

Abstract
Ribosomal repeats occupy 5% of a plant genome, yet there has been little study of their diversity in the modern age of genomics. Ribosomal copy number and expression variation present an opportunity to tap a novel source of diversity. In the present study, we estimated the ribosomal DNA (rDNA) copy number and ribosomal RNA (rRNA) expression for a population of maize inbred lines and investigated the potential role of rDNA and rRNA dosage in regulating global gene expression. Extensive variation was found in both ribosomal DNA copy number and ribosomal RNA expression among maize inbred lines. However, rRNA abundance was not consistent with the copy number of the rDNA. We have not found that the rDNA gene dosage has a regulatory role in gene expression; however, thousands of genes are identified to be coregulated with rRNA expression, including genes participating in ribosome biogenesis and other functionally relevant pathways. We further investigated the potential roles of copy number and the expression level of rDNA on agronomic traits and found that both correlated with flowering time but through different regulatory mechanisms. This comprehensive analysis suggested that rRNA expression variation is a valuable source of functional diversity that affects gene expression variation and field-based phenotypic changes.
Ribosomes are the indispensable machinery for protein synthesis in all domains of life. They are assembled from four kinds of ribosomal RNA (5S rRNA, 5.8S rRNA, 18S rRNA, and 25S rRNA in plants), around 80 ribosomal proteins, and many other factors functioning in processing, modifying, and transporting rRNA and preribosomal particles (Klinge et al. 2011; Rabl et al. 2011; de la Cruz et al. 2015). The 18S, 5.8S, and 25S rRNAs are produced as a long primary transcript by polymerase I from the 45S rRNA loci and processed into mature forms by removing intergenic spacers (ITS1 and ITS2), whereas 5S is transcribed by polymerase III from a separate locus. In many higher organisms, 5S and 45S are usually physically separated in the genome (Garcia et al. 2017). However, the fate of 5S and two other components (5.8S and 18S) are biologically connected, because they cooperatively assembly the large ribosomal subunit. Recently, the 5S and 45S loci were reported to have evolved in a concerted fashion in the human genome (Gibbons et al. 2015). In addition, rRNA genes (also termed rDNA) are tandemly organized with hundreds or thousands of nearly identical copies, accounting for >5% of plant genomes, for example, in Arabidopsis (Rogers and Bendich 1987). In spite of the high conservation level of their sequence and function, the copy number (CN) of rDNA is quite variable both between and within species (Gerlach and Bedbrook 1979; Rogers and Bendich 1987; Long et al. 2013). The biological benefits from CN variation (CNV) of rDNA are not well understood (Weider et al. 2005). Some argue that the rDNA tandem array structure may favor transcription efficiency for the large demand for rRNA (Prokopowich et al. 2003), while others propose that the amount of rDNA amount is actually far beyond the actual number required for rRNA transcription because not every copy of the rRNA genes are actively transcribed; e.g., some copies are epigenetically silenced in the human genome (McStay and Grummt 2008), and numerous rRNA genes are selectively silenced during development in Arabidopsis (Chandrasekhara et al. 2016).
Although our understanding of the potential significance of CNV of rRNA genes is incomplete, the change in CN of rRNA has been documented to have epigenetic effects on global chromatin regulation (Paredes and Maggert 2009) or gene expression (Paredes et al. 2011). Recently, a genome-wide study involving the 45S rRNA CN in a human population reported that expression level of 1371 genes was significantly associated with rDNA dosage. Furthermore, those genes are functionally enriched within the regulation of rDNA expression and ribosome biogenesis (Gibbons et al. 2014). This interesting observation inspired us to ask another basic question: How does rDNA dosage affect gene expression? Most associated genes were found to be functionally connected with rDNA expression, which is very similar to the situation of the coexpressed genes within the same pathway (herein, the ribosome biogenesis pathway); therefore, we hypothesized that the connection between rDNA CN and gene expression is a mimic of the association among rDNA CN, rRNA expression, and coexpressed genes. Two recent studies reported that an artificially enhanced 45S rRNA expression level could lead to an increase in expression by up to twofold of 37 genes, including a ribosomal protein gene and several transcription factors, with obvious effects on aboveground plant growth, reinforcing our speculation about the coregulation between rDNA and protein coding genes (Makabe et al. 2017a,b).
Maize (Zea mays L.), one of the most important cereals worldwide, provides valuable resources for human food, livestock feed, and industrial production. Maize is also an ideal genetic model to understand genetic variation because of the large and diverse set of available germplasms (Buckler et al. 2006). However, CN and the expression abundance of ribosomal repeats still represent unexploited sources of genetic variation in maize. In the present study, by analyzing the DNA resequencing data and RNA-seq data for a maize inbred population, we comprehensively explored the rDNA CNVs and rRNA expression variation; the inherent connections between rDNA dosage, rRNA expression level, and related gene expression; and the potential biological function of the natural variation in the CN of ribosomal RNA and its expression abundance.
Results
Estimation of ribosomal RNA gene CN using a maize inbred population
First, we estimated the ribosomal RNA gene CN for the maize 282 association panel population (282 panel) representing a maize germplasm with extensive phenotype and genotype diversity (Flint-Garcia et al. 2005) using the methods previously described (Gibbons et al. 2014) with some modifications (see Methods; Supplemental Fig. S1). Briefly, we estimated the sequencing depths for single-copy (SC) genomic regions (introns and exons) and repetitive DNA regions (e.g., rRNA), respectively, and then calculated the CN by calculating the ratio between them after normalization. In the maize B73 reference genome, 4459 SC exons and 2982 SC introns were identified and used to calculate background per-base read depth (bRD) (Supplemental Table S1). The read depth we estimated between SC exons and introns was highly correlated and nearly identical (Pearson's r = 0.987, P-value <2.2 × 10−16) (Supplemental Fig. S2). For 256 maize inbred lines (bRD > 1×), the 45S rRNA was estimated to be present in 1061 to 17,347 copies , and CN for 5S rRNA was estimated to be between 1005 and 8208 (Supplemental Dataset 1). Our estimate is comparable with previous results generated using different experimental approaches; e.g., 3300–23,000 (Buescher et al. 1984). The 45S CNV could contribute at most 138 Mb (∼7%) to the genome size variation among maize inbred lines, reinforcing the positive relationship between genome size and the CN of rRNA repeats (Prokopowich et al. 2003; Long et al. 2013). CNs for the three components of 45S (5.8S, 18S, and 25S) were also assayed separately and found to be highly correlated with each other, as expected (Supplemental Fig. S3).
We also performed this analysis workflow on the third-generation maize haplotype map (HapMap3) data set, which is a larger data set with over 900 maize lines; however, the data were collected from an international consortium over ∼10 yr (Bukowski et al. 2018). We found that different library preparations could dramatically affect the sequencing data and further caused different estimates of absolute CN (Supplemental Table S2). In contrast, different sequencing runs from the same library usually give rise to similar estimates (Supplemental Table S2). We found that some samples had quite abnormal estimates for the 5.8S CN (Supplemental Table S3). However, other closely connected components (18S or 25S) showed relative normal estimates; therefore, we suspected that this was likely caused by inefficient PCR amplification during library preparations owing to the high complex secondary structure of the rRNA (Gottschling and Plotner 2004). Forty-one HapMap3 lines have previously been used to estimate knob abundance using a different approach (Chia et al. 2012). Thus, we compared these results with ours and found that both estimates were highly consistent (Supplemental Fig. S4). This result provided additional indirect evidence to support the accuracy of our method for CN estimation of repetitive elements.
To further validate our CN estimate on rDNA, we conducted qPCR experiment to quantify relative rDNA CNs for 24 maize lines (see Methods). We found qPCR results are consistent with our expectations under certain conditions (Supplemental Fig. S5A). Although qPCR generated different absolute rDNA CNs for each line (Supplemental Table S4), both approaches can give relatively similar estimates (Pearson's r = 0.792, P-value = 4.098 × 10−6); the maize lines with highest CN estimate by NGS data also have significant higher CN based on qPCR analysis (Supplemental Fig. S5B).
Maize inbred lines can be roughly divided into subgroups (Flint-Garcia et al. 2005). To examine the population structure effect, we compared the CNs of 45S among different maize subgroups and found that the 45S repeats were significantly more abundant in tropical/subtropical (ts) lines compared with some temperate lines (stiff stalk [ss]; P < 0.01); however, there were not significantly different from non-ss (nss) and admixed lines (Fig. 1A). In addition, 5S (P < 0.001) and two other kinds of knob repeats (P < 0.0001) were also significantly more abundant in ts lines (Fig. 1B–D), which was consistent with previous observations of more repeat sequences and larger genome sizes in these populations (Chia et al. 2012; Diez et al. 2013).
Figure 1.

Comparisons of the copy number (CN) for 45S rRNA and other common repeat types among different subgroups of maize inbred lines. (A) 45S rRNA. Pairwise comparison showing that only the tropical/subtropical (ts) and stiff-stalk (ss) lines are significantly different (P = 0.025). (B) 5S rRNA. The ts are significantly different to the ss (0.0009) and nss lines (0.0011). (C,D) Two kinds of maize knob repeats. Knob 180 and Knob TR1 are significantly more abundant in the ts lines compared with any other subgroups (P < 0.0001). (nss) Non-ss lines. (*) P < 0.05, (***) P < 0.001, (****) P < 0.0001.
For the maize genome, only single 45S and 5S rRNA loci were identified on Chromosome 6 and Chromosome 2, respectively (Li and Arumuganathan 2001). We detected significant correlation between the 5S and 45S CN (Pearson's r = 0.240, P < 0.0001) (Fig. 2A), which was not as strong as that observed in humans (Gibbons et al. 2015). Similar to the finding in humans, 5S also showed a slightly stronger correlation with both 5.8S and 25S than with 18S (Fig. 2B–D), which has been viewed as further evidence for coevolution. We identified 10 taxa with abnormal CN estimates as outliers. After removing these outliers, similar results were obtained (Supplemental Fig. S6).
Figure 2.

Evidence for the coevolution of 5S with 45S rRNA. (A) Correlation analysis between the CN of 5S and 45S rRNA (P-value = 0.0001). (B–D) Correlations between the CN of 5S and three components of 45S, including 5.8S (P = 3.366 × 10−5), 18S (P = 0.0003), and 25S (P = 7.942 × 10−5). The population size was n = 256, and r is the Pearson's coefficient.
Inheritance of 45S rRNA genes
To understanding whether 45S rRNA could be stably transferred through generations, we estimated the narrow sense heritability (h2) for 45S CN using a mixed linear model (MLM; see Methods). Both 45S (h2= 0.780) and 5S (h2 = 0.788) were highly heritable (Supplemental Table S5). In addition, two kinds of knob repeats and centromere repeats also showed high heritability (0.565–0.902) (Supplemental Table S5).
To detect whether we could capture significant associations in cis for 45S copy dosage in maize, we first conducted a genome-wide association study (GWAS) to identify genomic loci associated with the 45S CN. However, GWAS could not identify the known 45S cluster but detected some significant SNPs across the genome (Supplemental Fig. S7A). Alternatively, it is still possible that these associated SNPs might have arisen from a false-positive association.
Studies in Arabidopsis concluded that the 45S CN is a bona fide case of “missing heritability” (Rabanal et al. 2017). To test this hypothesis, we used variance component analysis (Speed et al. 2012; Gusev et al. 2013) to estimate the total variance explained by all common SNPs by detecting the total contributions from all chromosomes (see Methods). The results showed that all variants could explain >98% of total 45S CNV, of which the known cluster (Chromosome 6) independently could explain ∼80% of the variance, and the other nine chromosomes together contributed ∼20% (Supplemental Fig. S7B). This result suggested that the largest contribution to 45S rDNA CN actually originated from 45S array on Chromosome 6; however, it could not be detected by GWAS when testing each SNP one at a time, which is consistent with the findings in Arabidopsis with natural and controlled populations (Long et al. 2013; Rabanal et al. 2017).
Quantifying 45S rRNA gene expression
Recently, we generated 3′mRNA-seq data for seven tissues among 299 maize lines (Kremling et al. 2018). Because this method used a poly(T) primer to perform specific reverse transcription on mRNA transcripts with poly(A) tails, rRNA read contamination should be quite low in these RNA-seq data. We found that 4%–15% of the total reads from different RNA-seq libraries could be mapped onto the 45S rRNA gene. We found >95% of the reads were originated from five regions across the 45S region (Fig. 3). Among the five regions, Region 2 (nt 4058–4144) had the most mapped reads (∼50% of total reads mapping on whole 45S region). We tested a total of 1923 RNA-seq libraries and observed the same landscape for all mapping patterns (Supplemental Figs. S8, S9), highlighting the excellent repeatability of rRNA sequencing.
Figure 3.

45S rRNA can be sequenced by 3′mRNA-seq. (A) 3′mRNA-seq data (from one B73 GRoot sample) mapped on the maize 45S rDNA reference sequence shows five obvious peaks. Each peak represents a region that could be highly amplified using poly(T) primers and sequenced. A higher peak indicates more reads generated from these regions, implying higher reverse transcription efficiency. (B) The entire 45 rDNA gene locus with intergenic spacer region (IGS). (C) The bar plot indicates the frequency of the poly(A)-like motif across the 45S region. The y-axis represents the count of the motif.
Next, we tested whether mapped reads number within Region 2 can be treated as RNA-seq reads mapped on mRNA and calculated normalized expression value to represent the expression of 45S rDNA (see below and Discussion). The 3′ mRNA sequencing approach is designed to avoid rRNA amplification and sequencing, because only mRNA with poly(A) tails can be sequenced (see Methods). Thus, we thought that mismatch between poly(T) primer and some regions across 45S rRNA is the reason for initializing the reverse transcription and finally being sequenced. Combined with sequence analysis, we found that poly(A) motifs (more than three continuous A nucleotides) occurred more frequently in the sequence upstream of the five regions (Fig. 3). Although the amplification rate of Region 2 should be much lower than mRNA because of imperfect match with poly(T) primer. However, if the amplification rate is consistent among different libraries (e.g., in each RNA-seq library, 50% 45S rRNA transcripts can be amplified and sequenced), the RPKM for Region 2 can be treated as relative expression abundance of rDNA and compared among samples. Despite lacking direct evidence, several other analyses have been used to indirectly validate whether our method to measure rDNA expression is possible. First, conserved upstream sequences across different RNA-seq libraries have been verified by checking for sequence variation. We found only two SNPs (without affecting the poly(A)-like motif) that were present in less than five maize lines around Region 2 (the 350-bp upstream region) based on the results of SNP screening across the whole 45S rDNA (Supplemental Fig. S10). Thus, the poly(A)-like genomic regions are identical in the different maize lines, which may ensure a similar PCR efficiency around Region 2.
Second, we observed that Region 2 shows high correlations with other peak regions in terms of mapped read number, which is another indicator that Region 2 is the most robust representative of the 45S expression level (Supplemental Fig. S11A). Third, we assumed that if the mapped read number from Region 2 is randomly produced across different libraries caused by random amplification (i.e., different libraries with different PCR amplification rates), we would expect to see a low correlation between the mapped reads number and the total reads number. However, we observe a high correlation between the mapped reads number and the total reads number, suggesting the mapped reads number was increased by the increase of read depth. We also compared this result with 1000 randomly chosen genes. These results showed that the differences in 45S expression mainly originated from differences among libraries, which was very similar to the results for most of the genes (Supplemental Fig. S11B). Taken together, we eventually decided to treat Region 2 as an mRNA gene and used the normalized read count per million (RPM) within Region 2 as a proxy for 45S expression levels in the seven different tissues for each maize inbred line (Supplemental Dataset 2).
rRNA abundance is highly developmentally dynamic
Our intra-species comparisons of expression revealed that maize 45S rRNA expression may not be as conserved as previously thought, but may be differentially expressed among tissues and is unexpectedly variable among maize individuals (Fig. 4A). Generally, L3Base, GRoot, and GShoot, which represent young tissues, expressed more rRNA than old tissues, consistent with the fact that during rapid developmental stages, a larger amount of rRNA is required for ribosome production. Moreover, along one single maize leaf gradient, the 45S rRNA level gradually decreased from the leaf base (young) to the leaf tip (mature) (Fig. 4B), similar to the expression pattern of hundreds of mRNAs (Li et al. 2010). Cluster analysis based on 45S rRNA expression shows that the Kernel and L3Tip showed the most similar expression pattern, perhaps because both are mature tissues (Supplemental Fig. S12). We further investigated the relationships among 45S rRNA expression levels in different tissues. The results showed that except for the leaf base and leaf tip (likely because of their physical connections), no significant correlations for 45S abundance were found among the tissues (Fig. 4C), which suggested that 45S rRNA expression may be regulated independently in each tissue.
Figure 4.

The rDNA expression pattern among tissues. (A) Variable expression of rDNA between tissues. (B) From the leaf base to the leaf tip, the 45S rDNA expression decreased gradually (tested with 26 samples). (C) Pearson's correlation analyses of 45S expression in different tissues. (GRoot) Germinated plant root; (GShoot) germinated plant shoot; (L3Base) basal part from the third flag leaf; (L3Mid) middle part from the third flag leaf; (L3Tip) top part from the third flag leaf; (Kernel) maturing kernel; (LMAD) mature leaf under daylight; (LMAN) mature leaf at night.
To investigate the relationship between 45S CN and 45S rRNA expression abundance, we analyzed 240 maize lines for their 45S CN and expression data. The results indicated that there is no correlation between 45S rRNA CN and expression in any tissues, either for the whole population (Fig. 5) or for the subgroups (Supplemental Figs. S13, S14), suggesting that maize lines with more rRNA copies might not have a higher overall rRNA expression level, possibly owing to the existence of many nonfunctional copies.
Figure 5.

The relationship between 45S rDNA CN and 45S rRNA expression. (A) Correlation of 45S CN and expression in seven tissues. (B) Spearman's correlation coefficients between 45S rRNA CN and expression data in seven different tissues.
Ribo-coregulated genes are abundant and functionally enriched in coherent gene sets
The 45S copy dosage was revealed to trans-regulate global gene expression in humans (Gibbons et al. 2014); thus, we expected to identify gene loci that could be regulated by 45S CN in the maize genome. However, only 32 gene expression variants (in seven tissues) were found to be significantly correlated (P < 1 × 10−5) with 45S CN changes (Supplemental Fig. S15A).
We then conducted an association test between 45S expression abundance and individual gene expression levels in the seven tissues. We identified 1198 genes that were highly coexpressed with rDNA in GRoot (P < 6.5 × 10−6; false-discovery rate [FDR] = 0.001, gene number [n] = 17,770) (Supplemental Fig. S15B). Based on the same criteria, we discovered 1231 genes in GShoot (n = 18,521), 819 in Kernel (n = 17,745), 815 in L3Base (n = 17,843), 1266 in L3Tip (n = 15,306), 44 in LMAD (n = 16,677), and 1610 in LMAN (n = 16,357) (Supplemental Table S6). Furthermore, 817 genes remained highly significant after stringent Bonferroni correction (P < 8.5 × 10−8; FDR = 0.0001). Consistent with our expectations, positively correlated genes were significantly more abundant than negatively correlated genes in most samples, except for LMAN (Supplemental Table S6). The important role of 45S rRNA in ribosome assembly prompted us to investigate whether these 45S-coregulated genes were functionally enriched in ribosome biogenesis. As expected, we identified 1438 coregulated genes (shared by two or more tissues) for gene functional enrichment analysis by excluding tissue-specific genes. To assign these 1438 genes into functional categories, we performed Gene Ontology (GO) analysis. The top five GO terms are exclusively related to protein synthesis or ribosome biogenesis (Fig. 6), suggesting that a considerable number of genes coexpressed with rRNA play roles in assisting rRNA into the ribosome complex. Further Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis also identified 112 rRNA-coexpressed genes belonging to the ribosomal protein (r-protein) gene family (Supplemental Table S7; Supplemental Fig. S16). This synchronous expression in both rRNA and r-protein genes implied that ribosome biogenesis is a highly regulated and coordinated biological pathway.
Figure 6.
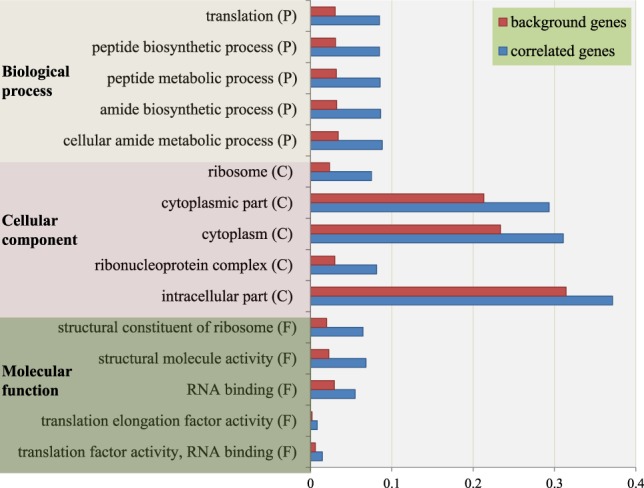
Gene Ontology (GO) enrichment analysis of 1438 coregulated genes. The top five GO terms in three different categories are exclusively enriched within ribosome biogenesis and protein synthesis.
Besides ribosome biogenesis, GO analysis revealed that rRNA-coexpressed genes were also enriched within other functional categories, yet these show great differences among tissues. In GRoot, the significantly enriched gene functional categories fall within the defense response. Moreover, we found that the establishment of the defense system is a complex process that involves a wide spectrum of functions, including response to wounding (16 correlated genes, P-value = 6.8 × 10−6), response to bacterium (26 genes, 7.1 × 10−6), response to biotic stimulus (41 genes, 7.7 × 10−6), response to acid chemicals (41 genes, 0.00027), and response to other organisms (39, 2.3 × 10−5) (Supplemental Table S8). The genes that were assigned to defensive function were mostly positively correlated with rRNA expression. Similarly, we found that genes that were positively correlated with rDNA in GShoot were also enriched in defensive functional groups (Supplemental Table S9). These results may indicate that in plants at very young stage, the primary requirement is to build a self-defense system for protection; thus, these categories of genes will be more active and productive at this stage. For maize kernels, however, unique functional categories of genes were found to be coexpressed with rDNA (Supplemental Table S10). Many genes involved with cellular macromolecule metabolic pathways and reproductive system development are highly expressed, which correlates with the fact that chemical compounds are synthesized and stored during the seed formation. In addition, we observed a considerable number of functionally enriched genes that have a role in gene regulation, including gene silencing and epigenetic regulation (Supplemental Table S10). These genes may regulate rDNA transcription by repressing its transcription or post-transcriptional degradation (e.g., GRMZM2G047949, which functions in RNA degradation).
We also observed some differences between L3Base and L3Tip by comparing the associated GO terms. In L3Base, most genes that positively correlated with rDNA were functionally enriched for translation and ribosome biogenesis (Supplemental Table S11), indicating the basic infrastructure (e.g., the ribosome) for protein translation is required in high amounts in very young cells. In L3Tip, however, ribosome assembly is not a priority pathway; instead, photosynthesis and starch export are the most important biological processes for the mature leaf cells (Supplemental Table S12). GO analysis in LMAN revealed a possible change during the shift between day and night. During the night, photosynthesis is weakened, while genes involved in response to external stimuli become more active, especially those related to adapting to changes in light and temperature (Supplemental Table S13).
Tissue-specific ribosome biogenesis
Our coexpression analyses revealed two features for the interaction between the transcription of 45S and r-protein genes. First, we identified 66, 11, 51, 98, and 106 r-protein genes in the GRoot, GShoot, Kernel, L3Base, and L3Tip libraries, respectively (Supplemental Table S14). However, no single gene was shared by all five tissues (Fig. 7A). Second, despite most ribosomal genes being positively correlated with the rRNA expression level, unexpectedly large numbers of ribosomal genes showed a negative correlation with rRNA in some tissues, especially in mature tissues. For example, 62 of 106 r-protein genes were negatively correlated with rDNA in L3Tip, and 40 of 51 genes showed a negative correlation with rDNA expression in the kernel. However, in very immature tissue, such as L3Base, almost all r-protein genes (97/98) showed a positive correlation with rDNA expression. Additionally, we identified 29 r-protein genes that were coexpressed with rDNA in both L3Base and L3Tip (Fig. 7B). Among these genes, 17 showed a positive correlation in both tissues, but the remaining 12 genes showed a negative correlation from L3Base to L3Tip (Supplemental Table S15). We observed an rRNA expression decrease from L3Base to L3Tip (Fig. 4B); therefore, we continued to investigate whether the increased expression of these 12 genes from L3Base to L3Tip is the reason for reverse direction of correlation. We therefore explored the pattern of expression alteration and found that the 29 shared r-protein genes, as well as rDNA, demonstrated an overall decrease in expression among most maize taxa during the transition from L3Base to L3Tip (Fig. 7C,D).
Figure 7.

Tissue-specific expression of r-protein genes. (A) Venn diagram shows shared r-protein genes among different tissues. (B) The expression pattern of 17 shared r-protein genes with same direction of correlation with rRNA between L3Base and L3Tip, suggesting that most genes in most maize lines show decreased expression from L3Base to L3Tip. (C) Twenty-nine r-protein genes were identified to be shared between L3Base and L3Tip. (D) The expression pattern of 12 shared r-protein genes, which show different directions of correlation with rRNA expression between L3Base and L3Tip. Similarly, these genes also showed decreased expression levels. Color key indicates log2 (E1/E2), where E1: rpm (reads per million) (L3Base); E2: rpm (L3Tip) for each gene. The blue bar indicates an expression level decrease, and the red bar indicates an expression level increase. Gene details can be found in Supplemental Table S15.
Tests on the potential role of 45S in phenotypic variation
To investigate whether 45S rRNA can cause phenotypic variation by CN variation or expression changes or both, we first investigated the relationship between the CN of 45S and phenotype measurements. CN of 45S showed no significant correlation with most measured agronomic traits, except for flowering time–related traits, which showed a weak positive correlation (Supplemental Fig. S17). We also tested this association for three other types of tandem repeats: Knob 180, Knob TR1, and CentC. As expected, the knob-related repeats had a very significant positive association with flowering time and the centromere repeats also showed a weak positive correlation (Supplemental Fig. S17). This result suggested that large amounts of repeat sequences within the maize genome might delay flowering by an unknown mechanism, as previously reported (Rayburn et al. 1994).
Correlation analysis between rRNA expression variation and phenotypic variation among maize taxa revealed a significant negative association between flowering time and rDNA expression level in leaves collected at night (LMAN) (Fig. 8). We hypothesized that rDNA might not regulate flowering time directly, but the genes that are coregulated with rDNA might play such a role. Therefore, we investigated the function of 1610 rDNA coregulated genes in LMAN and identified 40 genes that were functionally enriched in reproductive system development (related to flowering timing). Notably, all 40 genes showed positive correlations with rDNA expression. We further tested the correlation between flowering time and gene expression and showed that 22 of them have a significant negative correlation with days to silk (DTS) and days to pollen shed (DTA; Pearson's coefficient between −0.22 and −0.58) (Supplemental Table S15). We further tested the effects of expression variation of these genes in other tissues; however, no strong association was detected other than in LMAN, suggesting that these genes might specifically contribute to the genetic variation of maize flowering time under night conditions. KEGG analysis identified three genes (GRMZM2G107945, GRMZM2G172152, and GRMZM5G844173) that are involved in the blue light–triggered circadian rhythm pathway, which regulates the downstream flowering pathway (Supplemental Fig. S18). GRMZM2G107945 is homolog of a clock-controlled gene, FKF1, which regulates the transition to flowering in Arabidopsis. The fkf1 mutant shows a late-flowering phenotype (Nelson et al. 2000). GRMZM2G172152 is a homolog of CRY2, encoding cryptochrome-2, a photoreceptor regulated by blue light and involved in floral initiation (Liu et al. 2013). GRMZM5G844173 is a homolog of the GIGANTEA (GI) gene in Arabidopsis, which is a circadian clock–controlled gene that delays flowering under long day conditions when it is mutated (Fowler et al. 1999). Taken together, these multiple gene expression variants might play a role in controlling natural variation of flowering time among diverse maize lines.
Figure 8.

Flowering time is negatively correlated with the rDNA expression level and the coexpressed gene expression level among maize 282 panel (comprising 244 inbred lines). (A,B) Correlation between rDNA expression and best linear unbiased predictions (BLUPs) for days to silk (DTS) and days to pollen shed (DTA). The x-axes represent the CN of rRNA, and the y-axes represent phenotype measurements (BLUPs). (C–F) Correlation between BLUPs for DTS and three genes, and the mean expression value of 22 negatively correlated genes. The x-axes represent the RPM of individual genes, and the y-axes represent phenotype measurements (BLUPs).
Discussion
The reasonability of utilizing 3′mRNA-seq data to quantify rRNA expression
To use 3′mRNA-seq data to quantify 45S rRNA requires two facts: First, the total rRNA is entirely maintained in RNA samples for library preparations. This can be ensured with our 3′ mRNA sequencing technique, because there is no rRNA removal process and started with equal amount of total RNA. Second, the regions within 45S for generating reads can be repeatedly and robustly sequenced with the same efficiency to make the reads generated from different samples comparable. This is now difficult to be validated by experiments. However, in theory, several reasons are proposed to support this hypothesis.
First, we have proven these mapped reads are truly generated from rRNA. The reads, which can be mapped onto 45S gene locus, have two possible origins: one from genomic DNA contamination, the other from transcribed 45S rRNA. From the mapped positions of reads (Fig. 3), there are no reads mapped on the IGS (not transcribed intergenic spacer), excluding the possibility of DNA contamination.
Second, based on the methodology of 3′mRNA-seq, the most possible cause of unexpected 45S rRNA reads was the mismatch between poly(T) primer and some regions within 45S rRNA (Supplemental Fig. S19). Although we have no direct evidence, this mispriming should happen very often. For example, when trying to isolate mRNA with oligo(dT), a considerable amount of rRNA will be transferred together with mRNA. However, compared with poly(A) on mRNA, the poly(A)-like sequence regions are not as efficient as poly(A) and finally cause the unevenness of the sequencing coverage across the whole 45S. Actually, when checking the context of genomic sequence of rRNA, positive concordance between frequency of poly(A) like motif and read depth has been found, which is an indirect piece of evidence to support our hypothesis.
Third, this amplification by misguide of poly(T) is not accidental but repeatedly occurred among approximately 2000 samples. More importantly, we investigated the SNP pattern upstream of Region 2; there is no variation within all tested samples. That means the sequencing efficiency should be comparably similar among samples and made the normalized reads number comparable.
rRNA expression, not rDNA CN, affects gene expression in maize
In maize, we observed the lack of a correlation between the rRNA gene CN and the rRNA expression level, which is possible because rRNA transcription is regulated not only genetically but also epigenetically and because the expression level is potentially independent of the absolute number of 45S copies (Chen and Pikaard 1997; Zillner et al. 2015). A previous study also revealed there is not necessarily a positive correlation between CN and expression (Buescher et al. 1984). Another possible reason is that different maize inbred lines accumulate different proportional amounts of nonfunctional copies of 45S, which eventually leads to disruption of the correlation between CN and the expression level. First, many 45S copies have been found outside the cluster on Chromosome 6. These copies may gradually degrade into pseudogenes or silenced copies without the correction of gene conversion (Benevolenskaya et al. 1997; Ganley and Kobayashi 2007). Second, we found a considerable number of SNP types in 45S monomers that cannot be observed in 45S transcripts by comparing genomic data with the RNA-seq data (Supplemental Fig. S20). This observation directly supports the notion that a large number of 45S copies in the maize genome cannot be transcribed, at least under our tested conditions.
Our initial expectation was to detect the genes whose expression can be regulated by 45S CN in maize, as revealed in a human population (Gibbons et al. 2014); however, our results indicated that rRNA expression, not rDNA CN, plays such a role in regulating gene expression in maize. Actually, we suspected that the correlation detected between 45S CN and gene expression in humans might reflect the coexpression between rRNA and related genes because these correlated genes behaved as coexpressed genes and were enriched within the same pathway. We deduced that the reason why the investigators observed an association between rDNA CN and individual gene expression in humans was perhaps a reflection of a positive correlation between human rDNA CN and the rRNA expression level. However, this correlation is disrupted in maize, and no association between rRNA CN and gene expression could be observed.
Coregulated pathways between protein-coding genes and rRNA
Ribosome biogenesis requires the collaborative effort of cotranscribed rRNA and r-proteins (Nomura et al. 1984), but which one is the upstream regulator remains an open question. The rRNA and ribosomal proteins result from different transcription pathways; therefore, this cooperative behavior suggests that transcription via Pol I, Pol II, and Pol III may be well connected. In mammalian cells, rRNA transcription is the limiting step in ribosome biogenesis (Kopp et al. 2007), indicating the rRNA expression is an upstream regulator. After rRNA transcripts are generated, the r-proteins need to be translated to hierarchically bind on rRNA to protect it from degradation and eventually form a completely functional ribosome (Nomura et al. 1984; Kressler et al. 2010), implying that rRNA transcription precedes that of some r-protein genes.
In addition to genes participating in ribosome assembly, we also detected a large number of genes enriched within other functional groups that correlated with 45S expression. It is not well understood how coexpression between these genes and 45S rRNA is achieved. However, manipulation of rRNA transcription indeed affects gene expression (Derenzini et al. 2017; Makabe et al. 2017b). Notably, the coregulated genes are highly enriched within important functional groups, such as ribosome biogenesis in the developing leaf base, or photosynthesis in the mature leaf tip. Furthermore, a previous study measured the protein/mRNA ratios across a maize leaf gradient and found that protein/mRNA ratios are higher for proteins with critical functions at specific developmental stages (Ponnala et al. 2014), implying the genes coregulated with rRNA expression (or ribosome amount) maybe promote protein synthesis.
Methods
Maize data resources
The maize reference genome used in this study refers to the assembly B73 RefGene_v3. Genome sequences and the gene annotation GFF3 (v3.27) file were downloaded from ftp://ftp.ensemblgenomes.org/pub/plants/release-27/gff3/zea_mays. Whole-genome resequencing (WGS) data for the 282 panel were described on http://www.panzea.org/ and can be accessed in the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject) under accession number PRJNA389800. Phenotypic data for the 282 panel used in this study, including best linear unbiased predictions (BLUPs) for some traits, can be found at https://www.panzea.org/phenotypes. All 3′ mRNA sequencing data used in this study can be accessed in the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra) under accession number SRP115041 and in the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject) under accession number PRJNA383416. Methods and any other details about 3′ mRNA sequencing data can be found in our previous paper (Kremling et al. 2018).
Single-copy and multiple-copy data set
We initially identified SC exons and introns within the maize genome using the following procedures: (1) We extracted all exons and introns from the maize genome annotation from the longest mRNA for each gene model (300–5000 bp in length); (2) we conducted All-To-All BLASTN (E < 10−6) for exons and introns separately and kept the exons and introns if they could be aligned only to themselves (15,877 exons and 12,538 introns passed this filter); and (3) these potential SC exons and introns were aligned back to the reference genome (10 chromosomes and 511 scaffolds), and the exons and introns with only one hit (to themselves) were retained. Reference repeat sequences in the maize genome, including 5S rRNA, 45S rDNA, CentC (maize centromere repeat), and Knob 180 and Knob TR1 (two specific repeats enriched within maize knob heterochromatin regions), were downloaded from NCBI and verified by aligning with the B73 reference genome. Each kind of refined repeat sequence was then mapped onto the reference to recover all copies within the reference using BLASTN (E < 10−6). Repeat units <100 bp were excluded from repeat data set. Finally, several BED-formatted files were created to record the genomic positions of SC DNA and repeat DNAs. The 45S rRNA is too long (∼8 kb) to accurately identify every SC within the whole genome; therefore, structural elements, including 18S, ITS1, 5.8S, ITS2, and 25S, were aligned separately onto the B73 reference to determine the complete distribution of 45S rRNA. The custom Perl scripts for this part of the work can be accessed in Supplemental Text S1.
CN estimation
An overview of the pipeline to estimate the rDNA CN is illustrated in Supplemental Figure S1. This method has previously been used to calculate the 45S CN for a human population (Gibbons et al. 2014). Our method involved some modifications as follows: First, we started with whole-genome alignment BAM files, not the 45S rRNA reference alignment files. WGS reads were mapped onto the B73 reference sequence by using BWA with default parameters and allowed any reads with multiple best-quality hits to be randomly placed on the genome. We then identified all possible regions (repeat copies) that were aligned to retrieve all reads generated from a certain kind of repeat. This approach has the benefit of saving computing time and data storage space because you do not need to maintain large FASTQ files. Second, we used the multicov function in BEDTools (Quinlan and Hall 2010) to count the mapped read number and then calculated the read depth but did not use the mpileup function in SAMtools (Li et al. 2009) to directly calculate the read depth. This is because computing mpileup combined with a whole-genome BAM file is too expensive. The custom Perl scripts for this part of the work can be accessed in Supplemental Text S2. We removed 5% of the SC DNA with the maximum or minimum RD, because these SC DNA represent genomic regions either showing CNV among individual taxon or with greatest sequencing bias. This normalization resulted in a slight increase in the correlation between SC exons and introns. By using this approach, the CNs for 5S, 45S, Knob 180, Knob TR1, and CentC were estimated.
qPCR validation of rDNA CN results
We initially made a series of diluted genomic DNA templates (10–160 ng, by twofold increase) and tested whether qPCR result can reflect a twofold template increase in different samples. We collected seeds for 30 maize lines from 282 panel population (each of 10 have highest, medium, and lowest rDNA CN, respectively) and grew them in a climate control chamber. Of them, six maize lines had no germination, and genomic DNA for other 24 lines was extracted from the leaves from young plants (9 d after germination) by using a plant genomic DNA kit (catalog no DP305) provided by Tiangen Biotech. qPCR primers were designed for rDNA and SC gene in B73 reference (Supplemental Table S16). All PCR products were first checked with 1.5% agarose gel. After all primers were successfully validated, qPCR assays were conducted on a CFX real-time system with SsoFast EvaGreen supermix kit (Bio-Rad) following standard protocol. For each sample, three technique repeats were used to reduce the variation from the template input amount. The relative CN amount for rDNA was calculated based on the following equation:
where Ct0 is the mean of Ct value for rDNA, and Ct1 is the mean of Ct value for SC gene. The correlation analysis of CNs of rDNA between qPCR results and NGS results was used to measure the consistency of two methods.
GWAS and variance component analysis
Genome-wide association mapping of the 45S rDNA CN was implemented in TASSEL 5.0 (Bradbury et al. 2007) with the HapMap3.1 unimputed SNP data set. The details about the variance component analysis (or variance partitioning) were the same as those in a previous study (Rodgers-Melnick et al. 2016). We separately estimated the genetic contributions to the rDNA copy dosage explained by individual chromosomes. Maize 45S rDNA is mainly located on Chromosome 6; therefore, we first partitioned 50 Mb HapMap3.1 unimputed SNPs into two groups: SNPs from Chromosome 6 and SNPs from the other chromosomes. Each kinship matrix for the 282 panel population was calculated within the subgroup SNP data using the “Scaled_IBS” method in TASSEL 5.0. Partitioned variance between Chromosome 6 and the other chromosomes for the CN of 5.8S, 18S, and 25S rDNA was computed using the MultiBLUP method implemented in the LDAK package, version 4.5 (Speed and Balding 2014).
Estimate of the narrow sense heritability for the CN of repeats
Narrow sense heritability (h2) was calculated based on the following equation:
where represents additive variance, and represents environmental variance. For each trait, and were calculated by using TASSEL 5.0 with a MLM.
Identifying SNPs within 45S rDNA among maize inbred lines
WGS clean data (after removing adapters and low-quality reads) were aligned onto one B73 45S reference sequence using Bowtie 2 (Langmead and Salzberg 2012) with default parameters. The alignments were processed using the mpileup function in SAMtools with –d 1000000 to extract polymorphic information for each base for the 45S rDNA. In the 282 panel population, we counted the number of lines for each polymorphic site and summarized the density for different structural segments.
Quantifying 45S rDNA expression level
We selected 1923 RNA-seq data from GRoot (291 samples), GShoot (295), Kern (254), L3Base (302), L3Tip (295), LMAD (210), and LMAN (276) by matching taxa names with 282 panel data and mapped these reads onto the 45S and 5S B73 sequences using Bowtie 2. No reads from the 5S rDNA were found in our data. Thus, further research was only conducted on 45S rDNA expression. We counted mapped read numbers for each peak region using the idxstats function within SAMtools. To test the relationship between rDNA CN and expression, we matched two data sets by selecting taxa with both CN estimates and expression quantity.
Genome-wide association test between 45S rRNA and gene expression
Mapped read counts were generated for 39,625 maize reference gene models (including 153 miRNAs) (Kremling et al. 2018). To perform a correlation test between gene expression and rRNA expression, we first integrated rRNA read counts with gene read counts for each library and normalized the data using geometric mean implanted in the R package “DESeq2” (Love et al. 2014). Normalized gene expression data were used to perform the correlation analysis, with the requirement that any gene must have expression data for over 50 individuals. Gene expression data were first correlated with the CN of the rDNA and then correlated with rRNA expression data in seven tissues. Bonferroni correction was applied for multiple comparison P-value adjustments (Dunn 1961). Data analyses were performed using R (R Core Team 2016).
Coregulated gene function analysis
GO analysis were used to test the biological functions of the enriched genes using PlantRegMap (http://plantregmap.cbi.pku.edu.cn/go_result.php) (Jin et al. 2017). Biological pathway analysis was conducted using KOBAS 2.0 (http://kobas.cbi.pku.edu.cn/), which is an online tool developed for KEGG pathway analysis (Xie et al. 2011). For r-protein gene annotation, we refer to GO analysis results. Within this result, some r-protein gene could be assigned to very specific function, e.g., small subunit ribosomal protein S3e; some were just functionally annotated into r-protein groups.
Cluster analysis of r-protein genes from different tissues
Genes annotated as ribosomal protein coding genes from different tissues were clustered to identify shared genes. For the 29 shared genes between L3Base and L3Tip, we separated them into two subgroups (based on the correlation direction with the rRNA). To test the expression pattern of r-protein genes between L3Base and L3Tip, we calculated the ratio of expression between L3Base and L3Tip for each gene and generated heat maps to illustrate the gene expression patterns.
Supplementary Material
Acknowledgments
We thank Prof. GuangHui Hu, Prof. TianYu Wang, and Prof. ZhiWu Zhang for gathering maize seeds for validation experiments and Dr. YaFei Li for providing qPCR system for this project. This work was supported by the Young Visiting Scientist Project from China Academy of Sciences and the National Natural Science Foundation of China (grants no. 31401074 to B.L. and no. 31771409 to M.C.).
Author contributions: B.L. led this project, performed the data analyses, and wrote the manuscript; K.A.G.K. collected the samples, performed the 3′ mRNA library preparations, and provided guidance in analyzing the RNA-seq data; P.W. and R.B. helped to analyze the data; M.C.R. generated the resequencing data and provided guidance for the 282 panel data analysis; E.X. and B.L. conducted the qPCR experiments; E.S.B. conceived this project and modified the manuscript; and M.C. provided the instructions on data analysis, facility, and resources for this study.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.229716.117.
References
- Benevolenskaya EV, Kogan GL, Tulin AV, Philipp D, Gvozdev VA. 1997. Segmented gene conversion as a mechanism of correction of 18S rRNA pseudogene located outside of rDNA cluster in D. melanogaster. J Mol Evol 44: 646–651. [DOI] [PubMed] [Google Scholar]
- Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES. 2007. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23: 2633–2635. [DOI] [PubMed] [Google Scholar]
- Buckler ES, Gaut BS, Mcmullen MD. 2006. Molecular and functional diversity of maize. Curr Opin Plant Biol 9: 172–176. [DOI] [PubMed] [Google Scholar]
- Buescher PJ, Phillips RL, Brambl R. 1984. Ribosomal RNA contents of maize genotypes with different ribosomal RNA gene numbers. Biochem Genet 22: 923–930. [DOI] [PubMed] [Google Scholar]
- Bukowski R, Guo X, Lu Y, Zou C, He B, Rong Z, Wang B, Xu D, Yang B, Xie C. 2018. Construction of the third-generation Zea mays haplotype map. Gigascience 7: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrasekhara C, Mohannath G, Blevins T, Pontvianne F, Pikaard CS. 2016. Chromosome-specific NOR inactivation explains selective rRNA gene silencing and dosage control in Arabidopsis. Genes Dev 30: 177–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZJ, Pikaard CS. 1997. Transcriptional analysis of nucleolar dominance in polyploid plants: Biased expression/silencing of progenitor rRNA genes is developmentally regulated in Brassica. Proc Natl Acad Sci 94: 3442–3447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chia JM, Song C, Bradbury PJ, Costich D, de Leon N, Doebley J, Elshire RJ, Gaut B, Geller L, Glaubitz JC, et al. 2012. Maize HapMap2 identifies extant variation from a genome in flux. Nat Genet 44: 803–807. [DOI] [PubMed] [Google Scholar]
- de la Cruz J, Karbstein K, Woolford JL Jr. 2015. Functions of ribosomal proteins in assembly of eukaryotic ribosomes in vivo. Annu Rev Biochem 84: 93–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derenzini M, Montanaro L, Trere D. 2017. Ribosome biogenesis and cancer. Acta Histochem 119: 190–197. [DOI] [PubMed] [Google Scholar]
- Diez CM, Gaut BS, Meca E, Scheinvar E, Montes-Hernandez S, Eguiarte LE, Tenaillon MI. 2013. Genome size variation in wild and cultivated maize along altitudinal gradients. New Phytol 199: 264–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn OJ. 1961. Multiple comparisons among means. J Am Stat Assoc 56: 52–64. [Google Scholar]
- Flint-Garcia SA, Thuillet AC, Yu J, Pressoir G, Romero SM, Mitchell SE, Doebley J, Kresovich S, Goodman MM, Buckler ES. 2005. Maize association population: a high-resolution platform for quantitative trait locus dissection. Plant J 44: 1054–1064. [DOI] [PubMed] [Google Scholar]
- Fowler S, Lee K, Onouchi H, Samach A, Richardson K, Morris B, Coupland G, Putterill J. 1999. GIGANTEA: a circadian clock-controlled gene that regulates photoperiodic flowering in Arabidopsis and encodes a protein with several possible membrane-spanning domains. EMBO J 18: 4679–4688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganley AR, Kobayashi T. 2007. Highly efficient concerted evolution in the ribosomal DNA repeats: total rDNA repeat variation revealed by whole-genome shotgun sequence data. Genome Res 17: 184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia S, Kovarik A, Leitch AR, Garnatje T. 2017. Cytogenetic features of rRNA genes across land plants: analysis of the Plant rDNA database. Plant J 89: 1020–1030. [DOI] [PubMed] [Google Scholar]
- Gerlach WL, Bedbrook JR. 1979. Cloning and characterization of ribosomal RNA genes from wheat and barley. Nucleic Acids Res 7: 1869–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons JG, Branco AT, Yu S, Lemos B. 2014. Ribosomal DNA copy number is coupled with gene expression variation and mitochondrial abundance in humans. Nat Commun 5: 4850. [DOI] [PubMed] [Google Scholar]
- Gibbons JG, Branco AT, Godinho SA, Yu S, Lemos B. 2015. Concerted copy number variation balances ribosomal DNA dosage in human and mouse genomes. Proc Natl Acad Sci 112: 2485–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottschling M, Plotner J. 2004. Secondary structure models of the nuclear internal transcribed spacer regions and 5.8S rRNA in Calciodinelloideae (Peridiniaceae) and other dinoflagellates. Nucleic Acids Res 32: 307–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusev A, Bhatia G, Zaitlen N, Vilhjalmsson BJ, Diogo D, Stahl EA, Gregersen PK, Worthington J, Klareskog L, Raychaudhuri S, et al. 2013. Quantifying missing heritability at known GWAS loci. PLoS Genet 9: e1003993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Tian F, Yang DC, Meng YQ, Kong L, Luo J, Gao G. 2017. PlantTFDB 4.0: toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res 45 (Database issue): D1040–D1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinge S, Voigts-Hoffmann F, Leibundgut M, Arpagaus S, Ban N. 2011. Crystal structure of the eukaryotic 60S ribosomal subunit in complex with initiation factor 6. Science 334: 941–948. [DOI] [PubMed] [Google Scholar]
- Kopp K, Gasiorowski JZ, Chen D, Gilmore R, Norton JT, Wang C, Leary DJ, Chan EK, Dean DA, Huang S. 2007. Pol I transcription and pre-rRNA processing are coordinated in a transcription-dependent manner in mammalian cells. Mol Biol Cell 18: 394–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremling KAG, Chen SY, Su MH, Lepak NK, Romay MC, Swarts KL, Lu F, Lorant A, Bradbury PJ, Buckler ES. 2018. Dysregulation of expression correlates with rare-allele burden and fitness loss in maize. Nature 555: 520–523. [DOI] [PubMed] [Google Scholar]
- Kressler D, Hurt E, Bassler J. 2010. Driving ribosome assembly. Biochim Biophys Acta 1803: 673–683. [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Arumuganathan K. 2001. Physical mapping of 45S and 5S rDNA on maize metaphase and sorted chromosomes by FISH. Hereditas 134: 141–145. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Ponnala L, Gandotra N, Wang L, Si Y, Tausta SL, Kebrom TH, Provart N, Patel R, Myers CR, et al. 2010. The developmental dynamics of the maize leaf transcriptome. Nat Genet 42: 1060–1067. [DOI] [PubMed] [Google Scholar]
- Liu H, Wang Q, Liu Y, Zhao X, Imaizumi T, Somers DE, Tobin EM, Lin C. 2013. Arabidopsis CRY2 and ZTL mediate blue-light regulation of the transcription factor CIB1 by distinct mechanisms. Proc Natl Acad Sci 110: 17582–17587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long Q, Rabanal FA, Meng D, Huber CD, Farlow A, Platzer A, Zhang Q, Vilhjalmsson BJ, Korte A, Nizhynska V, et al. 2013. Massive genomic variation and strong selection in Arabidopsis thaliana lines from Sweden. Nat Genet 45: 884–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makabe S, Motohashi R, Nakamura I. 2017a. Growth increase of Arabidopsis by forced expression of rice 45S rRNA gene. Plant Cell Rep 36: 243–254. [DOI] [PubMed] [Google Scholar]
- Makabe S, Yamori W, Kong K, Niimi H, Nakamura I. 2017b. Expression of rice 45S rRNA promotes cell proliferation, leading to enhancement of growth in transgenic tobacco. Plant Biotechnol 34: 29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McStay B, Grummt I. 2008. The epigenetics of rRNA genes: from molecular to chromosome biology. Annu Rev Cell Dev Biol 24: 131–157. [DOI] [PubMed] [Google Scholar]
- Nelson DC, Lasswell J, Rogg LE, Cohen MA, Bartel B. 2000. FKF1, a clock-controlled gene that regulates the transition to flowering in Arabidopsis. Cell 101: 331–340. [DOI] [PubMed] [Google Scholar]
- Nomura M, Gourse R, Baughman G. 1984. Regulation of the synthesis of ribosomes and ribosomal components. Annu Rev Biochem 53: 75. [DOI] [PubMed] [Google Scholar]
- Paredes S, Maggert KA. 2009. Ribosomal DNA contributes to global chromatin regulation. Proc Natl Acad Sci 106: 17829–17834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paredes S, Branco AT, Hartl DL, Maggert KA, Lemos B. 2011. Ribosomal DNA deletions modulate genome-wide gene expression: “rDNA-sensitive” genes and natural variation. PLoS Genet 7: e1001376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponnala L, Wang Y, Sun Q, van Wijk KJ. 2014. Correlation of mRNA and protein abundance in the developing maize leaf. Plant J 78: 424–440. [DOI] [PubMed] [Google Scholar]
- Prokopowich CD, Gregory TR, Crease TJ. 2003. The correlation between rDNA copy number and genome size in eukaryotes. Genome 46: 48–50. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2016. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: http://www.R-project.org/. [Google Scholar]
- Rabanal FA, Nizhynska V, Mandakova T, Novikova PY, Lysak MA, Mott R, Nordborg M. 2017. Unstable inheritance of 45S rRNA genes in Arabidopsis thaliana. G3 (Bethesda) 7: 1201–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabl J, Leibundgut M, Ataide SF, Haag A, Ban N. 2011. Crystal structure of the eukaryotic 40S ribosomal subunit in complex with initiation factor 1. Science 331: 730–736. [DOI] [PubMed] [Google Scholar]
- Rayburn AL, Dudley JW, Biradar DP. 1994. Selection for early flowering results in simultaneous selection for reduced nuclear DNA content in maize. Plant Breed 112: 318–322. [Google Scholar]
- Rodgers-Melnick E, Vera DL, Bass HW, Buckler ES. 2016. Open chromatin reveals the functional maize genome. Proc Natl Acad Sci 113: E3177–E3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers SO, Bendich AJ. 1987. Ribosomal RNA genes in plants: variability in copy number and in the intergenic spacer. Plant Mol Biol 9: 509–520. [DOI] [PubMed] [Google Scholar]
- Speed D, Balding DJ. 2014. MultiBLUP: improved SNP-based prediction for complex traits. Genome Res 24: 1550–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speed D, Hemani G, Johnson MR, Balding DJ. 2012. Improved heritability estimation from genome-wide SNPs. Am J Hum Genet 91: 1011–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weider LJ, Elser JJ, Crease TJ, Mateos M, Cotner JB, Markow TA. 2005. The functional significance of ribosomal (r)DNA variation: impacts on the evolutionary ecology of organisms. Annu Rev Ecol Evol Syst 36: 219–242. [Google Scholar]
- Xie C, Mao X, Huang J, Ding Y, Wu J, Dong S, Kong L, Gao G, Li CY, Wei L. 2011. KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res 39(Web Server issue): W316–W322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zillner K, Komatsu J, Filarsky K, Kalepu R, Bensimon A, Németh A. 2015. Active human nucleolar organizer regions are interspersed with inactive rDNA repeats in normal and tumor cells. Epigenomics 7: 363. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.