

Abstract
Mycobacterium tuberculosis produces dideoxymycobactin-838 (DDM-838), a lipopeptide that potently activates T cells upon binding to the MHC-like antigen-presenting molecule CD1a. M. tuberculosis produces DDM-838 in only trace amounts and a previous solid-phase synthesis provided sub-milligram quantities. We describe a high yielding solution-phase synthesis of DDM-838 that features a Mitsunobu substitution that avoids yield-limiting epimerization at lysine during esterification, and amidation conditions that prevent double-bond isomerization of the Z-C20:1 acyl chain, and provides material with equivalent antigenicity to natural DDM-838. Isomers of DDM-838 that varied in stereochemistry at the central lysine and the C20:1 acyl chain were compared for their ability to be recognised by CD1a-restricted T cell receptors (TCRs). These TCRs, derived from unrelated human donors, exhibited a similar spectrum of reactivity towards the panel of DDM-838 isomers, highlighting the exquisite sensitivity of lipopeptide-reactive T cells for the natural DDM stereochemistry.
Keywords: immunology, peptidolipid, antigens, natural product, T cell
Entry for the Table of Contents
Sensing stereochemistry: M. tuberculosis dideoxymycobactin-838 (DDM-838) is an antigen involved in cell-mediated immunity. DDM-838 and stereoisomers epimeric in the depsipeptide backbone and isomeric in the C20:1 acyl-chain were synthesized. T cell receptors derived from unrelated human donors preferentially bind natural DDM-838 in complex with the antigen presenting molecule CD1a and display similar patterns of discrimination for DDM-838 stereoisomers.
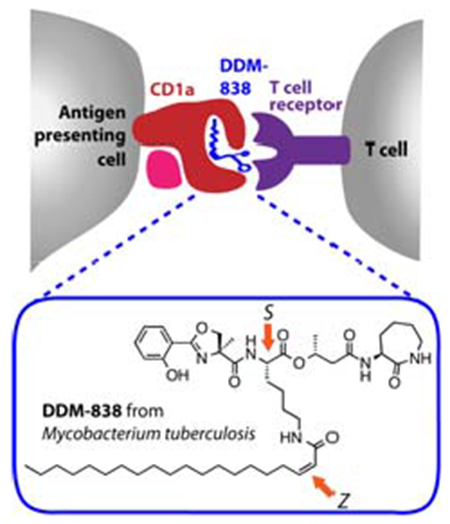
Introduction
Humans express four antigen-presenting proteins (CD1a, CD1b, CD1c and CD1d) belonging to the CD1 family, which present lipid-based molecules to T cells via their T cell receptors (TCRs).[1] CD1a differs from the other CD1 molecules based on its constitutive, high-density expression on dendritic cells, termed Langerhans cells, in the skin and mucosa.[2] Moreover, distinct from the other human CD1 molecules, CD1a possesse a cytoplasmic tail that lacks lysosomal targeting motifs. Thus, CD1a localizes predominantly at the cell surface where it can promiscuously capture foreign or exogenous lipids. The only extensively-characterized type of foreign lipidic molecule that activates CD1a-restricted T cells is a family of lipopeptides isolated from Mycobacterium tuberculosis, termed dideoxymycobactins (DDMs).[3]
The initial discovery of the DDMs was achieved through bioassay-guided fractionation using a single T cell clone (CD8-2) obtained through stimulation and expansion with a chloroform/methanol M. tuberculosis extract[4] A three-dimensional X-ray structure of CD1a with a silylated, DDM-like lipopeptide showed the fatty acyl chain buried in the A’ pocket of CD1a with the peptide protruding to the surface of CD1a, where it presumably contacts the TCR.[5] The recent development of CD1 a tetramers and dextramers that bind directly to polyclonal T cells in the ex vivo setting has allowed the detection and isolation of DDM-reactive T cell clones from small cohorts of tuberculosis patients.[6] This suggests that CD1a–DDM reactive T cells occur in unrelated individuals and that CD1a–DDM tetramers may be useful reagents for the ex vivo diagnosis of M. tuberculosis infection. Further, the existence of polyclonal T cells in small human cohorts raises the possibility that DDMs could be used to immunize or stimulate T cell responses in tuberculosis patients.[6] However, the key obstacle to these lines of new medical technology development is the absence of an abundant supply of DDM.
DDMs are extremely potent, but comprise less than 10 parts per million of the cell wall mass of M. tuberculosis, a pathogen, whose laboratory cultivation is slow and cumbersome because it must be grown in biosafety level 3 conditions. Thus, isolation in high yield from natural sources remains impractical.[3] The key challenge for chemical synthesis is that naturally occurring DDMs possess a complex depsipeptide backbone bearing a range of saturated and unsaturated fatty acyl groups linked through an amide to the ε-amino group of a lysine residue, and an unusual α-methylserine-derived angular methyl group. The structure of the most reactive compound, DDM-838 (1, Figure 1) was proposed through mass spectrometric and NMR analysis,[3a] as well as the preparation of a range of possible stereoisomers and matching of their spectroscopic data and biological activity to the natural product,[3b] and is consistent with the stereochemistry of the biosynthetically-related mycobactins, iron-chelating hydroxamic acid siderophores.
Figure 1.
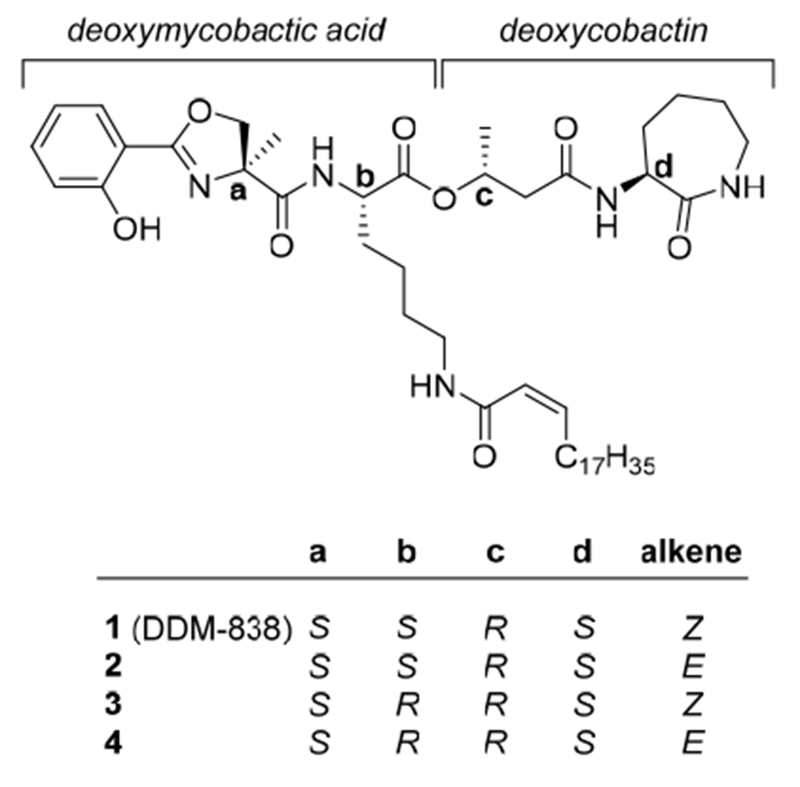
DDM-838 (1) and three stereoisomers.
A previous synthetic approach[3b] to DDM-838 used solid-phase peptide chemistry to provide sufficient material to help clarify the structure of the natural product, yet the poor overall yield provided only sub-milligram quantities that are insufficient for in vivo studies. Prior work demonstrated that the correct stereochemistry at positions a and c (Figure 1), and the angular methyl group at position a are necessary for activating the DDM-reactive T cell clone CD8-2, and that natural DDMs lacking the unsaturation in the acyl chain are at least 10-fold less potent for T cell activation.[3] In the present work we investigate solution-based peptide-coupling approaches for the preparation of milligram quantities of DDM-838. Through careful isolation of by-products produced in this process, we identify difficulties with the establishment of the key ester linkage that occurs with partial epimerization at lysine, a problem that may have occurred in the previous approach of Young and coworkers (unpublished). This and other problems were overcome by the development of a new route that creates the challenging ester linkage through a Mitsunobu coupling and allows the preparation of milligram quantities of DDM-838 (1). Through the isolation of stereochemical isomers of DDM-838 (2-4) we have explored structure-activity relationships in antigen recognition by the prototypical DDM-reactive T cell clone CD8-2, as well as new DDM-reactive clones, and demonstrate the importance of peptide and lipid stereochemistry for T cell receptor recognition and T cell activation.
Results and Discussion
Total synthesis of DDM-838
Young and co-workers reported the synthesis of DDM-838 and analogues, emphasizing solid-phase approaches[3b] For assembly of DDM-838 itself, a linear depsipeptide bearing the fatty acyl chain was assembled on Fmoc-Lys Sasrin resin. Following release of the depsipeptide from the resin, the terminal lysine was cyclized to form the caprolactam moiety. After extensive FIPLC purification, this approach yielded only trace quantities of DDM-838 (~95 μg or 2% overall yield), which we verified independently. As this synthesis was conducted on solid phase, it is difficult to identify the yield-limiting steps. In a solution-phase approach, used for the synthesis of analogues that lacked the distinguishing α,-methyl group, an ε-amino-protected deoxymycobactic acid was coupled to deoxycobactin, and the depsipeptide was deprotected prior to attaching the fatty acyl chain. This resulted in low yields for both attachment of the fatty acyl chain and for formation of the ester linkage. Moreover, unexpected doubling of signals in NMR spectra of advanced intermediates was attributed to the presence of rotational isomers, but could alternatively reflect the formation of undefined stereoisomers. With the above issues in mind, we initially sought to develop a solution-based synthesis of DDM-838 broadly inspired by this earlier study,[3b] but with the intention to fully characterize all intermediates and identify yield-limiting steps in order to identify an improved synthetic approach.
The required building block fragments deoxymycobactic acid 13, deoxycobactin 16, and Z-C20:1 fatty acid 20 were prepared by optimization of the previously described literature approaches.[3b] Oxazoline acid 11 was prepared from α-methyl-l-serine 5, by protection as the methyl ester 6, which was condensed with 2-benzyloxysalicylic acid 8 using COMU to afford amide 9 (Scheme 1a)[7] Cyclization to the oxazoline 10 proceeded smoothly with XtalFluor-M in CH2Cl2.[8] Saponification of the ester of 10 with LiOH afforded the O-benzyl-protected acid 11, which was coupled with NH2-l-Lys(Boc)OMe using EDC.FICI/FIOAt/DMAP. Saponification of 12 afforded deoxymycobactic acid 13. Next, deoxycobactin 16 was prepared by coupling of (R)-3-hydroxybutyric acid 14 and l-α-amino-ε-caprolactam hydrochloride 15 in the presence of EDC.HCI/HOAt/DMAP (Scheme 1b).[3b] Finally, the Z-C20:1 fatty acid 20 was synthesized using Ando’s reagent,[9] which enables a highly Z-selective Florner-Emmons-Wadsworth coupling. Treatment of octadecanol 17 with PCC in CH2Cl2 afforded stearylaldehyde 18, which condensed with Ando’s reagent[9–10] in the presence of DBU/Nal to afford a 9:1 Z:E ratio of alkenes 19 (use of the Still-Gennari reagent provided the alkenes in a 3:1 Z:E ratio) (Scheme 1c). These isomers could be separated by careful chromatography, prior to saponification to afford the acid 20.
Scheme 1.
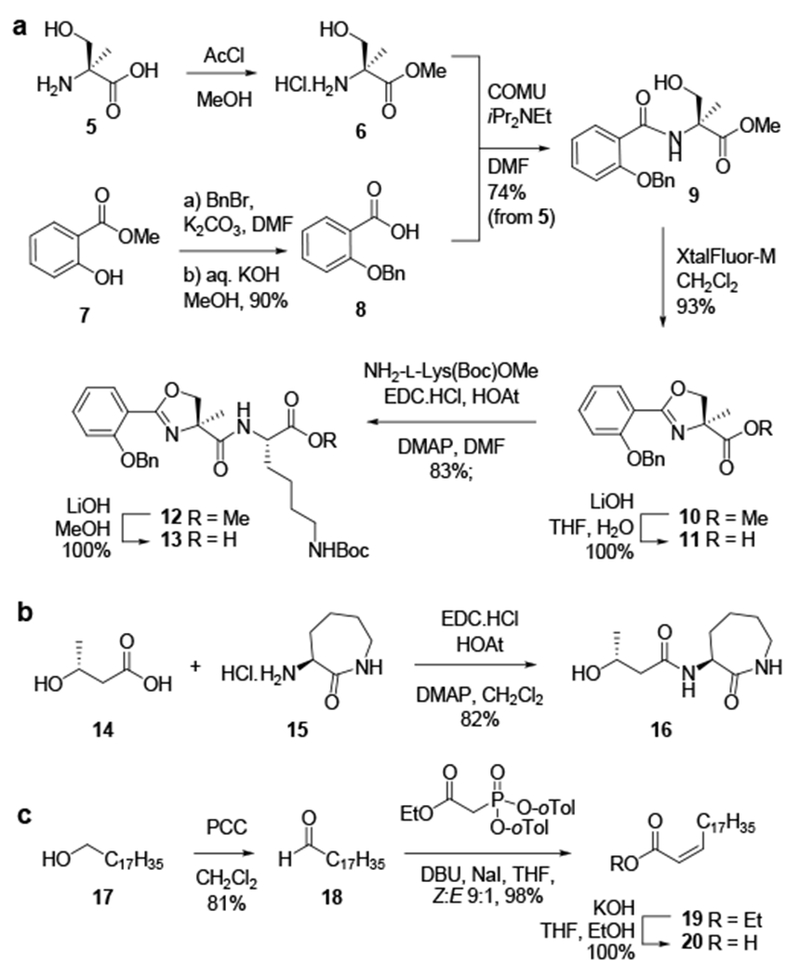
Synthesis of deoxymycobactic acid 13, deoxycobactin 16, and Z-C20:1 fatty acid 20.
Coupling of deoxymycobactic acid 13 with deoxycobactin 16 was explored under a range of esterification conditions using assorted peptide coupling reagents. In general, the esterification reactions were slow and low yielding, and occurred with varying amounts of epimerization at lysine. The best results were achieved using DIC/HOAt in the presence of 1.5 eq of DMAP, which afforded a 1:1 ratio of S-21/R-21 (Scheme 2). Importantly, the O-benzyl protected epimeric intermediates 21 were separable by preparative t.l.c., but stereochemical assignment was not possible at this stage. Each isomer was individually carried through to the lipopeptide by deprotection of benzyl and Boc groups (to give S- and R-22), then coupled with the Z-C20:1 acid 20 using EDC.HCl/HOAt/DMAP in the presence of iPr2NEt. Each product was formed as a mixture of E and Z isomers, which were separated by flash chromatography to afford four pure DDM isomers 1-4, and in some cases small amounts of a contaminating trifluoroacetamide. The less polar isomer 21 afforded material whose spectral data matched the natural product DDM-838 (1). At this stage the stereochemistry of the central lysine was not known, but established later (vide infra).
Scheme 2.
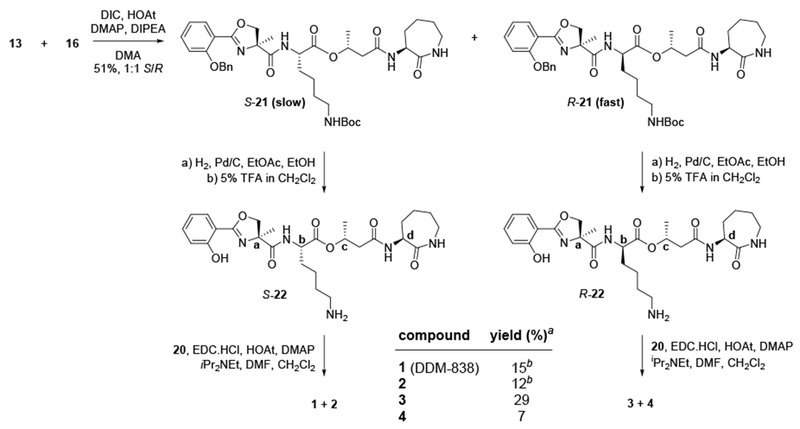
Preparation of DDM-838 (1) and stereoisomers 2-4. Slow = higher polarity product; fast = lower polarity product. a yield over 3 steps from (S)- or (R)-21. b analysis of 1H NMR of crude mixture revealed the formation of 1:2:trifluoroacetamide byproduct at a ratio of 6:3:4, presumably derived from residual trifluoroacetate anion carried over from step iii.
The above work, and an alternative approach utilizing a different order of assembly that gave improved yields yet also suffered from similar problems (see SI for details), highlighted two key problems that likely contributed to the low yield of the prior solid- and solution-based syntheses of DDM-838.[3b] The first is the difficulty of effecting the esterification of 3-hydroxybutyramide, which occurred at best in only moderate yield and was complicated by epimerization at the central lysine. The difficulty of this coupling likely arises from the sterically-congested nature of the linkage, between an α-branched acid, and a secondary alcohol. The propensity for epimerization at the central lysine during esterification of the 3-hydroxybutyramide group means that the stereochemistry at this position had not been unambiguously secured, although the biosynthetic relationship of DDM-838 to the mycobactins allows assumption of the l-configuration[11] The second problem was the unexpected isomerization of the unsaturated fatty acyl chain, and sometimes contamination with a trifluoroacetamide byproduct.
We initially focussed on solving the problem experienced with the unexpected formation of E and Z isomers upon acylation of S- and R-22 with Z-C20:1 acid 20, and the formation of the trifluoroacetamide by-product. By conversion of the trifluoroacetate salt of compound 22 to the free base, prior to acylation, we were able to avoid trifluoroacetamide formation. Treatment of a pure Z-C20:1-Lys model with iPr2NEt did not result in double bond isomerization, suggesting that isomerization was occurring to the activated acyl-intermediate. Attempts to acylate the model amine (Bz-l-Lys-OMe) using DIC/HOAt/DMAP, without iPr2NEt, or with pyridine, still led to double bond isomerization, suggesting that base-catalyzed isomerization is faster than acylation of lysine. Hocking has shown that double bond isomerization of cis-crotonoyl chloride during reaction with ammonia does not occur when ammonia is passed over the surface of an ethereal solution of the acid chloride.[12] Inspired by this precedent, we examined pre-forming the activated ester by combining DIC and Z-C20:1 20 in CH2Cl2, followed by addition of a CH2Cl2 solution of Bz-l-Lys-OMe, which afforded the cis-amide as the sole product.
To overcome the problem with epimerization at the central lysine, a Mitsunobu reaction was explored to form an ester bond and interchange the roles of the acid and alcohol to nucleophile and electrophile, respectively, thereby potentially alleviating the steric demands in forming this linkage. In related work, Miller and co-workers applied the Mitsunobu reaction in the synthesis of Mycobactins S and S2 through coupling of cobactin and mycobactic acid fragments in 49 and 50% yield, respectively.[13] In a recent article, Tsakos et al. reviewed a large number of challenging esterifications, and suggested that performing difficult esterification reactions on smaller fragments can often avoid yield-limiting side reactions by reducing the number of potentially interfering functional groups.[14]
Based on these considerations we devised a second linear approach to DDM-838. Coupling of (S)-3-hydroxybutyric acid ((S)-14) (rather than (R)-14) to l-α-amino-ε-caprolactam 15 using EDC.HCl/HOAt/DMAP and afforded the deoxycobactin 23 (Scheme 3).[3b] Mitsunobu coupling of 23 with Fmoc-l-Lys(Z)-OH in the presence of Ph3P/DIAD afforded the ester 24 with the stereochemistry of 3-hydroxybutyrate inverted, and an elimination by-product 25 inseparable by flash chromatography. While the yield of this reaction was modest, no epimerization at lysine was observed. Removal of the Fmoc group with piperidine allowed separation of the alkene 25 by chromatography and afforded pure amine 26 in 26% yield over two steps. Condensation of amine 26 and oxazoline acid 11 afforded the depsipeptide 27. Global deprotection of depsipeptide 27 with H2 over Pd/C gave enantiomerically-pure amine S-22, which was isolated as a free base and then coupled with the Z-C20:1 acid 20, this time using only DIC, and afforded DDM-838 1 as a pure Z isomer in 49% yield over two steps. The spectral data were in agreement with the natural product. By this approach we unambiguously established the stereochemistry of DDM-838 (1) as SSRS, as well as the configuration of the three new stereoisomers (2–4) (Schemes 2 and SI).
Scheme 3.
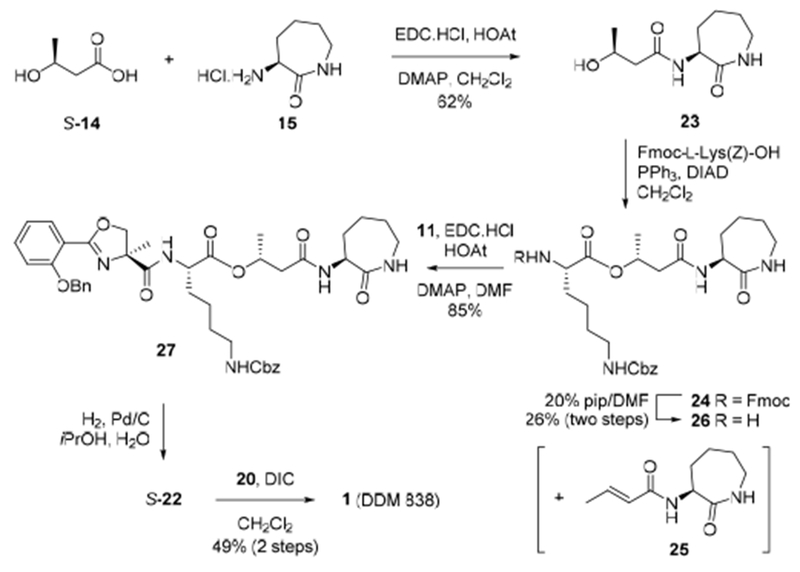
Mitsunobu route to DDM-838 (1).
Immunological investigation of CD1a-restricted T cell clones
We explored the ability of our synthetic compounds to mediate binding interactions between CD1a and TCRs using a series of immunological assays that measure either the biological potency of compounds in cellular assays (through the expression of activation marker CD69) or the mechanistic basis of TCR binding to CD1a-lipopeptide complexes (through tetramer staining and flow cytometry). We studied several CD1a-restricted TCR+ clones including autoreactive clones that recognize CD1a alone, and those recognizing CD1a presenting foreign DDM and related antigens. Initially, we compared the biological activity of synthetic DDM-838 (1) prepared here, with natural DDM-838 isolated from M. tuberculosis, and material synthesized through the solid phase route of Young and coworkers.[3b] Titration of lipids into cultures of the native CD8-2 T cell clone with all three materials and quantification of IFNγ release by ELISA provided dose-response curves for the two synthetic compounds that were essentially identical (Figure 2). The slightly lower potency of natural DDM can be explained by the presence of small amounts of naturally occurring variants in acyl chain length and saturation, which are known to be less potent than DDM-838.[3] This result established the chemical identity of the synthetic compounds, and concordant activity with natural DDM-838.
Figure 2.
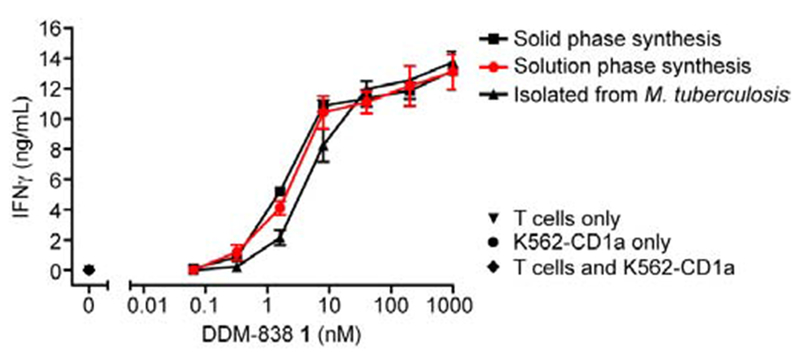
Synthetic and natural DDM-838 possess equivalent ability to activate the native CD8-2 T cell clone. Compound 1 prepared herein (solution phase); according to Young and co-workers (solid phase);[3b] and isolated from M. tuberculosis H37Ra, were incubated with the human T cell clone CD8-2 and CD1a-expressing K562 myeloid cells[15] over 24 h and IFNγ release measured by ELISA. DDM-838 isolated from M. tuberculosis contained 25% of the saturated analogue DDM-840,[3a] which is less stimulatory than DDM-838. Error bars indicate standard deviation among triplicates. Data representative of three independent experiments. Controls at 0 nM DDM-838 are for experiments including T cells alone, K562 cells alone, and both cell lines.
The BK6 clone[16] recognizes cellular CD1a when loaded with endogenous lipids and does not require DDM.[17] Jurkat cells expressing the BK6 TCR were stained brightly by CD1a loaded with endogenous lipids (endo, Figure 3) (these endogenous lipids are derived from the mammalian HEK293 GnT1− cells used for expression of CD1a monomers). By contrast, staining of Jurkat.BK6 cells was partially or completely inhibited by all four DDM analogs, suggesting that DDM lipids were loaded into CD1a and affected the TCR binding site. In contrast, Jurkat cells transfected with any of three TCRs derived from DDM-reactive T cell clones (CD8-2;[4] and Clone 8 and Clone 15, isolated from two different subjects with latent tuberculosis infection) stained more brightly with CD1a tetramers loaded with synthetic DDM-838, compared to CD1a-endo tetramers, indicating that synthetic lipopeptide enhances the interaction between CD1a and these TCRs. In comparing the heirarchy of CD8-2 staining, CD1a tetramers loaded with compound 2 (containing the E-C20:1 acyl lipid) stained less intensely than compound 1. Compound 3 (bearing the opposite stereochemistry at the central lysine residue compared to DDM-838), and compound 4 (with unnatural stereochemistry at lysine and the acyl lipid) showed minimal, if any staining of the DDM-838-reactive T cell clones. A negative control cell line expressing the MR1-restricted TCR, Jurkat.M33,[18] provided only background staining levels with all of the CD1a tetramers, indicating that the interactions observed with CD1a-DDM tetramers were due to specific TCR interactions. These data indicate a role of peptide and stereochemistry in T cell response, and the highly similar patterns seen among 3 distinct TCRs suggests a conserved mechanism of DDM recognition.
Figure 3.
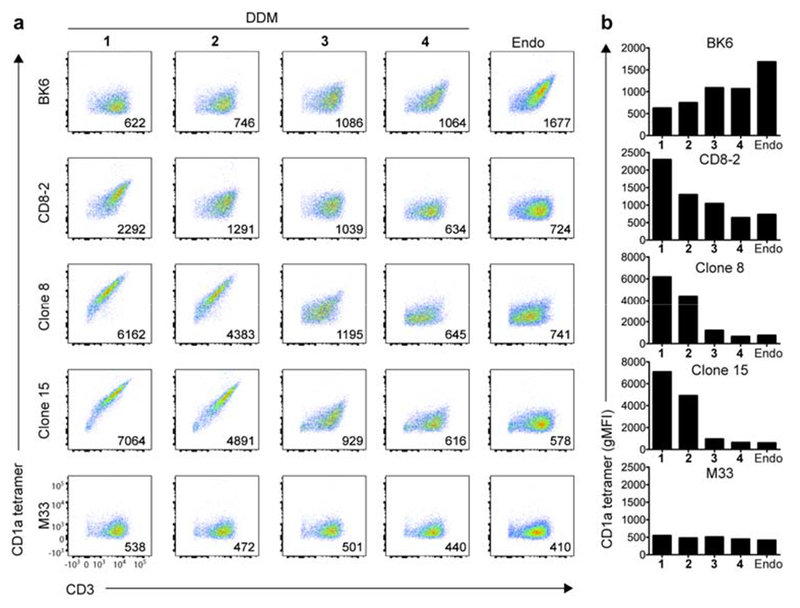
Reactivity of CD1a-restricted TCR+ Jurkat cell clones to DDM isomers, (a) Flow cytometry (left) of BK6, CD8-2, Clone-8, Clone-15 and M33 TCR expressing cells labeled with CD1a-endo tetramers or CD1a tetramers loaded with DDM-838 (cis-SSRS) 1, DDM trans-SSRS 2, DDM (cis-SRRS) 3 and DDM (trans-SRRS) 4, or endogenous lipid antigen (Endo). Numbers in the bottom right corner indicate geometric mean fluorescence intensity (gMFI) of staining with CD1a-DDM or CD1a-endo. (b) gMFI of each T cell line against endo and DDM isomers 1-4. Data are representative of three independent experiments.
To determine if synthetic DDMs could also activate cells, we next investigated if compounds 1–4 to could cause TCR-transduced Jurkat cells to express an activation marker, CD69(Figure 4). Titration of the DDM-reactive TCR+ CD8-2 cell line with compounds 1-4 resulted in a concentration-dependent increase in activation with the same pattern of potency as was observed in tetramer-based binding assays. CD1a-autoreactive BK6 cells showed no sign of activation by any of the compounds, and even showed a concentration-dependent reduction in activation. These data are consistent with lipopeptide loading into CD1a but reduced binding of CD1a-lipopeptide to the CD8-2 TCR for compounds 2-4. Titration of the DDM-reactive TCR+ clone 8 and clone 15 cell lines with 1-4 showed limited activation (data not shown).
Figure 4.
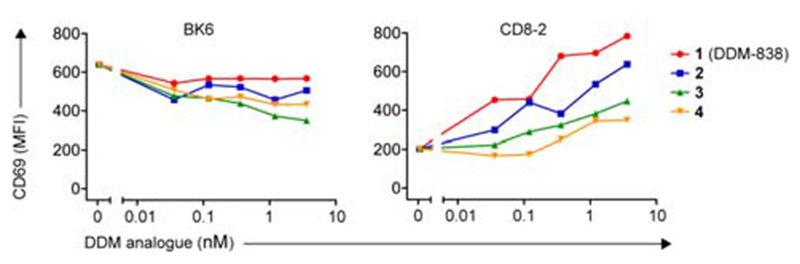
Activation of Jurkat cells expressing BK6 and CD8-2 TCRs by C1R cells transduced to express CD1a, measured by expression of the activation marker CD69. BK6 and CD8-2 expressing cells were co-cultured overnight with CD1a-expressing C1R cells in the presence of DDM isomers 1-4 at varying concentrations (0.036, 0.12, 0.36, 1.2, 3.6 nM). For BK6 and CD8-2 expressing cells with a high TCR to GFP ratio, the level of CD69 expression was measured as mean fluorescence intensity (MFI). Data is representative of three independent experiments.
Discussion
Although CD1a is broadly expressed on human Langerhans cells and myeloid dendritic cells, efforts to study foreign lipid-reactive CD1a-restricted T cells have been limited by the availability of the few known bacterial ligands. Although DDM-838 has been bound to CD1a and used in small-scale studies of tuberculosis patients,[6] this depsipeptidolipid has only been available in trace quantities from either natural sources or the reported synthetic route. The present work has investigated several synthetic approaches to DDM-838; demonstrated a new approach that markedly improves yield; and provides insight into a conserved mechanism of DDM-recognition by CD1a-restricted T cells. The new method utilizes the Mitsunobu reaction to construct the challenging ester linkage and provides access to milligram quantities of this important mycobacterial antigen. Notably, unlike direct esterification methods that caused epimerization at the central lysine, this approach generates the ester linkage of the depsipeptide in a substitution reaction without epimerization permitting an explicit assignment of the l-lysine stereochemistry of DDM-838.
The preparation of three diastereomers 2-4 of DDM-838 1 permitted investigation of structure-activity relationships for recognition by DDM-838 reactive CD1a-restricted TCRs, as well as the CD1a-autoreactive BK6 TCR. The BK6 TCR+ cell line is brightly stained by CD1a loaded with permissive self lipids[17] but less brightly stained with CD1a tetramers treated with compounds 1-4. Structural studies have revealed that the BK6 TCR binds to the A’ roof of CD1a, whereas non-permissive ligands such as sphingomyelin and sulfatide protrude from the A’ cleft and disrupt the BK6 TCR-CD1a contact zone.[17] An X-ray structure of a silylated mycobactin analogue revealed the alkyl chain inserted deep within the A’ pocket of the antigen-binding groove, with the peptide folded over in the F’ pocket and with both the caprolactam and salicyl moieties extending outward to the predicted plane of the TCR contact.[5] This binary structure, as well as molecular modelling studies of CD1a-DDM-838 complexes,[3] predict that the peptide moiety might block approach of the BK6 TCR to CD1a, based on the known docking mode of BK6 with CD1a.[17] This prediction is supported by our results, which also provides evidence that all four DDM analogs bind to CD1a. Furthermore, Jurkat cells expressing the CD8-2, Clone 8 and Clone 15 TCRs fail to bind to CD1a tetramers loaded with self lipids that are permissive to CD1a–BK6 TCR binding. This suggests an alternative TCR recognition mode compared to BK6 in which the peptide moiety likely protrudes from the groove to facilitate rather than block TCR binding.
Conclusions
Our data reveal a conserved spectrum of T cell recognition for the panel of DDM isomers for a set of three unrelated CD1a-DDM-reactive TCRs. The natural form, DDM-838 1 is the most potent, while compound 2, which varies in the lipid stereochemistry, maintained significant, albeit reduced, activity relative to 1. This result is reminiscent of the effect seen upon saturation of the acyl chain of DDM-838 to generate DDM-840,[3a] demonstrating the importance of the cis-Δ2-unsaturation of DDM-838 for its potent antigenicity. The stereochemistry and location of this unsaturation (at C2) is unusual and is generated by mycobactin synthase N (MbtN).[19] While the biochemical function of the unsaturation is unknown, this work confirms its importance for antigen recognition.
The inversion of stereochemistry at the lysine of the depsipeptide backbone compromises the activity of lipopeptide and this was reflected in the low reactivity of the DDM-reactive TCRs to CD1a-compound 3 tetramers, similar to prior studies of DDM-838 varying at either the oxazoline or butyrate moieties (positions a and c, Figure 1).[3b] The strong attenuation of TCR binding and T cell activation by compound 4, which is both epimerized at the central lysine and contains the trans acyl chain, reinforces these structure-activity relationships. Collectively, these results reveal that the stereochemistry of the depsipeptide backbone is important for DDM-recognition by CD1a-restricted TCRs, and that variation in stereochemistry or saturation in the lipid chain leads to partial attenuation of reactivity. The recognition of DDM-838 1 by three distinct T cell clones supports the biomedical development of these compounds to target the naturally occurring polyclonal T cells present in latent TB patients.[6] The ability to synthesize larger quantities of DDM-838 by the solution-phase scheme described here makes possible structural and functional studies of TCR recognition of CD1a–DDM-838 complexes, as well as in vivo studies that use DDM as a vaccine component or recall antigens for immunodiagnosis of TB.
Experimental Section
Expression and purification of CD1a
CD1a was expressed by recombinant methods in a mammalian expression system by cloning of constructs encoding CD1a and β2-microglobulin in a pHLsec vector[20] as carboxyterminal fusions with the leucine zippers Jun (CD1a) and Fos (β2-microglobulin) and a ‘post-zipper’ BirA-HiS6 tag for CD1a. Fusion proteins were expressed by cotransfection of HEK293 GnT1− cells with the plasmids pHLsec-CD1a-Jun-BirA-HiS6 and pHLsec-β2m-Fos.[20] Purification of CD1a-β2-microglobulin heterodimers was achieved by immobilized nickel affinity followed by size-exclusion chromatography.
Solubilization of DDM isomers
DDM SSRS Z-C20:1 1, DDM SSRS E-C20:1 2, DDM SRRS Z-C20:1 3 and DDM SRRS E-C20:1 4 were solubilized in a solution of 0.5% tyloxapol (Sigma) in Tris-buffered saline, pH 8.0 at 1 mg/mL. To ensure each lipopeptide was completely solubilized, 12 rounds of sonication (1 h, water bath temperature 25–45°C), vortex (10 sec) and freezing at −20°C (20 mins) were performed until the mixture was a homogenous suspension.
Production of CD1a-restricted Jurkat 76 cell lines
Parental Jurkat 76 cells lack endogenous TCR α-chains and β-chains[22] and therefore TCR specificity is conferred by the transgene. CD1a-restricted BK6, CD8-2, Clone 8, Clone 15 and control MR1-restricted M33.64[23] Jurkat 76 cells were generated as described[24] using retroviral transduction whereby green fluorescent protein and an antibody to CD3ε (anti-CD3ε) (UCHT1; BD Biosciences) signified successful transduction. These have recently been tested and confirmed to be free of Mycoplasma.
Loading of CD1a, production of CD1a tetramers and staining of Jurkat 76 cells with CD1a tetramers
Soluble mammalian CD1a samples were enzymatically biotinylated with BirA biotin ligase as previously described.[21] Biotinylated CD1a was loaded overnight with each DDM isomer at a molar ratio of 1:6. CD1a tetramers were prepared by mixture of streptavidin-phycoerythrin (BD Biosciences) with DDM-loaded biotinylated CD1a at a molar ratio of 1:4. CD1a-DDM tetramers (35 μL of stock CD1a-DDM tetramers at 2.5 μg/mL) were incubated overnight with 3 × 104 CD1a-restricted BK6, CD8-2, Clone 8, Clone 15 and control MR1-restricted M33.64 Jurkat 76 cells, generated as described above. CD1a tetramer–positive cells were stained with antibody to CD3ε (anti-CD3ε) (UCHT1; BD Biosciences) and were analyzed with an LSR Fortessa (BD Biosciences). Data were processed with FlowJo software (TreeStar).
Assay of activation of the human BK6 and CD8-2 Jurkat 76 cell lines
CD1a-restricted BK6 and CD8-2 Jurkat 76 cells were generated as described above. CD1a-expressing C1R cells were generated as previously described.[17] 3.33 × 103 CD1a-expressing C1 R cells were co-cultured overnight with 2 × 104 Jurkat cells (BK6 or CD8-2) and DDM isomers 1-4 at varying concentrations (0.036, 0.12, 0.36, 1.2, 3.6 nM). The cells were collected by centrifugation, the supernatant was removed and the cells were then stained with anti-CD3e (BV421, UCHT1, BD Biosciences), anti-CD69 (PE, FN50, BD Biosciences), anti-CD19 (APC/Cy7, SJ25C1, BD Biosciences) and 7-AAD (Sigma Aldrich). Activation of CD3+, GFP high (top 50%) BK6 and CD8-2 Jurkat 76 cells were assessed by upregulation of CD69. Cells were analyzed with an LSR Fortessa (BD Biosciences) and data were processed with FlowJo software (TreeStar).
Activation of human T cell clone CD8-2 by three sources of DDM-838 (1)
CD1a-expressing K562 myeloid cells have been described previously and are routinely tested by flow cytometry to confirm surface CD1a expression.[17] DDM-838 from solid-phase synthesis was provided by Dr David Young using the reported method.[3b] Natural DDM was purified from M. tuberculosis H37Ra as described previously[3b] and characterized by electrospray ionization mass spectrometry to be a mixture of DDM-838 (75%) and DDM-840 (25%). The concentrations of DDM-838 in natural DDM and of solution-phase DDM-838 were determined by UV spectroscopy absorbance at 306 nm, normalized to absorbance of 80 μM solid-phase DDM-838. 5 × 104 CD8-2 T cells were incubated with 2.5 × 104 CD1a-expressing K562 cells and serial dilutions of DDM-838 in 150 μl T cell media for up to 24 hrs at 37 °C. Secreted IFNγ in the undiluted cell culture media was quantified by standard sandwich ELISA using human IFNγ antibody clones 2G2 (Thermo-Fisher), biotin-conjugated B133.5 (Thermo-Fisher) and streptavidin-horse radish peroxidase (BD Pharmingen). Absorbance was measured at 405 nm using a VersaMax plate reader (Molecular Devices) and SoftMaxPro software. IFNγ concentrations were extrapolated from a standard curve in GraphPad Prism 6.
Supplementary Material
Acknowledgements
This work was supported by the Australian Research Council (DP130102763, DP160100597, FT130100103, CE140100011 and LE110100106). DGP is supported by an NHMRC ECF fellowship (1054431); DPF and DIG are supported by NPIMRC Senior Principal Research Fellowships (1027369, 1020770); JR is supported by an NPIMRC Australia Fellowship (AF50); DBM is supported by the Bill and Melinda Gates Foundation Vaccine Accelerator Award and NIH (AI R01049313 and U19111224).
References
- [1].Godfrey DI, Uldrich AP, McCluskey J, Rossjohn J, Moody DB, Nat. Immunol 2015, 16, 1114. [DOI] [PubMed] [Google Scholar]
- [2].a) Meunier L, Vian L, Lagoueyte C, Lavabre-Bertrand T, Duperray C, Meynadier J, Cano JP, Cytometry 1996, 26, 260; [DOI] [PubMed] [Google Scholar]; b) Plunger RE, Sieling PA, Ochoa MT, Sugaya M, Burdick AE, Rea TH, Brennan PJ, Belisle JT, Blauvelt A, Porcelli SA, Modlin RL, J. Clin. Invest 2004, 113, 701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a) Moody DB, Young DC, Cheng TY, Rosat JP, Roura-Mir C, O’Connor PB, Zajonc DM, Walz A, Miller MJ, Levery SB, Wilson IA, Costello CE, Brenner MB, Science 2004, 303, 527; [DOI] [PubMed] [Google Scholar]; b) Young DC, Kasmar A, Moraski G, Cheng TY, Walz AJ, Hu J, Xu Y, Endres GW, Uzieblo A, Zajonc D, Costello CE, Miller MJ, Moody DB, J. Biol. Chem 2009, 284, 25087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Rosat JP, Grant EP, Beckman EM, Dascher CC, Sieling PA, Frederique D, Modlin RL, Porcelli SA, Furlong ST, Brenner MB, J. Immunol 1999, 162, 366. [PubMed] [Google Scholar]
- [5].Zajonc DM, Crispin MD, Bowden TA, Young DC, Cheng TY, Hu J, Costello CE, Rudd PM, Dwek RA, Miller MJ, Brenner MB, Moody DB, Wilson IA, Immunity 2005, 22, 209. [DOI] [PubMed] [Google Scholar]
- [6].Kasmar AG, Van Rhijn I, Magalhaes KG, Young DC, Cheng TY, Turner MT, Schiefner A, Kalathur RC, Wilson IA, Bhati M, Gras S, Birkinshaw RW, Tan LL, Rossjohn J, Shires J, Jakobsen S, Altman JD, Moody DB, J. Immunol 2013, 191, 4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].El-Faham A, Funosas RS, Prohens R, Albericio F, Chem. Eur. J 2009, 15, 9404. [DOI] [PubMed] [Google Scholar]
- [8].a) Pouliot M-F, Angers L, Hamel J-D, Paquin J-F, Tetrahedron Lett 2012, 53, 4121; [Google Scholar]; b) Pouliot M-F, Angers L, Hamel J-D, Paquin J-F, Org. Biomol. Chem 2012, 10, 988. [DOI] [PubMed] [Google Scholar]
- [9].Ando K, J. Org. Chem 1997,62, 1934. [DOI] [PubMed] [Google Scholar]
- [10].Ando K, Oishi T, Hirama M, Ohno H, Ibuka T, J. Org. Chem 2000, 65, 4745. [DOI] [PubMed] [Google Scholar]
- [11].Madigan CA, Cheng TY, Layre E, Young DC, McConnell MJ, Debono CA, Murry JP, Wei JR, Barry CE 3rd, Rodriguez GM, Matsunaga I, Rubin EJ, Moody DB, Proc. Natl. Acad. Sci. USA 2012, 109, 1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hocking MB, Can. J. Chem 1968, 46, 466. [Google Scholar]
- [13].a) Maurer PJ, Miller MJ, J.Am. Chem. Soc 1983,105, 240; [Google Scholar]; b) Hu J, Miller MJ, J. Am. Chem. Soc 1997, 119, 3462. [Google Scholar]
- [14].Tsakos M, Schaffert ES, Clement LL, Villadsen NL, Poulsen TB, Nat. Prod. Rep 2015, 32, 605. [DOI] [PubMed] [Google Scholar]
- [15].de Jong A, Pena-Cruz V, Cheng TY, Clark RA, Van Rhijn I, Moody DB, Nat. Immunol 2010, 11, 1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Porcelli S, Brenner MB, Greenstein JL, Balk SP, Terhorst C, Bleicher PA, Nature 1989, 341, 447. [DOI] [PubMed] [Google Scholar]
- [17].Birkinshaw RW, Pellicci DG, Cheng TY, Keller AN, Sandoval-Romero M, Gras S, de Jong A, Uldrich AP, Moody DB, Godfrey DI, Rossjohn J, Nat. Immunol 2015, 16, 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Reantragoon R, Kjer-Nielsen L, Patel O, Chen Z, Illing PT, Bhati M, Kostenko L, Bharadwaj M, Meehan B, Hansen TH, Godfrey DI, Rossjohn J, McCluskey J, J. Exp. Med 2012, 209, 761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Madigan CA, Martinot AJ, Wei JR, Madduri A, Cheng TY, Young DC, Layre E, Murry JP, Rubin EJ, Moody DB, PLoS Pathog 2015, 11, e1004792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Aricescu AR, Lu W, Jones EY, Acta Crystallogr. D Biol. Crystallogr 2006, 62, 1243. [DOI] [PubMed] [Google Scholar]
- [21].O’Callaghan CA, Byford MF, Wyer JR, Willcox BE, Jakobsen BK, McMichael AJ, Bell JI, Anal. Biochem 1999, 266, 9. [DOI] [PubMed] [Google Scholar]
- [22].a) Shima EA, Le Beau MM, McKeithan TW, Minowada J, Showe LC, Mak TW, Minden MD, Rowley JD, Diaz MO, Proc. Natl. Acad. Sci. U.S.A 1986, 83, 3439; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Heemskerk MHM, Hoogeboom M, de Paus RA, Kester MGD, van der Hoorn MAWG, Goulmy E, Willemze R, Falkenburg JHF, Blood 2003, 102, 3530. [DOI] [PubMed] [Google Scholar]
- [23].Reantragoon R, Corbett AJ, Sakala IG, Gherardin NA, Furness JB, Chen Z, Eckle SBG, Uldrich AP, Birkinshaw RW, Patel O, Kostenko L, Meehan B, Kedzierska K, Liu L, Fairlie DP, Hansen TH, Godfrey DI, Rossjohn J, McCluskey J, Kjer-Nielsen L, J. Exp. Med 2013, 210, 2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Uldrich AP, Le Nours J, Pellicci DG, Gherardin NA, McPherson KG, Lim RT, Patel O, Beddoe T, Gras S, Rossjohn J, Godfrey DI, Nat. Immunol 2013, 14, 1137. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.