

ABSTRACT
Mesothelioma is an aggressive asbestos induced cancer with extremely poor prognosis and limited treatment options. Immune checkpoint blockade (ICPB) has demonstrated effective therapy in melanoma and is now being applied to other cancers, including mesothelioma. However, the efficacy of ICPB and which immune checkpoint combinations constitute the best therapeutic option for mesothelioma have yet to be fully elucidated. Here, we used our well characterised mesothelioma tumour model to investigate the efficacy of different ICBP treatments to generate effective therapy for mesothelioma. We show that tumour resident regulatory T cell co-express high levels of CTLA-4, OX40 and GITR relative to T effector subsets and that these receptors are co-expressed on a large proportion of cells. Targeting any of CTLA-4, OX40 or GITR individually generated effective responses against mesothelioma. Furthermore, the combination of αCTLA-4 and αOX40 was synergistic, with an increase in complete tumour regressions from 20% to 80%. Other combinations did not synergise to enhance treatment outcomes. Finally, an early pattern in T cell response was predictive of response, with activation status and ICP receptor expression profile of T effector cells harvested from tumour and dLN correlating with response to immunotherapy. Taken together, these data demonstrate that combination ICPB can work synergistically to induce strong, durable immunity against mesothelioma in an animal model.
Keywords: Mesothelioma, immune checkpoint blockade, immunotherapy, anti-CTLA-4, anti-OX40
Introduction
Malignant mesothelioma is an aggressive, asbestos induced cancer arising from mesothelial cells of the pleura, peritoneum, pericardium or tunica vaginalis.1 Mesothelioma has a poor prognosis; once diagnosed the median survival of untreated cases is 6–9 months and first line cisplatin-pemetrexed combination chemotherapy, which has remained unchanged since 2003, only extends median survival to about 12 months.2 Therefore, more effective therapies for mesothelioma, such as immunotherapies, are urgently needed. There is emerging evidence that immunotherapy may be a useful strategy in mesothelioma3 and a number of clinical trials are ongoing, including in chemo-immunotherapy.
The T cell immune response to cancer is initiated following antigen recognition by the T cell receptor and is followed by a stimulatory signal generated by the ligation of CD28 on T cells with CD80/CD86 on antigen presenting cells. The activity of T cells is further modulated by co-signalling via surface receptor immune checkpoint molecules (ICP). These ICP control the amplitude and duration of the immune response through co-inhibitory and co-stimulatory signals to regulate the balance of T cell activity. Immune checkpoint blockade (ICPB) utilises antibodies that target ICP receptors to alter immune cell activation, proliferation and effector function and promote anti-tumour immunity. Antibodies that block inhibitory ICP cytotoxic T-lymphocyte antigen 4 (CTLA-4) and programmed death-1 (PD-1) have proven effective in melanoma.4,5 and other solid cancers.6
There is a strong immunological rationale for using ICPB to treat mesothelioma. High levels of CD8 tumour infiltrating lymphocytes (TILs) observed in mesothelioma are positively correlated with tumour regression and improved survival.7 However, despite the infiltration of T effector (Teff) cells, tumours can escape elimination by the immune system through the involvement of inhibitory molecules,8 or the development of a suppressive tumour microenvironment (TME), characterised by high levels of regulatory T cells (Treg) and suppressive cytokines such as TGF-β and IL-1.9
The application of ICPB to mesothelioma has primarily focused on targeting CTLA-4 or PD-1. CTLA-4 opposes the stimulatory signals generated by CD28 co-stimulation early in the T cell immune response.10 Activation of CTLA-4 generates inhibitory signals, decreases cytokine secretion, arrests cell cycle progression11 and is associated with the suppressive function of Treg.12 PD-1 functions primarily in peripheral tissue to inhibit T cell activation, where the PD-1 ligands programmed death ligand-1 (PD-L1) and programmed death ligand-2 (PD-L2) are expressed.13,14 Similar to CTLA-4, PD-1 activation reduces T cell proliferation and secretion of effector cytokines.15 In preclinical studies, CTLA-4 blockade in murine mesothelioma tumour models delay tumour growth.16,17 Limited preclinical information on PD-1 blockade is published in mesothelioma, however, we and others have previously shown delayed tumour growth when ICPB was combined with adjuvant therapies in other preclinical models.17-20
Despite these encouraging pre-clinical results, a randomised clinical trial using the αCTLA-4 agent Tremelimumab failed to show any survival advantage over best supportive care.21 Results from a number of small studies using αPD-1 and αPD-L1 monotherapies in the second line setting have demonstrated modest response rates.3 Studies in other cancers combining αCTLA-4 and αPD-1 blockade reported an improved response rate and median progression free survival over monotherapy.22 As such, a number of phase II studies of combination treatment have reported in abstract form, and a large randomised phase III clinical trial testing combined αCTLA-4 and αPD-1 blockade in mesothelioma has completed accrual and is expected to report in 2019–2020 (NCT02899299).
Alternative immune checkpoints including inhibitory receptor T cell immunoglobulin 3 (TIM-3) and the stimulatory receptors OX40 and glucocorticoid induced tumour necrosis factor receptor (TNFR) related protein (GITR) are also in the early stages of clinical development and may be suitable alternative targets for ICPB in mesothelioma. TIM-3 is an inhibitory ICP that mediates immune tolerance23 and along with PD-1 expression, mark a dysfunctional population of CD8 T cells.24 Stimulatory OX40 and GITR promote the survival and proliferation of Teff and reduce the suppressive ability of Treg.25,26 Although ICPB shows promise for cancer therapy, only a small proportion of mesothelioma patients experience a durable response from the strategies tested clinically to date. Given the limited preclinical data on ICPB for mesothelioma, there is a unique opportunity to investigate the efficacy of targeting novel immune checkpoints or combination ICPB in mesothelioma-specific preclinical models.
Here we demonstrate that ICPB is an effective therapy for mesothelioma. Targeting any of CTLA-4, OX40 or GITR individually generated an effective response against while the combination of αCTLA-4 and αOX40 acted synergistically to significantly improve therapeutic outcome. Other combinations did not synergise to enhance treatment outcomes. We also show that the activation status and immune checkpoint receptor expression profile of Teff cells harvested from tumour and dLN correlated with therapeutic response. Taken together, these data demonstrate that combination ICPB can work synergistically to induce strong, durable immunity against mesothelioma in an animal model.
Results
ICPB therapy is effective against AB1-HA mesothelioma
ICPB targeting CTLA-4 and PD-1 has been shown to delay tumour growth and improve survival and can invoke durable clinical responses in a variety of different cancers.27 We used our well characterised syngeneic murine model of mesothelioma to determined tumour response to ICPB therapy. Mice bearing AB1-HA tumours were treated with antibodies targeting either CTLA-4, OX40, GITR, PD-1 or TIM-3 on days 10, 13 and 16 post tumour inoculation (Figure 1). We observed a significant improvement in survival for mice treated with either αCTLA-4 (p < 0.01), αGITR or αOX40 (p < 0.05) when compared to untreated (PBS) controls (Figure 1B). Interestingly, only αCTLA-4 treatment significantly delayed tumour growth compared to controls (p < 0.001; Figure 1C). The response to checkpoint blockade varied in individual mice and each of complete response (tumour regression), partial response (delayed tumour growth) or no response were seen within any single experiment (Figure 1D). The response to αCTLA-4 was the most pronounced with delayed tumour growth observed in 75% (6/8) mice, and complete tumour regression in a single mouse. Mice treated with either αGITR or αOX40 demonstrated mainly partial responses, with only one complete response observed in either treatment group. No change in survival or tumour growth relative to untreated controls was observed for treatment with αPD-1 or αTIM-3 monotherapy. These data indicated a survival benefit for monotherapy with αCTLA-4, αOX40 and αGITR, and delayed tumour growth for αCTLA-4.
Figure 1.
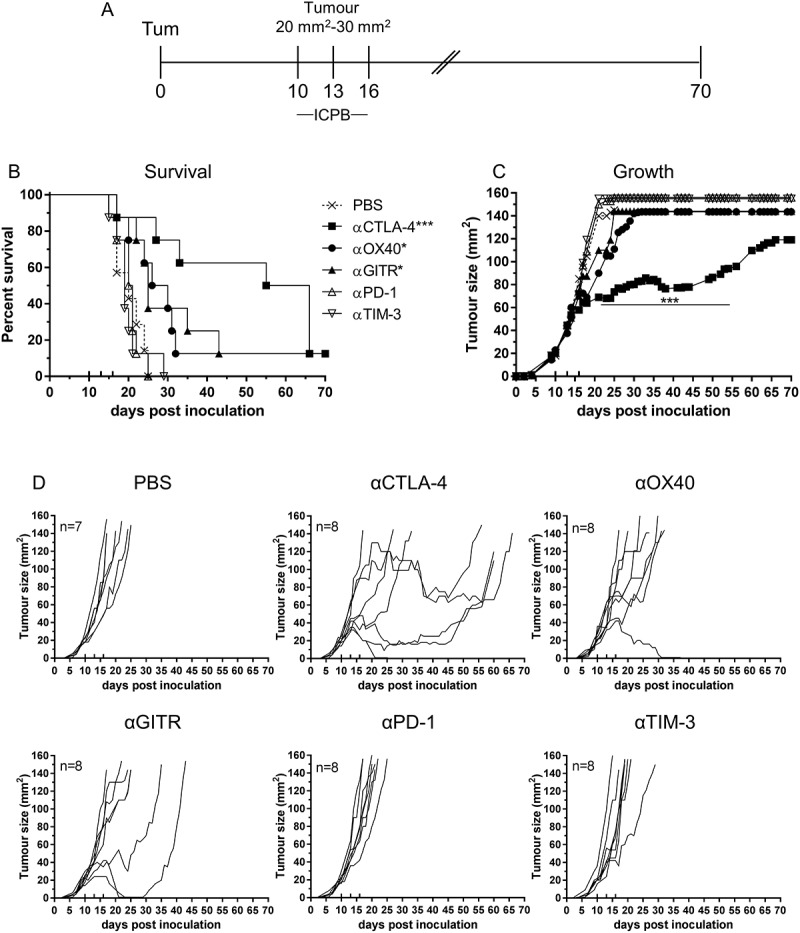
Effective Immune checkpoint blockade for mesothelioma.
BALB/c mice bearing AB1 HA tumours were treated with antibodies targeting individual immune checkpoint molecules on days 10, 13 and 16 post tumour inoculation. (A) Experimental design. (B) Survival and (C) tumour growth plots of mice treated with ICPB monotherapies. The horizontal line indicates significant delay in tumour growth over time compared to PBS. (D) Tumour growth curves for individual mice in each group compared to PBS controls. Combined data from two independent experiments analysed using Log rank (Mantel Cox) for survival curves or Mixed model ANOVA for mean tumour growth. * = p < 0.05, *** = p < 0.001, **** = p < 0.0001, ns = not significant.
Combining CTLA and αOX 40 synergistically improves AB1-HA anti-tumour immunity
We hypothesised that dual blockade therapy would enhance the anti-tumour immune response, improving survival and delaying tumour growth relative to monotherapy. BALB/c mice were inoculated with AB1-HA and treated with dual combinations of each ICPB antibodies (experimental design as for Figure 1A), and survival and tumour growth monitored (Figure 2).
Figure 2.
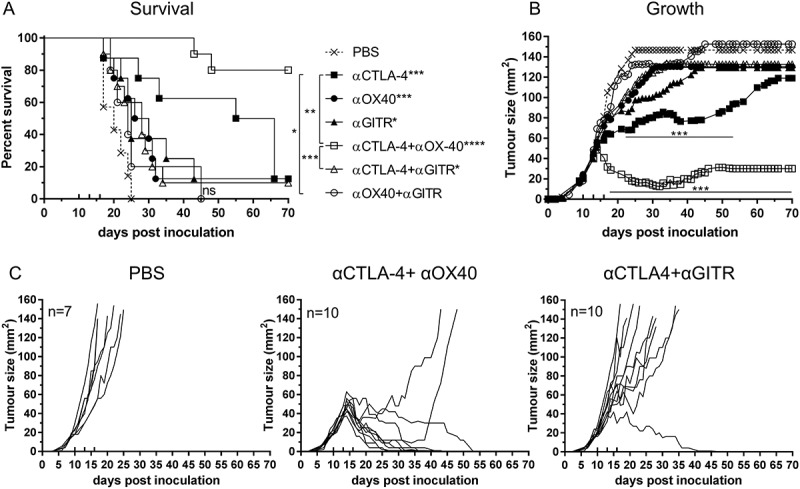
Effective combination Immune checkpoint blockade for mesothelioma.
BALB/c mice bearing AB1‑HA tumours were treated with antibodies targeting immune checkpoint molecules either alone or in combination on days 10, 13 and 16 post tumour inoculation. (A) Survival and (B) tumour growth plots for mice treated with combination ICPB therapy. Significant differences for groups relative to PBS control unless otherwise shown. Horizontal lines indicates significant delay in tumour growth over time for each group compared to PBS. (C) Tumour growth curves for PBS and combination groups. Each line represents a tumour bearing mouse. Combined data from two independent experiments analysed using Log rank (Mantel-Cox) for survival curves or Mixed model ANOVA for mean tumour growth. * = p < 0.05, *** = p < 0.001, **** = p < 0.0001, ns = not significant.
Combination αCTLA-4+αOX40 therapy significantly improved survival compared to untreated (PBS) controls (p < 0.0001, Figure 2A) or either monotherapy alone (αCTLA-4 or αOX40, p < 0.001). ICPB monotherapy with αCTLA-4 led to long term survival in 12.5% (1/8) of treated animals and was significantly improved (p < 0.0001) when administered concurrently with αOX40 (80%, 8/10 mice). A significant delay in tumour growth over time (p < 0.001) was observed after day 17 when comparing combination αCTLA-4+ αOX40 therapy to PBS controls and after day 20 (p < 0.001) when compared to either αCTLA-4 or αOX40 monotherapy (Figure 2B). Notably all mice responded to combination αCTLA-4+αOX40 therapy and displayed either complete (8/10), or partial (2/10 mice) tumour regression (Figure 2C). Combination therapy was also effective in an orthotopic tumour model in which AB1-HA cells were injected directly into the pleural cavity. In this experiment, 90% (9/10) of αCTLA-4+αOX40 treated mice remained healthy for the duration of the experiment, with minimal signs of disease while all PBS (untreated) mice had succumbed to disease by day 28 (Figure 3).
Figure 3.
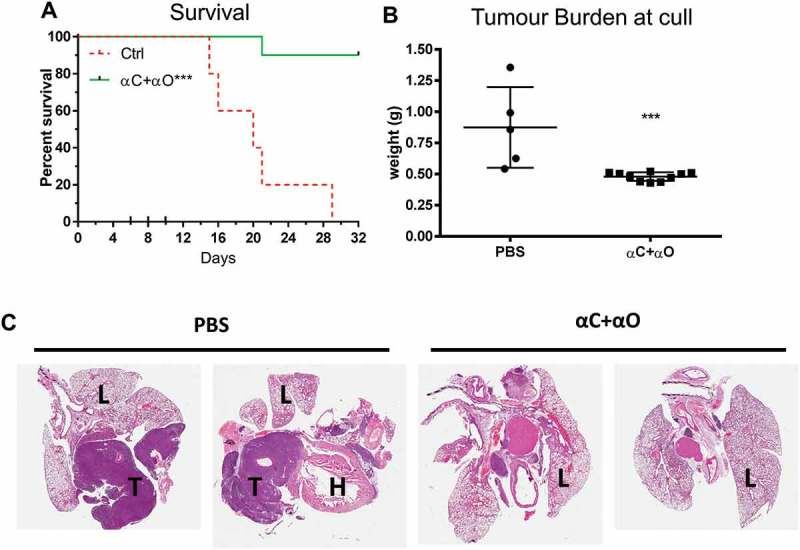
Combination immune checkpoint blockade is effective in an orthotopic mesothelioma model.
BALB/c mice (n = 5 PBS, n = 10 αCTLA4+ αOX40) were injected intrapleurally with 2 × 105 AB1-HA cells on day 0 and treated with PBS or ICPB on days 6, 8 and 10. (A) Overall survival. (B) Tumour burden (weight of heart + lungs + tumour) for individual mice on the day they were culled. (C) Representative histology (20 x Mag) showing haematoxylin and eosin (H&E) staining of pleural tissues taken from two mice randomly selected from each treatment group (L = lung; H = heart and T = tumour). ICPB (αC+ αO); 100 μg αCLTA-4 + 200 μg αOX40. Data shows mean ± SD. *** p < 0.001. Survival analysis: Log rank (Mantel-Cox). Tumour burden analysis: unpaired t-test (Mann-Whitney).
We observed no additional survival benefit or delay in tumour growth in the αCTLA-4+αGITR combination therapy group. Indeed, this combination negated the effect of αCTLA-4 monotherapy on both overall survival and tumour growth (Figure 2A). Complete tumour regression was only observed in 10% (1/10) of αCTLA-4+αGITR treated mice and no partial responses to therapy were observed (Figure 2C). Despite all ICPB therapy combinations being assessed (supplemental Figure 1), no other combination offered any significant improvement in survival or delay in tumour growth relative to either αCTLA-4, αOX40 or αGITR monotherapy. Interestingly, treatment with αTIM-3 or αPD-1, alone or in combination, mirrored PBS controls and failed to have any impact on tumour growth. Taken together, these data indicate that mesothelioma has a differential response to ICPB therapy and the success of individual monotherapy is not necessarily predictive of outcomes to combination therapy.
Immune checkpoint therapy is less effective against AB12 and line-1M tumours
We next assessed the efficacy of αCTLA-4, αOX40, αGITR and combination αCTLA-4+αOX40 treatments in AB12 and Line-1M tumour models (Figure 4). In mice bearing AB12 mesothelioma tumours, we observed no difference in overall survival or tumour growth for any ICPB treatment groups relative to PBS controls (Figure 4A and B). Assessment of individual tumour growth indicated that all groups failed to respond to ICPB, except for a single mouse that demonstrated a partial response to αCTLA-4 monotherapy (Figure 4C). In the Line-1M alveolar adenocarcinoma model, a significant survival benefit was observed for monotherapy groups αCTLA-4 (p < 0.001), αOX40 (p < 0.01), αGITR (p < 0.05) and αCTLA-4+αOX40 combination therapy (p < 0.001) relative to PBS controls (Figure 4D), an outcome that correlated with significant delay in tumour growth over time (p < 0.01) between days 17–35 in treated groups relative to controls (Figure 4E). However, despite the partial delay in tumour growth, complete regression of Line-1M tumours was only observed in a single mouse receiving αCTLA-4+αOX40 combination therapy. All other treated mice displayed a mixture of partial or non-response (Figure 4F). Taken together, these data indicate substantial variation among mesothelioma and lung carcinoma murine modes in response to ICPB therapy.
Figure 4.
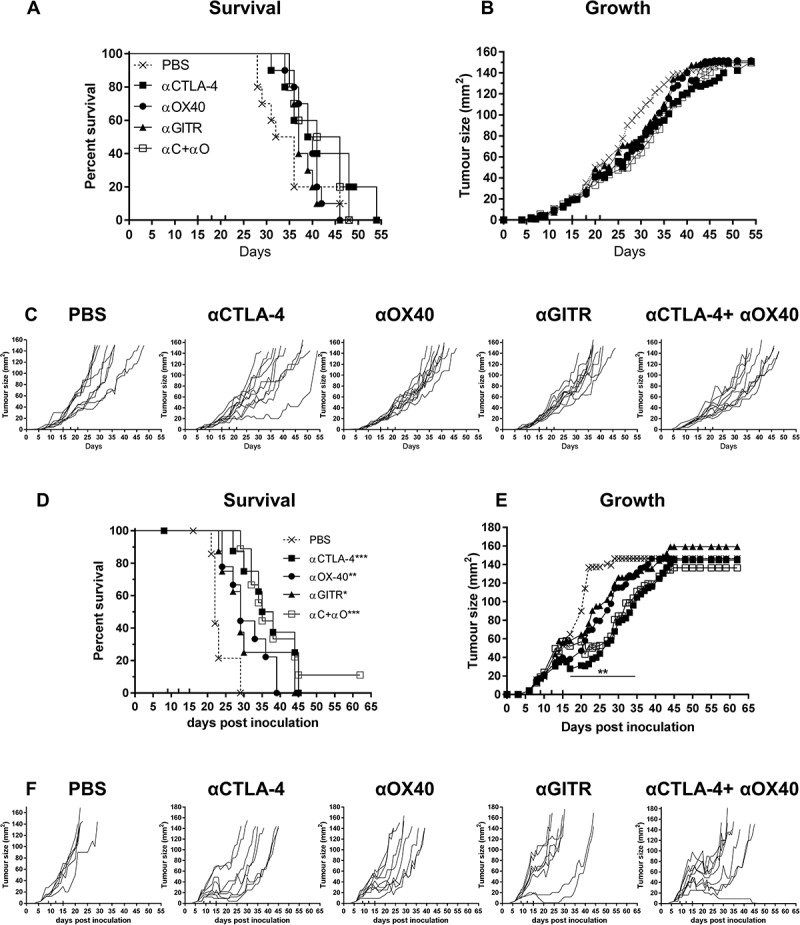
Immune checkpoint therapy is less effective in AB12 and Line-1M.
Tumour bearing BALB/c mice were treated with antibodies targeting selective immune checkpoint molecules either alone, or in combination when tumours were 25 mm2 to 30 mm2 (treatment days indicated by up ticks). Data shows survival and tumour growth plots for mice bearing AB12 (A-B) and Line-1M (D-E) tumours treated with indicated ICPB therapies. Tumour growth curves for individual mice in each group compared to PBS controls for AB12 (C) and Line-1M (F). Each line represents a tumour bearing mouse with significant differences for groups relative to PBS controls shown unless otherwise stated. Horizontal lines indicates significant delay in tumour growth over time for each group compared to PBS. Combined data from two independent experiments analysed using Log rank (Mantel-Cox) for survival curves or Mixed model ANOVA for mean tumour growth. * = p < 0.05, *** = p < 0.001, **** = p < 0.0001, ns = not significant.
Differential ICP molecule expression profiles between ICP sensitive and less sensitive tumours
We hypothesised that the ICP expression profile of anti-tumour immune cells would differ between ICPB sensitive and less sensitive tumours. To test our hypothesis we used flow cytometry to assess expression of the ICP molecules CTLA-4, GITR, PD-1, OX40, and TIM-3 on tumour infiltrating (TIL) and draining lymph node (dLN) resident Treg and CD4 and CD8 effector T cell populations harvested from mice bearing small AB1-HA, AB12 or Line-1M tumours (Figure 5); consistent with when ICPB treatment was first administered in the previous experiments (i.e. day 10, tumours ~ 25 mm2).
Figure 5.

AB1-HA, AB12 and Line-1M tumour and dLN lymphocyte expression of ICP receptors.
Flow cytometry was used to assess ICP expression profiles of tumour and draining lymph node resident immune cells harvested from mice bearing small (day 10, ~ 25 mm2) AB1-HA, AB12 or Line-1M tumours. (A) Experimental design. (B) Gating strategy for identification of CD4 (CD3+ CD4+ FoxP3-), Treg (CD4+ CD25+ FoxP3+) and CD8 (CD3+ CD8+) lymphocytes, each of which were further gated in histogram plots depicting MFI expression of CTLA-4, GITR, OX40, PD-1 and TIM-3. (C) Tumour CD3 frequency of total live cells; CD4, Treg and CD8 percentage frequency of CD3 population and frequency of TIL ICP receptor expression for CD4, Treg and CD8 T cells. (D) Tumour dLN CD3 percentage frequency of total live cells, and CD4, Treg and CD8 percentage frequency of CD3 population. Tumour dLN frequency of ICP receptor expression for CD4, Treg and CD8 T cells. CD4 (white), Treg (black) and CD8 (hatched) columns, n = 5–9 mice/group from two independent experiments, data are mean ± SEM; One Way ANOVA *p < 0.05, **p < 0.01 compared to respective AB1-HA population.
AB12 tumours had significantly more (p < 0.01) CD3 and CD8 TILs relative to AB1-HA and Line-1M, while little difference was observed between CD4 and Treg subsets between the three tumour types (Figure 5C). GITR expression was consistently high (> 97%) among all T cell subsets, in all tumour types, with the GITR mean fluorescence intensity (MFI) being 4 to 9-fold higher for Treg (11,999.3 ± 2737 SD) compared to either CD4 or CD8 effector T cell subsets (2,914.5 ± 455 SD and 1,243.5 ± 130.4 SD respectively; Supplementary Figure 2). Indeed, tumour resident Treg expressed the highest levels of CTLA-4, GITR and OX40 relative to CD4 or CD8 T cells in all tumours except AB12, in which OX40 expression was significantly reduced in CD4 and Treg populations (p < 0.01, Figure 5C). Similarly, expression of PD-1 was significantly reduced (p < 0.01) on AB12 CD4 and CD8 effector TILs, while TIM-3 was significantly lower (p < 0.01) on AB12 Treg relative to the other tumours (Figure 5C). Analysis of tumour dLN indicated similar levels of dLN resident immune cell subsets across all tumours. Consistent with the tumour data, Treg displayed the highest level of CTLA-4, OX40 and GITR expression relative to CD4 and CD8 T cells (Figure 5D).
These data demonstrate that prior to ICPB therapy, ICP expression profiles were relatively consistent between tumour and dLN resident T cell subsets isolated from ICPB sensitive AB1-HA and Line-1M tumours, while OX40 and PD-1 expression was significantly different in the less sensitive AB12 tumours.
Early TIL response to ICP therapy is predictive of tumour outcome
As a large proportion of patients who receive ICPB treatment fail to respond, early identification of non-responders to ICPB therapy would be beneficial. We next used a dual tumour model, in which each tumour responds to ICPB symmetrically,28 to assess whether differences in the frequency and activation/inhibition status of tumour (TIL) and dLN resident immune cell subsets correlated with response to ICPB therapy. We have previously shown that treatment-induced response was symmetrical, in a highly reproducible manner over multiple experiments.28
Mice bearing dual AB1-HA tumours (one on each flank) were treated with ICPB therapy targeting CTLA-4 and OX40, either alone or in combination on days 10, 13, and 16. Two days post-therapy (day 18) the right hand flank (RHF) tumour and its corresponding dLN were surgically excised and the remaining tumour (LHF) monitored for treatment response (Figure 6A). Mice treated with PBS or αOX40 monotherapy failed to respond to therapy. Conversely, all mice treated with either αCTLA-4 or αCTLA-4+αOX40 responded to therapy. Consistent with our previous data, complete tumour regression was observed in 70% (7/10) of αCTLA-4 and 90% (9/10) αCTLA-4+αOX40 treated mice, while later tumour relapse was observed in 30% (3/10) and 10% (1/10) of αCTLA-4 and αCTLA-4+αOX40 treated mice respectively (Figure 6B).
Figure 7.
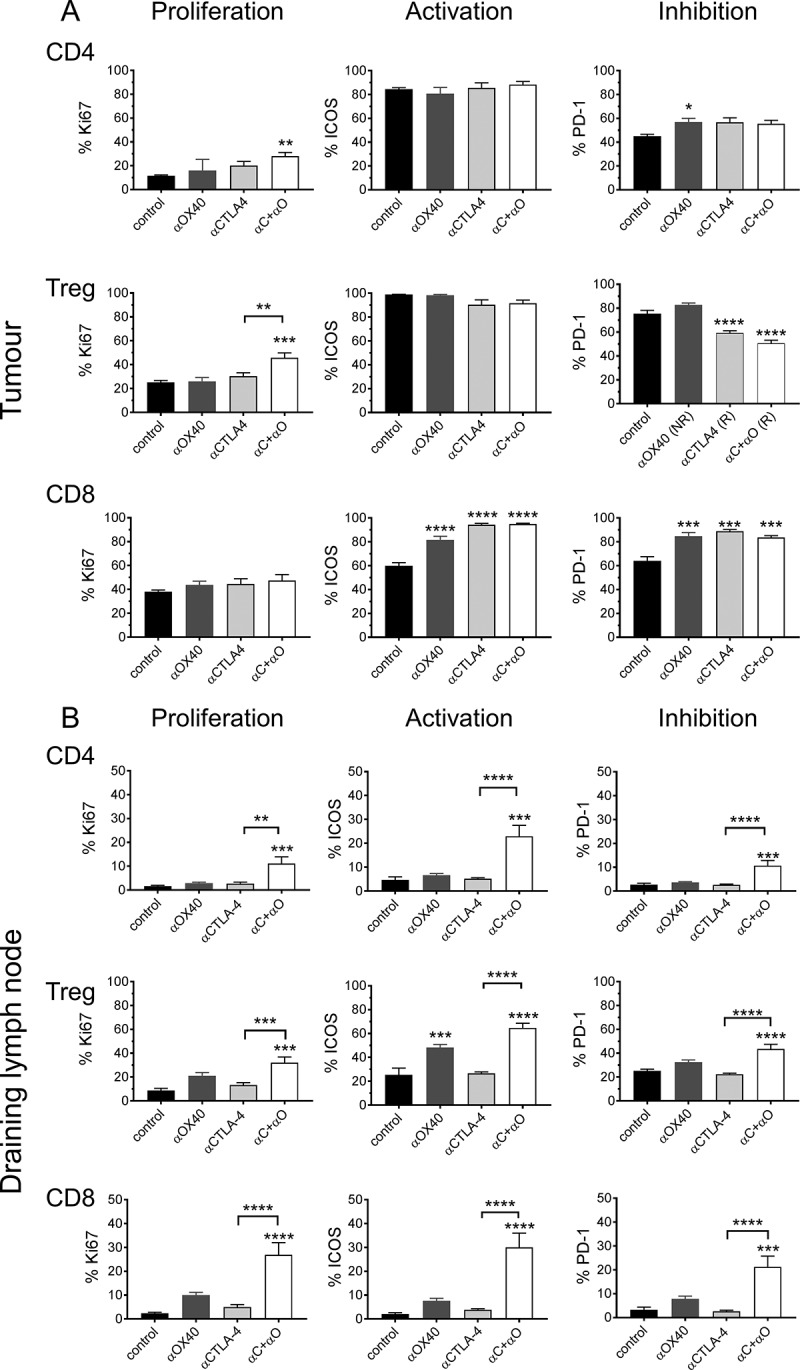
Response to ICPB is associated with distinct T cell profile for activation and inhibition markers.
Flow cytometry was used to assess the expression of cell surface markers denoting cellular proliferation (Ki67), activation ICOS and the inhibitory checkpoint molecule PD-1) on (A) tumour (TIL) and (B) tumour draining lymphnode (dLN) resident Treg and CD4 and CD8 effector T cells. Tumours are from mice described in Figure 6 (n = 10 mice/group). Data are mean ± SEM; All statistics are one way ANOVA with Bonferroni’s test for multiple comparisons; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 relative to control unless otherwise indicated.
Figure 6.
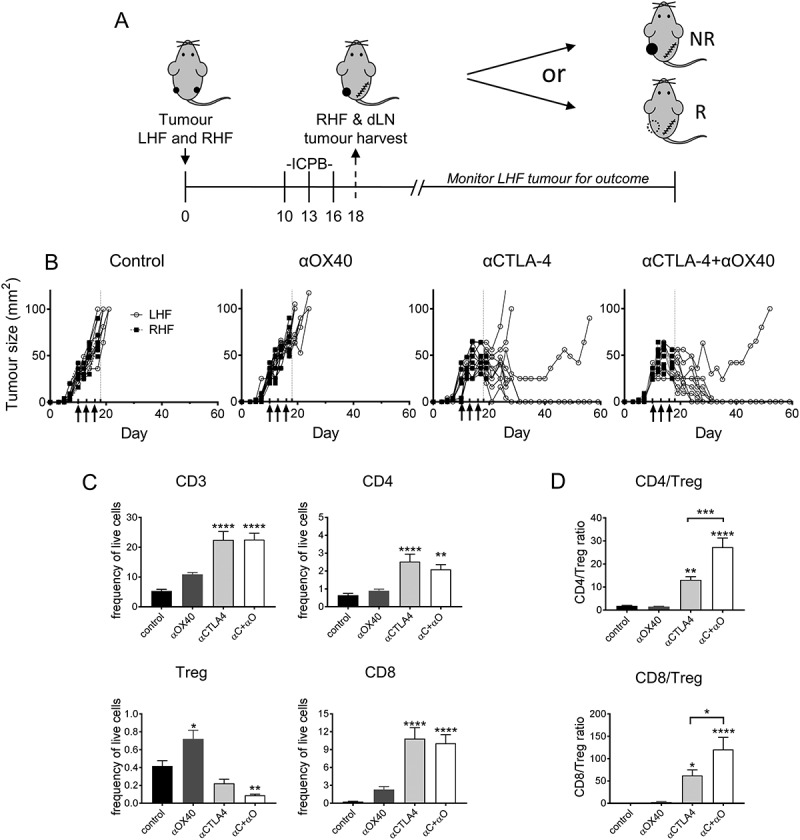
AB1-HA ICPB dual tumour growth.
Mice were s.c. inoculated on right (RHF) and left (LHF) flank with 5 × 105 AB1 HA cells and immune checkpoint immunotherapy administered i.p. on day 10, 13 and 16 (black arrows). Two days post therapy (day 18, dotted line) RHF tumours and their associated draining lymphnode were harvested for analysis. (A) Dual tumour experimental design. (B) Individual tumour growth curves for both RHF (squares, excised day 18) and LHF (circles) tumours. Data shown: n = 10 mice/group from two independent experiments. (C) Immune cell subset phenotype as a frequency of total live tumour and CD3 T cells. (D) CD8 and CD4 effector T cell to Treg cell ratio. (C D) Data are mean ± SEM; All statistics are one way ANOVA with Bonferroni’s test for multiple comparisons; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 relative to control unless otherwise indicated.
We next examined whether the frequencies of TIL subsets were altered in the different ICPB treatment groups (Figure 6C). We observed a significant increase (p < 0.0001) in CD3 and CD8 TILs, as a proportion of total tumour cells, in αCTLA-4, but not αOX40 monotherapy treated groups compared to controls. CD4 T cells as a percentage of total tumour cells were significantly elevated in αCTLA-4 treated groups (p < 0.01). Although Treg comprised a very small proportion of live tumour cells at day 18, tumour resident Treg were reduced in αCTLA-4 treated groups compared to controls, although only αCTLA-4+αOX40 treatment was significant (p < 0.01). Conversely, Treg were significantly elevated (p < 0.05) in αOX40 treated mice that failed to respond. Consistent with these data, the ratio of CD4 or CD8 effector T cells relative to Treg was significantly elevated after αCTLA-4 treatment (p < 0.5); especially after αCTLA-4+αOX40 combination therapy (Figure 6D).
ICPB treated groups also had significantly higher (p < 0.0001) CD8 TIL activation as indicated by increased ICOS and PD-1 expression in comparison to controls (Figure 7A). Analysis of the corresponding tumour dLN indicated that the frequency of Treg and CD8 T cells, as a percentage of total dLN cells, was consistent between all ICPB treatment groups and PBS controls, with Treg and CD8 T cells comprising ~ 5% and 20% of total dLN cells respectively (Figure 7B and data not shown). Interestingly, only mice that received combination αCTLA-4+αOX40 therapy had significantly higher activation (ICOS; p < 0.001), proliferation (Ki67; p < 0.001) and inhibitory marker (PD-1; p < 0.001) expression for both CD4 and CD8 effector T cells and Treg populations relative to controls and αCTLA-4 monotherapy. Taken together, these data indicate that effective ICPB therapy is associated with a decrease in tumour resident Treg in association with increased CD8 effector T cell activation.
Discussion
Despite the clinical success of ICPB in other cancers, only a small fraction of mesothelioma patients respond to αCTLA-4 or αPD-1 monotherapy.3,29 Therefore, the potential for combination therapy to enhance anti-tumour immunity, or the ability to target ICP molecules in addition to CTLA-4 or PD-1 may offer an alternative therapeutic strategy to improve ICPB efficacy in mesothelioma patients. However, given the cost (time, patients and financial) of exploring multiple combinations in the clinic, we investigated the efficacy of individual and dual ICPB therapy in subcutaneous models of murine mesothelioma and lung cancer in order to provide a robust preclinical rationale to guide selection of combinations for testing. Consistent with previous studies,16,18,20 αCTLA-4 monotherapy was sufficient to increase overall survival and delay tumour growth in a small number of mice compared to controls. In addition, we observed a similar survival benefit for αOX40 or αGITR monotherapy; demonstrating the efficacy of targeting the alternative checkpoint molecules OX40 or GITR in pre-clinical models of mesothelioma.
The limited response of mesothelioma to ICPB monotherapies was significantly improved by αCTLA-4+αOX40, but not αCTLA-4+αGITR combination therapy. Treatment with αCTLA-4+αOX40 induced a strong, durable response in ICPB treated mice, in both subcutaneous and orthotopic models. In the subcutaneous model, complete tumour regression was preceded by reduced tumour resident Treg and increased activation and proliferation of CD8 TILs. These data are consistent with studies in other solid cancer models in which combination αCTLA-4+αOX40 induced strong, durable anti-tumour immunity and tumour regression was ascribed to either eradication of tumour resident Treg30 or expansion of a T-bethigh/Eomeshigh granzyme B+ subset of CD8 effector T cells.31 A phase 1 multicentre, open-label clinical trial is currently recruiting to assess this ICPB combination in adults with advanced solid cancers (NCT02705482).
Conversely, combination αCTLA-4+αGITR therapy seemed to abrogate the efficacy of either monotherapy. GITR targeted therapy alone or in combination has been shown to induce tumour regression in a variety of other solid cancer models.32-35 Two studies using Meth A and CT26 tumour models demonstrated that combined αCTLA-4 and αGITR therapy was synergistic, delaying tumour growth and inducing tumour regression.36,37 While differences between models and treatment protocols make it difficult to directly compare our data, variations in antibody dose and schedule might explain the contrasting results. The Ko36 and Mitsui37 studies administered a single high dose (≥ 500 μg) αGITR, whereas we treated with low dose (50 μg) αGITR concurrently with αCTLA-4 on three consecutive occasions. Furthermore, it has been proposed that αGITR therapy may have stimulatory effects on the activity of Treg (reviewed in)38, raising the possibility that concurrent administration of αCTLA-4+αGITR antibodies may favour the Treg stimulatory activity of αGITR antibodies and promote an immunosuppressive TME, limiting any potential αCTLA-4 mediated Teff tumour destruction. Taken together, this highlights the importance of understanding the mechanisms that underpin ICPB response to better inform the development of successful ICPB combination in the clinical setting.
In contrast to the response observed in AB1-HA mesothelioma the same ICPB therapies failed against AB12 mesothelioma, suggesting that not all murine mesotheliomas are equally susceptible to ICPB. Although unexpected, this intriguing result may help us to understand the biological and immunological mechanisms that dictate ICPB responders from non-responders. Both the parental AB1 and AB12 cell lines were isolated from asbestos exposed BALB/c mice,39 and have been shown to be responsive to Treg-specific immunotherapy40 (unpublished data), indicating that AB12 tumours can be susceptible to immunotherapy. A recent report from our group demonstrated that the AB12 cell line had 6-fold fewer somatic mutations compared to AB1 (70 vs. 430 respectively),41 a finding that is consistent with studies demonstrating that patients with cancers that have a higher mutational burden tend to have better overall survival42 and are more likely to respond to ICPB.43-46 However, it is clear that some cancers with relatively low mutational burden can also respond to ICPB.6 Alternatively, lack of TILs or a highly immunosuppressive TME47 may limit response to ICPB. This explanation seems unlikely as we observed a similar proportion of tumour infiltrating immune cells for each tumour and little difference in the amount of the immunosuppressive cytokine TGF-β produced by AB1-HA and AB12 tumours (data not shown). However, we cannot exclude the potential for additional immunosuppressive mechanisms such as other immunosuppressive cytokines, or the presence/influence of myeloid derived suppressor cells or M2 macrophages in AB12 cells.
In conclusion, we demonstrate that combination ICPB can work synergistically to induce strong, durable immunity against mesothelioma in animal models. Importantly, the results of this preclinical study will provide information on which combinations of ICPB induce an anti-tumour response in murine mesothelioma, which then can be used to inform and guide the design of future clinical studies. Clearly, some combinations also have the potential to be antagonistic and hence caution should be used in clinical development of combinations. This study further indicates that successful strategies for ICP receptor response are dependent on the both the cellular and anatomical site of expression. In particular tumour expression of ICP receptors limited to Treg cells, and not CD8 T cells, was indicative of therapy response. Finally responders to αCTLA-4+αOX40 therapy expressed an immune profile in tumour and dLN that was indicative of a positive response to therapy.
Materials and methods
Mice
Female BALB/c mice (6–8 weeks) were obtained from the Animal Resource Centre (Murdoch, WA, Australia) and housed at the Harry Perkins Bio Resources Facility (Nedlands, WA, Australia). Mice aged 6–8 weeks or 10–12 weeks were used for subcutaneous and intrapleural tumour models respectively. All mice were maintained under standard specific pathogen free housing conditions and animal experiments were conducted in accordance with Australian code for the care and use of animals for scientific purposes 8th and The University of Western Australia animal ethics guidelines and protocols (Perkins Animal ethics protocols AE008, AE030).
Cell lines
The murine mesothelioma cell lines AB1 and AB12 were generated by inoculating crocidolite asbestos intraperitoneally (i.p.) in BALB/c mice, collecting the peritoneal exudate and passaging cells in vivo and in vitro until a stable, clonal cell line was obtained.39 The AB1 cell line was then transfected with influenza hemagglutinin (HA) from the Mt Sinai strain of PR8/34/H1N1 influenza virus to generate the well characterised AB1-HA cell line,48 which can be obtained from Cell Bank Australia. Line-1M cell line was derived from murine Line-1 lung alveolar adenocarcinoma cell line after excision of lung tumour that had naturally metastasized following subcutaneous tumour growth.49 All cells were maintained in R10 media (Gibco BRL, Invitrogen, Mulgrave VIC, Australia) supplemented with 20 mM HEPES (Sigma-Aldrich, Castle Hill, NSW, Australia), 0.05 mM 2 Mercaptoethanol (Sigma-Aldrich), 100 U/ml Benzylpenecillin (Sir Charles Gardiner Hospital Pharmacy, Nedlands,WA, Australia), 500 g/ml Gentamicin (Sir Charles Gardiner Hospital Pharmacy), 10% foetal calf serum (FCS; Life Technologies, Mulgrave, VIC, Australia) and 50 mg/ml Geneticin (G418; Life Technologies, Mulgrave, VIC, Australia). Adherent cells were trypsinised, washed two times in phosphate buffered saline (PBS) and counted. Only cell suspensions with a viability of > 95% as assessed by trypan blue exclusion assay were used for inoculation.
Tumour cell inoculation
For the subcutaneous tumour model, mice were inoculated subcutaneously (s.c.) into the shaved, right hand flank (and left hand flank for dual tumour model), with 5 × 105 AB1-HA, AB12 or or 1 × 106 Line-1M cells suspended in 100 µl PBS. Tumour size was monitored with callipers and calculated by multiplying width and length to determine tumour area in square millimetres (mm2). Mice were euthanized once tumour size exceeded 150 mm2 in accordance with animal ethics guidelines. For the intrapleural tumour model, mice were anaesthetised with isoflurane and inoculated with either 2 × 105 AB1-HA cells suspended in 200 µl PBS directly into the pleural space as described previously.50 Animals were monitored and euthanized when signs of terminal illness (i.e. laboured/rapid breathing, lethargy etc.) became apparent. Lungs, heart and tumour tissue were removed and weighed to determine the tumour burden and prepared for histological analysis.
Immune checkpoint blockade therapy
For the subcutaneous tumour model, prior to treatment, tumour area was measured and mice randomized into treatment groups to have balanced mean tumour sizes (20–30 mm2). Mice were randomised in all other experiments to treatment groups. Immune checkpoint antibodies (BioXcell, West Lebanon, NH) were administered, as indicated, by intraperitoneal injection (i.p.) in 100 µl of sterile PBS at the following doses unless otherwise stated; 100 μg anti-CTLA-4 (clone 9H10, Syrian Hamster IgG), 200 μg anti-OX40 (clone OX-86, Rat IgG1), 50 μg anti-GITR (clone DTA 1, Rat IgG2b), 200 μg anti-PD-1 (clone RMP1-14, Rat IgG2a) and 100 μg anti-TIM-3 (clone RMT 3–23, Rat IgG2a). Control mice received 100 µl PBS alone.
Preparation of single cell suspensions
Single cell suspensions were prepared from tumour and draining lymph node (dLN) upon harvest. All samples were centrifuged for 3 minutes at 625 x g unless otherwise stated. Tumour was digested using the GentleMACSTM Octo Dissociator (Miltenyi Biotec, Macquarie Park, NSW, Australia). Tumour tissue was cut into 2–4 mm pieces, placed into GentleMACSTM C tubes with 2.35 ml RPMI 1640, 100 µl ‘enzyme D’, 50 µl ‘enzyme R’ and 12.5 µl ‘enzyme A’ and run on program 37C_m_TDK_2 as per the manufacturer’s instructions. Once digested, samples were sequentially centrifuged, washed and filtered through 70 µm and 30 µm filters respectively. Cells were collected into a pellet by centrifugation and resuspended in PBS + 2% FCS. Tumour draining lymph nodes (dLN, inguinal) were mechanically digested between frosted slides and collected in PBS + 2% FCS as single cell suspensions. dLN were filtered into 30 µm filter top tubes, centrifuged and resuspended in PBS + 2% FCS.
Flow cytometry
100 µl of tumour or dLN cell suspensions were added to the appropriate wells of a 96 well plate. Prior to staining all samples were stained with live dead viability marker (ef780 or Zombie; eBioscience, San Diego CA) to differentiate between live and dead cells during analysis. 50 µl was added to each well and incubated at room temperature (RT) in the dark for 30 minutes. Samples were washed twice with PBS + 2% FCS before cell surface staining with α-mouse; CD3 (clone 145-2C11, Armenian Hamster IgG1), CD4 (clone RM4 5, Rat IgG2a), CD8 (clone 53–6.7, Rat IgG2a), CD25 (clone PC61, Rat IgG1), PD-1 (clone J43, Hamster IgG2a) and GITR (clone DTA-1), all BD Biosciences; OX40 (clone OX 86), CTLA-4 (clone UC10-4B9, Armenian Hamster IgG) and TIM-3 (clone B8.2C12), all BioLegend (San Diego, CA). Cells were fixed and permeabilised using the FOXP3 staining buffer set according to the manufacturer’s instructions (eBioScience), and stained with FoxP3 (clone FJK 16s) and Ki67 (clone SolA15) from eBioscience. Fluorescence minus-one (FMO) and single stain controls (compensation beads, OneComp eBeads, eBioscience) were used. Two antibody panels were used with antibodies conjugated to fluorochromes BUV395, BV510, BV421, BV786, BV711, AF700, APC, APC-Cy7, PE-CF594, PE, FITC and PE-Cy7. Flow cytometry was performed using the LSR Fortessa (BD Biosciences). Analysis was completed using Flow Jo version 9 (Tree Star, Ashland, Oregon).
Statistical analysis
Results are given as mean and standard error of the mean (± SEM) unless otherwise stated. Unpaired, nonparametric (Mann-Whitney) Student’s t-test was used to measure significance between two individual groups unless otherwise stated. Mixed model analysis of variance was used to analyse difference in tumour growth. Log rank (Mantel-Cox) analysis was performed on survival curves. All analysis was performed using Graph Pad Prism Software V6/7 (Graph Pad Software Inc., La Jolla, CA), or IBM® SPSS® statistics version 22 (Armonk, NY). Results were determined as significant when p < 0.05; *p < 0.05, **p < 0.01, *** p < 0.001, **** p < 0.0001.
Funding Statement
This project was supported, in part, by an iCARE dust diseases care competitive grant (2015-2017). The views expressed herein are those of the authors and are not necessarily those of iCARE dust diseases care. Additional support was provided in part by the Insurance Commission of Western Australia, the Government of Western Australia Department of Health and NHMRC Centre for Research Excellence (APP1107043).
Disclosure statement
No potential conflicts of interest were disclosed.
Supplementary Materials
Supplementary data can be accessed here
References
- 1.Robinson BW, Musk AW, Lake RA. Malignant mesothelioma. Lancet 2005; 366:397–408. doi: 10.1016/S0140-6736(05)67025-0 [DOI] [PubMed] [Google Scholar]
- 2.Vogelzang NJ, Rusthoven JJ, Symanowski J, Denham C, Kaukel E, Ruffie P, et al. Phase III study of pemetrexed in combination with cisplatin versus cisplatin alone in patients with malignant pleural mesothelioma. J Clin Oncol 2003; 21:2636–2644. doi: 10.1200/JCO.2003.11.136 [DOI] [PubMed] [Google Scholar]
- 3.Alley EW, Lopez J, Santoro A, Morosky A, Saraf S, Piperdi B, et al. Clinical safety and activity of pembrolizumab in patients with malignant pleural mesothelioma (KEYNOTE-028): preliminary results from a non-randomised, open-label, phase 1b trial. Lancet Oncol 2017; 18:623–630. doi: 10.1016/S1470-2045(17)30169-9 [DOI] [PubMed] [Google Scholar]
- 4.Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2015; 372:320–330. doi: 10.1056/NEJMoa1412082 [DOI] [PubMed] [Google Scholar]
- 5.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010; 363:711–723. doi: 10.1056/NEJMoa1003466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012; 366:2443–2454. doi: 10.1056/NEJMoa1200690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yamada N, Oizumi S, Kikuchi E, Shinagawa N, Konishi-Sakakibara J, Ishimine A, et al. CD8+ tumor-infiltrating lymphocytes predict favorable prognosis in malignant pleural mesothelioma after resection. Cancer Immunol Immunother 2010; 59:1543–1549. doi: 10.1007/s00262-010-0881-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A 2002; 99:12293–12297. doi: 10.1073/pnas.192461099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hegmans JP, Hemmes A, Hammad H, Boon L, Hoogsteden HC, Lambrecht BN. Mesothelioma environment comprises cytokines and T-regulatory cells that suppress immune responses. Eur Respir J 2006; 27:1086–1095. doi: 10.1183/09031936.06.00135305 [DOI] [PubMed] [Google Scholar]
- 10.Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med 1995; 182:459–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krummel MF, Allison JP. CTLA-4 engagement inhibits IL-2 accumulation and cell cycle progression upon activation of resting T cells. J Exp Med 1996; 183:2533–2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science 2008; 322:271–275. doi: 10.1126/science.1160062 [DOI] [PubMed] [Google Scholar]
- 13.Keir ME, Liang SC, Guleria I, Latchman YE, Qipo A, Albacker LA, et al. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J Exp Med 2006; 203:883–895. doi: 10.1084/jem.20051776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol 2001; 2:261–268. doi: 10.1038/85330 [DOI] [PubMed] [Google Scholar]
- 15.Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med 2000; 192:1027–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu L, Yun Z, Tagawa T, Rey-McIntyre K, De Perrot M. CTLA-4 blockade expands infiltrating T cells and inhibits cancer cell repopulation during the intervals of chemotherapy in murine mesothelioma. Mol Cancer Ther 2012; 11:1809–1819. doi: 10.1158/1535-7163.MCT-11-1014 [DOI] [PubMed] [Google Scholar]
- 17.Lesterhuis WJ, Salmons J, Nowak AK, Rozali EN, Khong A, Dick IM, et al. Synergistic effect of CTLA-4 blockade and cancer chemotherapy in the induction of anti-tumor immunity. PLoS One 2013; 8:e61895. doi: 10.1371/journal.pone.0061895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leshem Y, O’Brien J, Liu X, Bera TK, Terabe M, Berzofsky JA, et al. Combining local immunotoxins targeting mesothelin with CTLA-4 blockade synergistically eradicates murine cancer by promoting anticancer immunity. Cancer Immunol Res 2017; 5:685–694. doi: 10.1158/2326-6066.CIR-16-0330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mkrtichyan M, Najjar YG, Raulfs EC, Abdalla MY, Samara R, Rotem-Yehudar R, et al. Anti-PD-1 synergizes with cyclophosphamide to induce potent anti-tumor vaccine effects through novel mechanisms. Eur J Immunol 2011; 41:2977–2986. doi: 10.1002/eji.201141639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu L, Wu MO, De La Maza L, Yun Z, Yu J, Zhao Y, et al. Targeting the inhibitory receptor CTLA-4 on T cells increased abscopal effects in murine mesothelioma model. Oncotarget 2015; 6:12468–12480. doi: 10.18632/oncotarget.3487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maio M, Scherpereel A, Calabro L, Aerts J, Perez SC, Bearz A, et al. Tremelimumab as second-line or third-line treatment in relapsed malignant mesothelioma (DETERMINE): a multicentre, international, randomised, double-blind, placebo-controlled phase 2b trial. Lancet Oncol 2017; 18:1261–1273. doi: 10.1016/S1470-2045(17)30446-1 [DOI] [PubMed] [Google Scholar]
- 22.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 2015; 373:23–34. doi: 10.1056/NEJMoa1504030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sabatos CA, Chakravarti S, Cha E, Schubart A, Sanchez-Fueyo A, Zheng XX, et al. Interaction of tim-3 and tim-3 ligand regulates T helper type 1 responses and induction of peripheral tolerance. Nat Immunol 2003; 4:1102–1110. doi: 10.1038/ni988 [DOI] [PubMed] [Google Scholar]
- 24.Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med 2010; 207:2187–2194. doi: 10.1084/jem.20100643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Valzasina B, Guiducci C, Dislich H, Killeen N, Weinberg AD, Colombo MP.. Triggering of OX40 (CD134) on CD4(+)CD25+ T cells blocks their inhibitory activity: a novel regulatory role for OX40 and its comparison with GITR. Blood 2005; 105:2845–2851. doi: 10.1182/blood-2004-07-2959 [DOI] [PubMed] [Google Scholar]
- 26.Cohen AD, Schaer DA, Liu C, Li Y, Hirschhorn-Cymmerman D, Kim SC, et al. Agonist anti-gitr monoclonal antibody induces melanoma tumor immunity in mice by altering regulatory t cell stability and intra-tumor accumulation. PLoS ONE 2010; 5:e10436. doi: 10.1371/journal.pone.0010436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ott PA, Hodi FS, Kaufman HL, Wigginton JM, Wolchok JD. Combination immunotherapy: a road map. J Immunother Cancer 2017; 5:16. doi: 10.1186/s40425-017-0218-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lesterhuis WJ, Rinaldi C, Jones A, Rozali EN, Dick IM, Khong A, et al. Network analysis of immunotherapy-induced regressing tumours identifies novel synergistic drug combinations. Scientific Reports 2015; 5:12298. doi: 10.1038/srep12298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Calabro L, Morra A, Fonsatti E, Cutaia O, Fazio C, Annesi D, et al. Efficacy and safety of an intensified schedule of tremelimumab for chemotherapy-resistant malignant mesothelioma: an open-label, single-arm, phase 2 study. Lancet Respir Med 2015; 3:301–309. doi: 10.1016/S2213-2600(15)00092-2 [DOI] [PubMed] [Google Scholar]
- 30.Marabelle A, Kohrt H, Sagiv-Barfi I, Ajami B, Axtell RC, Zhou G, et al. Depleting tumor-specific Tregs at a single site eradicates disseminated tumors. J Clin Invest 2013; 123:2447–2463. doi: 10.1172/JCI64859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Redmond WL, Linch SN, Kasiewicz MJ. Combined targeting of costimulatory (OX40) and coinhibitory (CTLA-4) pathways elicits potent effector T cells capable of driving robust antitumor immunity. Cancer Immunol Res 2014; 2:142–153. doi: 10.1158/2326-6066.CIR-13-0031-T [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu P, Arias RS, Sadun RE, Nien YC, Zhang N, Sabzevari H, et al. Construction and preclinical characterization of Fc-mGITRL for the immunotherapy of cancer. Clin Cancer Res 2008; 14:579–588. doi: 10.1158/1078-0432.CCR-07-0940 [DOI] [PubMed] [Google Scholar]
- 33.Lu L, Xu X, Zhang B, Zhang R, Ji H, Wang X. Combined PD-1 blockade and GITR triggering induce a potent antitumor immunity in murine cancer models and synergizes with chemotherapeutic drugs. J Transl Med 2014; 12:36. doi: 10.1186/1479-5876-12-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramirez-Montagut T, Chow A, Hirschhorn-Cymerman D, Terwey TH, Kochman AA, Lu S, et al. Glucocorticoid-induced TNF receptor family related gene activation overcomes tolerance/ignorance to melanoma differentiation antigens and enhances antitumor immunity. J Immunol 2006; 176:6434–6442. [DOI] [PubMed] [Google Scholar]
- 35.Shimizu J, Yamazaki S, Takahashi T, Ishida Y, Sakaguchi S. Stimulation of CD25(+)CD4(+) regulatory T cells through GITR breaks immunological self-tolerance. Nat Immunol 2002; 3:135–142. doi: 10.1038/ni759 [DOI] [PubMed] [Google Scholar]
- 36.Ko K, Yamazaki S, Nakamura K, Nishioka T, Hirota K, Yamaguchi T, et al. Treatment of advanced tumors with agonistic anti-GITR mAb and its effects on tumor-infiltrating Foxp3+CD25+CD4+ regulatory T cells. J Exp Med 2005; 202:885–891.doi: 10.1084/jem.20050940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mitsui J, Nishikawa H, Muraoka D, Wang L, Noguchi T, Sato E, et al. Two distinct mechanisms of augmented antitumor activity by modulation of immunostimulatory/inhibitory signals. Clin Cancer Res 2010; 16:2781–2791. doi: 10.1158/1078-0432.CCR-09-3243 [DOI] [PubMed] [Google Scholar]
- 38.Shevach EM, Stephens GL. The GITR-GITRL interaction: co-stimulation or contrasuppression of regulatory activity? Nat Rev Immunol 2006; 6:613–618. doi: 10.1038/nri1867 [DOI] [PubMed] [Google Scholar]
- 39.Davis MR, Manning LS, Whitaker D, Garlepp MJ, Robinson BW.. Establishment of a murine model of malignant mesothelioma. Int J Cancer 1992; 52:881–886. [DOI] [PubMed] [Google Scholar]
- 40.Fisher SA, Aston WJ, Chee J, Khong A, Cleaver AL, Solin JN, et al. Transient treg depletion enhances therapeutic anti-cancer vaccination. Immun Inflamm Dis 2017; 5:16–28. doi: 10.1002/iid3.136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sneddon S, Patch AM, Dick IM, Kazakoff S, Pearson JV, Waddell N, et al. Whole exome sequencing of an asbestos-induced wild-type murine model of malignant mesothelioma. BMC Cancer 2017; 17:396. doi: 10.1186/s12885-017-3382-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brown SD, Warren RL, Gibb EA, Martin SD, Spinelli JJ, Nelson BH, et al. Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res 2014; 24:743–750. doi: 10.1101/gr.165985.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Efremova M, Finotello F, Rieder D, Trajanoski Z. Neoantigens Generated by Individual Mutations and Their Role in Cancer Immunity and Immunotherapy. Front Immunol 2017; 8:1679. doi: 10.3389/fimmu.2017.01679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and transcriptomic features of response to anti-pd-1 therapy in metastatic melanoma. Cell 2016; 165:35–44. doi: 10.1016/j.cell.2016.02.065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015; 348:124–128. doi: 10.1126/science.aaa1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Strickland KC, Howitt BE, Shukla SA, Rodig S, Ritterhouse LL, Liu JF, et al. Association and prognostic significance of BRCA1/2-mutation status with neoantigen load, number of tumor-infiltrating lymphocytes and expression of PD-1/PD-L1 in high grade serous ovarian cancer. Oncotarget 2016; 7:13587–13598. doi: 10.18632/oncotarget.7277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bai J, Gao Z, Li X, Dong L, Han W, Nie J. Regulation of PD-1/PD-L1 pathway and resistance to PD-1/PD-L1 blockade. Oncotarget 2017; 8:110693–110707. doi: 10.18632/oncotarget.22690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marzo AL, Lake RA, Robinson BW, Scott B. T-cell receptor transgenic analysis of tumor-specific CD8 and CD4 responses in the eradication of solid tumors. Cancer Res 1999; 59:1071–1079. [PubMed] [Google Scholar]
- 49.McLean M, Wallace HL, Sharma A, Hill HC, Sabel MS, Egilmez NK. A BALB/c murine lung alveolar carcinoma used to establish a surgical spontaneous metastasis model. Clin Exp Metastasis 2004; 21:363369. [DOI] [PubMed] [Google Scholar]
- 50.Lansley SM, Cheah HM, Varano Della Vergiliana JF, Chakera A, Lee YC. Tissue plasminogen activator potently stimulates pleural effusion via a monocyte chemotactic protein-1-dependent mechanism. Am J Respir Cell Mol Biol 2015; 53:105–112. doi: 10.1165/rcmb.2014-0017OC [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.