osteocyte, osteoporosis, thrombospondin, bone, parathyroid hormone, bone resorption, CD36, CD47, TSP-1
Introduction
Osteoclastogenesis, the formation of specialized multinucleated giant cells that resorb the inorganic and organic components of bone, is a complex multistep process. Under physiologic conditions, osteoclasts originate from cells in the monocyte/macrophage lineage. Committed osteoclast precursors initially express c-Fms, the receptor for CSF-1, followed by the expression of TNFRSF11A, the receptor for receptor activator of NF-κB ligand (RANKL)3 (1). RANKL then drives further differentiation of preosteoclasts. An important, but as yet incompletely understood, step in the formation of multinucleated osteoclasts is the fusion of osteoclast precursors to form the final differentiated cell. Under physiologic conditions, cells in the osteoblast-osteocyte lineage are thought to be the principal source for RANKL in bone, although it can be made by a variety of cells, including B and T cells (2, 3).
Although both RANKL and CSF-1 are required for normal osteoclastogenesis, other cytokines and growth factors contribute to this process (4). None of these other factors have been shown to be capable of inducing osteoclastogenesis in the genetic absence of RANKL or CSF-1, but they appear to have important ancillary roles. TNFα and IL-1 are both capable of stimulating osteoclastogenesis in vitro and in vivo and are thought to be important mediators of bone loss associated with inflammatory disorders (5). Immunoreceptor tyrosine-based activation motif (ITAM) costimulatory molecules Fc receptor γ and DAP12 associate with several cell-surface receptors expressed on osteoclasts and are important for normal osteoclastogenesis to occur (6). Dendritic cell-specific transmembrane protein (DC-STAMP) and osteoclast stimulatory transmembrane protein (OC-STAMP) have been shown to be critically important to cell–cell fusion during osteoclastogenesis (7, 8). The extent and types of cytokines and receptors that contribute to osteoclastogenesis under different physiologic and pathophysiologic conditions likely vary and are at present incompletely understood. One such condition is malignancy-associated bone loss.
Osteolysis is an important and serious complication of malignancy. Mechanisms that mediate osteoclastogenesis in malignancy are varied but are thought to mostly involve the RANK–RANKL system as a final common pathway. In certain malignancies, such as myeloma, it appears that cells besides marrow osteoclast precursors, specifically dendritic cells, may serve as cellular precursors for osteoclasts (9). We recently reported that when myeloma cells are cocultured with immature dendritic cells, the dendritic cells transdifferentiate to TRAP-positive bone-resorbing multinucleated giant cells in a process that requires cell–cell contact (9). In these studies, it was observed that thrombospondin-1 (TSP-1) was one of the most significantly up-regulated genes in dendritic cells after coculture with tumor cells. TSP-1 is a multifunctional extracellular glycoprotein that mediates a wide range of intercellular and cell matrix interactions. One of the binding partners for TSP-1, CD47, was found to be highly up-regulated in myeloma cells. The addition of TSP-1 antibody to dendritic cell/myeloma cell cocultures led to significant inhibition of osteoclast formation. Neutralizing TSP-1 also blocked osteoclastogenesis from monocyte osteoclast precursors. Consistent with this latter observation, we showed that parathyroid hormone-induced bone resorption could be markedly attenuated in vivo using the same neutralizing antibody to TSP-1 (9). This last observation indicated a role for TSP-1 in physiologic osteoclastogenesis as well as a possible role in myeloma bone disease. We therefore sought to further characterize the mechanism by which TSP-1 modulates normal osteoclastogenesis. In the present study, we focused on the role of TSP-1 in the differentiation of monocyte/macrophage precursors to mature osteoclasts.
Results
Neutralizing TSP-1 completely blocks murine osteoclastogenesis both in coculture and marrow osteoclast precursor cultures
In prior studies, we have shown that antibody-mediated blockade of TSP-1 led to inhibition of human osteoclast (OCL) formation when cultures of dendritic cells or monocytes were induced by myeloma cells or RANKL and CSF-1 (9). Therefore, we first analyzed whether this antibody had a similar effect in a murine model of physiologic osteoclastogenesis. During normal bone remodeling, it is thought that cells of the osteoblast lineage induce osteoclastogenesis from marrow precursors through a paracrine signaling cascade in which RANKL produced by osteoblasts engages RANK on osteoclast precursors. The coculture system in which bone marrow cells are cultured with osteoblasts recapitulates in vitro the essential features of normal osteoclastogenesis in vivo (10). 1,25-Dihydroxyvitamin D and prostaglandin E2 are added to the cocultures to induce RANKL expression from osteoblasts.
Using this assay, it was observed that neutralizing TSP-1 completely blocked osteoclastogenesis (Fig. 1A). In the experiment shown in Fig. 1A, anti-TSP-1 antibody was added on Days 2, 3, and 5 of the coculture. Panel c quantifies the data from the coculture assay illustrated in Fig. 1A. To determine whether the same findings were observed when murine marrow osteoclast precursors were driven to differentiate with CSF-1 and RANKL, isolated marrow cells were treated with 100 ng/ml RANKL and 100 ng/ml of CSF-1. As shown in Fig. 1B, we also observed complete inhibition of osteoclastogenesis with the neutralizing antibody to TSP-1. Fig. 1B, panel b, quantifies the data from the osteoclastogenesis assay. Together, these data support an important role for TSP-1 in physiologic osteoclastogenesis.
Figure 1.

Neutralizing TSP-1 prevents osteoclastogenesis in vitro. A, murine osteoblast/bone marrow cocultures were treated with a control isotype-matched IgG1 antibody (panel a) or a neutralizing antibody to TSP-1 (panel b) and stained for TRAP on Day 6 of culture. Antibody was added at Days 2, 3, and 5 of the coculture. Abundant large, multinucleated osteoclasts are seen in panel a, whereas none are seen in panel b. Panel c shows quantification of the effect of the anti-TSP-1 antibody on osteoclast formation in the murine coculture assays shown in panels a and b (for IgG1, n = 3;, for neutralizing antibody to TSP-1, n = 5; ***, p ≤ 0.0001). As described under “Experimental procedures,” the murine osteoblast/marrow coculture used for the studies summarized in A involves culturing primary murine osteoblasts with marrow-derived osteoclast precursors. Osteoclast-like cells are generated in these cultures after the addition of PGE2 and 1,25(OH)2-vitamin D3. B, panel a, murine marrow osteoclastogenesis cultures were treated with vehicle (Control; upper two images), control isotype-matched IgG1 antibody (middle two images), or the neutralizing antibody to TSP-1 (bottom two images). In these experiments, the TSP-1 antibody was added at Day 0 along with RANKL and CSF-1. Neutralizing TSP-1 completely abrogated osteoclastogenesis. The cultures were TRAP-stained on Day 5. Panel b quantifies the effect of the anti-TSP-1 antibody on osteoclast formation in the murine osteoclastogenesis assay shown in panel a (n = 3; †, p ≤ 0.0566). As described under “Experimental procedures,” osteoclasts in B were generated from murine marrow precursors driven to differentiate by adding RANKL and CSF-1 in the absence of osteoblasts. The scatter plots represent mean ± S.D. (error bars).
The genetic absence of CD47 does not prevent osteoclastogenesis
Prior studies indicated that CD47 is the TSP-1 binding partner that mediates osteoclast formation (9). CD47 has also been implicated in cell fusion, an essential step in the formation of OCLs from macrophages (11, 12). To evaluate the contribution of CD47 to osteoclastogenesis, WT osteoblasts from C57/BL6j mice were used as the source of osteoblasts so that the genetic absence of CD47 just in osteoclast precursors could be examined. As can be seen in Fig. 2A, panel a, osteoclast precursors from CD47−/− mice still formed osteoclasts when cocultured with WT osteoblasts, although the number of osteoclasts generated and their size were reduced (Fig. 2B). When osteoclast precursors, isolated from CD36−/− or double knockout (DKO) mice, were used in the coculture assay, mature osteoclasts still formed in vitro (Fig. 2A, panels b and c). As was the case when osteoclasts were generated from CD47−/− precursors, treating CD36−/− or DKO cultures with a neutralizing antibody to TSP-1 completely abrogated osteoclastogenesis (Fig. 2A, panels b and c, right-hand columns). Interestingly, osteoclasts formed from CD36−/− preosteoclasts were significantly larger in size than WT osteoclasts (Fig. 2B). Osteoclasts derived from DKO osteoclast precursors were significantly smaller than WT osteoclasts and intermediate in size between CD36−/− osteoclasts and CD47−/− osteoclasts (Fig. 2B). To determine whether these findings extended to a human model of osteoclastogenesis, siRNA was used to suppress expression of CD47 in human peripheral blood monocytes that were differentiated to osteoclast-like cells with RANKL and CSF-1. As shown in Fig. 2C, despite suppression of CD47 expression by nearly 70%, osteoclast formation still occurred, albeit at a reduced level. These data suggest that deficiency of CD47 alone does not fully recapitulate the effects of antibody-mediated blockade of TSP-1 in both murine and human cells.
Figure 2.

CD47 is not required for osteoclastogenesis in vitro. A, panel a shows representative images from cocultures of osteoblasts isolated from neonatal C57WT mice and bone marrow cells isolated from CD47−/− mice. The first two images are from cocultures treated with a control IgG antibody. Osteoclasts are present in both images, although in some areas the osteoclasts are small. The right-most image in panel a is from a coculture treated with the neutralizing antibody to TSP-1. The scatter plot to the right of panel a summarizes the effect of TSP-1 antibody on osteoclastogenesis in three cocultures using CD47−/− preosteoclasts (n = 3; **, p ≤ 0.003). In the absence of TSP-1 antibody, the number of osteoclasts formed was equivalent to that seen in WT mice when CD47−/− marrow was used as the source of osteoclast precursors. Panel b shows representative images from cocultures of osteoblasts isolated from neonatal C57/BL/6j mice and bone marrow cells isolated from CD36−/− mice. Again, the first two images are from cocultures treated with a control IgG antibody. Abundant osteoclasts formed, and this effect was completely blocked by neutralizing TSP-1 (right-most image in panel b). The scatter plot to the right in panel b summarizes the effects of the neutralizing antibody on osteoclast formation (n = 4; *, p ≤ 0.02). Panel c shows representative images from cocultures of osteoblasts isolated from neonatal C57/BL/6j mice and bone marrow cells isolated from mice in which the genes for both CD47 and CD36 were deleted (DKO). Osteoclast formation occurred when control IgG was added in the first two images but was completely blocked by anti-TSP-1 antibody (third image and scatter plot to the right). Panel c represents data from one pair of cocultures. Consequently, no formal statistical analyses of data in panel c were performed. B, mean area of mature osteoclasts differentiated from WT osteoclast precursors (C57/BL/6j; left column), osteoclast precursors isolated from CD36−/− mice (second column from the left), osteoclast precursors isolated from CD47−/− mice (third column from the left), and osteoclast precursors isolated from DKO mice (far right column). Data are derived from 200 osteoclasts (C57WT versus CD36−/−; **, p ≤ 0.0017; for C57WT versus CD47−/−, for CD47−/− versus CD36−/−, for CD36−/− versus DKO, and for CD47−/− versus DKO, *** indicates p < 0.0001; for C57WT versus DKO, * indicates p ≤ 0.0490). C, human osteoclast-like cells generated from peripheral monocytes were treated either with a scrambled RNA (NT) or an siRNA directed against CD47. Suppressing CD47 did not abrogate osteoclastogenesis (upper left), although the number of osteoclasts formed was reduced by roughly 50% (upper right) (n = 3; **, p ≤ 0.0063). The scatter plots represent mean ± S.D. (error bars). Lower left, Western blot analysis demonstrates that the siRNA suppressed CD47 expression by 60–70%. CD47 has an apparent molecular mass on SDS-PAGE of ∼55 kDa. In the upper left image, the top panel is a phase-contrast image, and the bottom panel is from a TRAP-stained culture. HPFs, high-power fields.
CD36 is another binding partner in TSP-1–mediated osteoclastogenesis
The above data also indicate that CD47 is not solely responsible for mediating the pro-osteoclastogenic action of TSP-1. Initial microarray studies in human monocytes cultured in the presence of RANKL and CSF-1 identified CD36 as one of the top genes up-regulated upon RANKL exposure (data not shown). CD36 is a well known TSP-1 binding partner and is also implicated in cell fusion (13). We therefore next decided to evaluate the contribution of CD36 to osteoclastogenesis. Before doing so, we first determined the changes in expression of TSP-1, CD47, and CD36 during osteoclastogenesis.
The TSP-1–CD47–CD36 signaling complex changes during osteoclast maturation
To determine whether the expression of TSP-1, CD47, and CD36 proteins changes as osteoclasts mature, we examined the expression of these molecules in early preosteoclasts and compared it with that of fully mature multinucleated osteoclasts. Specifically, we examined the expression of TSP-1, CD47, and CD36 in freshly isolated murine preosteoclasts (Day 0), early in the differentiation process (Day 2 of treatment with RANKL and CSF-1), and at peak osteoclast formation (which usually occurred on Day 4 of treatment with RANKL and CSF-1). As shown in Fig. 3A, TSP-1 expression was highest at Day 0 and was undetectable thereafter. Similar to TSP-1, expression of CD36 was only detected by Western analyses at Day 0. CD47 was first detected at Day 2 and persisted at Day 4. In addition, we observed that, although cell-surface expression of both CD36 and CD47 was detected in preosteoclasts, both disappeared from the surface of mature osteoclasts. Thus, as shown in Fig. 4, A and B, there was abundant expression of CD47 and CD36 on osteoclast precursors but no expression of either of these molecules on the surface of mature, freshly isolated osteoclasts, indicating that the level of expression and the cellular distribution of these two molecules were altered during osteoclastogenesis.
Figure 3.
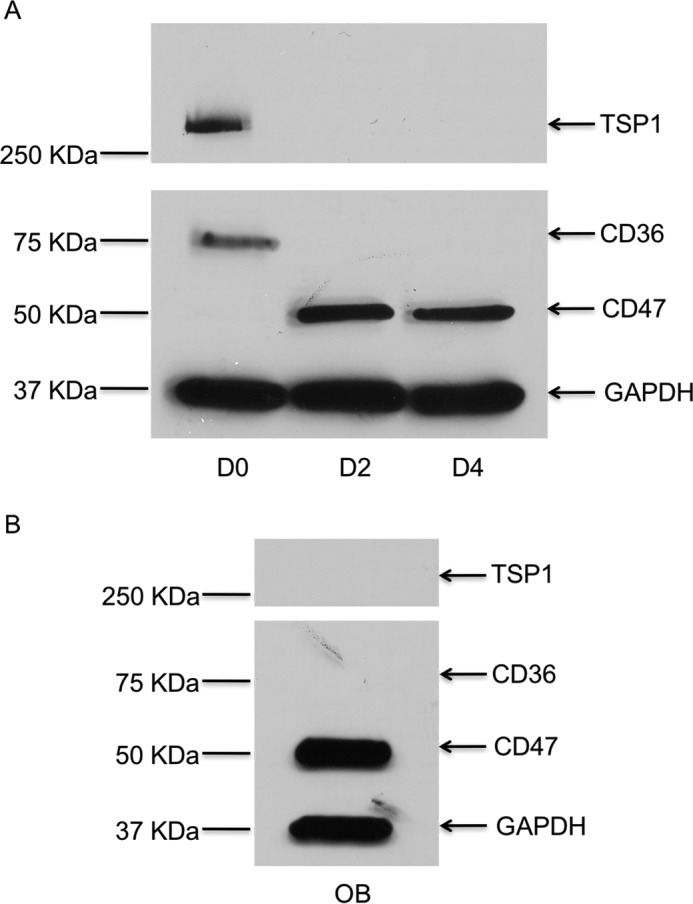
Level of protein expression for TSP-1, CD47, and CD36 in osteoblasts and osteoclasts. A, representative Western blot for TSP-1, CD47, and CD36 in whole-cell lysates prepared from primary murine osteoclasts at the indicated days of differentiation induced with RANKL and CSF-1. During osteoclast differentiation, TSP-1 protein is expressed at Day 0 and is not expressed at Day 2 or Day 4. CD47 is not expressed at Day 0 but is seen at Day 2 and Day 4. CD36 is only expressed at Day 0. B, osteoblasts express CD47 but do not express CD36 or TSP-1. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Figure 4.
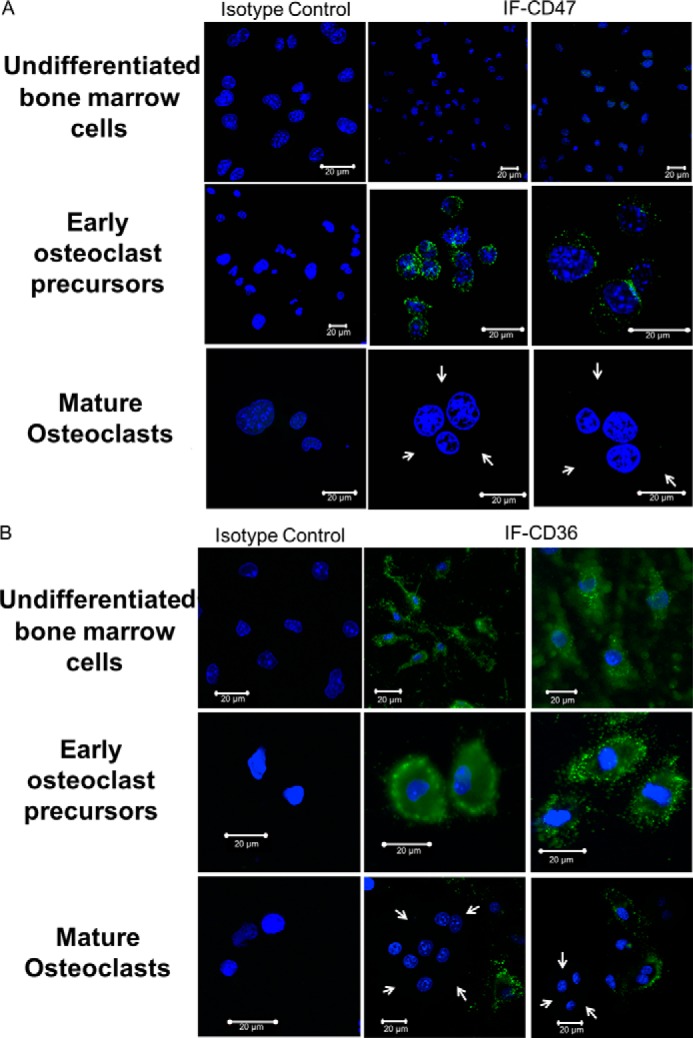
Cell-surface expression of CD47 and CD36 changes during osteoclastogenesis. A, cell-surface expression of CD47 changes during osteoclastogenesis. The top three images are undifferentiated bone marrow cells, the middle three images are early osteoclast precursors, and the bottom three images are mature osteoclasts. The first column represents the isotype control. There is abundant cell-surface expression of CD47 on osteoclast precursors (second row, middle and right images) but no expression of the molecule on the surface of mature, freshly isolated osteoclasts (third row, middle and right images). White arrows point to the cytoplasmic edge of the mature osteoclasts. B, cell-surface expression of CD36 changes during osteoclastogenesis. The top three images are undifferentiated bone marrow cells, the middle three images are early osteoclast precursors, and the bottom three images are mature osteoclasts. The first column represents the isotype control. There is abundant cell-surface expression of CD36 on osteoclast precursors (second row, middle and right images) but no expression of the molecule on the surface of mature, freshly isolated osteoclasts. White arrows point to the cytoplasmic edge of the mature osteoclasts (third row, middle and right images). IF, immunofluorescence.
We next sought to clarify the cellular sources of TSP-1, CD36, and CD47 in our model systems. As noted earlier, the coculture system most faithfully recapitulates the in vivo circumstance in which osteoblast/stromal cells support osteoclastogenesis by elaborating a variety of cytokines, most importantly RANKL and CSF-1. As shown in Fig. 3A, TSP-1, CD36, and CD47 were all detectable by Western blot analysis in osteoclast precursors early in differentiation. In primary murine osteoblasts (Fig. 3B), we also detected the expression of CD47. However, neither TSP-1 nor CD36 was expressed in osteoblasts.
Activation of CD36 contributes to osteoclastogenesis
To directly examine the contribution of CD36 to osteoclastogenesis, we used a selective peptide agonist for CD36, p907 (14). As shown in Fig. 5A, panels a and b, adding p907 to murine cocultures treated with anti-TSP-1 antibody restored osteoclastogenesis. Adding p907 or IgG alone to murine cocultures did not significantly influence the number of osteoclasts formed (Fig. 5A, third and fourth panels, respectively). In osteoclastogenesis experiments using human monocytes as precursors, suppressing CD36 inhibited osteoclastogenesis as shown in Fig. 5B. As shown in Fig. 5C, suppressing CD36 in human monocytes abrogated the ability of p907 to rescue the inhibitory effect of anti-TSP-1 on osteoclastogenesis. In contrast, p907 was still able to rescue the inhibitory effect of the anti-TSP-1 antibody on osteoclastogenesis when expression of CD47 was suppressed with an siRNA (Fig. 5D). Together, these data identify CD36 as a hitherto unappreciated but important participant in regulating TSP-1–mediated osteoclastogenesis.
Figure 5.

Activating CD36 rescues the anti-osteoclastogenic effect of TSP-1 antibody. A, panel a, representative images of Day 5 murine osteoblast/preosteoclast cocultures treated with anti-TSP-1 antibody (top image), anti-TSP-1 antibody plus p907 (second from top), p907 alone (third from top), or IgG alone (bottom image) as described under “Experimental procedures.” As can be seen, osteoclasts form in the presence of anti-TSP-1 antibody with the addition of p907, although some are small. Adding p907 or IgG alone to the cocultures had no effect on osteoclastogenesis. Panel b, quantification of data shown in panel a (n = 3 for anti-TSP-1 antibody and anti-TSP-1 antibody plus p907; *, p ≤ 0.0004; n = 2 for p907 alone and IgG alone). B, human osteoclast-like cells generated from peripheral blood monocytes were treated either with a control siRNA (NT) or an siRNA directed against CD36. As can be seen in the upper image, suppressing CD36 suppressed osteoclast formation as quantified in the lower left scatter plot. The top row of the upper left image is a phase-contrast photomicrograph. The bottom row of the upper left image shows staining for TRAP (n = 4; ***, p ≤ 0.0006). The lower right image shows that CD36 was suppressed by ∼60–70% in these experiments. CD36 has an apparent molecular mass on SDS-PAGE of ∼88 kDa. C, fewer numbers of osteoclasts were formed when human mononuclear cells in which CD36 has been suppressed were used as osteoclast precursors (filled and open circles). The filled and open triangles demonstrate that p907 cannot restore osteoclast numbers in cells in which CD36 was suppressed. The third set of data from the left (filled and open diamonds) demonstrates that neutralizing TSP-1 completely suppresses osteoclastogenesis regardless of whether CD36 is suppressed. The right-most set of data (filled and open squares) demonstrate that the ability of p907 to rescue the inhibitory effect of anti-TSP-1 was blocked when CD36 was suppressed (n = 3 for each data set; *, p < 0.035; **, p < 0.008). D, the p907 peptide was able to rescue the anti-osteoclastogenic effect of a TSP-1 antibody on human osteoclast precursors in which CD47 was suppressed. Left, photomicrographs. The upper two rows show phase-contrast photomicrographs; the lower two rows show TRAP staining. Right, scatter plot. The first two data sets (filled and open circles) show the effect of suppressing CD47 on osteoclast formation. The second two data sets (filled and open triangles) show the effect of p907 on osteoclast formation in the presence and absence of siRNA to CD47. The third set of data (filled and open diamonds) demonstrates that neutralizing TSP-1 completely suppressed osteoclastogenesis regardless of whether CD47 is suppressed. The fourth set of data (filled and open squares) show the effect of adding p907 to osteoclast precursors in which CD47 has been suppressed and then treated with anti-TSP-1. p907 was able to overcome the effect of the anti-TSP-1 antibody whether or not CD47 is suppressed (n = 3 for each data set; *, p < 0.02; ***, p ≤ 0.0008). The scatter plots represent mean ± S.D. (error bars). HPFs, high-power fields.
Nitric oxide and cGMP participate in TSP-1–mediated osteoclastogenesis
CD36 is known to suppress nitric oxide generation (14). Suppression of nitric oxide generation has been previously reported to stimulate osteoclastogenesis (15–17). We consequently determined whether part of the mechanism by which TSP-1 acts was via a nitric oxide/cGMP pathway. As shown in Fig. 6A, when neutralizing antibody to TSP-1 was added to murine cocultures on Day 5, just before osteoclasts appear in the cultures, there was a time-dependent, significant increase in the amount of nitric oxide accumulating in the cell layer. In concert with the increase in NO production, neutralizing TSP-1 in the same cultures led to a significant increase in cGMP generation (Fig. 6B). These data are consistent with the idea that TSP-1 suppresses nitric oxide and cGMP production as part of its pro-osteoclastogenic pathway. To test this hypothesis, we treated murine cocultures with the neutralizing antibody to TSP-1 and l-nitroarginine methyl ester (l-NAME), which was added at a final concentration of 100 nm. As shown in Fig. 6C, panel a, adding l-NAME to murine cocultures treated with antibody to TSP-1 completely rescued osteoclastogenesis. Fig. 6C, panel b, quantifies the data in panel a. To determine whether the effects of anti-TSP-1 to increase nitric oxide synthesis and cGMP were central to its anti-osteoclastogenic actions, we determined whether addition of the CD36 activator p907 would suppress anti-TSP-1–induced nitric oxide production. As shown in Fig. 6D, the induction of nitric oxide by the neutralizing antibody to TSP-1 was substantially suppressed by the addition of p907, although it did not suppress NO production to the level seen in the cultures treated with isotype-matched control antibody.
Figure 6.

Effect of neutralizing TSP-1 on NO and cGMP accumulation in murine cocultures. A, effect of neutralizing TSP-1 on nitric oxide production by murine cocultures. Nitric oxide was significantly elevated at 6 and 24 h after the addition of neutralizing antibody to TSP-1 (n = 3; ***, p ≤ 0.0003). B, effect of neutralizing TSP-1 on cGMP production by murine cocultures. Cell layers were extracted at 3, 6, and 24 h after the addition of antibody (Ab), and cGMP content was measured (n = 3). The two curves are statistically significantly different by one-way analysis of variance (p < 0.01). C, panel a, representative images of experiments in which l-NAME rescues the effect of neutralizing TSP-1 on osteoclastogenesis. l-NAME was added at a final concentration of 1 mm at Day 4, antibody was added at Day 5, and the cultures were fixed and stained for TRAP on Day 6. Abundant mature osteoclasts were seen in cultures treated with l-NAME despite the presence of neutralizing antibody to TSP-1. Panel b, quantification of data seen in panel a. A formal t test could not be performed because there were no osteoclasts present in the anti-TSP-1–treated cells. D, p907 suppresses nitric oxide production in the presence of neutralizing antibody to TSP-1. p907 was added on Days 4 and 5 at a final concentration of 5 μm, and media were harvested for NO measurement on Day 6. The p907 peptide significantly suppressed NO production in cocultures treated with neutralizing antibody to TSP-1 (compare first two data sets in the graph). Parallel cultures treated with isotype-matched IgG antibody served as controls (n = 5; *, p < 0.001, *** p < 0.0001). The scatter plots represent mean ± S.D. (error bars).
The hypercalcemic response to a continuous PTH infusion is attenuated in mice lacking CD36 and CD47
The above data indicate a role for both CD36 and CD47 in mediating the pro-osteoclastogenic effects of TSP-1. Our prior in vivo studies established a role for TSP-1 in contributing to the hypercalcemia induced by continuous PTH infusion. However, the acceptor molecules responsible for mediating the effects of TSP-1 in vivo were not clarified by that result. We hypothesized that both CD36 and CD47 mediate the actions of TSP, and, therefore, in the genetic absence of both molecules, the hypercalcemic response to PTH would also be attenuated. To directly test this hypothesis, we generated animals in which both CD36 and CD47 were genetically deleted and infused them with PTH as described under “Experimental procedures.” In these experiments, only female animals were studied.
Because generating the DKO mice required breeding CD36−/− and CD47−/− mice, we were able to separately analyze the skeletal phenotype of these animals. The deletion of only CD36 or CD47 in vivo led to somewhat divergent phenotypes. There was no significant change in trabecular bone volume to total volume fraction (BV/TV) or trabecular thickness in the CD47−/− mice. Trabecular BV/TV refers to the percentage of mineralized tissue in the total tissue volume analyzed in the trabecular compartment. Trabecular number was increased, whereas trabecular spacing was reduced in the CD47−/− mice (Fig. 7). Surprisingly, cortical BV/TV and thickness were both significantly reduced in CD47−/− mice (Fig. 8). The phenotype of the CD36−/− animals was more pronounced. Trabecular BV/TV, trabecular thickness (Fig. 7), and cortical BV/TV (Fig. 8) were all significantly higher in the CD36−/− animals. Surprisingly, trabecular number was reduced, and trabecular spacing was increased (Fig. 7). Compared with controls, there were no significant differences in body weight in either the CD36−/− or CD47−/− mice.
Figure 7.

Mice in which CD47, CD36, and/or CD36 and CD47 (DKO) are genetically deleted have altered spinal trabecular bone mass and microarchitecture. Mean bone density was determined by micro-CT in 12-week-old female CD47−/−, CD36−/−, and DKO mice and 12-week-old female WT controls. The data represent the trabecular envelope of the spine. In CD47−/− mice, there was no significant effect on trabecular bone mass, but trabecular number was significantly increased, and trabecular spacing was reduced (n = 10 for WT and n = 5 for CD47−/− mice; **, p ≤ 0.01; NS, nonsignificant). CD36−/− mice had a significantly increased trabecular bone mass. In addition, trabecular thickness was increased, although, somewhat surprisingly, trabecular number was reduced, and trabecular spacing was increased (n = 10 for WT and n = 6 for CD36−/−; **, p < 0.01; NS, nonsignificant). Trabecular number was significantly higher, and trabecular spacing was significantly lower in the DKO animals (n = 10 for WT and n = 8 for DKO; **, p < 0.005; NS, nonsignificant). The scatter plots represent mean ± S.D. (error bars).
Figure 8.

Mice in which CD47, CD36, or both CD36 and CD47 (DKO) are genetically deleted have altered femoral cortical bone mass or cortical parameters. Mean bone density was determined by micro-CT in 12-week-old female CD47−/−, CD36−/−, and DKO mice and 12-week-old female WT controls. In CD47−/− mice, cortical bone volume and cortical thickness were both significantly reduced (n = 10 for WT and n = 5 for CD47−/− mice; **, p ≤ 0.01; ***, p < 0.0001; NS, nonsignificant). Cortical bone mass was significantly increased in the CD36−/− mice (n = 10 for WT and n = 11 for CD36−/−; **, p < 0.01; NS, nonsignificant). Cortical thickness was significantly reduced, and endosteal circumference was significantly increased in the DKO mice. Because periosteal circumference did not change but endosteal circumference increased, per force cortical thickness was reduced (n = 10 for WT and n = 8 for DKO; *, p < 0.05; **, p < 0.005; NS, nonsignificant). The scatter plots represent mean ± S.D. (error bars).
The phenotype of the DKO mice reflected the differing skeletal changes observed in the CD36−/− and CD47−/− animals. Thus, trabecular BV/TV tended to be higher with an accompanying significant increase in trabecular number and a significant decrease in trabecular spacing (Fig. 7). There was no change in cortical BV/TV in the DKO animals (Fig. 8). These data are consistent with a role for both CD47 and CD36 in regulating skeletal homeostasis, likely, in part, by modulating the actions of TSP-1 in bone. In keeping with that notion, TSP-1 knockout mice have been reported to have a low bone density (18).
As described in the Introduction, we postulated that TSP-1, acting through CD47 and CD36, participates in the bone resorptive response to a PTH infusion. Consistent with this hypothesis, when female DKO animals were infused with PTH, serum C-terminal type 1 collagen cross-links (CTx) and serum calcium were both significantly lower in the double knockout animals as compared with controls. CTx is a degradation product of mature cross-linked type 1 collagen, and serum levels are a systemic indicator of the rate of bone resorption. In contrast, when PTH was infused into either CD47−/− or CD36−/− mice, there were no significant differences in either the hypercalcemic response or the increase in CTx in these animals compared with WT controls (Fig. 9).
Figure 9.

DKO mice have a blunted hypercalcemic response to a PTH infusion. 12-week-old female DKO, CD47−/−, CD36−/−, and control mice (C57WT) were infused with PTH for 5 days as described under “Experimental procedures.” Serum calcium and serum CTx were no different at baseline in WT and DKO animals (first two data sets in the upper and lower left graphs; calcium: n = 4 for WT, n = 5 for DKO; CTx: n = 11 for WT, n = 9 for DKO; NS, nonsignificant). At Day 5 of the PTH infusion, serum calcium and serum CTx were both significantly lower in the DKO animals compared with controls (second data set in the left two panels; calcium: n = 9 for WT, n = 9 for DKO; CTx: n = 5 for WT, n = 7 for DKO; *, p < 0.02). Both serum calcium and CTx were significantly higher in the PTH-infused control animals compared with vehicle-infused animals (***, p ≤ 0.0001). There was also a significant increase in serum calcium and serum CTx in the PTH-infused DKO animals compared with the vehicle-infused DKO mice (calcium: **, p ≤ 0.006; CTx: ***, p ≤ 0.0003). The two middle graphs summarize data from vehicle- and PTH-infused 12-week-old female CD47−/− and control mice. At Day 5 of vehicle infusion, there were no differences in mean serum calcium and serum CTx in control and CD47−/− animals (calcium: n = 4 for WT, n = 3 for CD47−/−; CTx: n = 3 for WT, n = 3 for CD47−/−; NS, nonsignificant). At Day 5 of the PTH infusion, serum calcium and serum CTx were both equivalently elevated in the CD47−/− mice and normal controls (calcium: n = 6 for WT, n = 6 for CD47−/−; CTx: n = 6 for WT, n = 6 for CD47−/−; NS, nonsignificant). Serum calcium was significantly higher in the PTH-infused control mice compared with animals receiving vehicle infusion. Mean serum CTx also rose with PTH infusion in the control mice, but the difference between PTH- and vehicle-infused mice was not statistically significantly different (calcium: ***, p ≤ 0.0002; CTx: NS, nonsignificant). In the CD47−/− mice, both serum calcium and CTx were significantly higher in the PTH-infused mice compared with vehicle-infused animals (calcium: ***, p ≤ 0.0002; CTx: **, p ≤ 0.009). The data in the right two graphs represent data from 12-week-old female control and CD36−/− mice. Serum calcium and serum CTx were no different at baseline in WT and CD36−/− animals (calcium: n = 4 for WT, n = 5 for CD36−/−; CTx: n = 5 for WT, n = 5 for CD36−/−; NS, nonsignificant). At Day 5 of the PTH infusion, serum calcium and serum CTx were both significantly and equivalently elevated in the CD36−/− mice and normal controls (calcium: n = 6 for WT, n = 6 for CD36−/−; CTx: n = 6 for WT, n = 6 for CD36−/−; NS, nonsignificant). The serum calcium was higher in both the PTH-infused CD36−/− animals compared with their respective vehicle-infused controls (**, p ≤ 0.0026 and ***, p ≤ 0.0001, respectively). After 5 days of PTH infusion, there was no significant difference in mean CTx values in the PTH-infused CD36−/− and control animals. Although CTx was higher in both the CD36−/− and control animals infused with PTH compared with vehicle-infused animals, the differences were not significant for either genotype. The scatter plots represent mean ± S.D. (error bars).
Impaired resorptive activity of DKO osteoclasts
The in vitro osteoclastogenesis assays did not indicate there was defective osteoclastogenesis in the genetic absence of CD47 and CD36 (Fig. 2A, panel c). Therefore, to better understand the cellular basis for the impaired hypercalcemic response and the attenuated increase in CTx in response to PTH in the DKO animals, bone histomorphometry was performed in PTH-infused control and DKO animals (Table 1). Surprisingly, the DKO animals evidenced a greater number of osteoclasts in bone during PTH infusion than did WT controls. OcS/BS was more than 2-fold greater in the DKO animals as was the number of osteoclasts (NOc)/TAR. The NOc/bone perimeter (BPm) was also significantly elevated, nearly 3-fold greater in the PTH-infused DKO animals. Thus, consistent with our in vitro osteoclastogenesis assay data, a defect in osteoclastogenesis in vivo was not the explanation for the impaired resorptive response to a PTH infusion. Next, in vitro resorption assays were performed to examine whether the DKO osteoclasts had defective resorptive capability. As shown in Fig. 10, the area resorbed per osteoclast was 27% lower in the DKO as compared with C57BL/6j (C57WT) animals. In each of four experiments, the DKO osteoclasts resorbed less matrix than the C57WT cells. However, because of the variability in the absolute resorbed area in each experiment, the mean difference did not reach statistical significance.
Table 1.
Bone histomorphometry in PTH-infused control and DKO animals
ObS, osteoblast surface.
| Group | BV/TV | ObS/OS | OcS/BS | NOc/TAR | NOc/BPm |
|---|---|---|---|---|---|
| Control | 25.27 ± 2.51 | 58.77 ± 5.29 | 1.09 ± 0.29 | 3.53 ± 0.67 | 0.35 ± 0.06 |
| DKO | 30.81 ± 4.60 | 70.09 ± 4.61 | 2.94 ± 0.31 | 10.02 ± 0.26 | 0.88 ± 0.10 |
Figure 10.
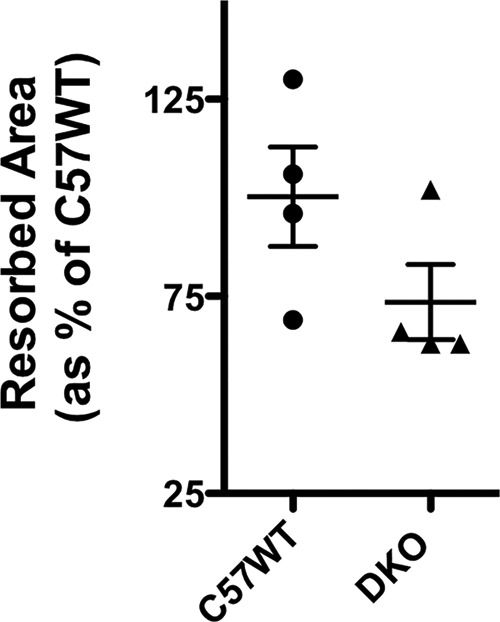
Resorptive activity of control and DKO osteoclasts quantified on OsteoAssay plates. Resorbed area was quantified as pixels2. The mean area resorbed by C57WT cells was taken as 100%. The resorbed area by the DKO osteoclasts in each of the four experiments was calculated and expressed as a percentage of the mean area resorbed by C57WT cells. Three separate wells were analyzed for each genotype in each experiment. Resorbed area was quantified in 25 fields at ×100 magnification for each genotype as detailed under “Experimental procedures.” The scatter plots represent mean ± S.D. (error bars).
Discussion
The current work identifies TSP-1 and its binding partners, CD36 and CD47, as having important and interacting roles in osteoclastogenesis, in part by regulating nitric oxide signaling. Inhibiting TSP-1 in three in vitro osteoclastogenesis models uniformly led to marked inhibition of osteoclast formation. The murine coculture system perhaps most closely recapitulates osteoclastogenesis in vivo because the production of RANKL is regulated by cells in the osteoblast lineage, which are thought to be a key source of endogenous RANKL in bone. In addition, osteoblasts provide other pro-osteoclastogenic factors, such as CSF-1 and IL-6. In the two culture systems lacking an endogenous source of RANKL and CSF-1, the murine marrow and the human monocyte osteoclastogenesis assay, in which RANKL and CSF-1 are added in pharmacologic amounts, neutralizing TSP-1 still abrogated osteoclastogenesis. Our prior in vivo work, demonstrating that neutralizing TSP-1 attenuates PTH-induced hypercalcemia, supports the idea that the actions of TSP-1 on osteoclast precursors is physiologically relevant. The stage or stages in osteoclast formation where TSP-1 acts are not yet clear. In some TSP-1 neutralizing experiments, mononuclear TRAP-positive cells were present, suggesting that perhaps TSP-1 plays a role in fusion of mononuclear precursors to form mature osteoclasts. Conversely, it is possible that TSP-1 acts at multiple steps during osteoclast differentiation. Regardless of the steps at which TSP-1 acts, it appears that both CD47 and CD36 are important mediators of TSP-1's actions in osteoclast precursors as well as mature osteoclasts.
However, deleting CD47, CD36, or both in osteoclast precursors did not completely phenocopy the effects of neutralizing TSP-1 in vitro because osteoclasts still formed. The impact of suppressing CD47 or its genetic absence varied, depending on the cell model used. In human osteoclastogenesis cultures, suppressing CD47 led to a significant inhibition of osteoclast formation. In murine osteoblast cocultures using CD47−/− osteoclast precursors, the number of mature osteoclasts formed varied, but in general, the cells appeared smaller. Consistent with our findings, Maile et al. (19) recently reported that osteoclast formation was impaired when murine CD47−/− marrow was used as the source for osteoclast precursors.
Suppressing CD36 in human marrow cells also suppressed osteoclastogenesis. Importantly, the effects of neutralizing TSP-1 could be partially reversed with the CD36 peptide agonist p907 in WT murine marrow cells and in murine marrow cells isolated from CD47−/− mice or human monocyte cultures in which CD47 had been suppressed. p907 has been shown to activate CD36, and when CD36 was suppressed in monocytes, p907 could no longer rescue the anti-osteoclastogenic effect of TSP-1. These data support a role for CD36 in modulating TSP-1–induced osteoclastogenesis.
Although, as noted above, CD36 has been reported to play a role in macrophage fusion, Helming et al. (13) reported that osteoclast fusion was unimpaired when bone marrow macrophages from CD36−/− mice were treated with RANKL and CSF-1. However, this study did not provide data on absolute numbers of osteoclasts formed. Kevorkova et al. (20) have reported a low-bone-mass phenotype in CD36−/− mice. This phenotype was seen primarily in trabecular bone. Cortical bone was largely unaffected. Reduced numbers of osteoblasts in vivo and a reduced bone formation rate seemed to underlie this phenotype. These animals had a significantly reduced body mass, which could have influenced these findings. Our findings in the CD36−/− mice differ from these. Body mass was identical in our CD36−/− and WT controls, and, in contrast to the findings of Kevorkova et al. (20), trabecular bone mass was significantly increased in these animals. Our findings in the CD47−/− mice are more consistent with prior reports. Thus, Maile et al. (19) reported a reduction in trabecular bone volume in CD47−/− mice due to impairment in osteoblast function. As noted, the osteoclastogenic potential of bone marrow precursors isolated from CD47−/− animals was also reduced compared with WT controls (19, 21). More recently, Koskinen et al.(22) reported reduced trabecular bone mass and impaired bone formation rates in CD47−/− animals. There have been no prior reports of the skeletal phenotype in mice with genetic deletion of both CD36 and CD47. The skeletal phenotype in the DKO mice was complex with a trend toward higher bone density and a more robust trabecular microarchitecture when compared with controls. In particular, trabecular number was significantly higher, and trabecular spacing was significantly lower in animals in which both CD36 and CD47 were genetically absent. In contrast, cortical thickness was significantly lower, and consistent with that, endosteal circumference was significantly higher, whereas periosteal circumference was not changed in these animals. These findings may reflect the combined effects of deleting CD36 and CD47 in all skeletal cells, including osteoblasts, osteoclasts, and osteocytes. Thus, for example, TSP-1 has also been shown to inhibit cells in the osteoblast lineage by altering the activity of transforming growth factor-β (23).
Our in vivo studies in the DKO mice are consistent with our earlier work and support the conclusion that CD36/CD47 participate in mediating PTH-induced hypercalcemia (9). The fact that animals in which CD36 and CD47 were deleted showed a diminished resorptive response to PTH strongly supports the physiologic relevance of these two molecules in modulating osteoclast activity in vivo. It may be that the TSP-1/CD36/CD47 signaling system is most relevant in accelerated or pathologic osteoclast-mediated bone resorption. Interestingly, osteoclastogenesis was not impaired in the PTH-infused DKO mice. Rather, DKO osteoclasts showed less resorptive activity in an in vitro resorption assay, indicating that this was the likely mechanism for the attenuated resorptive response to PTH in the DKO mice. This suggests that, in addition to playing a role in osteoclast formation, CD36 and CD47 play an important role in mediating mature osteoclast function.
Recently, Amend et al. (18) reported in vivo and in vitro characterization of mice in which TSP-1 had been genetically deleted. Consistent with our earlier in vivo data suggesting an important role for TSP-1 in skeletal homeostasis, these investigators reported that bone mass was increased in TSP-1−/− mice. Amend et al. (18) also found that serum CTx values were significantly lower in these animals, suggesting a defect in osteoclast function, which is consistent with both our in vitro and in vivo findings. Markers of osteoblast differentiation, markers of osteoblast activity in vivo (P1NP and osteocalcin), and osteoblast number were not different in WT and TSP-1−/− mice, suggesting that the primary mechanism for the increased bone mass was impaired osteoclast function. Our data are consistent with these findings of Amend et al. (18) and help to clarify the mechanism of action of TSP-1 in bone by demonstrating that both CD36 and CD47 are targets for this ligand in modulating osteoclast biology.
Engagement of CD36 by TSP-1 leads to suppression of nitric oxide synthesis and cGMP accumulation (14). Consistent with that, we found that neutralizing TSP-1 led to a rise in nitric oxide synthesis and cGMP production. That these effects are important to the anti-osteoclastogenic actions of the TSP-1 antibody is supported by the fact that adding back l-NAME in the presence of a neutralizing TSP-1 antibody reversed the effect of the antibody, establishing that TSP-1 regulates osteoclastogenesis via the NO/cGMP pathway. Importantly, suppressing NO synthesis has previously been reported to induce osteoclastogenesis (16). Interestingly, Amend et al. (18) found that treatment with l-NAME in vivo stimulated a rise in CTx in TSP-1−/− mice but not in WT controls.
Our expression studies suggest that TSP-1/CD47/CD36 signaling occurs mostly during osteoclast formation, at least as judged by the fact that expression levels for all three molecules are markedly reduced in fully formed osteoclasts when compared with osteoclast precursors. However, the impaired resorptive activity of the mature DKO osteoclasts indicates a lasting effect of these earlier signaling events. The mechanisms underlying the altered cellular localization of CD47 and CD36 during osteoclastogenesis are not entirely clear, although for CD36 they may be mediated by post-translational modifications (24).
Although our in vitro data establish a critical role for TSP-1 in osteoclastogenesis, the selective deletion of either or both of its two binding partners, CD36 and CD47, did not prevent osteoclastogenesis; nonetheless DKO osteoclasts were significantly smaller than WT cells. The impaired resorptive response of these cells in vitro provides a mechanistic understanding for why there was an attenuated calcemic response to PTH infusion in the DKO mice. In the setting of defective osteoclastic resorptive activity, it is not uncommon to see a compensatory increase in the number of osteoclasts in bone, perhaps in an attempt to compensate for the defective cellular machinery (25). This appears to be the case in the PTH-infused DKO animals. Osteoclast number was increased in the DKO animals in response to PTH infusion relative to the numbers seen in WT animals despite that their resorptive activity was impaired. This likely explains the observed in vivo differences in response to PTH. Whether the TSP-1/CD47/CD36 signaling system plays a role in regulating the resorptive activity of mature osteoclasts beyond impacting cellular size is a question not addressed by our findings.
Our data support one interpretation of the signaling cascade entrained by the TSP-1, CD47, and CD36 axis, namely that this signaling system is acting in an autocrine fashion (Fig. 11). In support of this hypothesis is the fact that two key proteins, TSP-1 and CD36, are expressed in osteoclast precursors but not in osteoblasts. The cellular events entrained by the interactions of these three molecules clearly are complex and affect multiple steps in osteoclast differentiation and differentiated osteoclast function. Thus, the number and/or appearance of mature osteoclasts generated in the genetic absence of CD47, CD36, or both, coupled with the defect in resorptive activity of DKO osteoclasts, suggests that this signaling system modulates both osteoclastogenesis and mature osteoclast function.
Figure 11.

One model for the TSP-1/CD47/CD36 signaling cascade in osteoclasts. Prior to induction, only TSP-1 protein is expressed in preosteoclasts (left). During differentiation (right), perhaps under the control of RANKL and CSF-1 binding to their cognate receptors (RANK and c-Fms, respectively) as well as other pro-osteoclastogenic cytokines, CD47 and CD36 are expressed on committed preosteoclasts. TSP-1 binding to CD47 and CD36 leads to suppression of NO production, which helps to drive differentiation to mature osteoclasts. The TSP-1/CD36/CD47 signaling system also modulates mature osteoclast function.
Alternative explanations for how this system might operate in vivo obviously exist. Thus, TSP-1, expressed in osteoclast precursors, could act via CD47 expressed on osteoblasts, resulting in paracrine signaling in which osteoblasts stimulated by TSP-1 elaborate signals that help to induce osteoclastogenesis. Osteoclastogenesis studies using CD47−/− osteoblasts might help to test this model.
The expression pattern of TSP-1, CD36, and CD47 in osteoclasts may help explain the complex phenotypes of the CD47−/−, CD36−/−, and DKO mice. The skeletal phenotype of the CD36−/− mouse is consistent with the observation that this molecule is primarily expressed on osteoclasts. Thus, impaired osteoclast function would be expected to lead to a high-bone-mass phenotype, which is what we observed. In contrast, the phenotype of the CD47−/− animals was more complex and might reflect the effect of this molecule both on osteoclasts and osteoblasts, again consistent with the pattern of protein expression for this molecule. The DKO mice had bone densitometric findings that could be viewed as intermediate between those of the CD36−/− and CD47−/− mice.
In conclusion, TSP-1 and its binding partners, CD47 and CD36, play important complimentary roles in mediating osteoclast function by regulating NO signaling. The present work adds CD36 as an important component of this axis and a potential target for novel therapies regulating osteoclast formation.
Experimental procedures
Materials
1,25(OH)2-Vitamin D was from Wako Pure Chemical Industries, Ltd. (Chuo-ku, Osaka, Japan). Prostaglandin E2 and l-NAME were from Sigma-Aldrich. l-NAME is a competitive nitric-oxide synthase inhibitor that requires hydrolysis of the methyl ester group in cells to become functional. Its Ki values range between 15 and 40 nm. Murine RANKL, CSF-1, and TNFα were from R&D Systems (Minneapolis MN). The neutralizing antibody to TSP-1 and the isotype-matched control IgG1 were from Thermo Scientific (catalogue numbers MS-420-P and 553447, respectively). The antibody to TSP-1 targets an epitope on the G-module without targeting other TSP-1 domains, such as the heparin-binding domain or those implicated in platelet agglutination and angiogenesis (26). The antibodies to human CD36 and human CD47 were from Santa Cruz Biotechnology (sc-9154 and sc-7059, respectively; Santa Cruz, CA). Horseradish peroxidase–conjugated β-actin (5125S; Cell Signaling Technology, Boston, MA) was used as an internal loading control in Western blotting of human cell lysates. For Western analyses of murine lysates, the anti-TSP-1 antibody was from Thermo Scientific (Waltham, MA; MS-420-p). The anti-CD36 mAb was from Abcam (Cambridge, MA; ab133625). Amino acids 350–450 comprise the epitope against which the anti-CD36 antibody is directed. The monoclonal anti-CD47 antibody (Abcam; ab108415) is directed against a fragment of CD47 that extends from amino acid 250 to the C terminus. Cell culture media were from Sigma. Collagenase A and Dispase II, used in isolating primary murine calvarial osteoblasts, were from Roche Applied Science. Fetal bovine serum was from Atlanta Biologicals (Lawrenceville, GA). Ficoll-PaqueTM Plus was from GE Healthcare. The peptide p907 was a generous gift from Dr. David D. Roberts (Laboratory of Pathology, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD). p907 is a TSP-1–derived peptide with the sequence 434GDGVITRIR442 (where I (Ile) is the d isomer). This peptide was published, and its agonist activity was described by Isenberg et al. (Ref. 14, see Fig. 1D).
Animals
CD1 and C57WT were purchased from Charles River Laboratories International, Inc. (Wilmington, MA). CD47−/− mice (stock number 003173) were purchased from The Jackson Laboratory (Bar Harbor, ME). CD36−/− mice were kindly provided by Dr. Roy Silverstein (Medical College of Wisconsin) as described previously (27). Mice in which both CD47 and CD36 were deleted were generated using standard breeding protocols. Genotyping was accomplished by PCR. CD1 mice were used for the experiment shown in Fig. 1 only. CD47−/− and CD36−/− mice were on a C57BL/6j background. C57BL/6j mice matched for age, sex, and weight were used as controls for all experiments using CD47−/−, CD36−/−, and DKO mice. The use of all strains of animals as well as all of the experimental studies conducted in vitro and in vivo were reviewed and approved by the Yale Institutional Animal Care and Use Committee.
Murine osteoclast-like cells generated by coculture
Cocultures were prepared as described previously (10). Bone marrow cells were prepared as reported earlier (10). Primary murine osteoblasts were obtained by serial collagenase/Dispase digestion of neonatal mouse calvariae (10). Calvariae were dissected from neonatal mice, pooled, and subjected to sequential digestion with 0.1% bacterial collagenase and 0.2% Dispase. Digestions were for 10 min at 37 °C with rapid shaking. Digests 2–5 were pooled, plated into 100-mm dishes, allowed to come to confluence, trypsinized, and cryopreserved at a concentration of 2 × 106 cells/ml. For use in the coculture system, osteoblasts were plated at an initial density of 2.5 × 104 cells/cm2 18 h before the addition of marrow. Marrow cells (at an initial density of 1.5 × 105 cells/cm2) were then added, and the mixture was cocultured for 6–7 days in the presence of 10−8 m 1,25(OH)2-vitamin D and 10−6 m prostaglandin E2 with a media change every other day. This results in a preparation in which ∼90% of the cellular material comprises solely OCLs. OCLs possess the important phenotypic and biochemical markers of authentic osteoclasts. They stain strongly for tartrate-resistant acid phosphatase, express calcitonin receptors, and, most importantly, can resorb bone (28).
Osteoclastogenesis assay using murine bone marrow osteoclast precursors
Preparation of bone marrow cells and the osteoclastogenesis assay were described previously (29). Briefly, unfractionated bone marrow cells were isolated from 8-week-old C57BL/6j mice and centrifuged for 10 min at 1000 × g. The cell pellet was resuspended, and the cells were plated overnight in 10 ml of α-MEM containing 10% FBS and 25 ng/ml CSF-1 in 10-cm dishes. Nonadherent cells were then collected and layered on 10 ml of Ficoll-Paque Plus and centrifuged for 20 min at 707 × g. Cells at the interface were collected, washed with PBS, and plated in 24-well plates at a density of 2.5 × 105 cells/well in α-MEM containing 10% FBS and 25 ng/ml CSF-1 for 24 h at which point various treatments were begun. Media were changed every other day. To quantify osteoclast numbers, cells were fixed at Day 6 of culture, and TRAP staining was performed using the Acid Phosphatase, Leukocyte (TRAP) kit (Sigma-Aldrich). For Western blotting analyses of TSP-1, CD36, and CD47 expression in osteoclastogenesis, preosteoclasts were cultured in CSF-1 alone for 48 (instead of 24) h before differentiation was induced with both RANKL and CSF-1.
Tartrate-resistant acid phosphatase staining
Tartrate-resistant acid phosphatase activity in osteoclast cultures was assessed using the leukocyte acid phosphatase kit (Sigma-Aldrich) following the manufacturer's recommended protocol. Cells were photographed with a Zeiss AXIO microscope (Zeiss AxioVision v4.8.2.0).
Osteoclastogenesis assay using human peripheral blood mononuclear cells
CD14+ monocytes were separated from peripheral blood as described previously (9). Monocytes (1 × 106 cells/ml) were cultured in α-MEM supplemented with 10% fetal bovine serum, gentamicin, 50 ng/ml RANKL, and 25 ng/ml CSF-1 in 6-well plates for 9–10 days. Cytokines were replenished every 3 days.
Quantifying osteoclast resorption
The resorbing potential of OCLs generated from osteoclast precursors isolated from control and DKO mice was quantified using 24-well OsteoAssay® plates. Unfractionated bone marrow cells were isolated from control and DKO male mice and centrifuged for 10 min at 1000 × g. The cell pellet was resuspended, and the cells were plated overnight in α-MEM containing 10% FBS and 30 ng/ml CSF-1. Nonadherent cells were then collected and layered on Ficoll-Paque and centrifuged for 20 min at 707 × g. Cells at the interface were collected, washed twice with ice-cold PBS, and plated on OsteoAssay plates at a density of 2.5 × 105/well in α-MEM containing 10% FBS, 50 ng/ml CSF-1, and 75 ng/ml RANKL. Cells were maintained at 37 °C in a humidified incubator with a media change every other day. On day 7 of culture, media were aspirated, and 600 μl of 10% sodium hypochlorite was added to each well for 5 min at room temperature. The wells were then aspirated, washed twice with 900 μl of deionized water, and allowed to dry completely at room temperature. Three separate wells were analyzed for each genotype. Resorbed area was quantified in 25 fields at ×100 magnification for each genotype using a template published previously (29), and the mean value for all fields was compared. The entire experiment was repeated four times using marrow from four DKO and four C57WT mice studied in parallel.
Effect of siRNA-mediated suppression of CD36 and CD47 on in vitro osteoclastogenesis from human osteoclast precursors
CD14+ cells were isolated as described previously (9), washed with Opti-MEM I (Invitrogen), and resuspended in Opti-MEM I. A total of 4 × 106 CD14+ cells were electroporated with 20 μg of CD36 or CD47 siRNA (siGENOME SMARTpool, Dharmacon, Thermo Fisher Scientific, Rockford, IL; catalogue number MU-010206-01-0010 for CD36 and catalogue number MU-019505-01-0010 for CD47) or nontargeting siRNA (siCONTROL Nontargeted siRNA, Dharmacon, Thermo Fisher Scientific; catalogue number D-001206-13-50) in a 4-mm electroporation cuvette (Bio-Rad) by use of an ECM 830 Square Wave pulse of 500 V for 0.5 ms twice (1–3). The electroporated mononuclear cells were immediately resuspended in α-MEM complete medium and supplemented with RANKL and CSF-1, which were replenished every 3 days. After 9–10 days of culture, cells were used for TRAP staining or RNA or protein isolation.
Western blotting
Western analyses were performed using standard methodology as reported previously (29).
NO measurements in conditioned media
NO was measured in conditioned media of cocultures using a Sievers 270B NO analyzer (GE Analytical Instruments, Boulder, CO). For experiments where the effect of l-NAME was assessed, antibody with or without l-NAME at a final concentration of 1 mm was added at Day 4, and the cocultures were analyzed at Day 6.
cGMP assay
cGMP was measured in cell lysates using a commercially available enzyme immunoassay kit (Cayman Chemical Co., Ann Arbor, MI; catalogue number 581021). On Day 5, isobutylmethylxanthine at a final concentration of 0.5 mm was added to all cultures 15 min prior to the addition of antibody. At 3, 6, and 24 h after the addition of antibody, the cell layer was washed, and 1 ml of 0.2 m HCI was added for every 35 cm2 of surface area. Plates were incubated at room temperature for 20 min with gentle rocking. The cell layer was removed by gentle scraping with a rubber policeman. The extract was triturated using a Pasteur pipette and centrifuged at 1000 × g for 10 min at 4 °C. The resulting supernatant was frozen at −80 °C until analyzed.
PTH infusion protocol
Human PTH(1–34) (Bachem, Torrance, CA) was infused at a rate of 0.67 pmol/g of body weight/h for 5 days using ALZET minipumps. PTH was prepared and minipumps were placed as described previously (9). Vehicle infusions consisted of the carrier for PTH, which was 10 mm acetic acid containing 2% heat-inactivated mouse serum. At the end of 5 days, blood was drawn for measurement of serum calcium and CTx. All animals that became hypercalcemic (>10.0 mg/dl) during the infusions were considered to have successfully received the hormone and were included in the analysis.
Measurement of CTx
Serum levels of CTx were measured as reported previously using a commercially available ELISA (Immunodiagnostic Systems Inc., Gaithersburg, MD) (30).
Measurement of serum calcium
Serum calcium was measured using a Wasserman Autoanalyzer as we have reported previously (30).
Micro-CT
Femora were stripped of soft tissue and stored in 70% EtOH at 4 °C for subsequent microcomputed tomographic analyses. The bones were scanned using a Scanco μCT-35 instrument (Scanco, Brutissellen, Switzerland) as described previously (29). Briefly, volumetric regions for trabecular analyses, selected within the endosteal borders of the distal femoral metaphysis to include the secondary spongiosa located 1 mm from the growth plate and extending 1 mm proximally, were scanned at 12-μm resolution. Cortical morphometry was quantified and averaged volumetrically through 50 serial cross-sections (600 μm) extending distally from the diaphyseal midpoint between proximal and distal growth plates. We used a customized thresholding technique (Scanco) that provided the best segmentation of the bone tissue. Both 2D and 3D microcomputed tomographic data included BV/TV and trabecular number, thickness space, and connectivity density. Cortical thickness averaged for both cortices was also quantified.
Histomorphometric analyses
Histomorphometry was performed as reported previously (31, 32) in a ∼2.5-mm2 area of distal femoral cancellous bone containing only secondary spongiosa and located 0.5–2.5 mm proximal to the epiphyseal growth cartilage. Femora were harvested from PTH-infused control and DKO animals at Day 5. Accepted nomenclature was used to report results (33, 34).
Statistical analyses
All statistical analyses were performed using unpaired t tests except for the data summarized in Fig. 6B that were analyzed by one-way analysis of variance. Data are represented using scatter plots with standard deviations.
Author contributions
S. V. K., B.-h. S., J. M. W., M. Z., and C. S. data curation; S. V. K., B.-h. S., J. M. W., M. Z., C. S., M. D., and K. L. I. formal analysis; S. V. K., J. M. W., C. S., M. D., and K. L. I. validation; S. V. K., B.-h. S., J. M. W., M. Z., C. S., M. D., and K. L. I. investigation; S. V. K., B.-h. S., J. M. W., M. D., and K. L. I. methodology; J. M. W. software; J. M. W., M. D., and K. L. I. visualization; J. M. W., M. D., and K. L. I. writing-original draft; J. M. W., M. D., and K. L. I. project administration; J. M. W., M. D., and K. L. I. writing-review and editing; M. D. and K. L. I. conceptualization; M. D. and K. L. I. resources; M. D. and K. L. I. supervision; M. D. and K. L. I. funding acquisition.
This work was supported by NIAMS, National Institutes of Health Grant AR060322 (to K. L. I. and M. D.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- RANKL
- receptor activator of NF-κB ligand
- RANK
- receptor activator of NF-κB
- CD
- cluster of differentiation
- TSP-1
- thrombospondin-1
- PTH
- parathyroid hormone
- DKO
- double knockout
- l-NAME
- l-nitroarginine methyl ester
- TNF
- tumor necrosis factor
- IL
- interleukin
- TRAP
- tartrate-resistant acid phosphatase
- OCL
- osteoclast
- BV
- bone volume
- TV
- total volume
- CTx
- C-terminal type 1 collagen cross-links
- OcS
- osteoclast surface
- BS
- bone surface
- nOc
- number of osteoclasts
- TAR
- tissue area
- BPm
- bone perimeter
- C57WT
- C57BL/6j
- α-MEM
- minimum essential medium Eagle α
- FBS
- fetal bovine serum
- micro-CT
- microcomputed tomography.
References
- 1. Arai F., Miyamoto T., Ohneda O., Inada T., Sudo T., Brasel K., Miyata T., Anderson D. M., and Suda T. (1999) Commitment and differentiation of osteoclast precursor cells by the sequential expression of c-Fms and receptor activator of nuclear factor κB (RANK) receptors. J. Exp. Med. 190, 1741–1754 10.1084/jem.190.12.1741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yeo L., Toellner K. M., Salmon M., Filer A., Buckley C. D., Raza K., and Scheel-Toellner D. (2011) Cytokine mRNA profiling identifies B cells as a major source of RANKL in rheumatoid arthritis. Ann. Rheum. Dis. 70, 2022–2028 10.1136/ard.2011.153312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Josien R., Wong B. R., Li H. L., Steinman R. M., and Choi Y. (1999) TRANCE, a TNF family member, is differentially expressed on T cell subsets and induces cytokine production in dendritic cells. J. Immunol. 162, 2562–2568 [PubMed] [Google Scholar]
- 4. Miyamoto T. (2011) Regulators of osteoclast differentiation and cell-cell fusion. Keio J. Med. 60, 101–105 10.2302/kjm.60.101 [DOI] [PubMed] [Google Scholar]
- 5. Pfeilschifter J., Chenu C., Bird A., Mundy G. R., and Roodman G. D. (1989) Interleukin-1 and tumor necrosis factor stimulate the formation of human osteoclastlike cells in vitro. J. Bone Miner. Res. 4, 113–118 10.1002/jbmr.5650040116 [DOI] [PubMed] [Google Scholar]
- 6. Koga T., Inui M., Inoue K., Kim S., Suematsu A., Kobayashi E., Iwata T., Ohnishi H., Matozaki T., Kodama T., Taniguchi T., Takayanagi H., and Takai T. (2004) Costimulatory signals mediated by the ITAM motif cooperate with RANKL for bone homeostasis. Nature 428, 758–763 10.1038/nature02444 [DOI] [PubMed] [Google Scholar]
- 7. Miyamoto H., Suzuki T., Miyauchi Y., Iwasaki R., Kobayashi T., Sato Y., Miyamoto K., Hoshi H., Hashimoto K., Yoshida S., Hao W., Mori T., Kanagawa H., Katsuyama E., Fujie A., et al. (2012) Osteoclast stimulatory transmembrane protein and dendritic cell-specific transmembrane protein cooperatively modulate cell-cell fusion to form osteoclasts and foreign body giant cells. J. Bone Miner. Res. 27, 1289–1297 10.1002/jbmr.1575 [DOI] [PubMed] [Google Scholar]
- 8. Oursler M. J. (2010) Recent advances in understanding the mechanisms of osteoclast precursor fusion. J. Cell. Biochem. 110, 1058–1062 10.1002/jcb.22640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kukreja A., Radfar S., Sun B. H., Insogna K., and Dhodapkar M. V. (2009) Dominant role of CD47-thrombospondin-1 interactions in myeloma-induced fusion of human dendritic cells: implications for bone disease. Blood 114, 3413–3421 10.1182/blood-2009-03-211920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Insogna K. L., Sahni M., Grey A. B., Tanaka S., Horne W. C., Neff L., Mitnick M., Levy J. B., and Baron R. (1997) Colony-stimulating factor-1 induces cytoskeletal reorganization and c-src-dependent tyrosine phosphorylation of selected cellular proteins in rodent osteoclasts. J. Clin. Investig. 100, 2476–2485 10.1172/JCI119790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Han X., Sterling H., Chen Y., Saginario C., Brown E. J., Frazier W. A., Lindberg F. P., and Vignery A. (2000) CD47, a ligand for the macrophage fusion receptor, participates in macrophage multinucleation. J. Biol. Chem. 275, 37984–37992 10.1074/jbc.M002334200 [DOI] [PubMed] [Google Scholar]
- 12. Xing L., Xiu Y., and Boyce B. F. (2012) Osteoclast fusion and regulation by RANKL-dependent and independent factors. World J. Orthop. 3, 212–222 10.5312/wjo.v3.i12.212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Helming L., Winter J., and Gordon S. (2009) The scavenger receptor CD36 plays a role in cytokine-induced macrophage fusion. J. Cell Sci. 122, 453–459 10.1242/jcs.037200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Isenberg J. S., Jia Y., Fukuyama J., Switzer C. H., Wink D. A., and Roberts D. D. (2007) Thrombospondin-1 inhibits nitric oxide signaling via CD36 by inhibiting myristic acid uptake. J. Biol. Chem. 282, 15404–15415 10.1074/jbc.M701638200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kasten T. P., Collin-Osdoby P., Patel N., Osdoby P., Krukowski M., Misko T. P., Settle S. L., Currie M. G., and Nickols G. A. (1994) Potentiation of osteoclast bone-resorption activity by inhibition of nitric oxide synthase. Proc. Natl. Acad. Sci. U.S.A. 91, 3569–3573 10.1073/pnas.91.9.3569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Collin-Osdoby P., Rothe L., Bekker S., Anderson F., and Osdoby P. (2000) Decreased nitric oxide levels stimulate osteoclastogenesis and bone resorption both in vitro and in vivo on the chick chorioallantoic membrane in association with neoangiogenesis. J. Bone Miner. Res. 15, 474–488 10.1359/jbmr.2000.15.3.474 [DOI] [PubMed] [Google Scholar]
- 17. Brandi M. L., Hukkanen M., Umeda T., Moradi-Bidhendi N., Bianchi S., Gross S. S., Polak J. M., and MacIntyre I. (1995) Bidirectional regulation of osteoclast function by nitric oxide synthase isoforms. Proc. Natl. Acad. Sci. U.S.A. 92, 2954–2958 10.1073/pnas.92.7.2954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Amend S. R., Uluckan O., Hurchla M., Leib D., Novack D. V., Silva M., Frazier W., and Weilbaecher K. N. (2015) Thrombospondin-1 regulates bone homeostasis through effects on bone matrix integrity and nitric oxide signaling in osteoclasts. J. Bone Miner. Res. 30, 106–115 10.1002/jbmr.2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Maile L. A., DeMambro V. E., Wai C., Lotinun S., Aday A. W., Capps B. E., Beamer W. G., Rosen C. J., and Clemmons D. R. (2011) An essential role for the association of CD47 to SHPS-1 in skeletal remodeling. J. Bone Miner. Res. 26, 2068–2081 10.1002/jbmr.441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kevorkova O., Martineau C., Martin-Falstrault L., Sanchez-Dardon J., Brissette L., and Moreau R. (2013) Low-bone-mass phenotype of deficient mice for the cluster of differentiation 36 (CD36). PLoS One 8, e77701 10.1371/journal.pone.0077701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Uluçkan O., Becker S. N., Deng H., Zou W., Prior J. L., Piwnica-Worms D., Frazier W. A., and Weilbaecher K. N. (2009) CD47 regulates bone mass and tumor metastasis to bone. Cancer Res. 69, 3196–3204 10.1158/0008-5472.CAN-08-3358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Koskinen C., Persson E., Baldock P., Stenberg Å., Bostrom I., Matozaki T., Oldenborg P. A., and Lundberg P. (2013) Lack of CD47 impairs bone cell differentiation and results in an osteopenic phenotype in vivo due to impaired signal regulatory protein α (SIRPα) signaling. J. Biol. Chem. 288, 29333–29344 10.1074/jbc.M113.494591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bailey Dubose K., Zayzafoon M., and Murphy-Ullrich J. E. (2012) Thrombospondin-1 inhibits osteogenic differentiation of human mesenchymal stem cells through latent TGF-β activation. Biochem. Biophys. Res. Commun. 422, 488–493 10.1016/j.bbrc.2012.05.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hoosdally S. J., Andress E. J., Wooding C., Martin C. A., and Linton K. J. (2009) The human scavenger receptor CD36: glycosylation status and its role in trafficking and function. J. Biol. Chem. 284, 16277–16288 10.1074/jbc.M109.007849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Marzia M., Sims N. A., Voit S., Migliaccio S., Taranta A., Bernardini S., Faraggiana T., Yoneda T., Mundy G. R., Boyce B. F., Baron R., and Teti A. (2000) Decreased c-Src expression enhances osteoblast differentiation and bone formation. J. Cell Biol. 151, 311–320 10.1083/jcb.151.2.311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dixit V. M., Haverstick D. M., O'Rourke K. M., Hennessy S. W., Grant G. A., Santoro S. A., and Frazier W. A. (1985) Effects of anti-thrombospondin monoclonal antibodies on the agglutination of erythrocytes and fixed, activated platelets by purified thrombospondin. Biochemistry 24, 4270–4275 10.1021/bi00337a003 [DOI] [PubMed] [Google Scholar]
- 27. Febbraio M., Abumrad N. A., Hajjar D. P., Sharma K., Cheng W., Pearce S. F., and Silverstein R. L. (1999) A null mutation in murine CD36 reveals an important role in fatty acid and lipoprotein metabolism. J. Biol. Chem. 274, 19055–19062 10.1074/jbc.274.27.19055 [DOI] [PubMed] [Google Scholar]
- 28. Akatsu T., Tamura T., Takahashi N., Udagawa N., Tanaka S., Sasaki T., Yamaguchi A., Nagata N., and Suda T. (1992) Preparation and characterization of a mouse osteoclast-like multinucleated cell population. J. Bone Miner. Res. 7, 1297–1306 10.1002/jbmr.5650071109 [DOI] [PubMed] [Google Scholar]
- 29. Kawano T., Zhu M., Troiano N., Horowitz M., Bian J., Gundberg C., Kolodziejczak K., and Insogna K. (2013) LIM kinase 1 deficient mice have reduced bone mass. Bone 52, 70–82 10.1016/j.bone.2012.09.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhu M., Sun B. H., Saar K., Simpson C., Troiano N., Dallas S. L., Tiede-Lewis L. M., Nevius E., Pereira J. P., Weinstein R. S., Tommasini S. M., and Insogna K. L. (2016) Deletion of Rac in mature osteoclasts causes osteopetrosis, an age-dependent change in osteoclast number, and a reduced number of osteoblasts in vivo. J. Bone Miner. Res. 31, 864–873 10.1002/jbmr.2733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Insogna K. L., Stewart A. F., Vignery A. M., Weir E. C., Namnum P. A., Baron R. E., Kirkwood J. M., Deftos L. M., and Broadus A. E. (1984) Biochemical and histomorphometric characterization of a rat model for humoral hypercalcemia of malignancy. Endocrinology 114, 888–896 10.1210/endo-114-3-888 [DOI] [PubMed] [Google Scholar]
- 32. Yao G. Q., Wu J. J., Ovadia S., Troiano N., Sun B. H., and Insogna K. (2009) Targeted overexpression of the two colony-stimulating factor-1 isoforms in osteoblasts differentially affects bone loss in ovariectomized mice. Am. J. Physiol. Endocrinol. Metab. 296, E714–E720 10.1152/ajpendo.90631.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Parfitt A. M., Drezner M. K., Glorieux F. H., Kanis J. A., Malluche H., Meunier P. J., Ott S. M., and Recker R. R. (1987) Bone histomorphometry: standardization of nomenclature, symbols, and units. Report of the ASBMR Histomorphometry Nomenclature Committee. J. Bone Miner. Res. 2, 595–610 10.1002/jbmr.5650020617 [DOI] [PubMed] [Google Scholar]
- 34. Dempster D. W., Compston J. E., Drezner M. K., Glorieux F. H., Kanis J. A., Malluche H., Meunier P. J., Ott S. M., Recker R. R., and Parfitt A. M. (2013) Standardized nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J. Bone Miner. Res. 28, 2–17 10.1002/jbmr.1805 [DOI] [PMC free article] [PubMed] [Google Scholar]