

ABSTRACT
Cancer-induced myeloid-derived suppressor cells (MDSC) play an important role in tumor immune evasion. MDSC programming or polarization has been proposed as a strategy for leveraging the developmental plasticity of myeloid cells to reverse MDSC immune suppressive functions, or cause them to acquire anti-tumor activity. While MDSC derived ex vivo from murine bone marrow precursor cells with tumor-conditioned medium efficiently suppressed T cell proliferation, MDSC derived from conditioned medium in presence of TGF-β1 (TGFβ-MDSC) acquired a novel immune-stimulatory phenotype, losing the ability to inhibit T cell proliferation and acquiring enhanced antigen-presenting capability. Altered immune function was associated with SMAD-2 dependent upregulation of maturation and costimulatory molecules, and downregulation of inducible nitric oxide synthase (iNOS), an effector mechanism of immunosuppression. TGFβ-MDSC also upregulated FAS-ligand expression, leading to FAS-dependent killing of murine human papillomavirus (HPV)-associated head and neck cancer cells and tumor spheroids in vitro and anti-tumor activity in vivo. Radiation upregulated FAS expression on tumor cells, and the combination of radiotherapy and intratumoral injection of TGFβ-MDSC strongly enhanced class I expression on tumor cells and induction of HPV E7 tetramer-positive CD8 + T cells, leading to clearance of established tumors and long-term survival. TGFβ-MDSC derived from human PBMC with tumor conditioned medium also lost immunosuppressive function and acquired tumor-killing activity. Thus, TGFβ1 mediated programming of nascent MDSC leads to a potent anti-tumor phenotype potentially suitable for adoptive immunotherapy.
Keywords: MDSC, TGF-beta, CD86, tumor killing, radiotherapy, caspase-3, myeloid-derived suppressor cells (written out), SMAD2, adoptive cellular therapy
Introduction
Immunotherapy has become an emerging standard of care for numerous cancers, with agents such as chimeric antigen receptor (CAR) T cells and checkpoint inhibitors targeting programmed death receptor/ligand 1 (PD-1/PD-L1) and cytotoxic T lymphocyte associated antigen 4 (CTLA-4) showing significant activity against hematopoietic malignancies and solid tumors, respectively.1–4 However, while a fraction of patients treated with highly active immunotherapies such as checkpoint inhibition have dramatic responses, the majority of patients treated do not receive durable benefit.5–7 One major barrier to effective immunotherapy is the highly immunosuppressive tumor immune-microenvironment (TIME). Cancers induce an immunologically unfavorable TIME primarily by inducing intratumoral inflammation and the subsequent recruitment of immunosuppressive immunocytes. Myeloid derived suppressor cells (MDSC) are a major cellular component of the TIME8-10 that promotes immune evasion and tumor growth by preventing T-cell infiltration and activation. In conditions of pathologic inflammation, such as cancer, MDSC are recruited from myeloid precursor cells in the bone marrow, circulate in the blood of cancer patients at levels many times greater than in healthy controls, and traffic into the TIME where they suppress endogenous and treatment-induced anti-tumor immune responses.11–13 Thus, therapeutic approaches that target the induction of MDSC from myeloid precursor cells have the potential to both reverse a major cellular mediator of immune suppression and promote the induction of beneficial myeloid cell types.
Several strategies have been proposed to modulate the differentiation of MDSC into functional myeloid cells that have reduced immunosuppressive activity, increased anti-tumor effects, or both. Trans-retinoic acid (ATRA) has been used as a treatment option to differentiate MDSC into mature myeloid cells expressing increased levels of HLA-DR,8,14 leading to decreases in tumor growth and increased in CD8 T cell responses.9 ATRA has also been used in combination with anti-CD25 with the intent of targeting MDSCs and Tregs simultaneously10 leading to depletion of MDSC in ATRA-treated patients with high (> 150 ng/mL) but not low (< 135 ng/mL) plasma concentrations of ATR.15 Other differentiating agents like Vitamin D3 have also been used to mature MDSC and induce anti-tumor activity16 . Therapeutic interventions targeting MDSCs effector molecules like inducible nitric oxide synthase (iNOS), arginase, and reactive oxygen specie17,(18,19 have also been tested, but as yet these approaches remain experimental.
Since MDSC have many properties similar to myeloid precursor cells which differentiate to M1 (tumoricidal) and M2 (tumor promoting) macrophages20,21 another approach targeting MDSC is directing their repolarization into a M1/tumorcidal myeloid cells by inhibiting the paired immunoglobulin-like receptor B (PIR-B) signaling pathwa.22 As yet, no approaches targeting MDSC have entered clinical practice, emphasizing the need for novel therapeutic approaches that directly address the hostile tumor microenvironment by depleting, inactivating, or repolarizing MDSCs. In our current study, we describe a unique approach to MDSC repolarization, in which exposing myeloid precursor cells to TGF-β1 during their development into MDSC leads to loss of immunosuppressive function and acquisition of immune-stimulating and anti-tumor activity.
TGF-β1 is a pleiotropic cytokine which has wide-ranging effects on differentiation and function of numerous cell types, including myeloid cells;23 and is known to mediate cell fate decisions and differentiation of hematopoietic cell populations.24,25 Most prior literature describing TGF-β1 immune effects in cancer focuses on its immunosuppressive functions, including its ability to prevent T-cell proliferation and activation, drive the development of inducible regulatory T cells (Tregs), and its well-described role in polarizing macrophages towards the tumor-promoting M2 phenotype.26–28 Despite the predominantly immunosuppressive role of TGF-β1 in the TIME, a previous report found that MDSC generated ex vivo from human peripheral blood mononuclear cells (PBMC) in the presence of TGF-β1 had reduced ability to suppress T cell proliferation.29 We also previously reported that inhibition of the MDSC-promoting inflammatory molecule inducible nitric oxide synthase (iNOS) led simultaneously to TGF-β1 release in the TIME and reduced MDSC function, suggesting a possible relationship between these findings.30
In the present study, we describe unexpected immune-stimulating and anti-tumor effects of MDSC derived ex vivo in the presence of TGF-β1 (TGFβ-MDSC). We further demonstrate that these cells are capable of inducing in vivo tumor clearance and long-term survival of mice bearing established tumors in combination with radiotherapy. Our results suggest a novel role for TGF-β1 programmed MDSC as a highly active adoptive cellular therapy suitable for combination with radiotherapy and other therapeutic approaches.
Results
MDSC induced in the presence of TGF-β1 acquire a unique phenotype
To evaluate the effect of TGF-β1 on the generation of MDSCs from myeloid precursor cells, bone marrow from immunocompetent C57bl/6 mice was co-cultured ex vivo with MEER tumor-conditioned medium in the presence or absence of TGF-β1 for 5 days. MEER cells, a syngeneic tumor line derived from mouse pharyngeal epithelial cells transformed with Ras and the E6 and E7 oncogenes of HPV 16, produce modest levels of TGF-β1 (Supplementary Figure S1), so the major source of TGF-β1 in culture is exogenous addition. Following culture, cells were stained for myeloid markers and profiled by multicolor flow cytometry. Bone marrow cells cultured with tumor conditioned medium with or without TGF-β1 showed similar increases in the percentages of CD11b+/Gr-1+ double-positive MDSC (Figure 1A, top). Geimsa stain after cytospin of CD11b+ sorted cells showed an abundance of neutrophil-like cells in all tested culture conditions. Control (tumor-conditioned media alone) cultured MDSCs showed a 10-fold decrease in macrophage-like myeloid cells compared to untreated bone marrow cells, while addition of TGF-β1 induced a striking 20-fold increase of macrophage-like and monocyte-like cells (Figure 1A bottom and B; p < 0.001) compared to untreated (no treatment) cells. Addition of TGF-β1 to bone marrow cells in the absence of tumor conditioned medium had no effect on myeloid cell morphology (data not shown). To characterize protein and signaling pathway alterations in TGFβ-MDSC, we performed reverse phase protein array (RPPA) on control and TGFβ-MDSC. RPPA is an antibody-based targeted proteomic platform that enables quantification of both total and phospho-protein expression.31 Principal component analysis (PCA) of the RPPA data confirmed clear separation of control and TGFβ-MDSC (Figure 1C), with 93% RPPA variation explained by the first principal component. Of the 211 signaling proteins quantified in the RPPA platform, we identified 16 proteins or phosphoproteins whose expression was significantly (p-value < 0.05, fold change > 1.5 or < 0.67) altered in TGFβ-MDSC compared to control MDSC (Figure 1D). From the heat-map generated from RPPA, it was evident that p-SMAD2 was the most significantly changed protein, upregulated 5-fold in TGFβ-MDSC (Figure 1D). Since SMAD2 is an important downstream mediator of TGF-β¹ signaling, we validated p-SMAD2 protein expression in MDSC by flow cytometry and found that TGFβ-MDSC had 4-fold increased p-SMAD2 expression compared to control MDSC. (Figure 1E).
Figure 1.

TGFβ-MDSC acquire more macrophage like phenotype and upregulate maturation markers.
Naïve bone marrow cells cultured in medium only (no treatment) or bone marrow cells cultured with MEER tumor conditioned medium alone (Control) or in the presence of TGF-β1 (TGF-β1) were used to generate MDSC. After 5 days in culture MDSC were evaluated by flow cytometry or Giemsa staining. A) Representative flow cytometry plots showing percent of total MDSC (n = 16) along with their respective Giemsa images depicting morphology of CD11b+ cells B) Quantitation of different cellular fractions expressed as a stacked bar graph (n = 8–12/group). C-D) Protein lysates from control or TGF-β MDSC were profiled with Reverse Phase Protein Assay (RPPA). C) Principal Component analysis (PCA) of the RPPA data. x, y, and z axes represent three major principal components. Control MDSC samples are represented as red dots; TGFβ-MDSC samples as blue dots D) Heat map of proteins (n = 16) most differentially-expressed between control and TGFβ-MDSC (p-value < 0.05, fold change > 1.5 or < 0.8). E) Representative flow cytometric histogram overlay of p-SMAD2 expression of control and TGFβ-MDSC (top), and bar graph showing cumulative data from atleast 3 experiments. F) Representative flow cytometric histogram overlay (top) showing cell surface expression of MHC II, CD86, Flt-3 and Fas-L on MDSC generated from no treatment, control and TGF-β conditioned tumor medium with summary bar graph below showing mean fluorescence intensity (n = 5–9 per graph). G) Summary of MHC Class II, CD86 and Fas-L expression in TGFβ¯ MDSC generated in the presence or absence of 3 µM of SMAD-2 inhibitor, SB431542 (n = 12). Error bars indicate standard error of the mean (SEM). *p ≤ 0.05. Each graph summarizes data obtained from at least 3 experiments. Error bars indicate standard error of mean (SEM). *p ≤ 0.05, ** p ≤ 0.01
To further characterize the altered MDSC phenotype induced by TGF-β1, we assessed changes in markers of maturation and co-stimulation namely MHC II, Flt-3, Fas-L and CD86 by flow cytometry. TGFβ-MDSC showed a 2-fold or greater upregulation of all myeloid maturation and co-stimulation markers compared to control MDSC (Figure 1F). Further, inhibition of SMAD2 during the development of TGFβ-MDSC led to > 50% decrease in expression of MHC II, CD86 and FAS-L, confirming a role for TGF-β1-induced SMAD2 signaling in mediating increased maturation of TGFβ-MDSC and upregulation of co-stimulatory molecule expression (Figure 1G). These data together suggest that while TGF-β1 does not quantitatively affect MDSC induction, it induces a more macrophage-like MDSC phenotype, with enhanced co-stimulatory molecule expression and myeloid maturation markers.
MDSC induced in the presence of TGF-β1 lose the ability to suppress T cell proliferation, and acquire enhanced antigen-presenting and T cell costimulatory capability
A hallmark of MDSC function is their potent ability to inhibit T cell proliferation by diverse mechanisms including upregulation of iNOS, arginase, production of nitric oxide (NO)/reactive oxygen species (ROS), and upregulation of programmed death ligand-1 (PD-L1)32–36 . To determine whether induction in the presence of TGFβ-1 altered MDSC functional markers, control and TGFβ-MDSC were profiled by multicolor flow cytometry (Figure 2A). We observed no change in expression of arginase, or ROS production, but observed a 3-fold decrease in expression of iNOS and NO by TGFβ-MDSC compared to control MDSC. We observed no change in iNOS or NO expression levels in myeloid cells cultured in presence of TGF-β1 without tumor-conditioned medium (data not shown).
Figure 2.
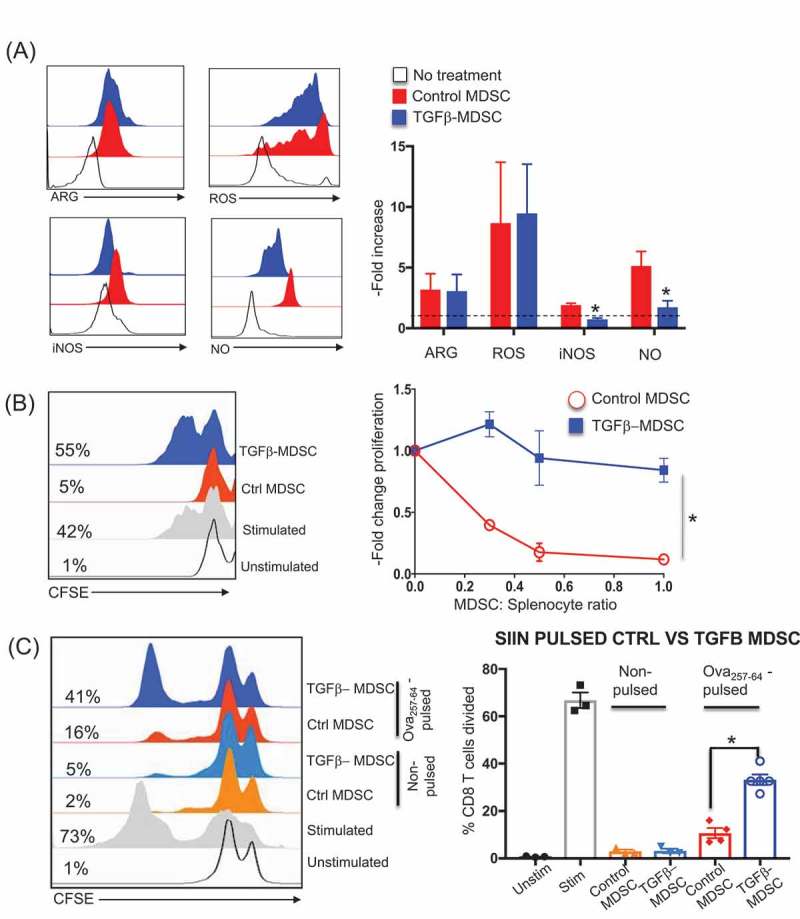
TGFβ-MDSC lose immunosuppressive function, promote T cell proliferation and present antigen.
Control and TGFβ¯MDSC were isolated and screened for functional markers. A) Histogram overlay and corresponding bar graphs (normalized to no treatment) showing mean fluorescent intensity (MFI) of iNOS, Nitric oxide (NO), Reactive oxygen species (ROS), and Arginase (ARG) in MDSC from no treatment, control or TGFβ-MDSC (n = 9). Control or TGFβ-MDSC were then co-cultured at different ratios with CFSE labelled naïve splenocytes (activated with soluble anti-CD3 and anti-CD28 antibodies). After 72 hours, CFSE dilution profile of CD8 T cells were obtained by flow cytometry B) Representative CFSE plots for unstimulated; anti-CD3- and anti-CD28- stimulated splenocytes, plus control or TGFβ¯ MDSC (left). Relative proliferation of CD8 + T cells co-cultured with either control or TGFβ- MDSC normalized to T cells stimulated with anti-CD3 and anti-CD28 alone across various MDSC: splenocyte cell ratios (n = 6) (right). C) Representative histogram plot overlay showing CD8 T proliferation (expressed as a percentage of CD8 T cells that have divided) using CFSE dilution analysis, obtained with either non-pulsed or ova 257–64 pulsed control or TGFβ¯MDSC (left); bar graph depicts aggregate data (n = 4) Each graph summarizes data obtained from at least 3 experiments. Error bars indicate standard error of the mean (SEM). *p ≤ 0.05.
To assess the effects of TGF-β1 on MDSC function, CD8 + T-cell proliferation was evaluated by CFSE dilution analysis of anti-CD3 and anti-CD28 activated splenocytes co-cultured with ex-vivo derived control or TGFβ-MDSC. While control MDSC inhibited CD8 + T-cell proliferation in a dose dependent manner, TGFβ-MDSC failed to inhibit proliferation across all MDSC:CD8 ratios (Figure 2B). A similar decrease in T cell suppression ability, associated with loss of iNOS/NO expression, was also observed when TGFβ-MDSC were generated using conditioned medium derived from the MT-RET murine melanoma tumor cell line (Supplementary Figure S2), which we have previously shown to be a strong inducer of MDSC.33 From these data, we infer that TGF-β1 reverses MDSC T-cell suppressive function, associated with downregulation of iNOS and NO. Since we observed increased expression of costimulatory molecules on TGFβ-MDSC, we tested their ability to present antigen to and induce antigen-specific T cell activation (Figure 2C). Both control and TGF-β MDSC were pulsed with the antigen Ova 257–264 (SIINFEKL) and co-cultured with CFSE labeled MHC Class I restricted OT1 SIINFEKL–specific CD8 + T cells. We observed that unpulsed MDSC (both control and TGF-β1) failed to induce proliferation of OT1 cells. While the extent of proliferation was less than peptide-pulsed CD8 T cells alone, antigen-pulsed TGFβ-MDSC induced a > 2.5-fold increase in OT1 proliferation compared to control MDSC, demonstrating enhanced antigen-presenting and T cell stimulating capability.
Tgfβ¯mdsc acquire tumor killing activity
In addition to suppression of T-cell function, MDSCs promote cancer growth and survival through non-immune mechanisms, such as enhanced angiogenesis and protection of tumor cells from apoptosis.37,38 We determined the direct effects of TGFβ-MDSC on tumor survival and growth by co-culturing control or TGFβ-MDSC with MEER cells at different ratios for 48 hours prior to viability assessment. MEER cells co-cultured with TGFβ-MDSC at a 1:1 ratio experienced > 4-fold greater levels of tumor cell death compared to control MEER cells co-cultured with control MDSC, as measured by LDH release (Figure 3A) and a > 2-fold greater expression of the apoptotic marker annexin V (Figure 3B). (Co-culture flow cytometry gating strategy is presented in Supplementary Figure S3). Since apoptosis induced by Fas-ligand (Fas-L) activation of Fas is a well-characterized mechanism of tumor cell killing utilized by cytotoxic immune cells,39 and since TGFβ-MDSC upregulated Fas-L by > 2-fold compared to control MDSC (Figure 1F), we determined the effect of blocking mAb to Fas-L on tumor killing in co-culture Ant-Fas-L antibody reduced TGFβ-MDSC-induced tumor killing by nearly 2-fold (Figure 3C), indicating that Fas-L upregulation by TGFβ-MDSC strongly contributes to its tumor-killing phenotype.
Figure 3.

TGFβ¯MDSC-mediate tumor killing both ex vivo and in vivo.
MEER tumor cells were co-cultured in vitro with control or TGF-β MDSC at a 1:1 ratio for 48 hours after which cell viability and tumor cytotoxicity were measured in tumor cells. A) Percent cytoxicity as measured by LDH release of MEER cancer cells co-cultured with control or TGFβ-MDSC (n = 9–10 per group). B) Percentage of annexin positive MEER tumor cells after 48 hours co-culture with control or TGFβ-MDSC (n = 9). C) Cumulative bar graph showing percentage of annexin positive (n = 6) tumor cells co-cultured with control or TGFβ¯ MDSC, at a 1:1 ratio, either in the presence of FAS-L neutralizing antibody or isotype control. D) Representative images of H&E or Ki-67 and Caspase-3 IHC in 3D spheroids co-cultured with control or TGFβ¯MDSC for 72h (left) along with quantification of Ki-67 and Caspase-3 levels (expressed as mean fluorescence) shown to the right (n = 10). Control or TGFβ¯MDSC were intratumorally injected on days 4, 8, and 12 following tumor cell inoculation alone or in combination with 30 Gy radiation delivered as 2 × 15 Gy doses to MEER tumors. E) Tumor growth curves (n = 9–10 mice per group). F) Representative H&E and IHC of Ki-67 and Caspase 3 in tumor sections at day 20 after tumor cell inoculation shown on top. Quantitation expressed as mean fluorescence below. Error bars indicate standard error of the mean (SEM). *p ≤ 0.05, ** p ≤ 0.01.
Tumor killing by TGFβ-MDSC was further tested in a 3-dimensional MEER tumor spheroid model, in which MEER cells and purified CD11b+ MDSC were co-cultured in a “hanging drop” suspension and allowed to form spheroids containing both cell types. We found that spheroids created with TGFβ-MDSC, had a pronounced central zone of tumor cell clearing when compared to those created with control MDSC (Figure 3D). Further staining and analysis of TGFβ-MDSC/MEER spheroids revealed a 2-fold increase in the expression of pro-death marker cleaved caspase-3 and a significant decrease in tumor cell proliferation measured by Ki-67, compared to control MDSC co-cultured spheroids (Figure 3D).
To determine whether adoptive transfer of TGFβ-MDSC into the tumor microenvironment might lead to direct killing of cancer cells in vivo, mice bearing palpable MEER tumors were intratumorally injected with ex vivo derived control or TGFβ-MDSC on days 4,8 and 12 after tumor inoculation. While mice treated with TGFβ-MDSC showed only modest reduction in tumor growth compared to those injected with control MDSC (Figure 3E), histology of resected tumors revealed marked zones of central necrosis in tumors injected with TGFβ-MDSC, similar to what we observed in TGFβ-MDSC treated spheroids (Figure 3F). These zones of clearance featured > 2-fold decrease in Ki-67 expression and a comparable increase in Caspase-3 immunostaining, indicating decreased tumor cell proliferation and higher levels of apoptosis in vivo, respectively. Collectively these data suggest that TGFβ¯MDSC directly induce death of tumor cells in vitro and in vivo.
Combination of TGFβ-mdsc adoptive transfer and radiotherapy leads to regression and long-term tumor control in vivo
The observations above demonstrate that TGFβ-MDSC not only lose tumor-protective immune suppression capabilities but also acquire functions (antigen presentation and tumor killing) which might be leveraged for cancer treatment. Since TGFβ-MDSC mediated direct tumor killing is Fas/Fas-L dependent (Figure 3C), and FAS expression on tumor cells is known to be upregulated by radiation treatment,40 we sought to further enhance the anti-tumor efficacy of TGFβ-MDSC by delivering them in combination with external beam radiation. We first demonstrated that reatment of MEER tumor cells with radiation in vitro upregulated Fas expression in a dose-dependent fashion (Figure 4A). We then treated established MEER tumors in vivo with two 15 Gy doses of tumor-directed radiation (spaced 8 days apart) delivered concurrently with three intratumoral injections of control or TGFβ-MDSC, or sham (PBS) injection control. Treatment with radiation alone partly suppressed tumor growth, and approximately 20% of treated mice were alive at 150 days post tumor inoculum. The beneficial effects of radiotherapy were completely reversed by concurrent injection of control MDSC, which induced only a slight growth delay compared to tumors in untreated mice and led to 0% surviving mice by day 60 post tumor inoculum (Figure 4B, C & D). However, radiotherapy combined with intratumoral injection of TGFβ-MDSC significantly decreased tumor growth (Figure 4B & C), and resulted in approximately 70% of treated mice alive at 150 days post tumor inoculum (Figure 4D). In order to evaluate whether combination therapy (15 Gy X 2 + TGFβ-MDSC) conferred a memory response against the tumor, mice which had cleared tumors, 150 days post tumor inoculation (3 mice) were further re-challenged with 8–106 MEER tumor cells in the right flank (opposite side from the initial site of tumor inoculum). 3/3 mice failed to develop palpable tumors by 4 weeks after rechallenge despite the high tumor inoculum (data not shown) indicating development of a protective memory immune response. A similarly striking response to combination therapy with TGFβ-MDSC and radiotherapy was also seen in the aggressive B16-F10 melanoma tumor model, in which tumor-bearing mice treated with radiotherapy and TGFβ-MDSC experienced significantly decreased tumor growth and 40% of mice completely rejected their tumors (Supplementary Figure S4). These data strongly support the therapeutic potential of intratumoral injection of TGFβ-MDSC combined with radiotherapy.
Figure 4.

Intratumoral TGFβ-MDSC in combination with radiotherapy leads to durable tumor clearance in vivo.
A) Flow cytometry histogram overlay and corresponding MFI bar graph showing FAS expression in MEER tumor cells 48 hours after exposure to increasing doses of radiation (N = 2). B) Individual mouse tumor growth curves showing tumor size in mm2 for mice receiving no treatment (control), radiation alone (15 Gy x 2), radiation with intratumoral control MDSC (15 Gy x 2 + control MDSC), or radiation with intratumoral TGFβ-MDSC (15 x 2 + TGFβ-MDSC) (n = 8–10 mice per group). Each line represents tumor growth curve of an individual mouse. The individual graphs also show the ratio of surviving mice on day 60. The treatment schedule is depicted as a timeline above the graph C) Tumor growth curves depicting cumulative tumor sizes in mm2 from mice receiving no treatment or control, 15 Gy x 2 alone, 15 Gy x 2 + Control MDSC and 15 Gy x 2 + TGFβ-MDSC. D) Kaplan-Meier survival plots for each treatment group (n = 8–10 mice per group). Colored p-value symbols indicate the line to which the group was compared. Each experiment was performed at least 2 times. Error bars indicate standard error of mean (SEM). *p ≤ 0.05.
Combination of TGFβ- MDSC and radiotherapy in vivo induces activation of the tumor-immune microenvironment and enhanced antigen-specific immune response
The strong memory immune response implied by resistance to rechallenge of mice that cleared tumor after treatment with TGFβ-MDSC and radiotherapy suggested that initial tumor clearance may be also occur through immune mechanisms In order to further characterize effects of intratumoral injection of TGFβ-MDSC on the TIME, a single dose of 15 Gy radiation was delivered to established MEER tumor-bearing mice followed by 2 intratumoral injections of control or TGFβ-MDSC. Tumors were harvested 2 days later and were of comparable sizes in the different treatment groups, ensuring lack of bias by tumor size. The tumor-infiltrating leukocyte (TIL)-free tumor fraction was evaluated to determine tumor cell expression of the pro-apoptotic marker cleaved Caspase-3/7 and for their expression of MHC class I. We found that, while single treatments had little effect, TGFβ-MDSC in combination with radiotherapy induced a nearly 2-fold increase in the expression of both Caspase-3/7 (Figure 5B) and MHC Class I (Figure 5C). When we examined the induction of tumor antigen-specific T cells, we found that both TGFβ-MDSC and (to a lesser degree) tumor-directed radiation were capable of increasing the numbers of tumor-infiltrating HPV16 E7 tetramer-positive CD8 + T cells, with the most striking effect (> 5-fold upregulation) seen for TGFβ-MDSC in combination with radiotherapy. From these data, we infer that the injection of TGFβ-MDSC on its own is capable of inducing an antigen-specific CD8 + T cell response, but tumor directed radiation is required to sensitize tumor cells to killing through upregulation of Fas (direct TGFβ-MDSC-mediated killing), and MHC I (sensitization to T cell-mediated killing).
Figure 5.

Intratumoral TGFβ-MDSC and radiotherapy activates the tumor immune microenvironment.
15 Gy external beam radiation was administered as a single dose directly to the MEER tumor in the left flank of mice on day 20 after initial tumor inoculum. This was followed by intratumoral administration of control or TGFβ¯MDSC on days 21 and 22. Tumor was harvested on day 24 and the tumor infiltrating leukocyte fraction (TIL) and TIL- free (tumor-containing) fractions were stained for E7 tetramers, and Caspase-3 and MHC Class I respectively A) Schematic representation of the treatment schedule. B-C) Representative histogram overlay and corresponding bar graph depicting MFI of B) Caspase-3/7 and C) MHC Class I (n = 6–8 mice per group). D) Representative FACS plots and corresponding bar graph depicting percentages of CD8 + T cells expressing E7 tetramer (n = 6–8 mice per group). Error bars indicate standard error of the mean (SEM). ***p ≤ 0.001, **** p ≤ 0.0001.
TGFβ-mdsc derived from human PBMC share key features of murine TGFβ-mdsc
To determine whether human MDSC derived in the presence of TGF-β1 acquire a unique phenotype similar to that observed for murine TGFβ-MDSC, MDSC were generated from human peripheral blood mononuclear cells (PBMC) co-cultured with SCC47 (squamous carcinoma cell line producing relatively low amounts of TGF-β1, Supplementary Figure S1) tumor conditioned medium ± TGF-β1. Addition of TGF-β1 resulted in a 3-fold greater accumulation of CD33+, CD11b+, HLA-DRlow/- MDSCs than in its absence (Figure 6A). We then evaluated human MDSC for their ability to suppress autologous T-cell proliferation on a per-cell basis. Control or TGFβ-MDSC were co-cultured with cell trace violet labeled CD3 + T cells and stimulated with plate-bound anti-CD3 antibody for 72 hours. While control MDSC suppressed T cell proliferation even at 1:8 (MDSC: T cell) ratio, TGFβ-MDSC were only significantly suppressive at much higher (1:1) ratios (Figure 6B). Consistent with murine data, we found that both control and TGFβ¯MDSC failed to stimulate T cell proliferation in the absence of exogenous antigen, human TGFβ-MDSC pulsed with a mixture of CMV, EBV, and flu (CEF) peptides exhibited 3-fold greater T cell proliferation as compared to control MDSC (Figure 6C). Unlike murine MDSC, we did not observe significant upregulation of MHC II (HLA-DR) expression in human MDSC after induction in the presence of TGF-β1 (Figure 6A). However, expression of the co-stimulatory molecule CD86 was significantly (3-fold) upregulated in human TGFβ-MDSC compared to control MDSC (Figure 6D). We further examined expression of molecules important to MDSC function (iNOS, ROS and PD-1 ligand [PD-L1]) in control and TGFβ-MDSC. We observed no change in iNOS or ROS (data not shown); however, expression of PD-L1 was significantly (two-fold) decreased in TGFβ-MDSC compared to control-MDSC (Figure 6E). Next, we examined the effect of in vitro co-culture of human TGFβ-MDSC with human head and neck cancer cells on tumor viability. After 72 hours co-culture, we observed a 1.5-fold increase in annexin expression in SCC47 HPV-positive human squamous cell carcinoma cells when co-cultured with human TGFβ-MDSC (Figure 6F), consistent with the increased tumor-killing ability we noted in murine TGFβ-MDSC. In parallel experiments using D-HEp3 cells (HPV-negative squamous cell carcinoma) we found that 48hour co-culture with TGFβ-MDSC significantly decreased the number of viable tumor cells (Supplementary Figure S5). Together, these data show that key features of murine TGFβ-MDSC are recapitulated by MDSC derived from human cells in the presence of TGF-β1, including loss of T cell suppression, upregulation of CD86, and acquisition of tumor cell killing ability.
Figure 6.

Human TGFβ¯MDSC lose ability to suppress T cell proliferation and acquire enhanced antigen presentation ability.
Control or TGFβ¯MDSC were derived from human PBMCs by culture with SCC47 tumor conditioned medium ± TGF-β1 prior to functional characterization. A) Representative flow cytometry plot showing induction of CD11b+ CD33+ HLA-DRlow/- MDSC in the absence or presence of TGF-β1. B) Representative histogram and analysis of T cell proliferation (using anti-CD3/anti-CD28 + IL-2 stimulation) in the presence of control MDSC or TGFβ-MDSC across the indicated MDSC:T cell ratios (n = 5). Percentages indicate % of proliferating T cells. Cumulative analysis of fold change in T cell proliferation across various MDSC: CD3 T cell ratio is presented in the right panel. C) Representative CD3 T cell proliferation histogram overlay from unstimulated, CEF-pulsed, CEF-pulsed control or TGFβ-MDSC and non-pulsed control or TGFβ-MDSC along with corresponding bar graph showing-fold change of CD3 T cell proliferation from CEF pulsed control and TGFβ-MDSC groups (n = 8). D-F) Representative flow cytometry histogram and quantitative bar graph showing-fold MFI change in D) CD86 expression and E) PD-L1 expression in TGFβ-MDSC compared to control MDSC (n = 4). F) Changes in Annexin+ SCC47 cells following 48 hours co-culture with either control or TGFβ-MDSC. In all cases, -fold change is relative to the control MDSC condition (n = 5). *p ≤ 0.05, ** p ≤ 0.01.
Discussion
The developmental plasticity of myeloid cells provides opportunities for their “reprogramming” or directed polarization towards anti-tumor phenotypes. While the classical split between M1 (tumoricidal) and M2 (tumor-promoting “alternatively polarized”) macrophages41,42 is best-understood, MDSC are also potential targets of reprogramming/polarization-based therapeutic approaches.43,44 In the present study, we propose a novel use of TGF-β1 during the induction of MDSC from myeloid precursors, to polarize/program these cells to adopt an immune-stimulating and anti-tumor phenotype. We further propose that TGF-β¹-programmed MDSC can be utilized for adoptive cellular immunotherapy, leading to striking anti-tumor effects when combined with radiotherapy. Unlike CAR T cells or adoptive T cell transfer, the anti-tumor effect of TGFβ-MDSC is antigen- and haplotype-independent, facilitating application to diverse solid tumors and ease of administration.
The role of TGF-β¹ in promoting the development/differentiation of immunosuppressive immune cells and an immunologically hostile tumor microenvironment has been well described.26 A key facet of TGF-β1 is its ability to regulate hematopoietic stem/progenitor cell self-renewal by affecting proliferation, survival, and differentiation.28 With respect to MDSC, TGF-β¹ has generally been shown to enhance MDSC induction or to act as an MDSC effector mechanism.45 However, prior studies have not specifically investigated the effects of TGF-β1 on cancer-induced development of MDSC from precursor hematopoietic cells under defined conditions. Since we previously observed an association between TGF-β¹ release and loss of MDSC immunosuppressive function in tumor treated with a small-molecule iNOS inhibitor30 we set out to determine the specific effects of TGF-β1 on myeloid cells destined to become MDSC after exposure to tumor-conditioned medium or tumor-associated cytokines. The exposure of nascent MDSC to TGF-β1ex vivo led to the acquisition of a unique myeloid cell phenotype with immune-stimulating and tumor killing properties (TGFβ-MDSC). Although CD11b-sorted TGFβ-MDSC acquired a more macrophage-like morphology, these cells did not clearly fall into either the M2 (tumor-promoting) or M1 (tumoricidal) macrophage categories.44,46 TGFβ-MDSC share attributes of both M1 (Cd86hi, MHC class II hi) and M2 (F4/80 hi, MHC class II hi, iNOS low, Cd206 hi [data not shown]) macrophages. Despite their anti-tumor activity, TGFβ-MDSC fail to acquire many markers of M1-polarized macrophages, and in fact down-regulate iNOS, a classical M1 marker. Thus, we consider them a form of alternatively polarized “macrophage like” MDSC, rather than a tumoricidal M1 macrophage. The distinct phenotype differentiating TGFβ-MDSC from conventional MDSC is supported by our principal component analysis of signal transduction protein expression profiles determined by reverse-phase protein array (RPPA) which separated conventional and TGFβ-MDSC into distinct clusters and identified TGF-β1-induced SMAD2 activation as a key feature of TGFβ-MDSC.
We found that both murine and human MDSC precursors developed cells with reduced immunosuppressive and enhanced anti-tumor activity when induced in the presence of TGF-β1. However, induction of human and murine TGFβ-MDSC involved modulation of different effector mechanisms: downregulation of iNOS and NO in the murine TGFβ-MDSC and down regulation of PD-L1 in human TGFβ-MDSC. Murine but not human TGFβ-MDSC upregulated class II MHC expression. Conversely, both murine and human TGFβ-MDSC also upregulated the co-stimulatory molecule CD86, conferring enhanced T cell stimulating properties. Thus, both common and species-specific mechanisms drive the switch from an immune suppressive to an immune stimulating and anti-tumor phenotype when MDSC are induced in the presence of TGF-β1. Based on these findings, we propose a model (Figure 7) where TGF-β1 plays a central role in the generation of reprogrammed MDSC (TGFβ-MDSC) that have very different properties from conventional MDSC. These findings have potential therapeutic implications, since these cells acquire both pro-immune and tumor-killing properties.
Figure 7.
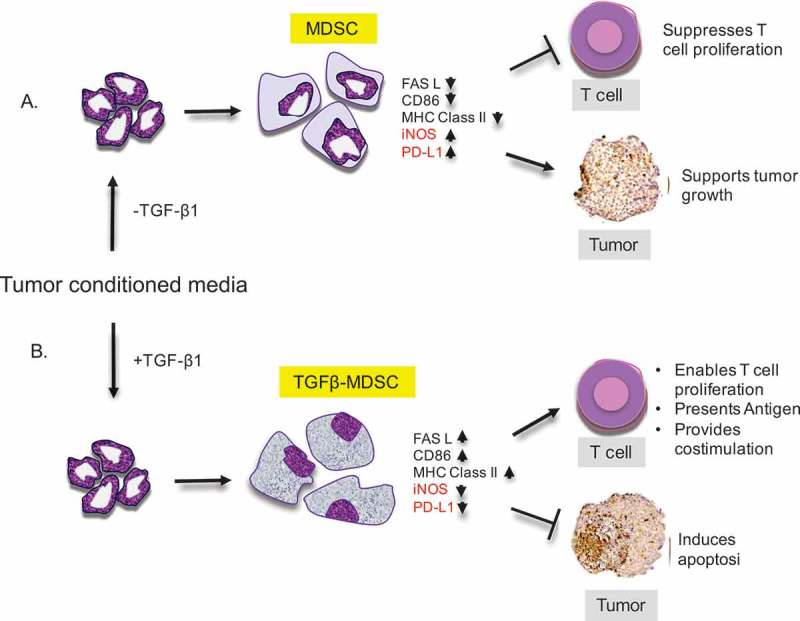
Working Model.
Myeloid precursor cells when A) co-cultured with tumor enriched conditioned medium in the absence of TGF-β1, give rise to conventional myeloid derived suppressor cells (MDSC) which have increased levels of iNOS and PD-L1. They suppress T cell proliferation and promote tumor growth. When precursor cells are co-cultured with B) tumor conditioned medium in the presence of TGF-β1, they give rise to a distinct population of MDSCs (TGFβ-MDSC) that are macrophage-like and have upregulated levels of FAS-L, CD86 and MHC Class II. They lose ability to suppress T cell proliferation, acquire enhanced T – cell stimulation activity, and mediate FAS-FAS-L driven apoptosis in tumor cells.
TGF-β1 has been frequently implicated in apoptosis via Fas-L upregulation or through signaling through SMAD protein47.48 In studies conducted by Chatzaki et al TGF-β1 upregulated Fas-L expression in the endometrial stromal cells leading to apoptosis in during embryonic remodeling. Our data therefore is consistent with above studies, and consistent with a pivotal role for TGF-β1-induced SMAD2 signaling in the upregulation of Fas-L in TGFβ-MDSC. A role for SMAD2 signaling in the upregulation of Fas-L is also supported by decreased Fas-L expression in the presence of a SMAD2 inhibitor. Antibody-mediated neutralization of Fas-L decreased TGFβ-MDSC tumor killing activity, while conversely, external beam radiation further increased Fas expression on tumor cells, thus increasing their sensitivity to TGFβ-MDSC-mediated killing. Together, these data point to a central role for Fas-L/Fas-mediated apoptosis in the anti-tumor activity of TGFβ-MDSC.
Although TGFβ-MDSC induced FAS- dependent tumor killing in vitro, they had modest anti-tumor effect in vivo as a monotherapy. Since FAS is upregulated by radiotherapy, we tested the combination of TGFβ-MDSC and tumor-directed radiation, a standard-of-care therapeutic approach for head and neck squamous cell carcinoma and many other solid tumors. We observed in two murine solid tumor lines (MEER HPV-associated head and neck squamous carcinoma cells and B16-Melanoma) that intratumoral injection of conventional TGFβ-MDSC in combination with radiotherapy leads to regression and long-term clearance of established tumors. In contrast, intratumoral injection of conventional MDSC reverses the beneficial effects of radiotherapy. The striking anti-tumor effects of TGFβ-MDSC in combination with radiotherapy are shown to be a combination of direct MDSC-mediated killing of FAS-upregulated cancer cells, and enhanced antigen-specific antitumor CD8+ response.
Limitations of this approach include the lack of in vivo tumor-killing data with human TGFβ¯MDSC and the unknown feasibility of their production in numbers suitable for human therapy, since they only constitute a fraction of CD33+ cells. However, it is encouraging that induction in the presence of TGF-β1 increases the CD33+ MHCIIlo “MDSC” fraction. TGFβ-MDSC, like other granulocytic cells tend to be short-lived (48–72 hours- data not shown) and multiple intratumoral injections were required for a succcessful anti-tumor response in vivo. We also have not yet investigated the ability of TGFβ-MDSC to home to tumors when administered through IV or IP routes, rather than direct intratumoral injection. Future studies will investigate the in vivo anti-tumor activity of human TGFβ¯MDSC, as well as the persistence and homing potential human and murine TGFβ¯MDSC in vivo.
In summary, we have shown that MDSC induced in the presence of TGF-β1 (TGFβ¯MDSC) acquire a novel phenotype characterized by loss of T cell suppression ability and acquisition of enhanced antigen-presenting and tumor killing functions. The striking anti-tumor effects of TGFβ-MDSC in vivo, particularly in combination with radiotherapy, strongly support their further investigation as a novel strategy for antigen- and haplotype-independent adoptive cellular therapy of solid tumors. These data underscore the functional plasticity of tumor-induced myeloid cells, and the potential benefits of continued development of specific approaches to leveraging this plasticity for therapeutic benefit.
Materials and methods
Mice and tumor model
C57BL/6 mice were obtained from the center for comparative medicine housed in Baylor College of Medicine animal facility under specific pathogen-free conditions. C57BL/6-Tg(TcraTcrb)1100Mjb/J (“OT-1”) transgenic mice (stock number 003831) were obtained from Jackson Laboratories. All animal experiments were performed in accordance with the regulations of the local Institutional Animal Care and Use Committee. Stable lines of MEER murine pharyngeal epithelial cells expressing HPV16 E6 and E7, and hRas, were obtained from Dr. John Lee and Dr. Chad Spanos at the Sanford Research Center/University of South Dakota and maintained in E-media as previously described49 and used as a transplantable flank tumor. 1 × 106 MEER cells were injected subcutaneously into male C57BL/6 mice, since we have observed that tumors grow poorly in female mice.
Ex vivo generation of murine mdscs
Bone marrow was harvested from femur of naïve C57BL/6 mice and made into a single cell suspension using RPMI 1640 media with 10% FBS and 1% Pen/Strep. 3 × 106 bone marrow cells were seeded in a 6-well plate and co-cultured with 30% v/v of MEER tumor conditioned medium (derived from near confluent flask) ± TGF-β1-10ng/ml (Millipore GF439) for 5 days. For p-SMAD2 inhibition, MDSCs were derived in the presence of 3 μM of TGF-β RI Kinase Inhibitor VI, SB431542 (Calbiochem- 301836–41-9). At the end of culture conditions, either unfractionated cells were harvested or CD11b+ cells were further enriched using positive selection with CD11b+ magnetic beads (Miltenyi Biotec; 130–049-601).
Morphology of MDSC
Wright-Giemsa staining was performed on cytospin preparations of CD11b+ cells to assess the morphology of control and TGFβ-MDSC. Images were acquired and evaluated using a Nikon Eclipse E400 microscope.
Histology and immunohistochemistry
Slides were prepared from murine tumor tissues or spheroids (murine and human) for histology using hematoxylin and eosin stains from paraffin embedded blocks. Immunohistochemistry was also performed on slides using Ki-67 (EMD Millipore AB9260) and caspase-3 antibodies (Abcam ab4051).
Tumor harvest and digestion
Tumors were dissected from euthanized mice, cut into 1-2mm cubes and digested in 10X digestion cocktail (collagenase I, 10 mg/mL; collagenase IV, 2500 U/mL; and DNase I, 200 U/mL, all from Sigma-Aldrich); in base RPMI-1640 media without FBS) for 1 h at 37° while lightly shaking at 60–100 rpm on an orbital plate shaker. After 1 h, the collagenase treatment was neutralized by adding 1 mL of RPMI-1640 media supplemented with 5% FBS and 2 mM EDTA to each well. Tumor pieces were mechanically disaggregated by pushing them through a strainer (Falcon) with a 10 ml syringe plunger (Falcon) and collected into 10 ml RPMI complete media. Tumor infiltrating leukocytes were derived from the unfractionated tumor using a ficoll gradient, washed twice and used for cellular characterization by flow cytometry. The TIL-free tumor-enriched fraction was also obtained and stained for flow cytometry analysis.
Surface and intracellular antibodies
Cells were surface-stained with mouse anti-Gr-1, CD11b, F4/80, MHC Class II, CD86, Flt-3, FAS-L antibodies (all from eBioscience/Thermo Fisher, Waltham, MA) and E6/E7 tetramer (obtained from the NIH tetramer core-at Emory University), and then intracellularly stained with iNOS, Caspase-3(Molecular probes), MHC Class I (eBioscience), or Arginase (R&D) or pSmad2 (BD Phosflow pS465/pS467) antibodies according to manufacturer’s instructions.
Murine MDSC T-cell suppression assay
Control MDSC or TGFβ-MDSC were obtained as described earlier and added to CFSE (carboxyfluorescein diacetate succinimidyl ester)-labeled wild-type (WT) splenocytes at different ratios. All conditions (except unstimulated control) were activated with soluble anti-CD3 (0.5 μg/mL) + anti-CD28 (0.5 μg/mL) antibodies for 72 hours at 37°C. Cells were harvested and stained with anti-CD4 and -CD8 antibodies. All the above antibodies were from eBioscience. CFSE dilution was measured by flow cytometry. Fold proliferation was calculated as % T cell division in the presence of MDSC/% T cell division with anti CD3+ anti CD28 alone.
NO/ROS measurement
Ex vivo-generated control or TGFβ-MDSC were surface stained for Gr-1 and CD11b after which they were washed and resuspended in 30 μM DAF-DA FM (Millipore; 251520) or 10 μM DCF-DA FM (Sigma-Aldrich; D6883-50MG) in buffer according to manufacturer’s instructions. Cells were then resuspended in FACs buffer for flow cytometry analysis.
Antigen presentation assay
Murine control or TGFβ¯MDSC were pulsed for 2–3 hours with 1µg/ml of Ova 257–64 (S7951 Sigma), washed 3 times with PBS and co-cultured with CFSE labeled CD8 T cells purified from OT-1 mice, at a 1:1 ratio of T cells: MDSC for 3 days. After 3 days, cells were harvested and proliferation measured by CFSE dilution. Human control or TGFβ-MDSC were pulsed with 2 µg/ml of CEF peptide (Mabtech, 3615–1) overnight after which they were washed 3 times and co-cultured with cell trace violet-labeled allogeneic CD3 T cells (positively selected using Militeyi Biotec beads) from PBMCs) at a 1:1 ratio. At the end of 4 days, cells were harvested and CD3 T cell proliferation was measured by cell trace violet dilution by flow cytometry.
RPPA analysis
Reverse phase protein array assays were carried out as described previously with minor modifications.50 Protein lysates prepared from ex vivo derived murine MDSC were used for RPPA assay. 211 antibodies were used to generate data. Validated antibodies for RPPA represent key cell signaling and functional proteins relevant to cancer biology. A complete list of validated antibodies can be found with the following link: https://www.bcm.edu/centers/cancer-center/research/shared-resources/cprit-cancer-proteomics-and-metabolomics/reverse-phase-proteinarray). The median of triplicate experimental values (normalized signal intensity) was taken for each sample for subsequent statistical analysis. Significantly changed proteins were determined by employing Student’s t-test (significant for p < 0.05) specifying a fold change of at least 1.5X.
Tumor MDSC co-culture assays
Control MDSC or TGFβ- MDSC were derived as described earlier and co-incubated with MEER tumor cells at equal numbers for 2–3 days at 37°C. Cells were then harvested and stained with anti-CD45, Gr-1, CD11-b and FAS-L as mentioned above. In some cases, surface stained cells were further stained with annexin and PI (eBioscience Annexin/PI staining kit) per manufacturer’s instructions. Samples were analyzed using by FACS.
3D spheroid cultures
20,000 tumor cells (MEER) mixed with equal numbers of control or TGFβ-MDSC were grown as a hybrid 3D spheroid as previously described51 with slight modifications using the hanging drop method for 3 days after which they were transferred to an agarose coated 24 well plate and incubated for 48 hours . At the end of the culture period, the spheroids were collected and submitted for histology and immunochemistry.
Intratumoral injection of control or TGFβ¯mdsc
MEER tumors were established as described previously, and tumor growth was determined by caliper measurement. Once tumors became established (> 25 mm2), 0.8-1x106 CD11b+ enriched control or TGFβ-MDSCs were administered intratumorally in a volume of 50 µl at the base, mid and closest regions of the tumor (of the tumor bearing mice) on days 4,8 and 12 after initial tumor inoculum. For experiments involving radiotherapy in combination with TGFβ-MDSC, mice received a 30 Gy total radiation dose administered as 2 × 15 Gy doses on days 8 and 16. Radiation was directed to the flank tumor by using lead shields to protect non-tumor tissues (Braintree Scientific, Braintree, MA). 3 total intratumoral injections of control or TGFβ-MDSC or PBS control were performed as described above concomitantly with radiation treatment (days 10, 15, and 17). Tumors were measured regularly and mice were euthanized when tumors reached 150 mm2.
Induction of human mdscs from normal donor peripheral blood cells
Blood was obtained from healthy donors under the local institutional review board protocol H-15152. PBMCs were harvested from fresh blood obtained by venipuncture via Ficoll centrifugation. Tumor-conditioned medium from SCC-47 head and neck squamous cell carcinoma cells (a generous gift from Dr. Susanne M. Gollin at University of Pittsburgh Graduate School of Public Health) was used to derive MDSCs. PBMCs (at 4 × 106 cells/mL) were cultured in tumor conditioned medium (complete-RPMI-1640 media with 30% v/v tumor supernatants, 10% FBS, 1% Pen/Strep) ± TGF-β1-10ng/ml (GF439 from EMD Millipore) for 7 days with media being replaced with fresh tumor conditioned medium ± TGF-β1 on days 3 and 5. Resultant MDSCs were harvested and analyzed by multi-color flow cytometry for CD33 (Clone WM53), CD14 (Clone rmC5-3), HLA-DR (Clone G46-6), CD11b (Clone ICRF44). MDSCs were defined as CD33+ CD11b+ HLA-DR low/-.52 In addition to the above markers, MDSCs were also stained for CD86 (Clone 2331) and PD-L1(Clone MIH1). All antibodies were purchased from BD Biosciences. MDSCs were purified by magnetic selection via CD33 magnetic microbeads (Miltenyi Biotec), per manufacturer’s instructions, for downstream applications.
Human MDSC T-cell suppression assay
1x105 cell trace violet-labeled autologous T cells (CD3+ cells enriched from PBMCs) were plated onto 96-well plates in the presence of plate-bound 1µg/mL anti-CD3 (OKT3; NCI) and 1 µg/mL anti-CD28 (Clone CD28.2, BD Biosciences) antibodies with 50 IU/mL IL-2 in the presence of control or TGFβ-MDSC. After 4 days of incubation, T cells were harvested, labeled with CD3 and cell trace violet dye dilution determined by flow cytometry. Fold proliferation was calculated as described earlier.
Tgf-β1 ELISA
Both total and bioactive (free) TGFβ1 in tumor culture conditioned medium was determined by ELISA (Promega TGFβ1Emax ImmunoAssay Systems) as per the manufacturer’s instructions.
Statistics
GraphPad Prism version 7.00 for Mac (La Jolla, CA) was used for graph generation and statistical analysis. Normal data distribution was verified by Shapiro-Wilk test prior to comparisons. Two-way comparisons (i.e. control vs TGFβ-MDSC) were statistically compared using a 2-tailed, unpaired student’s t-test assuming unequal variance. Multiple comparisons (i.e. tumor growth curves, TIME cellular markers analysis) were performed using a 2-way ANOVA with Bonferroni-corrected multi-comparison. For all comparisons, a p-value less than 0.05 was considered statistically significant. For Kaplan Meir analyses, log rank test was used to assess significance.
Funding Statement
This work was supported by the Cancer Research Institute Clinical strategy Team Grant - Targeting the Tumor Immune Microenvironment to Enhance Immune-stimulating Effects of Chemoradiotherapy (PI Sikora); Cytometry and Cell Sorting Core at Baylor College of Medicine with funding from the NIH (P30 AI036211, P30 CA125123, and S10 RR024574) and the expert assistance of Joel M. Sederstrom; 2) The antibody-based proteomics core was funded by the CPRIT Core Facility Award RP170005 (DPE, SH) and NCI P30 CA123125 Cancer Center Support Grant for the Dan L. Duncan Comprehensive Cancer Center (DPE, SH). JMN acknowledges financial support from the National Institute of General Medical Sciences T32 predoctoral training grant (T32GM088129) and the National Institute of Dental & Craniofacial Research F31 NRSA training grant (F31DE026682) both of the National Institutes of Health. This content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Acknowledgments
We thank Qianxing Mo, Fuli Jia MS and Danli Wu PhD from Baylor College of Medicine, Antibody-based Proteomics Core/Shared Resource for performing RPPA.
We thank M. Sayeeduddin, Shahida Salar, Zahida Sayeeduddin and Patricia Castro, PhD from Baylor College of Medicine, Pathology and Histology Core (HTAP).
References
- 1.Rosenberg SA. Decade in review—cancer immunotherapy: entering the mainstream of cancer treatment. Nat Rev Clin Oncol. Internet. 2014; 11: 630–632. Available from: http://www.nature.com/doifinder/10.1038/nrclinonc.2014.174 doi: 10.1038/nrclinonc.2014.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Topalian SL, Weiner GJ, Pardoll DM. Cancer immunotherapy comes of age. J Clin Oncol. Internet. 2011; 29: 4828–4836. Available from: http://jco.ascopubs.org/content/29/36/4828.short doi: 10.1200/JCO.2011.38.0899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vanneman M, Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer. Internet. 2012; 12: 237–251. Available from: http://www.nature.com/doifinder/10.1038/nrc3237 doi: 10.1038/nrc3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vanneman M, Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer. Internet. 2012; 12: 237–251. Available from: doi: 10.1038/nrc3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Postow MA, Callahan MK, Wolchok JD. Immune Checkpoint Blockade in Cancer Therapy. J Clin Oncol. Internet. 2015; 33: JCO.2014.59.4358- Available from: http://jco.ascopubs.org/content/early/2015/01/20/JCO.2014.59.4358.full doi: 10.1200/JCO.2014.59.4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Webster RM. The immune checkpoint inhibitors: where are we now?. Nat Rev Drug Discov. Internet. 2014; 13: 883–884. Available from: http://www.nature.com/doifinder/10.1038/nrd4476 doi: 10.1038/nrd4476. [DOI] [PubMed] [Google Scholar]
- 7.Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell. 2015; 27(4): 451–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kusmartsev S, Su Z, Heiser A, Dannull J, Eruslanov E, Kübler H, Yancey D, Dahm P, Vieweg J. Reversal of myeloid cell-mediated immunosuppression in patients with metastatic renal cell carcinoma. Clin Cancer Res. Internet. 2008; 14: 8270–8278. Available from:: http://www.ncbi.nlm.nih.gov/pubmed/19088044 doi: 10.1158/1078-0432.CCR-08-0165. [DOI] [PubMed] [Google Scholar]
- 9.Kusmartsev S, Cheng F, Yu B, Nefedova Y, Sotomayor E, Lush R, Gabrilovich D. All-trans-retinoic acid eliminates immature myeloid cells from tumor-bearing mice and improves the effect of vaccination. Cancer Res. 2003; 63: 4441–4449. [PubMed] [Google Scholar]
- 10.Weiss T, Vitacolonna M, Zöller M. The efficacy of an IL-1α Vaccine depends on IL-1RI availability and concomitant myeloid-derived suppressor cell reduction. J Immunother. 2009; 32: 552–564. doi: 10.1097/CJI.0b013e31819b7b9e. [DOI] [PubMed] [Google Scholar]
- 11.Umansky V, Blattner C, Gebhardt C, Utikal J. The Role of Myeloid-Derived Suppressor Cells (MDSC) in Cancer Progression. Vaccines. Internet. 2016(4): 36 Available from:: http://www.mdpi.com/2076-393X/4/4/36 doi: 10.3390/vaccines4040036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol. Internet. 2009; 182: 4499–4506. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19342621%5Cnhttp://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC2810498 doi: 10.4049/jimmunol.0802775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wesolowski R, Markowitz J, Carson WE. Myeloid derived suppressor cells – a new therapeutic target in the treatment of cancer. J Immunother Cancer. Internet. 2013; 1: 10 Available from:: http://jitc.biomedcentral.com/articles/10.1186/2051-1426-1-10 doi: 10.1186/2051-1426-1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hengesbach LM, Hoag KA. Physiological concentrations of retinoic acid favor myeloid dendritic cell development over granulocyte development in cultures of bone marrow cells from mice. J Nutr. 2004; 134: 2653–2659. doi: 10.1093/jn/134.10.2653. [DOI] [PubMed] [Google Scholar]
- 15.Mirza N, Fishman M, Fricke I, Dunn M, Neuger AM, Frost TJ, Lush RM, Antonia S, Gabrilovich DI. All-trans-retinoic acid improves differentiation of myeloid cells and immune response in cancer patients. Cancer Res. 2006; 66: 9299–9307. doi: 10.1158/0008-5472.CAN-06-1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wiers KM, Ridley Lathers DM, Wright MA, Young MRI. Vitamin D3 treatment to diminish the levels of immune suppressive CD34+ cells increases the effectiveness of adoptive immunotherapy. J Immunother. 2000; 23: 115–124. doi: 10.1097/00002371-200001000-00014. [DOI] [PubMed] [Google Scholar]
- 17.Serafini P, Meckel K, Kelso M, Noonan K, Califano J, Koch W, Dolcetti L, Bronte V, Borrello I. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J Exp Med. Internet. 2006; 203: 2691–2702. Available from: http://www.jem.org/lookup/doi/10.1084/jem.20061104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Molon B, Ugel S, Del Pozzo F, Soldani C, Zilio S, Avella D, De Palma A, Mauri P, Monegal A, Rescigno M, et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J Exp Med. Internet. 2011; 208: 1949–1962. Available from: http://www.jem.org/lookup/doi/10.1084/jem.20101956 doi: 10.1084/jem.20110118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ahmad R, Raina D, Meyer C, Triterpenoid KD. CDDO-methyl ester inhibits the Janus-activated kinase-1 (JAK1)→signal transducer and activator of transcription-3 (STAT3) pathway by direct inhibition of JAK1 and STAT3. Cancer Res. 2008; 68: 2920–2926. doi: 10.1158/0008-5472.CAN-07-3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Umemura N, Saio M, Suwa T, Kitoh Y, Bai J, Nonaka K, Ouyang GF, Okada M, Balazs M, Adany R, et al. Tumor-infiltrating myeloid-derived suppressor cells are pleiotropic-inflamed monocytes/macrophages that bear M1- and M2-type characteristics. J Leukoc Biol. Internet. 2008; 83: 1136–1144. Available from: http://www.jleukbio.org/cgi/doi/10.1189/jlb.0907611 doi: 10.1189/jlb.1107745. [DOI] [PubMed] [Google Scholar]
- 21.Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006; page 605–12. doi: 10.1158/0008-5472.CAN-05-4005. [DOI] [PubMed] [Google Scholar]
- 22.Ma G, Pan PY, Eisenstein S, Divino CM, Lowell CA, Takai T, Chen S-H. Paired immunoglobin-like receptor-B regulates the suppressive function and fate of myeloid-derived suppressor cells. Immunity. 2011; 34: 385–395. doi: 10.1016/j.immuni.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment Trends Immunol. 2016; 37(3): 208–220. doi: 10.1016/j.it.2016.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Derynck R, Akhurst RJ. Differentiation plasticity regulated by TGF-beta family proteins in development and disease. Nat Cell Biol. 2007; 9: 1000–1004. doi: 10.1038/ncb434. [DOI] [PubMed] [Google Scholar]
- 25.Moustakas A, Pardali K, Gaal A, Heldin CH. Mechanisms of TGF-beta signaling in regulation of cell growth and differentiation. Immunol Lett. 2002; 82: 85–91. doi: 10.1016/S0165-2478(02)00023-8. [DOI] [PubMed] [Google Scholar]
- 26.Bierie B, Moses HL. Transforming growth factor beta (TGF-beta) and inflammation in cancer. Cytokine Growth Factor Rev. Internet. 2010; 21: 49–59. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20018551 doi: 10.1016/j.cytogfr.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sica A, Schioppa T, Mantovani A, Allavena P. Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: potential targets of anti-cancer therapy. Eur J Cancer. 2006; 42: 717–727. doi: 10.1016/j.ejca.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 28.Bierie B, Moses HLTGF-B. and cancer. Cytokine Growth Factor Rev. 2006; 17: 29–40. doi: 10.1016/j.cytogfr.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 29.Lechner MG, Liebertz DJ, Epstein AL. Characterization of cytokine-induced myeloid-derived suppressor cells from normal human peripheral blood mononuclear cells. J Immunol. 2010; 185: 2273–2284. doi: 10.4049/jimmunol.1000901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jayaraman P, Alfarano MG, Svider PF, Parikh F, Lu G, Kidwai S, Xiong H, Sikora AG. iNOS expression in CD4+T cells limits treg induction by repressing TGFβ1: combined iNOS inhibition and treg depletion unmask endogenous antitumor immunity. Clin Cancer Res. 2014; 20: 6439–6451. doi: 10.1158/1078-0432.CCR-13-3045. [DOI] [PubMed] [Google Scholar]
- 31.Creighton CJ, Huang S. Reverse phase protein arrays in signaling pathways: a data integration perspective. Drug Des Devel Ther. 2015; 9: 3519–3527. doi: 10.2147/DDDT.S38375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.CA Corzo, MJ Cotter, Cheng P, Cheng F, Kusmartsev S, Sotomayor E, Padhya T, McCaffrey TV, McCaffrey JC, Gabrilovich DI. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol. Internet. 2009; 182: 5693–5701. Available from:: http://www.ncbi.nlm.nih.gov/pubmed/19380816%5Cnhttp://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC2833019 doi: 10.4049/jimmunol.0802775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jayaraman P, Parkh F, Lopez-Rivera E, Hailemichael Y, Clark A, Ma G, Cannan D, Ramacher M, Kato M, Overwijk WW, et al. Tumor-expressed iNOS controls induction of functional myeloid derived suppressor cells (MDSC) through modulation of VEGF release. October. 2008; 141: 520–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Corzo CA, Condamine T, Lu L, Cotter MJ, Youn J-I, Cheng P, Cho H-I, Celis E, Quiceno DG, Padhya T, et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med. Internet. 2010; 207: 2439–2453. Available from: http://www.jem.org/lookup/doi/10.1084/jem.20100587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trikha P, Carson WE. Signaling pathways involved in MDSC regulation. Biochim Biophys Acta Rev Cancer. 2014; 55–65. doi: 10.1016/j.bbcan.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Raber P, Ochoa AC, Rodríguez PC. Metabolism of L-Arginine by Myeloid-Derived Suppressor Cells in Cancer: mechanisms of T cell suppression and Therapeutic Perspectives. Immunol Invest. Internet. 2012; 41: 614–634. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3519282&tool=pmcentrez&rendertype=abstract doi: 10.3109/08820139.2012.680634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schmid MC, Varner JA. Myeloid Cells in the Tumor Microenvironment: modulation of Tumor Angiogenesis and Tumor Inflammation. J Oncol. Internet. 2010; 2010: 1–10. Available from: http://www.hindawi.com/journals/jo/2010/201026/ doi: 10.1155/2010/201026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer. Internet 2008; (8): 618–631. Available from: http://www.nature.com/doifinder/10.1038/nrc2444. [DOI] [PubMed] [Google Scholar]
- 39.Waring P, Müllbacher A. Cell death induced by the Fas/Fas ligand pathway and its role in pathology. Immunol. Cell Biol.. 1999; 312–317. doi: 10.1046/j.1440-1711.1999.00837.x. [DOI] [PubMed] [Google Scholar]
- 40.Reap EA, K R, Maynor K, Borrero M, J B, Cohen PL. Radiation and stress-induced apoptosis: a role for Fas/Fas ligand interactions. Proc Natl Acad Sci U S A. Internet. 1997; 94: 5750–5755. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=9159145 doi: 10.1073/pnas.94.11.5750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rhee I. Diverse macrophages polarization in tumor microenvironment. Arch. Pharm. Res.. 2016; page 1588–96. doi: 10.1007/s12272-016-0820-y. [DOI] [PubMed] [Google Scholar]
- 42.Elliott LA, Doherty GA, Sheahan K, Ryan EJ. Human tumor-infiltrating myeloid cells: phenotypic and functional diversity. Front. Immunol; 2017: doi: 10.3389/fimmu.2017.00086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang W-C, Ma G, Chen S-H, Pan P-Y. Polarization and reprogramming of myeloid-derived suppressor cells. Supplementary Data. J Mol Cell Biol. Internet. 2013; 5: 207–209. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3695657&tool=pmcentrez&rendertype=abstract doi: 10.1093/jmcb/mjt009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. Identification of Two Distinct Macrophage Subsets with Divergent Effects Causing either Neurotoxicity or Regeneration in the Injured Mouse Spinal Cord. J Neurosci. Internet. 2009; 29: 13435–13444. Available from: http://www.jneurosci.org/cgi/doi/10.1523/JNEUROSCI.3257-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shvedova AA, Kisin ER, Yanamala N, Tkach A V., Gutkin DW, Star A, Shurin GV, Kagan VE, Shurin MR. MDSC and TGF?? are required for facilitation of tumor growth in the lungs of mice exposed to carbon nanotubes. Cancer Res. 2015; 75: 1615–1623. doi: 10.1158/0008-5472.CAN-14-3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes Trends Immunol. 2002; 23(11): 549–555 [DOI] [PubMed] [Google Scholar]
- 47.Jang C-W, Chen C-H, Chen -C-C, Chen J, Su Y-H, Chen R-H TGF-Β. induces apoptosis through Smad-mediated expression of DAP-kinase. Nat Cell Biol. Internet. 2002; 4: 51–58. Available from: http://www.nature.com/doifinder/10.1038/ncb731. [DOI] [PubMed] [Google Scholar]
- 48.Schuster N, Krieglstein K. Mechanisms of TGF-??-mediated apoptosis. Cell Tissue Res. 2002; page 1–14. doi: 10.1007/s00441-001-0479-6. [DOI] [PubMed] [Google Scholar]
- 49.Spanos WC, Nowicki P, Lee DW, Hoover A, Hostager B, Gupta A, Anderson ME, Lee JH. Immune response during therapy with cisplatin or radiation for human papillomavirus-related head and neck cancer. Arch Otolaryngol Head Neck Surg. Internet. 2009; 135: 1137–1146. Available from:: http://www.ncbi.nlm.nih.gov/pubmed/19917928 doi: 10.1001/archoto.2009.159. [DOI] [PubMed] [Google Scholar]
- 50.Chang C-H, Zhang M, Rajapakshe K, Coarfa C, Edwards D, Huang S, Huang S, Rosen JM. Mammary Stem Cells and Tumor-Initiating Cells Are More Resistant to Apoptosis and Exhibit Increased DNA Repair Activity in Response to DNA Damage. Stem Cell Reports. Internet. 2015; 5: 378–391. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4618454&tool=pmcentrez&rendertype=abstract doi: 10.1016/j.stemcr.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Foty R, Simple Hanging A. Drop Cell Culture Protocol for Generation of 3D Spheroids. J Vis Exp. Internet. 2011; Available from: http://www.jove.com/index/Details.stp?ID=2720 doi: 10.3791/2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bronte V, Brandau S, Chen S-H, Colombo MP, Frey AB, Greten TF, Mandruzzato S, Murray PJ, Ochoa A, Ostrand-Rosenberg S, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. Internet. 2016; 7: 12150 Available from: http://www.nature.com/doifinder/10.1038/ncomms12150. [DOI] [PMC free article] [PubMed] [Google Scholar]