

Abstract
Colloidal dispersions of cellulose nanofibrils (CNFs) are viable alternatives to cellulose II dissolutions used for filament spinning. The porosity and water vapor affinity of CNF filaments make them suitable for controlled breathability. However, many textile applications also require water repellence. Here, we investigated the effects of postmodification of wet-spun CNF filaments via chemical vapor deposition (CVD). Two organosilanes with different numbers of methyl substituents were considered. Various surface structures were achieved, either as continuous, homogeneous coating layers or as three-dimensional, hairy-like assemblies. Such surface features reduced the surface energy, which significantly affected the interactions with water. Filaments with water contact angles of up to 116° were obtained, and surface energy measurements indicated the possibility of developing amphiphobicity. Dynamic vapor sorption and full immersion experiments were carried out to inquire about the interactions with water, whether in the liquid or gas forms. Mechanical tests revealed that the wet strength of the modified filaments were almost 3 times higher than that of the unmodified precursors. The hydrolytic and mechanical stabilities of the adsorbed layers were also revealed. Overall, our results shed light on the transformation of aqueous dispersions of CNFs into filaments that are suited for controlled interactions with water via concurrent hydrolysis and condensation reactions in CVD, while maintaining the moisture buffering capacity and breathability of related structures.
Keywords: cellulose nanofibrils, filaments, surface structuring, hydrophobization, organosilanes, chemical vapor deposition, wet spinning
Introduction
The scarcity of fossil resources, climate change, and sustainability are some of the current megatrends that have triggered a renewed interest in biobased materials. Within this category, cellulosic materials occupy a prime position owing to the unique properties of cellulose, which is widely abundant in nature. For example, the high moisture sorption capacity of cellulose1−3 can be beneficial in wearable textiles, which are often designed to optimize breathability. Conventionally, cellulosic textiles are made of native or regenerated cellulose fibers. More recently, alternative methods have been introduced for obtaining textile filaments from cellulose nanofibrils (CNFs) through spinning.4−17 Compared to filaments from native and regenerated cellulose, those from CNFs have the potential to develop advanced moisture sorption profiles, given their high surface area and colloidal size, which can be exploited to obtain an optimal porosity, depending on the spinning conditions.
In addition to breathability, repellence to liquid water is desired, especially in wearable textiles for use outdoors. Conventionally, water-repellent but breathable fabrics are mainly produced using microporous or hydrophilic membranes or coatings or such closely woven structures that only individual water molecules can pass through.18,19 Often, these approaches rely on synthetic fibers as well as membranes or coatings, that may include fluorinated polymers.18,19 As a more environmentally sound option, nonfluorinated organosilanes have been shown to be effective for hydrophobizing surfaces.20 Organosilane modification through a solvent-free process has also been demonstrated on cellulosic substrates.21−23
Earlier, we reported highly moisture-sorbing filaments wet-spun from CNFs.10 Herein, we report a solvent-free chemical modification of CNF filaments by using nonfluorinated organosilanes. We show how this approach can tailor the surface chemistry and morphology of the filaments, turning them water-repellent although still maintaining their moisture sorption capacity.
Experimental Section
Materials
Never-dried bleached birch kraft wood fibers were refined in a Voith LR 40 laboratory refiner for 12 min at a solids content of 3 wt % and a refiner speed of 200 rpm, using a specific edge load of 0.5 J·m–1, a net specific energy of 294 kWh·t–1, and a net refining power of 1.39 kW. The refined wood fibers were fluidized for six passes through a high-pressure microfluidizer (Microfluidics Corp., Newton, MA) at a solids content of 2 wt %. CNFs from the same fiber source and prepared by the same protocols were imaged via atomic force microscopy and reported earlier by us.10 Polydispersed fibril sizes were determined with an average thickness of ∼5 nm. Prior to the experiments, the obtained pristine CNF hydrogel was ultracentrifuged to obtain suspensions of 5 wt % solids. On the basis of our previous experience, this solids content produced the most flexible filaments (strain at break >8%),10 which is a desirable property in the design of wearable materials.
Trichloromethylsilane (TC), dimethyldichlorosilane (DC), formamide, diiodomethane, and potassium sulfate were purchased from Sigma-Aldrich and used as received.
Wet Spinning of the CNF Hydrogel
The CNF hydrogel (5 wt % solids) was wet-spun through a needle (diameter of 1.3 mm and length of 10.5 cm) into an acetone bath at a speed of 7.5 m·min–1 (10 mL·min–1). The spun filaments were coagulated in the bath for approximately 5 min. After coagulation, they were dried in air with both ends fixed in order to prevent longitudinal contraction. The diameters of the filaments thus prepared were in the range 150–250 μm. Optical micrographs of the filaments were acquired using a Leica DM750 optical microscope operating under ambient conditions.
Hydrophobization of CNF Filaments
The surface of the CNF filaments was modified by a simple chemical vapor deposition (CVD) using either TC or DC (three or two chlorine substituents, respectively; Figure 1). In brief, filaments cut into ca. 2 cm lengths (weighing about 6 mg in total) were placed in a small Teflon dish and then introduced to a large container. A given volume of organosilane, ranging from 5 to 20 μL (Table 1), was then placed in a small aluminum cup inside the same container. Subsequently, the container was closed with a lid and put in a water bath at 55 °C for 30 min. At the end, the Teflon dish containing the filaments was placed inside a desiccator, which was then connected to a water pump for 30 min. This latter procedure removed any excess of reagent, and HCl formed as a byproduct of the chemical modification. The identification of the filaments prepared is given in Table 1.
Figure 1.
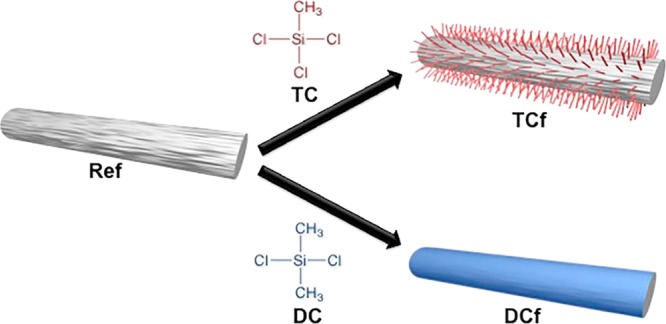
Schematic illustration of the chemical modification of a CNF filament with TC and DC.
Table 1. Identification of the Filament Samples Prepared in This Work.
| volume of organosilane (μL) | TC | DC |
|---|---|---|
| 0 | ref | ref |
| 5 | TCf5 | DCf5 |
| 10 | TCf10 | DCf10 |
| 15 | TCf15 | DCf15 |
| 20 | TCf20 | DCf20 |
As a supplementary test, CNF filaments were successively modified with both organosilanes using the same setup mentioned above but following two different routes: (i) filaments were first modified with 10 μL of TC and then 10 μL of DC; (ii) filaments were first modified with 10 μL of DC and then 10 μL of TC.
Spectroscopy Analyses
Fourier transform infrared (FTIR) spectra were recorded using a Nicolet 380 FTIR spectrometer, equipped with a Smart Orbit single-reflection attenuated-total-reflectance (ATR) diamond system. The acquisition conditions were 32 scans and 8 cm–1 resolution.
X-ray photoelectron spectroscopy (XPS) was applied for surface chemical analysis of the filaments. The data were recorded with an AXIS Ultra electron spectrometer (Kratos Analytical) and analyzed with CasaXPS software. Samples were mounted with carbon tape on the sample holder as a filament bundle and preevacuated overnight to ensure stable ultrahigh-vacuum conditions. Measurements were performed at several locations, but away from the end of the filaments, using low-dose monochromated Al Kα irradiation at 100 W; no sample damage was observed during the measurements. The analysis area in this setup was nominally 400 × 800 μm, and the XPS analysis depth was less than 10 nm. Elemental surface compositions were determined from low-resolution survey spectra, while high-resolution scans of C 1s were utilized for a more detailed chemical analysis. A pure cellulose specimen was used as reference.
Field-Emission Scanning Electron Microscopy (FE-SEM)
FE-SEM was conducted with a JEOL JSM-7500FA high-resolution microscope operating at 2 kV. The dry filaments were fixed on metal stubs using carbon tape and coated with a ca. 4 nm layer of a gold/palladium alloy using a LEICA EM ACE600 sputter coater.
Contact-Angle Measurements
Contact angles of water, formamide, and diiodomethane on the CNF filaments were measured using a Sigma 70 force tensiometer, equipped with a COHU solid-state CCD monochrome camera, according to the dynamic Wilhelmy method. The contact angle was calculated from the measured forces and the known geometry (perimeter) of the filament and surface tension of the testing liquid. For this purpose, filament probes were brought vertically into contact with a reservoir containing the given liquids. The tensiometer recorded the change in forces Ftotal from the moment the solid filament first contacted the liquid (zero depth of immersion) until a given depth of immersion (1 mm):
| 1 |
where Fw is the wetting force, Fp is the weight of the probe, and Fb is the buoyancy. Because the unit tare is the weight of the probe and one can remove the buoyancy effects (by extrapolating the force–distance plot to zero depth of immersion), the determined wetting force can be used to calculate the contact angle θ according to eq 2:
| 2 |
where γL stands for the liquid’s surface tension (Table 2) and P is the perimeter of the filament, determined assuming a round cross section. Contact angles were obtained for a minimum of eight filaments, and the average values reported have an associated error in the range 1–10°. On the basis of the contact angles measured with the three different liquids, the surface energy was calculated following the Owens–Wendt geometric mean equation (3):
| 3 |
where γLD and γL are the dispersive and polar components of the liquid’s surface tension, respectively. Similarly, γSD is the dispersive and γS the polar component of the surface energy of the solid. γL, γLD, and γL of the applied liquids are compiled in Table 2. Because these parameters are known and θ was measured for three different liquids, γSP and γS were solved from eq 3, and their sum was used to report the total surface energy of the solid, γS.
Table 2. Surface Tension and Dispersive and Polar Components of the Liquids Used To Determine the Contact Angle and Surface Energy of the Filaments.
| liquid | γL (mJ·m–2) | γLD (mJ·m–2) | γLP (mJ·m–2) |
|---|---|---|---|
| water | 72.8 | 21.8 | 51 |
| formamide | 58.0 | 39 | 19 |
| diiodomethane | 50.8 | 50.8 | 0 |
Dynamic Vapor Sorption (DVS)
Water sorption isotherms were determined with a DVS intrinsic apparatus (Surface Measurement Systems, London, U.K.). CNF filaments were cut into ca. 5 mm pieces to fit into the sample pan, which was loaded with approximately 4 mg of the sample and hung from a microbalance in a climate-controlled chamber. The relative humidity (RH) inside the chamber was first decreased to 0% until the sample weight stabilized (change in mass below 0.002%·min–1 over a period of 10 min). After this, the RH was increased stepwise up to 95%, and the equilibrium moisture content was recorded at RH = 10, 20, 30, 40, 50, 60, 70, 80, 90, and 95%.
Mechanical Testing
The mechanical properties of the filaments were measured on an Instron 5944 (500 N load cell). Prior to testing in dry conditions, the ends of the filaments (with ca. 24 mm length) were stuck on thick pieces of paper using an epoxy glue and then conditioned for 48 h at 23 °C and RH = 50%. The filaments were tested in tensile mode using a gauge length of 12 mm and a strain rate of 10%·min–1. The measurements in wet conditions were performed in a similar way, except that prior to testing the filaments were soaked in water for 1 h (note: the surface-modified filaments were forced to sink because otherwise they would remain on the surface). The gauge length varied in the range 10–12 mm. The diameters of the filaments were assessed with an optical microscope, and three or four specimens of each type of filament were measured.
Stability Tests
For the dimensional stability tests, cuts (∼2.5 cm) of unmodified and modified filaments (with 20 μL of organosilanes) were placed in a small Teflon dish and then inserted into a container with a saturated K2SO4 salt solution at 25 °C, which was tightly closed to attain RH = 97%. The filaments were kept under these conditions for up to 1 week. The diameters of the filaments were measured by optical microscopy images before and after 24 h and 1 week in the humid environment. For the hydrolytic stability test, pieces (∼2.5 cm) of unmodified and modified filaments (with 20 μL of organosilanes) were placed in a plastic container with 15 mL of deionized water and sonicated three times over 30 s at 50% amplitude. For the mechanical stability test, pieces (∼2.5 cm) of unmodified and modified filaments (with 20 μL of organosilanes) were manually rubbed with tweezers 10 times and then rotated 90° and rubbed again 10 more times. The surface morphologies of both the initial and rubbed filaments were analyzed using FE-SEM.
Results and Discussion
Surface Chemistry and Morphology of the Filaments
The organosilane gas–solid reaction via CVD was effective for surface modification of the filaments by condensation reactions, as has been reported between cellulose surface hydroxyl groups and Si–Cl moieties.21 Any in-depth change was assumed to be insignificant. The variables studied included the volume of organosilane used in CVD, which ranged from 0 to 20 μL (Table 1). The success of the chemical modification was assessed by ATR-FTIR by monitoring the emergence of bands at ∼1260 and ∼800 cm–1, attributed to the Si–CH3 symmetric deformation and rocking modes, respectively (Figure 2).24 As shown in Figure 2a, the extent of surface modification for TC-treated samples increased with the volume of the organosilane used. Such an increase was less noticeable in the for DC-modified filaments (Figure 2b).
Figure 2.
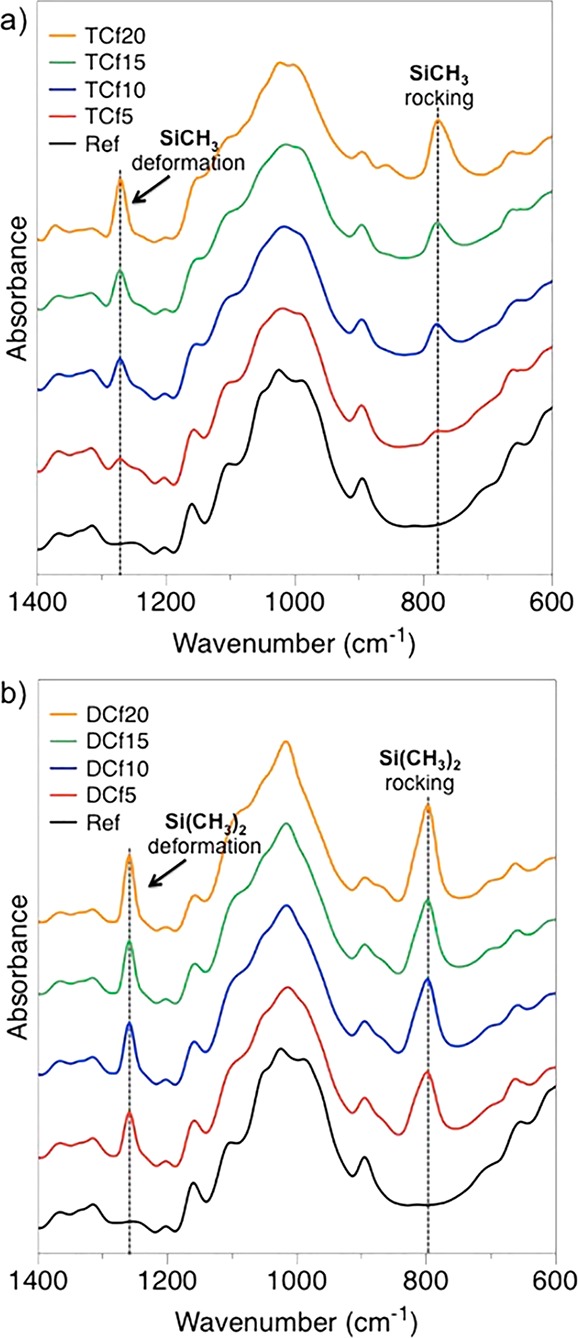
ATR-FTIR spectra of the CNF filaments modified with (a) trichloromethylsilane (TCf) and (b) dimethyldichlorosilane (DCf) in comparison to the unmodified filaments (ref).
The typical vibration of Si–O–C moieties, arising from coupling between the organosilane reagents and cellulose OH groups, usually in the range 1080–1110 cm–1,24 was not readily detected by ATR-FTIR because it overlaps with the large and intense cellulose C–O stretching band centered at ca. 1026 cm–1.24 Likewise, the vibration frequencies of silanol moieties (Si–OH) and Si–O–Si bridges, resulting from moisture-induced hydrolysis and condensation of the Si–Cl functions, typically around 950 and 1100 cm–1, respectively,24 could not be detected.
The surfaces of both unmodified and modified filaments were characterized with XPS using pure cellulose as an in situ reference. A summary of the XPS results is included in Figure 3. In the case of unmodified filaments, the cellulose signatures of C–O and O–C–O (originating from carbon atoms with one and two oxygen neighbors at 286.7 and 288 eV) were strong and well-resolved in the high-resolution C 1s signal.25 The noncellulosic component in C 1s, i.e., C–C, consisting of carbon atoms without oxygen neighbors was also significant in the wet-spun CNF filaments compared with the 100% cellulose reference. This may be a result of the spinning process; however, this kind of surface passivation has been frequently observed for CNFs because carbonaceous surface layers form when the highly hydrophilic nanocellulose surface is exposed to a dry environment.26 In the case of the modified filaments, the organosilane treatments completely changed the surface chemistry. In wide spectra, strong silicon intensities (Si 2p and Si 2s) were observed in addition to the C 1s and O 1s signals. A detailed analysis revealed that the binding energy of Si 2p agreed well with that for silanes.25 Furthermore, the high-resolution data for carbon indicated that the cellulosic component became negligible after the 5 μL organosilane treatment and fully vanished for samples treated with 20 μL of organosilane. Instead, the CC and CSi components dominated the C 1s region for all spectra of the modified filaments.
Figure 3.
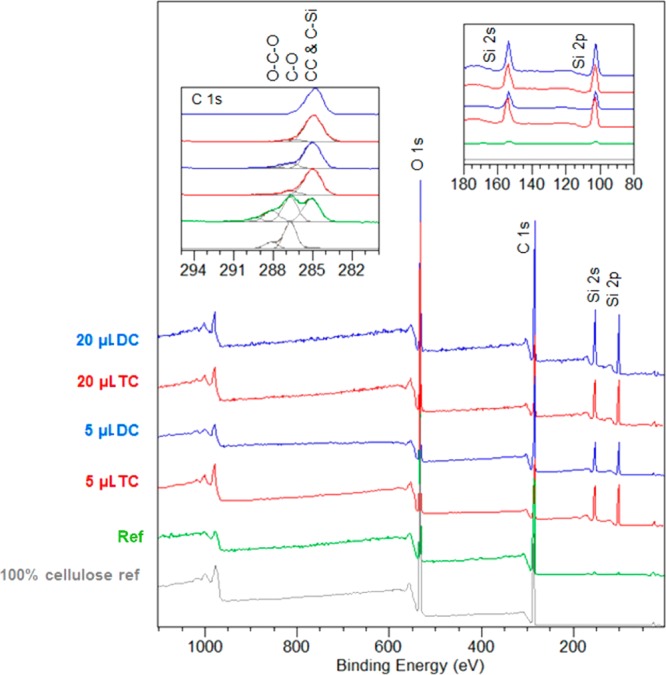
Normalized XPS wide spectra for unmodified and silane-treated CNF filaments, together with a pure cellulose reference. Insets: component-fitted C 1s high-resolution regions (left) and magnified image of the wide spectra at the Si 2s and 2p regions.
The presence of silane-containing moieties on the modified filaments was confirmed by thermogravimetric analysis. A slightly more complex degradation behavior in the thermograms of the modified filaments was observed compared to that of the unmodified sample. The changes were attributed to decomposition of the installed methyl groups. In addition, the modified filaments yielded a higher residual mass, ascribed to the inorganic component present on the surface (Figure S1).
The surface morphology of the filaments was evaluated using FE-SEM. The micrographs of the unmodified filament (Figure 4a,d) showed a surface morphology that included “longitudinal fissures” of micrometer scales. These fissures had a preferential axial orientation, induced by the spinning process, as was already observed in previous studies.5,10 After chemical modification with TC, hairy features, not thicker than 1 μm and as long as 40 μm, emerged on the surface of the filaments (Figure 4b,e). These likely resulted from the concurrent hydrolysis and condensation reactions typical of reactive silane compounds, which led to the formation of three-dimensional structures.20 These hairy features comprise Si–O–Si tridimensional networks formed by TC self-assembly and vertical polymerization.21 Such features were reported earlier for other systems.27,28 For instance, a study dealing with the superhydrophobization of textile materials with TC indicated the appearance of hairy features, therein described as nanofilaments.27 Within a different scope, Korhonen et al.28 implemented a controlled system to grow hollow polysiloxane nanofilaments from silicon wafer surfaces using TC as the precursor and provided detailed insight into the growing mechanisms.
Figure 4.

SEM micrographs at different magnifications of unmodified and organosilane-treated CNF filaments: (a and d) ref, (b and e) TCf20, and (c and f) DCf20 at (a–c) 100× and (d–f) 500× magnification (see Table 1 for the nomenclature).
After chemical modification with DC, the surface morphology of the filaments became smoother compared to that of the precursor filament (Figure 4c,f). This observation agrees with a linear polymerization of DC moieties, leading to a two-dimensional coating, rather than the three-dimensional structures observed for TC.20 It is well-known that DC is one of the precursors of poly(dimethylsiloxane) (PDMS), which, unless cross-linked, is a fluid substance. This suggests that a freshly polymerized PDMS-like coating can, in principle, cover the filament surface and mold to (or fill-in) “surface imperfections”, in this particular case the “longitudinal fissures”, thus smoothing the surface. Additionally, a magnified image of the surface revealed signs of a wrinklelike patterning (Figure 4f). This may be an artifact derived from the interaction of the strong electron beam from SEM with the soft PDMS-like coating on the DCf samples. In fact, previous studies have shown that a stiff skin can be formed on the surface of PDMS-based materials upon exposure to a focused ion beam, leading to surface wrinkles.29
The reagent amount did not seem to influence significantly the surface morphology of the DC-modified filaments (homogeneous coatings in all samples), contrary to the case of TC. This is in line with the chemical fingerprints assessed by ATR-FTIR. Accordingly, samples modified with the lowest amount of TC (TCf5) held a few randomly distributed areas containing short hairy features, while those modified with the highest amount (TCf20) displayed a more extended surface coverage with, overall, longer hairy features (Figure S2). However, the surface coverage in the latter case was still not complete or as homogeneous as that for DC-modified filaments.
Clearly, when the type of organosilane is changed, distinctively different surface morphologies were obtained on the CNF filaments. To further explore this observation, the two organosilanes were consecutively deposited on the surface of the filaments (CVD) and the order of deposition was tested. The effect of such an approach on the surface morphology of the filaments is shown in Figure 5. When chemical deposition of TC preceded that of DC, the surface appeared irregular, owing to the deposition of a continuous fluidlike coating onto the hairy features previously grown from the surface of the CNF filament. When the reverse modification protocol was applied, the hairy structures seemed to be shorter, more individualized, and more homogeneously spread on the surface of the filament (which was initially turned smooth via DC deposition).
Figure 5.

SEM micrographs at different magnifications of CNF filaments treated consecutively with the two organosilanes: (a and b) TC followed by DC and (c and d) DC followed by TC at (a and c) 100× and (b and d) 500× magnification. The inset in part c shows a 1000× magnification micrograph.
Interactions with Water
So far, it was demonstrated that the chemical modification of the CNF filaments with different organosilanes led to significant changes, not only on the surface chemistry but also on the surface morphologies. To assess the impact of these changes on the wettability of the filaments, a simple test was carried out. Unmodified and modified filaments were placed in direct contact with liquid water. Whereas the unmodified filament showed the typical behavior of a hydrophilic material and sank immediately, the modified filaments remained atop the water surface (Figure 6a), denoting their hydrophobic character. Moreover, menisci were clearly observed for the modified filaments on water, owing to the effect of surface tension (Figure 6a) and resembling the legs of some insects such as water striders.
Figure 6.

(a) Behavior of unmodified and organosilane-modified CNF filaments in contact with water: ref and TCf20. Contact angles with (b) water and (c) diiodomethane as well as (d) the surface energies of unmodified and organosilane-modified CNF filaments (see Table 1 for nomenclature).
In order to quantify the hydrophobicity of the modified filaments, water contact angles (WCAs) were estimated by means of tensiometry, following the dynamic Wilhelmy method. As depicted in Figure 6b, after chemical modification, the WCA of the filaments became >90°, i.e., increased substantially (by about 30–60°) compared to that of the precursor sample (57°). This confirms not only the hydrophobic character of the modified filaments but also the drastic change that such a simple surface treatment produced. It is important to note that, whereas the WCA seemed not to depend strongly on the volume of DC used (unchanged WCA of modified filaments of ca. 95°), it scaled with the volume of TC used in the respective filament modification, with WCAs of up to 116° (Figure 6b). These results strongly correlate with the SEM observations discussed above and can be attributed to the homogeneity/quality and extent of surface coverage upon chemical modification (Figure 4). Moreover, the observations suggest that variation in the extent of DC coverage did not play an important role in terms of the water wettability because, even with the lowest DC volume (5 μL), a hydrophobic character was attained.
Contact angles were also measured with liquids of different polarities, namely, formamide (γLV= 58.0 mJ·m–2) and diiodomethane (γLV= 50.8 mJ·m–2; Table S1). Contact angles with diiodomethane, the least polar of the three liquids tested, showed a trend similar to that for the WCA. After chemical modification, diiodomethane contact angles increased from 61°, in the unmodified filaments, to up to 100°, in the modified counterparts (Figure 6c). Once more, the effect of organosilane was only noticed for the TC-modified filaments. It must be highlighted that some of the TC-modified filaments (TCf15 and TCf20) showed both hydrophobic and lipophobic (diiodomethane contact angle >90°) character and can thus be categorized as amphiphobic materials.
The values of the total surface energy (γS) of the modified samples were low, in the range 10–20 mJ·m2, compared with ∼50 mJ·m2 for the unmodified filaments (Figure 6d). As observed in Figure 6d, this remarkable reduction in the surface energy was caused mostly by the contribution of the polar component, although the dispersive counterpart also decreased. Similar surface energy values were previously reported for other cellulosic substrates modified with organosilanes.21 The change in the surface properties can be rationalized in terms of the presence of Si–CH3 moieties at the surface of the modified filaments, which are known to promote water repellency20 and, in the specific case of the TC-modified samples, also the emergence of the new methylsiloxane-based hairy features, which provided additional surface roughness. The combination of low surface energy and increased surface roughness explains why, overall, TC-modified samples attained higher hydrophobicity than the DC-modified counterparts.
The equilibrium moisture contents of the unmodified filaments and those modified with 5 and 20 μL organosilanes were also assessed at different RHs by means of DVS measurements. Only the TCf20 sample showed a slight decrease in the moisture content compared to the precursor filament, irrespective of the RH (Figure 7a). The other modified samples presented an equilibrium moisture content very similar to that of the reference sample in the whole range of RHs studied (Figure 7a). The final equilibrium moisture content reached by these filaments at RH = 95% (∼23%) was similar to that reported earlier for unmodified CNF filaments.
Figure 7.

(a) DVS isotherms [equilibrium moisture content (EMC) as a function of the RH] of unmodified and organosilane-modified CNF filaments. (b) Optical micrographs of unmodified and organosilane-modified CNF filaments before and after 24 h and 1 week of exposure to RH = 97%.
From the DVS results, it can be hypothesized that the CNF filament–water interactions highly depend on the physical state of water, whether as a liquid or a gas. Thus, while surface modification with organosilanes containing methyl groups (−CH3) was effective in decreasing the filament affinity with liquid water, it still allowed the filaments to maintain a high level of gas-phase water sorption capacity. A similar behavior has been reported in other contexts.30−35 This is beneficial if the filaments are used in applications, such as outdoor clothing, where they should repel liquid water but retain their breathability.
To understand the changes of filaments in contact with moisture, a dimensional stability study at RH = 97% was performed. As observed in the optical micrographs of Figure 7b, the diameter of the filaments slightly increased after 24 h in contact with moisture; after 1 week, it increased further. However, it is worth noting that the measured diameters may be, in fact, smaller than the actual values because some drying may have occurred during the imaging process. Interestingly, the increase in the diameter was more pronounced for the unmodified filament and the filament modified with DC, with variations in the diameter by 12 and 10 μm, respectively. These results correlate with the DVS data because the filament modified with TC sorbed the least amount of moisture (lowest variation in the diameter), even though from the SEM micrographs it was observed that the surface coverage for these filaments was not as homogeneous as that for the DC-modified counterparts.
It is worth mentioning that the condensation reaction among TC and DC molecules also leads to some polar silanol groups (Si–OH), which may have been engaged in interactions with water. This issue can be investigated further by capping the silanol groups using additional chemical deposition, for example, with a monofunctional organosilane (chain terminator), such as trimethylchlorosilane.20
Mechanical Performance
The mechanical performance of the filaments was assessed by means of uniaxial tensile testing (Table S2). In dry conditions, the tensile strength and Young’s modulus of the unmodified CNF filaments were ca. 160 MPa and 10 GPa, respectively. Similar values were obtained in previous studies.10 After chemical modification with the lowest volume of TC and DC (5 μL), the mechanical properties were maintained, as can be observed in Figure 8a. However, when the amount of organosilane increased to 20 μL, the mechanical performance was slightly compromised, and reductions as high as 35 MPa and 1.5 GPa for the tensile strength and Young’s modulus, respectively, were noted compared to those for the precursor filament (Figure 8a). Thus, it is likely that deterioration of the filaments occurred upon contact with HCl released as a byproduct of the chemical modification.
Figure 8.
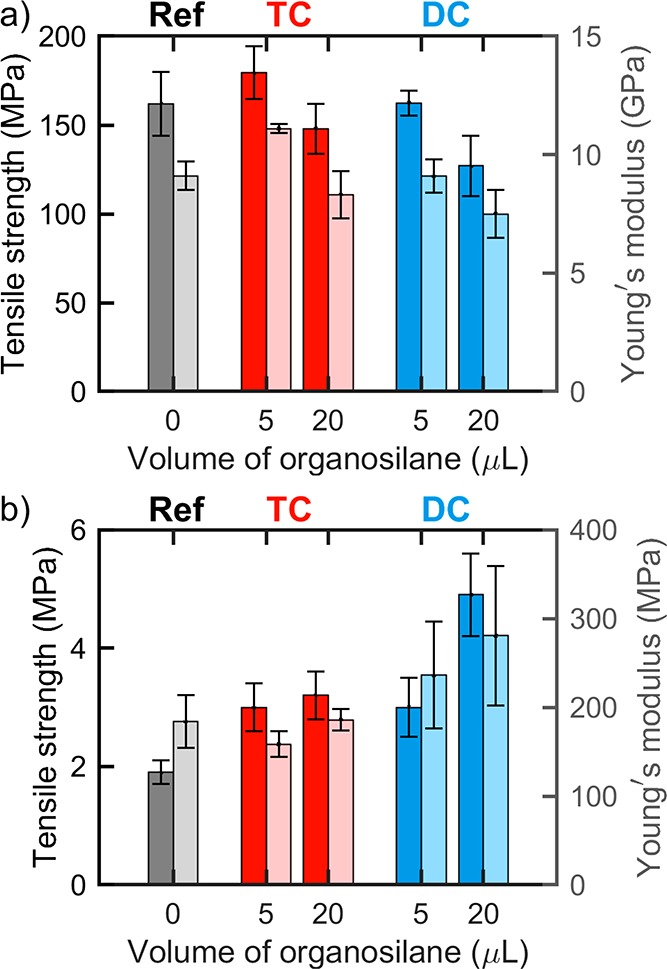
Tensile strength (darker color, left axis) and Young’s modulus (lighter color, right axis) of unmodified and organosilane-modified CNF filaments in (a) dry and (b) wet conditions.
In wet conditions, after the immersion of unmodified filaments in water for 1 h, the mechanical strength was dramatically compromised, and the tensile strength and Young’s modulus of the unmodified filaments decreased to ca. 2 and 185 MPa, respectively (Figure 8b). This represents a significant decline in the mechanical strength (80 and 50 times for the tensile strength and Young’s modulus, respectively). The effect of water was less pronounced for the modified filaments, especially those modified with DC. For instance, compared to the unmodified filament, those modified with 20 μL of DC (DCf20) presented 2.5 and 1.5 times higher wet tensile strength and Young’s modulus (ca. 5 and 280 MPa), respectively. This can be explained by a homogeneous coverage of the hydrophobic layer on the surface of the filaments, as observed by SEM. Again, the physical state of water plays an important role in the filament–water interactions. When exposed to liquid water for a prolonged time, the TC-modified filaments, which generally presented higher hydrophobicity and lower moisture sorption, were the ones that underwent the highest reduction in strength. This can be explained by the fact that their surface coverage was not homogeneous (as assessed by SEM); i.e., they were subjected to more extensive interaction with water via the more accessible and unprotected hydroxyl groups at the surface of the filaments. Conversely, in the DC-modified analogues, the more homogeneous coating hindered (or slowed down) liquid water absorption. The results from the hydrolytic stability presented below corroborate this hypothesis.tensile strength and Young’s modulus (ca. 5 and 280 MPa), respectively. This can be explained by abetter and more homogeneous surface coverage of the filaments, as observed by SEM. Again, the physical state of water plays an important role in the filament–water interactions. When exposed to liquid water for a prolonged time, the TC-modified filaments, which generally presented higher hydrophobicity and lower moisture sorption, were the ones that underwent the highest reduction in strength. This can be explained by the fact that their surface coverage was not homogeneous (as assessed by SEM); i.e., they were subjected to more extensive interaction with water via the more accessible and unprotected hydroxyl groups at the surface of the filaments. Conversely, in the DC-modified analogues, the more homogeneous coating hindered (or slowed down) liquid water absorption. The results from the hydrolytic stability presented below corroborate this hypothesis. better and more homogeneous surface coverage of the filaments
Hydrolytic and Mechanical Stabilities
Both unmodified and modified filaments (with 20 μL of organosilanes) were introduced in a plastic container with water and submitted to sonication to assess their hydrolytic stability. The tests were repeated at least three times, and a reproducible behavior was observed: As depicted in Figure 9, the unmodified filament fragmented into several small pieces, indicating a poor stability in water. In contrast, the DC-modified filament did not suffer fragmentation. An intermediate behavior was observed for the TC-modified filament, which fragmented in half. The observations support the results of mechanical tests carried out in wet conditions. As far as the hydrolytic stability, one needs to consider the benefit of chemical modification and its homogeneity: the surface-homogeneous DC-modified filament and the more hydrophobic but heterogeneous morphology of the TC-modified filaments.
Figure 9.

Photographs of unmodified and organosilane-modified CNF filaments in deionized water (a–c) before and (d–e) after sonication. SEM micrographs at 100× magnification of (g and h) ref, (i and j) TCf20, and (k and l) DCf20 (g, i, and k) before and (h, j, and l) after rubbing (see Table 1 for nomenclature). The insets are micrographs at 500× magnification. The DC-modified filament remained intact after sonication and sunk to the bottom of the container (noted with the arrow added in part f). In contrast, the unmodified filament was fragmented upon sonication (noted by the arrows added in part d).
The mechanical stability of the filaments and, more specifically, the surface-bound organosilane layer was investigated by rubbing the filaments with a pair of tweezers. The surface of unmodified and TC-modified filaments became smoother, suggesting the removal of surface layers, and in the latter case, the hairy features were destroyed (Figure 9h,j). On the other hand, no apparent differences were observed in the morphology of the DC-modified filaments (Figure 9k,l), denoting the better resiliency of the PDMS-like coating on such filaments.
The relatively new types of cellulosic filaments considered here have recently attracted the attention of the cellulose community but have remained challenging as far as their surface properties. The adoption of known modifications has allowed us to highlight their potential for gaining control on the cellulose–water interactions, as shown by the results from a unique combination of experimental approaches. The discussed mechanical and hydrolytic stabilities of the coated filaments and the effect of multiple chemical modification and combination of TC and DC treatments were attempted here for the first time. The results show promise in the utilization of naturally based filaments, for example, in wearable materials, given their hydrophobic but breathable character.
Conclusions
Hydrophobization of wet-spun CNF filaments was achieved by a simple CVD using organosilanes differing in the number of methyl substituents. Hydrophobic DC-modified CNF filaments presented a rather homogeneous surface coverage, characterized by a smooth and soft PDMS-like coating. In contrast, amphiphobic TC-modified filaments included hairy structures self-assembled on their surfaces. Both types of filaments presented improved stability in water but retained a similar level of moisture sorption compared to the unmodified precursors. The results suggest that the modified filaments could function in applications demanding water repellence combined with breathability, such as wearable designs for outdoor use. An appropriate level of wet strength remains a challenge for CNF-based filaments.
Acknowledgments
This work has been funded by Business Finland through a strategic opening entitled Design Driven Value chains in the World of Cellulose and supported by the Academy of Finland’s Centers of Excellence program (Project 264677, HYBER). This work made use of the facilities of Aalto University’s Nanomicroscopy Center. The authors acknowledge Dr. Lauri Rautkari for discussions regarding DVS analysis.
Glossary
Abbreviations
- ATR-FTIR
attenuated-total-reflectance Fourier transform infrared
- CNF
cellulose nanofibrils
- CVD
chemical vapor deposition
- DC
dimethyldichlorosilane
- DCf
dimethyldichlorosilane-functionalized filament
- DVS
dynamic vapor sorption
- EMC
equilibrium moisture content
- FE-SEM
field-emission scanning electron microscopy
- PDMS
poly(dimethylsiloxane)
- RH
relative humidity
- TC
trichloromethylsilane
- TCf
trichloromethylsilane-functionalized filament
- WCA
water contact angle
- XPS
X-ray photoelectron spectroscopy
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsanm.8b01268.
Thermogravimetric analysis, SEM micrographs of filaments modified with different amounts of organosilanes, contact angles with different liquids, and tensile testing results (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Klemm D.; Kramer F.; Moritz S.; Lindström T.; Ankerfors M.; Gray D.; Dorris A. Nanocelluloses: A New Family of Nature-Based Materials. Angew. Chem., Int. Ed. 2011, 50, 5438–5466. 10.1002/anie.201001273. [DOI] [PubMed] [Google Scholar]
- Moon R.; Martini A.; Nairn J.; Simonsen J.; Youngblood J. Cellulose Nanomaterials Review: Structure, Properties and Nanocomposites. Chem. Soc. Rev. 2011, 40, 3941–3994. 10.1039/c0cs00108b. [DOI] [PubMed] [Google Scholar]
- Benitez A.; Torres-Rendon J.; Poutanen M.; Walther A. Humidity and Multiscale Structure Govern Mechanical Properties and Deformation Modes in Films of Native Cellulose Nanofibrils. Biomacromolecules 2013, 14, 4497–4506. 10.1021/bm401451m. [DOI] [PubMed] [Google Scholar]
- Iwamoto S.; Isogai A.; Iwata T. Structure and Mechanical Properties of Wet-Spun Fibers Made from Natural Cellulose Nanofibers. Biomacromolecules 2011, 12, 831–836. 10.1021/bm101510r. [DOI] [PubMed] [Google Scholar]
- Walther A.; Timonen J. V. I; Díez I.; Laukkanen A.; Ikkala O. Multifunctional High-Performance Biofibers Based on Wet-Extrusion of Renewable Native Cellulose Nanofibrils. Adv. Mater. 2011, 23, 2924–2928. 10.1002/adma.201100580. [DOI] [PubMed] [Google Scholar]
- Håkansson K. M. O.; Fall A. B.; Lundell F.; Yu S.; Krywka C.; Roth S. V.; Santoro G.; Kvick M.; Prahl Wittberg L.; Wågberg L.; Söderberg D. Hydrodynamic Alignment and Assembly of Nanofibrils Resulting in Strong Cellulose Filaments. Nat. Commun. 2014, 5, 4018. 10.1038/ncomms5018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Rendon J. G.; Schacher F. H.; Ifuku S.; Walther A. Mechanical Performance of Macrofibers of Cellulose and Chitin Nanofibrils Aligned by Wet-Stretching: A Critical Comparison. Biomacromolecules 2014, 15, 2709–2717. 10.1021/bm500566m. [DOI] [PubMed] [Google Scholar]
- Hooshmand S.; Aitomäki Y.; Norberg N.; Mathew A. P.; Oksman K. Dry-Spun Single-Filament Fibers Comprising Solely Cellulose Nanofibers from Bioresidue. ACS Appl. Mater. Interfaces 2015, 7, 13022–13028. 10.1021/acsami.5b03091. [DOI] [PubMed] [Google Scholar]
- Mertaniemi H.; Escobedo-Lucea C.; Sanz-Garcia A.; Gandía C.; Mäkitie A.; Partanen J.; Ikkala O.; Yliperttula M. Human Stem Cell Decorated Nanocellulose Threads for Biomedical Applications. Biomaterials 2016, 82, 208–220. 10.1016/j.biomaterials.2015.12.020. [DOI] [PubMed] [Google Scholar]
- Lundahl M. J.; Cunha A. G.; Rojo E.; Papageorgiou A. C.; Rautkari L.; Arboleda J. C.; Rojas O. J. Strength and Water Interactions of Cellulose I Filaments Wet-Spun from Cellulose Nanofibril Hydrogels. Sci. Rep. 2016, 6, 30695. 10.1038/srep30695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y.; Orelma H.; Sneck A.; Kataja K.; Salmela J.; Qvintus P.; Suurnäkki A.; Harlin A. High Velocity Dry Spinning of Nanofibrillated Cellulose (CNF) Filaments on an Adhesion Controlled Surface with Low Friction. Cellulose 2016, 23, 3393–3398. 10.1007/s10570-016-1044-5. [DOI] [Google Scholar]
- Mohammadi P.; Toivonen M. S.; Ikkala O.; Wagermaier W.; Linder M. B. Aligning Cellulose Nanofibril Dispersions for Tougher Fibers. Sci. Rep. 2017, 7, 11860. 10.1038/s41598-017-12107-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuoriluoto M.; Orelma H.; Lundahl M.; Borghei M.; Rojas O. J. Filaments with Affinity Binding and Wet Strength can be Achieved by Spinning Bifunctional Cellulose Nanofibrils. Biomacromolecules 2017, 18, 1803–1813. 10.1021/acs.biomac.7b00256. [DOI] [PubMed] [Google Scholar]
- Ghasemi S.; Tajvidi M.; Bousfield D. W.; Gardner D. J.; Gramlich W. M. Dry-Spun Neat Cellulose Nanofibril Filaments: Influence of Drying Temperature and Nanofibril Structure on Filament Properties. Polymers 2017, 9, 392. 10.3390/polym9090392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Huang S.; Lu X.; Xu Z.; Zhao Y.; Li J.; Wang X. Wet-Spinning of Highly Conductive Nanocellulose-Silver Fibers. J. Mater. Chem. C 2017, 5, 9673–9679. 10.1039/C7TC03217J. [DOI] [Google Scholar]
- Clemons C. Nanocellulose in Spun Continuous Fibers: A Review and Future Outlook. J. Renewable Mater. 2016, 4, 327–339. 10.7569/JRM.2016.634112. [DOI] [Google Scholar]
- Lundahl M. J.; Klar V.; Wang L.; Ago M.; Rojas O. J. Spinning of Cellulose Nanofibrils into Filaments: A Review. Ind. Eng. Chem. Res. 2017, 56, 8–19. 10.1021/acs.iecr.6b04010. [DOI] [Google Scholar]
- Holmes D. A.Waterproof Breathable Fabrics. Handbook of Technical Textiles; Woodhead Publishing: Cambridge, U.K., 2000; pp 282–315. [Google Scholar]
- Mukhopadhyay A.; Midha V. K. A Review on Designing the Waterproof Breathable Fabrics Part I: Fundamental Principles and Designing Aspects of Breathable Fabrics. J. Ind. Text. 2008, 37, 225–262. 10.1177/1528083707082164. [DOI] [Google Scholar]
- Gao L.; McCarthy T. J. Wetting 101°. Langmuir 2009, 25, 14105–14115. 10.1021/la902206c. [DOI] [PubMed] [Google Scholar]
- Cunha A. G.; Freire C.; Silvestre A.; Neto C. P.; Gandini A.; Belgacem M. N.; Chaussy D.; Beneventi D. Preparation of Highly Hydrophobic and Lipophobic Cellulose Fibers by a Straightforward Gas-Solid Reaction. J. Colloid Interface Sci. 2010, 344, 588–595. 10.1016/j.jcis.2009.12.057. [DOI] [PubMed] [Google Scholar]
- Johansson L.-S.; Campbell J. M. Reproducible XPS on Biopolymers: Cellulose Studies. Surf. Interface Anal. 2004, 36, 1018–1022. 10.1002/sia.1827. [DOI] [Google Scholar]
- Owens D. K.; Wendt R. C. Estimation of the Surface Free Energy of Polymers. J. Appl. Polym. Sci. 1969, 13, 1741–1747. 10.1002/app.1969.070130815. [DOI] [Google Scholar]
- Bellamy L. J.The Infrared Spectra of Complex Molecules, 3rd ed.; Chapman and Hall: London, 1975. [Google Scholar]
- Beamson G.; Briggs D. High Resolution XPS of Organic Polymers: The Scienta ESCA300 Database. J. Chem. Educ. 1993, 70, A25. 10.1021/ed070pA25.5. [DOI] [Google Scholar]
- Johansson L.-S.; Tammelin T.; Campbell J. M.; Setälä H.; Österberg M. Experimental Evidence on Medium Driven Cellulose Surface Adaptation Demonstrated Using Nanofibrillated Cellulose. Soft Matter 2011, 7, 10917–10924. 10.1039/c1sm06073b. [DOI] [Google Scholar]
- Zimmermann J.; Reifler F. A.; Fortunato G.; Gerhardt L.-C.; Seeger S. A Simple, One-Step Approach to Durable and Robust Superhydrophobic Textiles. Adv. Funct. Mater. 2008, 18, 3662–3669. 10.1002/adfm.200800755. [DOI] [Google Scholar]
- Korhonen J. T.; Huhtamäki T.; Verho T.; Ras R. H. A. Hollow Polysiloxane Nanostructures Based on Pressure-Induced Film Expansion. Surf. Innovations 2014, 2, 116–126. 10.1680/si.13.00047. [DOI] [Google Scholar]
- Moon M.-W.; Lee S. H.; Sun J. Y.; Oh K. H.; Vaziri A.; Hutchinson J. W. Wrinkled Hard Skins on Polymers Created by Focused Ion Beam. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 1130–1133. 10.1073/pnas.0610654104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peydecastaing J.; Vaca-Garcia C.; Borredon E. Interactions with Water of Mixed Acetic-Fatty Cellulose Esters. Cellulose 2011, 18, 1023–1031. 10.1007/s10570-011-9530-2. [DOI] [Google Scholar]
- Cunha A. G.; Zhou Q.; Larsson P. T.; Berglund L. A. Topochemical Acetylation of Cellulose Nanopaper Structures for Biocomposites: Mechanisms for Reduced Water Vapour Sorption. Cellulose 2014, 21, 2773–2787. 10.1007/s10570-014-0334-z. [DOI] [Google Scholar]
- Lozhechnikova A.; Bellanger H.; Michen B.; Burgert I.; Österberg M. Surfactant-Free Carnauba Wax Dispersion and Its Use for Layer-by-Layer Assembled Protective Surface Coatings on Wood. Appl. Surf. Sci. 2017, 396, 1273–1281. 10.1016/j.apsusc.2016.11.132. [DOI] [Google Scholar]
- Solala I.; Bordes R.; Larsson A. Water Vapor Mass Transport Across Nanofibrillated Cellulose Films: Effect of Surface Hydrophobization. Cellulose 2018, 25, 347–356. 10.1007/s10570-017-1608-z. [DOI] [Google Scholar]
- Lozhechnikova A.; Vahtikari K.; Hughes M.; Österberg M. Toward Energy Efficiency Through an Optimized Use of Wood: The Development of Natural Hydrophobic Coatings That Retain Moisture-Buffering Ability. Energy Buildings 2015, 105, 37–42. 10.1016/j.enbuild.2015.07.052. [DOI] [Google Scholar]
- Forsman N.; Lozhechnikova A.; Khakalo A.; Johansson L.-S.; Vartiainen J.; Österberg M. Layer-by-Layer Assembled Hydrophobic Coatings for Cellulose Nanofibril Films and Textiles, Made of Polylysine and Natural Wax Particles. Carbohydr. Polym. 2017, 173, 392–402. 10.1016/j.carbpol.2017.06.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.