

Abstract
Pseudoachondroplasia (PSACH), a severe short-limbed dwarfing condition, is associated with life-long joint pain and early onset osteoarthritis. PSACH is caused by mutations in cartilage oligomeric matrix protein (COMP), a pentameric matricellular protein expressed primarily in cartilage and other musculoskeletal tissues. Mutations in COMP diminish calcium binding and as a result perturb protein folding and export to the extracellular matrix. Mutant COMP is retained in the endoplasmic reticulum (ER) of growth plate chondrocytes resulting in massive intracellular COMP retention. COMP trapped in the ER builds an intracellular matrix network that may prevent the normal cellular clearance mechanisms. We have shown that accumulation of intracellular matrix in mutant-COMP (MT-COMP) mice stimulates intense unrelenting ER stress, inflammation and oxidative stress. This cytotoxic stress triggers premature death of growth plate chondrocytes limiting long-bone growth. Here, we review the mutant COMP pathologic mechanisms and anti-inflammatory/antioxidant therapeutic approaches to reduce ER stress. In MT-COMP mice, aspirin and resveratrol both dampen the mutant COMP chondrocyte phenotype by decreasing intracellular accumulation, chondrocyte death and inflammatory marker expression. This reduction in chondrocyte stress translates into an improvement in long-bone growth in the MT-COMP mice. Our efforts now move to translational studies targeted at reducing the clinical consequences of MT-COMP and painful sequelae associated with PSACH.
Keywords: Anti-inflammatory, Antioxidant, Matricellular protein
1. Pseudoachondroplasia – the skeletal dysplasia
The first clinical and radiographic description of pseudoachondroplasia (PSACH) was reported in 1959 [1]. Since then, numerous studies of PSACH provide a comprehensive understanding of the natural history of the disorder [2–9]. PSACH babies are indistinguishable from other newborns during the first year of life because they have a normal birth length and weight. Diminished linear growth and/or a waddling gait are the first signs that alert the health care practitioner and/or parents that there is a growth problem. Radiographic examination leads to a diagnosis by the age of 18–24 months based on characteristic x-ray findings including shortening of all the long bones, small abnormal epiphyses, widened and irregular metaphyses, small, underossified capital femoral epiphyses and platyspondyly [3,7,9–11]. During childhood, limb shortening, brachydactyly, widened joints and joint laxity become obvious and lower limb abnormalities develop, ranging from genu varus to genu valgum or a combination of both [9,11]. Lower extremity abnormalities generally require surgical interventions (osteotomies); the timing of the procedures depends on the extent of joint laxity and the degree of deformity. The average adult height is 3′9″–3′11″ which is equivalent to the height of an average 6 year old (https://ghr.nlm.nih.gov/condition/pseudoachondroplasia). However, stature is variable with some being as tall as 4′10″. Early onset osteoarthritis occurs in young adults and produces significant discomfort. This affects all the major joints necessitating joint replacements usually starting with hip replacements in the second to third decades [4,7,11,12]. A recent natural history study found that pain starts in early childhood and is a significant problem for which there is no systematic or standard pain treatments [9,13]. Chronic pain, the most debilitating feature of PSACH, compromises mobility ultimately limiting physical activity and quality of life [7].
PSACH is an autosomal dominant disorder, that occurs as a (de novo) new event in 70–80% of families with the remaining cases being inherited from an affected parent [10,13,14]. Although autosomal recessive inheritance was reported based on recurrence in siblings of unaffected parents, these cases were subsequently shown to result from germline mosaicism. Affected individuals have a 50% risk of passing the mutation to their offspring in each pregnancy and prenatal diagnosis is available using molecular testing. Prenatal ultrasound will not detect PSACH since skeletal abnormalities develop postnatally overtime. Prenatal molecular diagnosis will establish affection status for familal cases.
2. Mutations in cartilage oligomeric matrix protein (COMP) cause PSACH
PSACH was first described as an rough endoplasmic reticulum (rER) storage disorder in 1972 based on electron micrography studies of iliac crest biopsies [3,15,16]. These studies revealed retention of a lamellar-appearing material in massively dilated ER cisternae of growth plate chondrocytes [17–24]. In 1995, mutations in COMP were shown to cause PSACH and the stored ER material was identified as COMP [10, 15]. Since then, more than 200 mutations have been identified with ≅99% found in the the highly conserved calcium-binding repeat domains indicating that this domain is extremely sensitive to genetic alterations (LOVD Mendelian genes https://grenada.lumc.nl/LOVD2/mendelian_genes/variants_statistics.php) [9,25–27]. Approximately 30% of cases result from deletion of one of five sequential aspartic acid residues at position 469–473 and is denoted as the D469del mutation [10].
COMP is a homopentameric protein that has a bouquet appearance on rotary shadowing with the N-terminal domain joining the five subunits [28]. Each COMP monomer has four distinct domains: N-terminal pentamerization domain, epidermal growth factor (EGF)-like domain (four repeats), a type 3 calcium-binding domain (7 repeats) and a C-terminal lectin-like globular region [29]. Mutations in the calcium-binding domain interfere with the number of bound calciums, which is predicted to disrupt protein folding of each arm thereby having a dominant negative effect on the protein [30]. Chondrocytes from three PSACH patients with different mutations, [D469del, G427E and D511Y], all show similar intracellular retention of COMP in the endoplasmic reticulum [31,32]. Crystallographic studies show that the type 3 calcium binding domain wraps around the calcium metal scaffold in a unique 3D structure and mutations in this domain are predicted to cause a local collapse of the 3D structure [33]. Indeed, functional studies confirm that mutations disrupt calcium binding and protein folding with the mutant arms measuring longer that the wild type arms [28].
3. Function of COMP
COMP was first isolated from cartilage and thought to be cartilage-specific but was later shown to be relatively abundant in other musculoskeletal tissues such as tendon, ligament and smooth muscle [18,29, 34–43]. COMP is a secreted glycoprotein found in extracellular matrices (ECM) and is best characterized in the pericellular and territorial matrices surrounding growth plate and articular chondrocytes [21,40]. COMP, the fifth member of the thrombospondin gene family; also designated as, TSP-5, binds to a number of proteins in the ECM with the C-terminal domain a hub for interaction(s) with other ECM proteins [44–46]. For example, COMP binds to matrillins-1, –3 and –4 (MATN) [19,45], interacts with glycosaminoglycan (GAG) side chains of aggrecan within the type 3 repeats and the C-terminal domain [47]. The C-terminal globular domain binds to types I, II, IX, XII and XIV collagens [44,46,48,49] and may enhance mechanical strength of the ECM and serve as anchor plaques/points for protein complexes. COMP has also been shown, in vitro, to increase the speed at which type II collagen fibrils assemble by potentially positioning free collagen molecule close to each other [50]. Other work suggests that the interaction of COMP with collagens organizes the collagen ECM network (fibril morphology and density) and that COMP is critical for collagen secretion [51]. Interestingly, this observation is consistent with iliac crest growth plate and ligament PSACH biopsies showing varied collagen fibril structure, diameters [52] and fused fibrils [53]. Moreover, a recent study of COMP null mice showed showing alterations in collagen fibrils in skin and tendon [51] while an earlier study did not [54]. These studies support the idea that collagen assembly in the matrix is affected by the absence of COMP in the matrix either by knocking it out or because of retention within the chondrocytes.
Fibronectin binds to integrins, which are anchored in the cellular membrane, and fibronectin also interacts with COMP at the C-terminal domain [55]. COMP directly facilitates chondrocyte attachment through interaction specifically with α5β1 and α5β3 integrins [56]. The interaction of these integrins with the RGD sequence in the calcium binding repeats are critical for chondrocyte attachment [56]. Granulin epithelin precursor (GEP), a growth factor, interacts with the EGF domain of COMP and this interaction stimulates chondrocyte proliferation [57]. The precise function of the EGF domain in this context is unclear but the domain includes six cysteine residues involved in disulphide bonds which are common in secreted proteins (http://smart.embl.de/smart/do_annotation.pl?DOMAIN=SM00181). Collectively these findings suggests that COMP may be a structural component of cartilage also involved in regulating chondrocyte function.
The specific role of COMP in different tissues is not well defined. Weight bearing equine tendons produce more COMP than non-weight bearing ones suggesting that COMP may play a role in withstanding mechanical stress consistent with the presence of a mechanosensitive region in the promoter [58]. In contrast, COMP null mice manifest only minor alterations in articular cartilage with exercise [59]. However, COMP null mice have a diminished skin fibrotic response during healing [51] and enlarged atherosclerotic plaques [60]. Moreover, COMP expression is up-regulated during human skin healing of photodamage and many fibrotic conditions of the skin, liver and lung suggesting that COMP may be an important component in fibrosis regardless of whether the fibrotic process is a component of healing or pathology [51,61–67].
4. Towards understanding the PSACH chondrocyte pathology
4.1. Mutant (MT-COMP) mouse
The bigenic MT-COMP or (wild-type) WT-COMP mice express COMP in response to doxycycline (DOX) administration and were generated from breeding two mouse lines (Fig. 1). The TRE-COMP transgenic mouse line contains the coding sequence of human COMP gene with FLAG-tag driven by the tetracycline responsive element (TRE) promoter. The TET-On-Col II mouse expresses the transactivator protein (rtTA) driven by a type II collagen promoter [68]. Western blot analysis confirmed protein expression in cartilage (COMP and FLAG antibodies) and tissue specificity of expression was validated using RTPCR [69].
Fig. 1.

MT-COMP transgenic mouse. (A) MT-COMP bigenic mouse line was generated using the Tet-On system with the tetracycline responsive element (TRE) driving human D469del-MT-COMP expression. The type II collagen promoter (Col IIa) drives rtTA (Tet-On) expression in chondrocytes. (B) In the presence of doxycycline (DOX), the bigenic MT-COMP mouse expresses mutant human D469del-COMP in growth plate and articular chondrocytes as visualized using human-specific COMP antibody. MT-COMP (hCOMP in red) expression is detected in the murine growth plate chondrocytes at 4 weeks with administration of DOX from conception to 4 weeks (C-4wk) (C), whereas mouse COMP (mCOMP in green) is primarily extracellular (B and C). Similarly, MT-COMP is found in articular cartilage chondrocytes of 12 week mice administered DOX from 8 to 12 weeks (E) but not in controls (D). The articular cartilage border is marked by a dashed line. DAPI staining of nuclei are shown in blue (D and E).
Limb length and intracellular retention of MT-COMP in growth plate chondrocytes was assessed in MT-COMP mice administered DOX (500 ng/ml) pre- and postnatally [69]. Consistent with the human PSACH phenotype, newborn MT-COMP and control mice have normal birth parameters. By 1 week postnatal life, MT-COMP mice are smaller than controls and the difference in size becomes more evident with age with obvious short limbs by 2 weeks of age (Fig. 2) [70]. In contrast, WT-COMP mice show no phenotypic difference from control mice and limb lengths are equivalent (Fig. 3). MT-COMP mice remain smaller than control mice throughout adulthood [71]. Intracellular retention of MT-COMP in growth plate chondrocytes is a progressive process beginning in the prenatal period (E15), when only a small number of growth plate chondrocytes showing MT-COMP in the ER [72]. Intracellular retention increases over time, peaking at P21 with almost every chondrocyte containing MT-COMP [72]. The progressive nature of intracellular retention and the accompanying chondrocyte death in the growth plate mirror the progressive loss of long-bone growth.
Fig. 2.

MT-COMP causes skeletal growth retardation. MT-COMP and control (C57BL\6) mice are compared in all panels. Skeletons were stained with alizarin (red) and alcian blue to visualize bone and cartilage, respectively. MT-COMP mice are shorter than controls by P7 (Panel 1) and remain shorter into adulthood [71] (Panels 2–4). All long bones and paws are markedly reduced (G–N Panels 1–4). Skull lengths are reduced primarily due to a decrease in the snout length. Adapted from Posey 2014 [70]. (Bar = 0.1 cm).
Fig. 3.
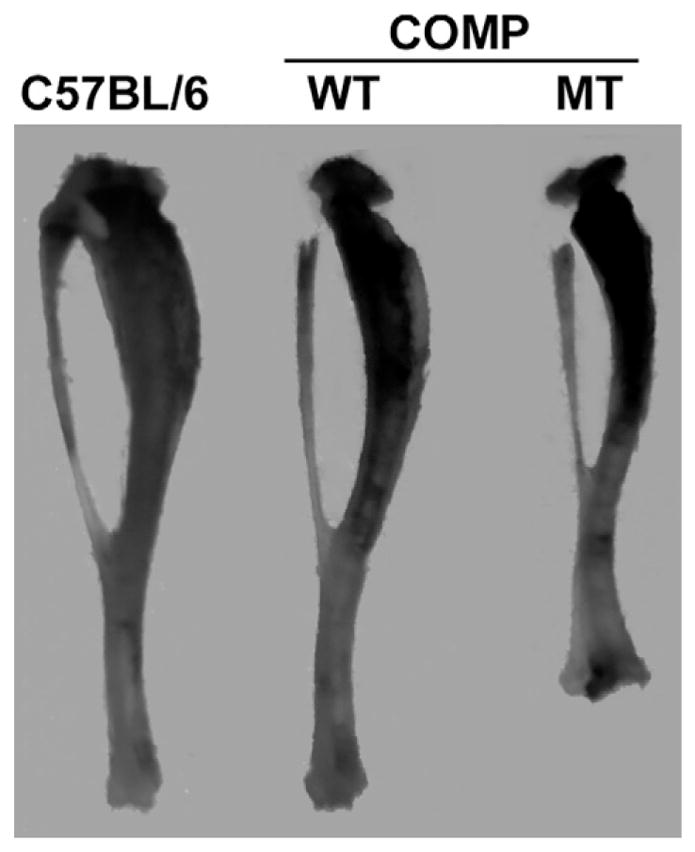
MT-COMP tibias are abnormally modeled. MT-COMP tibias are short and have metaphyseal flaring similar to that seen in PSACH. Overexpression of WT-COMP does not affect long bone morphology or length.
4.2. COMP mutations cause inflammatory processes that triggers chondrocyte death
WT-COMP monomeric subunits are immediately assembled into a pentameric protein followed by rapid folding of the five arms and transport to Golgi for posttranslational modification and finally export to ECM [73]. In contrast, pentameric proteins with mutant subunits cannot fold correctly and are retained in the ER creating dominant negative effects [17,20]. Mutant COMP stalled in the ER appears to participate in premature intracellular assembly of extracellular matrix composed of MT-COMP, types IX and II collagens and matrilin-3 (MATN3) as well as other ECM proteins [32,69,74]. Adding a BM40 signal peptide hastens the rate at which mutant COMP moves through the ER, decreasing intra-cellular accumulation and resulting in a very mild phenotype in mice [75–77]. It is likely that the unique repertoire of proteins synthesized by chondrocytes promotes intracellular matrix formation. Interestingly, some studies have suggested that MT-COMP is secreted in tendon, ligament and COS-7 (green monkey kidney cells) which would likely alter the pathological consequences of MT-COMP and manifest in poor matrix quality [17,18,23,78]. In chondrocytes, the stalled COMP and intracellular COMP matrix activates the unfolded protein response (UPR), which functions to either refold or degrade the misfolded protein [17, 20]. Misfolded proteins in the ER that cannot be refolded should be transported to the cytosol by retrotranslocation where ubiquitin is added and the protein is degraded by the proteasome [79] (Fig. 4A). We have previously shown that this mechanism fails [32,69]. This failure likely results because the MT-COMP intracellular matrix complex may not be able to fit through the pore in the ER preventing clearance of MT-COMP from the ER (Fig. 4B). Using deconvolution microscopy, we have shown intracellular matrix in the ER of MT-COMP mice and human PSACH chondrocytes indicating that this complex may be blocking translocation from the ER (Fig. 4C). Moreover, the reduction of ubiquitination in the MT-COMP growth plate supports this theory (Fig. 4D–E). The presence of intracellular matrix may explain why the UPR clearance mechanisms are unable to prevent the massive intracellular protein accumulation in PSACH growth plate chondrocytes [3,9,11, 12,18,20–22].
Fig. 4.

MT-COMP promotes the assembly of intracellular ER network in chondrocytes. (A) Schematic model showing how misfolded proteins in the ER are exported to the cytosol by retrotranslocation where ubiquitin (Ub) is added signaling proteasomal degradation [79]. (B) Model depicting how retained MT-COMP interacts with other matrix proteins in the ER generating a matrix network that is likely not able to fit through the retrotranslocation ER pore that leads to the cytosol. (C) Deconvolution microscopy shows intracellular matrix in chondrocyte ER composed of MT-COMP (green), matrilin-3 (blue), types 2 (yellow) and 9 (red) collagens. Adapted from Posey 2009 [69]. Ubiquitin immunohistochemistry of control (D), MT-COMP (E) and resveratrol treated MT-COMP growth plate chondrocytes (F).
We have demonstrated that CHOP/DDIT3 (CCAAT-enhancer-binding protein homologous protein/DNA-Damage-Inducible Transcript 3), a component of the UPR, plays an essential role in MT-COMP chondrocyte pathology [72,80]. CHOP is a multifunctional transcription factor involved in sensing ER stress [81]. CHOP is stimulated through the activation of PRKR-like ER kinase (PERK) one of the three branches of the unfolded protein response (UPR) [82]. Chronic activation of CHOP results in pathophysiological conditions that promote cell death by restarting protein translation and generating reactive oxygen species (ROS) [81]. CHOP restarting protein translation can generate additional stress in the ER [81]. MT-COMP expressed in rat chondrosarcoma cells (RCS) stimulated CHOP and restarted protein translation as monitored by changes in the phosphorylation state of translation initiation factor 2 (eIF2α) [80]. CHOP also activates an ER oxidase (ERO1), which generates hyper-oxidizing environment and stimulates oxidative stress [81, 83]. In the MT-COMP mice, a number of mRNAs involved in oxidative stress including ERO1 are upregulated and we have demonstrated that RCS cells expressing MT-COMP have excessive oxidative stress by measuring the conversion of a fluorescent probe by ROS [72,80]. In the MT-COMP mice, the restart of general protein translation stimulates additional ER stress and the oxidizing environment of the ER leads to more ER stress, oxidative stress and an inflammatory process.
The inflammatory processes, in the MT-COMP mice, were identified and characterized by transcriptome profiling, RTPCR analysis and immunostaining [70–72]. In the MT-COMP growth plate, IL1 and TNFα are induced by IL16 and IL18 [70]. The pro-inflammatory cytokine, IL-16, attracts eosinophils in cooperation with CCR5 [84,85] and this is the origin of the increase in eosinophil-related mRNAs in the MT-COMP growth plate [70]. Our findings confirm similar reports showing that inflammation can induce both oxidative and ER stress [83,86,87]. As shown in Fig. 5, chronic ER stress, inflammation, oxidative stress and DNA damage drive MT-COMP growth plate chondrocytes to necroptosis, a programmed form of necrosis stimulated by inflammation [72]. This becomes a self-perpetuating pathological process with each cellular stress exacerbating the others [83,88–91]. The relentless stress depletes the pool of growth plate chondrocytes necessary for long-bone growth and results in a dwarfed phenotype in the MT-COMP mice similar to that observed in humans.
Fig. 5.

PSACH chondrocyte death results from intense cellular stress. Schematic showing that MT-COMP intracellular ER retention stimulates ER stress through CHOP-mediated UPR generating an inflammatory response. Prolonged ER stress also produces excessive ROS (reactive oxygen species) causing oxidative stress that further contributes to the inflammatory process and creates a self-perpetuating stress loop in chondrocytes. This results in the death of growth plate chondrocytes, which translates into diminished long-bone growth and skeletal dysplasia.
4.3. Therapeutic interventions
Our first therapeutic approach to treat PSACH in our MT-COMP mouse was administration of ER stress reducing, drugs valproate, lithium and phenylbutyric acid from birth until 4 weeks after birth. While all three drugs decreased intracellular MT-COMP retention, chondrocyte death and inflammatory markers, these drugs were not well tolerated by either control or MT-COMP mice and caused reduced limb length and in some cases reduced viability [70]. Given the negative side effects of ER stress reduction drugs, we next focused on interrupting the pathological loop between inflammation, ER and oxidative stresses in MT-COMP chondrocytes [92]. This treatment approach is supported by other work that achieved successful reduction of ER stress with antioxidant therapy (butylated hydroxyanisole) [87] and NSAIDS treatments (diclofenac, indomethacin, ibuprofen, aspirin or ketoprofen) [93]. MT-COMP mice were treated with aspirin, ibuprofen, resveratrol, GSE, turmeric or CoQ10 from birth to 4 weeks [92]. Each of these therapies produced dampening of the MT-COMP chondrocyte phenotype [92]. Aspirin and resveratrol treatment dramatically reduced intracellular retention of MT-COMP in growth plate chondrocytes (Fig. 6A–D) resulting in less chondrocyte death as demonstrated by decreased TUNEL staining (Fig. 6E–H) and decreased TNFα, IL-1β, OSM (oncostatin M) and IL-16, all markers of inflammatory process associated with MT-COMP chondrocyte pathology (Fig. 6I–X). IL-16 has been implicated in the risk for knee OA [94] and OSM expression is associated with OA [95] suggesting that this inflammatory process elicited by MT-COMP may play a role in early joint erosion associated with PSACH. These antioxidant/anti-inflammatory treatments restored chondrocyte proliferation in MT-COMP growth plates, and most importantly, femoral length increased with aspirin or resveratrol treatment compared to the untreated MT-COMP mouse [92]. Grape see extract (GSE), turmeric and CoQ10 (antioxidants) reduced intracellular retention of MT-COMP to a lesser extent than aspirin or resveratrol [92]. Interestingly, some extracellular MT-COMP was observed with resveratrol and turmeric treatments [92] suggesting that decreases in retention with drug treatments are in part due to some export of MT-COMP. Treatments with GSE, CoQ10, and turmeric reduced TUNEL staining to a lesser extent than aspirin or resveratrol treatments in the MT-COMP growth plates but caused disorganization in control growth plates compared to untreated controls [92]. The alteration in chondrocyte organization makes these therapies less desirable than aspirin or resveratrol.
Fig. 6.

Anti-inflammatory and antioxidant treatments normalize MT-COMP growth plates. MT-COMP and control mice were treated with either aspirin or resveratrol from birth to 4 weeks and hind limbs were collected for analysis. Human-specific COMP antibody assessed ER retention in mouse growth plates chondrocytes. C57BL\6 growth plate chondrocytes were negative control because transgenic human MT-COMP is not present (A). Untreated MT-COMP growth plate chondrocytes show intracellular human COMP (B) and retention is decreased by aspirin or resveratrol (C–D). TUNEL assessment was used to assess cell death in growth plates. Basal level of apoptosis in the C57BL\6 growth plate is limited to a few hypertrophic chondrocytes (E). Most MT-COMP chondrocytes are TUNEL positive in the absence of treatment (F) but aspirin or resveratrol treatments showed reduced the number of TUNEL positive (green) chondrocytes (G and H). Inflammation markers, TNFα, IL-1β, OSM and IL-16, were assessed by immunostaining. TNFα is low in controls (I), elevated in MT-COMP growth plates (J) and decreased by aspirin and to a lesser degree resveratrol treatments (K and L). IL-1β signal shows a similar pattern of expression, low levels in control (M) and treated MT-COMP (O and P) and elevated in untreated MT-COMP (N). OSM, a cytokine associated with OA, is elevated in untreated (R) and decreased in control (Q) and MT-COMP treated mice (S and T). IL-16, showed similar findings as other inflammatory markers (U–X). Adapted from Posey 2014 [70,71].
In a second approach, we tested whether systemic administration would deliver COMP targeted antisense oligonucleotides (ASO) to growth plate chondrocytes and decrease MT-COMP growth plate chondrocyte pathology [96]. We found that ASO1 was successfully delivered reducing steady-state levels of cartilage oligomeric matrix protein mRNA dampening intracellular retention of MT-COMP. Interestingly, although ASO1 was designed against human COMP, mutant human COMP mRNA was reduced 38% and wild-type mouse COMP mRNA was reduced 60% [96]. Reduction in COMP retention in chondrocytes, led to a reduction of inflammatory markers, interleukin-16 (IL-16) and chitinase-like 3 (Chil3/YM1 a macrophage protein), the number of TUNEL positive chondrocytes, and a partial restoration of DNA proliferation [96]. These exciting novel results demonstrate that ASOs can be delivered and knock down mRNAs in growth plate and articular cartilages and open the door for a new antisense treatment approach for PSACH and potentially other human cartilage disorders.
PSACH is caused by different heterozygous COMP mutations (the vast majority in the type 3 calcium binding repeats) that all initiate and perpetuate dominant negative effect. Based on our findings that MT-COMP intracellular accumulation elicits chronic ER stress stimulating inflammation and oxidative stress and thereby driving the chondrocytes to necroptosis [70,72], we have identified avenues for a mechanistic-based therapeutic approach in the MT-COMP mice [92]. We show two different anti-inflammatory/antioxidant therapeutics that interrupt the MT-COMP stimulated feed-forward stress loop in growth plate chondrocytes. Suppressing cellular stress in chondrocytes improves chondrocyte longevity most likely by reducing the pressure on the ER thereby permitting normal UPR clearance mechanisms to function. This exciting therapeutic approach using over-the-counter medications, which are easy to access, administer and are well tolerated have the potential to greatly improve the quality of life for PSACH individuals. Importantly, the time between preclinical studies and clinical use is abbreviated for over-the-counter medications especially those that are well tolerated. This is an advantage for future trials in PSACH. Additionally using a knock-down approach, we demonstrate a reduction in PSACH chondrocyte pathology by eliminating the source of the dominant negative effect. The latter approach is novel and demonstrates that ASOs can reach the relatively avascular growth plate. Using multiple approaches, we show that reduction of PSACH chondrocyte pathology can be accomplished in our murine model. These therapeutic approaches are a significant advance towards developing human PSACH treatment targeted at alleviating the chronic joint pain that diminishes quality of life and mobility. Our findings set the foundation for testing a wide variety of drugs and therapeutic approaches including antisense technology for PSACH and other cartilage-related conditions.
Acknowledgments
Research supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under Award Number RO1-AR057117-05 and the Leah Lewis Family Foundation. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- 1.Maroteaux P, Lamy M. Pseudo-achondroplastic forms of spondyloepiphyseal dysplasias. Presse Med. 1959;67(10):383–386. [PubMed] [Google Scholar]
- 2.Briggs MD, Chapman KL. Pseudoachondroplasia and multiple epiphyseal dysplasia: mutation review, molecular interactions, and genotype to phenotype correlations. Hum Mutat. 2002;19(5):465–478. doi: 10.1002/humu.10066. [DOI] [PubMed] [Google Scholar]
- 3.Cooper RR, Ponseti IV, Maynard JA. Pseudoachondroplastic dwarfism. A rough-surfaced endoplasmic reticulum storage disorder. J Bone Joint Surg Am. 1973;55(3):475–484. [PubMed] [Google Scholar]
- 4.Hall JG. Pseudoachondroplasia. Birth Defects Orig Artic Ser. 1975;11(6):187–202. [PubMed] [Google Scholar]
- 5.Heselson NG, Cremin BJ, Beighton P. Pseudoachondroplasia, a report of 13 cases. Br J Radiol. 1977;50(595):473–482. doi: 10.1259/0007-1285-50-595-473. [DOI] [PubMed] [Google Scholar]
- 6.Langer LO, Jr, Schaefer GB, Wadsworth DT. Patient with double heterozygosity for achondroplasia and pseudoachondroplasia, with comments on these conditions and the relationship between pseudoachondroplasia and multiple epiphyseal dysplasia, Fairbank type. Am J Med Genet. 1993;47(5):772–781. doi: 10.1002/ajmg.1320470535. [DOI] [PubMed] [Google Scholar]
- 7.McKeand J, Rotta J, Hecht JT. Natural history study of pseudoachondroplasia. Am J Med Genet. 1996;63(2):406–410. doi: 10.1002/(SICI)1096-8628(19960517)63:2<406::AID-AJMG16>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 8.Stevens JW. Pseudoachondroplastic dysplasia: an Iowa review from human to mouse. Iowa Orthop J. 1999;19:53–65. [PMC free article] [PubMed] [Google Scholar]
- 9.Unger S, Hecht JT. Pseudoachondroplasia and multiple epiphyseal dysplasia: new etiologic developments. Am J Med Genet. 2001;106(4):244–250. [PubMed] [Google Scholar]
- 10.Hecht JT, Nelson LD, Crowder E, Wang Y, Elder FF, Harrison WR, Francomano CA, Prange CK, Lennon GG, Deere M, Lawler J. Mutations in exon 17B of cartilage oligomeric matrix protein (COMP) cause pseudoachondroplasia. Nat Genet. 1995;10(3):325–329. doi: 10.1038/ng0795-325. [DOI] [PubMed] [Google Scholar]
- 11.Posey KL, Hayes E, Haynes R, Hecht JT. Role of TSP-5/COMP in pseudoachondroplasia. Int J Biochem Cell Biol. 2004;36(6):1005–1012. doi: 10.1016/j.biocel.2004.01.011. [DOI] [PubMed] [Google Scholar]
- 12.Posey KL, Hecht JT. The role of cartilage oligomeric matrix protein (COMP) in skeletal disease. Curr Drug Targets. 2008;9(10):869–877. doi: 10.2174/138945008785909293. [DOI] [PubMed] [Google Scholar]
- 13.Ferguson HL, Deere M, Evans R, Rotta J, Hall JG, Hecht JT. Mosaicism in pseudoachondroplasia. Am J Med Genet. 1997;70(3):287–291. [PubMed] [Google Scholar]
- 14.Napiontek M, Bernardczyk K, Kruczynski J. Familial occurrence of pseudoachondroplasia. Chir Narzadow Ruchu Ortop Pol. 1986;51(1):47–51. [PubMed] [Google Scholar]
- 15.Briggs MD, Hoffman SM, King LM, Olsen AS, Mohrenweiser H, Leroy JG, Mortier GR, Rimoin DL, Lachman RS, Gaines ES. Pseudoachondroplasia and multiple epiphyseal dysplasia due to mutations in the cartilage oligomeric matrix protein gene. Nat Genet. 1995;10(3):330–336. doi: 10.1038/ng0795-330. [DOI] [PubMed] [Google Scholar]
- 16.Maynard JA, Cooper RR, Ponseti IV. A unique rough surfaced endoplasmic reticulum inclusion in pseudoachondroplasia. Lab Investig. 1972;26(1):40–44. [PubMed] [Google Scholar]
- 17.Maddox BK, Keene DR, Sakai LY, Charbonneau NL, Morris NP, Ridgway CC, Boswell BA, Sussman MD, Horton WA, Bachinger HP, Hecht JT. The fate of cartilage oligomeric matrix protein is determined by the cell type in the case of a novel mutation in pseudoachondroplasia. J Biol Chem. 1997;272(49):30993–30997. doi: 10.1074/jbc.272.49.30993. [DOI] [PubMed] [Google Scholar]
- 18.Hecht JT, Deere M, Putnam E, Cole W, Vertel B, Chen H, Lawler J. Characterization of cartilage oligomeric matrix protein (COMP) in human normal and pseudoachondroplasia musculoskeletal tissues. Matrix Biol. 1998;17(4):269–278. doi: 10.1016/s0945-053x(98)90080-4. [DOI] [PubMed] [Google Scholar]
- 19.Hecht JT, Hayes E, Haynes R, Cole WG. COMP mutations, chondrocyte function and cartilage matrix. Matrix Biol. 2005;23(8):525–533. doi: 10.1016/j.matbio.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 20.Hecht JT, Hayes E, Snuggs M, Decker G, Montufar-Solis D, Doege K, Mwalle F, Poole R, Stevens J, Duke PJ. Calreticulin, PDI, Grp94 and BiP chaperone proteins are associated with retained COMP in pseudoachondroplasia chondrocytes. Matrix Biol. 2001;20(4):251–262. doi: 10.1016/s0945-053x(01)00136-6. [DOI] [PubMed] [Google Scholar]
- 21.Hecht JT, Makitie O, Hayes E, Haynes R, Susic M, Montufar-Solis D, Duke PJ, Cole WG. Chondrocyte cell death and intracellular distribution of COMP and type IX collagen in the pseudoachondroplasia growth plate. J Orthop Res. 2004;22(4):759–767. doi: 10.1016/j.orthres.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 22.Dinser R, Zaucke F, Kreppel F, Hultenby K, Kochanek S, Paulsson M, Maurer P. Pseudoachondroplasia is caused through both intra- and extracellular pathogenic pathways. J Clin Invest. 2002;110(4):505–513. doi: 10.1172/JCI14386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen TL, Stevens JW, Cole WG, Hecht JT, Vertel BM. Cell-type specific trafficking of expressed mutant COMP in a cell culture model for PSACH. Matrix Biol. 2004;23(7):433–444. doi: 10.1016/j.matbio.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 24.Hashimoto Y, Tomiyama T, Yamano Y, Mori H. Mutation (D472Y) in the type 3 repeat domain of cartilage oligomeric matrix protein affects its early vesicle trafficking in endoplasmic reticulum and induces apoptosis. Am J Pathol. 2003;163(1):101–110. doi: 10.1016/S0002-9440(10)63634-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Briggs MD, Mortier GR, Cole WG, King LM, Golik SS, Bonaventure J, Nuytinck L, De Paepe A, Leroy JG, Biesecker L, Lipson M, Wilcox WR, Lachman RS, Rimoin DL, Knowlton RG, Cohn DH. Diverse mutations in the gene for cartilage oligomeric matrix protein in the pseudoachondroplasia-multiple epiphyseal dysplasia disease spectrum. Am J Hum Genet. 1998;62(2):311–319. doi: 10.1086/301713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deere M, Sanford T, Ferguson HL, Daniels K, Hecht JT. Identification of twelve mutations in cartilage oligomeric matrix protein (COMP) in patients with pseudoachondroplasia. Am J Med Genet. 1998;80(5):510–513. doi: 10.1002/(sici)1096-8628(19981228)80:5<510::aid-ajmg14>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 27.Deere M, Sanford T, Francomano CA, Daniels K, Hecht JT. Identification of nine novel mutations in cartilage oligomeric matrix protein in patients with pseudoachondroplasia and multiple epiphyseal dysplasia. Am J Med Genet. 1999;85(5):486–490. doi: 10.1002/(sici)1096-8628(19990827)85:5<486::aid-ajmg10>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 28.Chen H, Deere M, Hecht JT, Lawler J. Cartilage oligomeric matrix protein is a calcium-binding protein, and a mutation in its type 3 repeats causes conformational changes. J Biol Chem. 2000;275(34):26538–26544. doi: 10.1074/jbc.M909780199. [DOI] [PubMed] [Google Scholar]
- 29.Adams JC, Tucker RP, Lawler J. The Thrombospondin Gene Family. R.G. Landes Co; Austin, Tex., U.S.A: 1995. [Google Scholar]
- 30.Hou J, Putkey JA, Hecht JT. Delta 469 mutation in the type 3 repeat calcium binding domain of cartilage oligomeric matrix protein (COMP) disrupts calcium binding. Cell Calcium. 2000;27(6):309–314. [Google Scholar]
- 31.Merritt TM, Alcorn JL, Haynes R, Hecht JT. Expression of mutant cartilage oligomeric matrix protein in human chondrocytes induces the pseudoachondroplasia phenotype. J Orthop Res. 2006;24(4):700–707. doi: 10.1002/jor.20100. [DOI] [PubMed] [Google Scholar]
- 32.Merritt TM, Bick R, Poindexter BJ, Alcorn JL, Hecht JT. Unique matrix structure in the rough endoplasmic reticulum cisternae of pseudoachondroplasia chondrocytes. Am J Pathol. 2007;170(1):293–300. doi: 10.2353/ajpath.2007.060530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kvansakul M, Adams JC, Hohenester E. Structure of a thrombospondin C-terminal fragment reveals a novel calcium core in the type 3 repeats. EMBO J. 2004;23(6):1223–1233. doi: 10.1038/sj.emboj.7600166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.DiCesare PE, Morgelin M, Mann K, Paulsson M. Cartilage oligomeric matrix protein and thrombospondin 1. Purification from articular cartilage, electron microscopic structure, and chondrocyte binding. Eur J Biochem. 1994;223(3):927–937. doi: 10.1111/j.1432-1033.1994.tb19070.x. [DOI] [PubMed] [Google Scholar]
- 35.Bornstein P. Thrombospondins: structure and regulation of expression. FASEB J. 1992;6(14):3290–3299. doi: 10.1096/fasebj.6.14.1426766. [DOI] [PubMed] [Google Scholar]
- 36.Bornstein P. Thrombospondins as matricellular modulators of cell function. J Clin Investig. 2001;107(8):929–934. doi: 10.1172/JCI12749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Adams JC. Thrombospondins: multifunctional regulators of cell interactions. Annu Rev Cell Dev Biol. 2001;17:25–51. doi: 10.1146/annurev.cellbio.17.1.25. [DOI] [PubMed] [Google Scholar]
- 38.Adams JC, Lawler J. The thrombospondins. Int J Biochem Cell Biol. 2004;36(6):961–968. doi: 10.1016/j.biocel.2004.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li QY, Lennon GG, Brook JD. The identification of exons from the MED/PSACH region of human chromosome 19. Genomics. 1996;32(2):218–224. doi: 10.1006/geno.1996.0108. [DOI] [PubMed] [Google Scholar]
- 40.Hedbom E, Antonsson P, Hjerpe A, Aeschlimann D, Paulsson M, Rosa-Pimentel E, Sommarin Y, Wendel M, Oldberg A, Heinegard D. Cartilage matrix proteins. An acidic oligomeric protein (COMP) detected only in cartilage. J Biol Chem. 1992;267(9):6132–6136. [PubMed] [Google Scholar]
- 41.Urban JP, Maroudas A, Bayliss MT, Dillon J. Swelling pressures of proteoglycans at the concentrations found in cartilaginous tissues. Biorheology. 1979;16(6):447–464. doi: 10.3233/bir-1979-16609. [DOI] [PubMed] [Google Scholar]
- 42.Kempson GE, Freeman MA, Swanson SA. Tensile properties of articular cartilage. Nature. 1968;220(172):1127–1128. doi: 10.1038/2201127b0. [DOI] [PubMed] [Google Scholar]
- 43.Schmidt MB, Mow VC, Chun LE, Eyre DR. Effects of proteoglycan extraction on the tensile behavior of articular cartilage. J Orthop Res. 1990;8(3):353–363. doi: 10.1002/jor.1100080307. [DOI] [PubMed] [Google Scholar]
- 44.Holden P, Meadows RS, Chapman KL, Grant ME, Kadler KE, Briggs MD. Cartilage oligomeric matrix protein interacts with type IX collagen, and disruptions to these interactions identify a pathogenetic mechanism in a bone dysplasia family. J Biol Chem. 2001;276(8):6046–6055. doi: 10.1074/jbc.M009507200. [DOI] [PubMed] [Google Scholar]
- 45.Mann HH, Ozbek S, Engel J, Paulsson M, Wagener R. Interactions between the cartilage oligomeric matrix protein and matrilins. Implications for matrix assembly and the pathogenesis of chondrodysplasias. J Biol Chem. 2004;279(24):25294–25298. doi: 10.1074/jbc.M403778200. [DOI] [PubMed] [Google Scholar]
- 46.Thur J, Rosenberg K, Nitsche DP, Pihlajamaa T, Ala-Kokko L, Heinegard D, Paulsson M, Maurer P. Mutations in cartilage oligomeric matrix protein causing pseudoachondroplasia and multiple epiphyseal dysplasia affect binding of calcium and collagen I, II, and IX. J Biol Chem. 2001;276(9):6083–6092. doi: 10.1074/jbc.M009512200. [DOI] [PubMed] [Google Scholar]
- 47.Chen FH, Herndon ME, Patel N, Hecht JT, Tuan RS, Lawler J. Interaction of cartilage oligomeric matrix protein/thrombospondin 5 with aggrecan. J Biol Chem. 2007;282(34):24591–24598. doi: 10.1074/jbc.M611390200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kalchishkova N, Furst CM, Heinegard D, Blom AM. NC4 domain of cartilage-specific collagen IX inhibits complement directly due to attenuation of membrane attack formation and indirectly through binding and enhancing activity of complement inhibitors C4B-binding protein and factor H. J Biol Chem. 2011;286(32):27915–27926. doi: 10.1074/jbc.M111.242834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Agarwal P, Zwolanek D, Keene DR, Schulz JN, Blumbach K, Heinegard D, Zaucke F, Paulsson M, Krieg T, Koch M, Eckes B. Collagen XII and XIV, new partners of cartilage oligomeric matrix protein in the skin extracellular matrix suprastructure. J Biol Chem. 2012;287(27):22549–22559. doi: 10.1074/jbc.M111.335935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Halasz K, Kassner A, Morgelin M, Heinegard D. Comp acts as a catalyst in collagen fibrillogenesis. J Biol Chem. 2007;282(43):31166–31173. doi: 10.1074/jbc.M705735200. [DOI] [PubMed] [Google Scholar]
- 51.Schulz JN, Nuchel J, Niehoff A, Bloch W, Schonborn K, Hayashi S, Kamper M, Brinckmann J, Plomann M, Paulsson M, Krieg T, Zaucke F, Eckes B. COMP-assisted collagen secretion - a novel intracellular function required for fibrosis. J Cell Sci. 2016;129(4):706–716. doi: 10.1242/jcs.180216. [DOI] [PubMed] [Google Scholar]
- 52.Stoss H, Pesch HJ. Structural changes of collagen fibrils in skeletal dysplasias. Ultra-structural findings in the iliac crest. Virchows Arch A Pathol Anat Histol. 1985;405(3):341–364. doi: 10.1007/BF00710070. [DOI] [PubMed] [Google Scholar]
- 53.Pirog KA, Jaka O, Katakura Y, Meadows RS, Kadler KE, Boot-Handford RP, Briggs MD. A mouse model offers novel insights into the myopathy and tendinopathy often associated with pseudoachondroplasia and multiple epiphyseal dysplasia. Hum Mol Genet. 2010;19(1):52–64. doi: 10.1093/hmg/ddp466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Svensson L, Aszodi A, Heinegard D, Hunziker EB, Reinholt FP, Fassler R, Oldberg A. Cartilage oligomeric matrix protein-deficient mice have normal skeletal development. Mol Cell Biol. 2002;22(12):4366–4371. doi: 10.1128/MCB.22.12.4366-4371.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Di Cesare PE, Chen FS, Moergelin M, Carlson CS, Leslie MP, Perris R, Fang C. Matrix-matrix interaction of cartilage oligomeric matrix protein and fibronectin. Matrix Biol. 2002;21(5):461–470. doi: 10.1016/s0945-053x(02)00015-x. [DOI] [PubMed] [Google Scholar]
- 56.Chen FH, Thomas AO, Hecht JT, Goldring MB, Lawler J. Cartilage oligomeric matrix protein/thrombospondin 5 supports chondrocyte attachment through interaction with integrins. J Biol Chem. 2005;280(38):32655–32661. doi: 10.1074/jbc.M504778200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xu K, Zhang Y, Ilalov K, Carlson CS, Feng JQ, Di Cesare PE, Liu CJ. Cartilage oligomeric matrix protein associates with granulin-epithelin precursor (GEP) and potentiates GEP-stimulated chondrocyte proliferation. J Biol Chem. 2007;282(15):11347–11355. doi: 10.1074/jbc.M608744200. [DOI] [PubMed] [Google Scholar]
- 58.Amanatullah DF, Lu J, Hecht J, Posey K, Yik J, Di Cesare PE, Haudenschild DR. Identification of a 3Kbp mechanoresponsive promoter region in the human cartilage oligomeric matrix protein gene. Tissue Eng A. 2012;18(17–18):1882–1889. doi: 10.1089/ten.tea.2011.0497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Posey KL, Hankenson K, Veerisetty AC, Bornstein P, Lawler J, Hecht JT. Skeletal abnormalities in mice lacking extracellular matrix proteins, thrombospondin-1, thrombospondin-3, thrombospondin-5, and type IX collagen. Am J Pathol. 2008;172(6):1664–1674. doi: 10.2353/ajpath.2008.071094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bond AR, Hultgardh-Nilsson A, Knutsson A, Jackson CL, Rauch U. Cartilage oligomeric matrix protein (COMP) in murine brachiocephalic and carotid atherosclerotic lesions. Atherosclerosis. 2014;236(2):366–372. doi: 10.1016/j.atherosclerosis.2014.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kobayashi M, Kawabata K, Kusaka-Kikushima A, Sugiyama Y, Mabuchi T, Takekoshi S, Miyasaka M, Ozawa A, Sakai S. Cartilage oligomeric matrix protein increases in photodamaged skin. J Investig Dermatol. 2016;136(6):1143–1149. doi: 10.1016/j.jid.2016.02.802. [DOI] [PubMed] [Google Scholar]
- 62.Inui S, Shono F, Nakajima T, Hosokawa K, Itami S. Identification and characterization of cartilage oligomeric matrix protein as a novel pathogenic factor in keloids. Am J Pathol. 2011;179(4):1951–1960. doi: 10.1016/j.ajpath.2011.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vuga LJ, Milosevic J, Pandit K, Ben-Yehudah A, Chu Y, Richards T, Sciurba J, Myerburg M, Zhang Y, Parwani AV, Gibson KF, Kaminski N. Cartilage oligomeric matrix protein in idiopathic pulmonary fibrosis. PLoS One. 2013;8(12):e83120. doi: 10.1371/journal.pone.0083120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Agarwal P, Schulz JN, Blumbach K, Andreasson K, Heinegard D, Paulsson M, Mauch C, Eming SA, Eckes B, Krieg T. Enhanced deposition of cartilage oligomeric matrix protein is a common feature in fibrotic skin pathologies. Matrix Biol. 2013;32(6):325–331. doi: 10.1016/j.matbio.2013.02.010. [DOI] [PubMed] [Google Scholar]
- 65.Rice LM, Ziemek J, Stratton EA, McLaughlin SR, Padilla CM, Mathes AL, Christmann RB, Stifano G, Browning JL, Whitfield ML, Spiera RF, Gordon JK, Simms RW, Zhang Y, Lafyatis R. A longitudinal biomarker for the extent of skin disease in patients with diffuse cutaneous systemic sclerosis. Arthritis Rheum. 2015;67(11):3004–3015. doi: 10.1002/art.39287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Andreasson K, Hesselstrand R, Saxne T, Holmberg A, Norrgren H, Jonsson G. Cartilage oligomeric matrix protein: a new promising biomarker of liver fibrosis in chronic hepatitis C. Infect Dis. 2015;47(12):915–918. doi: 10.3109/23744235.2015.1075659. [DOI] [PubMed] [Google Scholar]
- 67.Otteby KE, Holmquist E, Saxne T, Heinegard D, Hesselstrand R, Blom AM. Cartilage oligomeric matrix protein-induced complement activation in systemic sclerosis. Arthritis Res Ther. 2013;15(6):R215. doi: 10.1186/ar4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tsumaki N, Tanaka K, Arikawa-Hirasawa E, Nakase T, Kimura T, Thomas JT, Ochi T, Luyten FP, Yamada Y. Role of CDMP-1 in skeletal morphogenesis: promotion of mesenchymal cell recruitment and chondrocyte differentiation. J Cell Biol. 1999;144(1):161–173. doi: 10.1083/jcb.144.1.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Posey KL, Veerisetty AC, Liu P, Wang HR, Poindexter BJ, Bick R, Alcorn JL, Hecht JT. An inducible cartilage oligomeric matrix protein mouse model recapitulates human pseudoachondroplasia phenotype. Am J Pathol. 2009;175(4):1555–1563. doi: 10.2353/ajpath.2009.090184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Posey KL, Coustry F, Veerisetty AC, Liu P, Alcorn JL, Hecht JT. Chondrocyte-specific pathology during skeletal growth and therapeutics in a murine model of pseudoachondroplasia. J Bone Miner Res. 2014;29(5):1258–1268. doi: 10.1002/jbmr.2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Posey KL, Alcorn JL, Hecht JT. Pseudoachondroplasia/COMP - translating from the bench to the bedside. Matrix Biol. 2014;37:167–173. doi: 10.1016/j.matbio.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Posey KL, Coustry F, Veerisetty AC, Liu P, Alcorn JL, Hecht JT. Chop (Ddit3) is essential for D469del-COMP retention and cell death in chondrocytes in an inducible transgenic mouse model of pseudoachondroplasia. Am J Pathol. 2012;180(2):727–737. doi: 10.1016/j.ajpath.2011.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Delot E, Brodie SG, King LM, Wilcox WR, Cohn DH. Physiological and pathological secretion of cartilage oligomeric matrix protein by cells in culture. J Biol Chem. 1998;273(41):26692–26697. doi: 10.1074/jbc.273.41.26692. [DOI] [PubMed] [Google Scholar]
- 74.Hecht JT, Montufar-Solis D, Decker G, Lawler J, Daniels K, Duke PJ. Retention of cartilage oligomeric matrix protein (COMP) and cell death in redifferentiated pseudoachondroplasia chondrocytes. Matrix Biol. 1998;17(8–9):625–633. doi: 10.1016/s0945-053x(98)90113-5. [DOI] [PubMed] [Google Scholar]
- 75.Schmitz M, Niehoff A, Miosge N, Smyth N, Paulsson M, Zaucke F. Transgenic mice expressing D469Delta mutated cartilage oligomeric matrix protein (COMP) show growth plate abnormalities and sternal malformations. Matrix Biol. 2008;27(2):67–85. doi: 10.1016/j.matbio.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 76.Holden P, Keene DR, Lunstrum GP, Bachinger HP, Horton WA. Secretion of cartilage oligomeric matrix protein is affected by the signal peptide. J Biol Chem. 2005;280(17):17172–17179. doi: 10.1074/jbc.M411716200. [DOI] [PubMed] [Google Scholar]
- 77.Hansen U, Platz N, Becker A, Bruckner P, Paulsson M, Zaucke F. A secreted variant of cartilage oligomeric matrix protein carrying a chondrodysplasia-causing mutation (p.H587R) disrupts collagen fibrillogenesis. Arthritis Rheum. 2011;63(1):159–167. doi: 10.1002/art.30073. [DOI] [PubMed] [Google Scholar]
- 78.Weirich C, Keene DR, Kirsch K, Heil M, Neumann E, Dinser R. Expression of PSACH-associated mutant COMP in tendon fibroblasts leads to increased apoptotic cell death irrespective of the secretory characteristics of mutant COMP. Matrix Biol. 2007;26(4):314–323. doi: 10.1016/j.matbio.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 79.Tsai B, Ye Y, Rapoport TA. Retro-translocation of proteins from the endoplasmic reticulum into the cytosol. Nat Rev Mol Cell Biol. 2002;3(4):246–255. doi: 10.1038/nrm780. [DOI] [PubMed] [Google Scholar]
- 80.Coustry F, Posey KL, Liu P, Alcorn JL, Hecht JT. D469del-COMP retention in chondrocytes stimulates caspase-independent necroptosis. Am J Pathol. 2012;180(2):738–748. doi: 10.1016/j.ajpath.2011.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, Nagata K, Harding HP, Ron D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004;18(24):3066–3077. doi: 10.1101/gad.1250704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kaufman RJ. Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev. 1999;13(10):1211–1233. doi: 10.1101/gad.13.10.1211. [DOI] [PubMed] [Google Scholar]
- 83.Malhotra JD, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxid Redox Signal. 2007;9(12):2277–2293. doi: 10.1089/ars.2007.1782. [DOI] [PubMed] [Google Scholar]
- 84.Lynch EA, Heijens CA, Horst NF, Center DM, Cruikshank WW. Cutting edge: IL-16/CD4 preferentially induces Th1 cell migration: requirement of CCR5. J Immunol. 2003;171(10):4965–4968. doi: 10.4049/jimmunol.171.10.4965. [DOI] [PubMed] [Google Scholar]
- 85.Ferland C, Flamand N, Davoine F, Chakir J, Laviolette M. IL-16 activates plasminogen-plasmin system and promotes human eosinophil migration into extracellular matrix via CCR3-chemokine-mediated signaling and by modulating CD4 eosinophil expression. J Immunol. 2004;173(7):4417–4424. doi: 10.4049/jimmunol.173.7.4417. [DOI] [PubMed] [Google Scholar]
- 86.Malhotra JD, Kaufman RJ. The endoplasmic reticulum and the unfolded protein response. Semin Cell Dev Biol. 2007;18(6):716–731. doi: 10.1016/j.semcdb.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Malhotra JD, Miao H, Zhang K, Wolfson A, Pennathur S, Pipe SW, Kaufman RJ. Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc Natl Acad Sci U S A. 2008;105(47):18525–18530. doi: 10.1073/pnas.0809677105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gotoh T, Endo M, Oike Y. Endoplasmic reticulum stress-related inflammation and cardiovascular diseases. Int J Inflamm. 2011;2011:259462. doi: 10.4061/2011/259462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Salminen A, Kauppinen A, Suuronen T, Kaarniranta K, Ojala J. ER stress in Alzheimer’s disease: a novel neuronal trigger for inflammation and Alzheimer’s pathology. J Neuroinflammation. 2009;6:41. doi: 10.1186/1742-2094-6-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cullinan SB, Diehl JA. Coordination of ER and oxidative stress signaling: the PERK/ Nrf2 signaling pathway. Int J Biochem Cell Biol. 2006;38(3):317–332. doi: 10.1016/j.biocel.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 91.Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140(6):900–917. doi: 10.1016/j.cell.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Posey KL, Coustry F, Veerisetty AC, Hossain M, Alcorn JL, Hecht JT. Antioxidant and anti-inflammatory agents mitigate pathology in a mouse model of pseudoachondroplasia. Hum Mol Genet. 2015;24(14):3918–3928. doi: 10.1093/hmg/ddv122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yamazaki T, Muramoto M, Oe T, Morikawa N, Okitsu O, Nagashima T, Nishimura S, Katayama Y, Kita Y. Diclofenac, a non-steroidal anti-inflammatory drug, suppresses apoptosis induced by endoplasmic reticulum stresses by inhibiting caspase signaling. Neuropharmacology. 2006;50(5):558–567. doi: 10.1016/j.neuropharm.2005.10.016. [DOI] [PubMed] [Google Scholar]
- 94.Luo SX, Li S, Zhang XH, Zhang JJ, Long GH, Dong GF, Su W, Deng Y, Liu Y, Zhao JM, Qin X. Genetic polymorphisms of interleukin-16 and risk of knee osteoarthritis. PLoS One. 2015;10(5):e0123442. doi: 10.1371/journal.pone.0123442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hu K, Xu L, Cao L, Flahiff CM, Brussiau J, Ho K, Setton LA, Youn I, Guilak F, Olsen BR, Li Y. Pathogenesis of osteoarthritis-like changes in the joints of mice deficient in type IX collagen. Arthritis Rheum. 2006;54(9):2891–2900. doi: 10.1002/art.22040. [DOI] [PubMed] [Google Scholar]
- 96.Posey KL, Coustry F, Veerisetty AC, Hossain M, Gattis D, Booten S, Alcorn JL, Seth PP, Hecht JT. Antisense reduction of mutant COMP reduces growth plate chondrocyte pathology. Mol Ther. 2017 doi: 10.1016/j.ymthe.2016.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]